

Soleimani-Meigooni et al. compare antemortem 18F-flortaucipir (FTP) tau PET and autopsy findings in 20 patients with neurodegenerative diseases. FTP-PET reliably detects advanced Alzheimer’s tau pathology, but cannot reliably detect tau in non-Alzheimer’s tauopathies or differentiate these from tau-negative conditions.
Keywords: tau, neurofibrillary tangle, Alzheimer’s disease, positron emission tomography, neuropathology
Abstract
Few studies have evaluated the relationship between in vivo18F-flortaucipir PET and post-mortem pathology. We sought to compare antemortem 18F-flortaucipir PET to neuropathology in a consecutive series of patients with a broad spectrum of neurodegenerative conditions. Twenty patients were included [mean age at PET 61 years (range 34–76); eight female; median PET-to-autopsy interval of 30 months (range 4–59 months)]. Eight patients had primary Alzheimer’s disease pathology, nine had non-Alzheimer tauopathies (progressive supranuclear palsy, corticobasal degeneration, argyrophilic grain disease, and frontotemporal lobar degeneration with MAPT mutations), and three had non-tau frontotemporal lobar degeneration. Using an inferior cerebellar grey matter reference, 80–100-min 18F-flortaucipir PET standardized uptake value ratio (SUVR) images were created. Mean SUVRs were calculated for progressive supranuclear palsy, corticobasal degeneration, and neurofibrillary tangle Braak stage regions of interest, and these values were compared to SUVRs derived from young, non-autopsy, cognitively normal controls used as a standard for tau negativity. W-score maps were generated to highlight areas of increased tracer retention compared to cognitively normal controls, adjusting for age as a covariate. Autopsies were performed blinded to PET results. There was excellent correspondence between areas of 18F-flortaucipir retention, on both SUVR images and W-score maps, and neurofibrillary tangle distribution in patients with primary Alzheimer’s disease neuropathology. Patients with non-Alzheimer tauopathies and non-tau frontotemporal lobar degeneration showed a range of tracer retention that was less than Alzheimer’s disease, though higher than age-matched, cognitively normal controls. Overall, binding across both tau-positive and tau-negative non-Alzheimer disorders did not reliably correspond with post-mortem tau pathology. 18F-flortaucipir SUVRs in subcortical regions were higher in autopsy-confirmed progressive supranuclear palsy and corticobasal degeneration than in controls, but were similar to values measured in Alzheimer’s disease and tau-negative neurodegenerative pathologies. Quantification of 18F-flortaucipir SUVR images at Braak stage regions of interest reliably detected advanced Alzheimer’s (Braak VI) pathology. However, patients with earlier Braak stages (Braak I–IV) did not show elevated tracer uptake in these regions compared to young, tau-negative controls. In summary, PET-to-autopsy comparisons confirm that 18F-flortaucipir PET is a reliable biomarker of advanced Braak tau pathology in Alzheimer’s disease. The tracer cannot reliably differentiate non-Alzheimer tauopathies and may not detect early Braak stages of neurofibrillary tangle pathology.
Introduction
Aggregation of hyper-phosphorylated tau characterizes various neurodegenerative diseases, collectively known as tauopathies (Lee et al., 2001; Gibbons et al., 2019). Alzheimer’s disease is the most common tauopathy, and neurofibrillary tangles (NFTs) consisting of paired helical filaments and straight filaments of hyperphosphorylated tau are one of the pathologic hallmarks of this disease (Fitzpatrick et al., 2017). Other tauopathies show disease-specific tau deposits, with distinct neuronal and glial vulnerabilities, tau ultrastructural conformations, and regional distributions in the brain. Tauopathies can be classified by the number of repeats of the tau microtubule-binding domain [i.e. three-repeat (3R), four-repeat (4R), or mixed (3R/4R)] and ultrastructural conformation (Fitzpatrick et al., 2017; Falcon et al., 2018, 2019; Goedert et al., 2018; Scheres et al., 2020, Zhang et al., 2020). Alzheimer’s disease inclusions contain 3R/4R tau isoforms in paired helical filaments. Inclusions in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease primarily contain 4R tau isoforms in straight filaments (Komori, 1999; Takahashi et al., 2002; Zhukareva et al., 2002; Arima, 2006). Pick’s disease inclusions consist of predominantly 3R tau isoforms in straight and twisted filaments (Murayama et al., 1990; Komori, 1999; King et al., 2001; Arima, 2006).
PET imaging tracers have been developed to enable in vivo imaging of pathological tau. One of the most widely used tau PET tracers, 18F-flortaucipir (FTP) (formerly 18F-AV1451 and 18F-T807), has been shown by autoradiography to bind selectively to paired helical filament tau versus amyloid-β, α-synuclein, and TAR DNA-binding protein 43 (TDP-43) deposits in post-mortem human brain tissue (Xia et al., 2013; Marquié et al., 2015; Lowe et al., 2016; Sander et al., 2016). FTP-PET accurately differentiates clinically diagnosed Alzheimer’s disease from healthy controls and other neurodegenerative diseases (Chien et al., 2013; Cho et al., 2016; Ossenkoppele et al., 2018). In non-Alzheimer tauopathies, in vitro autoradiography studies demonstrate absent-to-low binding in non-Alzheimer tauopathies consisting of straight tau filaments (Marquié et al., 2015, 2017a; Lowe et al., 2016; Sander et al., 2016; Ono et al., 2017). In vivo, FTP shows tracer uptake in regions expected to contain tau pathology, which is less intense than in Alzheimer’s disease and partly overlaps with areas of off-target (i.e. non-tau-related) binding (Cho et al., 2017a,b; Coakeley et al., 2017; Passamonti et al., 2017; Schonhaut et al., 2017; Smith et al., 2017a, b; Jones et al., 2018; Tsai et al., 2019). Furthermore, low-level binding is also seen in patients expected to harbour tau-negative, TDP-43 pathology based on clinical phenotype or presence of a disease-causing mutation (Bevan-Jones et al., 2018a, b; Makaretz et al., 2018; Smith et al., 2019a; Tsai et al., 2019). Further FTP-to-autopsy correlation is needed in Alzheimer’s disease, non-Alzheimer tauopathies, and non-tau frontotemporal lobar degeneration (FTLD).
In vivo FTP retention patterns match the topography of Braak NFT staging, suggesting that FTP could be used as a surrogate marker of tau burden and severity in Alzheimer’s disease (Schöll et al., 2016; Schwarz et al., 2016; Marquié et al., 2017b). Recently, Smith et al. (2019b) showed strong correlation between FTP retention and post-mortem tau pathology in a patient with Alzheimer’s disease caused by presenilin 1 (PSEN1) mutation. Lowe et al. (2019) showed that patients with primary pathological diagnoses of Alzheimer’s disease, who had Braak NFT stages IV–VI, exhibited elevated FTP retention in a temporal composite meta-region of interest, and found strong correlation between FTP binding and quantitative phosphorylated tau immunohistochemistry at autopsy. Additionally, a larger autopsy study, using a binary visual classification scheme to interpret antemortem FTP scans, found that positively read scans accurately identified patients with advanced tau (Braak NFT stages V–VI) and Alzheimer’s disease [high Alzheimer’s disease neuropathological change (ADNC)] pathology (Fleisher et al., 2020).
Our primary aims in this study were to: (i) compare FTP-PET to pathology in patients with a primary autopsy diagnosis of Alzheimer’s disease; (ii) compare FTP-PET to pathology in patients with a primary autopsy diagnosis of non-Alzheimer tauopathy or tau-negative FTLD; and (iii) evaluate whether FTP-PET detects early-stage (Braak I–IV) neurofibrillary changes of Alzheimer’s disease.
Materials and methods
Participants
Twenty consecutive participants enrolled in research at the University of California, San Francisco (UCSF) Memory and Aging Center underwent antemortem FTP-PET and brain autopsy (Table 1). One patient underwent FTP-PET as part of a clinical trial sponsored by Avid Radiopharmaceuticals (Flortaucipir PET Imaging in Subjects with Frontotemporal Dementia, ClinicalTrials.gov Identifier: NCT03040713), while all other patients were enrolled in UCSF-specific protocols. Three patients had FTP-PET and autopsy results described in previous clinical studies (Schonhaut et al., 2017; Tsai et al., 2019). All participants underwent antemortem history, neurological exam, neuropsychological testing, genetic testing, brain MRI, and FTP-PET. Clinical diagnoses were made by consensus application of standard research criteria (Albert et al., 2011; Gorno-Tempini et al., 2011; McKhann et al., 2011; Rascovsky et al., 2011; Armstrong et al., 2013; Höglinger et al., 2017; McKeith et al., 2017). Seventeen participants underwent antemortem 11C-Pittsburgh compound-B (PiB) PET and one participant underwent 18F-florbetapir PET for in vivo determination of brain amyloid-β status. Written informed consent was obtained from participants or their designated surrogate decision-makers. Institutional review boards at UCSF, University of California, Berkeley, and Lawrence Berkeley National Laboratory (LBNL) approved the study.
Table 1.
Patient characteristics
| Autopsy diagnosis category | ID | Autopsy diagnosis (primary, contributing) | Clinical diagnosis | Gender | Age at PET | Time: PET-to- autopsy, months | MMSE at PET | Amyloid PET | Amyloid PET quant. (centiloid SUVR) | Pathology: Braak stage; ABC score; ADNC level |
|---|---|---|---|---|---|---|---|---|---|---|
| Alzheimer’s disease | A1 | AD, LBD | AD (logopenic PPA) | Female | 53 | 36 | 16 | n/a | n/a | VI; A3, B3, C3; High |
| A2 | AD, LBD | AD/PD | Male | 71 | 8 | 22 | Positive | 109 | VI; A3, B3, C3; High | |
| A3 | AD, LBD | AD (logopenic PPA) | Male | 73 | 38 | 22 | Positive | 115 | VI; A3, B3, C3; High | |
| A4 | AD | AD | Male | 68 | 31 | 8 | Positive | 106 | VI; A3, B3, C3; High | |
| A5 | AD | AD (logopenic PPA) | Female | 60 | 59 | 27 | Positive | 89 | VI; A3, B3, C3; High | |
| A6 | AD | AD (PCA) | Female | 59 | 40 | 9 | Positive | 124 | VI; A3, B3, C3; High | |
| A7 | AD | AD (logopenic PPA) | Female | 63 | 25 | 28 | Positive | 172 | VI; A3, B3, C3; High | |
| A8 | AD | AD (PCA) | Male | 53 | 44 | 26 | Positive | 96 | VI; A3, B3, C3; High | |
| Non-Alzheimer tauopathies | 1 | PSP | MCI (mixed) | Male | 60 | 12 | 26 | Negative | 0 | II; A1, B1, C1; Low |
| 2 | PSP | Non-fluent PPA | Female | 74 | 29 | 27 | Negative | −6 | II; A1, B1, C0; Low | |
| 3 | PSP | PSP | Male | 76 | 50 | 24 | Negative | 18 | II; A1, B1, C0; Low | |
| 4 | PSP, AD | PSP | Male | 72 | 49 | 26 | Positive | 69 | III; A3, B2, C2; Intermediate | |
| 5 | CBD | PSP | Female | 63 | 9 | 16 | Negative | −1 | I; A0, B1, C0; Not ADNC | |
| 6 | CBD, HS (left), TDP-43 unclassifiable | bvFTD | Male | 46 | 43 | 22 | Negative | −4 | III; A0, B2, C0; Not ADNC | |
| 7 | FTLD-tau (MAPT P301L), AD | FTLD (possible TES) | Male | 67 | 36 | 5 | Positive | 69 | IV; A3, B2, C3; Intermediate | |
| 8 | FTLD-tau (MAPT S301I), HS (right) | bvFTD | Female | 37 | 26 | 4 | Negative | 12 | 0; A0, B0, C0; Not ADNC | |
| 9 | AGD, vascular | bvFTD | Male | 68 | 4 | 29 | Negative | −4 | III; A0, B2, C0; Not ADNC | |
| Non-tau FTLD | 10 |
TDP-43 type A (GRN NM_002087: c.708 + 6_708 + 9del) |
bvFTD | Male | 68 | 18 | 21 | Negative | −11 | I; A1, B1, C0; Low |
| 11 | TDP-43 type B (C9orf72) | bvFTD | Female | 48 | 20 | 26 | Negative | −6 | I; A1, B1, C0; Low | |
| 12 | FTLD-FUS with MND | bvFTD | Male | 34 | 12 | 22 | n/a | n/a | 0; A0, B0, C0; Not ADNC | |
| Summary statistics |
Male: 12 Female: 8 |
61 | 30 | 20 |
ABC = Amyloid Braak CERAD; AD = Alzheimer’s disease; AGD = argyrophilic grain disease; bvFTD = behavioural variant frontotemporal dementia; HS = hippocampal sclerosis; LBD = Lewy body disease; MCI = mild cognitive impairment; MMSE = Mini-Mental State Examination; MND = motor neuron disease; n/a = not applicable (these patients did not undergo antemortem amyloid PET); PCA = posterior cortical atrophy; PD = Parkinson’s disease; PPA = primary progressive aphasia; TES = traumatic encephalopathy syndrome.
Genetic assessment
All participants received targeted sequencing of the most common causative genes for Alzheimer’s disease and FTLD, including PSEN1 and PSEN2, amyloid precursor protein (APP), microtubule-associated protein tau (MAPT), chromosome 9 open reading frame 72 (C9orf72), transactive response DNA binding protein 43 kDa (TARDBP), and progranulin (GRN) (Kim et al., 2018).
MRI acquisition and preprocessing
T1-weighted magnetization prepared rapid gradient echo (MPRAGE) MRI sequences were acquired at UCSF, either on a 3 T Siemens Tim Trio (n = 15) or a 3 T Siemens Prisma Fit (n = 5) scanner. Detailed MRI acquisition parameters are described in the Supplementary material.
MRIs were segmented and parcellated using Freesurfer 5.3 (https://surfer.nmr.mgh.harvard). Statistical Parametric Mapping (SPM12; Wellcome Trust Center for Neuroimaging, London, UK, https://www.fil.ion.ucl.ac.uk/spm) was used to process PET data as described below.
PET acquisition and preprocessing
FTP-PET images were acquired on a Siemens Biograph PET/CT scanner at LBNL (n = 19) or a GE Discovery STE/VCT PET/CT scanner at the UCSF Department of Radiology and Biomedical Imaging at China Basin (n = 1). We analysed PET data acquired 80–100 min (four 5-min frames) after injection of ∼10 mCi of FTP. A low-dose CT scan was performed for attenuation correction, and data were reconstructed using an ordered subset expectation maximization algorithm with weighted attenuation and smoothed with a 4 mm Gaussian kernel with scatter correction.
PET frames were realigned, averaged, and coregistered onto their corresponding MRI. Standardized uptake value ratio (SUVR) images were created in native space using MRI-defined inferior cerebellum grey matter as a reference region (Maass et al., 2017).
Eighteen patients also underwent amyloid PET with 11C-PiB (n = 17) or 18F-florbetapir (n = 1). Acquisition, preprocessing, and analysis of FTP and amyloid PET images are described in detail in the Supplementary material.
Neuropathology
All participants underwent a standardized post-mortem assessment at the UCSF Neurodegenerative Disease Brain Bank (Schonhaut et al., 2017; Kim et al., 2018). Pathological assessment was performed blinded to FTP-PET. The fresh brains were fixed, and tissue blocks were obtained from neurodegenerative disease-related neuroanatomical regions of interest. Microvacuolation, astrogliosis, and neuronal loss were scored on haematoxylin and eosin stained sections. Immunohistochemistry was performed using antibodies against hyperphosphorylated tau (CP-13, S202, mouse, 1:250, courtesy of Dr P. Davies), amyloid-β (1–16, clone DE2, mouse, 1:500, Millipore), α-synuclein (LB509, mouse, 1:5000, courtesy of Drs J. Trojanowski and V. Lee), and TDP-43 (rabbit, 1:4000, Proteintech Group).
Neuropathological diagnosis followed currently accepted guidelines (Litvan et al., 1996; McKeith et al., 1996; Dickson et al., 2002; Ferrer et al., 2008; MacKenzie et al., 2010; Montine et al., 2012). Pathologies were designated as primary, contributing, or incidental, based on the neuropathologist’s interpretation of their contribution to the patient’s clinical syndrome. ADNC was rated according to the latest ‘ABC’ score criteria, which includes Braak staging for neurofibrillary changes (Braak and Braak, 1991; Montine et al., 2012). Typically, intermediate-to-high ADNC was deemed a primary or contributing pathology associated with some component of the clinical syndrome, whereas low ADNC was always considered incidental in this cohort.
Experimental design and statistical analyses
FTP-PET W-score maps
To obtain maps of abnormally elevated FTP-PET at the individual patient level (i.e. distinguish tracer binding beyond background level), we computed voxelwise FTP-PET W-score maps (Z-score maps adjusted for age) from SUVR images, which are also referred to as W-maps (O’Brien and Dyck, 1995; Jack et al., 1997; La Joie et al., 2012). See the Supplementary material for additional information on W-map analysis.
FTP-PET region of interest analyses
Using Freesurfer segmentation, based on the Desikan atlas (Desikan et al., 2006), the average cortical SUVR value, weighted by subregion volume, was extracted from each patient in a set of a priori regions of interest corresponding to Braak NFT stages (Maass et al., 2017). Entorhinal cortex (Braak I) was used instead of a Braak I/II region of interest, because exclusion of the hippocampus was necessary to reduce signal spill-in due to off-target FTP retention in the adjacent choroid plexus (Marquié et al., 2015, 2017c; Schöll et al., 2016; Lowe et al., 2016; Lemoine et al., 2018; Baker et al., 2019). The Braak III/IV and V/VI regions of interest have been previously described (Maass et al., 2017).
SUVRs were also extracted from each patient at seven regions of interest (precentral gyrus, postcentral gyrus, putamen, globus pallidus, subthalamic nucleus, substantia nigra, dentate nucleus) corresponding to neuroanatomical regions that often have elevated tau burden in PSP and CBD (Hauw et al., 1994; Dickson et al., 2002). The tools used to define these cortical and subcortical regions of interest are described in the Supplementary material.
As a standard for tau-negativity, mean and standard deviation (SD) of SUVR levels were calculated at each region of interest for 14 young (mean age 26, SD 5 years), non-autopsy, cognitively normal controls who underwent FTP-PET on the same scanner as the patients. Using these data, a mean + 2 SD SUVR threshold was created for each region of interest. Any FTP-PET SUVR above the threshold was considered potentially distinguishable from controls (i.e. higher than noise and off-target binding seen in tau-negative controls) at the region of interest. The FTP-PET entorhinal cortex and Braak stage region of interest data also underwent partial volume correction (PVC) based on the geometric transfer matrix approach tailored for FTP-PET (Rousset et al., 1998; Baker et al., 2017, 2019). The other regions of interest underwent PVC using a method combining geometric transfer matrix and geometric transfer matrix-derived region-based voxel-wise approaches to enhance accuracy given the inclusion of small subcortical regions of interest (Thomas et al., 2011). All analyses were repeated with PVC data.
Data availability
All data used in this study are available for review upon request.
Results
Participants and neuropathological diagnoses
Table 1 summarizes demographic, clinical, and pathological characteristics. The patients were predominantly male (n = 12), relatively young (mean age 61, SD 12 years), and most had dementia at the time of FTP-PET imaging. The time gap between FTP-PET and autopsy ranged from 4 to 59 months, with a median of 30 months. All patients with clinical syndromes that predict Alzheimer’s disease, including four patients with logopenic variant primary progressive aphasia and two with posterior cortical atrophy, had high ADNC (A3, B3, C3; Braak VI) pathology at autopsy. Patients who met clinical criteria for PSP had pathological diagnoses of PSP or CBD at autopsy. Patients who met clinical diagnostic criteria for behavioural variant frontotemporal dementia or non-fluent primary progressive aphasia, had either tau (PSP, CBD, argyrophilic grain disease, FTLD-tau due to MAPT mutation), TDP-43, or FUS pathology at autopsy.
Comparison of FTP retention to pathology in patients with a primary neuropathological diagnosis of Alzheimer’s disease
On visual inspection of SUVR images and W-maps, all patients with primary Alzheimer’s disease autopsy diagnosis had intense tracer retention in the parietal lobes, especially the precuneus, and posterior cingulate (Fig. 1 and Supplementary Fig. 1). Three of four patients with logopenic variant primary progressive aphasia had asymmetric, left greater than right hemisphere tracer retention (Supplementary Fig. 1). Both patients with posterior cortical atrophy had parieto-occipital tracer retention (Fig. 1 and Supplementary Fig. 1). The location of tracer retention mirrored the clinical and neuroanatomical variability of the different Alzheimer’s disease phenotypes.
Figure 1.

FTP-PET SUVR and W-score map images. (A) Example SUVR and W-map images are shown for patients with primary Alzheimer’s disease (AD) neuropathological diagnosis. One patient had a typical amnestic clinical phenotype and the other patient had an atypical posterior cortical atrophy (PCA) phenotype. Patients with primary Alzheimer’s disease pathology had marked FTP uptake, and high SUVR and W-map thresholds were chosen to highlight the full range of tracer retention in these patients. Images for all Alzheimer’s disease patients are shown in Supplementary Fig. 1. (B) Example SUVR and W-map images are shown for patients with primary autopsy diagnoses of non-Alzheimer tauopathies, FTLD-TDP, and FTLD-FUS. These patients had lower FTP uptake than patients with primary Alzheimer’s disease autopsy diagnosis, and lower SUVR and W-map thresholds needed to be used to highlight the areas of tracer retention in these patients. To allow for comparison with the Alzheimer’s disease cases, the W-map upper threshold (W > 3.10) is the same as the lower threshold used in patients with primary Alzheimer’s disease pathology. Images for all patients with non-Alzheimer’s autopsy diagnoses are shown in Supplementary Fig. 2.
All patients with a primary neuropathological diagnosis of Alzheimer’s disease had areas of tracer retention that corresponded to the distribution and severity of NFT pathology in both typical and atypical presentations of Alzheimer’s disease (Fig. 2, Supplementary Fig. 1 and Supplementary Table 1). For example, compared to typical amnestic Alzheimer’s disease, the patients with posterior cortical atrophy had more NFT pathology in the calcarine cortex and less NFT pathology in the hippocampus (Fig. 2).
Figure 2.

Comparison of FTP-PET SUVR images to tau immunohistochemistry in Alzheimer’s disease. SUVR images are shown for two patients with autopsy diagnoses of Alzheimer’s disease. Both patients were assigned antemortem clinical diagnoses of Alzheimer’s disease, but one patient had a typical amnestic phenotype and the other patient had an atypical posterior cortical atrophy (PCA) phenotype. CA1, calcarine cortex, and middle frontal gyrus are indicated on each patient’s SUVR images (arrows), and the corresponding tau (CP-13 antibody) immunohistochemistry is shown. Scale bars = 250 µm in the CA1 micrographs and 25 µm in the other micrographs.
Compared to patients with other neuropathological diagnoses at autopsy, Alzheimer’s disease patients had more intense tracer binding throughout the cortex, which necessitated the use of higher upper thresholds to visualize the full range of tracer binding (Fig. 1 and Supplementary Fig. 1). Altogether, the distribution and intensity of tracer uptake in patients with a primary Alzheimer’s disease autopsy diagnosis allowed for qualitative differentiation of these patients from those with other primary neurodegenerative pathologies at the single patient level on both SUVR images and W-maps (Fig. 1 and Supplementary Figs 1 and 2).
Comparison of FTP retention to pathology in patients with non-Alzheimer primary neuropathological diagnoses
Progressive supranuclear palsy
All four patients with a primary neuropathological diagnosis of PSP showed tracer uptake in basal ganglia (particularly the globus pallidus), frontal white matter, and midbrain. Three patients showed significant tracer uptake in the cerebellar dentate nucleus (Fig. 1 and Supplementary Fig. 2). Though many of these regions overlap with areas of off-target FTP binding, W-maps confirmed the intensity of binding was greater than expected for age based on normative data from amyloid-negative controls. The intensity of binding was lower than in Alzheimer’s disease.
At autopsy, all patients showed midbrain pathological changes typical of PSP, including tufted astrocytes, globose tangles, and oligodendroglial coiled bodies. The globus pallidus was examined in all four patients, and three patients had tufted astrocytes and two patients had coiled bodies in this region. The cerebellar dentate nucleus was examined in three patients, and all had globose tangles and glial coiled bodies (Fig. 3 and Supplementary Tables 10–13). Comparing two patients (Patients 1 and 4) who had tau pathology in the globus pallidus and cerebellar dentate, the patient with the higher burden of tau pathology in these regions (Patient 4) had higher tracer retention in these regions (Fig. 3). Both patients had low FTP retention in the motor cortex, and there was less tau pathology in this region compared to globus pallidus and cerebellar dentate. Patient 2 had extensive PSP tau pathology in the inferior frontal gyrus, primary motor cortex, and brainstem, with relative sparing of the globus pallidus and dentate nucleus (the areas with lower tau pathology lacked FTP retention; Supplementary Fig. 2). Affected regions showed significant astrogliosis. Tracer retention in the frontal white matter did not correspond to tau pathology on stained tissue sections.
Figure 3.

Comparison of FTP-PET SUVR images with tau immunohistochemistry in PSP. SUVR images are shown for two patients with autopsy diagnoses of PSP. The cerebellar dentate nucleus, globus pallidus, and motor cortex are indicated on each patient’s SUVR images (arrows), and the corresponding tau (CP-13 antibody) immunohistochemistry is shown. Superior frontal gyrus is also shown for one of the patients. Scale bars = 25 µm.
Temporal and limbic neuroanatomical structures contained moderate levels of PSP tau pathology and also NFTs. Patient 4 had contributing Alzheimer’s disease pathology, with intermediate ADNC and Braak stage III, and the other patients had incidental Braak stage II. Despite the presence of both PSP tau pathology and NFTs of Alzheimer-type, there was no discernible tracer retention in the entorhinal cortex, hippocampus, or amygdala on either SUVR images or W-maps. Most of the temporal tracer retention localized to the white matter (Supplementary Fig. 2).
Corticobasal degeneration
The two patients with CBD at autopsy had asymmetric tracer retention, more prominent in one hemisphere, mostly involving the perirolandic cortex, inferior frontal gyrus, frontal white matter, basal ganglia (especially the globus pallidus), thalamus, substantia nigra, and brainstem (Fig. 1 and Supplementary Fig. 2). On inspection of SUVR images and W-maps, these patients had lower tracer retention than patients with primary Alzheimer’s disease pathology, but they generally had more tracer retention than other sporadic non-Alzheimer tauopathies, FTLD-TDP, or FTLD-FUS.
The areas of highest tracer retention (perirolandic cortex, frontal white matter, basal ganglia, substantia nigra, and brainstem) corresponded to regions of high CBD tau pathology burden (astrocytic plaques, coiled bodies, and white matter threads; Supplementary Tables 14 and 15). Patient 5 had incidental Braak I NFT pathology, but there was no entorhinal cortex tracer retention on the SUVR image or W-map. This patient had elevated FTP-PET signal in the temporal white matter (Fig. 1 and Supplementary Fig. 2), which corresponded to underlying CBD tau pathology. Patient 6 had incidental Braak III NFT pathology and left hippocampal sclerosis due to TDP-43 pathology. This patient showed FTP tracer retention in left greater than right mesial temporal lobes and temporal white matter on the SUVR image, which was also apparent on the W-map (Supplementary Fig. 2). The mesial temporal lobes were atrophied and contained CBD tau pathology, NFTs, and TDP-43 pathology, making it difficult to attribute the elevated temporal FTP retention to a specific pathology.
Argyrophilic grain disease
The patient with primary argyrophilic grain disease neuropathology showed tracer retention limited to basal ganglia, substantia nigra, and right temporal lobe (right inferior temporal gyrus, hippocampus) on the SUVR image, which was not significant on the W-map compared to age-matched controls (Supplementary Fig. 2). Tau-positive pretangles, neuropil thread, and grains were found in the entorhinal cortex, CA1/subiculum, CA2, and amygdala, with sparse pre-tangles present in the inferior temporal gyrus (Supplementary Table 18). Incidental Braak III NFT pathology was also present. Very little tau pathology was observed in the basal ganglia, substantia nigra, or other neocortical regions assessed at autopsy.
FTLD-tau pathology due to MAPT mutations
Both patients with FTLD-tau pathology due to MAPT mutations exhibited increased tracer retention in the temporal lobes, temporal white matter, and basal ganglia (Fig. 1 and Supplementary Fig. 2). The patient carrying a P301L mutation was PiB-positive and showed elevated FTP-PET signal in right greater than left temporal and occipital cortex, as well as small clusters of elevated retention in posterior cingulate and lateral parietal cortex. At autopsy, the patient had intermediate ADNC (A3, B2, C3; Braak IV NFT pathology) and extensive FTLD-tau pathology (neuronal cytoplasmic inclusions, bushy astrocytes) throughout the hippocampus, amygdala, cortex, and basal ganglia (Supplementary Tables 1 and 16).
The patient carrying an S305I mutation showed marked tracer retention in the frontal and temporal white matter. At autopsy, FTLD-tau pathology was abundant in cortical and subcortical regions with features reminiscent of diffuse neocortical argyrophilic grain disease with granular tau-positive neuronal cytoplasmic inclusions, often with a perinuclear ring-like pattern, coiled oligodendroglial cytoplasmic inclusions, threads, and grains. There was extensive thread and grain pathology in the subcortical white matter (Fig. 4 and Supplementary Table 17). The frontal and temporal tracer retention was localized in areas in which there was considerable FTLD-tau pathology (Fig. 4). There was no Alzheimer-type NFT pathology (Braak 0). Hippocampal sclerosis was observed in the absence of TDP-43 inclusions.
Figure 4.

Comparison of FTP-PET SUVR images with tau immunohistochemistry in patient with FTLD-tau due to MAPT S305I mutation. The cerebellar dentate nucleus, entorhinal cortex, globus pallidus, inferior frontal gyrus, middle frontal gyrus (cortex and white matter), and motor cortex are indicated on the patient’s SUVR images (arrows), and the corresponding tau (CP-13 antibody) immunohistochemistry is shown. Scale bars = 10 µm.
FTLD TDP-43 type A due to GRN mutation
The patient with FTLD TDP-43 type A due to GRN (NM_002087: c.708 + 6_708 + 9del) mutation exhibited a moderate level of tracer retention throughout the frontal cortex (superior frontal gyrus, middle frontal gyrus, orbitofrontal cortex, and frontal pole), frontal white matter, basal ganglia (especially, globus pallidus and putamen), lateral temporal cortex, and temporal poles (Fig. 1 and Supplementary Fig. 2). At autopsy, tau NFTs were limited to the entorhinal cortex (Braak I). Notably, tau immunohistochemistry was negative in the sampled cortical regions, which had high FTP signal (Fig. 5, Supplementary Tables 1 and 19). Extensive TDP-43 immunoreactive neuronal cytoplasmic inclusions and dystrophic neurites were present throughout the frontal lobes, temporal lobes, and basal ganglia.
Figure 5.
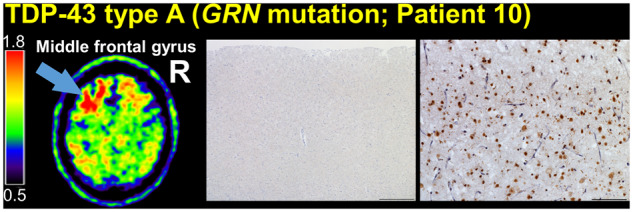
Comparison of FTP-PET SUVR images with tau and TDP-43 immunohistochemistry in a patient with FTLD TDP-43 type A due to GRN (NM_002087: c.708 + 6_708 + 9del) mutation. The left middle frontal gyrus is indicated on the patient’s SUVR image (arrow). Left middle frontal gyrus tau immunohistochemistry (CP-13 antibody, haematoxylin counterstain, left) was negative, whereas TDP-43 immunohistochemistry (pan-TDP-43 antibody, right) showed short, thin neurites, crescentic neuronal cytoplasmic inclusions, and other typical features of FTLD-TDP type A, consistent with the patient’s known mutation in GRN. Scale bars = 500 µm and 100 µm for the tau and TDP-43 immunohistochemistry micrographs, respectively.
FTLD TDP-43 type B due to C9orf72 expansion
The patient with FTLD TDP-43 type B due to a C9orf72 expansion exhibited small clusters of elevated tracer uptake in the frontal white matter, striatum, thalamus, substantia nigra, and right half of the pons (Fig. 1 and Supplementary Fig. 2). At autopsy, there was a small amount of tau pathology in the middle frontal gyrus, and extensive TDP-43 inclusions throughout the frontal lobes (Supplementary Table 20). The frontal tau pathology did not correspond to FTP-PET signal in the frontal lobes. The striatum, thalamus, and substantia nigra only contained TDP-43 inclusions. The right half of the pons did not undergo immunohistochemistry.
FTLD-FUS
The patient with FTLD-FUS showed minimal tracer retention in the frontal lobes and frontal white matter, which corresponded to areas of significant atrophy (Fig. 1 and Supplementary Fig. 2). Tracer retention was significantly higher than amyloid-negative healthy controls in the left caudate, as seen in the W-map, but this region was not sampled for pathological analysis. FUS pathology was present in the frontal lobes, and tau pathology was absent in this area. Braak stage was 0. Neuropil threads were present in the entorhinal cortex, dorsal raphe, and locus coeruleus but no tracer retention was evident in these areas (Supplementary Table 21).
Evaluating FTP-PET SUVR in precentral gyrus, postcentral gyrus, and subcortical regions of interest
All patients with PSP and CBD showed elevated FTP-PET SUVR levels in the putamen, globus pallidus, and subthalamic nucleus regions of interest that were above the mean + 2 SD SUVR threshold for these regions of interest in young, cognitively normal controls (Fig. 6). Patients with Alzheimer’s disease, other non-Alzheimer tauopathies, and FTLD-TDP pathology had similarly elevated tracer retention in these regions compared to controls. SUVR in precentral and postcentral gyrus were highest in patients with Alzheimer’s disease, and tracer retention in these regions were similar in patients with CBD, PSP, and controls. SUVR in the dentate nucleus region of cerebellum was similar across tauopathies and in controls. PVC was performed to account for the potential loss of sensitivity due to atrophy, which can influence FTP uptake measured in the regions of interest. PVC data yielded similar results (Supplementary Fig. 3).
Figure 6.

FTP-PET SUVR quantification at precentral gryus, postcentral gyrus, and subcortical regions of interest. SUVR quantification, not corrected for partial volume effects, was performed at precentral gyrus, postcentral gyrus, putamen, globus pallidus, subthalamic nucleus, substantia nigra, and dentate nucleus regions of interest. Each patient is represented by a single point and coded by time from PET-to-autopsy (shape) and primary neuropathological diagnosis (x-axis). The dotted line represents the threshold for significance, which is calculated from the mean SUVR plus two standard deviations for the young, cognitively normal (CN), non-autopsy controls. Points crossing the significance threshold are highlighted with a black outline. AD = Alzheimer’s disease; AGD = argyrophilic grain disease.
Evaluating the Braak neurofibrillary tangle stages detectable by FTP-PET quantification
All patients with primary Alzheimer’s disease autopsy diagnosis, who had Braak VI NFT pathology, showed markedly elevated FTP-PET SUVR levels in the Braak stage regions of interest compared to young, cognitively normal controls (Fig. 7). Patient 4, who had primary PSP pathology and contributing Alzheimer’s disease pathology [intermediate ADNC (A3, B2, C2), Braak III] showed elevated FTP-PET SUVR in the entorhinal cortex (Braak I) and Braak III/IV regions of interest. Patient 7, who had primary FTLD-tau pathology due to MAPT P301L mutation and contributing Alzheimer’s disease pathology [intermediate ADNC (A3, B2, C3), Braak IV], did not show elevated FTP-PET SUVR in the entorhinal cortex or Braak III/IV regions of interest. Patient 8, who had primary FTLD-tau pathology due to MAPT S305I showed elevated FTP-PET SUVR in the entorhinal cortex region of interest, but this patient did not have any NFT pathology. Patients with primary non-Alzheimer autopsy diagnoses and Braak I–III NFT incidental pathology (low ADNC or not ADNC) did not have FTP-PET SUVR levels that were distinguishable from young, cognitively normal controls (Fig. 7). Overall, this quantitative method detected advanced Alzheimer’s disease pathology (Braak VI) but did not reliably detect early (Braak I–IV) NFT pathology.
Figure 7.
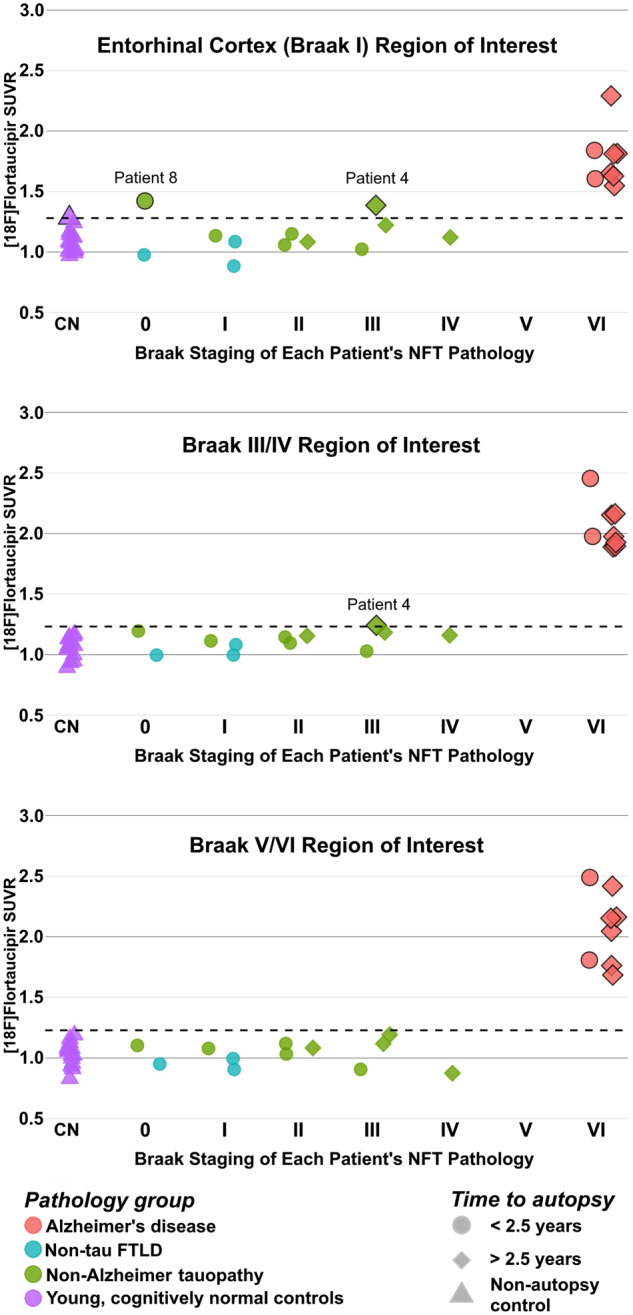
FTP-PET SUVR quantification of Braak stage regions of interest. SUVR quantification, not corrected for partial volume effects, was performed at entorhinal cortex (Braak I), Braak III/IV, and Braak V/VI regions of interest. Each patient is represented by a single point and coded by their primary autopsy diagnosis (colour), time from PET-to-autopsy (shape), and Braak neurofibrillary tangle stage (x-axis). The dotted line represents the threshold for significance, which is calculated from the mean SUVR plus two standard deviations for the young, cognitively normal (CN), non-autopsy controls. Points crossing the significance threshold are highlighted with a black outline.
PVC recovered signal in Braak III/IV regions of interest in the CBD patient with incidental Braak III NFT pathology (Patient 6) and the MAPT P301L patient with contributing Alzheimer’s disease pathology and Braak IV NFTs (Patient 7) (Supplementary Fig. 4). However, PVC also led to several false positive values in patients with non-Alzheimer tauopathies.
Discussion
We assessed the relationship between antemortem FTP-PET and autopsy in 20 patients with a broad range of primary neurodegenerative pathologies including Alzheimer’s disease, non-Alzheimer tauopathies, FTLD-TDP, and FTLD-FUS. First, we found an excellent correspondence between areas of FTP retention and NFT pathology in patients with primary Alzheimer’s disease pathology. Second, patients with non-Alzheimer tauopathies showed a broad range of tracer retention that was elevated compared to controls on W-maps (even within known regions of ‘off-target’ tracer binding in the basal ganglia and midbrain), though lower in intensity compared to the level of retention characteristic of Alzheimer’s disease. This signal usually (but not always) corresponded to locations of non-Alzheimer tau pathology found at autopsy. Patients with FTLD-TDP had a similar level and range of tracer retention compared to patients with non-Alzheimer tauopathies, but the tracer retention was in tau-negative regions. Patients with FTLD-FUS also showed small foci of tracer retention in tau-negative regions. FTP-PET SUVR in subcortical regions susceptible to tau pathology in PSP and CBD were similar in these conditions compared to other neurodegenerative pathologies. Therefore, FTP appears to have poor sensitivity and specificity for non-Alzheimer tauopathies. Third, quantification of FTP-PET SUVR in Braak stage regions of interest could reliably detect increased tracer retention in patients with Braak VI NFT pathology, but there was lower sensitivity for detection of earlier Braak stage (I–IV) pathology, at least in this cohort of patients who died during the later stages of neurodegenerative disease.
Patients with primary Alzheimer’s disease pathology had intense FTP retention in the parietal lobes and posterior cingulate on SUVR images, and W-maps showed significant increases in these areas compared to amyloid-negative healthy controls. Patients with typical and atypical Alzheimer’s disease phenotypes had patterns of tracer retention that mirrored the clinical and neuroanatomical variability of these phenotypes (Ossenkoppele et al., 2016), and NFT pathology was found in the areas of tracer retention. All Alzheimer’s disease patients had high ADNC (Braak VI) pathology, which was reliably detected on visual (SUVR images and W-maps) and quantitative assessment of FTP-PET.
CBD was associated with asymmetric tracer retention involving the frontal white matter, perirolandic cortex, basal ganglia, and substantia nigra. These areas contained CBD tau pathology. These findings are consistent with previous case reports, which found good PET-to-autopsy comparison for FTP in CBD (Josephs et al., 2016; McMillan et al., 2016; Schonhaut et al., 2017). Patients with CBD showed lower tracer retention than patients with primary Alzheimer’s disease pathology, but more extensive tracer retention than other sporadic non-Alzheimer neurodegenerative pathologies (Smith et al., 2017b). However, the pattern of uptake in CBD was very similar to the pattern observed in a patient with FTLD-TDP type A (Fig. 1), suggesting that FTP-PET has limited ability to differentiate CBD from FTLD-TDP pathology.
PSP was associated with tracer retention in the frontal white matter, globus pallidus, midbrain, and cerebellar dentate nucleus that corresponded with underlying tau pathology. These results are consistent with previous case reports (Marquié et al., 2017a; Passamonti et al., 2017). Interestingly, comparison of two PSP patients with tau pathology in the globus pallidus and cerebellar dentate revealed that the patient with higher tau burden in these regions also had higher corresponding tracer retention. Altogether, there is some concordance between the regional tau burden and FTP retention in PSP, but this may not be a clinically meaningful marker of pathological burden and disease severity. Compared to CBD, patients with PSP usually had more restricted tracer retention on SUVR images and restricted significance on W-maps. The low overall intensity, limited spatial extent, and significant overlap with regions showing ‘off-target’ tracer uptake in healthy controls suggests that FTP-PET has limited utility for imaging tau pathology in PSP.
Patients with FTLD-tau due to MAPT P301L and S305I mutations showed increased tracer retention in several common regions including the temporal lobes, temporal white matter, and basal ganglia. While attribution of tracer uptake in the patient with a P301L mutation was challenging given significant Alzheimer’s disease co-pathology, this signal corresponded to regions showing tau inclusion pathology in the patient with the S305I mutation. Mutations in MAPT can yield different combinations of tau isoforms and different forms of tau fibrils (Jones et al., 2018; Tsai et al., 2019). Previous studies showed convincing elevated FTP binding in patients with the R406W or V337M mutations, which yield ‘Alzheimer’s-like’ paired helical tau filaments with a mix of 3R/4R isoforms (Smith et al., 2016; Spina et al., 2017; Jones et al., 2018; Tsai et al., 2019). Here we report for the first-time elevated binding in the S305I mutation, corresponding to 4R tau, and showing pathological features similar to argyrophilic grain disease (Kovacs et al., 2008; Tacik et al., 2017). Notably, the burden of post-mortem tau pathology in this case was very high, corresponding to clearly elevated antemortem FTP retention.
Patients with FTLD-TDP and FTLD-FUS showed a range of tracer binding in areas containing non-tau FTLD pathology. These findings in the FTLD-TDP patients are consistent with previous reports of FTP binding in patients with semantic variant primary progressive aphasia and GRN and C9orf72 mutation carriers (Bevan-Jones et al., 2018a, b; Makaretz et al., 2018; Smith et al., 2019a; Tsai et al., 2019). The substrate of tracer binding in these cases remains unclear. FTP may show low-level binding to TDP-43 or FUS aggregates in vivo that does not persist in vitro following tissue preparations for autoradiography (Marquié et al., 2015; Lowe et al., 2016; Sander et al., 2016). Alternatively, the tracer could bind to other local pathological changes, such as monoamine oxidase B expressed in reactive astrocytes or iron (or associated proteins) in degenerating axons (Lockhart et al., 2017; Lemoine et al., 2018; Baker et al., 2019; Drake et al., 2019). We also cannot exclude the possibility that such cross-reactivity underlies tracer binding in non-Alzheimer tauopathies; however, the broad range of binding observed in patients with a similar degree of neurodegeneration (prominent in some, minimal in others) suggests that FTP binding is not simply a non-specific correlate of neurodegeneration in non-Alzheimer conditions.
Our findings are consistent with a recent large, multi-site study that demonstrated that FTP-PET accurately discriminates Alzheimer’s disease from other neurodegenerative disorders (Ossenkoppele et al., 2018), while variable uptake is seen in clinical syndromes associated with both tau-positive and tau-negative neuropathologies. Another 3R/4R tau disorder, chronic traumatic encephalopathy, has modest binding with lower SUVR values than Alzheimer’s disease patients (Lesman-Segev et al., 2019; Stern et al., 2019), and a recent case report found only moderate correlations with autopsy (Mantyh et al., 2020). There are several factors that may contribute to difficulties differentiating non-Alzheimer tauopathies from other pathologies and controls. First, FTP autoradiography studies have reported absent-to-low, displaceable binding in non-Alzheimer tauopathies and FTLD-TDP, with much lower binding than observed to tau NFTs in Alzheimer’s disease under the same conditions (Marquié et al., 2015; Lowe et al., 2016; Sander et al., 2016). Notably, we found that FTP-PET SUVR in regions susceptible to PSP and CBD was not elevated in patients with these primary autopsy diagnoses compared to other tauopathies and FTLD-TDP. Recent cryo-electron microscopy studies have uncovered unique tau ultrastructural differences in Alzheimer’s disease, Pick’s disease, CBD, and chronic traumatic encephalopathy (Fitzpatrick et al., 2017; Falcon et al., 2018, 2019; Scheres et al., 2020, Zhang et al., 2020), and tau ultrastructure may be a key reason for differential binding of the same tracer across tauopathies. Second, the density and overall mass of tau pathology affects the level of FTP retention. Sporadic 4R tauopathies, in particular PSP, are typically characterized by relatively low overall tau burden compared to Alzheimer’s disease (Ono et al., 2017). Notably, FTP signal was quite high in the patient with the MAPT S305I mutation, corresponding to a higher burden of tau pathology than in other cases with 4R tau pathology. Lastly, there is a great degree of variability in cortical, white matter, and basal ganglia FTP retention, which could be related to off-target tracer retention (Baker et al., 2019). It will be interesting to see how second-generation tau PET tracers perform across the spectrum of tau neurodegenerative diseases, given their different binding characteristics and pharmacokinetic properties (Leuzy et al., 2019).
All patients with primary Alzheimer’s disease autopsy diagnosis, who had Braak VI NFT pathology (high ADNC), showed significantly elevated FTP-PET SUVR levels across Braak stage regions of interest. This quantitative method could reliably detect advanced Alzheimer’s disease pathology, but there was difficulty detecting Braak I–III NFT pathology, even after correcting for partial volume effects. Given there was only one patient in our series with Braak IV and no patients with Braak V NFTs, we could not assess the tracer in these clinically meaningful stages. Using a different quantitative method (temporal lobe region of interest), Lowe et al. (2019) reliably detected patients who had Braak V and VI NFT pathology. Their study included two patients with Braak IV NFT pathology (one with primary Alzheimer’s pathology and another with secondary Alzheimer’s pathology) who narrowly crossed their significance threshold. Similar to our results, they were unable to detect Braak I–III NFTs. Additionally, a large autopsy study, with mean PET-to-autopsy time of 2.6 months, found that visual interpretation of FTP scans accurately detected advanced tau (Braak NFT stages V–VI) and Alzheimer’s disease (high ADNC) pathology. Patients with earlier Braak NFT pathology on autopsy (Braak NFT stages I–IV) were often read as negative (Fleisher et al., 2020). Based on these results, the United States Food and Drug Administration (FDA) recently approved the use of FTP ‘to estimate the density and distribution of aggregated tau neurofibrillary tangles in adult patients with cognitive impairment who are being evaluated for Alzheimer's disease’. The FDA specified that FTP is not indicated for the evaluation of patients with chronic traumatic encephalopathy (FDA News Release, 2020). Our findings further suggest that FTP should not be used in the evaluation of other non-Alzheimer tauopathies in the FTLD spectrum. In interpreting results, clinicians should also be aware of potential low sensitivity for lower Braak stages of Alzheimer’s disease that may still be clinically meaningful. Fluid biomarkers, such as CSF and plasma phosphorylated tau (pTau181, pTau217) could aid in earlier detection of tau in Alzheimer’s disease, and could also differentiate Alzheimer’s disease from non-Alzheimer tauopathies and other neurodegenerative diseases (Barthélemy et al., 2020; Janelidze et al., 2020; Mattsson-Carlgren et al., 2020; Thijssen et al., 2020).
The key strength of our study was the broad sample of well-characterized patients who had diverse underlying neuropathologies. A major difference between our sample and a recently published case series is the large number of patients with tau and non-tau FTLD pathology (Lowe et al., 2019). Additionally, many patients had Braak I–IV incidental or contributing NFT pathology, even though they had other primary neurodegenerative disorders, which allowed for examination of FTP-PET tracer retention in these early Braak stages.
There were several limitations to this study. First, our sample size was modest, though more diverse compared to previous reports. Second, the quantitative technique used to detect in vivo early stage (Braak I–IV) NFT pathology can be affected by the density of NFTs and the size of regions of interest, which probably limits detection of low-level NFT pathology in atrophic brain tissue contained within the large, bilateral Braak stage regions of interest. Third, we attempted to detect low-level NFT pathology in patients who had other pathological changes and atrophy; thus, our quantitative method may produce different results in other potential applications (e.g. detecting early Braak stage pathology in cognitively normal subjects being evaluated for pre-symptomatic tau accumulation). Developing more sensitive methods and defining the threshold of detection of tau in Alzheimer’s disease are important goals for future research. Fourth, PET-to-autopsy interval was variable, but on average, over 2 years. Additional neurodegenerative pathology could have accumulated between imaging and autopsy, which could impact the comparison between regions of tracer retention and autopsy pathology. Fifth, we did not perform quantitative immunohistochemistry, which could be helpful to understand the correlation between the density of different tau pathologies and FTP retention, and also to identify potential non-tau related binding targets (e.g. monoamine oxidase A or B, iron, or metal-associated proteins) (Lockhart et al., 2017; Baker et al., 2019; Drake et al., 2019). Finally, we did not compare FTP binding across tauopathies to binding of other tau tracers that may show different selectivity patterns (Okamura et al., 2018; Betthauser et al., 2019; Leuzy et al., 2019; Lohith et al., 2019; Mormino et al., 2020). Comparing the sensitivity across tracers both in vitro and in vivo is another important area for future research.
In conclusion, FTP reliably detected high ADNC (Braak VI) pathology on SUVR images, W-maps, and SUVR quantification of Braak stage regions of interest. FTP-PET had poor sensitivity and specificity for non-Alzheimer tauopathies. Quantification of FTP-PET SUVR in Braak stage regions of interest had low sensitivity for early Braak stage NFT pathology.
Supplementary Material
Acknowledgements
We would like to thank all faculty and staff at the UCSF Alzheimer’s Disease Research Center, especially the Neurodegenerative Disease Brain Bank and autopsy coordinators. We would also like to thank our participants for their generous contributions toward advancing our understanding of neurodegenerative diseases. Avid Radiopharmaceuticals enabled use of the 18F-flortaucipir tracer for the UCSF/LBNL scans by providing precursor.
Funding
Funding sources include: National Institute on Aging grants (R01-AG045611), (P50-AG023501), (P30-AG062422), (U54-NS092089), (R01-AG038791), (K08-AG052648), (K24-AG053435); Alzheimer’s Association (AACSF-19-617663); Rainwater Charitable Foundation; The Bluefield Project to Cure FTD; The Association for Frontotemporal Dementia; John Douglas French Alzheimer's Foundation; Michael J. Fox Foundation; Avid Radiopharmaceuticals A19 study (NCT03040713).
Competing interests
D.N.S., L.I., R.L.J, V.B., L.E., R.E., M.G., M.J., J.H.K., O.H.L., T.M., B.L.M., J.P., H.J.R., S.S., W.W.S., and A.S. report no competing interests. S.L.B. has served as a consultant to Genentech. A.L.B. has received research support from Avid Radiopharmaceuticals, Biogen, BMS, C2N, Cortice, Eisai, Eli Lilly, Forum, Genentech, Janssen, Novartis, Pfizer, Roche, and TauRx, and has served as a consultant for Aeton, Abbvie, Alector, AGTC, Amgen, Arkuda, Arvinas, Asceneuron, Eisai, Ionis, Lundbeck, Novartis, Passage BIO, Sangamo, Samumed, Third Rock, Toyama, and UCB. W.J.J. has served as a consultant to Genentech, Novartis, and Bioclinica. L.T.G. has research support from Avid Radiopharmaceuticals and Eli Lilly and has served as a consultant for CurSen Incorporated (in last 12 months). G.D.R. has research support from Avid Radiopharmaceuticals, GE Healthcare, Eli Lilly, and Life Molecular Imaging and has served as a consultant for GE Healthcare and Eisai (in the last 12 months) and an Associate Editor for JAMA Neurology.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- ADNC =
Alzheimer’s disease neuropathological change
- CBD =
corticobasal degeneration
- FTLD =
frontotemporal lobar degeneration
- FTP =
18F-flortaucipir
- NFT =
neurofibrillary tangle
- PSP =
progressive supranuclear palsy
- PVC =
partial volume correction
- SUVR =
standardized uptake value ratio
- TDP-43 =
TAR DNA-Binding Protein 43
Contributor Information
David N Soleimani-Meigooni, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA; Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
Leonardo Iaccarino, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Renaud La Joie, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Suzanne Baker, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
Viktoriya Bourakova, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Adam L Boxer, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Lauren Edwards, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Rana Eser, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Maria-Luisa Gorno-Tempini, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
William J Jagust, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA; Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA, USA; Helen Wills Neuroscience Institute, University of California, Berkeley, CA, USA.
Mustafa Janabi, Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
Joel H Kramer, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Orit H Lesman-Segev, Department of Diagnostic Imaging, Sheba Medical Center, Tel Hashomer, Ramat Gan, Israel.
Taylor Mellinger, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Bruce L Miller, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Julie Pham, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Howard J Rosen, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Salvatore Spina, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
William W Seeley, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Amelia Strom, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Lea T Grinberg, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA.
Gil D Rabinovici, Memory and Aging Center, Department of Neurology, University of California, San Francisco, CA, USA; Molecular Biophysics and Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA, USA; Helen Wills Neuroscience Institute, University of California, Berkeley, CA, USA; Department of Radiology and Biomedical Imaging, University of California, San Francisco, CA, USA.
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s. Dement 2011; 7: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima K. Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 2006; 26: 475–83. [DOI] [PubMed] [Google Scholar]
- Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80: 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Harrison TM, Maaß A, La Joie R, Jagust W.. Effect of off-target binding on 18F-Flortaucipir variability in healthy controls across the lifespan. J Nucl Med 2019; 60: 1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SL, Maass A, Jagust WJ.. Considerations and code for partial volume correcting [18F]-AV-1451 tau PET data. Data Brief 2017; 15: 648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med 2020; 26: 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betthauser TJ, Cody KA, Zammit MD, Murali D, Converse AK, Barnhart TE, et al. In vivo characterization and quantification of neurofibrillary tau PET radioligand 18 F-MK-6240 in humans from Alzheimer disease dementia to young controls. J Nucl Med 2019; 60: 93–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan-Jones RW, Cope TE, Jones SP, Passamonti L, Hong YT, Fryer T, et al. AV-1451 binding is increased in frontotemporal dementia due to C9orf72 expansion. Ann Clin Transl Neurol 2018. a; 5: 1292–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan-Jones WR, Cope TE, SimonJones P, Passamonti L, Hong YT, Fryer TD, et al. AV-1451 binding in vivo mirrors the expected distribution of TDP-43 pathology in the semantic variant of primary progressive aphasia. J Neurol Neurosurg Psychiatry 2018. b; 89: 1032–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su M-Y, et al. Early clinical PET imaging results with the novel PHF-Tau Radioligand [F-18]-T807. J Alzheimers Dis 2013; 34: 457–68. [DOI] [PubMed] [Google Scholar]
- Cho H, Baek MS, Choi JY, Lee SH, Kim JS, Ryu YH, et al. 18F-AV-1451 binds to motor-related subcortical gray and white matter in corticobasal syndrome. Neurology 2017. a; 89: 1170–8. [DOI] [PubMed] [Google Scholar]
- Cho H, Choi JY, Hwang MS, Lee JH, Kim YJ, Lee HM, et al. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016; 87: 375–83. [DOI] [PubMed] [Google Scholar]
- Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS, et al. Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord 2017. b; 32: 134–40. [DOI] [PubMed] [Google Scholar]
- Coakeley S, Cho SS, Koshimori Y, Rusjan P, Harris M, Ghadery C, et al. Positron emission tomography imaging of tau pathology in progressive supranuclear palsy. J Cereb Blood Flow Metab 2017; 37: 3150–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006; 31: 968–80. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002; 61: 935–46. [DOI] [PubMed] [Google Scholar]
- Drake LR, Pham JM, Desmond TJ, Mossine AV, Lee SJ, Kilbourn MR, et al. Identification of AV-1451 as a weak, nonselective inhibitor of monoamine oxidase. ACS Chem Neurosci 2019; 10: 3839–46. [DOI] [PubMed] [Google Scholar]
- Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018; 561: 137–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019; 568: 420–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA News Release. FDA Approves First Drug to Image Tau Pathology in Patients Being Evaluated for Alzheimer’s Disease [Internet]. United States Food and Drug Administration 2020[cited 2020 Jul 5] Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-image-tau-pathology-patients-being-evaluated-alzheimers-disease.
- Ferrer I, Santpere G, Van Leeuwen FW.. Argyrophilic grain disease. Brain 2008; 131: 1416–32. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017; 547: 185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Pontecorvo MJ, Devous MD, Lu M, Arora AK, Truocchio SP, et al. Positron Emission Tomography Imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol 2020; 77: 829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons GS, Lee VMY, Trojanowski JQ.. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol 2019; 76: 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Falcon B, Zhang W, Ghetti B, Scheres SHW.. Distinct conformers of assembled tau in Alzheimer’s and Pick’s diseases. Cold Spring Harb Symp Quant Biol 2018; 83: 163–71. [DOI] [PubMed] [Google Scholar]
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76: 1006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, et al. Preliminary NINDS neuropathologic criteria for steele-richardson-olszewski syndrome(progressive supranuclear palsy). Neurology 1994; 44: 2015–9. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord 2017; 32: 853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Petersen RC, Xu YC, Waring SC, O’Brien PC, Tangalos EG, et al. Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. Neurology 1997; 49: 786–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med 2020; 26: 379–86. [DOI] [PubMed] [Google Scholar]
- Jones DT, Knopman DS, Graff-Radford J, Syrjanen JA, Senjem ML, Schwarz CG, et al. In vivo 18 F-AV-1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology 2018; 90: e947–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Tacik P, Duffy JR, Senjem ML, Tosakulwong N, et al. 18F]AV-1451 tau-PET uptake does correlate with quantitatively measured 4R-tau burden in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016; 132: 931–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E-J, Brown JA, Deng J, Hwang J-HL, Spina S, Miller ZA, et al. Mixed TDP-43 proteinopathy and tauopathy in frontotemporal lobar degeneration: nine case series. J Neurol 2018; 265: 2960–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King ME, Ghoshal N, Wall JS, Binder LI, Ksiezak-Reding H.. Structural analysis of Pick’s disease-derived and in vitro-assembled tau filaments. Am J Pathol 2001; 158: 1481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T. Tau-positive dial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol 1999; 9: 663–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Pittman A, Revesz T, Luk C, Lees A, Kiss E, et al. MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol 2008; 116: 103–18. [DOI] [PubMed] [Google Scholar]
- La Joie R, Perrotin A, Barré L, Hommet C, Mézenge F, Ibazizene M, et al. Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J Neurosci 2012; 32: 16265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ.. Neurodegenerative tauopathies. Annu Rev Neurosci 2001; 24: 1121–59. [DOI] [PubMed] [Google Scholar]
- Lemoine L, Leuzy A, Chiotis K, Rodriguez-Vieitez E, Nordberg A.. Tau positron emission tomography imaging in tauopathies: the added hurdle of off-target binding. Alzheimer’s Dement Diagnosis. Alzheimer's Dement 2018; 10: 232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesman-Segev OH, Joie R, Stephens ML, Sonni I, Tsai R, Bourakova V, et al. Tau PET and multimodal brain imaging in patients at risk for chronic traumatic encephalopathy. NeuroImage Clin 2019; 24: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, Rodriguez-Vieitez E, et al. Tau PET imaging in neurodegenerative tauopathies—Still a challenge. Mol Psychiatry 2019; 24: 1112–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, et al. Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 1996; 55: 97–105. [DOI] [PubMed] [Google Scholar]
- Lockhart SN, Ayakta N, Winer JR, La Joie R, Rabinovici GD, Jagust WJ.. Elevated 18F-AV-1451 PET tracer uptake detected in incidental imaging findings. Neurology 2017; 88: 1095–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohith TG, Bennacef I, Vandenberghe R, Vandenbulcke M, Salinas CA, Declercq R, et al. Brain imaging of Alzheimer dementia patients and elderly controls with 18 F-MK-6240, a PET tracer targeting neurofibrillary tangles. J Nucl Med 2019; 60: 107–14. [DOI] [PubMed] [Google Scholar]
- Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 2016; 4: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Lundt ES, Albertson SM, Min H-K, Fang P, Przybelski SA, et al. Tau-positron emission tomography correlates with neuropathology findings. Alzheimer’s Dement 2019; 46: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage 2017; 157: 448–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie IRA, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010; 119: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makaretz SJ, Quimby M, Collins J, Makris N, McGinnis S, Schultz A, et al. Flortaucipir tau PET imaging in semantic variant primary progressive aphasia. J Neurol Neurosurg Psychiatry 2018; 89: 1024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh WG, Spina S, Lee A, Iaccarino L, Soleimani-Meigooni D, Tsoy E, et al. Tau positron emission tomographic findings in a former US football player with pathologically confirmed chronic traumatic encephalopathy. JAMA Neurol 2020; E1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquié M, Normandin MD, Meltzer AC, Siao Tick Chong M, Andrea NV, Antón-Fernández A, et al. Pathological correlations of [F-18]. Ann Neurol 2017. a; 81: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]. Ann Neurol 2015; 78: 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquié M, Siao Tick Chong M, Antón-Fernández A, Verwer EE, Sáez-Calveras N, Meltzer AC, et al. F-18]-AV-1451 binding correlates with postmortem neurofibrillary tangle Braak staging. Acta Neuropathol 2017. b; 134: 619–28. [ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquié M, Verwer EE, Meltzer AC, Kim SJW, Agüero C, Gonzalez J, et al. Lessons learned about [F-18]-AV-1451 off-target binding from an autopsy-confirmed Parkinson’s case. Acta Neuropathol Commun 2017. c; 5: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson-Carlgren N, Andersson E, Janelidze S, Ossenkoppele R, Insel P, Strandberg O, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer’s disease. Sci Adv 2020; 6: eaaz2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 2017; 89: 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996; 47: 1113–24. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement 2011; 7: 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan CT, Irwin DJ, Nasrallah I, Phillips JS, Spindler M, Rascovsky K, et al. Multimodal evaluation demonstrates in vivo 18F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016; 132: 935–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 2012; 123: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Toueg TN, Azevedo C, Castillo JB, Guo W, Nadiadwala A, et al. Tau PET imaging with 18F-PI-2620 in aging and neurodegenerative diseases. Eur J Nucl Med Mol Imaging 2020; 1–12. doi: 10.1007/s00259-020-04923-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama S, Mori H, Ihara Y, Tomonaga M.. Immunocytochemical and ultrastructural studies of Pick’s disease. Ann Neurol 1990; 27: 394–405. [DOI] [PubMed] [Google Scholar]
- O’Brien PC, Dyck PJ.. Procedures for setting normal values. Neurology 1995; 45: 17–23. [DOI] [PubMed] [Google Scholar]
- Okamura N, Harada R, Ishiki A, Kikuchi A, Nakamura T, Kudo Y.. The development and validation of tau PET tracers: current status and future directions. Clin Transl Imaging 2018; 6: 305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017; 140: 764–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016; 139: 1551–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R, Rabinovici GD, Smith R, Cho H, Scholl M, Strandberg O, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA 2018; 320: 1151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passamonti L, Rodríguez PV, Hong YT, Allinson KSJ, Williamson D, Borchert RJ, et al. 18F-AV-1451 positron emission tomography in Alzheimer’s disease and progressive supranuclear palsy. Brain 2017; 140: 781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011; 134: 2456–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset OG, Ma Y, Evans AC.. Correction for partial volume effects in PET: principle and validation. J Nucl Med 1998; 39: 904–11. [PubMed] [Google Scholar]
- Sander K, Lashley T, Gami P, Gendron T, Lythgoe MF, Rohrer JD, et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimer’s Dement 2016; 12: 1116–24. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Zhang W, Falcon B, Goedert M.. Cryo-EM structures of tau filaments. Curr Opin Struct Biol 2020; 64: 17–25. [DOI] [PubMed] [Google Scholar]
- Schöll M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, et al. PET imaging of tau deposition in the aging human brain. Neuron 2016; 89: 971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhaut DR, McMillan CT, Spina S, Dickerson BC, Siderowf A, Devous MD, et al. 18F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: a multicenter study. Ann Neurol 2017; 82: 622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, et al. Regional profiles of the candidate tau PET ligand 18 F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016; 139: 1539–50. [DOI] [PubMed] [Google Scholar]
- Smith R, Puschmann A, Schöll M, Ohlsson T, Van Swieten J, Honer M, et al. 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016; 139: 2372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Santillo AF, Waldö ML, Strandberg O, Berron D, Vestberg S, et al. 18 F-Flortaucipir in TDP-43 associated frontotemporal dementia. Sci Rep 2019. a; 9:6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Schöll M, Honer M, Nilsson CF, Englund E, Hansson O.. Tau neuropathology correlates with FDG-PET, but not AV-1451-PET, in progressive supranuclear palsy. Acta Neuropathol 2017. a; 133: 149–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Schöll M, Widner H, Van Westen D, Svenningsson P, Hägerström D, et al. In vivo retention of 18 F-AV-1451 in corticobasal syndrome. Neurology 2017. b; 89: 845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Wibom M, Pawlik D, Englund E, Hansson O.. Correlation of in vivo [18F]Flortaucipir with postmortem Alzheimer disease tau pathology. JAMA Neurol 2019. b; 76: 310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina S, Schonhaut DR, Boeve BF, Seeley WW, Ossenkoppele R, O’Neil JP, et al. Frontotemporal dementia with the V337M MAPT mutation. Neurology 2017; 88: 758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern RA, Adler CH, Chen K, Navitsky M, Luo J, Dodick DW, et al. Tau Positron-Emission Tomography in Former National Football League players. N Engl J Med 2019; 380: 1716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacik P, Sanchez-Contreras M, DeTure M, Murray ME, Rademakers R, Ross OA, et al. Clinicopathologic heterogeneity in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) due to microtubule-associated protein tau (MAPT) p. P301L mutation, including a patient with globular glial tauopathy. Neuropathol Appl Neurobiol 2017; 43: 200–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Weidenheim KM, Dickson DW, Ksiezak-Reding H. Morphological and biochemical correlations of abnormal tau filaments in progressive supranuclear palsy. J Neuropathol Exp Neurol 2002; 61: 33–45. [DOI] [PubMed] [Google Scholar]
- Thijssen EH, La Joie R, Wolf A, Strom A, Wang P, Iaccarino L, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med 2020; 26: 387–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BA, Erlandsson K, Modat M, Thurfjell L, Vandenberghe R, Ourselin S, et al. The importance of appropriate partial volume correction for PET quantification in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2011; 38: 1104–19. [DOI] [PubMed] [Google Scholar]
- Tsai RM, Bejanin A, Lesman-Segev O, LaJoie R, Visani A, Bourakova V, et al. 18F-flortaucipir (AV-1451) tau PET in frontotemporal dementia syndromes. Alzheimers Res Ther 2019; 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C-F, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, et al. 18F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease]. Alzheimer’s Dement 2013; 9: 666–76. [DOI] [PubMed] [Google Scholar]
- Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, et al. Novel tau filament fold in corticobasal degeneration. Nature 2020; 580: 283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukareva V, Shah K, Uryu K, Braak H, Del Tredici K, Sundarraj S, et al. Biochemical analysis of τ proteins in argyrophilic grain disease, Alzheimer’s disease, and Pick’s disease: a comparative study. Am J Pathol 2002; 161: 1135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study are available for review upon request.