

Abstract
The fronto-striatal circuitry, involving the nucleus accumbens, ventral tegmental area, and prefrontal cortex, mediates goal-directed behavior and is targeted by both drugs of abuse and HIV-1 infection. Acutely, both drugs and HIV-1 provoke increased dopamine activity within the circuit. However, chronic exposure to drugs or HIV-1 leads to dysregulation of the dopamine system as a result of fronto-striatal adaptations to oppose the effects of repeated instances of transiently increased dopamine. Specifically, chronic drug use leads to reduced dopaminergic tone, upregulation of dopamine transporters, and altered circuit connectivity, sending users into an allosteric state in which goal-directed behaviors are dysregulated (i.e., addiction). Similarly, chronic exposure to HIV-1, even with combination antiretroviral therapy (cART) treatment, dysregulates dopamine and dopamine transporter function and alters connectivity of the fronto-striatal circuit, contributing to apathy and clinical symptoms of HIV-1 associated neurocognitive disorders (HAND). Thus, in a drug user also exposed to HIV-1, dysregulation of the fronto-striatal dopamine circuit advances at an exacerbated rate and appears to be driven by mechanisms unique from those seen with chronic drug use or HIV-1 exposure alone. We posit that the effects of drug use and HIV-1 infection on microglia interact to drive the progression of motivational dysfunction at an accelerated rate. The current review will therefore explore how the fronto-striatal circuit adapts to drug use (using cocaine as an example), HIV-1 infection, and both together; emphasizing proper methods and providing future directions to develop treatments for pathologies disrupting goal-directed behaviors and improve clinical outcomes for affected patients.
Keywords: Addiction, HIV-1, Dopamine, Microglia, Apathy, HIV-1 Associated Neurocognitive Disorder
Graphical Abstract:
Drug use and HIV-1 in the fronto-striatal circuit. Drugs of abuse and HIV-1 infection both target the fronto-striatal circuit which mediates goal-directed behavior. Acutely, drugs and HIV-1 increase dopamine activity; in contrast chronic exposure produces circuit adaptions leading to dysregulation, addiction and/or apathy. Comorbid drug use and HIV-1 infection may interact with microglia to exacerbate motivational dysregulation.
I. Introduction
The fronto-striatal circuit mediates learning about rewarding environments and therefore contributes to the assembly of goal-directed behaviors. Addictive drugs target this system via multiple mechanisms to drive chronic drug users into a cycle of drug-directed behaviors. Similarly, HIV-1 infection targets the fronto-striatal circuit, despite treatment with antiretroviral therapies to provoke apathy, or a decline in goal-driven behaviors, in patients with chronic HIV-1 exposure. While acute drug use and HIV-1 infection increase dopamine (DA) levels, chronic exposure drives fronto-striatal adaptations which decrease DA tone, resulting in motivational alterations, including addiction and apathy. The current review focuses on fronto-striatal circuit adaptations that occur with either chronic drug use or chronic HIV-1 infection, as well as the neurocognitive and behavioral consequences of comorbid drug abuse with HIV-1 infection. Given the increased prevalence of cocaine use in patients with HIV-1 (Heaton et al. 2010; McIntosh et al. 2015) and evidence that, more than other drugs, cocaine increases HIV-1 seroprevalence, exacerbating the progression of HIV-1 symptoms and neurocognitive decline (Buch et al. 2011), cocaine will serve as the primary example. Part I of the current review examines how the central nervous system (CNS) adapts to drugs of abuse; producing a “pathologic” state which deviates further from homeostasis with continued drug use. Part II examines how the CNS responds to HIV-1 infection alone and in the context of pre-existing fronto-striatal adaptations prior to drug use (see Fig 1). We posit that microglia activation may play a key role in advanced motivational decline seen in patients with comorbid drug dependence and HIV-1 infection. In Part III of the current review, we provide future directions for studying the unique mechanisms of neuronal dysfunction following drug exposure, HIV-1 exposure, or both and summarize why understanding the etiology of these alterations may be crucial to developing effective HAND treatments. A supplemental focus of the current review is on appropriate methods by which to study neural adaptations to chronic toxin (i.e., drug or HIV-1 infection) exposure, as the mechanisms and consequences of chronic exposure are usually unique from those of acute exposure and inappropriate interpretation of such results can mislead directions for future research.
Fig1.

The progression of HIV-1 associated neurocognitive disorders (HAND) are greatly exacerbated in HIV-1 infected patients that have a history of using drugs such as cocaine. Part I of the current review focuses on cocaine effects on the fronto-striatal circuit in a non-seropositive individual, in which acute cocaine elicits a transient but robust increase in dopamine (DA) release to the nucleus accumbens (NAc) which promotes learning about the drug context. While continued cocaine use still elicits DA release, overall DA function within the fronto-striatal circuit becomes weakened. Additionally, afferents from the amygdala (A) and hippocampus (Hipp) are strengthened, contributing to withdrawal related stress and drug craving, respectively, while afferents from the prefrontal cortex (PFC) are weakened, reducing top-down control over goal-directed behaviors. Part II first discusses the effects of HIV-1 infection on the brain, including decreases in DA transporter (DAT) function and DA concentrations in addition to altered dendritic spine morphology and disrupted fronto-striatal connectivity, which is present despite treatment with combination antiretroviral therapies (cART). Because fronto-striatal dysfunction is brought by both cocaine and HIV-1 infection, chronic cocaine users infected with HIV-1 may experience advanced progression to HAND with dopamine dysfunction and deterioration of the neurocircuitry. Exploring the interactions between drug and HIV-1 exposure may provide important insights into the mechanisms which contribute to the progression of apathy and/or HAND.
II. PART I:
II.I. Reward-related circuitry: The Fronto-Striatal Circuit
Preclinical research has contributed greatly to the current understanding of the brain circuitry that mediates goal-directed behavior, a process central to animal learning and memory (Olds and Milner 1954; Kornetsky and Esposito 1979; Wise and McDevitt 2018). Projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) and the prefrontal cortex (PFC) make up the fronto-striatal circuit and are important, interconnected brain regions associated with the function of drugs as reinforcers. The role of instrumental and Pavlovian learning in the transition from recreational use to the maintenance of drug dependence is emphasized in contemporary theories of drug dependence. Therefore, exploring components of fronto-striatal circuitry and how this circuitry adapts to prolonged drug use is important to understand how drug addiction is maintained. The current section will describe (1) the basic tenets of reinforcement and Pavlovian learning as related to contemporary drug dependence theory; (2) the neuroanatomy of the fronto-striatal circuit, and (3) functional changes of this circuitry that occur with acute and repeated cocaine exposure.
II.II. Learning Theory- Reinforcement and Conditioning
Contemporary theories of drug dependence attribute the maintenance of chronic drug-taking behavior and drug seeking, in part, to learning processes. Goal-directed behavior includes both instrumental and Pavlovian conditioning processes (see Rescorla 1987). Explicitly, positive reinforcement occurs when an appetitive consequence is presented quickly after a particular response. The term “positive” signifies that the consequence is added or presented following the response; however, the term does not describe the quality of the consequence per se. “Reinforcement” signifies that the experience of the response and consequence occurring consistently and contingently increases the likelihood that the response will occur again (Skinner 1938). When the reinforcer is cocaine, for example, the euphoric affective response occurs quickly and reliably. Therefore, the frequency of cocaine self-administration and cocaine-directed behaviors will likely increase. Learning that taking cocaine produces positive hedonic outcomes is an important part of the initiation of cocaine dependence (Wise and Koob 2014). In negative reinforcement, another powerful source of learning and motivation, a particular response removes the presentation of an aversive consequence (Skinner 1938). Successfully responding in order to prevent an emotionally or physically painful event increases the likelihood that the response will occur again (Herrnstein and Hineline 1966; Blume 2012). Individuals experiencing emotional and/or physical withdrawal when drug taking ceases may prevent the aversive consequences of withdrawal by self-administering the drug (Van Dyke and Byck 1982; Koob 2009; Blume 2012). Negative reinforcement is therefore a prominent behavioral contingency hypothesized to maintain drug self-administration in individuals who have transitioned from recreational use to drug dependence (Wise and Koob 2014). Therefore, contemporary theories of drug dependence emphasize a role for both forms (i.e., positive and negative) of reinforcement learning in the maintenance of pathological drug use.
Pavlovian conditioning also provides an important source of motivation during goal-directed behavior (Robinson and Berridge 2008; Robinson et al. 2018). Through repeated associations of a conditional stimulus (CS) with an unconditional stimulus (US), the CS alone can eventually elicit a conditioned response (CR) that prepares the animal for the occurrence of the US. Thus, during drug self-administration, stimuli that precede the onset of the drug effect are associated with drug taking, and these conditional stimuli have the ability to attract the individual and command their attention at a later time (Berridge 2007); a form of motivation that can contribute to the maintenance of drug taking and promote relapse during times of abstinence (Robinson et al. 2018). Examples include seeing, holding, or feeling drug paraphernalia including a glass pipe, syringe, lighter or vape pen, tasting or smelling the drug, as well as being in a particular context in which the drug is regularly taken.
II.III. Mesocortical projections between the NAc and VTA primarily mediate reinforcement learning and provide incentive salience during acute drug use
Phasic DA release in the VTA plays a role in reinforcement learning. Experiments quantifying phasic DA release from the VTA during Pavlovian learning trials indicate that firing patterns of DA neurons reflect how well the CS predicts the outcome of the US (Schultz et al. 1997). During initial pairing of a neutral tone stimulus with food (US), phasic DA firing occurs in concert with the consumption of the US. This “positive reward prediction error” is suggested to favor learning regarding the tone’s ability to predict the US. Thus, following repeated tone and food pairings the phasic DA release is shifted in response to the presentation of the tone (Schultz et al. 1997; Mohebi et al. 2019), indicating that this stimulus is predictive of the US (i.e., the tone functions as a CS). On subsequent trials, when the tone is presented in the absence of the food, phasic DA release occurs to the CS and DA firing is inhibited during the time the US usually occurs. This negative reward prediction error is suggested to play a role in learning that a CS is no longer predictive of an US (i.e., extinction learning). DA release in response to the Pavlovian CS does not represent a signal for each occurrence of an US; rather, reward prediction errors indicate that phasic DA release from the VTA plays an important role in predicting the occurrence of available reinforcers (Schultz 2016; Wise and McDevitt 2018). Given that cocaine produces excessive synaptic DA tone in relation to natural reinforcers (e.g., food), it is hypothesized (Schultz, 2016) that modulation of reward prediction errors produced by DA signaling promotes aberrant reinforcement learning, thus contributing to the development of drug dependence.
The Incentive Salience hypothesis of NAc DA highlights the motivational aspects of Pavlovian learning in relation to rewarding stimuli such as food and drugs (Robinson and Berridge 2008). According to Incentive Salience theory, mesolimbic circuitry is hypothesized to attribute salience, or importance, to the rewarding stimulus during instrumental conditioning by providing a neural representation of motivational value in relation to the CS associated with reward. The motivational function of NAc DA therefore is to sub-serve ‘wanting’, and thus, the CS elicits a desire to obtain the reward through associative learning. CS-elicited wanting is contrasted with ‘liking’, or the hedonic, pleasurable effects related to reward, mediated by neurotransmitter systems other than DA (Berridge 2007). Drugs which produce acutely elevated levels of DA (e.g., cocaine), relative to naturalistic stimuli (e.g.., food), are proposed to result in a neural representation of exaggerated salience (Schultz 2016), and thus are represented in memory with high incentive value. Thus, Pavlovian learning makes drug use increasingly prescient upon future exposure to drug-associated cues. With extended drug use, the drug-associated cues provide increased incentive to notice, approach and handle the drug stimulus. Accordingly, individuals who are increasingly sensitive to CSs associated with drug reward (i.e., sign-trackers) and who also exhibit poor decision-making skills (e.g., impulsivity) may be more vulnerable to the development of drug dependence (Robinson et al. 2018).
II.IV. Mesolimbic neurocircuitry involved in the initial rewarding effects of acute drug use and incentive salience
How DA actions are mediated within the mesolimbic circuit is critical for understanding the neural etiology of drug dependence. DA modulation by cells of the VTA is regulated through “short” and “long” DA-mediated feedback loops (Fig 2). Presynaptic D2 autoreceptors are activated by extracellular DA released from VTA cells to directly inhibit further DA release from these presynaptic cells (i.e. “short” feedback loop; Ding et al. 2009). Extracellular DA released from VTA cells activate postsynaptic D1 or D2 receptors to elicit gamma aminobutyric acid (GABA) release from medium spiny neurons (MSNs) of the NAc (Ding et al. 2009), which acts on DA cells of the VTA to indirectly prevent further DA release (i.e. “long” feedback loop; Yang et al. 2018). Notably, medial MSNs of the NAc also target non-DA cells of the VTA to disinhibit DA release from this region (Yang et al. 2018). DA release into the NAc from VTA cells is, thus, regulated by inputs to local GABA and glutamatergic (GLU) cells of the VTA. MSNs are also the primary target of GLU release from the medial PFC (Fig 2). The GLU input to GABA cells of the VTA and NAc elicits indirect inhibition of DA release (Sesack and Pickel 1992), serving as a negative feedback system to modulate DA activity.
Fig2.
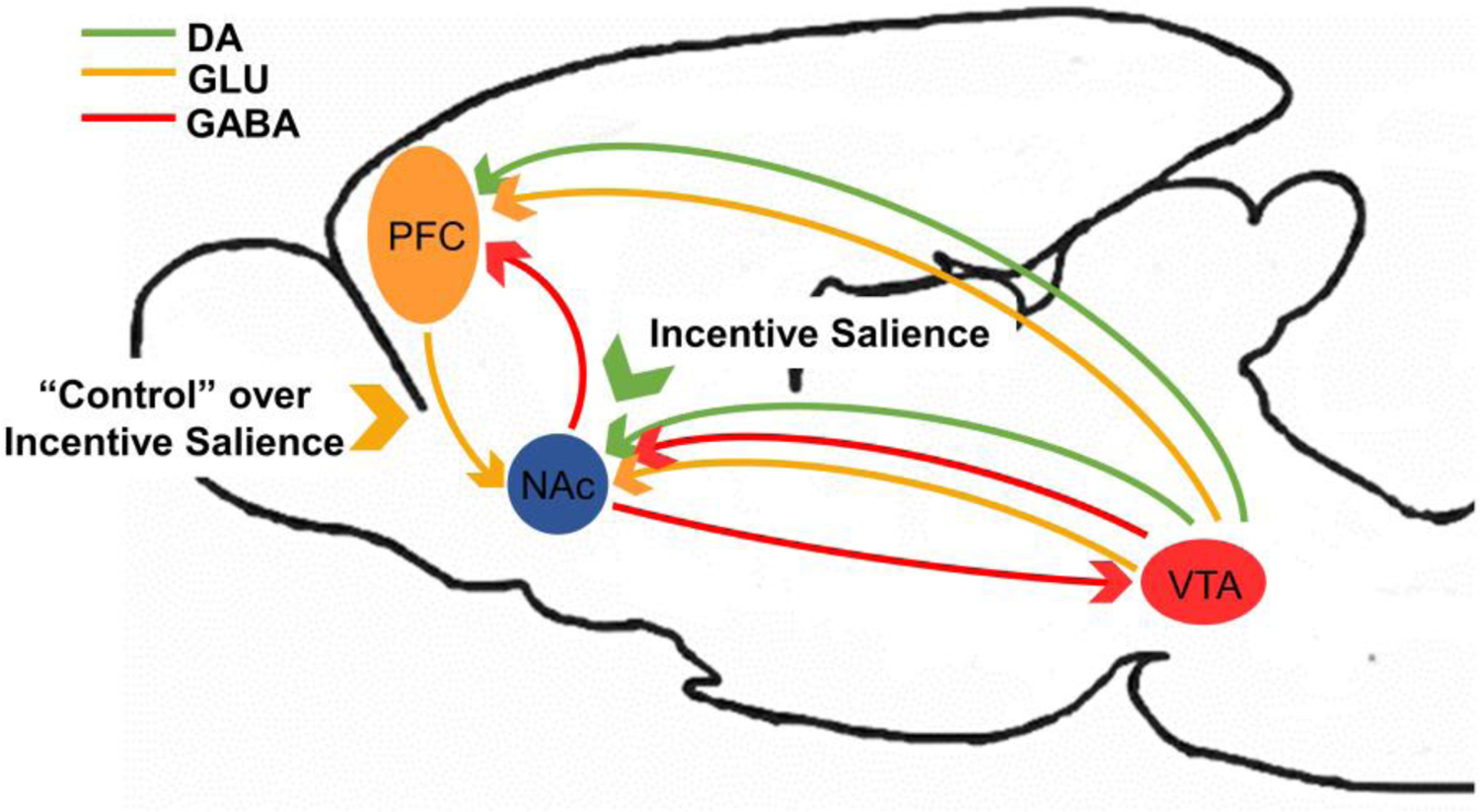
Dopaminergic (DA; green), glutamatergic (GLU; orange) and GABAergic (GABA; red) activity within the fronto-striatal circuit primarily mediates reinforcement learning and incentive salience processing. Unpredicted reinforcers provoke DA release from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) and prefrontal cortex (PFC) to promote learning. Specifically, salience is attributed to the reinforcer and cues/contexts related to the reinforcer so that actions associated with obtaining the reinforcer are more likely to occur in the future. DA activity within the NAc, and likewise, reinforcement learning and incentive salience, are further modulated by GLU inputs from the PFC, thought to provide top-down “control” over behaviors motivated by incentive salience, as well as from feedback loops between the NAc and VTA.
Acute exposure to psychostimulant drugs profoundly increases release of DA from the VTA to activate D1 and D2 receptors on MSNs. For example, amphetamine and methamphetamine act acutely to release DA from presynaptic terminals, whereas drugs like cocaine and methylphenidate (Ritalin) bind to DA transporters (DAT) and prevent DA reuptake to produce increased synaptic DA levels. Specifically, within 1 minute following cocaine administration, 2 – 4-fold increases in extracellular DA and dose-dependent increases in DA uptake in the NAc have been recorded (Oleson et al. 2009). As previously stated, accumbal DA is hypothesized to drive reward learning and support the attribution of excessive incentive salience to drug-associated cues; two processes that set the stage for escalation of drug self-administration behavior and dependence. DA activation of D1-expressing MSNs is increased in response to cocaine and promotes the formation of cocaine reward-context associations (Calipari et al. 2016). Increased extracellular DA in the NAc has been observed within approximately 1.4 – 2.2 seconds following a response for cocaine or following presentation of a learned cocaine-related cue in male rats trained to bar press for intravenous (IV) cocaine (0.33 mg/kg/infusion; Phillips et al. 2003). Additionally, increased spine densities in the NAc core and shell following cocaine administration are correlated with increased preference to be in the location with which cocaine is commonly paired (i.e. cocaine-induced place preference; Marie et al. 2012). These results suggest that cocaine-induced changes in MSNs regulate approach to contextual cues commonly paired with the US effects of cocaine. Overall, the elevation of extracellular DA in the NAc following initial drug exposures is necessary for the development of drug dependence (George et al. 2012).
II.V. The effects of repeated cocaine use on fronto-striatal circuitry and how it contributes to the “pathological state” of addiction
A large body of evidence attributes drug dependence to the acutely exaggerated mesolimbic DA response. However, the immediate DA response to acute drug exposure per se is not sufficient to produce the neural and, consequently, behavioral dysregulation which characterizes drug dependence. Indeed, it is the neural adaptations within and outside the fronto-striatal circuitry that occur in response to chronically escalated DA activity to drive escalation of drug use and the development of drug withdrawal symptoms (Wise and Koob 2014). Specifically, synaptic connectivity between the NAc and regions related to stress (amygdala), memory (hippocampus), and decision making (PFC) are altered with repeated drug use, playing a large role in the processes which lead to the development of drug dependence. Such neural adaptations mediate a transition in motivational states such that drug self-administration is initially maintained by a positive reinforcement contingency and later maintained by a negative reinforcement contingency (Koob and Le Moal 2001).
Drug dependence progresses across three stages: Binge/Intoxication, Withdrawal/Negative Affect, and Preoccupation/Anticipation (Koob and Le Moal, 2001). During the Binge/Intoxication phase (Fig 3) recreational drug use reflects responding for drug on a positive reinforcement contingency, in which drug use is intermittent and highly rewarding. Initial drug-taking behavior is promoted by elevated DA in the NAc and PFC, exaggerated reward learning, and excessive attribution of incentive salience (as described above), in combination with the positive hedonic effects of cocaine reward (i.e., positive reinforcement). In response to repeated exposure to cocaine and these consequent processes, system adaptations occur such that the hedonic effects of the drug are neutralized both by mechanisms of metabolic tolerance and by the influence of ‘anti-reward’ systems (Koob and Le Moal 2008) emerging as opponent processes (Solomon and Corbit 1974). Thus, despite the importance of elevated DA during acute drug-taking episodes, both preclinical and clinical studies show that transient increases in mesolimbic DA immediately following cocaine use are not sufficient to maintain drug use, as DA throughout this system actually decreases during escalated drug self-administration (George et al. 2012). For example, phasic DA release in the ventromedial striatum is decreased in rats that display escalated cocaine intake. Importantly, restoration of DA by L-DOPA treatment prior to self-administration sessions dose-dependently decreases cocaine intake to pre-escalation levels, indicating the observed reduction in DA was necessary for escalation of cocaine self-administration (Willuhn et al. 2014). Moreover, postsynaptic D2 receptor availability and striatal responses to DA-enhancing drugs are decreased in cocaine abusers (Volkow et al. 1990, 1997, respectively). Upregulation of DAT is also reported following a history of cocaine use (Oleson et al. 2009). While not the focus of the current review, it should be noted that chronic cocaine treatment elicits reductions in accumbal GLU from the PFC (McFarland et al. 2003) as well as GLU transporters in the NAc core (Fischer et al. 2013). Importantly, accumbal GLU input from the PFC is increased in cocaine reinstatement, an experimental paradigm utilized to model key aspects of drug relapse in humans.
Fig3.

Much research inappropriately attributes the development of cocaine dependence to transient increases in dopamine (DA) that occurs with acute cocaine exposure. However, tolerance mechanisms are enacted with repeated cocaine use, leading to a marked decrease in DA activity within the nucleus accumbens (NAc). Additionally, chronic cocaine use alters the connectivity between the NAc and areas involving stress (amygdala; A), memory (hippocampus; Hipp), and decision-making (prefrontal cortex; PFC) which may contribute to withdrawal-related stress, craving, and loss of control over drug-directed behaviors, respectively. As a whole, these neural adaptations propel drug-using individuals into a Binge/Intoxication phase that contributes to circuit alterations. These alterations establish a state in which drug use is maintained on a negative reinforcement contingency during the Withdrawal/Negative Affect phase; moreover, top-down control over drug taking cannot overcome DA responses to drug and drug-related cues in the Preoccupation/Anticipation phase.
In the Withdrawal/Negative Affect stage, the recruitment and sensitization of anti-reward systems drive the emergence of negative affective states (Fig 3), leading to drug withdrawal in the absence of the drug and maintenance of drug taking according to a negative reinforcement contingency. This negative adaptation is discussed by Wise and Koob (2014) as a between systems recruitment of increased corticotrophin-releasing factor, dynorphin, and norepinephrine release throughout the central nucleus of the amygdala, bed nucleus of the stria terminalis, and the shell of the NAc. Additionally, chronic cocaine stimulates neuroplasticity/spine growth and thus alters the connectivity between the accumbens and other regions. For instance, cells of the basolateral amygdala projecting to D1-expressing MSNs exhibit increased spine density following just five days of cocaine exposure (Barrientos et al. 2018), likely contributing to withdrawal-related stress. Moreover, when drug-taking behavior is maintained through negative reinforcement, the individual becomes preoccupied with securing the drug and anticipating the removal of negative affective states, characteristic of the Preoccupation/Anticipation stage of dependence. Increases in spine density on ventral hippocampal cells terminating on D1 containing MSNs (Barrientos et al. 2018) likely contribute to enhanced salience of drugs and drug-related contexts, leading to preoccupation with the drug as well as to drug craving (Hitchcock and Lattal 2018). In contrast, projections from pyramidal cells of the prelimbic cortex to D1 MSNs as well as projections from the PFC to both D1 and D2 MSNs display reduced spine density following five days of cocaine exposure (Barrientos et al. 2018), suggesting that “control” over incentive salience (see Fig 2) is diminished. Likewise, drug users’ performance under high cognitive demand and high incentive conditions suggest an overactive incentive salience system as well as reduced control over decision-making due to striatal dominance over top-down control systems (Bechara 2005). Further, DA responses to reward and punishment are likely impacted by drugs of abuse and consequently alter learning to influence dependent individuals to choose drug despite negative outcomes. There is evidence that individuals with a history of cocaine abuse display reduced modulation of reward prediction error signals, as measured by electroencephalogram feedback negativity, in response to an unpredicted loss compared to a predicted loss (Parvaz et al. 2015) and as measured by single-cell firing in response to repeated reward trails (Takahshi et al. 2019).
A key element connecting the cocaine-induced disturbances in spines to altered circuit function is cellular injury occurring from pro-inflammatory responses, such as activated microglia (Kohno et al. 2019). Microglial activation elicited by cocaine exposure has been observed in both in vivo and in vitro experimental models (Guo et al. 2015; Periyasamy et al. 2018). Chronic cocaine use results in an increase in IL-6 (Fox et al. 2012) and increases the ratio of pro-inflammatory to anti-inflammatory markers (Moreira et al. 2016), once again emphasizing the critical importance of chronic exposure models for studies of drug dependence. Reactive microglia may therefore play an important role in cocaine dependence and supports the use of anti-inflammatory drugs in treating drug dependence. The interactions between microglia and chronic drug use will be further discussed in Part II which considers the condition of the fronto-striatal DA system when a chronic drug user becomes HIV-1 positive.
III. PART II: Fronto-striatal alterations seen in HIV-1 infection and interactions with drug history
With the advent of cART, the mortality rate has declined, and clinical outcomes have improved for patients living with HIV-1. However, cART is only successful at suppressing the HIV-1 virus in the periphery, whereas in the CNS, HIV-1 viral proteins and other products continue to impact neural processes (Heaton et al. 2010). Thus, even with effective treatment with cART, HIV-1 targets the fronto-striatal circuit, dysregulates DA, and eventually leads to the appearance of HIV-1 associated neurocognitive disorders (HAND); characterized by alterations in neurocognitive function (e.g., attention, memory, executive function; Cysique et al. 2004; Heaton et al. 2010), Parkinsonian symptoms, and altered motivation (i.e., apathy; Levy and Dubois, 2006; Kamat et al. 2012; Bertrand et al. 2018). The focus of the current review is on the mechanisms by which chronic HIV-1 infection drives a form of motivational dysfunction termed apathy - derived from the Greek “pathos” - meaning a lack of passion. Currently, a commonly accepted heuristic and operational definition of apathy is the quantitative reduction of self-generated voluntary and purposeful (goal-directed) behaviors (Levy and Dubois 2006). It should be noted, however, that apathy is a distinct clinical entity from depression, with different neurobiological mechanisms (Levy et al. 1998). Although apathy can be one of the clinical expressions of a depressed state, it is not a consequence of depression as it appears in a variety of neurological diseases (Levy and Dubois 2006). According to Levy and Dubois (2006), apathy often results from fronto-striatal damage. Specifically, apathy can emerge from an inability to link emotional-affective signals to ongoing or initiating behavior (due to damage to the orbital-medial PFC and its connections with the ventral striatum), disruptions in cognitive processes involved in planning behaviors (due to damage to the lateral PFC and the dorsal caudate nuclei), and/or an inability to initiate internally driven behaviors (due to damage to dorsal-medial aspect of the PFC or the basal ganglia).
The current section first explores the status of the DA system as a clinically relevant target in both acute and chronic HIV-1 infection and proposes that adaptations to the acute effects of HIV-1 are the neurochemical mechanisms underlying the development of motivational dysregulation and apathy in HAND. It is important to note that the results of chronic HIV-1 infection described in the current review are within the context of suppressed viral replication, either through cART or as a characteristic of the missing gag and pol genes in HIV-1 transgenic (Tg) rodents. As such, the specific influences cART may have on a DA system dysregulated by drug use and acute HIV infection are not discussed directly. Second, because HIV-1+ patients exhibit high rates of comorbid psychostimulant dependence and apathy (McIntosh et al. 2015), and because this comorbidity appears to exacerbate progression of HAND (Buch et al. 2011), the neural dysfunction which results from HIV-1 infection in the presence of cocaine use and dependence will be discussed.
III.I. DA dysfunction results from acute and chronic HIV-1 exposure via unique mechanisms
Within the CNS, HIV-1 viral proteins, Tat and gp120, target areas of the fronto-striatal circuit including the NAc and PFC to produce dysfunction of the DA system (Fitting et al. 2015). It is well appreciated that the DAT protein is critical for DA homeostasis, maintaining stable synaptic DA concentrations, and regulating executive/frontal disease (Nieoullon 2002) in addition to reward/striatal processes resembling those described in Part I. Table 1 summarizes how the DAT differentially responds to acute and chronic HIV-1 proteins (see Nath et al. 2001; Rippeth et al. 2004; Carey et al. 2006; Chang et al. 2005; Ferris et al. 2008; Purohit et al. 2011). As first documented in our own in vitro work, acute exposure to the HIV-1 viral protein, Tat, blocks DAT activity (Zhu et al. 2009), consequently increasing extracellular DA levels (Ferris et al. 2009) functionally evidenced by increased acute cocaine-induced locomotor activity when Tat is injected into the accumbens of rats (Harrod et al. 2008). Following chronic, long-term, exposure to HIV-1 proteins, DA activity is decreased (Denton et al. 2019), which is consistent with PET imaging in HIV-1 Tg rats (Sinharay et al. 2017) and human HIV-1 PET imaging (Chang et al. 2008) and may be due to viral protein actions on DAT (Wang et al., 2004) and vesicular monoamine transporter function (Midde et al. 2012). Furthermore, decreases in DA circuit neurotransmission occur despite maximal DAT function (Bertrand et al. 2018), suggesting that therapeutics which only target DAT in HIV-1+ humans are unlikely to be successful in treating long-term HIV-1 protein exposure, in which circuit adaptations have occurred.
Table 1.
Divergent Effects of HIV-1 Proteins with Chronic Cocaine Exposure
| Acute HIV-1 | Chronic HIV-1 | Cocaine + Chronic HIV-1 | |
|---|---|---|---|
| Activity | ↓ DAT | ↓ DAT | ↓ DAT |
| Levels | ↔ | ↓ DAT | ↑ DAT |
| Concentration | ↑ DA | ↓ DA | ↓ DA |
| Spines/Connectivity | ↓ Spine Length | ↑ Stubby Spines ↓ Thin Spines |
? |
Thus, alterations in DA levels results from both acute (increase) and chronic (decrease) HIV-1 protein exposure, with differing mechanisms. Chronic HIV-1 infection elicits connectivity alterations within the fronto-striatal circuitry and appears to drive DA dysfunction, as dendritic spines of accumbal MSNs shift from a thin, more “plastic” spine to an inactive, stubby spine type (Roscoe et al. 2014). Alterations to accumbal and frontal white matter tracts (Paul et al. 2005; Hoare et al. 2010; Kamat et al. 2014) contribute to motivational dysfunction in ways similar to that seen with chronic cocaine exposure. A recent study demonstrated a strong relationship between apathy and white matter abnormalities of HIV-1+ patients with lower CD4 cell counts (Kamat et al. 2014). Indeed, apathy is one of the most frequent behavioral changes associated with diseases or lesions affecting either the PFC or the basal ganglia (van Reekum et al. 2005; Levy and Dubois 2006). Decreased expression of the presynaptic protein, synaptophysin, and the dendritic microtubule activation protein 2 is observed throughout the frontal cortex of patients with latent HIV-1 infection (Desplats et al. 2013). In single protein (Tat) Tg mice, decreased spine density and synaptic dysfunction are found in hippocampal pyramidal CA1 neurons (Fitting et al. 2013). Similarly, HIV-1 Tg rats display HIV-1-induced alterations in a key component of the DA circuitry: the MSNs of the NAc (Roscoe et al. 2014; McLaurin et al. 2018). Specifically, DiOlistic labeling and confocal microscopy revealed a profound reduction in branching complexity on MSNs of the NAc. Additionally, compared to F344/N animals, HIV-1 Tg rats exhibit a shift in spine morphology with increased relative frequency of stubby spines on more proximal branches of MSNs. Because the more proximal branches receive DA afferents from the VTA and GLU afferents from the PFC, a shift from thin and mushroom spines to stubby spines suggest disruption of these inputs (McLaurin et al., 2018). Understanding the effects of chronic HIV-1 exposure will be needed to restore fronto-striatal circuit connectivity and to achieve functional recovery following long-term exposure to HIV-1 proteins in the brain. The discussed evidence also suggests that studies of a single mediator (i.e. DAT) are unlikely to be informative to divergent effects or provide effective therapeutics for long-term HIV-1 exposure. Circuit level analyses of connectivity systems will be needed to determine the mechanisms of DA circuit level dysfunction, which appears with HIV-1.
It is not surprising that HIV-1infection leads to reduced DA concentrations. Although other neural systems may be involved in neurocognitive dysfunction, the mapping of clinical presentations of HAND symptoms onto evidence of DA system dysfunction is quite striking (Ferris et al. 2008). Some of the earliest lines of evidence that chronic HIV-1 infection disrupts the DA system provided findings that HIV-1+ individuals exhibit lower cerebrospinal fluid DA (Berger et al. 1994), symptoms common in Parkinson’s disease, sensitivity to DA receptor antagonists, and abnormalities in basal ganglia structure and function (Berger and Nath 1997). Since then, human imaging (Ipser et al. 2015; Plessis et al. 2014), ultrasound scans (Obermann et al. 2009), and post-mortem examinations (Silvers et al. 2006; Kumar, et al. 2009) have provided further evidence that DA circuitry is compromised and a clinically relevant HAND target. Specifically, significant reductions in DA levels in the substantia nigra are correlated with poor performance on learning and memory tasks in HIV-1+ individuals (Kumar et al. 2011). Altogether, in humans, HIV-1 ultimately produces a hypodopaminergic state.
The DA hypofunction described as occurring with chronic HIV-1 exposure can lead to motivational dysregulation and reductions in goal-directed behaviors pertinent to maintaining employment, medication adherence, and interpersonal functioning in persons living with HIV (Gorman et al. 2009; Shapiro et al. 2013). Difficulties conducting instrumental activities of daily living are positively correlated with apathy in persons with HIV (Shapiro et al. 2013; Kamat et al. 2016). Apathy was identified as a neuropsychiatric consequence of HIV-1 infection and AIDS early in the epidemic (Navia et al. 1986) but is now even more recognized as a common motivational alteration in HIV-1+ individuals, affecting between 30–60% of the population, despite the great success of cART (van Reekum et al. 2005). As such, there is a highly significant clinical need to identify a clear neurobiological mechanism fundamental for treatment development.
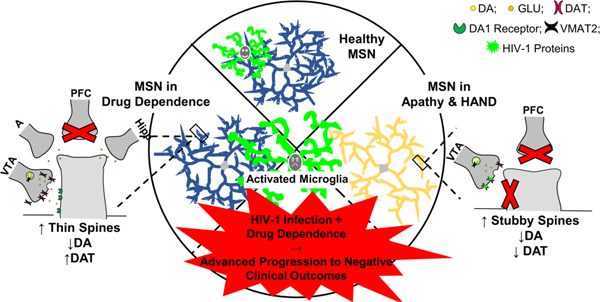
In addition to the direct products of the HIV-1 virus, microglia are putative major players in HIV neuropathogenesis (Garvey et al. 2014; Hong and Banks 2015) and have long been recognized as the primary cell type in the brain to be productively infected with HIV-1 (Michaels et al. 1988; Dickson et al. 1993; Garden 2002). During HIV infection, metabolic shifts have been reported in immune cells (Palmer et al. 2016) and metabolic shifts outside of HIV infection have been shown to regulate activation, polarization, and senescence-related changes in microglia and in turn, their inflammatory behavior (Chen et al. 2017; Holland et al. 2018). While the specific mechanisms which relate metabolism and microglia have not been specifically tested in vitro or in biological systems utilized to model key aspects of HIV-1 infection and HAND, cerebrospinal fluid levels of energy metabolites among people with HIV have been shown to be significantly associated with either improving or worsening temporal changes in neurocognitive state (Dickens et al. 2015). Specifically, accumulation of citrate, indicative of increased glycolysis, was associated with decreased neurocognitive function and increased lactate, indicative of anaerobic glycolysis, was associated with improved neurocognitive function. Despite cART, microglia and perivascular macrophages in the brain become productively infected early during HIV-1 infection (Garvey et al. 2014; Hong and Banks 2015), leading to chronic cellular activation that damages neuronal structures, including dendritic spines (Tremblay et al. 2013), reflecting the synaptodendritic simplification and synapse loss characteristic of HAND. Additionally, a recent single-cell RNA sequencing study showed that “microglia-like” myeloid cells, present in low numbers in the cerebrospinal fluid of persons living with HIV, share gene expression signatures with neurodegenerative-disease-associated microglia (Farhadian et al. 2018). Thus, the likelihood is high that metabolic reprogramming also plays a role in neuro-inflammation and neurocognitive decline in people with HIV since microglial activation occurs despite suppressive cART. Cocaine also enhances HIV-1 replication in CNS glia and promotes gliosis via astrocyte and microglial activation and proliferation, as well as microglial migration (Cai et al. 2016) indicating that these cells may contribute to the unique DA profile observed in HIV-1 patients with a history of drug use (Fig 4).
Fig4.

The hypothetical relationship between microglia, medium spiny neurons (MSNs) of the nucleus accumbens (NAc), and dopaminergic system function is illustrated as a function of the absence (HIV-1 seronegative, A) or presence (HIV-1 seropositive, B) of HIV-1 viral proteins. Presence of HIV-1 viral proteins decreases dendritic branching complexity in MSNs and increases the relative frequency of stubby spines on more distal branches relative to HIV-1 seronegative controls. In sharp contrast, HIV-1 seronegative controls exhibit an increased relative frequency of mushroom spines on more distal branches. Morphological characteristics commonly associated with stubby and mushroom spines may reflect the activity of dopaminergic (DA) afferents from the VTA which target the spine neck. Specifically, the absence of a spine neck suggests the dendritic spines are not receiving sufficient dopamine from the VTA, leading to dysfunctional dopamine neurotransmission compared to systems not exposed to HIV-1 viral proteins; effects which are exacerbated by the use of drugs of that also alter these systems. Furthermore, microglia (shown as activated, frowning face, in seropositive individuals), which play a key role in pruning developing synapses, regulating synaptic plasticity, and synaptic connectivity may represent a key element underlying synaptic dysfunction in HIV and may be another contributor to exacerbated dopamine dysfunction in the presence of both chronic drug and HIV-1 exposure.
III.II. Interactions between the effects of drug use and HIV-1 infection leads to accelerated disease progression
Many of the mechanisms driving neurocognitive impairment in patients with HIV-1 may be potentiated by drugs of abuse as both agents target the fronto-striatal DA system. Behaviorally, patients with HIV-1 who also use cocaine display reduced medication adherence, likely contributing to increased neurocognitive decline (Meade et al. 2011). However, neurobiological, rather than behavioral, interactions between the effects of chronic cocaine use and HIV-1 exposure may drive the progression of neurocognitive impairment and apathy in these patients. When a history of drug use is present, HIV-1 infection occurs in the context of decreased DA concentrations and altered DA and DAT expression, leading to marked DA dysfunction with mechanisms distinct from those seen in patients with HIV infection alone (see Table 1). Compared to drug-naïve cells, in which DAT levels and spine length increase following cocaine exposure, cocaine actions on the DA system appear to be unique when HIV-1 is present. In particular, the cocaine-dependent increases in DA concentration and spines are not seen (Javadi-Paydar et al. 2017; Bertrand et al. 2018). More work is needed to determine the roles DA and other mechanisms play in chronic drug abuse and HIV-1 interactions. White matter damage (Tang et al. 2015; Alakkas et al. 2019), and iron deficiency (Drakesmith et al. 2005; Ersche et al. 2017) are symptoms of cocaine use that have also been associated with HAND and therefore may be promising targets for treatment development. Overall, we posit that chronic drug use and chronic HIV-1 exposure produce a hypodopaminergic microenvironment, activating microglia to drive circuit/spine dysfunction, resulting in the clinical presentations of HAND and apathy.
IV. PART III: Working Models and Future Directions
The current review has discussed how DA dysfunction occurs within the fronto-striatal circuit following both cocaine exposure and HIV-1 exposure, while emphasizing several key points. The fronto-striatal circuitry is the primary mediator of goal-driven behavior and dysfunction of this circuit can lead to pathologies related to addiction and/or apathy. The effects and mechanisms of drug use and HIV-1 infection differ, depending on whether exposure was acute or chronic. Further, acute exposure is not sufficient for observing adaptations which occur with chronic exposure nor circuit-level changes to connectivity, and therefore studies which do not appropriately model chronic exposure cannot generalize nor be translated to the clinical disease.
A lack of understanding of common pharmacological techniques has led to the misinterpretation of how DA contributes to the positive hedonic effects of drugs and how this drives the development of addiction. As such, it is important to review proper methods to accurately model the development and effects of cocaine addiction in the CNS. Initially, in vitro experiments are very useful for examining the cellular mechanism of drug exposure. However, cell culture experiments are not fully capable of replicating the complexity of neuronal circuitry and the adaptations that occur with chronic drug use. Additionally, acute cellular responses do not always predict the long-term adaptive response of the brain and may not accurately reflect the clinical profile seen in drug dependent patients with HAND. For example, our studies in cell culture indicate that DAT is inhibited following exposure to the HIV-1 protein, Tat, (Aksenova et al. 2006); results which were supported by animal models with acute exposure to Tat and other viral proteins (Ferris et al. 2009). However, recent studies using more contemporary methods (Fast scan cyclic voltammetry; FSCV) found that the DAT is actually more active in the surviving synapses/circuitry of cells chronically (months to years) exposed to HIV-1 proteins (Javadi-Paydar et al. 2017), suggesting a homeostatic adaptation. While technically demanding, FSCV can be conducted in intact neural systems of anesthetized or freely moving animals, thereby providing the opportunity to observe the function of cells impacted by various circuits in brains (Denton et al. 2019), and a reliable method to characterize adaptations of the brain chronically exposed to drugs or HIV-1 infection.
In preclinical studies, certain experimental conditions provide models by which to study the neurological underpinnings of behavioral changes that characterize drug dependence in humans, such as neural sensitization, an increase in DA responsiveness to acute drug use, which underlies escalation of drug self-administration in the Binge/Intoxication phase (Robinson and Berridge, 2008; Ahmed et al. 2002). As a part of ensuring that the neural system has the opportunity to sufficiently express adaptations, it is also important to consider which methods of drug administration most closely mimic the effects of drug in humans. Cocaine is used by humans most commonly by inhalation, IV injection, or insufflation (Dunn and Laranjeira 1999) routes of administration. In humans the pharmacokinetics and pharmacodynamics of cocaine inhalation and IV injection are similar. IV models provide near instantaneous distribution throughout the vascular system and to the brain (Bystrowska et al. 2012). Peak arterial concentrations occur within 15 – 30 seconds of injection in humans (Evans et al. 1996) as well as animal models (Booze et al. 1997; Parlaman et al. 2007). This rapidly peaking pharmacokinetic profile of cocaine drives the euphoric effects/addiction processes described above. Importantly, both the concentration and regional distribution of drug within the brain over time via IV administration is distinct from that found following subcutaneous (SC), intraperitoneal (IP), or oral routes of cocaine administration in animal models. Relative to the IV pharmacokinetics, the other routes have much lower peak levels and have been shown to have effects on different brain structures. For example, IP administration of cocaine does not activate the mesocorticolimbic circuitry, relative to the more widespread DA activation following IV dosing (Porrino 1993). The SC route of administration produces skin lesions, thereby limiting the use of this route (Bruckner et al. 1982). In sum, a lack of knowledge on pharmacological techniques has led neuroscientists to misinterpret the acute effects of certain neuroactive agents, such as cocaine and HIV-1, as effects of chronic exposure and as a result are misleading the development of effective treatments. Reviewing the current literature with these key points in mind has revealed several future directions to overcome current therapeutic challenges associated with chronic drug abusers who become HIV-1 infected:
First, divergent neurochemical and neurocognitive effects are present following HIV-1 infection when there is a history of drug abuse. It is clear that pharmaceutical targets may be best identified in studies of chronic drug dependence, and not be designed based primarily on acute effects. Therapeutically decreasing the activity of the DAT, for example, is unlikely to provide an effective treatment approach if the DA circuitry is disrupted and residual DAT is already functioning maximally. Understanding the impact of HIV-1 on neural circuitry in a chronically-drug exposed, dependent, intact brain is critical, as demonstrated by our studies concerning DAT, for development of effective therapeutics.
Second, it is well-recognized that the factor of biological sex plays a prominent role in the progression of cocaine addiction (Becker 2016), HAND (McLaurin et al. 2019), and HIV-induced synaptic dysfunction in MSNs (McLaurin et al. 2018). Specifically, women begin using cocaine at an earlier age, exhibit faster escalation to addiction, and display a more severe addiction relative to males (Kosten et al. 1993; Becker 2016); observations which have been translationally modeled in rodents (e.g., Roberts et al. 1989; Lynch and Carroll 1999). In HAND, profound sex differences in neurocognitive impairments have been reported (for review, Rubin et al. 2019); these sex differences are characterized both by different effect functions, evidenced by greater deficits dependent upon neurocognitive domain, in one sex (e.g., temporal processing; McLaurin et al. 2016; Royal et al. 2016) as well as sex specific effects (e.g., McLaurin et al. 2019). The role of biological sex on neurobehavioral alterations, including apathy, however, has not yet been systematically evaluated in the context of long-term HIV-1 viral protein exposure. Furthermore, evidence suggests that the mechanism underlying alterations in synaptic connectivity in MSNs in the HIV-1 Tg rat are dependent upon biological sex (McLaurin et al. 2018). In combination, results support the critical need for future research to consider how biological sex may influence the progression of DA dysfunction with chronic drug use and/or HIV-1 infection.
Third, effective therapeutics need to address the adaptations that occur in the brain. The current review has discussed how the acute effects of cocaine use and/or HIV-1 infection diverge from the chronic effects of cocaine use and/or HIV-1 infection, respectively. While studying the acute effects of drugs and/or HIV-1 infection is helpful for understanding how these neuroactive agents directly impact the CNS, it is critical to remember that the brain is dynamic and adapts to chronic exposure to these elements over time. A highly relevant model, therefore, would utilize a longitudinal approach to assess whether impairments in goal-directed behavior are progressive in the context of HAND and cocaine abuse.
Fourth, in both drug addiction and HAND, synaptodendritic injury is most clearly associated with neurocognitive impairments, including preoccupation, withdrawal, and craving in drug abuse, as well as apathy in HAND. As synaptic damage is potentially reversible, early diagnosis and treatment of motivational deficits would be beneficial and may be facilitated by repair of synaptic injury/dysfunction and restoration of neural circuitry.
Finally, further investigating the role of activated microglia in circuitry dysfunction in comorbid psychostimulant use and HAND is needed, as work in these areas has potential impact for novel drug discovery. Questions remain as to the role of microglia in regulation of dendritic spine architecture, and the interplay between chronic cocaine and HIV-1 effects on microglial in progressive impairments in HAND. However, this area may be very promising for discovering novel therapeutic approaches to the issue of HIV-1 infection in the drug dependent population.
Although the present review has focused on cocaine as it is widely used in the population of HIV-1+ drug users (Heaton et al. 2010), the progression of drug use through the stages of binge/intoxication, to negative affective states, to anticipation/preoccupation, as proposed by Koob and colleagues, is not specific to cocaine (Koob and Le Moal 2001; George et al. 2012). All addictive drugs reinforce drug-taking behavior and promote escalation of self-administration in vulnerable individuals (positive reinforcement; Wise and Koob 2014). According to this model, any drug of abuse that is chronically self-administered will further produce within-circuit adaptations (e.g., decreased mesocorticolimbic DA) and between-circuit adaptations (e.g., increased corticotrophin-releasing factor and dynorphin). The progressive departure from homeostasis and the negatively affective withdrawal states that are manifest in the absence of the drug together provide a source of motivation to continue drug self-administration (i.e., negative reinforcement; Koob 2009; Blume, 2012; George et al., 2012). In sum, divergent neurochemical and neurocognitive effects are present following long-term HIV-1+ in the context of chronic drug abuse. Specific, targeted, therapeutic approaches are needed to address this vulnerable population which may be based on circuit adaptations and restoration of homeostatic mechanisms not present in acute disease models or initial clinical presentations.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- Ahmed S, Kenny P, Koob G, & Markou A (2002) Neurobiological evidence for hedonic allostasis associated with escalating cocaine use. Nat Neurosci 5(7): 625–626. [DOI] [PubMed] [Google Scholar]
- Aksenova M, Silvers J, Aksenov M, Nath A, Ray P, Mactutus C, & Booze R (2006) HIV-1 Tat neurotoxicity in primary cultures of rat midbrain fetal neurons: Changes in dopamine transporter binding and immunoreactivity. Neurosci Lett 395(3): 235–239. [DOI] [PubMed] [Google Scholar]
- Alakkas A, Ellis R, Watson C, Umlauf A, Heaton R, Letendre S et al. (2019) White matter damage, neuroinflammation, and neuronal integrity in HAND. J Neurovirol 25(1): 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos C, Knowland D, Wu M, Lilascharoen V Huang K, Malenka R, Lim B (2018) Cocaine-Induced Structural Plasticity in Input Regions to Distinct Cell Types in Nucleus Accumbens. Biol Psychiatry 84(12): 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A (2005) Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci 8(11): 1458–1463. [DOI] [PubMed] [Google Scholar]
- Becker JB (2016) Sex differences in addiction. Dialogues Clin Neurosci 18(4): 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger J, Nath A (1997) HIV dementia and the basal ganglia. Intervirology 40(2–3): 122–131. [DOI] [PubMed] [Google Scholar]
- Berger J, Kumar M, Kumar A, Fernandez J, Levin B (1994) Cerebrospinal fluid dopamine in HIV-1 infection. AIDS 8(1): 67–71. [DOI] [PubMed] [Google Scholar]
- Berridge K (2007) The debate over dopamine’s role in reward: the case for incentive salience. Psychopharmacology 191(3): 391–431. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Mactutus C, Harrod S, Moran L, Booze R (2018) HIV-1 proteins dysregulate motivational processes and dopamine circuitry. Sci Rep 8: 7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume A (2012) Negative reinforcement and substance abuse: using a behavioral conceptualization to enhance treatment. Behav Anal Today 2(2): 86–90. [Google Scholar]
- Booze RM, Lehner AF, Wallace DR, Welch MA, Mactutus CF (1997) Dose-response cocaine pharmacokinetics and metabolite profile following intravenous administration and arterial sampling in unanesthetized, freely moving male rats. Neurotoxicol Teratol 19(1): 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner J, Jiang W, Ho B, Levy B (1982) Histopathological evaluation of cocaine-induced skin lesions in the rat. J Cutan Pathol 9(2): 83–95. [DOI] [PubMed] [Google Scholar]
- Buch S, Yao H, Guo M, Mori T, TP S, Wang J (2011) Cocaine and HIV-1 interplay: molecular mechanisms of action and addiction. J Neuroimmune Pharmacol 6(4): 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrowska B, Adamczyk P, Moniczewski A, Zaniewska M, Fuxe K, Filip M (2012) LC/MS/MS evaluation of cocaine and its metabolites in different brain areas, peripheral organs and plasma in cocaine self-administering rats. Phamracol Rep 64(6): 1337–1349. [DOI] [PubMed] [Google Scholar]
- Cai Y, Yang L, Callen S, Buch S (2016) Multiple Faceted Roles of Cocaine in Potentiation of HAND. Curr HIV Res 14(4). [DOI] [PubMed] [Google Scholar]
- Calipari E, Bagot R, Purushothaman I, Davidson T, Yorgason J, Pena C, et al. (2016) In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci USA 113(10): 2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey C, Woods S, Rippeth J, Gonzalez R, Heaton R, Grant I (2006) Additive deleterious effects of methamphetamine dependence and immunosuppression on neuropsychological functioning in HIV infection. AIDS Behav 10(2): 185–190. [DOI] [PubMed] [Google Scholar]
- Chang L, Wang G, Volkow N, Ernst T, Telang F, Logan J, Fowler J (2008) Decreased brain dopamine transporters are related to cognitive deficits in HIV patients with or without cocaine abuse. Neuroimage 42(2): 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Ernst T, Speck O, Grob C (2005) Additive effects of HIV and chronic methamphetamine use on brain metabolite abnormalities. Am J Psychiatry 162(2): 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Partridge A, Sell C, Torres C, Martin-Garcia J (2017) Fate of microglia during HIV-1 infection: From activation to senescence? Glia 65(3): 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cysique LA, Maruff P, Brew BJ (2004) Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: A combined study of two cohorts. J Neurovirol 10(6): 350–357. [DOI] [PubMed] [Google Scholar]
- Denton A, Samaranayake S, Kirchner K, Roscoe R, Berger S, Harrod S et al. (2019) Selective monoaminergic and histaminergic circuit dysregulation following long-term HIV-1 protein exposure. J Neurovirol 25(4): 540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Dumaop W, Smith D, Adame AE, Letendre S, Ellis R, et al. (2013) Molecular and pathologic insights from latent HIV-1 infection in the human brain. Neurology 80(15): 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens AM, Anthony DC, Deutsch R, Mielke MM, Claridge TD, Grant I, et al. (2015) Cerebrospinal fluid metabolomics implicate bioenergetic adaption as a neural mechanism regulating shifts in cognitive states of HIV-infected patients. AIDS 29(5): 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson D, Lee S, Mattiaca L, Yen S, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7(1): 75–83. [DOI] [PubMed] [Google Scholar]
- Ding Z, Liu W, Engleman EA, Rodd ZA, McBride WJ (2009) Differential effects of dopamine D2 and GABAA receptor antagonists on dopamine neurons between the anterior and posterior ventral tegmental area of female Wistar rats. Pharmacol Biochem Be 92: 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakesmith H, Chen N, Ledemann H, Screaton G, Townsend A, Xu X (2005) HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proc Natl Acad Sci USA 102(31): 11017–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn J, Laranjeira R (1999) Transitions in the route of cocaine administration--characteristics, direction and associated variables. Addiction 94(6): 813–824. [DOI] [PubMed] [Google Scholar]
- Ersche K, Acosta-Cabronero J, Jones P, Ziauddeen H, van Swekm R, Laarakkers C, et al. (2017) Disrupted iron regulation in the brain and periphery in cocaine addiction. Transl Psychiatry 7(2): e1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S, Cone E, Henningfield J (1996) Arterial and venous cocaine plasma concentrations in humans: relationship to route of administration, cardiovascular effects and subjective effects. J Pharmacol Exp Ther 279(3): 1345–1356. [PubMed] [Google Scholar]
- Farhadian S, Mehta S, Zografou C, Robertson K, Price RP, Chiarella J, Hafler D (2018) Single-cell RNA sequencing reveals microglia-like cells in cerebrospinal fluid during virologically suppressed HIV. JCI Insight 3(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris M, Frederick-Duus D, Fadel J, Mactutus C, Booze R (2009). The human immunodeficiency virus-1-associated protein, Tat1–86, impairs dopamine transporters and interacts with cocaine to reduce nerver terminal function: a no-net-flux microdialysis study. Neuroscience 159(4),: 1292–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris M, Mactutus C, Booze R (2008) Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: Current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev 32(5): 883–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KD, Houston ACW, Rebec GV (2013) Role of the major glutamate transporter GLT1 in nucleus accumbens core versus shell in cue-induced cocaine-seeking behavior. J Neurosci 33(22): 9319–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Booze R, Mactutus C (2015) HIV-1 Proteins, Tat and gp120, Target the Developing Dopamine System. Curr HIV Res 13(1): 21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska B, Bull C, Skoff R, Lichtman A, Wise L, et al. (2013) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry 73(5): 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox H, D’Sa C, Kimmerling A, Siedlarz K, Tuit K, Stowe R, Sinha R (2012) Immune system inflammation in cocaine dependent individuals: implications for medications development. Hum Psychopharmacol 27(2): 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden G (2002) Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 40(2): 240–251. [DOI] [PubMed] [Google Scholar]
- Garvey L, Pavese N, Politis M, Ramlackhansingh A, Brooks D, Taylor-Robinson S, Winston A (2014) Increased microglia activation in neurologically asymptomatic HIV-infected patients receiving effective ART. AIDS 28(1): 67–72. [DOI] [PubMed] [Google Scholar]
- George O, Le Moal M, Koob G (2012) Allostasis and addiction: role of the dopamine and corticotrophin-releasing factor systems. Physiol Behav 106(1): 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman A, Foley J, Ettenhofer M, Hinkin C, van Gorp W (2009) Functional Consequences of HIV-Associated Neuropsychological Impairment. Neuropsychol Rev 19(2): 186–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Liao K, Periyasamy P, Yang L, Cai Y, Callen S, Buch S (2015) Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 11(7): 995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrod SB, Mactutus CF, Fitting S, Hasselrot U, Booze RM (2008) Intra-accumbal Tat1–72 alters acute and sensitized responses to cocaine. Pharmacol Biochem Behav 90(4): 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton R, Clifford D, Franklin DJ, Woods S, Ake C, Vaida F, et al. (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 75(23): 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrnstein RJ, Hineline PN (1966). Negative reinforcement as shock-frequency reduction. J Exp Anal Behav 9(4): 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock L, Lattal K (2018) Involvement of the dorsal hippocampus in expression and extinction of cocaine-induced conditioned place preference. Hippocampus 28(3): 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare J, Fouche J, Spottiswoode B, Joska J, Schoeman R, Stein D, Carey P (2010) White matter correlates of apathy in HIV-positive subjects: a diffusion tensor imaging study. J Neuropsychiatry Clin Neurosci 22(3): 313–320. [DOI] [PubMed] [Google Scholar]
- Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, et al. (2018). Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun 68: 183–196. [DOI] [PubMed] [Google Scholar]
- Hong S, Banks W (2015) Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun 45: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipser J, Brown G, Bischoff-Grethe A, Connolly C, Ellis R, Heaton R, et al. (2015) HIV Infection Is Associated with Attenuated Frontostriatal Intrinsic Connectivity: A Preliminary Study. J Int Neuropsychol Soc 21(3): 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadi-Paydar M, Roscoe RJ, Denton A, Mactutus C, Booze R (2017) HIV-1 and cocaine disrupt dopamine reuptake and medium spiny neurons in female rat striatum. PLoS One 12(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat R, Brown G, Bolden K, Fennema-Notestein C, Archibald S, Marcotte T, et al. (2014) Apathy is associated with white matter abnormalities in anterior, medial brain regions in persons with HIV infection. J Clin Exp Neuropsychol 36(8): 854–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat R, Woods S, Cameron M, Iudicello J, The HIV Neurobehavioral Research Program Group (2016) Apathy is associated with lower mental and physical quality of life in persons infected with HIV. Psychol Health Med 21(7): 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat R, Woods SP, Marcotte TD, Ellis RJ, Grant I (2012) Implications of apathy for everyday functioning outcomes in persons living with HIV infection. Arch Clin Neuropsych 27(5): 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno M, Link J, Dennis L, McCready H, Huckans M, Hoffman W, Loftis J (2019) Neuroinflammation in addiction: A review of neuroimaging studies and potential immunotherapies. Pharmacol Biochem Behav 179: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G, (2009) Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology 56: 1873–7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G, Le Moal M (2008) Addiction and the brain antireward system. Annu Rev Psychol 59: 29–53. [DOI] [PubMed] [Google Scholar]
- Koob G, Le Moal M (2001) Drug Addiction, Dysregulation of Reward, and Allostasis. Neuropsychopharmacology 24(2): 97–129. [DOI] [PubMed] [Google Scholar]
- Kornetsky C Esposito RU(1979) Euphorigenic drugs: effects on the reward pathways of the brain. Fed Proc 38: 2473–2476. [PubMed] [Google Scholar]
- Kosten TA, Gawin FH, Kosten TR, Rounsaville BJ (1993) Gender differences in cocaine use and treatment response. J Subst Abuse Treat 10(1): 63–66. [DOI] [PubMed] [Google Scholar]
- Kumar A, Fernandez J, Singer E, Commins D, Waldrop-Valverde D, Ownby R, Kumar M (2009) Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol 15(3): 257–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Ownby R, Waldrop-Valverde D, Fernandez B, Kumar M (2011) Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J Neurovirol 17(1): 26–40. [DOI] [PubMed] [Google Scholar]
- Levy M, Cummings J, Fairbanks L, Masterman D, Miller B, Craig A, et al. (1998) Apathy is not depression. J Neuropsychiatry Clin Neurosci 10(3): 314–319. [DOI] [PubMed] [Google Scholar]
- Levy R, Dubois B (2006) Apathy and the Functional Anatomy of the Prefrontal Cortex-Basal Ganglia Circuits. Cereb Cortex 16(7): 916–928. [DOI] [PubMed] [Google Scholar]
- Lynch WJ, Carroll ME (1999) Regulation of intravenously self-administered nicotine in rats. Exp Clin Psychopharm 7(3): 198–207. [DOI] [PubMed] [Google Scholar]
- Marie N, Canestrelli C, Noble F (2012) Transfer of neuroplasticity from nucleus accumbens core to shell is required for cocaine reward. PLoS One 7(1): e30241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish C, Kalivas P (2003) Prefrontal Glutamate Release into the Core of the Nucleus Accumbens Mediates Cocaine-Induced Reinstatement of Drug-Seeking Behavior. J Neurosci 23(8): 3531–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh S, Sexton T, Pattison L, Childers S, Hemby S (2015) Neuropathological sequelae of Human Immunodeficiency Virus and apathy: a review of neuropsychological and neuroimaging studies. Neurosci Biobehav Rev 55: 147–164. [DOI] [PubMed] [Google Scholar]
- McLaurin KA, Booze RM, Mactutus CF (2016) Progression of temporal processing deficits in the HIV-1 transgenic rat. Scient Rep 6 32831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin K, Cook A, Li H, League A, Mactutus C, Booze R (2018) Synaptic Connectivity in Medium Spiny Neurons of the Nucleus Accumbens: A Sex-Dependent Mechanism Underlying Apathy in the HIV-1 Transgenic Rat. Front Behav Neurosci 12: 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaurin KA, Li H, Booze RM, Mactutus CF (2019) Disruption of timing: NeuroHIV progression in the post-cART era. Scient Rep 9(1): 827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade CS, Conn NA, Skalski LM, Safren SA (2011) Neurocognitive impairment and medication adherence in HIV patients with and without cocaine dependence. J Behav Med 34(2): 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels J, Price R, Rosenblum M (1988) Microglia in the giant cell encephalitis of acquired immune deficiency syndrome: proliferation, infection and fusion. Acta Neuropathol 76(4): 373–379. [DOI] [PubMed] [Google Scholar]
- Midde NM, Gomez AM, Zhu J (2012) HIV-1 Tat protein decreases dopamine transporter cell surface expression and vesicular monoamine transporter-2 function in rat striatal synaptosomes. J Neuroimmune Pharmacol 7(3): 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohebi A, Pettibone JR, Hamis AA, Wong JT, Vinson LT, Patriarchi T, et al. (2019) Dissociable dopamine dynamics for learning and motivation. Nature, 570: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira F, Medeiros J, Lhullier A, Souza L, Jansen K, Portela L, et al. (2016) Cocaine abuse and effects in the serum levels of cytokines IL-6 and IL-10. Drug Alcohol Depend 158: 181–185. [DOI] [PubMed] [Google Scholar]
- Nath A, Maragos W, Avison M, Schmitt F, Berger J (2001) Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol 7(1): 66–71. [DOI] [PubMed] [Google Scholar]
- Navia BA, Jordan BD, Price RW (1986) The AIDS Dementia complex .1. Clinical Features. Ann Neurol 19(6): 517–524. [DOI] [PubMed] [Google Scholar]
- Nieoullon A (2002) Dopamine and the regulation of cognition and attention. Prog Neurobiol 67(1): 53–83. [DOI] [PubMed] [Google Scholar]
- Obermann M, Kuper M, Kastrup O, Yaldizli O, Esser S, Theirmann J, German Competence Network HIV/AIDS (2009) Substantia nigra hyperechogenicity and CSF dopamine depletion in HIV. J Neurol 256(6): 948–953. [DOI] [PubMed] [Google Scholar]
- Olds J, Milner P (1954) Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427. [DOI] [PubMed] [Google Scholar]
- Oleson E, Talluri S, Childers S, Smith J, Roberts D, Bonin K, Budygin E (2009) Dopamine uptake changes associated with cocaine self-administration. Neuropsychopharmacology 24(5): 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Cherry CL, Sada-Ovalle I, Singh A, Crowe SM (2016) Glucose Metabolism in T Cells and Monocytes: New Perspective in HIV Pathogenesis. EBioMedicine 6, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlaman J, Thompson B, Levitt P, Stanwood G (2007) Pharmacokinetic profile of cocaine following intravenous administration in the female rabbit. Eur J Pharmacol 563(1–3): 124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvaz MA, Konova AB, Proudfit GH, Dunning JP, Malaker P, Moeller SJ et al. (2015) Impaired neural response to negative prediction errors in cocaine addiction. J Neurosci 35(5): 1872–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Brickman AN, Hinkin C, Malloy P, Jefferson A, Cohen R, et al. (2005) Apathy is associated with volume of the nucleus accumbens in patients infected with HIV. J Neuropsychiatry Clin Neurosci 17(2): 167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periyasamy P, Liao K, Kook Y, Niu F, Callen S, Guo M, Buch S (2018) Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol Neurobiol 55(4): 3196–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P, Stuber G, Heien M, Wightman R, Carelli R (2003) Subsecond dopamine release promotes cocaine seeking. Nature 422(6932): 614–618. [DOI] [PubMed] [Google Scholar]
- Plessis S, Vink M, Joska J, Koutsilieri E, Stein D, Emsley R (2014) HIV infection and the fronto- striatal system: a systematic review and meta- analysis of fMRI studies. AIDS 28(6): 803–811. [DOI] [PubMed] [Google Scholar]
- Porrino L (1993) Functional consequences of acute cocaine treatment depend on route of administration. Psychopharmacology 112(2–3): 343–351. [DOI] [PubMed] [Google Scholar]
- Purohit V, Rapaka R, Shurtleff D (2011) Drugs of Abuse, Dopamine, and HIV-Associated Neurocognitive Disorders/HIV-Associated Dementia. Mol Neurobiol 44(1): 102–110. [DOI] [PubMed] [Google Scholar]
- Rescorla R (1987) A Pavlovian Analysis of Goal-Directed Behavior. American Psychologist 42(2): 119–129. [Google Scholar]
- Rippeth J, Heaton R, Carey C, Marcotte T, Moore D, Gonzalez R, The HNRC Group (2004) Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc 10(1): 1–14. [DOI] [PubMed] [Google Scholar]
- Roberts DCS, Bennett SAL, Vickers GJ (1989) The estrous cycle affects cocaine self-administration on a progressive ratio schedule in rats. Psychopharmacol 98(3): 408–411. [DOI] [PubMed] [Google Scholar]
- Robinson T, Berridge K (2008) The Incentive Sensitization Theory of Addiction: Some Current Issues. Philos Trans R Soc Lond B Biol Sci 363(1507): 3137–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson T, Carr C, Kawa A (2018) The Propensity to Attribute Incentive Salience to Drug Cues and Poor Cognitive Control Combine to Render Sign-Trackers Susceptible to Addiction In Morrow J & Tomie A (eds) Sign Tracking and Drug Addiction, Michigan Publishing, Ann Arbor. [Google Scholar]
- Roscoe R, Mactutus C, Booze R (2014) HIV-1 Transgenic Female Rat: Synaptodendritic Alterations of Medium Spiny Neurons in the Nucleus Accumbens. J Neuroimmune Pharmacol 9: 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royal W III, Cherner M, Burdo TH, Umlauf A, Letendre SL, Jumare J, et al. (2016) Associations between cognition, gender and monocyte activation among HIV infected individuals in Nigeria PLoS ONE 11(2): e0147182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin LH, Neigh GN, Sundermann EE, Xu YX, Scully EP, Maki PM (2019) Sex differences in neurocognitive function in adults with HIV: Patterns, Predictors, and Mechanisms. Curr Psychiatry Rep 21(10): 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W (2016) Dopamine reward prediction error coding. Dialogues Clin Neurosci 18(1): 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague P (1997) A neural substrate of prediction and reward. Science 275(5306): 1593–1599. [DOI] [PubMed] [Google Scholar]
- Sesack S, Pickel V (1992) Prefrontal Cortical Efferents in the Rat Synapse on Unlabeled Neuronal Targets of Catecholamine Terminals in the Nucleus Accumbens Septi and on Dopamine Neurons in the Ventral Tegmental Area. J Comp Neurol 320(2): 145–160. [DOI] [PubMed] [Google Scholar]
- Shapiro M, Mahoney J, Zingman B, Pogge D, Verghese J (2013) Apathy correlates with cognitive performance, functional disability, and HIV RNA plasma levels in HIV-positive individuals. J Clin Exp Neuropsychol 35(9): 934–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvers J, Aksenov M, Aksenova M, Beckley J, Olton P, Mactutus C, Booze R (2006) Dopaminergic marker proteins in the substantia nigra of human immunodeficiency virus type 1-infected brains. J Neurovirol 12(2): 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinharay S, Lee D, Shah S, Muthusamy S, Papadakis GZ, Zhang X, et al. (2017) Cross-sectional and longitudinal small animal PET shows pre and post-synaptic striatal dopaminergic deficits in an animal model of HIV. Nucl Med Biol 55: 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner B (1938) The Behavior of Organisms: An Experimental Analysis New York: Appleton-Century. [Google Scholar]
- Solomon RL, Corbit JD (1974) An opponent-process theory of motivation: 1. Temporal dynamics of affect. Psychol Rev 81: 119–145. [DOI] [PubMed] [Google Scholar]
- Takahashi YK, Stalnaker TA, Marrero-Garcia Y, Rada RM, Schenbaum G (2019) Expectancy-related changes in dopaminergic error signals are impaired by cocaine self-administration. Neuron 101(2): 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang V, Lang D, Giesbrecht C, Panenka W, Willi T, Procyshyn R, et al. (2015) White matter deficits assessed by diffusion tensor imaging and cognitive dysfunction in psychostimulant users with comorbid human immunodeficiency virus infection. BMC Res Notes 8(515). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M, Marker D, Puccini J, Muly E, Lu S, Gelbard H (2013) Ultrastructure of microglia-synapse interactions in the HIV-1 Tat-injected murine central nervous system. Commun Integr Biol 6(6): e27670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke C, Byck R (1982) Cocaine. Sci Am 246(3): 128–140. [DOI] [PubMed] [Google Scholar]
- van Reekum R, Stuss D, Ostrander L (2005) Apathy: why care? J Neuropsychiatry Clin Neurosci 17(1): 7–19. [DOI] [PubMed] [Google Scholar]
- Volkow N, Fowler J, Wolf A, Schlyer D, Shiue C, Alpert R, et al. (1990) Effects on Chronic Cocaine Abuse on Postsynaptic Dopamine Receptors. Am J Psychiatry 147(6): 719–724. [DOI] [PubMed] [Google Scholar]
- Volkow N, Wang G, Fowler J, Logan J, Gatley S, Hitzemann R, et al. (1997) Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature 386(6627): 830–833. [DOI] [PubMed] [Google Scholar]
- Wang G, Chang L, Volkow N, Telang F, Logan J, Ernst T, Fowler J (2004) Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 127(11): 2452–2458. [DOI] [PubMed] [Google Scholar]
- Willuhn I, Burgeno L, Groblewski P, Phillips P (2014) Excessive cocaine use results from decreased phasic dopamine signaling in the striatum. Nat Neurosci 17(5): 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise R, Koob G (2014) The Development and Maintenance of Drug Addiction. Neuropsychopharmacology 39(2): 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise R, McDevitt R (2018) Drive and Reinforcement Circuitry in the Brain: Origins, Neurotransmitters, and Projection Fields. Neuropsychopharmacology 43(4): 680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, de Jong J, Tak Y, Peck J, Bateup H, Lammel S (2018) Nucleus Accumbens Subnuclei Regulate Motivated Behavior via Direct Inhibition and Disinhibition of VTA Dopamine Subpopulations. Neuron 97(2): 434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mactutus C, Wallace D, Booze R (2009) HIV-1 Tat Protein-Induced Rapid and Reversible Decrease in [H-3]Dopamine Uptake: Dissociation of [H-3]Dopamine Uptake and [H-3]2 beta-Carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) Binding in Rat Striatal Synaptosomes. J Pharmacol Exp Ther 329(3): 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]