

By analysing 1638 antemortem brain scans from 557 autopsied patients with Alzheimer’s disease neuropathological changes, Josephs et al. show that high TDP-43 stage is associated with accelerated brain atrophy early in the course of both Alzheimer’s disease and primary age-related tauopathy.
Keywords: TDP-43, Braak stage, longitudinal, rate of atrophy, PART
Abstract
Alzheimer’s disease is characterized by the presence of amyloid-β and tau deposition in the brain, hippocampal atrophy and increased rates of hippocampal atrophy over time. Another protein, TAR DNA binding protein 43 (TDP-43) has been identified in up to 75% of cases of Alzheimer’s disease. TDP-43, tau and amyloid-β have all been linked to hippocampal atrophy. TDP-43 and tau have also been linked to hippocampal atrophy in cases of primary age-related tauopathy, a pathological entity with features that strongly overlap with those of Alzheimer’s disease. At present, it is unclear whether and how TDP-43 and tau are associated with early or late hippocampal atrophy in Alzheimer’s disease and primary age-related tauopathy, whether either protein is also associated with faster rates of atrophy of other brain regions and whether there is evidence for protein-associated acceleration/deceleration of atrophy rates. We therefore aimed to model how these proteins, particularly TDP-43, influence non-linear trajectories of hippocampal and neocortical atrophy in Alzheimer’s disease and primary age-related tauopathy. In this longitudinal retrospective study, 557 autopsied cases with Alzheimer’s disease neuropathological changes with 1638 ante-mortem volumetric head MRI scans spanning 1.0–16.8 years of disease duration prior to death were analysed. TDP-43 and Braak neurofibrillary tangle pathological staging schemes were constructed, and hippocampal and neocortical (inferior temporal and middle frontal) brain volumes determined using longitudinal FreeSurfer. Bayesian bivariate-outcome hierarchical models were utilized to estimate associations between proteins and volume, early rate of atrophy and acceleration in atrophy rates across brain regions. High TDP-43 stage was associated with smaller cross-sectional brain volumes, faster rates of brain atrophy and acceleration of atrophy rates, more than a decade prior to death, with deceleration occurring closer to death. Stronger associations were observed with hippocampus compared to temporal and frontal neocortex. Conversely, low TDP-43 stage was associated with slower early rates but later acceleration. This later acceleration was associated with high Braak neurofibrillary tangle stage. Somewhat similar, but less striking, findings were observed between TDP-43 and neocortical rates. Braak stage appeared to have stronger associations with neocortex compared to TDP-43. The association between TDP-43 and brain atrophy occurred slightly later in time (∼3 years) in cases of primary age-related tauopathy compared to Alzheimer’s disease. The results suggest that TDP-43 and tau have different contributions to acceleration and deceleration of brain atrophy rates over time in both Alzheimer’s disease and primary age-related tauopathy.
Introduction
Alzheimer’s disease is a neurodegenerative disorder characterized by the accumulation of two abnormal proteins, amyloid-β and tau (3 + 4 repeat paired helical filament tau), that are detected in post-mortem brain tissue. Hippocampal atrophy is one of the characteristic features of typical Alzheimer’s disease, and cross-sectional MRI histological studies of Alzheimer’s disease have found smaller hippocampal volumes to be associated with amyloid-β (Zarow et al., 2005) and tau deposition (Jack et al., 2002; Zarow et al., 2005). Over time, subjects with Alzheimer’s disease almost invariably show increased rates of hippocampal atrophy (Jack et al., 1998). Intriguingly, no relationship was identified between hippocampal neuronal cellular organelle activity and neurofibrillary tangle (NFT) burden (Salehi et al., 1995). Furthermore, two longitudinal MRI pathological studies have assessed whether rates of hippocampal atrophy are associated with amyloid-β and tau, and neither study found an association with either protein (Silbert et al., 2003; Erten-Lyons et al., 2013).
Transactive response DNA binding protein 43 (TDP-43) (Ou et al., 1995) accumulation occurs in almost 75% of brains of subjects with Alzheimer’s disease neuropathological changes (ADNC) (Arai et al., 2009; Josephs et al., 2015; McAleese et al., 2017). TDP-43 is also associated with clinical features including memory loss (Josephs et al., 2008, 2014b; James et al., 2016) and genetic risk factors for Alzheimer’s disease, such as the apolipoprotein epsilon 4 allele (APOE4) (Josephs et al., 2017a; Wennberg et al., 2018; Yang et al., 2018). In neuroimaging studies, TDP-43 in the presence of ADNC has also been associated with smaller hippocampal volume on MRI closest to death (Josephs et al., 2008, 2014b; Bejanin et al., 2019) and faster rates of hippocampal atrophy prior to death (Josephs et al., 2017a; Buciuc et al., 2020). TDP-43 has also been associated with smaller hippocampal volumes in primary age-related tauopathy (PART) (Josephs et al., 2019b); a pathological entity that strongly overlaps with Alzheimer’s disease (Crary et al., 2014; Duyckaerts et al., 2015).
There are many biological unknowns linking proteins, including TDP-43, to brain atrophy over time. These unknowns are important knowledge gaps to be investigated. First, it is unknown whether TDP-43 associated rate of atrophy in the hippocampus is linear (constant) over time, including over the disease course, whether there is evidence for acceleration or deceleration, or whether faster rates of hippocampal atrophy is a relatively early or later feature. Second, it is unknown whether TDP-43 is associated with rates of brain atrophy or acceleration of rates of atrophy, in neocortical regions specifically associated with these diseases and how the relationships between TDP-43 and the hippocampus compare to the relationships between TDP-43 and neocortical regions. To address these unknowns, we performed an MRI-histological-Bayesian analysis allowing for the potential of non-linearity of volume loss over time. We hypothesize that TDP-43 would be associated with acceleration of rates of atrophy of hippocampi and neocortex, and that the association of TDP-43 with hippocampal atrophy would be an early, rather than a late, feature of the Alzheimer’s disease neurodegenerative process, as previously hypothesized (Josephs et al., 2014b).
Materials and methods
Subjects
We identified all subjects who had been prospectively followed in the NIH-funded Mayo Clinic Alzheimer’s Disease Research Center or Mayo Clinic Alzheimer’s Disease Patient Registry/Study of Aging, had died with a brain autopsy between 1 January 1992 and 31 December 2015, had ADNC according to diagnostic criteria (Group, 1997; Montine et al., 2012), available paraffin blocks of brain tissue for TDP-43 analysis and at least one usable antemortem volumetric head MRI. Cases with a pathological diagnosis of a neurodegenerative disease other than ADNC, including frontotemporal lobar degeneration (FTLD), amyotrophic lateral sclerosis, or a primary tauopathy such as corticobasal degeneration or progressive supranuclear palsy were excluded (n = 114). For FTLD, specifically, we excluded all cases with degeneration of the frontal and/or temporal lobes, on gross examination or histologically on haematoxylin and eosin showing microvacuolation, neuronal loss and astrogliosis prominently in laminar II or transcortical, in the presence of low ADNC, as previously described (Josephs et al., 2019c). These FTLD exclusion criteria are based on published criteria for diagnosing FTLD (Cairns et al., 2007; Mackenzie et al., 2009). We identified 557 autopsied cases meeting these criteria with a total of 1638 usable MRI scans, spanning 1.0–16.8 years of disease duration prior to death. As part of the enrolment protocol, APOE genotyping (Corder et al., 1993; Schmechel et al., 1993) was performed. Demographic features of the 557 cases are shown in Table 1.
Table 1.
Demographics of subjects by TDP-43 stages
| T0 (n = 309) | T1 (n = 59) | T2 (n = 74) | T3 (n = 52) | T4 (n = 63) | Total (n = 557) | |
|---|---|---|---|---|---|---|
| Female sex | 138 (45%) | 29 (49%) | 48 (65%) | 34 (65%) | 35 (56%) | 284 (51%) |
| Education | 14 (12, 16) | 15 (12, 16) | 15 (12, 16) | 14 (12, 16) | 14 (12, 16) | 14 (12, 16) |
| APOE4 carrier | 121 (39%) | 36 (61%) | 44 (60%) | 29 (56%) | 32 (50%) | 261 (47%) |
| Age at first scan | 78 (71, 83) | 78 (70, 82) | 80 (76, 85) | 79 (74, 83) | 81 (77, 85) | 79 (72, 84) |
| Age at last scan | 81 (73, 87) | 80 (76, 85) | 83 (78, 87) | 82 (76, 86) | 84 (81, 88) | 82 (75, 87) |
| Age at death | 84 (76, 91) | 84 (78, 88) | 88 (83, 92) | 86 (81, 92) | 89 (85, 94) | 86 (79, 91) |
| No. scans/subject | 2 (1, 4) | 3 (2, 4) | 2 (1, 4) | 3 (2, 5) | 2 (1, 4) | 3 (1, 4) |
| First to last scan, years | 2 (0, 4) | 3 (1, 4) | 2 (0, 4) | 3 (1, 5) | 2 (0, 4) | 2 (0, 4) |
| First scan to death, years | 6 (4, 8) | 6 (4, 9) | 7 (5, 10) | 8 (6, 11) | 8 (4, 11) | 6 (4, 9) |
| Last scan to death, years | 3 (2, 5) | 3 (1, 4) | 4 (2, 7) | 4 (2, 7) | 5 (2, 7) | 4 (2, 6) |
| MMSE at first scana | 26 (23, 28) | 26 (23, 28) | 25 (21, 28) | 25 (20, 27) | 24 (22, 27) | 26 (22, 28) |
| MMSE at last scana | 24 (17, 28) | 23 (17, 26) | 21 (15, 26) | 20 (16, 24) | 21 (16, 25) | 23 (16, 27) |
| Braak stage | ||||||
| B1 | 71 (23%) | 11 (19%) | 3 (4%) | 2 (4%) | 7 (11%) | 94 (17%) |
| B2 | 95 (31%) | 10 (17%) | 9 (12%) | 10 (19%) | 14 (22%) | 138 (25%) |
| B3 | 143 (46%) | 38 (6%) | 62 (84%) | 40 (77%) | 42 (67%) | 325 (58%) |
| CERAD score | ||||||
| C0 | 61 (20%) | 11 (19%) | 3 (4%) | 4 (8%) | 10 (16%) | 89 (16%) |
| C1 | 52 (17%) | 4 (7%) | 9 (12%) | 4 (8%) | 5 (8%) | 74 (13%) |
| C2 | 68 (22%) | 15 (25%) | 17 (23%) | 9 (17%) | 18 (29%) | 127 (23%) |
| C3 | 128 (41%) | 29 (49%) | 45 (61%) | 35 (67%) | 30 (48%) | 267 (48%) |
Summaries are median (Q1, Q3) for continuous variables, n (%) for categorical variables. MMSE = Mini-Mental State Examination.
MMSE scores are shown at the median time of the first MRI scans which is 6 years from death and at the median time of the last MRI scan, which is 3 years from death. Hence, the interval between the first-to-last MMSE is ∼3 years.
This study was approved by the Mayo Clinic Institutional Review Board. All subjects or their proxies had provided written consent for brain autopsy examination, which was reaffirmed upon death.
Pathological analyses
All 557 cases had undergone standardized neuropathological examination including tissue sampling and semiquantitation of Alzheimer’s disease pathology according to the recommendations of the National Institute on Aging and Alzheimer’s Association (NIA-AA) criteria (Hyman et al., 2012; Montine et al., 2012). Each case had been assigned a Braak NFT stage using anti-tau antibodies (clone AT8, 1:1000 dilution; Endogen) (Braak and Braak, 1991). For this study we followed the recommendations of the NIA-AA for the staging of NFT deposition (Hyman et al., 2012; Montine et al., 2012). Hence, all cases were classified as NFT stages B1, B2 or B3 based on the following: B1 (transentorhinal stage) = Braak stages I + II; B2 (limbic stage) = Braak stages III + IV; and B3 (isocortical stage) = Braak stages V + VI. We had only a few cases of B0 and, hence, they were not included in the study. Each case was also assigned a score for neuritic plaque burden in the neocortex using thioflavin-S microscopy, modified Bielschowsky silver impregnation and antibodies to amyloid-β (clone 6F/3D, 1:10 dilution; Novocastra Vector Labs). Here again we followed the recommendations of the NIA-AA for the Consortium to Establish a Registry for Alzheimer’s disease (CERAD) (Mirra et al., 1991) scoring of neuritic plaques in neocortex: C0 = no neuritic plaques; C1 = sparse neuritic plaques; C2 = moderate neuritic plaques; and C3 = frequent neuritic plaques (Hyman et al., 2012; Montine et al., 2012). These schemes were specifically selected given the design of our study with focus on hippocampal, temporal and frontal neocortical atrophy. To adjust for vascular disease severity, we created a five-point vascular composite score (0–4) based on published guidelines (Skrobot et al., 2016) and accounting for the presence or severity of four vascular lesion types: arteriolosclerosis, cerebral amyloid angiopathy (CAA), cortical microinfarcts and lacunar or large infarcts (Deramecourt et al., 2012). A score of 0 = no vascular lesions present; 1 = mild arteriolosclerosis or CAA only; 2 = moderate-severe arteriolosclerosis or CAA only; 3 = presence of cortical microinfarcts without lacunar or large infarcts; and 4 = presence of lacunar or large infarcts.
Diagnostic subclassification
All 557 cases with ADNC were subclassified (grouped) by utilizing the NIA-Reagan criteria (Hyman and Trojanowski, 1997) and Braak stage and CERAD score recommendations from the NIA-AA criteria (Hyman et al., 2012; Montine et al., 2012). We also incorporated the autopsy criteria for PART (Crary et al., 2014; Jellinger et al., 2015) to account for cases without amyloid-β neuritic plaques (CERAD = 0). This allowed us to cover the entire spectrum of ADNC cases. As a result, all cases were subclassified into high probability Alzheimer’s disease (high-ADNC), low-intermediate probability Alzheimer’s disease (low-ADNC) and PART: high-ADNC = cases with C3/B3 combinations (n = 244; MRI scans = 800); low-ADNC = C1/C2 with all B-stages; C3/B1B2 combinations (n = 224; MRI scans = 633); and PART = C0/B1–B3 combinations (n = 89; MRI scans = 205).
Assessment of TDP-43
TDP-43 appears to be first deposited in the amygdala (stage 1), followed by the subicular region of the hippocampus and entorhinal cortex (stage 2), then the dentate gyrus of the hippocampus and occipitotemporal cortex (stage 3), then ventral striatum, basal forebrain, insular cortex and inferior temporal cortex (stage 4), then brainstem regions (stage 5) and ultimately middle frontal cortex and basal ganglia (stage 6) (Hu et al., 2008; Josephs et al., 2014a, 2016). This staging scheme has been independently validated (Tan et al., 2015) and supported by consensus recommendation for research purposes (Nelson et al., 2019,).
For this study, all 557 cases were assessed for the presence of TDP-43 as previously described (Josephs et al., 2014b). Briefly, amygdala blocks were sectioned and immunostained for TDP-43 using a polyclonal antibody (MC2085, from Professor Leonard Petrucelli) (Zhang et al., 2009) that recognizes a peptide sequence in the 25-kDa C-terminal fragment on a Dako-Autostainer (Dako-Cytomaton) and 3,3′-diaminobenzidine as the chromogen. Sections were lightly counterstained with haematoxylin. Amygdala sections were screened (by D.W.D., M.E.M. and K.A.J.) to assess for the presence of any TDP-43 immunoreactive neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear inclusions, perivascular inclusions, fine neurites in hippocampus (Hatanpaa et al., 2008) or NFT-associated TDP-43 (TDP-43 type-β) (Josephs et al., 2019d) (Fig. 1). We screened the amygdala as it has been shown to be first affected in Alzheimer’s disease (Higashi et al., 2007; Hu et al., 2008; Arai et al., 2009; Josephs et al., 2014a, 2016). For all cases in which TDP-43 was observed in the amygdala, we subsequently assessed 14 additional brain sections for the presence of TDP-43 in the hippocampus (subiculum, CA1 and dentate fascia), entorhinal, occipitotemporal, inferior temporal, basal forebrain, insular, ventral striatum and middle frontal cortices, as well as basal ganglia and brainstem regions, as previously described (Josephs et al., 2014a, 2016). All cases were then classified to generate a TDP-43 stage to best capture associations with our regions of interest (hippocampus and temporal and frontal neocortices) as follows: T0 (those without any TDP-43 immunoreactivity = TDP-43 stage 0); T1 (TDP-43 restricted to the amygdala = TDP-43 stage 1); T2 (TDP-43 extending into hippocampus but not beyond = TDP-43 stages 2 + 3); T3 (TDP-43 extending into temporal cortex but not frontal lobe = TDP-43 stage 4 + 5); and T4 (TDP-43 extending into frontal cortex = TDP-43 stage 6). This categorization was based on a modification of the full TDP-43 staging scheme (Josephs et al., 2016) and is in line with recent recommendations to use this scheme given that these regions are commonly obtained at autopsy in aged individuals (Nelson et al., 2019,).
Figure 1.
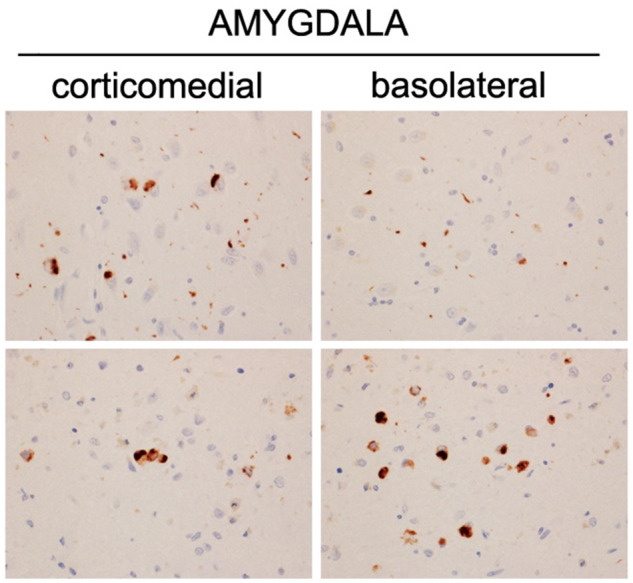
Histological assessment of TDP-43 immunoreactive inclusions in two cases with Alzheimer’s disease neuropathological changes (ADNC). Both cases were Braak NFT stage VI. TDP-43 inclusions included small, round and grain-like neuronal cytoplasmic inclusions, short dystrophic neurites, pre-inclusions, and NFT-associated inclusions. TDP-43 immunoreactivity magnification ×20.
MRI analyses
All MRIs were performed using a standardized protocol that included a T1-weighted 3D volumetric sequence (Jack et al., 2008a). All subjects were scanned with one of two GE scanners (DISCOVER MR750 or Signa HDxt) using the same standardized protocol. There were no significant differences between TDP-43 groups as to which GE model was used and all of our scanners undergo a standardized quality control calibration procedure daily, which monitors geometric fidelity over a 200 mm volume along all three cardinal axes, signal-to-noise, and transmit gain, and maintains the scanner within a tight calibration range. While some subjects had been scanned at 1.5 T and some at 3 T (Table 1), longitudinal analyses were always run using sets of serial scans performed at the same field strength. That is, subjects with 1.5 and 3.0 T scans were considered to have two independent series. All images underwent preprocessing that included corrections for gradient non-linearity and intensity inhomogeneity using both the N3 bias correction (Sled et al., 1998) followed by the SPM12-based bias correction.
Serial volumes were calculated using FreeSurfer version 5.3.0 (http://surfer.nmr.mgh.harvard.edu/) (Fischl et al., 2002). All scans were first run through the FreeSurfer cross-sectional pipeline, which includes intensity normalization and skull-stripping, automated Talairach transformation, segmentation, tessellation of the grey-white matter boundary, automated topology correction, surface deformation, and registration to a spherical atlas to match cortical geometry across subjects. Once these procedures were completed, an unbiased template was created using all time-points for each subject, and the longitudinal FreeSurfer pipeline was run using the unbiased template and 9 degrees of freedom registration to account for scaling fluctuations. For subjects with only one MRI, volumes were calculated from the cross-sectional FreeSurfer pipeline.
Statistical analyses
The broad goal of this analysis was to describe and compare volume, early rate of atrophy and a modification to the early rate of atrophy (acceleration/deceleration) across brain regions in an autopsy cohort with TDP-43, as well as ADNC data, and to describe the relationships between TDP-43 (TDP-43 stage), as well as tau (Braak NFT stage) and neuritic amyloid-β neocortical burden (CERAD plaque score) with volume and atrophy. To do this, we fit two bivariate outcome Bayesian models that jointly model the longitudinal change in two regions simultaneously, which allowed us to directly compare volume, rate of atrophy, and acceleration in rate of atrophy in the hippocampus to those in the inferior temporal and middle frontal regions. Bayesian methods are well suited to address complex questions such as these by using the idea of shrinkage and partial pooling of variances to obtain more stable and thus generalizable estimates while managing the collinearity when multiple observations (regions and scans) come from a single experimental unit (subject), as well as addressing the problem of multiple comparisons by creating comprehensive hierarchical models that can answer many questions simultaneously (Gelman and Hill, 2006; Gelman et al., 2014).
Each bivariate outcome model included the hippocampus and either the inferior temporal or middle frontal region. Both model formulations were analogous; the outcome in each model was volume expressed in terms of standard deviations of young, healthy volume predicted by T (TDP-43), B (tau) and C (neuritic plaques) stages or scores, including region-specific adjustments for vascular disease (vascular composite score), total intracranial volume (TIV), sex, age at death, and field strength. The models also included person-specific regional intercepts and person-specific slopes. The decision to use two bivariate-outcome models rather than a single trivariate-outcome model was based on computational tractability; these pairwise models used ∼1-week of computation time each due to the large number of subjects and scans available in this cohort.
Of particular interest in these models was how volume (intercept), early atrophy rate (slope), and modification of this early atrophy rate (acceleration) were associated with TDP-43 stage. As a secondary assessment we also looked at how these measures are associated with ADNC group. For interpretability, we assessed these relationships for three groups: high-ADNC, low-ADNC and PART.
To identify overarching patterns of interest, we included regional intercepts and region-specific terms for time to death using a restricted cubic spline with knots 7, 5, and 3 years from death (approximately quartiles in our data) (Harrell, 2015). This restricted cubic spline allowed for a smooth transition between what we term an ‘early’ rate, the linear rate of atrophy in our cohort 7 years or more before death, and a second rate add-on term, or acceleration/attenuation, that when added to the ‘early rate’ resulted in a ‘late rate’, i.e. a rate in the last 3 years of life. We then included region-specific intercept and rate terms interacted with continuous predictors of TDP-43 stage, Braak stage and CERAD score and two-way interactions between these staging variables. We also included terms for TDP-43 stage, Braak stage and CERAD score that modified each regional acceleration term. These terms can be thought of as overall patterns of change in volume, early atrophy, and late atrophy associated with unit changes in TDP-43 stage and ADNC group. To account for the imbalanced distributions across these stage variables in this cohort, we included pairwise interactions between each protein stage and allow these interactions to modify the intercept and early atrophy rate terms, which accounted for the fact that Braak stage and CERAD score may be dependent and simultaneous changes in Braak stage and CERAD score may not result in strictly additive effects. Thus, the basis of our model was formed by predicting standardized volume by main effects for intercept, rate, and acceleration terms, continuous protein stage modifications of these main effects, and interactions between proteins modifying the overall intercept and rate terms. The model also included regional volume adjustments for age at death, sex, total intracranial volume, field strength and vascular composite score, and age at death and vascular composite score were also allowed to modify the early atrophy rate term. The final terms in the model, person-and-region deviations of the overall intercept and person-specific rate terms, were included as random effects. See the online Supplementary material for additional statistical methods, including calculation of a standard outcome across regions, mathematical notation of the model formulation, and specification of prior distributions.
The models were fit using Markov Chain Monte Carlo simulation to estimate a posterior distribution for each parameter using R version 3.5.2 and rjags version 4–6 running JAGS version 4.3. This process included up to 100 iterations of initial adaptation, 10 000 discarded burn-in samples, and 10 000 traced samples thinned (again for computational efficiency) to every tenth value across 50 parallel chains. Results were based on 50 000 posterior draws for each parameter and the median and quantile intervals were used to summarize results. The Gelman Rubin diagnostic was ∼1 and visual assessment of the trace plots indicated convergence of all parameters in both pairwise models. Additionally, to assess agreement of the models, hippocampal estimates from both models were overlaid and qualitatively followed identical distributions, also indicating stable results.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Subject and MRI demographics
Demographic and clinical features of the 557 subjects stratified by TDP-43 stage (T-stage) are shown in Table 1. Of the 557 cases, 284 (51%) were female. Mini-Mental State Examination score at baseline MRI was median 26 (Q1, Q2 = 22, 28) and 23 (16, 27) at the time of last MRI prior to death. Two hundred and forty-eight cases (45%) were TDP-43 positive. The number of MRI scans available for analysis segregated by TDP-43 stage, Braak NFT stage and CERAD score are shown in Fig. 2. As seen, almost 50% of the MRI scans were performed on subjects in the B3C3 cell, which represents cases having the highest burden and distributions of both NFTs and neuritic plaques, respectively, and hence the highest probability of having Alzheimer’s disease (Group, 1997; Hyman et al., 2012; Montine et al., 2012). Some cells had only a few subjects with MRI scans (e.g. B1C3 and B3C0) due the fact that such combinations of neuritic plaque burden and NFT distribution are very rare and represent the <1% extremes of the proposed staging schemes for ADNC (Group, 1997; Hyman et al., 2012; Montine et al., 2012). The low frequencies in these cells are therefore not reflective of bias in our sample, but instead highlight the fact that such combinations are rare. The strong relationship between Braak stage and CERAD scores was one of the main drivers behind the classification scheme in the study (i.e. high-ADNC, low-ADNC and PART).
Figure 2.

Data distribution by TDP-43 stage within combinations of Braak stage and CERAD score. The label in each panel indicates number of scans [number of subjects] (per cent of all scans) for each combination of Braak NFT stage and CERAD score (across all TDP-43 stages) present in the data. This distribution prompted us to form three ADNC strata: high-ADNC, low-ADNC, and PART.
Trajectories of brain atrophy over the disease course
Figure 3 shows that 15 years from death the volumes of all three regions of the subjects with ADNC are at the lower end of values seen in young middle-aged healthy control subjects. The figure also demonstrates a natural ordering of how regions are affected based on the severity of the atrophy—the most affected region is the hippocampus, followed by the inferior temporal cortex and finally the middle frontal cortex. Our model allows for non-linear rates of atrophy. There was strong evidence for the inclusion of non-linear terms in each region in the model, with acceleration of atrophy observed in the middle frontal neocortex and deceleration of atrophy observed in the hippocampus. However, the total volume loss and rate of atrophy in the neocortex never ‘catches up’ to that of the hippocampus.
Figure 3.
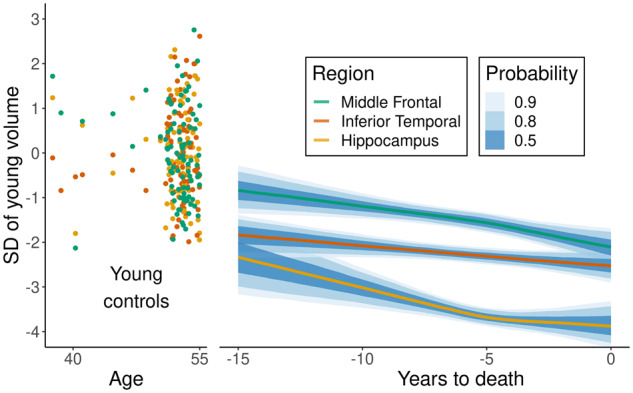
Z-scored volume of 87 young controls (35–55 years old) across three regions and mean volume on the z-scored scale as a function of years from death. The interval estimates for the mean volumes represent 50%, 80%, and 90% confidence intervals/Bayesian credible intervals. Fitted volumes represent a 65-year-old female with total intracranial volume of 1.4 l who died at age 80 and was scanned using 1.5 T field strength. SD = standard deviation.
Associations between TDP-43 and brain atrophy
Figure 4 shows the associations between TDP-43 stage and volumes (shifted intercept; −5 years from death), early atrophy rate (early rate; −15 to −7 years from death), and late atrophy rate (early rate plus acceleration; −3 years until death) within each of three ADNC diagnoses.
Figure 4.

The three main components of the model in each ADNC group by TDP-43 stage, across regions. Top: The estimated volume in z-score units across TDP-43 stages (T-stages); Middle: the early atrophy rates in z-score units per year; and Bottom: the late atrophy rates in z-score units per year (defined as the early atrophy rate term plus the acceleration/modification term from the model). In each row, the median posterior estimate is represented by a circle, with the thick and thin bars representing 80% and 90% intervals of the posterior distribution. Comparisons between adjacent T-stages within a region and ADNC group are represented as follows: a black asterisk overlaid on the median estimate indicates strong evidence (probability > 95%) of a difference between that estimate and the immediately preceding TDP-43 stage, while a white cross indicates moderate evidence (probability > 90%) of a difference. For example, in the middle panel and focusing on PART (third column), early atrophy in the inferior temporal (second row), the black asterisk in T1 indicates strong evidence the early atrophy rate in T1 was faster than in T0. The white cross in T4 indicates moderate evidence the atrophy rate in T4 was faster than in T3. Because no symbol is seen on the T3 estimate, we say there was little evidence that T3 early atrophy was faster than T2 early atrophy. SD = standard deviation.
For volume −5 years from death
Higher TDP-43 stage was strongly associated with smaller volumes across all three ADNC groups in both the hippocampus and the inferior temporal region. In the middle frontal region, there was moderate evidence of a similar association in the high-ADNC group, with less clear associations in the low-ADNC and PART groups.
For early atrophy rate (−15 to −7 years from death)
We found strong evidence that higher TDP-43 stage was associated with faster early rates of hippocampal atrophy across the three diagnostic groups. In the inferior temporal and middle frontal regions, there was little evidence of any association between TDP-43 stage and early rate of atrophy in high-ADNC, while there was some evidence (in lower TDP-43 stages) that early rates of atrophy increase with increases in TDP-43 stage in low-ADNC, with a more clear association across TDP-43 stages in PART.
For late atrophy rate (−3 years until death)
In the hippocampus, there was little evidence of an association between TDP-43 stage and late atrophy rates in all three ADNC groups. In the inferior temporal region, there was no evidence of an association between TDP-43 stage and late atrophy rates in high-ADNC, but some evidence for an association in low-ADNC and PART. In the middle frontal region, there was again little evidence of a TDP-43 stage association with late atrophy rates in high-ADNC, while there was moderate evidence of an association in the low-ADNC and PART groups.
Figure 5 shows how the volume shift, early atrophy rate, and late atrophy rate combine to create trajectories of regional volumes in the three ADNC groups for each TDP-43 stage, with volume differences represented as vertical differences across TDP-43 stage and rate and acceleration represented by angle of the slopes. Incorporating what we learned in Fig. 4, in the hippocampus we observe that higher TDP-43 stage is associated with lower volumes in high-ADNC and low-ADNC throughout the last 15 years of life, although in the 5 years before death, rates appear to slightly accelerate in low TDP-43 stage cases and decelerate in the high TDP-43 stage cases. In PART, the association is similar, although smaller sample sizes preclude drawing strong conclusions, and there is a lack of acceleration in those with low TDP-43 stage. In the inferior temporal region, a similar association is much less marked across the three pathological diagnoses. In the middle frontal region, in high-ADNC there was only a minor difference in volume anywhere over the last 15 years of life associated with differences in TDP-43 stage, while rate of atrophy accelerated slightly in the 5 years before death in all TDP-43 stages. In low-ADNC, early atrophy rates were affected by differences in TDP-43 stages, with greatest rates in cases with high TDP-43 stage, without large volume shifts or systemic changes in late atrophy rate (i.e. early atrophy rate is mostly constant for 15 years) associated with changes in TDP-43 stage. In PART, early atrophy rate was affected by TDP-43 stage changes, resulting in large volume differences 15 years before death, with high TDP-43 stage having both the largest volumes 15 years before death and the fastest atrophy rates. Proximal to death, all TDP-43 stages seem to converge in terms of total volume in PART.
Figure 5.

Estimated mean volume by ADNC group (columns) and region (rows), demonstrating how volume shift, early atrophy rate, and late atrophy rate are combined over the last 15 years of life. Volumes across TDP-43 stage (T-stage) are represented by colour gradient ranging from low TDP-43 stage (green) to high TDP-43 stage (orange). Dashed lines indicate a grouping of ADNC group and TDP-43 stage with <2% representation in our cohort, by scan count. Higher transparency also indicates groups with less representation in our cohort, with 10% or greater representation being opaque. SD = standard deviation.
Relationships with Braak and CERAD
Supplementary Figs 1 and 2 show more granularity of the data by revealing how TDP-43 stage estimates vary by different combinations of Braak stage and CERAD score (Supplementary Fig. 1) and how Braak stage estimates vary by different combinations of CERAD score and TDP-43 stage (Supplementary Fig. 2). This also allows an indirect comparison of TDP-43 stage versus Braak stage associations. There was little evidence that Braak stage modified the associations between TDP-43 stage and atrophy (Supplementary Fig. 1). However, an observation for the hippocampus is that an acceleration in rates in the 5 years before death in low TDP-43 stage was predominantly observed in cases with high CERAD score (C3), and the deceleration of rates in high TDP-43 stage was most striking in high Braak stage (B3) (Supplementary Fig. 1). Higher Braak stage was associated with smaller hippocampal and inferior temporal volumes across the 15 years before death (Supplementary Fig. 2). Braak stage was associated with rate of atrophy in the low TDP-43 stages. In fact, in the low TDP-43 stages the cases with higher Braak stage showed acceleration of rates in the 5 years before death. Braak stage did not show any association with middle frontal volumes or rates (Supplementary Fig. 2).
Discussion
In this study we describe associations between high pathologic TDP-43 stage and antemortem volume, faster rates of atrophy, and acceleration of atrophy in the hippocampus and neocortex (inferior temporal and middle frontal gyri) in subjects with ADNC. We identified strong associations between TDP-43 (T) stage and hippocampal atrophy in high and low-ADNC and PART. The evidence suggests that TDP-43 is associated with changes early in the atrophic process with strong relationships throughout the last 15 years of life. We also identified less strong associations between TDP-43 stage and neocortical atrophy. Similar to the hippocampus, higher TDP-43 stage was associated with smaller volumes of temporal and frontal lobes across all ADNC groups. However, TDP-43 appears to have a role in rates of neocortical atrophy more so in PART and those with low-ADNC compared to those with high-ADNC, where higher TDP-43 stages are associated with faster rates of atrophy.
Our analysis modelled the trajectories of volume loss of the hippocampus and neocortex across ADNC for at least 15 years prior to death. It is clear that 15 years prior to death, regional volumes in subjects with ADNC tend to be much smaller than volumes in young controls, suggesting that a significant amount of atrophy already has occurred. In other words, the data captured do not explain the entire end of life atrophy occurring in ADNC. It also appears that, regardless of the aetiologies of atrophy, the hippocampus is affected to a greater degree earlier in the disease course compared to the inferior temporal and middle frontal gyri, and continues to be most severely affected up to the time of death. There was some evidence for acceleration in rates in neocortical regions, although the acceleration was subtle when modelled over 15 years. Previous clinical studies have reported that rates of hippocampal atrophy accelerate during the early clinical phases of ADNC (Chan et al., 2003; Jack et al., 2008b; Leung et al., 2013). The findings from our study add to those previous studies by demonstrating that there is a subsequent slowing, or deceleration, of hippocampal atrophy rate close to death. It is possible that this slowing reflects burnout of the hippocampus, with little volume left to lose late in the disease. Unlike in the hippocampus, deceleration may have not been observed in the temporal and frontal lobes if atrophy in those regions is occurring relatively later in the disease compared to the hippocampus (Whitwell et al., 2007), as both regions would be expected to have relatively more volume left to lose.
The most striking link between TDP-43 and brain regions is in the hippocampus. We found higher TDP-43 stages to be clearly associated with smaller hippocampal volumes, as we and others have previously reported in high-ADNC (Josephs et al., 2008, 2014b, 2017a; Buciuc et al., 2020; Yu et al., 2020), as well as faster rates of hippocampal atrophy in high-ADNC (Josephs et al., 2017a; Buciuc et al., 2020). In addition, we have also reported similar cross-sectional associations in PART (Josephs et al., 2019b). While there are no studies to date that have reported on the influence of TDP-43 on volume, rates of atrophy and acceleration in low-ADNC, or rates of atrophy and acceleration in PART as distinct groups, we previously have assessed associations between TDP-43 and volume loss closest to death in PART (Josephs et al., 2017b, 2019b). In the original study on asymptomatic definite PART (no amyloid-β plaques present), we found no TDP-43-associated cross-sectional volume loss (Josephs et al., 2017b). However, in a subsequent study that included cases with asymptomatic and symptomatic definite and possible PART (scant non-neuritic plaques present) we found TDP-43-associated hippocampal volume loss (Josephs et al., 2019b). The findings from this study show that TDP-43 is also associated with faster rates of atrophy in PART. Hence, the association between TDP-43 and faster rates of hippocampal atrophy is not specific to cases with amyloid-β deposition, but is a feature across all forms of ADNC, whether one agrees that PART should or should not be a component of Alzheimer’s disease (Crary et al., 2014; Duyckaerts et al., 2015; Jellinger et al., 2015).
One of the novelties of this study is the determination of whether TDP-43 is associated with acceleration of atrophy in ADNC. An interesting trend, while not statistically significant, was that for a one-unit change in TDP-43 stage, in high-ADNC, there was slowing of hippocampal atrophy rates late in the disease in higher TDP-43 stages. On the other hand, in lower TDP-43 stages, atrophy appears to start more slowly and later accelerates proximal to death. Similar trends, although less striking, were observed with the inferior temporal lobe. These results point to an early influence of TDP-43 in hippocampal atrophy with TDP-43 contributing to severe loss of volume and likely burnout later in the disease course. The late acceleration of rates in cases with low TDP-43 stage would suggest that other factors are contributing to the acceleration in rates 5 years before death. The behaviours of the trajectories of hippocampal atrophy in PART were similar to those that we observed with high-ADNC and low-ADNC although it appears as though TDP-43-associated atrophy in PART may be occurring later (∼3 years later) in the disease course compared to when atrophy begins in those with high-ADNC and low-ADNC. The reason for a later association in PART is not clear but may be related to lower tau burden or to the absence of amyloid-β, which would support the argument that amyloid-β influences other processes in Alzheimer’s disease, early in the degenerative process (Gordon et al., 2018; McDade et al., 2018). In the inferior temporal lobe, no atrophy occurs in the 15 years before death in the absence of TDP-43. This is not unexpected given that PART cases have low Braak stages with absent to minimal tau effect on lateral temporal lobe. Volumes in PART were also generally larger than those observed in high and low-ADNC, particularly for the hippocampus, early over the 15-year period. It is possible that this difference in volume reflects lower NFT stage and/or tau burden in PART (Janocko et al., 2012), and/or reflects more focal patterns of regional degeneration in PART. Indeed, we have previously shown that Braak NFT stage is related to volume loss of the head of the hippocampus in PART (Josephs et al., 2017b).
Another novelty of this study is the assessment of relationships between TDP-43 and neocortical regional volumes, and comparing TDP-43 associations with hippocampus to TDP-43 associations with neocortex. In one previous cross-sectional study we found some evidence for an association between TDP-43 stage and lateral temporal and orbitofrontal volume loss (Bejanin et al., 2019). In the current study with a larger sample size, we confirm the lateral temporal association and show that TDP-43 stage is also related to smaller lateral (middle) frontal volumes. In addition, we observed TDP-43 stage to be associated with faster rates of atrophy in both neocortical regions for low-ADNC and PART cases. The lack of association in those with high-ADNC could reflect the fact that tau plays a bigger role in the frontal lobe in high-ADNC cases compared to the low-ADNC and PART cases where tau is not present in the frontal lobes. These findings would suggest that TDP-43 may have widespread effects, outside of the hippocampus and limbic regions (Bejanin et al., 2019; Josephs et al., 2019a). However, when comparing across regions (Fig. 6), the association between TDP-43 and volume and between TDP-43 and early rate of atrophy is clearly stronger in the hippocampus compared to the neocortical regions.
Figure 6.

Regions are placed next to each other to allow for better visualization and interpretation of differences in volumes, early atrophy rate and acceleration between the three regions. As in Fig. 4, a black asterisk overlaid on the point estimate indicates strong evidence (probability > 95%) of a difference between that estimate and the immediately preceding TDP-43 stage (T-stage), while a white cross indicates moderate evidence (probability > 90%) of a difference, similar to Fig. 4.
The temporal profile of the atrophy trajectories of hippocampus, inferior temporal lobe, and middle frontal lobe in subjects with PART was intriguing. In PART, it appears that volume loss of higher TDP-43 stages is comparable with volume loss of lower TDP-43 stages for hippocampus around 12 years prior to death, for inferior temporal around 10 years prior to death, and for middle frontal lobe around 4 years prior to death. This phenomenon, of ‘spread’ beginning with the hippocampus, followed by the inferior temporal lobe and then the middle frontal lobe, mirrors the TDP-43 staging scheme showing TDP-43 affecting hippocampus prior to inferior temporal and lastly affecting the frontal lobe (Josephs et al., 2014a, 2016; Tan et al., 2015). For the neocortex, TDP-43 stage seems to be associated with early atrophy rates and acceleration in PART (and perhaps in low-ADNC), whereas there is less evidence that TDP-43 stage plays a substantial role in the neocortex for high-ADNC.
While this study focuses on the influence of TDP-43 on rates of atrophy, we also assessed the influence of NFTs with the Braak stage surrogate. Consistent with previous studies (Jack et al., 2002; Whitwell et al., 2008; Apostolova et al., 2015; Josephs et al., 2017b; Quintas-Neves et al., 2019), we found that increasing Braak stage was associated with reduced volumes of the hippocampus and inferior temporal lobe, regardless of TDP-43 stage. We did not see strong evidence for an association between Braak stage and rate of hippocampal or rate of inferior temporal atrophy, except in those cases with low TDP-43 stage where higher Braak stage was associated with faster rates of atrophy and acceleration in hippocampal rates late in the disease. It, therefore, appears as though tau in NFTs—as summarized by Braak stage—may, at least in part, be the explanation for why cases with low TDP-43 stage show accelerating rates of atrophy ∼5 years before death. Although we did not directly compare the influence of TDP-43 stage and Braak stage, it seems that TDP-43 has an earlier relationship with the hippocampus compared to tau, with higher TDP-43 stage being associated with faster rates of atrophy 15 years before death, with rates slowing close to death, and higher Braak stage associated with acceleration in rates close to death. We did not detect an association between Braak stage and rate of hippocampal atrophy in the presence of high TDP-43 stage, except possibly in cases with low or absent amyloid. We also did not find much association between Braak stage and middle frontal atrophy. This may not be unexpected given that tau pathology is predominantly associated with atrophy of the temporal and parietal lobes in Alzheimer’s disease (Whitwell et al., 2008). Indeed, the middle frontal lobe showed relatively less atrophy than the hippocampus and inferior temporal lobe in our cohort.
The strengths of the study are the large number of autopsy-confirmed cases and large number of MRI studies available to analyse. Another is the wide window of MRI scans completed prior to death that allowed us to observe what may be happening up to 16 years prior to death. One of the limitations of this and any autopsy study is that not all research subjects agree to autopsy, which may limit generalizability of the results. In addition, we had a small number of subjects with specific combinations of Braak stage, CERAD score and TDP-43 stage. This was not a bias of the study, as noted previously, but highlights the fact that certain combinations of pathologies are rare. Lastly, we were unable to determine whether TDP-43 associations differ by TDP-43 type α/β given that type and stage are not independent (Josephs et al., 2019c) and we did not assess associations with specific TDP-43 inclusions (e.g. neuronal cytoplasmic inclusions, dystrophic neurites and intranuclear inclusions).
Funding
This study was funded by the US National Institutes of Heath (NIA) grants R01 AG037491 (PI: K.A.J.), P30 AG062677 (PI: R.C.P.), U01 AG006786 (PI: R.C.P.), R35 AG11378 (PI: C.R.J.) and R01 AG041851 (PI: C.R.J.). The funding bodies had no role in the study.
Competing interests
The authors report no competing interests. K.A.J., D.W.D., L.P., R.C.P., C.R.J., M.E.M. and J.L.W. receive research support from the National Institute of Health. L.P. has a U.S. patent #9,448,232 entitled ‘Methods and materials for detecting C9ORF72 hexanucleotide repeat expansion positive frontotemporal lobar degeneration or C9ORF72 hexanucleotide repeat expansion positive amyotrophic lateral sclerosis’. D.S.K. served on a Data Safety Monitoring Board for Lilly Pharmaceuticals; serves on a Data Safety Monitoring Board for Lundbeck Pharmaceuticals and for the DIAN study; served as a consultant to TauRx Pharmaceuticals ending in November 2012; was an investigator in clinical trials sponsored by Baxter and Elan Pharmaceuticals in the past 2 years; is currently an investigator in a clinical trial sponsored by TauRx; and receives research support from the NIH. R.C.P. serves as a consultant for Roche, Inc., Merck, Inc., Genentech, Inc., Biogen, Inc., Eli Lilly and Company and receives research support from the NIH. C.R.J. serves as a consultant for Janssen, Bristol-Meyer-Squibb, General Electric, and Johnson & Johnson; is involved in clinical trials sponsored by Allon and Baxter, Inc.; and receives research support from Pfizer, Inc., the NIA, and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
- ADNC =
Alzheimer’s disease neuropathological changes
- NFT =
neurofibrillary tangle
- PART =
primary age-related tauopathy
- TDP-43 =
transactive response DNA binding protein 43
References
- Apostolova LG, Zarow C, Biado K, Hurtz S, Boccardi M, Somme J, et al. Relationship between hippocampal atrophy and neuropathology markers: a 7T MRI validation study of the EADC-ADNI Harmonized Hippocampal Segmentation Protocol. Alzheimers Dement 2015; 11: 139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K, et al. Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol 2009; 117: 125–36. [DOI] [PubMed] [Google Scholar]
- Bejanin A, Murray ME, Martin P, Botha H, Tosakulwong N, Schwarz CG, et al. Antemortem volume loss mirrors TDP-43 staging in older adults with non-frontotemporal lobar degeneration. Brain 2019; 142: 3621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Buciuc M, Wennberg AM, Weigand SD, Murray ME, Senjem ML, Spychalla AJ, et al. Effect Modifiers of TDP-43-associated hippocampal atrophy rates in patients with Alzheimer's disease neuropathological changes. J Alzheimers Dis 2020; 73: 1511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007; 114: 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D, Janssen JC, Whitwell JL, Watt HC, Jenkins R, Frost C, et al. Change in rates of cerebral atrophy over time in early-onset Alzheimer's disease: longitudinal MRI study. Lancet 2003; 362: 1121–2. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993; 261: 921–3. [DOI] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128: 755–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deramecourt V, Slade JY, Oakley AE, Perry RH, Ince PG, Maurage CA, et al. Staging and natural history of cerebrovascular pathology in dementia. Neurology 2012; 78: 1043–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Braak H, Brion JP, Buee L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease. Acta Neuropathol 2015; 129: 749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erten-Lyons D, Dodge HH, Woltjer R, Silbert LC, Howieson DB, Kramer P, et al. Neuropathologic basis of age-associated brain atrophy. JAMA Neurol 2013; 70: 616–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002; 33: 341–55. [DOI] [PubMed] [Google Scholar]
- Gelman A, Carlin JB, Stern HS, Dunson DB, Vehtari A, Rubin DB.. Bayesian data analysis. 3rd edn. Boca Raton, FL: Chapman and Hall; 2014. [Google Scholar]
- Gelman A, Hill JL.. Data analysis using regression and multilevel/hierarchical models. Cambridge, UK: Cambridge University Press; 2006. [Google Scholar]
- Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol 2018; 17: 241–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group W. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997; 18 (4 Suppl): S1–2. [PubMed] [Google Scholar]
- Harrell F. Regression modelling stratergies. New York: Springer-Verlag New York; 2015. [Google Scholar]
- Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC, et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol 2008; 67: 271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res 2007; 1184: 284–94. [DOI] [PubMed] [Google Scholar]
- Hu WT, Josephs KA, Knopman DS, Boeve BF, Dickson DW, Petersen RC, et al. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol 2008; 116: 215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012; 8: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Trojanowski JQ.. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 1997; 56: 1095–7. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 2002; 58: 750–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008. a; 131: 665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Petersen RC, Xu Y, O'Brien PC, Smith GE, Ivnik RJ, et al. Rate of medial temporal lobe atrophy in typical aging and Alzheimer's disease. Neurology 1998; 51: 993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Weigand SD, Shiung MM, Przybelski SA, O'Brien PC, Gunter JL, et al. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology 2008. b; 70: 1740–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA.. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain 2016; 139: 2983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janocko NJ, Brodersen KA, Soto-Ortolaza AI, Ross OA, Liesinger AM, Duara R, et al. Neuropathologically defined subtypes of Alzheimer's disease differ significantly from neurofibrillary tangle-predominant dementia. Acta Neuropathol 2012; 124: 681–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 2015; 129: 757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L, et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer's disease: a longitudinal retrospective study. Lancet Neurol 2017. a; 16: 917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Mackenzie I, Frosch MP, Bigio EH, Neumann M, Arai T, et al. LATE to the PART-y. Brain 2019. a; 142: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Knopman DS, Petersen RC, et al. Brain atrophy in primary age-related tauopathy is linked to transactive response DNA-binding protein of 43 kDa. Alzheimers Dement 2019. b; 15: 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB, et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathol 2019. c; 137: 227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 2017. b; 133: 705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP-43 pathology in Alzheimer's disease. Acta Neuropathol 2014. a; 127: 441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol 2016; 131: 571–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008; 70: 1850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Liesinger AM, et al. TAR DNA-binding protein 43 and pathological subtype of Alzheimer's disease impact clinical features. Ann Neurol 2015; 78: 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol 2014. b; 127: 811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Zhang YJ, Baker M, Rademakers R, Petrucelli L, Dickson DW.. C-terminal and full length TDP-43 specie differ according to FTLD-TDP lesion type but not genetic mutation. Acta Neuropathol Commun 2019. d; 7: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KK, Bartlett JW, Barnes J, Manning EN, Ourselin S, Fox NC, et al. Cerebral atrophy in mild cognitive impairment and Alzheimer disease: rates and acceleration. Neurology 2013; 80: 648–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 2009; 117: 15–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J.. TDP-43 pathology in Alzheimer's disease, dementia with Lewy bodies and ageing. Brain Pathol 2017; 27: 472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade E, Wang G, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 2018; 91: e1295–e306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479–86. [DOI] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 2019; 142: 1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB.. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 1995; 69: 3584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Neves M, Teylan MA, Besser L, Soares-Fernandes J, Mock CN, Kukull WA, et al. Magnetic resonance imaging brain atrophy assessment in primary age-related tauopathy (PART). Acta Neuropathol Commun 2019; 7: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Ravid R, Gonatas NK, Swaab DF.. Decreased activity of hippocampal neurons in Alzheimer's disease is not related to the presence of neurofibrillary tangles. J Neuropathol Exp Neurol 1995; 54: 704–9. [DOI] [PubMed] [Google Scholar]
- Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 9649–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbert LC, Quinn JF, Moore MM, Corbridge E, Ball MJ, Murdoch G, et al. Changes in premorbid brain volume predict Alzheimer's disease pathology. Neurology 2003; 61: 487–92. [DOI] [PubMed] [Google Scholar]
- Skrobot OA, Attems J, Esiri M, Hortobagyi T, Ironside JW, Kalaria RN, et al. Vascular cognitive impairment neuropathology guidelines (VCING): the contribution of cerebrovascular pathology to cognitive impairment. Brain 2016; 139: 2957–69. [DOI] [PubMed] [Google Scholar]
- Sled JG, Zijdenbos AP, Evans AC.. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 1998; 17: 87–97. [DOI] [PubMed] [Google Scholar]
- Tan RH, Kril JJ, Fatima M, McGeachie A, McCann H, Shepherd C, et al. TDP-43 proteinopathies: pathological identification of brain regions differentiating clinical phenotypes. Brain 2015; 138: 3110–22. [DOI] [PubMed] [Google Scholar]
- Wennberg AM, Tosakulwong N, Lesnick TG, Murray ME, Whitwell JL, Liesinger AM, et al. Association of apolipoprotein E epsilon4 with transactive response DNA-binding protein 43. JAMA Neurol 2018; 75: 1347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology 2008; 71: 743–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Przybelski SA, Weigand SD, Knopman DS, Boeve BF, Petersen RC, et al. 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer's disease. Brain 2007; 130: 1777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HS, Yu L, White CC, Chibnik LB, Chhatwal JP, Sperling RA, et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community-based cohort study. Lancet Neurol 2018; 17: 773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Boyle PA, Dawe RJ, Bennett DA, Arfanakis K, Schneider JA.. Contribution of TDP and hippocampal sclerosis to hippocampal volume loss in older-old persons. Neurology 2020; 94: e142–e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C, Vinters HV, Ellis WG, Weiner MW, Mungas D, White L, et al. Correlates of hippocampal neuron number in Alzheimer's disease and ischemic vascular dementia. Ann Neurol 2005; 57: 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci USA 2009; 106: 7607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.