

Summary
Background:
Aspirin is associated with decreased risk of colorectal cancer (CRC), potentially by modulating the gut microbiome.
Aim:
To evaluate the effect of aspirin on the gut microbiome in a double-blinded, randomised placebo-controlled pilot trial.
Methods:
Healthy volunteers aged 50-75 received a standard dose of aspirin (325 mg, N = 30) or placebo (N = 20) once daily for 6 weeks and provided stool samples every 3 weeks for 12 weeks. Serial measurements of gut microbial community composition and bacterial abundance were derived from 16S rRNA sequences. Linear discriminant analysis of effect size (LEfSe) was tested for between-arm differences in bacterial abundance. Mixed-effect regression with binomial distribution estimated the effect of aspirin use on changes in the relative abundance of individual bacterial taxa via an interaction term (treatment × time).
Results:
Over the study period, there were differences in microbial composition in the aspirin vs placebo arm. After treatment, four taxa were differentially abundant across arms: Prevotella, Veillonella, Clostridium XIVa and Clostridium XVIII clusters. Of pre-specified bacteria associated with CRC (n = 8) or aspirin intake (n = 4) in published studies, interactions were significant for four taxa, suggesting relative increases in Akkermansia, Prevotella and Ruminococcaceae and relative decreases in Parabacteroides, Bacteroides and Dorea in the aspirin vs placebo arm.
Conclusion:
Compared to placebo, aspirin intake influenced several microbial taxa (Ruminococcaceae, Clostridium XIVa, Parabacteroides and Dorea) in a direction consistent with a priori hypothesis based on their association with CRC. This suggests that aspirin may influence CRC development through an effect on the gut microbiome. The findings need replication in a larger trial.
1. INTRODUCTION
Aspirin, an accessible and affordable drug, is one of the most intriguing prospects for cancer prevention. In 2016, the U.S. Preventive Services Task Force recommended using low-dose aspirin (81 mg) to reduce cardiovascular disease (CVD) and colorectal cancer (CRC) risk among adults aged 50-59 with a 10% increased CVD risk.1 Currently, more than 30% of adults over the age of 40 reported taking low-dose aspirin for primary or secondary CVD prevention; this translates into almost 39 million aspirin users in the U.S.2 In addition to preventing CVD, aspirin use may protect against CRC development. In randomised clinical trials, aspirin use was associated with a reduction in CRC risk by 24%-38%.3 However, aspirin has not been recommended for population-based prevention in healthy people because of aspirin’s side effects (eg gastrointestinal bleeding and cerebral haemorrhage) in some individuals and lack of understanding about population subgroups in which aspirin decreases CRC risk but does not cause adverse reactions.4,5 Tailoring aspirin to individuals who will especially benefit from using this drug for CRC prevention is one of the current priorities in cancer research in the U.S.6
Aspirin modulates inflammation and immune response via cyclooxygenase (COX)-dependent and -independent mechanisms.7–9 However, the exact mechanisms through which aspirin reduces risk of colorectal neoplasia and the molecular targets of aspirin in the context of CRC prevention have not been established. Data from animal studies have revealed that aspirin influences the gut microbiome either indirectly via an immune mechanism or directly via a local effect.10–14 Moreover, it was shown in humans that aspirin and other salicylate may inhibit the growth of pro-inflammatory bacteria in a dose-dependent manner.15 Despite these findings, surprisingly little research has been conducted in humans. An observational study in Finland suggested that aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) may alter the composition of the intestinal microbiome and partially counteract an increase in unfavourable bacterial taxa inhabiting the gut at older age.16 In addition, in a cross-sectional study of 150 individuals taking NSAIDs, four taxa (Prevotella species, Bacteroides species, Barnesiella species and Ruminococcaceae family) discriminated aspirin users from non-users (AUC = 0.96; 95% CI 0.84–1.00).17 These studies provide compelling evidence for aspirin’s role in modulating gut bacteria, but have been limited by their cross-sectional design.
In the present study, we addressed these gaps using a randomised placebo-controlled design to test the extentto which aspirin changes gut microbial composition. Given aspirin’s anti-inflammatory effect and its protective role in CRC, we hypothesised that aspirin alters the composition of gut microbiome in a way consistent with decreased inflammation and reduced CRC risk. Specifically, aspirin intake may lead to lower relative abundance of pro-inflammatory gut bacteria associated with CRC (e.g., Streptococcus) and higher abundance of anti-inflammatory/anti-CRC bacteria (eg butyrate-producing bacteria). Thus, we conducted a double-blinded, placebo-controlled, randomised trial of 50 healthy volunteers who took aspirin or placebo for 6 weeks and collected stool samples before, during and after the intervention. The objective of this pilot trial was to compare the between-arm changes in microbiome composition and pre-specified bacterial taxa associated with CRC and with inflammation in previous studies.17,18
2. METHODS
The trial was registered at www.clinicaltrials.gov NCT02761486. The study protocol was approved by the Institutional Review Board at the University of Minnesota, and all participants provided written informed consent.
2.1. Study design
This randomised, placebo-controlled, double-blinded study targeted 50 healthy subjects, between 50 and 75 years old who lived in the greater Twin Cities area. Participants were recruited from 1056 individuals who had previously given consent to be contacted for future studies after participating in two CRC-related studies: Evaluation of SEPT9 Biomarker Performance for Colorectal Cancer Screening (PreSEPT, NCT00855348) and Validation and Comparison of Biomarkers for the Early Detection of Colorectal Adenocarcinoma (BCCD, NCT01511653). Of these, 350 were willing to participate, but only 50 met eligibility criteria (Figure 1). Exclusion criteria for this study included: use of any antibiotic prescription in the last 3 months; use of any NSAIDs > 2 times a week in the last 3 months; use of antiplatelet or anticoagulant medication, medications for diabetes or hypertension within the past 30 days; gastrointestinal (GI) cancer or any serious GI condition or surgery within 6 months; any serious active medical (cancer, CVD) or psychiatric illness; BMI ≥ 40 or ≤ 17 kg/m2; unexplained change in weight of > 4.5 kg within the past 6 months; or major changes in eating habits within the past 3 months. Eligibility was further confirmed at baseline visit (Visit 1). At Visit 1, 50 eligible subjects signed a consent form and were randomised to the aspirin (N = 30) or placebo (N = 20) arm according to a 5-block randomisation scheme. We used unequal between-arm allocation to increase precision in the aspirin arm, because within-arm changes over time were deemed more important in aspirin than in the placebo arm. The duration of treatment (6 weeks) was based on trial of mesalazine for irritable bowel syndrome (IBS), in which mesalazine taken for 4 weeks altered microbiome composition, with this effect being reversed after a 4-week washout.19 We considered that our relatively healthy participants may have a microbial community more resistant to change than those with IBS, and thus we extended the duration of treatment (including placebo) and washout to 6 weeks.
FIGURE 1.
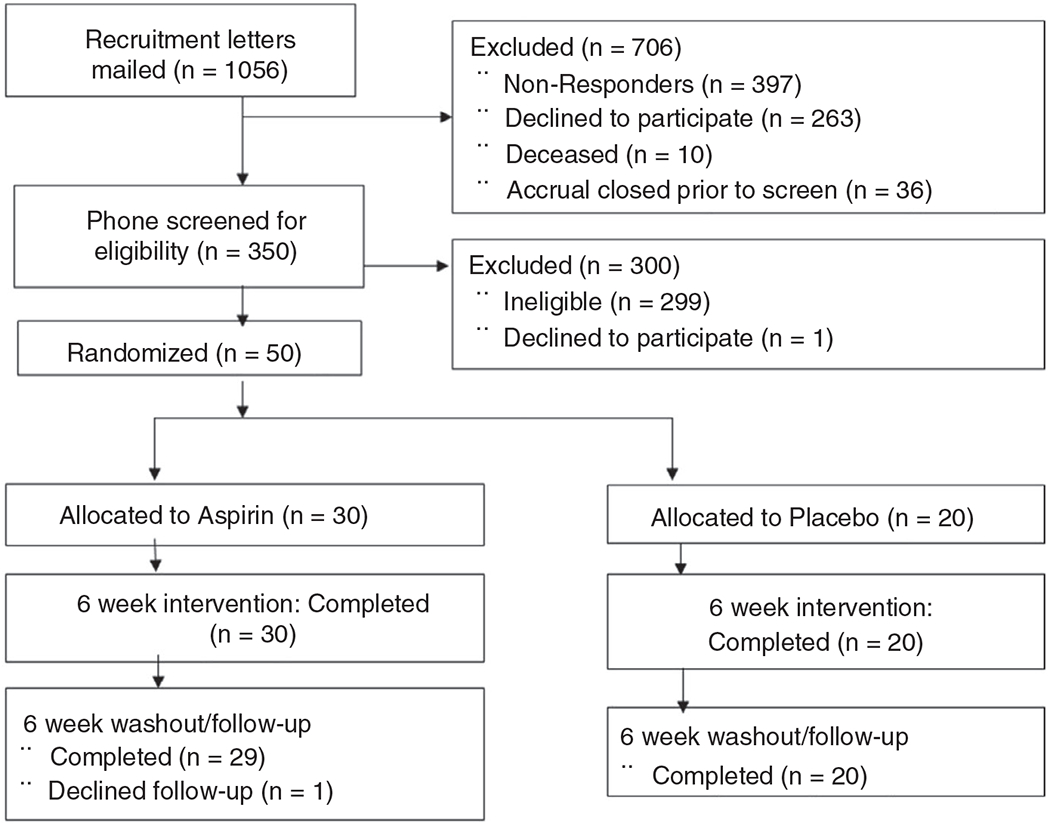
ASMIC study flow chart. Screening, enrolment and study completion
Additionally, at baseline (Visit 1), a trained technician measured subjects’ height and weight and asked a series of questions about health, medication use, and diet. The dietary questions were similar to the brief dietary questionnaire used in the Human Microbiome Project20 (shown in Table S1). The same questions were repeated after the 6-week treatment period (Visit 2). Visits were followed up with five phone calls at 3-week intervals (Figure 2) to ask about changes in health status and possible adverse events and to discuss subjects’ upcoming stool collection. Each participant collected five stool samples (every 3 weeks) and two urine samples, one before (baseline) and one after treatment (Week 6). Forty-nine subjects completed the study and provided five stool samples; one subject quit after completing the treatment phase and providing three stool samples. Study participants and all study staff (except the study statistician and pharmacist) were blinded to the treatment given; the pharmacist and the statistician knew the contents of medication bottles through a blind code.
FIGURE 2.
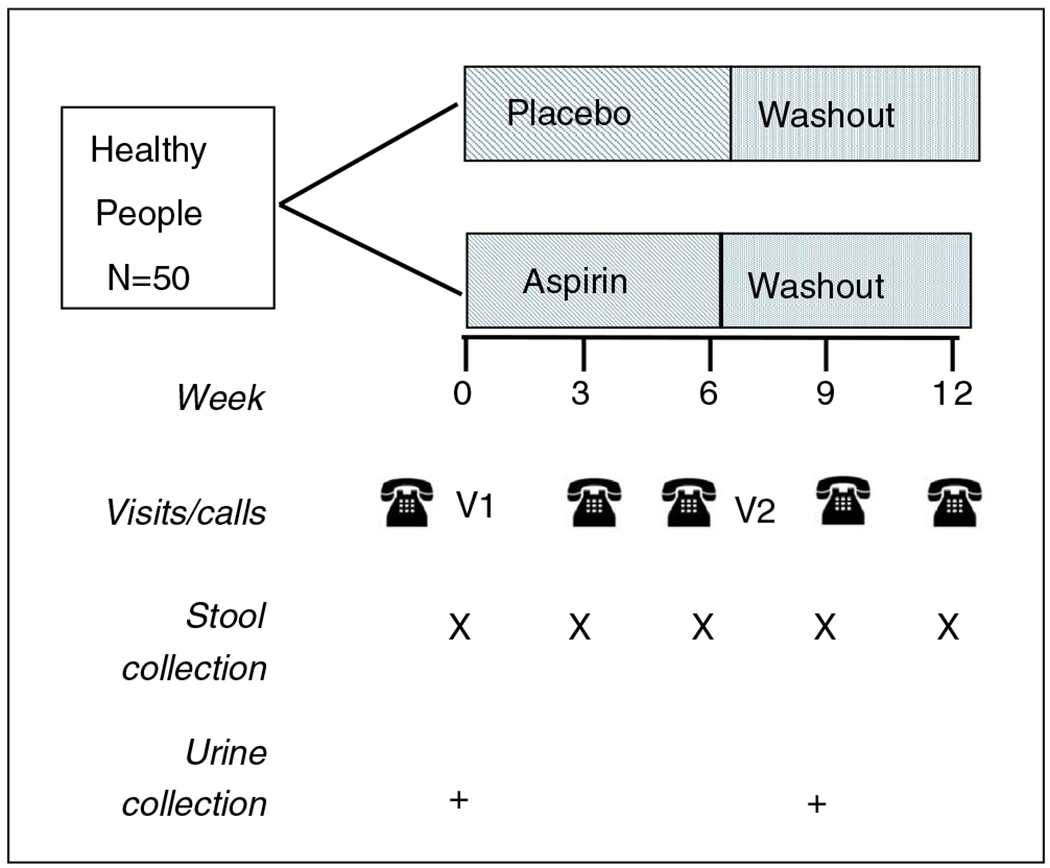
Diagram of interventions and sample collection. Of note, V1 is Visit 1 (baseline); V2 is Visit 2 (6 weeks)
2.2. Sample collection and laboratory analysis
Urine and stool samples were collected at home by participants. Stool collection kits contained 95% ethanol. Both sample types were put on ice and transported by local courier within 72 hours, before being put into −80°C freezer until needed for analysis. The samples from all collections were processed in one batch. DNA was extracted from 250-500 mg of stool using the PowerSoil DNA Isolation Kit (MoBio/Qiagen) following the manufacturer’s instructions. The V4 hypervariable region of the 16S rRNA gene was amplified and sequenced on a MiSeq (2 × 300 Paired-end; Illumina) using the 515F-806R primer set,21 containing dual indices by the University of Minnesota Genomics Center (UMGC).22 Negative (sterile water) controls were included and did not produce amplicons. Sequence data were deposited in the Sequence Reach Archive of the National Center for Biotechnology Information under BioProject accession number SRP127801.
We also measured urinary PGE-M, the urinary metabolite of prostaglandin E2. PGE-M is an inflammatory biomarker shown to be increased in CRC.23–25 Urinary PGE-M levels are reduced in aspirin users because PGE-M reflects the production of systemic PGE2, which is suppressed by aspirin.26–28 Pre-post treatment change in PGE-M levels not only allowed us to estimate the correlation between changes in PGE-M and gut bacteria due to aspirin intake, but also to check for compliance with aspirin intake. PGE-M was analysed using high-performance liquid chromatography/mass spectrometry (HPLC/MS)29 and normalised for creatinine levels measured using a test kit from Enzo Life Sciences, thus expressed in ng/mg creatinine. Both PGE-M and urinary creatinine were measured in the Eicosanoid Core Laboratory at Vanderbilt University Medical Center.
2.3. Medication preparation and treatment compliance
Aspirin and placebo (lactose) capsules were compounded and packaged by Fairview Investigational Drug Services (IDS), according to Good Manufacturing Practices. Blind-coded study bottles of 50 pills containing either aspirin or placebo were prepared fora 6-week daily treatment according to the randomisation scheme. Compliance was assessed in two ways: (a) we compared the number of pills returned after treatment completion to the expected number based on treatment duration, and (b) we assessed between-arm difference in urinary PGE-M levels and their change after treatment.
2.4. Bioinformatics analysis
Sequence data were processed and analysed using Mothur ver. 1.35.1.30–32 The total number of sequences generated for the analysis was 11 159 032, and the median number of sequences per sample was 39 555. High-quality sequences were aligned against the SILVA database ver. 123,33 and chimeric sequences were identified and removed using UCHIME software.34 Samples were rarefied to 8800 sequence reads per sample to reduce bias in comparisons. To estimate the proportion of the gut microbiome operational taxonomic units (OTUs) represented in our samples, the mean Good’s coverage among baseline samples was calculated.
Clustering of OTUs was performed at 97% identity. Taxonomic classification was performed against the version 16 data release from the Ribosomal Database Project.35
2.5. Statistical analysis
Using intention-to-treat (ITT), our main analysis tested how aspirin affected (a) the microbiome community in general and (b) the relative abundance of pre-specif ied bacterial taxa that were identified in previous studies.17,18
2.5.1. Analysis of aspirin’s effect on the gut microbiome community
The properties of the global gut microbiome community in aspirin vs placebo arms at different time points were compared using Mothur ver. 1.35.1. Alpha diversity, a global measure of within-person microbial community diversity, was calculated using the Shannon index for five stool collections. The microbial composition of serial samples to the baseline sample was compared using SourceTracker (version 0.9.8).36 SourceTracker uses a Bayesian approach to infer the similarity of subsequent samples (called “sink” sample) to the baseline sample (called “source”) based on the posterior probabilities of sink OTUs coming from the source. The similarity of composition of a stool sample during treatment and washout to baseline sample was also compared using ANOVA with Tukey’s post-hoc comparisons.
To determine bacterial taxa with differential abundance across the aspirin and placebo arms at different collection times, we used linear discriminant analysis (LDA) of effect size (LEfSe).37 The significance level was set at LDA ≥ 2.0 and P < 0.05 for all LEfse analyses.
2.5.2. Analysis of aspirin’s effect on individual pre-specified bacteria
Individual bacterial taxa were tested if they were present in >10% of subjects at the study baseline and if they were associated with (a) CRC status in the meta-analysis of eight cross-sectional studies of stool microbiome in CRC cases and cancer-free individuals by Shah et al.18 or (b) aspirin in a cross-sectional study of NSAIDs and stool microbiome by Rogers et al.17 (Table S2). In total, we analysed 12 bacterial taxa. Eight genera were identified in the meta-analysis and included Akkermansia, Blautia, Dialister, Dorea, Faecalibacterium, Parabacteroides, Ruminococcus and Streptococcus.18 Four taxa discriminated aspirin-users from non-users in the study of Rogers et al. and included the genera Prevotella, Bacteroides and Barnesiella and the family Ruminococcaceae.17 Finally, we tested individual genera that were identified in our LEfSe analysis.
The main focus in the analysis was differences in between-arm changes of relative abundance (assessed in percent) of each individual taxon over time. Mixed-effects regression with binomial distribution incorporating an interaction term (treatment × time) was used to estimate pre-post treatment changes in the relative abundance of each pre-specified bacterium in separate arms and to test whether the changes in the aspirin arm differed from those in the placebo arm. The mixed-effects model included a random effect for person and a fixed effect for samples times at 0, and 6 weeks. For each bacterial taxon, an absolute count of that taxon was included as a weight in the model. Regression coefficients (β) for the interaction term and Wald p-values were estimated for the abundance of pre-specified faecal bacterial taxa in aspirin vs placebo arms after 3 weeks and 6 weeks of treatment vs baseline. Positive β coefficients indicate a larger increase in the relative abundance of the taxon in the aspirin arm or a smaller decrease in the aspirin arm as compared to the placebo arm for week 6 (or week 3) vs baseline, while negative β coefficients indicate a larger decrease or smaller increase in taxon abundance in the aspirin compared to placebo.
This was a pilot study and hence of small size, and because we expected a modest effect, we decided to focus on a small number of individual bacterial taxa identified in previous studies17,18 and chose not to apply corrections for multiple comparisons in the microbiome analysis. Thus, in all analyses of individual taxa, a two-sided P-value < 0.05 was considered statistically significant. All the analyses were conducted using R (version 3.4.3).
For 30 subjects in the aspirin group and 20 in the control group, we had 80% power (two-sided α = 0.05) to detect between-arm difference of 8%, 17% and 40% in relative abundance of taxa with standard deviation of 0.1, 0.2 and 0.5%, respectively.38
2.5.3. Analysis of aspirin’s effect on the urinary inflammatory biomarker – PGE-M
We estimated the effect of aspirin on PGE-M by fitting an ANCOVA type model: the change in PGE-M was the outcome, the treatment group was an explanatory variable, and the model was adjusted for baseline (pre-treatment) PGE-M levels. We also estimated correlations between the changes in pre-specified bacterial taxa and PGE-M using Pearson correlation and p-values.
3. RESULTS
3.1. Patient characteristics at baseline
Successful randomisation of 30 subjects to aspirin and 20 to placebo is evidenced by the fact that there were no statistically significant differences in gender, age, BMI or PGE-M in the aspirin vs placebo arms after controlling for multiple comparisons (Table 1).
TABLE 1.
Participants’characteristics at baseline, the ASMIC study
| Participant characteristics at baseline | Aspirin N = 30 | Placebo N = 20 | P-valuea |
|---|---|---|---|
| Age, Mean (SD) y | 62.2 (5.1) | 61.2 (5.2) | 0.74 |
| Gender, Female; n (%) | 23 (76.7%) | 9 (45.0%) | 0.19 |
| BMI, Mean (SD), kg/m2 | 27.3 (4.3) | 28.2 (4.8) | 0.74 |
| Urinary PGE-Mb (adjusted for creatinine) Mean (SD) y | 11.82 (13.59) | 12.94 (7.39) | 0.74 |
P-value were adjusted for multiple inference using the PROC MULTTEST function based on the raw P-values for the continuous variable and the Yates corrected p-value for the categorical P-value.
PGE-M - metabolite of prostaglandin E2.
3.2. Treatment compliance
Treatment compliance was high as measured by number of returned pills and changes in PGE-M. The mean number of pills taken by participants was 42.3 (SD = 3.49); 94% (47 participants) had at least 90% pill compliance, with 60% of those achieving 100% compliance. The lowest compliance reported in the study was 74%. Treatment compliance was also verified by testing levels of urinary PGE-M pre-and post-treatment. As expected, mean levels of PGE-M were similar across the arms at baseline; however, after treatment, PGE-M levels decreased in the aspirin arm (p = 0.001), while stayed the same in the placebo arm (Table 2).
TABLE 2.
Urinary prostaglandin E metabolite (PGE-M) adjusted for creatinine [mean (standard deviation)] before and after treatment in the aspirin and placebo arm. [Correction added on August 28, 2020 after first online publication: The 1st column heading was revised.]
| Time of sample collection | Aspirin | Placebo | P-value | |
|---|---|---|---|---|
| PGE-Ma per creatinine, ng/mg | Before treatment | 12.29 (13.82) | 12.94 (7.39) | 0.74 |
| After treatment | 6.38 (4.77) | 13.30 (6.63) | 0.001 | |
| Change after treatment | −5.91 (11.15) | 0.36 (6.98) | 0.07 |
PGE-M - metabolite of prostaglandin E2.
3.3. Gut microbiome composition before, during and after treatment with aspirin and placebo
The mean Good’s coverage among all baseline samples was 98.6 ± 0.1%, suggesting that the majority of the known bacterial community was captured. Alpha diversity as measured by Shannon’s index did not vary significantly by treatment or sample collection (p = 0.32) (Table S3).
Figure 3 depicts the similarity of gut microbiome composition from serial stool samples to the baseline collection using SourceTracker analysis. The microbiome samples in the placebo arm maintained a significantly greater similarity to the baseline sample compared to the samples in the aspirin arm (ANOVA p = 0.009). Likewise, the microbiome composition from samples collected during and after treatment (weeks 3 and 6) was more similar to baseline in the placebo than aspirin arm (p = 0.01). Thus, aspirin is likely to drive divergence in the microbiome from baseline. During mid-wash-out, the microbiome composition (9 weeks) still differed between arms, but at the end of washout (week 12), the composition of the gut microbiome was similar in the aspirin and placebo arms, suggesting that the effect of treatment was short-term. Though similar at 12 weeks, the post-washout composition differed from the baseline microbiome composition in both arms.
FIGURE 3.
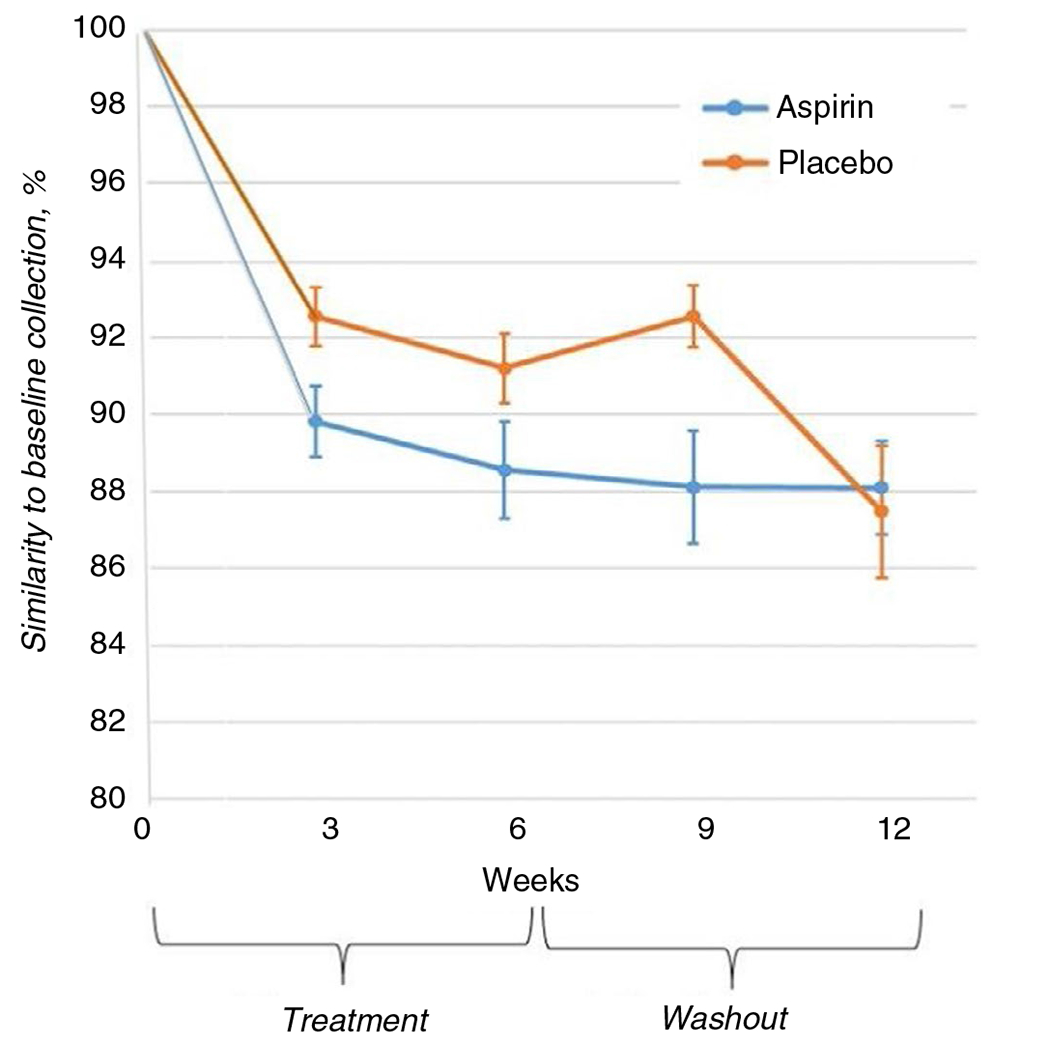
Similarity of the microbiome compositions between serial stool samples and the baseline sample using SourceTracker analysis36
The LEfSe analysis at the end of treatment (week 6) found several bacterial genera that were differentially abundant between arms: Prevotella, Akkermansia, Clostridium XIVa cluster and Clostridium XVIII cluster (Table 3). When LEfSe analysis was applied to baseline, before treatment, only one genus – Phascolarctobacterium – was differently abundant between arms (LDA = 4.40, p = 0.01, not shown in tables). The higher relative abundance of baseline Phascolarctobacterium was found in the aspirin vs. the placebo arm, most likely because of imperfect randomisation; however, the LEfSe analysis did not show any difference in the abundance of this bacterium after treatment.
TABLE 3.
Linear discriminant analysis (LDA) effect size (LEfSe) in samples collected after treatment (Week 6)
| Bacterial taxa | Comparison of aspirin to placebo arm, Week 6 | ||
|---|---|---|---|
| Class | LDA | P-value | |
| Clostridium XIVa | Aspirina | 3.66 | 0.035 |
| Clostridium XVIII | Placeboa | 3.49 | 0.005 |
| Prevotella | Aspirin | 3.38 | 0.004 |
| Veillonella | Placebo | 3.41 | 0.047 |
Class “Aspirin” means that the bacterium was more prevalent in the aspirin arm, and class. “Placebo” means that bacterium was more prevalent in the placebo arm.
3.4. Pre-specified bacteria before, during and after treatment with aspirin and placebo
Of the eight pre-specified genera associated with CRC in the meta-analysis,18 in the mixed-effects analysis, regression coefficients (β estimates) for the interaction term were significant at three and six weeks of treatment vs baseline for two genera: Parabacteroides (β = −0.43, P < 0.0001 for week 3 vs baseline) and Akkermansia (β = 0.30, P = 0.009 for week 6 vs baseline) (Table 4). The regression coefficients were also significant for Dorea (β = −1.57, P = 0.02) and Ruminococcus (β = −0.63, P = 0.03) at week 6 vs baseline (but not at week 3) and marginally significant for Faecalibacterium (β = 0.21, P = 0.05) at week 3 vs baseline.
TABLE 4.
Regression coefficients (β) and P-values for the interaction term comparing the abundance of pre-specified faecal bacterial taxa in aspirin to placebo arm after 3 weeks (upper row) and 6 weeks (lower row) of treatment vs baseline, the ASMIC study
| Bacterial taxa | Regression coefficient βa | P-value | Association with CRC reported in literature |
|---|---|---|---|
| Bacterial taxa differentially abundant in CRC cases and controls in the meta-analysis18 | |||
| Akkermansia | 0.30b 0.22 |
0.002 0.009 |
↑18,50 |
| Blautia | 0.15 0.31 |
0.40 0.10 |
↑18 |
| Dialister | 0.13 0.52 |
0.65 0.27 |
↑18 |
| Dorea | −0.16 −1.57c |
0.77 0.02 |
↑18 |
| Faecalibacterium | 0.21c 0.16 |
0.05 0.16 |
↓18 |
| Parabacteroides | −0.42c −0.43c |
0.0003 0.0002 |
↑18,40 |
| Ruminococcus | |||
| 1 | 0.01 −0.63 c |
0.96 0.03 |
↓18,40 |
| 2 | −0.42 −0.30 |
0.39 0.56 |
|
| Streptococcus | 0.59 0.41 |
0.38 0.49 |
↑18,50,51 |
| Bacterial taxa that discriminated aspirin and non-aspirin users17 (direction was not reported) | |||
| Bacteroides | −0.31b,c −0.39 |
<2.0 e-16 <2.00e-16 |
↑↓18,40,50,51 |
| Barnesiella | 0.23 0.12 |
0.37 0.61 |
Unclear17 |
| Prevotella | 0.87 bc 0.51 |
< 2.0e-16 4.66e-10 |
↑↓18,40 |
| Ruminococcaceae | 0.25c 0.33 |
<6.16e-13 < 2e-16 |
↓18,40 |
| Bacterial taxa that were differentially abundant in the aspirin vs placebo in our study | |||
| Clostridium XlVa | 0.44 b | 0.0008 | ↓39 |
| Clostridium XVIII | 0.27b | 0.70 | ↓64 |
| Prevotella | 0.51b,c | 4.66e-10 | ↑↓18,40 |
| Veillonella | Model did not converge | ↑65 | |
Positive regression coefficient (β) implies an increase in the abundance of the bacterial taxon, while negative, a decrease in the taxon abundance for 6 weeks (or three weeks) of treatment vs baseline in the aspirin compared to the placebo arm.
The association between the taxon and aspirin in our study is in the direction opposite to the association between that taxon and CRC status as reported in previous studies.
The association between the taxon and aspirin in our study is in the direction consistent with the association between the taxon and CRC status as reported in previous studies.
Three of four bacterial taxa that were previously associated with aspirin in the cross-sectional analysis17 also changed differentially in aspirin compared to placebo in our trial: Ruminococcaceae family (β = 0.33, P < 0.0001) and Bacteroides (β = −0.39; P < 0.0001) and Prevotella genera (β = 0.51; P < 0.0001); all the estimates are for week 6 compared to baseline (Table 4). Lastly, the mixed-effects analysis confirmed the associations for two bacteria, Clostridium XIVa cluster and Prevotella, that were differentially abundant after treatment, as shown by the LEfSe analysis (Table 3).
The bar plots showing relative abundances and 95% confidence intervals (CI) of pre-specified bacterial taxa significantly associated with treatment are presented in Figure 4. Unexpectedly, the relative abundance of several taxa (Bacteroides, Clostridium XIVa cluster, Parabacteroides, Prevotella and Ruminococcaceae) changed in the placebo even more markedly than in the aspirin group. These findings suggest that either aspirin counteracted the change in these bacteria or the changes (eg decrease) in the relative abundance of some bacteria in the placebo arm may be driven by changes in other taxa (increase) in the aspirin arm. Lastly, no correlation was found between the changes in the pre-specified bacteria and PGE-M (not shown).
FIGURE 4.
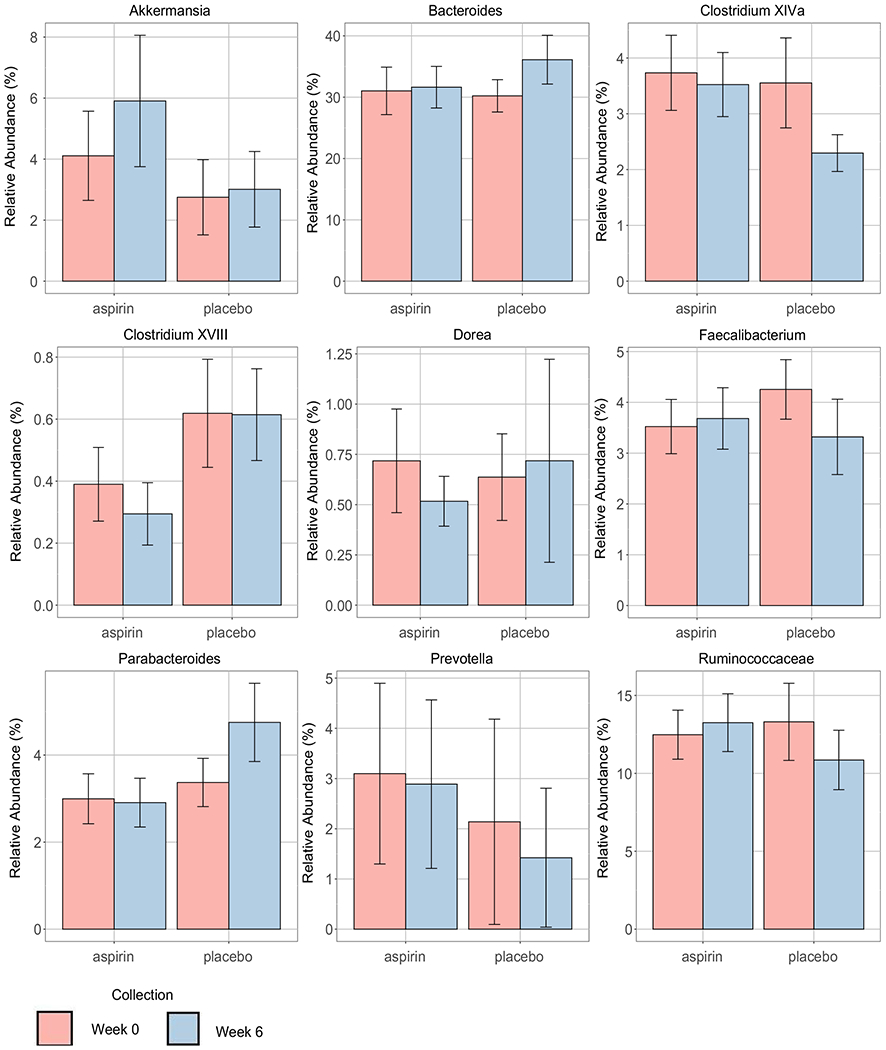
The mean relative abundance (in percent) and 95% confidence intervals (CI) of pre-specified taxa for post- vs pre-treatment in the aspirin and placebo arms. Post-treatment is at Week 6 (blue colour), and pre-treatment, is at baseline Week 0 (pink colour)
4. DISCUSSION
This double-blinded, placebo-controlled, randomised pilot trial of aspirin vs placebo found that, overall, bacterial community composition in stool samples collected during and after treatment was more similar to the pre-treatment samples in the placebo than in the aspirin. This finding suggests that aspirin induces changes in the gut microbiome.
Furthermore, the post-treatment relative abundance of four bacterial taxa differed between the aspirin and placebo arms with the associations for the Clostridium XIVa cluster and Prevotella being confirmed by mixed-effects analysis. An additional finding that gives credibility to our results is that we detected changes in three of four taxa that discriminated aspirin users from non-users in a previous cross-sectional study of NSAIDs use.17 Finally, in our analysis of individual pre-specified bacteria, the direction of changes for Ruminococcaceae, Parabacteroides, Clostridium XlVa and Dorea agree with the associations reported in the meta-analysis of individuals with and without CRC in terms of depicting a lower abundance of Ruminococcaceae and Clostridium XIVa cluster and a greater abundance of Parabacteroides and Dorea for those with CRC.18,39,40 These findings are concordant with our hypothesis that aspirin may decrease CRC risk partially through its effect on bacteria. In addition, we showed that the abundance of Akkermansia increased more in the aspirin compared to the placebo group, which is in line with anti-inflammatory role of this bacterium in cancer-free individuals.41–43
Our findings that aspirin alters the gut microbiome in humans are biologically plausible. They are consistent with animal studies that showed that aspirin impacts bacterial growth at concentrations within the range utilised for human therapy (5 mg/kg of body weight).13,14 Additional support comes from recent animal studies that found that two other NSAIDs, celecoxib44 and indomethacin45, induced changes in gut microbiota and their metabolites. In a pathway analysis of metabolites, the decrease in pro-carcinogenic bacteria in mice treated with celecoxib contributed to reduced intestinal polyp burden in normal and APCMm/+ mice.44 The authors concluded that NSAIDs’ effect on the gut microbiome may at least partially explain the anti-carcinogenic effect of NSAIDs.
The functional roles of bacteria explaining their associations with inflammation and CRC are postulated for some, but not all, of the bacteria mentioned above. For instance, bacteria in the Clostridium XIVa cluster participate in maintaining gastrointestinal functions by producing short-chain fatty acids and inducing colonic regulatory T (Treg) cells, both of which are known to be protective in inflammation and CRC development and progression.39,46 The Clostridium XlVa cluster and Ruminococcaceae are also reduced in those with inflammatory bowel disease (IBD) compared to those without disease,39,47 which agrees with their protective role in the gut inflammation and an anti-inflammatory effect of aspirin on the gut microbiome. Moreover, members of the family Ruminococcaceae degrade several types of polysaccharides in the lower gastrointestinal tract and facilitate the production of protective short-chain fatty acids 47,48 In contrast, one of the Ruminococcaceae family members, Ruminococcocus gnavus, was shown to be increased in IBD,48 which could explain an increased abundance of the Ruminococcocus genus in the aspirin vs placebo arm observed in our study. Additionally, a change in Faecalibacterium (although marginally statistically significant only at week 3 vs baseline), may be driven by the anti-inflammatory species Faecalibacterium prausnitzi.49
In addition to the hypothesised changes in the gut bacteria discussed above, we detected changes in other bacteria for which published findings were less consistent. Our findings for Bacteroides agree with data from some human40,50,51 and mouse studies,52,53 in which Bacteroides members (such as Bacteroides fragilis) have been linked to mucin degradation, intestinal inflammation and colorectal carcinogenesis. The differential associations for Bacteroides and Prevotella in various studies may be attributed to the large size of these genera that are characterised by high genomic diversity of strains and different functions of species.54
Additionally, findings from previous studies suggest that Akkermansia may play a different role in individuals with and without CRC. For instance, our findings for Akkermansia are consistent with data for cancer-free individuals that showed that Akkermansia may stimulate host mucosal anti-inflammatory pathways and improve epithelial barrier integrity, and thus decrease inflammation.41–43,55 However, the meta-analysis comparing gut microbiome in CRC individuals to those without, showed higher levels in Akkermansia muciniphila among those with CRC.18 It is possible that increased levels of Akkermansia muciniphila among CRC patients compared to cancer-free individuals may be related to an increased production of mucus which serves as a major nutrient source for this bacterium.56 The importance of mucus for Akkermansia muciniphila supports our finding of a greater increase in Akkermansia’s abundance in aspirin vs placebo users, because aspirin users, who do not have severe upper Gl complications, are likely to have increased mucin levels that contribute to host defense and potentially to a decrease in inflammation.57,58 Another possibility is that the inconsistent associations across various studies may reflect the complex nature of the microbiome in which detected changes in certain bacterial taxa may reflect changes in the whole system that we are unable to assess. For instance, in our study, because we examined the relative abundance of bacteria, some unexpected changes in the placebo arm (eg decrease in certain taxa) may be observed due to real changes (ie increase) in the relative abundance of other taxa in the aspirin arm. Aspirin could influence bacterial taxa via a systemic mechanism, local mechanism or both. A systemic mechanism of aspirin might involve the inactivation of cyclooxygenase, the suppression of prostaglandins, and the production of anti-inflammatory lipoxins, leading to the clearance of inflammatory bacteria by macrophages and other immune cells.59 A local mechanism may be explained by the deacetylation of aspirin and formation of salicylic acid in the liver and stomach and its permeation into human gastrointestinal tissue, where it comes in contact with enteric bacteria.12 If aspirin exerts its effect mainly through a local mechanism (ie influences bacteria through the absorption of salicylic acid into the tissue), it is understandable why we did not observe correlations between changes in PGE-M levels and bacterial taxa, since the urinary PGE-M levels reflect a decrease in systemic inflammation caused by aspirin.10 It is also possible that we did not see correlations with PGE-M because of variations in time between collecting stool and urine samples and taking aspirin across subjects. Noteworthy, the PGE-M levels increased in five of 30 people after taking aspirin, and four of those five individuals had high aspirin compliance (93%-100%), as derived from the number of returned pills. Thus, an increase in PGE-M among those individuals cannot be explained by low compliance and may reflect upper gastrointestinal inflammation caused by aspirin. The mechanism of aspirin’s effect on the gut microbiome should be examined in future studies with controlled time between sample collection and aspirin intake.
A major strength of our study is its novelty and robust design. For the first time, we provide evidence of aspirin-induced changes in the microbiome in a randomised study of healthy individuals with repeated measures of microbial composition. Inclusion of a control arm allowed us to reduce the influence of unmeasured confounders and account for normal changes over the study period that may affect the gut microbiome, because some bacteria may fluctuate even within several weeks, although, in general, the microbiome is considered stable within a period of 1–2 months.60–62 The main limitations of our study is the small number of subjects and modest duration that most likely account for small bacterial changes observed in our study. It also possible that sample size of 50 participants or a 6-week follow-up could be insufficient to observe modest aspirin-induced changes in some individual bacteria. Another limitation is that we cannot exclude that some changes in individual bacteria could have occurred due to temporal dietary changes. Unfortunately, we collected limited dietary information at baseline only and asked the participants not to make major dietary changes such as switching to vegetarian or other specific diet (eg carbohydrate or protein). Although participants reported no major dietary changes during follow-up, changes in individual dietary products cannot be excluded. In addition, given our healthy sample, we were unable to examine certain gut bacteria that are encountered mainly in CRC patients, such as Fusobacterium nucleatum, but rarely observed in heathy people.18,50,51 However, our main objective was to evaluate the influence of aspirin on bacteria predisposing to CRC, and we found that aspirin changed several bacterial taxa in a way consistent with reduced CRC risk. Finally, our placebo capsules consisted of lactose, and lactose could potentially affect the gut microbiome. Although it is highly unlikely that the amount of lactose in the placebo capsule (525 mg lactose per capsule) could have contributed to the observed changes in gut microbiota in the placebo arm because the amount of lactose in the standard American diet (12 gram per cup of milk) markedly exceeds the lactose amount in the placebo pill.63 In addition, aspirin capsules also contained lactose (162 mg). Finally, we lacked capacity to conduct metagenomics, which precluded examining bacteria at the species or strain levels. However, because this is a novel pilot study, we considered 16S RNA as an appropriate first step that will motivate a larger follow-up study that will use metagenomics to analyse gut microbiome.
In conclusion, our double-blind, randomised, placebo-controlled pilot trial suggests that aspirin induces changes in the gut microbiome, including changes in several bacteria previously shown to be associated with CRC or inflammation. Furthermore, due to normal changes in gut microbiome composition over time, our study demonstrated the critical importance of having a control arm in studies of changes in the gut microbiome. Although our findings for aspirin-induced changes in individual bacteria are preliminary, our results have potential public health implications given the large percentage (30%) of aspirin users.2 Our findings may inform the design of a large follow-up clinical trial that will examine the effect of different doses of aspirin on gut microbiome in individuals at high risk for CRC.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to study coordinators, Jennifer Stromberg and Allison Iwan, and other staff (Jeremiah Menk, Lori Strayer and Frank Strahan) who helped with the study. We thank the University of Minnesota Genomics Center for conducting the genetic analyses (sequencing data were processed and analysed using the resources of the Minnesota Supercomputing Institute); Eicosanoid Core Laboratory, University of Vanderbilt (Ginger Milne) for measuring urine biomarkers; Fairview Investigational Drug Services (Luke Darlette) for preparing treatment capsules; and Epidemiology Clinical Research Center (Margaret Krieser) for providing space for the recruitment of the study subjects. We are also grateful to Michael Franklin, a medical writer, for reviewing the paper. The study was registered using Clinical Trials Registration NCT02761486.
Funding information
The research was supported by the University of Minnesota Grant-in-Aid of Research, Artistry, and Scholarship program and the National Institutes of Health’s National Center for Advancing Translational Sciences, grant UL1TR002494. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health’s National Center for Advancing Translational Sciences.
APPENDIX A
AUTHORS’ COMPLETE AFFILIATIONS
Anna E. Prizment: Division of Hematology, Oncology&Transplantation, University of Minnesota, Minneapolis, MN, USA and Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA; Christopher Staley: Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA and Department of Surgery, University of Minnesota, Minneapolis, MN, USA; Guillaume C. Onyeaghala: Division of Epidemiology & Community Health, University of Minnesota, Minneapolis, MN, USA; Sithara Vivek: Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN, USA; Bharat Thyagarajan: Department of Laboratory Medicine and Pathology, University of Minnesota, Minneapolis, MN, USA; Robert J. Straka: Department of Experimental & Clinical Pharmacology, University of Minnesota, Minneapolis, MN, USA; Ryan T. Demmer: Division of Epidemiology & Community Health, University of Minnesota, Minneapolis, MN, USA and Department of Epidemiology, Columbia University, New York, NY, USA; Dan Knights: College of Biological Sciences, University of Minnesota, Minneapolis, MN, USA and College of Science and Engineering, University of Minnesota, Minneapolis, MN, USA; Katie A. Meyer: Department of Nutrition, University of North Carolina at Chapel Hill, NC, USA; Aasma Shaukat: Minneapolis VA Medical Center, Minneapolis, MN, USA, Division of Gastroenterology, University of Minnesota, Minneapolis, MN, USA and Division of Environmental Health Sciences, University of Minnesota, Minneapolis, MN, USA; Michael J. Sadowsky: Biotechnology Institute, University of Minnesota, Minneapolis, MN, USA and Department of Soil, Water, & Climate, University of Minnesota, Minneapolis, MN, USA; Timothy R. Church: Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA and Division of Environmental Health Sciences, University of Minnesota, Minneapolis, MN, USA.
Footnotes
Declaration of personal interests: DK serves as CEO of CoreBiome, a company involved in the commercialization of microbiome analysis. CoreBiome is now a wholly owned subsidiary of OraSure. These interests have been reviewed and managed by the University of Minnesota in accordance with its conflict-of-interest policies. All other authors do not have personal interests.
The Handling Editor for this article was Dr Colin Howden, and it was accepted for publication after full peer-review.
SUPPORTING INFORMATION
Additional supporting information will be found online in the Supporting Information section.
REFERENCES
- 1.USPSTF. Final Update Summary: Aspirin Use to Prevent Cardiovascular Disease and Colorectal Cancer: Preventive Medication.: U.S. Preventive Services Task Force; April 2016. https://www.uspreventiveservicestaskforce.org/Page/Document/UpdateSummaryFinal/aspirin-to-prevent-cardiovascular-disease-and-cancer?ds=1&s=aspirin; Accessed 10/01/2016 [Google Scholar]
- 2.Stuntz M, Bernstein B. Recent trends in the prevalence of low-dose aspirin use for primary and secondary prevention of cardiovascular disease in the United States, 2012-2015. Prev Med Rep. 2017;5:183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan AT, Arber N, Burn J, et al. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev Res (Phila). 2012;5:164–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng SL, Roddick AJ. Association of aspirin use for primary prevention with cardiovascular events and bleeding events: a systematic review and meta-analysis. JAMA. 2019;321:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Journal of the American College of Cardiology. 2019;74(10):1376–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaffee EM, Van Dang C, Agus DB, et al. Future cancer research priorities in the USA: a Lancet Oncology Commission. The Lancet Oncology. 2017;18(ll):e653–e706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang N, Bermudez EA, Ridker PM, Hurwitz S, Serhan CN. Aspirin triggers antiinflammatory 15-epi-lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci USA. 2004;101:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–1601. [DOI] [PubMed] [Google Scholar]
- 9.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW. Aspirin use and risk of fatal cancer. Cancer Research. 1993;53:1322–1327. [PubMed] [Google Scholar]
- 10.Stables MJ, Newson J, Ayoub SS, Brown J, Hyams CJ, Gilroy DW. Priming innate immune responses to infection by cyclooxygenase inhibition kills antibiotic-susceptible and -resistant bacteria. Blood. 2010;116:2950–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Upreti R, Kannan A, Pant AB. Experimental impact of aspirin expo-sure on rat intestinal bacteria, epithelial cells and cell line. Human Exp Toxicol. 2010;29:833–843. [DOI] [PubMed] [Google Scholar]
- 12.Creamer KE, Ditmars FS, Basting PJ, et al. Benzoate- and salicylate-tolerant strains of Escherichia coli K-12 lose antibiotic resistance during laboratory evolution. Appl Environ Microbiol. 2017:83:063271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cederlund H, Mardh PA. Antibacterial activities of non-antibiotic drugs. J Antimicrob Chemother. 1993;32(3):355–365. [DOI] [PubMed] [Google Scholar]
- 14.Nicolau D, Freeman C, Nightingale C, et al. Reduction of bacterial titers by low-dose aspirin in experimental aortic valve endocarditis. Infection and immunity. 1993;61:1593–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang WH, Wong WM, Dailidiene D, et al. Aspirin inhibits the growth of Helicobacter pylori and enhances its susceptibility to anti-microbial agents. Gut. 2003;52:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makivuokko H, Tiihonen K, Tynkkynen S, Paulin L, Rautonen N. The effect of age and non-steroidal anti-inflammatory drugs on human intestinal microbiota composition. Br J Nutr. 2010;103:227–234. [DOI] [PubMed] [Google Scholar]
- 17.Rogers MAM, Aronoff DM. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin Microbiol Infect. 2016;22:178.el71–178.el79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah MS, DeSantis TZ, Weinmaier T, et al. Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut. 2018;67:882–891. [DOI] [PubMed] [Google Scholar]
- 19.Andrews CN, Griffiths TA, Kaufman J, Vergnolle N, Surette MG, Rioux KP. Mesalazine (5-aminosalicylic acid) alters faecal bacterial profiles, but not mucosal proteolytic activity in diarrhoea-predominant irritable bowel syndrome. Aliment Pharmacol Ther. 2011;34:374–383. [DOI] [PubMed] [Google Scholar]
- 20.Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the lllumina HiSeq and MiSeq plat-forms. ISMEJ. 2012;6:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gohl DM, Vangay P, Garbe J, et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol. 2016;34:942–949. [DOI] [PubMed] [Google Scholar]
- 23.Maresso KC, Vilar E, Hawk ET. Urinary PGE-M in colorectal cancer: predicting more than risk? Cancer Prev Res. 2014;7:969–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bezawada N, Song M, Wu K, et al. Urinary PGE-M levels are associated with risk of colorectal adenomas and chemopreventive response to anti-inflammatory drugs. Cancer Prev Res. 2014;7:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrubsole MJ, Cai Q, Wen W, et al. Urinary prostaglandin E2 metabolite and risk for colorectal adenoma. Cancer prevention research. 2012;5(2):336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garland LL, Guillen-Rodriguez J, Hsu C-H, et al. Effect of intermittent versus continuous low-dose aspirin on nasal epithelium gene expression in current smokers: a randomized, double-blinded trial. Cancer Prev Res. 2019;12:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seyberth H, Sweetman B, Frolich J, Oates J. Quantification of the major urinary metabolite of the E prostaglandins by mass spectrometry: Evaluation of the method’s application to clinical studies. Prostaglandins. 1976;ll(2):381–397. [DOI] [PubMed] [Google Scholar]
- 28.Wang D, DuBois RN. Urinary PGE-M: a promising cancer biomarker. Cancer Prev Res. 2013;6:507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris PG, Zhou XK, Milne GL, et al. Increased levels of urinary PGE-M, a biomarker of inflammation, occur in association with obesity, aging, and lung metastases in patients with breast cancer. Cancer Prev Res (Phila). 2013;6:428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: opensource, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Staley C, Gould TJ, Wang P, Phillips J, Cotner JB, Sadowsky MJ. Evaluation of water sampling methodologies for amplicon-based characterization of bacterial community structure. J Microbiol Methods. 2015;114:43–50. [DOI] [PubMed] [Google Scholar]
- 32.Aronesty E. Comparison of sequencing utility programs. Open Bioinform J. 2013;7:1–8. [Google Scholar]
- 33.Pruesse E, Quast C, Knittel K, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England). 2011;27:2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole J, Wang Q, Fish J, et al. Ribosomal database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Research. 2014;42:D633–D642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knights D, Kuczynski J, Charlson ES, et al. Bayesian community-wide culture-independent microbial source tracking. Nat Methods. 2011;8:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dupont WD, Plummer WD. PS power and sample size program available for free on the Internet. Controlled Clin Trials. 1997;18:274. [Google Scholar]
- 39.Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog. 2013;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borges-Canha M, Portela-Cidade JP, Dinis-Ribeiro M, Leite-Moreira AF, Pimentel-Nunes P. Role of colonic microbiota in colorectal carcinogenesis: a systematic review. Rev Esp Enferm Dig. 2015;107:659–671. [DOI] [PubMed] [Google Scholar]
- 41.Ooi CY, Syed SA, Rossi L, et al. Impact of CFTR modulation with ivacaftor on gut microbiota and intestinal inflammation. Sci Rep. 2018;8:17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Passel MW, Kant R, Zoetendal EG, et al. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PloS ONE. 2011;6:el6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopetuso LR, Petito V, Graziani C, et al. Gut microbiota in health, diverticular disease, irritable bowel syndrome, and inflammatory bowel diseases: time for microbial marker of gastrointestinal disorders. Dig D/s. 2018;36:56–65. [DOI] [PubMed] [Google Scholar]
- 44.Montrose DC, Zhou XK, McNally EM, et al. Celecoxib alters the intestinal microbiota and metabolome in association with reducing polyp burden. Cancer Prev Res. 2016;9:721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang X, Bittinger K, Li X, Abernethy DR, Bushman FD, FitzGerald GA. Bidirectional interactions between indomethacin and the murine intestinal microbiota. Elife. 2015;4:e08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science (New York, NY). 2011;331(6015):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.La Reau AJ, Meier-Kolthoff JP, Suen G. Sequence-based analysis of the genus Ruminococcus resolves its phylogeny and reveals strong host association. Microb Genom. 2016;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchesi JR, Dutilh BE, Hall N, et al. Towards the human colorectal cancer microbiome. PLoS ONE. 2011;6:e20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brennan CA, Garrett WS. The gut microbiome, inflammation, and colorectal cancer. Annu Rev Microbiol. 2016;70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou S, Fang L, Lee M-H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol Rep. 2017;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014;2:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bloom SM, Bijanki VN, Nava GM, et al. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ley RE. Gut microbiota in 2015: Prevotella in the gut: choose carefully. Nat Rev Gastroenterol Hepatol. 2016;13:69–70. [DOI] [PubMed] [Google Scholar]
- 55.Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science (New York, NY). 2012;338:120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bresalier RS. Intestinal mucin and colorectal cancer: it’s not just goo. Gastroenterology. 2002;123:648–649. [DOI] [PubMed] [Google Scholar]
- 57.lijima K, Ara N, Abe Y, et al. Association of gastric acid and mucus secretion level with low-dose aspirin-induced gastropathy. J Gastroenterol. 2012;47:150–158. [DOI] [PubMed] [Google Scholar]
- 58.Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol. 2007;73:7767–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prescott D, McKay D. Aspirin-triggered lipoxin enhances macrophage phagocytosis of bacteria while inhibiting inflammatory cytokine production. Am J Physiol Gastrointest Liver Physiol. 2011;301:G487–497. [DOI] [PubMed] [Google Scholar]
- 60.Faith JJ, Guruge JL, Charbonneau M, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu BC, Randolph TW, Lim U, et al. Temporal variability and stability of the fecal microbiome: the Multiethnic Cohort Study. Cancer Epidemiol Prev Biomark. 2019;28:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12:R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.U.S. Department of Agriculture, Agricultural Research Service. USDA Food and Nutrient Database for Dietary Studies 2011-2012. 2014. Food Surveys Research Group Home Page, http://www.ars.usda.gov/ba/bhnrc/fsrg. Accessed 01/18/2020 [Google Scholar]
- 64.Narushima S, Sugiura Y, Oshima K, et al. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut microbes. 2014;5:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geng J, Song Q, Tang X, et al. Co-occurrence of driver and passenger bacteria in human colorectal cancer. Gut pathogens. 2014;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.