

Abstract
Rationale:
Over 2,700 e-cigarette, or vaping, product use-associated lung injury (EVALI) cases have been reported to the Centers for Disease Control and Prevention (CDC) during August 2019-February 2020. Bronchoalveolar lavage (BAL) fluid samples from 51 EVALI and 99 non-EVALI cases were analyzed for toxicants including petroleum distillates. We describe a novel method to measure petroleum distillates in BAL fluid using gas chromatography-mass spectrometry (GC/MS).
Methods:
n−Hexane, n−heptane, n−octane, methylcyclopentane, and cyclohexane were measured in BAL fluid specimens by headspace solid-phase microextraction /gas chromatography/mass spectrometry. We created and characterized BAL fluid pools from non-EVALI individuals to determine assay accuracy, precision, linearity, limits of detection, and analytical specificity. All measurements were conducted in accordance with the CDC’s Division of Laboratory Sciences rigorous method validation procedures.
Results
Matrix validation experiments showed that calibration curves in BAL fluid and saline had similar slopes, with differences less than 5%. Assay precision ranged from 1.98% - 18%. In addition, the limits of detection for the five analytes ranged from 0.05 – 0.10 μg/L, and their linearity was confirmed with R2 values >0.99. The analysis of selected petroleum distillates in BAL fluid analysis was shown to be comparable to their analysis in blood in which the 95th percentiles are below detection.
Conclusions:
We developed and validated a method to quantify petroleum distillates in BAL fluid specimens using GC/MS. The assay provided precise and accurate analyses of EVALI and non-EVALI BAL fluid specimens in support of CDC’s EVALI response. This method is applicable to the determination of a broad range of VOCs in BAL fluid specimens.
Keywords: Bronchoalveolar lavage fluid, GC/MS, EVALI, petroleum distillates
Rationale
A total of 2,807 hospitalized e-cigarette, or vaping, product use-associated lung injury (EVALI) cases have been reported to the Centers for Disease Control and Prevention (CDC) from 50 states, the District of Columbia, and two U.S. territories (Puerto Rico and U.S. Virgin Islands) as of February 18, 20201. Of these case, 68 deaths have been reported in 29 states, and the District of Columbia1. National data and active case reporting from state health departments around the country show a sharp rise in symptoms or cases of EVALI in August 2019, a peak in September 2019, and a gradual, but persistent decline since then. Blount et al. showed that vitamin E acetate, an additive in some THC-containing e-cigarette, or vaping, products, is strongly linked with EVALI2,3. In that study, bronchoalveolar lavage (BAL) fluid samples from 51 EVALI cases from 16 states and a comparison group of samples from 99 individuals without EVALI were analyzed for toxicants, including vitamin E acetate, plant oils, medium chain triglyceride (MCT) oil, coconut oil, petroleum distillates, and diluent terpenes. BAL specimens are obtained by injecting normal saline into the lung and applying mild suction to retrieve a fraction of that saline. Petroleum distillates toxicants are volatile organic compounds (VOCs), and can be analyzed using gas chromatography-mass spectrometry (GC/MS).
Our laboratory developed a method to quantify VOCs in blood4 in support of the National Centers for Health Statistic’s National Health and Nutrition Examination Survey (NHANES)5. Specifically, we provide measurements of VOCs in blood to obtain nationally representative estimates of the U.S. population’s exposure to VOCs6. To support CDC’s response to the EVALI crisis, the existing method was modified and validated to quantify VOCs in BAL fluid samples, focusing on selected petroleum distillate biomarkers from the existing method. We describe method performance parameters (e.g., accuracy, linearity, selectivity) for measuring the petroleum distillate biomarkers, n−hexane, n−heptane, n−octane, methylcyclopentane, and cyclohexane in BAL fluid. Our laboratory currently measures these five analytes in blood, and they were deemed to be representative of petroleum distillates posited to have been used to adulterate e-cigarette liquids associated with the EVALI crisis.
Methods
The target analytes were measured in BAL fluid specimens by headspace solid-phase microextraction (SPME)/gas chromatography/mass spectrometry, based on previously described methods4,7. We used SPME in this application for isolation of the specimen matrix from the GC/MS and preconcentration of the VOC. The SPME conditions used are for general VOC collection with water solubilities ranging from 1.6 to 210,000 mg/L and were not optimized for these specific alkanes. All glassware, headspace vial septa, vials, and reagent water were cleaned and verified to be free of those VOCs being analyzed8. After cleaning, glassware and septa were stored in a vacuum oven to prevent recontamination. All method parameters were examined in accordance with the CDC’s Division of Laboratory Sciences rigorous method validation procedures, which are based on standard practices9.
Standards preparation
Primary stock solutions were prepared from neat materials diluted with either purge and trap grade methanol or HPLC grade acetone. Lower concentration primary stock solutions involved only a single serial dilution of the highest concentration stock. Intermediate levels were formulated from the primary stock solutions in purge and trap grade methanol using only a single dilution step. The primary stock solution concentrations were based on the gravimetric measure of mass transferred to the volumetric flasks. Working standards (standards 0–7) were prepared in buffered saline to achieve eight concentrations ranging from (0.013 to 59.2 μg/mL) as listed in Table 1 using a single dilution of the corresponding intermediate levels. The highest concentration calibrators are at least an order of magnitude below the compounds’ water solubilities at 25°C, which are given in Table 1. The saline was tested to ensure that VOCs were below detectable levels for the analytes of interest using a blank produced using low-VOC water. Aqueous working standards were formulated in volumes of 25-mL quantities with buffered saline and added internal standard. Each of the aqueous working standards (3.0 mL) was transferred into cleaned 10-mL headspace vials using a gas-tight glass barrel/PTFE plunger pipetter. The vials were immediately sealed with recently cleaned caps and grouped by concentration in separate wide mouth sample jars to prevent cross contamination. The standard set was stored in a dedicated refrigerator at 2–6 °C and analyzed as part of an analytical run within one week.
Table 1.
Standard concentration levels (μg/L).
| Compound | Standard Level (μg/L) | Water Solubility at 25°C (μg/L) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||
| n−Hexane | 0.0317 | 0.0549 | 0.127 | 0.275 | 1.11 | 3.78 | 11.6 | 45.0 | 9500 |
| n−Heptane | 0.0234 | 0.0500 | 0.0937 | 0.252 | 0.771 | 2.90 | 8.63 | 39.8 | 3400 |
| n−Octane | 0.0331 | 0.0626 | 0.132 | 0.315 | 1.12 | 4.25 | 15.9 | 58.9 | 660 |
| Methylcyclopentane | 0.0141 | 0.0264 | 0.0564 | 0.129 | 0.437 | 1.47 | 4.64 | 17.6 | 42000 |
| Cyclohexane | 0.0128 | 0.0232 | 0.0513 | 0.116 | 0.403 | 1.42 | 4.49 | 17.6 | 55000 |
Preparation of isotopically labeled internal standard solutions
Primary isotopically labeled internal standard stock solutions were made by dilution of the neat compound into purge and trap grade methanol. Isotopic compound analogs were labeled fully with deuterium and of adequate chemical and isotopic purity (>98%) to produce levels needed for accurate quantitation; any impurities did not interfere with analyses of the other VOC analytes. Concentrations of the primary labeled internal standard stock solutions ranged from 0.7 to 12 mg/mL. The primary isotopically labeled internal standard stock solutions were stored in a freezer below −60 °C. Secondary isotopically labeled internal standard stock solutions were made by combining primary stock solutions and diluting to concentrations between standard levels 2 and 5. Stock solutions in ampoules were stored in a freezer below −60 °C. Working isotopically labeled internal standard solution was prepared daily from the secondary stock solution. The secondary stock solution was added to the standard formulations, water blanks, quality control samples, and specimen samples. Calibration was performed and samples quantified using the peak area ratios of quantitation ion to internal standard ion.
Preparation of quality control materials
Quality control (QC) materials were prepared in 1.5 liter batches at three concentration levels in bovine serum. After fortification with VOCs, 7 mL were pipetted into 10 mL cryules, which were flamed sealed with an oxyhydrogen torch and stored at approximately 70°C. The concentration homogeneity across the batch was evaluated by comparing samples prepared at the beginning, middle, and end of each concentration batch. Any variability across a concentration batch of more than 25% for any analyte resulted in reformulation of the affected batch.
Instrumentation and operation
Sample analysis was performed using a PAL system (CTC Analytics, Switzerland) autosampler, coupled to an Agilent 7890A gas chromatograph and a 5975C mass selective detector (Agilent, Santa Clara CA). Ionization was performed by electron ionization in the positive ion mode at an electron voltage of 70 eV. Quantitation was performed using selected ion monitoring mode of each primary quantitation ion, confirmation ion, and internal standard ion using a dwell times between 20 and 30 ms for each. Ions used are given in Table 2. Samples were queued on an autosampler tray and maintained at 15 ± 0.5 °C until they were analyzed. During analysis the samples were transferred to an agitating incubator set to 500 rpm and 40 ± 1 °C as the headspace is sampled with a 75-μm Carboxen-PDMS coated SPME fiber (Supelco, Bellefonte PA) for about 15 min. The SPME fiber was then immediately transferred into the injection port which was fitted with a glass liner with an i.d. of 2 mm and held at 250 ± 0.5 °C. The sample was introduced into an Agilent DB-VRX column (40 m × 0.18 mm × 1 μm film) via pulsed splitless injection set at 50 psi. After 1.5 min the injection port pressure was then dropped to maintain a constant flow of 1.0 ± 0.1 mL/min of helium. In-line, after the injection port, is a cryogenic trap. At the start of the GC run the cryotrap is set to −100 °C for 1 min then ballistically heated to approximately 215 °C (13.0 °C/sec). The GC oven temperature is programmed to ramp from 0 °C (1.5 min hold) at 7 °C/min to 140 °C, then 40 °C/min to 220 °C (8.5 min hold). During the analytical run, the SPME fiber remained in the GC injection port until ready to collect the next sample and was not exposed to the laboratory air for more than 1 min to reduce ambient air contamination. Carryover was evaluated for all analytes by running a blank sample after the highest calibrator in which no carryover was detected above the LLRs. If a sample had a concentration above the highest calibrator, a new sample was prepared from the specimen by diluting it with low VOC water to be within the calibration range.
Table 2.
Ions used for the quantitation of compounds and internal standards.
| Compound | Quantitation (m/z) | Confirmation (m/z) | Internal Standard (m/z) |
|---|---|---|---|
| n−Hexane | 57 | 41 | 66 |
| n−Heptane | 71 | 70 | 116 |
| n−Octane | 85 | 114 | 66 |
| Methylcyclopentane | 69 | 84 | 96 |
| Cyclohexane | 84 | 69 | 96 |
Calibration and calibration verification
All calibration standards were prepared in phosphate buffered saline because it was difficult to consistently obtain reduced VOC background levels in pooled BAL fluid specimen matrixes below detectable levels. Spike matrix experiments (matrix validation) were performed to verify that calibration curves in BAL fluid and saline had the same slope (Table 3). Experiments were performed on 15 mL pooled BAL fluid specimens. Because the available quantity of BAL fluid was not sufficient to perform all experiments, the pooled BAL was diluted with low VOC water. For this dilution 1.5 mL was used for sample analysis and 1.5 mL of low-VOC water was added to the matrix to achieve a total volume of nominally 3.0 mL. Characterization was performed once using at least six different concentrations distributed across the analytical range.
Table 3.
Matrix comparison (matrix validation) for selected petroleum distillates in BAL fluid with water.
| Compound | Difference Between Slopes (%) |
|---|---|
| n-Hexane | 1.09 |
| n-Heptane | 0.86 |
| n-Octane | 4.83 |
| Methylcyclopentane | 3.79 |
| Cyclohexane | 2.63 |
Accuracy, precision, linearity, limits of detection, analytical specificity
Recovery accuracy was evaluated by measuring nine different spiked levels in pooled BAL fluid specimen matrix. These spiked BAL levels were compared with similarly prepared reference standards in low VOC water as a means to compensate for handling biases associated with spiking low volumes of low concentration nonpolar compounds. Final concentrations ranged from 0.044 to 59 μg/L. Because the available quantity of BAL fluid was not sufficient to prepare the number of samples needed, we diluted the BAL with low VOC water. For this analysis 1.5 mL BAL fluid sample was used, and 1.5 mL of low-VOC water was added to achieve nominally 3.0 mL (Table 4).
Table 4.
Comparison of recovery accuracy for the measurement of select petroleum distillates in BAL fluid relative to low VOC water.
| Analyte | Level | H2O (μg/L) |
BAL fluid (μg/L) | % Error | |
|---|---|---|---|---|---|
| n-Hexane | 2 | 0.128 | 0.134 | 4.4 | |
| 2.5 | 0.227 | 0.226 | −0.6 | ||
| 3 | 0.319 | 0.315 | −1.0 | ||
| 4 | 0.932 | 0.915 | −1.8 | ||
| 4.5 | 1.94 | 1.86 | −4.1 | ||
| 5 | 3.07 | 3.01 | −2.0 | ||
| 6 | 10.4 | 10.3 | −0.8 | ||
| 6.5 | 24.8 | 27.6 | 11.6 | ||
| 7 | 62.3 | 62.2 | −0.1 | ||
| n-Heptane | 2 | 0.0584 | 0.0680 | 16.6 | |
| 2.5 | 0.111 | 0.109 | −1.4 | ||
| 3 | 0.165 | 0.186 | 12.5 | ||
| 4 | 0.531 | 0.557 | 4.9 | ||
| 4.5 | 1.15 | 1.20 | 3.9 | ||
| 5 | 1.86 | 1.86 | 0.1 | ||
| 6 | 6.94 | 7.17 | 3.2 | ||
| 6.5 | 19.4 | 21.3 | 9.9 | ||
| 7 | 73.4 | 62.2 | −15.3 | ||
| n-Octane | 2 | 0.101 | 0.147 | 46.1 | |
| 2.5 | 0.212 | 0.199 | −6.29 | ||
| 3 | 0.331 | 0.329 | −0.60 | ||
| 4 | 1.09 | 1.01 | −6.55 | ||
| 4.5 | 2.36 | 2.27 | −3.72 | ||
| 5 | 3.73 | 3.58 | −4.09 | ||
| 6 | 13.8 | 12.7 | −7.71 | ||
| 6.5 | 34.7 | 32.6 | −5.87 | ||
| 7 | 72.3 | 66.1 | −8.62 | ||
| Methylcyclopentane | 1 | 0.0253 | 0.0240 | −4.9 | |
| 2 | 0.0484 | 0.0496 | 2.5 | ||
| 2.5 | 0.0880 | 0.0862 | −2.0 | ||
| 3 | 0.135 | 0.129 | −4.2 | ||
| 4 | 0.366 | 0.384 | 4.8 | ||
| 4.5 | 0.747 | 0.780 | 4.5 | ||
| 5 | 1.20 | 1.23 | 2.3 | ||
| 6 | 4.36 | 4.38 | 0.6 | ||
| 6.5 | 10.9 | 12.2 | 12.0 | ||
| 7 | 26.9 | 28.2 | 5.1 | ||
| Cyclohexane | 2 | 0.0450 | 0.0515 | 14.4 | |
| 2.5 | 0.0800 | 0.0920 | 15.0 | ||
| 3 | 0.114 | 0.129 | 13.2 | ||
| 4 | 0.330 | 0.361 | 9.3 | ||
| 4.5 | 0.703 | 0.745 | 6.0 | ||
| 5 | 1.12 | 1.17 | 4.8 | ||
| 6 | 4.03 | 4.13 | 2.6 | ||
| 6.5 | 10.5 | 11.3 | 7.4 | ||
| 7 | 25.2 | 25.8 | 2.2 | ||
Precision was evaluated by repetitive analysis of standards. Six separate standards of the lowest calibrator (with a signal-to-noise of at least 5), a medium calibrator and a high calibrator were analyzed within the same acquisition. We calculated the standard deviation (SD) and the coefficient of variation (CV) for the results (Table 5). We determined the linearity of the assay by creating individual analyte calibration curves of at least six calibrators in buffered saline. These solutions were prepared and analyzed six times to evaluate variability in calibration curve linearity (Figure 1). Error bars are not shown in Figure 1 because they are small and obscured the symbol. Limits of detection (LOD, Table 6) were calculated using the three times the standard deviation at zero concentration (3S0)10 method where the mean concentration of the blank was set to zero. The lowest level reported (LLR, Table 6) was set above the LOD and above at least one standard level to ensure results are bracketed by at least one standard level. Below LLR results are reported as <LLR. In addition, analytical specificity was established by confirming similarity (within 25%) of analyte levels using the quantitation ion for quantification. Quantitation and confirmation ion retention times relative to the ISTD ion retention time are to be within 2 s of the retention time differences established using the standards. This retention time requirement is also to be applied to integrated peaks that fall below the LLR.
Table 5.
Precision of quality control materials.
| Analyte | Low Concentration (STD1, μg/L) |
Medium Concentration (STD3, μg/L) |
High Concentration (STD5, μg/L) |
|||
|---|---|---|---|---|---|---|
| Mean (STDEV) | % CV | Mean (STDEV) | % CV | Mean (STDEV) | % CV | |
| n-Hexane | 0.062 (0.007) | 10.6 | 0.250 (0.010) | 3.82 | 3.56 (0.114) | 3.21 |
| n-Heptane | 0.058 (0.010) | 18.1 | 0.220 (0.021) | 9.73 | 2.89 (0.301) | 10.4 |
| n-Octane | 0.073 (0.007) | 9.88 | 0.334 (0.007) | 1.98 | 4.73 (0.257) | 5.44 |
| Methylcyclopentane | 0.023 (0.002) | 6.58 | 0.122 (0.006) | 5.30 | 1.49 (0.050) | 3.34 |
| Cyclohexane | 0.019 (0.001) | 6.04 | 0.109 (0.002) | 1.98 | 1.39 (0.038) | 2.75 |
STDEV: Standard deviation
Figure 1.
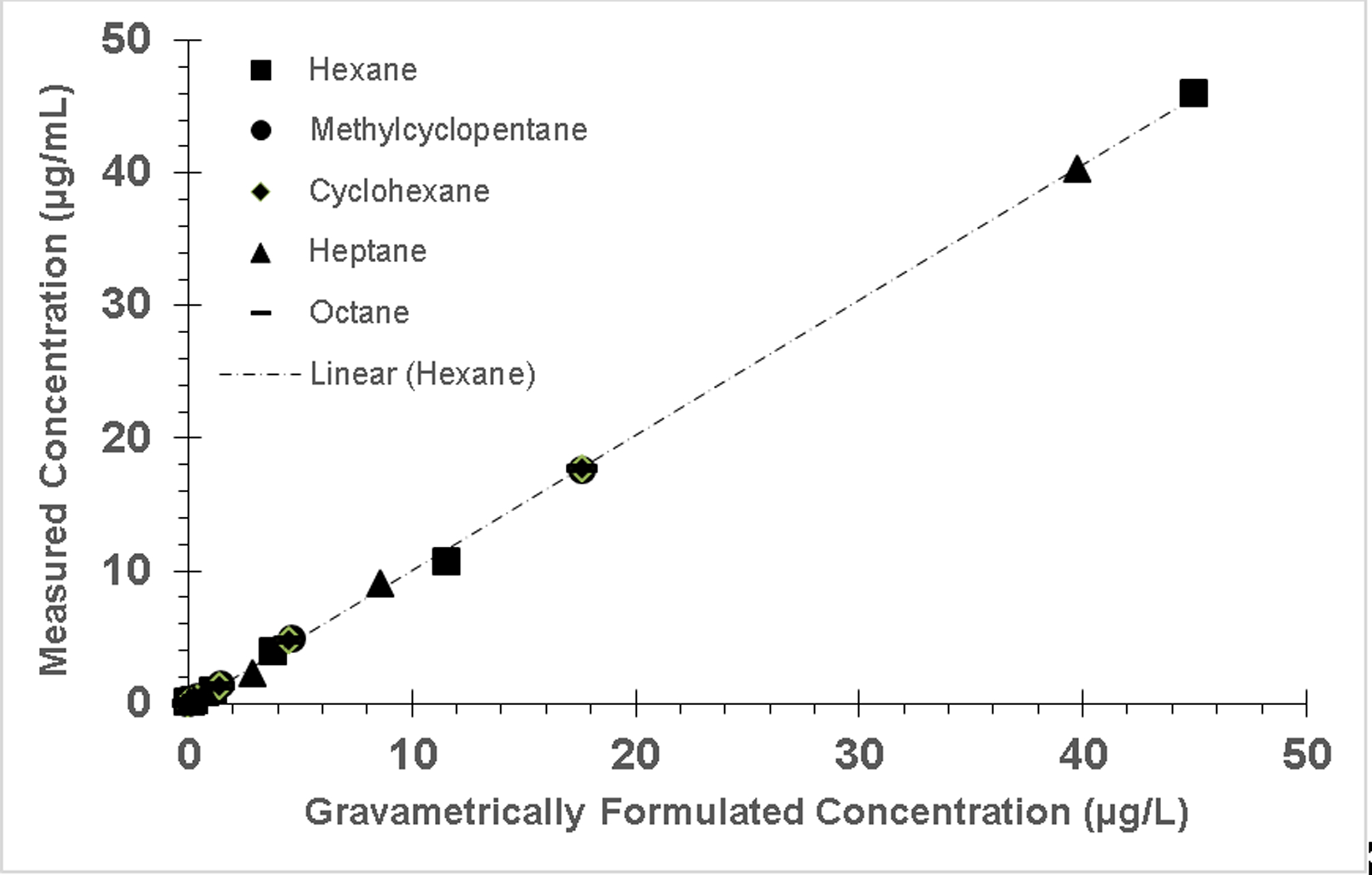
Calibration curves of target alkenes demonstrating linear trending response with increase in concentration. Error bars are not shown because they are small and obscured the symbol.
Table 6.
Limits of detection (μg/L) for selected petroleum distillates in BAL saline.
| Limit of Detection | Lowest Level Reported | |
|---|---|---|
| n-Hexane | 0.0230 | 0.100 |
| n-Heptane | 0.0282 | 0.100 |
| n-Octane | 0.0270 | 0.100 |
| Methylcyclopentane | 0.00228 | 0.020 |
| Cyclohexane | 0.00378 | 0.020 |
VOCs can diffuse from specimens collected and stored in non-hermetic containers such as cryovials and should be collected and analyzed as soon as possible. Although we do not have long-term stability data on storage of VOCs in BAL fluid because of limited available quantities, we performed long-term stability tests on alkanes spiked in bovine serum stored hermetically in vacutainers at 4°C. Stability of alkanes in these tests ranged from 6 (heptane) to 12 (methylcyclopentane) months. Loss is likely attributed to diffusion of VOC into the vacutainer stoppers rather than decomposition because alkanes are nonreactive compounds.
Analysis of BAL fluid specimens
Prior to analysis, all BAL fluid specimens were mixed by a rotating mixer for at least 30 min. BAL fluid specimens, QCs, and water blank samples were transferred to standard 10-mL headspace vials as 3-mL aliquots via separate 5-mL Luer lock syringes fitted with disposable needles. Specimens with insufficient quantity were diluted no more than a factor of 15 requiring at least 0.2 mL. For these samples, LLR was raised proportionally. Each sample was immediately spiked with the working internal standard solution, and capped. Sample quantities were verified gravimetrically.
Results
Chromatographic separation
We spiked pooled BAL fluid samples with the target analytes to examine their chromatographic separation in matrix (Figure 2). This was done to ascertain that there were no overlapping peaks, and to optimize sample injection volume and temperature gradient. All target analytes eluted from 11 – 18 minutes. In addition, we show the chromatogram obtained from an actual case control (Figure 3).
Figure 2.
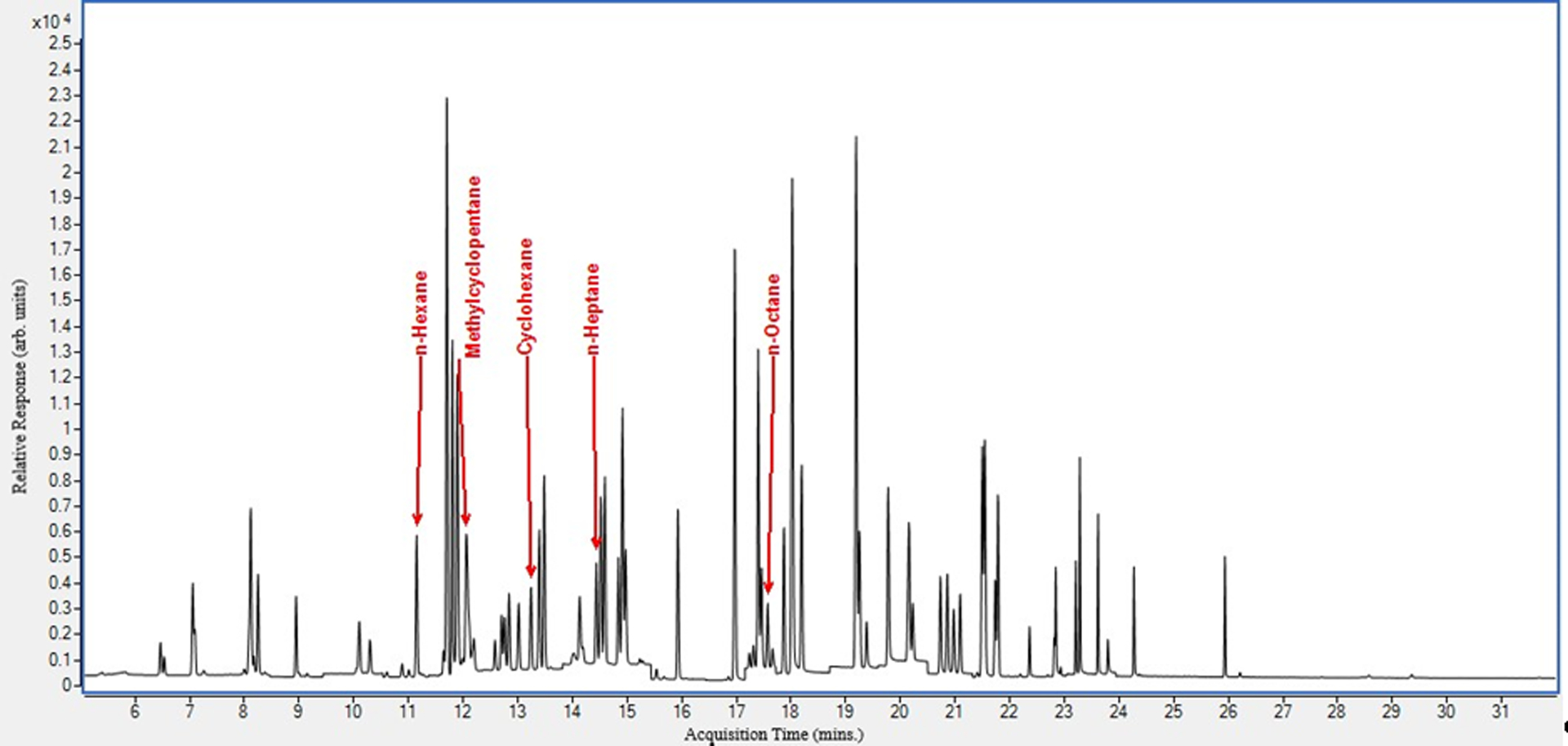
Spiked pooled BAL fluid sample chromatogram.
Figure 3.
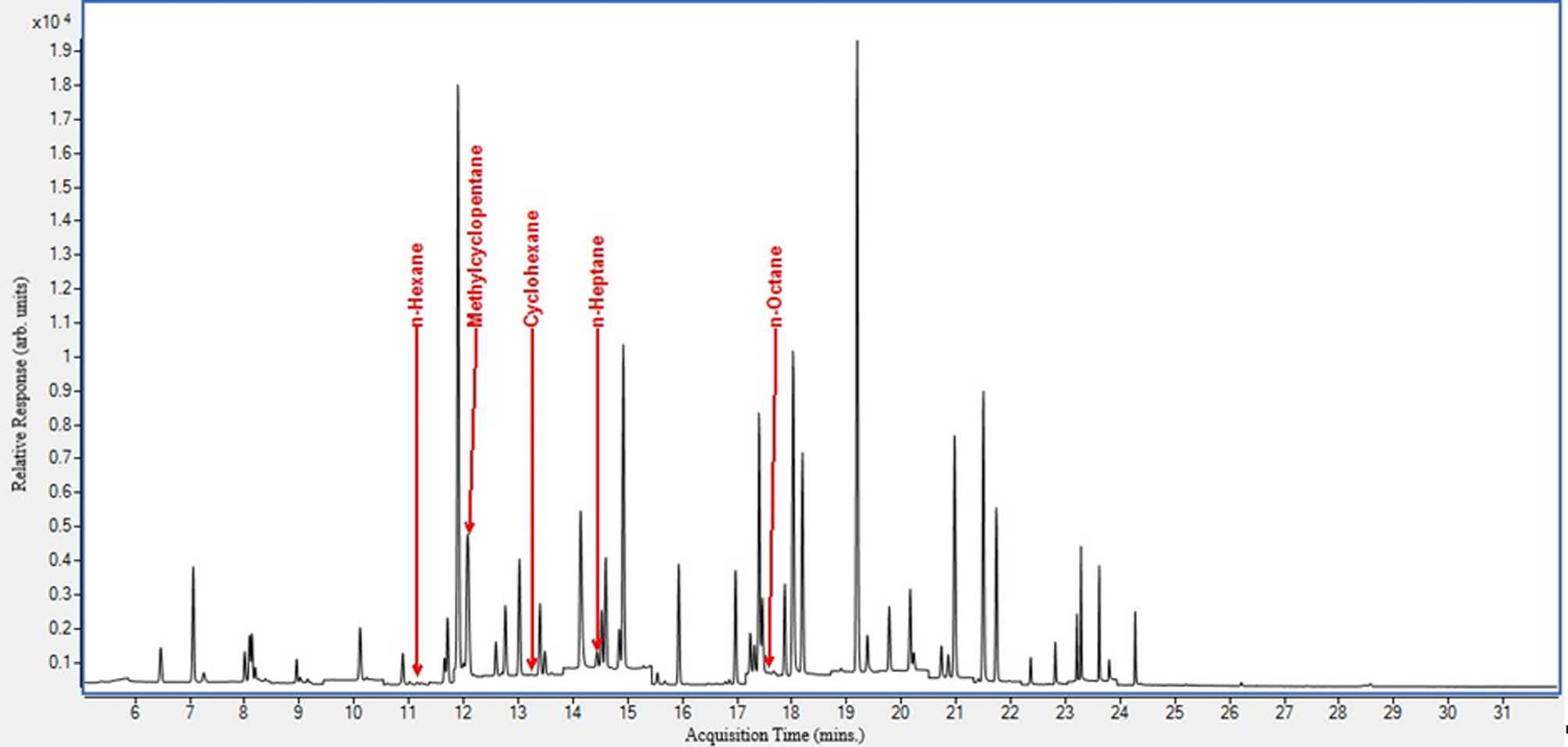
EVALI case control BAL fluid specimen chromatogram.
Matrix validation
We performed matrix validation experiments to verify that calibration curves in BAL fluid and water had the same slope (Table 3). Characterization was performed once at nine different concentrations distributed across the analytical range. Acceptance criteria included slope differences less than 5%. In addition, absolute internal standard response for these two matrices were within 20% ranging from 0.81 (cyclohexane, water solubility = 55,000 μg/L) to 1.00 (octane, water solubility = 660 μg/L), which is expected for nonpolar compounds.
ccuracy
Recovery accuracy was evaluated by comparison of nine different spiked levels in pooled BAL fluid specimen matrix with spiked water reference standards in which both samples were prepared similarly and at concentrations ranging from 0.044 to 59 μg/L (Table 4). The internal standard is used to adjust for any SPME vial headspace concentration biases caused by changes in matrix composition that effect compound solubility and absorption competition effects involving the SPME fiber coating.11 Because the pooled BAL fluid specimen matrix had background levels above LLR for some of the analytes, background correction was performed.
Precision
Precision within a run was evaluated by repetitive analysis of separately prepared saline matrix standards that were analyzed independent of the calibration curve standards at three concentration levels. For this analysis, six low (with concentrations that yielded a signal-to-noise of at least 5), medium and high concentration standards were analyzed within the same acquisition (Table 5). The method precision demonstrated with this experiment ranged from 2.75 to 18.1%.
Linearity
We created individual analyte calibration curves using at least six calibrators (phosphate-buffered saline) and analyzed them six times. R2 was ≥0.993. Acceptance criteria were R2 values >0.99. Calibration curves trended linearly as shown in the example given in Figure 1. Error bars are not shown in Figure 1 because they are small and obscured the symbols.
Limits of detection, analytical specificity
Limits of detection are presented in Table 6. In addition, analytical specificity was established by confirming similarity (within 25%) of quantified amount for the quantitation and confirmation ions. Additional steps taken to achieve analytical specificity involved removing interfering compounds from the sample analysis system. Interferences that have their source in the measurement apparatus itself were examined by measuring instrument blanks. Moreover, we checked for interferences in at least six BAL fluid specimens. All analytes passed the interference check.
Discussion
We successfully developed and validated a method to quantify petroleum distillates in BAL fluid specimens using GC/MS. The assay, based on a previously published method4,7, provided accurate and robust analyses of controls and case study BAL fluid specimens in support of CDC’s EVALI response. Case study results are available in a published report by Blount et al2.
Interpreting measurements of chemicals in BAL fluid presents several challenges. The collected volume of BAL fluid specimens obtained from a patient can vary according to technique of bronchial washing utilized. Thus, expressing concentrations per volume of BAL fluid may not be acceptable to quantitatively compare results among individuals. Furthermore, some of our targeted analytes have relatively poor solubility in normal saline, and thus collected BAL fluids may partition into multiple phases and complicate precise measurement. Other than normal saline, BAL fluid should contain the epithelial lining fluid and anything that is in it. BAL fluid that is contaminated with blood will be red tinged and interpretation of BAL fluid concentrations must consider blood as a potential source for analytes. In addition, BAL fluid specimen collection and handling prior to analysis must address the volatile nature of the target analytes. Since BAL fluid specimens are not typically assayed for the presence of VOCs, the specimens may not have been hermetically sealed upon collection. In addition, this method was developed to support a public health emergency, thus specimens were handled following existing protocols. These protocols, as described above, may not specifically address the need to keep hermetic conditions throughout the collection and shipping process. We showed, however, that the target analytes can be detected in BAL fluid when handled appropriately.
This analytical approach is applicable for the determination of other volatile petroleum distillate VOCs in BAL fluid specimens with detection limits in the parts-per-trillion range. Of note, the chosen analytes only represent a fraction of the wide variety of VOCs present in petroleum distillates. Other VOCs (e.g., aromatic hydrocarbons) were not examined. The analysis of petroleum distillate VOCs in BAL fluid at parts-per-trillion levels is an extremely complex measurement. However, the analysis of common petroleum distillates in BAL fluid was shown to be comparable to their analysis in blood in which the 95th percentiles are below detection12. There are no alternative analysis approaches that achieve the combined sensitivity and specificity for the compounds described in this method.
Footnotes
Disclaimer: The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Use of trade names in for identification only and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service, or the U.S. Department of Health and Human Services.
References
- 1.Outbreak of Lung Injury Associated with the Use of E-Cigarette, or Vaping, Products. 2019; https://www.cdc.gov/tobacco/basic_information/e-cigarettes/severe-lung-disease.html?s_cid=osh-stu-home-spotlight-006. Accessed November 22, 2019.
- 2.Blount BC, Karwowski MP, Shields PG, et al. Vitamin E Acetate in Bronchoalveolar-Lavage Fluid Associated with EVALI. N Engl J Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blount BC, Karwowski MP, Morel-Espinosa M, et al. Evaluation of Bronchoalveolar Lavage Fluid from Patients in an Outbreak of E-cigarette, or Vaping, Product Use-Associated Lung Injury - 10 States, August-October 2019. MMWR Morb Mortal Wkly Rep. 2019;68(45):1040–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blount BC, Kobelski RJ, McElprang DO, et al. Quantification of 31 volatile organic compounds in whole blood using solid-phase microextraction and gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;832(2):292–301. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey. https://www.cdc.gov/nchs/nhanes/.
- 6.U.S. Centers for Disease Control and Prevention. National Report on Human Exposure to Environmental Chemicals; Update Tables, January 2019. In:2019.
- 7.Chambers DM, Blount BC, McElprang DO, Waterhouse MG, Morrow JC. Picogram measurement of volatile n-alkanes (n-hexane through n-dodecane) in blood using solid-phase microextraction to assess nonoccupational petroleum-based fuel exposure. Analytical Chemistry. 2008;80(12):4666–4674. [DOI] [PubMed] [Google Scholar]
- 8.Chambers DM, McElprang DO, Mauldin JP, Hughes TM, Blount BC. Identification and elimination of polysiloxane curing agent interference encountered in the quantification of low-picogram per milliliter methyl tert-butyl ether in blood by solid-phase microextraction headspace analysis. Anal Chem. 2005;77(9):2912–2919. [DOI] [PubMed] [Google Scholar]
- 9.Westgard JO, Barry PL, Hunt MR, Groth T. A multi-rule Shewhart chart for quality control in clinical chemistry. Clin Chem. 1981;27(3):493–501. [PubMed] [Google Scholar]
- 10.Taylor JK, Tranter RL. Quality assurance of chemical measurements. Lewis Publishers; 1987. [Google Scholar]
- 11.Pawliszyn J Solid phase microextraction : theory and practice. New York: Wiley-VCH; 1997. [Google Scholar]
- 12.CDC. Fourth Report on Human Exposure to Environmental Chemicals. In. Vol 2. Atlanta, GA: Department of Health and Human Services, Centers for Disease Control and Prevention; 2019. [Google Scholar]