

Abstract
Wolbachia are widespread intracellular bacteria that mediate many important biological processes in arthropod species. In this study, we identified 210 conserved single-copy genes in 33 genome-sequenced Wolbachia strains in the A–F supergroups. Phylogenomic analyses with these core genes indicate that all 33 Wolbachia strains maintain the supergroup relationship, which was classified previously based on the multilocus sequence typing (MLST) genes. Using an interclade recombination screening method, 14 inter-supergroup recombination events were discovered in six genes (2.9%) among 210 single-copy orthologs. This finding suggests a relatively low frequency of intergroup recombination. Interestingly, they have occurred not only between A and B supergroups (nine events) but also between A and E supergroups (five events). Maintenance of such transfers suggests possible roles in Wolbachia infection-related functions. Comparisons of strain divergence using the five genes of the MLST system show a high correlation (Pearson correlation coefficient r = 0.98) between MLST and whole-genome divergences, indicating that MLST is a reliable method for identifying related strains when whole-genome data are not available. The phylogenomic analysis and the identified core gene set in our study will serve as a valuable foundation for strain identification and the investigation of recombination and genome evolution in Wolbachia.
Keywords: phylogenomics, Wolbachia, evolution, recombination, multilocus sequence typing (MLST), intracellular Alphaproteobacteria
Significance
For the intracellular symbiont Wolbachia, evolutionary analysis and supergroup classification were previously based on the multilocus sequence typing (MLST) genes. We performed phylogenomic analyses of genome-sequenced Wolbachia strains from six supergroups. In total, 210 single-copy protein coding genes were identified in these strains, and our interclade recombination screening method discovered 14 inter-supergroup recombination events, including A–E events which were not described before. We conclude that recombination between supergroups occurred in at least 2.9% of the core genes. We also observed almost perfect correlation in evolutionary divergence between genome sequences and MSLT genes, suggesting that MLST remains a useful tool in strain identification and evolutionary relationship analysis, before speedy and affordable genome sequencing and assembly approaches are readily available for single arthropods.
Introduction
The obligate intracellular bacteria Wolbachia commonly infect arthropods and filarial nematodes (Werren 1997; Fenn and Blaxter 2006; Werren et al. 2008). In particular, more than half of the arthropod species are infected by Wolbachia (Hilgenboecker et al. 2008; Zug and Hammerstein 2012), possibly representing a dynamic equilibrium between gain and loss on a global scale (Werren and Windsor 2000; Bailly-Bechet et al. 2017; Klopfstein et al. 2018). The Wolbachia–host interaction generally spans a range from reproductive parasitism to mutualism. Wolbachia can alter the host reproduction to enhance their own transmission in different ways, such as feminization of genetic males, male-killing, parthenogenetic induction, and cytoplasmic incompatibility (Stouthamer et al. 1999; Werren et al. 2008). Other effects of Wolbachia can include viral suppression (Hedges et al. 2008) and nutritional mutualism (Hosokawa et al. 2010). In nematodes, Wolbachia appear to have evolved a long-standing mutualistic relationship (Fenn and Blaxter 2006). Wolbachia strains have been found to move between species by horizontal (infectious) transmission, even between distantly related hosts, and by hybrid introgression between closely related species (Werren et al. 1995; Heath et al. 1999; Raychoudhury et al. 2009). Wolbachia pipientis have been divided into supergroups (A–H) based on 16S ribosomal RNA sequences and other sequence information, including six supergroups (A, B, and E–H) primarily identified in arthropods and two supergroups (C and D) commonly found in filarial nematodes (Werren et al. 2008). However, it has been proposed that supergroup G be decommissioned, as it is based primarily on recombinant wsp sequences and cluster with A supergroup based on five multilocus sequence typing genes (Baldo, Dunning Hotopp, et al. 2006; Baldo and Werren 2007), eight supergroups (A–H) are still widely used in the research community. A MLST system based on five house-keeping genes, (coxA, gatB, hcpA, ftsZ, and fbpA) has been developed for Wolbachia (Baldo, Dunning Hotopp, et al. 2006) and is widely used for strain typing and to characterize strain variation within Wolbachia. However, the increasing number of genome sequences for Wolbachia allows for more detailed characterization of their diversity, including inter-strain recombination events.
Genomic studies of Wolbachia started with the first complete genome of the A-Wolbachia parasite of Drosophila melanogaster (wMel) published in 2004 (Wu et al. 2004) and followed by the complete genome of D-Wolbachia (wBm) in nematode Brugia malayi in 2005 (Foster et al. 2005). Many more genomes have been published in the last decade, and a list of sequenced whole genomes of Wolbachia is summarized in supplementary table S1, Supplementary Material online.
Because of its endosymbiotic nature, multiple different Wolbachia strains can be present in the same host cells, allowing the potential for homologous recombination between strains (Jiggins et al. 2001; Jiggins 2002). Studies have observed recombination across strains and supergroups (Werren and Bartos 2001; Baldo et al. 2005; Duron et al. 2005), which may be mediated by bacteriophages and lead to mosaic genomes in Wolbachia (Klasson et al. 2009; Kent et al. 2011; Duplouy et al. 2013). Although coinfection of different strains exists in the same arthropod host, with recombination particularly in associated phage (Chafee et al. 2010), the supergroups may still remain genetically distinct clades (Ellegaard et al. 2013). Recombination events in Wolbachia have been discovered in wsp (Werren and Bartos 2001) and other genes in Crustaceans (Verne et al. 2007), mites (Ros et al. 2012) and various arthropod species (Werren and Bartos 2001; Reuter and Keller 2003; Baldo et al. 2005; Baldo, Bordenstein, et al. 2006; Ilinsky and Kosterin 2017). No inter-strain recombination has been reported in the filarial nematode Wolbachia strains (Foster et al. 2011).
Most of the previous research on recombination has focused on five MLST genes, Wolbachia surface protein (wsp), and 16S rRNA, or for a subset of genomes from the A–D and F supergroups (Lindsey et al. 2016). Therefore, whole-genome analyses in a large number of Wolbachia strains of all supergroups are needed to identify additional homologous recombination events among Wolbachia across the different supergroups. In this study, we performed phylogenomic analyses on 33 annotated Wolbachia genomes and analyzed the individual gene trees to identify potential recombination events across the supergroups. Relatively low frequencies of inter-supergroup recombination events were found, indicating a general genetic cohesiveness of supergroups. However, between supergroup recombination is still evident and could play a role in Wolbachia adaptation.
Results
Phylogenomic Analysis of Annotated Wolbachia Genomes
To identify a core gene set for phylogenomic analysis of Wolbachia strains, we initially compared 34 publicly available and annotated Wolbachia genomes as of November 2019, which include 16 A-group, 12 B-group, two C-group, two D-group, and one for E- and F-group strains from diverse host species (supplementary table S1, Supplementary Material online). Single-gene ortholog clusters were generated using the procedure described in the Materials and Methods. A total of 210 single-gene ortholog clusters (listed in supplementary data S1, Supplementary Material online) were identified that are shared among the 34 Wolbachia genomes. This is a smaller set than the 496 Wolbachia gene orthologs detected in Lindsey et al. (2016) for 16 Wolbachia strains, but ours included a larger strain set (34 Wolbachia strains), and we restricted our analysis to single-copy orthologs across all of the genomes.
Based on the concatenated coding nucleotide and protein sequences of this core gene set, maximum-likelihood (ML) phylogenetic trees of 34 Wolbachia genomes confirmed the separation of different supergroups A (wSuzi, wSpc, wRi, wHa, wAu, wMel, wMelPop, wGmm, wUni, wDacA, wNfe, wNpa, wNfla, wNleu, wVitA, and wOneA1), B (wAlbB, wStri, wDi, wNo, wTpre, wDacB, wVitB, Ob_Wba, wBol1, wPip_Mol, and wPip), C (wOo, wOv), D (wBm, wWb), E (wFol), and F (wCle) with 100% bootstrap support (supplementary fig. S1 and data S2 and S3, Supplementary Material online). One of the B-Wolbachia, wCon, appeared to be phylogenetically distant from other B strains (supplementary fig. S1, Supplementary Material online). However, its genome size is 2.11 Mb, almost double the B-Wolbachia average (1.288 Mb). Further examination of the genome assembly suggested that wCon is potentially a mixed assembly of one A- and one B-Wolbachia genomes. Therefore, we excluded wCon and reconstructed the ML tree using the rest 33 Wolbachia nucleotide sequences (fig. 1 and supplementary data S2 and S3, Supplementary Material online). For comparisons of nucleotide and protein phylogenies, we also constructed an ML phylogenetic tree of concatenated protein sequences from these core genes using RAxML (Stamatakis 2014). The protein ML phylogenetic tree (supplementary fig. S2 and data S4 and S5, Supplementary Material online) matched well with the nucleotide coding sequence ML tree, having the same 100% bootstrap for the same clades in A and D supergroups, and a very few variations in bootstrap values but same clustering patterns in other supergroups (fig. 1).
Fig. 1.

Phylogenomic relationships of 33 Wolbachia strains. The phylogenetic tree was constructed using ML method from a concatenated nucleotide sequence alignment of 210 single-copy orthologous genes among 33 genome-sequenced Wolbachia strains. Numbers on the branches represent the support from 1,000 bootstrap replicates. Branch transformation and rerooting were performed in FigTree 1.4.4. The assembly names were color-coded based on supergroup identity (A–F). Host taxonomic classifications and species common names were labeled.
As expected, our genomic analyses support extensive horizontal movement of Wolbachia strains between divergent host species. For example, wOneA1, which is an A-supergroup bacterium in the parasitoid Nasonia oneida (Wang et al. 2019), is more closely related to a subset of A-Wolbachia found in Drosophila (wHa, wRi, wSpc, and wSuzi) than to wVitA and wUni in closely related parasitoid wasps. This pattern was previously observed using MLST genes in Wolbachia (Raychoudhury et al. 2009) but is now supported by a much larger data set. The B-supergroup mosquito Wolbachia wAlb in Aedes albopictus gives another example of obvious major host shift (fig. 1 and supplementary fig. S3, Supplementary Material online).
Identification of Inter-Supergroup Recombination Events
Our focus in this study is to evaluate between supergroup recombination in Wolbachia. We therefore developed a prescreening method to detect candidate between supergroup recombination events and applied it to the 210 single-copy ortholog gene set. For each Wolbachia strain on an individual gene nucleotide tree, we computed the branch length distance to all other strains. We defined recombination candidates if their the nearest neighboring strain belongs to a different supergroup based on the concatenated gene tree (see Materials and Methods). The interclade recombination score (IR score) can range from 0 to 100. This method works for detecting recombinants within supergroups containing more than one genome-sequenced strain (see Materials and Methods). Five genes with IR > 65 were chosen as the cutoff for further investigation. We identified recombination events between A and B, and A and E supergroups. We also examined all 210 RAxML gene trees with both the corresponding protein and nucleotide sequence alignments. Both tree topologies and bootstrap values support the recombination events detected by the screening method (supplementary data S3 and S5, Supplementary Material online). One additional recombination event was found for an A-group strain that contains an E-group version dnaK gene (table 1).
Table 1.
List of Six Wolbachia Genes with Interclade Recombination Events
| Gene Name | Gene Description | Intragenic Recombination Breakpoint | Species with Interclade Recombination | Interclade Recombination Score | Nucleotide Tree Shown in |
|---|---|---|---|---|---|
| ftsH | ATP-dependent metalloprotease FtsH | 816 bp, P = 0.002 | wAu (B-in-A) | 99.9 | Figure 2A |
| rplU | 50S ribosomal protein L21 | None | wDacA (B-in-A) | 71.0 | Figure 2B |
| coxB | Cytochrome c oxidase subunit II | None |
wAlbB (A-in-B) wFol (A-in-E) |
92.3 NA |
Figure 3A |
| WONE_04820 | Hypothetical protein | None |
wDi (A-in-B) wAlbB (A-in-B) wTpre (A-in-B) wFol (A-in-E) |
91.3 82.6 67.1 NA |
Figure 3B |
| argS | Arginine-tRNA ligase | 1–561 bp |
wDi (A-in-B) wNo (A-in-B) wFol (A-in-E) |
99.9 43.9 NA |
Figure 4A |
| 562–1,707 bp | wDi (A-in-B) wDacA (E-in-A) |
23.3 NA |
Figure 4B | ||
| dnaK | Chaperone protein DnaK | None | wDacA (E-in-A) | NA | Figure 5 |
A total of five genes (2.4%) with nine recombination events were identified between A and B supergroups, including B-supergroup genes FtsH (ATP-dependent metalloprotease) and rplU (50S ribosomal protein L21) in A-supergroup strains wAu (fig. 2A) and wDacA (fig. 2B), respectively (B-in-A events in table 1), and seven A-in-B recombination events in coxB (cytochrome c oxidase subunit II), WONE_04820 (hypothetical protein), and argS (arginine-tRNA ligase) (table 1 and supplementary table S2, Supplementary Material online). GARD algorithm (Kosakovsky Pond et al. 2006) was used to detect intragenic recombination in these events and identify recombination breakpoints if intragenic recombination is involved. Two breakpoint positions among the identified genes were detected by GARD, including one breakpoint position at 816 bp in ftsH gene with a P value of 0.0002, another breakpoint position at 561 bp in argS gene with a P value of 0.0006.
Fig. 2.

Inter-supergroup recombination events of B-supergroup genes fstH and rplU in A-Wolbachia strains. (A, B) Nucleotide ML trees reveal IR events, in which genes from an A-Wolbachia clusters with B supergroup. The supergroup identities are labeled using the same color code as in figure 1. Bootstrap values >50 are shown in the figure. (C, D) Supergroup informative SNP positions are plotted for all strains (green: A; blue: C; yellow: G; and pink: T). These SNPs showed the general pattern of recombination, whether entire genes between clades or between clade recombination within genes.
Here, we describe the recombination events in more detail. There are two cases of A-Wolbachia strains that contain a B-Wolbachia gene transfer. For ftsH, the A-Wolbachia wAu strain gene clusters with B-Wolbachia strains with an IR score of 99.9, and this recombination event is supported in the nucleotide tree with a bootstrap value of 100 (fig. 2A and supplementary data S3 and S5, Supplementary Material online). As a universally conserved gene in bacteria, ftsH is known to be crucial for the proteolytic degradation of specific integral membrane proteins and cytoplasmic proteins, and it also targets soluble signaling factors like heat-shock sigma factor σ32 and transcriptional activator λ-CII (Wolfgang 1999). A second B into A recombination event involves a B-group rplU gene that has inserted into the A-Wolbachia wDacA (IR = 71), which is also supported with a bootstrap value of 100 in the corresponding nucleotide tree (fig. 2B and supplementary data S3 and S5, Supplementary Material online). Less is known about the function of rplU, except for its interaction with 23S rRNA (Vladimirov et al. 2000).
Three additional genes reveal recombination events of individual A-Wolbachia genes into B-Wolbachia strains. The coxB gene from an A-Wolbachia was transferred to B-Wolbachia wAlbB (IR = 92), supported by the corresponding nucleotide and trees with a bootstrap value of 99 (fig. 3A and supplementary data S3 and S5, Supplementary Material online). The coxB protein is a component of the electron transport chain which drives oxidative phosphorylation. The second case of an A to B transfer involves the hypothetical protein WONE_04820 gene. An A-Wolbachia gene is present in three B-Wolbachia strains wDi, wAlbB, and wTpre (IR = 91, 83, and 67, respectively). The corresponding nucleotide tree supports the general pattern with a bootstrap value of 74 (fig. 3B and supplementary data S3 and S5, Supplementary Material online). Based on the concatenated tree topology, it is difficult to resolve whether these indicate a single or independent transfer events, given that the three strains are not monophyletic within the B supergroup (fig. 1). The function of this gene is currently unknown.
Fig. 3.

Inter-supergroup recombination events of A-supergroup genes coxB and WONE_04820 in B-Wolbachia and E-Wolbachia strains. (A) Nucleotide ML trees reveal IR events, in which coxB genes from wAlbB (B-Wolbachia) and wFol (E-Wolbachia) cluster with A supergroup, with a bootstrap support of 99. (B) Nucleotide ML trees reveal IR events, in which hypothetical protein WONE_04820 from wAlbB, wDi, wTpre, (B-Wolbachia), and wFol (E-Wolbachia) clusters with A supergroup, with a bootstrap support of 74. (C, D) Supergroup informative SNP positions are plotted for all strains (green: A; blue: C; yellow: G; and pink: T). Bootstrap values >50 are shown in the figure.
In each case for the above examples, the complete gene was recombined into a different supergroup. However, recombination events can also occur within genes, as has been documented for the highly recombinogenic wsp gene (Baldo et al 2005). For argS, we found evidence for intragenic recombination (fig. 4A and B and supplementary data S3 and S5, Supplementary Material online), with significantly different topologies between the 5′ region (positions 1–561 bp) compared with the rest of the gene (positions 562–1707 bp). Intragenic recombination is supported by GARD, which identified the breakpoint at 561 (P value = 0.0006). As a member of the class I aminoacyl-tRNA synthetase family, expression of argS is reported to increase the aminoacyl-tRNA synthetase activity in bacteria (Oguiza et al. 1993). In addition, there is also an apparent A–B recombinant event in the coxB gene of wDacA based on a stretch of five A–B diagnostic single-nucleotide polymorphisms (SNPs; position 151, 194, 226, 245, and 285 in fig. 3C).
Fig. 4.

Intragenic recombination event between supergroups in argS gene. Intragenic recombination event was detected by the GARD method in argS (P value = 0.0006), and the inferred breakpoint is at 561 bp position in this gene. (A, B) Nucleotide ML trees for the 5′ region (1–561 bp) and 3′ region (positions 562–1,707 bp), respectively. argS genes from wDi, wNo, (B-Wolbachia), and wFol (E-Wolbachia) cluster with A supergroup (A). The supergroup classifications follow the color code in previous figures. Bootstrap values above 50 are shown in the figure. (A) argS from starting site to 561 bp, B-Wolbachia wDi, wNo, and E-Wolbachia wFol cluster with A supergroup with 19 bootstrap support, whereas (B) argS from 562 bp to stop site, wDi (B-Wolbachia) clusters with A supergroup with 79 bootstrap support, and wDacA (A-Wolbachia) clusters with E-Wolbachia with 35 bootstrap support, indicating intragenic recombination events; (C) nucleotides at selected positions (1–561 bp in argS) support the tree topology in (A); (D) nucleotides at selected positions (562–1,707 bp in argS) supported the tree topology in (B).
For the E supergroup, there is only one released genome (wFol). Nevertheless, we also found some evidence for recombination events between A and this single representative of the E supergroup. For instance, wFol genes cluster with A-Wolbachia in coxB, WONE_04820, and argS (figs. 3A and B and 4A; supplementary data S3 and S5, Supplementary Material online). Given the high similarity among most sequenced A-Wolbachia, it is not possible to confidently identify which is the likely source. In addition, there appear to be two E-group genes that have transferred into the A-Wolbachia strain wDacA, argS, and DnaK (figs. 4B and 5; supplementary data S3 and S5, Supplementary Material online). A better understanding of the evolutionary history of these transfers will be gained with additional E-supergroup genome sequences.
Fig. 5.

The nucleotide ML tree reveals recombination event where A-Wolbachia cluster with E-Wolbachia in dnaK gene. The supergroup classifications follow the color code in earlier figures. Bootstrap values above 50 are shown in the figure. Nucleotides at selected positions are shown in the right panels. wDacA (A-Wolbachia) clusters with wFol (E supergroup) with 90 bootstrap support. (A) The supergroup classifications follow the color code in previous figures. Bootstrap values above 50 are shown in the figure. (B) Supergroup informative SNP positions in dnaK are plotted for all strains (green: A; blue: C; yellow: G; and pink: T).
Taken together, 97% of the single-copy orthologs agree with the supergroup classification in Wolbachia, with a few cases of likely recombination events between Wolbachia strains of different supergroups. The recombination between A and B supergroups in gene coxB was reported by a previous study of six Wolbachia strains (Ellegaard et al. 2013), and the remaining identified inter-supergroup recombination events are novel findings in our study. The finding also indicates that these recombination events involve relatively small regions, rather than large recombination events involving many genes. The frequent gene order rearrangements observed in Wolbachia may make larger recombination tracks between supergroups less successful, as they are more likely to involve vital gene losses due to lack of synteny.
Concordance of MLST Genes and Whole-Genome Divergence
The MLST system (Baldo, Dunning Hotopp, et al. 2006) has been variously used for strain typing of Wolbachia, identification of related strains, recombination within genes (e.g., the wsp locus), and for phylogenetic inferences among strains. Recently, reliability of the MLST system has been criticized (Bleidorn and Gerth 2018), with whole-genome sequencing stated to be preferred. Although whole-genome data sets would always be desirable, the number of Wolbachia whole-genome sequences is small compared with the many hundreds of MLST sequences currently available for comparative analyses. We therefore undertook to compare genetic divergence based on the MLST to our set of 211 genes in 34 different Wolbachia strains.
The MLST performed very well in both identifying closely related strains and in genetic divergence among strains compared with the genome-wide data set. The Pearson correlation coefficient of estimated evolutionary divergence with core gene set and gatB, fbpA, hcpA, coxA, and ftsZ is 0.96, 0.9, 0.97, 0.92, and 0.97, respectively with P value < 2.2 × 10−16 (table 2 and supplementary data S6, Supplementary Material online). The Pearson correlation coefficient of estimated evolutionary divergence with core gene set and the concatenated MLST set is 0.98 with P value < 2.2 × 10−16 (fig. 6). Therefore, MLST is a reliable method for strain identification and close relationships among strains, even when similar strains occur in very different hosts, such as wDacB in a hemipteran (a true bug) and wVitB in a hymenopteran (a parasitoid wasp), or wBol1 in a lepidopteran (butterfly) and wPip in a dipteran (a mosquito) (fig. 1 and supplementary fig. S3, Supplementary Material online). Eventually, whole-genome data sets will supplant the MLST system. However, with over 1,900 isolates in the Wolbachia MLST database, this will likely take some time, and until then, MLST remains a reliable method for identifying closely related Wolbachia strains and their host associations. Furthermore, closely related Wolbachia strains identified by MLST, that differ in host type of phenotypic effects on hosts (e.g., cytoplasmic incompatibility, feminization, male-killing, parthenogenesis, and viral suppression), can be used for targeted whole-genome sequencing to reveal possible mechanisms involved in host and phenotypic shifts.
Table 2.
Correlation of Evolutionary Divergence Estimates between Wolbachia Species Using the 210 Core Gene Set and Five MLST Genes
| Correlation Coefficient (ρ) | Core Gene Set | gatB | fbpA | hcpA | coxA | ftsZ a |
|---|---|---|---|---|---|---|
| Core gene set | 1 | 0.96 | 0.90 | 0.97 | 0.92 | 0.97 |
| gatB | 1 | 0.86 | 0.91 | 0.90 | 0.94 | |
| fbpA | 1 | 0.87 | 0.84 | 0.92 | ||
| hcpA | 1 | 0.89 | 0.96 | |||
| coxA | 1 | 0.92 | ||||
| ftsZ a | 1 |
Estimates of evolutionary divergence using ftsZ gene were only conducted among 31 Wolbachia species excluding wBm, wWb, and wCon, because of the inability to correctly annotate ftsZ in these three species.
Fig. 6.
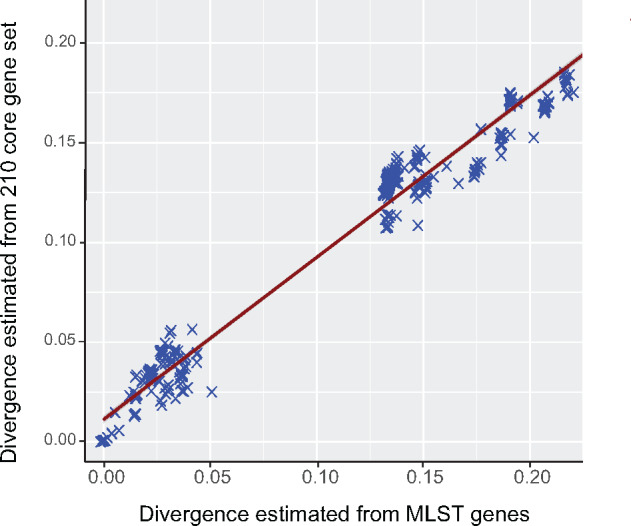
Correlation of evolutionary divergence estimated by core gene set and the five concatenated MLST genes. Pearson correlation coefficient = 0.98, P value < 2.2 × 10−16.
Discussion
The phylogenomic analysis of 33 annotated Wolbachia genomes in our study is the most comprehensive phylogenomic and evolutionary analysis conducted in Wolbachia strains to date. By including almost all available Wolbachia genomes in NCBI, we confirmed at the genome level that these Wolbachia strains group into distinct clusters (A–F supergroups) and different Wolbachia coinfected in the same host kept strain boundaries (Ellegaard et al. 2013). In total, 204 of the 210 single-gene trees are consistent with the strain tree. Six gene trees have major rearrangements among Wolbachia groups (figs. 2–5), indicating potential recombination events between strains. We estimated that recombination events between supergroups occurred in at least 2.9% of the core genes in the Wolbachia genomes, and recombination may be one of the evolutionary forces shaping the Wolbachia genomes.
In total, there are a total of 14 recombination events detected in six genes. Nine of these involve A–B recombination in five genes. The five genes with distinct tree structure differences from the consensus Wolbachia tree include ftsH, rpIU, coxB, hypothetical protein WONE_04820, and argS. In addition, five events were detected between the A and E supergroups. Most recombination events involved the entire gene, whereas a single intragenic event was found in argS. A second intragenic event may also be present in coxB (fig. 3C) although it was not detected by the IR or GARD methods. We conclude that inter-supergroup recombination is uncommon among the set of 210 core single ortholog genes used in this study. Recombination may be more frequent in other genes and clearly is so in phage-associated genes (Bordenstein and Bordenstein 2016; Wang et al. 2016) and the surface protein wsp (Baldo, Bordenstein, et al. 2006). Furthermore, within supergroup, recombination is also likely to be more common, although also more difficult to quantify due to the greater similarity within these groups.
Among the 14 recombination events observed, argS (five events) and WONE_04820 (four events) appear to be particularly prone to inter-supergroup recombination (table 1). argS is a class I aminoacyl-tRNA synthetases which catalyzes the ligation of arginine to its transfer RNA, whereas the function of WONE_04820 is not clear. WONE_04820 is conserved in Wolbachia and no known functional domains could be identified. In addition, two bacterial strains appear to be more prone to inter-supergroup recombination (wDacA and wDi). Notably, both are found in hemipterans. More sequencing of Wolbachia from different insect orders is needed, as the current set are predominantly from Diptera and Hymenoptera.
Recombination events among A- and B-Wolbachia supergroups have been documented in previous studies, and we identified addition cases through the phylogenomic analysis among 33 sequenced genomes. Interestingly, we also discovered recombination events between A and E supergroups, which was not known previously. The E-group Wolbachia is found in springtails (Vandekerckhove et al. 1999; Czarnetzki and Tebbe 2004; Fountain and Hopkin 2005). A recent study characterized the Wolbachia in 11 collembolan species by MLST and found that nearly all are E-group Wolbachia that are monophyletic, based on phylogenetic reconstruction using MLST genes (Ma et al. 2017). Our genome analysis of the single collembolan Wolbachia genome reveals a number of candidate recombination events, including intergroup recombination between A and E in coxB, dnaK, WONE_04820, and argS. Targeted sequencing of these genes in the additional collembolan species or additional genome sequencing will help reveal the origins and directions of these events. We further speculate that selective maintenance of such transfers could suggest a possible role in E-Wolbachia function, such as parthenogenesis induction found in this springtail (Ma et al. 2017). The focus of this study has been on recombination between supergroups, where the phylogenetic signal to noise ratio is much stronger. However, although more difficult to document, intra-supergroup recombination is likely to be more extensive than between supergroups and is a topic worthy of future study.
It has been recently argued that MLST genotyping has little utility in phylogenetic analyses and should be supplanted by genomic studies (Bleidorn and Gerth 2018). When the MLST system was developed, it was pointed out by the authors that the system would be most useful for identifying relatively closely related Wolbachia, due to potential recombination among more divergent strains (Baldo and Werren 2007). However, our comparison on genome sequence indicates that MLST typing is largely valid, both for supergroup identification and detection of closely related strains. Related Wolbachia based on MLST results are also closely related in the genome-wide analysis. This suggests that, until Wolbachia genome sequencing becomes much less expensive and can be readily performed on single arthropods, that MLST will remain a useful tool for identification of strains, their relationships, and host affinities. Nevertheless, caution should be exercised due to some documented recombination events within MLST genes and among them (Raychoudhury et al. 2009). Therefore, topologies should be compared among genes for evidence of discordance, rather than simply relying of phylogenetic reconstructions of concatenated sequences.
Materials and Methods
Phylogenomic Analysis of Annotated Wolbachia Genomes
To examine the phylogeny of Wolbachia at the genome level, we conducted phylogenomic analysis using 34 annotated Wolbachia genomes (GenBank accession numbers and reference papers listed in supplementary table S1, Supplementary Material online). Homologous genes and ortholog clusters among all 34 Wolbachia genomes were determined by using OrthoFinder v1.1.8 (Emms and Kelly 2015) with default settings. In total, 210 single-copy ortholog groups were identified, and gene IDs in each of the ortholog groups were used to extract the corresponding nucleotide and protein sequences from 34 Wolbachia genomes. The 210 core single-copy genes in all 34 Wolbachia genomes were aligned with MAFFT (Katoh and Standley 2014) at the protein sequence level. PAL2NAL (Suyama et al. 2006) was used to check the consistency between the nucleotide and protein sequences, and all inconsistent nucleotide sequences downloaded from GenBank were manually corrected. In total, 210 core single-copy genes were identified for the subsequent analysis; their accession numbers are listed in supplementary data S1, Supplementary Material online. These single-gene alignments were concatenated into one alignment to use in the subsequent phylogenetic analysis. A ML tree was constructed with the GTRGAMMA model and 1,000 bootstrap replicates by RAxMLv8.2 (Stamatakis 2014) using the concatenated nucleotide sequence alignment of the core gene set. For phylogenetic analysis of protein sequences from the core gene set, the best-fit model of protein evolution was searched by ProtTest 3 (Darriba et al. 2011). The final ML phylogenetic tree was inferred by using RAxML v8.2 (Stamatakis 2014) with the FLU protein model (best-fit model identified by ProtTest 3) and 1,000 rapid bootstrap replicates.
The single-gene ML trees for all 210 core genes were constructed with their corresponding nucleotide sequence alignments using the GTRGAMMA model and 1,000 bootstrap replicates by RAxML v8.2 (Stamatakis 2014). We also constructed protein trees for these identified genes with their corresponding protein sequence alignments using the best-fit protein model detected by ProtTest 3 (Darriba et al. 2011) and 1,000 rapid bootstrap replicates by RAxML v8.2 (Stamatakis 2014). The gene trees and protein trees were visualized using FigTree v1.4.4 (Rambaut 2018). For better viewing of short branches, transformation and rerooting were performed in FigTree to generate the main figures. The original gene trees were shown in supplementary figure S4, Supplementary Material online.
Identification of Individual Gene Trees with Intergroup Recombination Events
To search for IR events, we developed a prescreening tool for the identification of specific gene/protein recombinants that move a particular gene/protein outside its respective supergroup. Based on the concatenated strain phylogeny, we assign a supergroup identity for each strain. For every gene, we calculate the branch length between all strain combinations and then determine the nearest neighbor based on shortest branch length. Candidate recombination events are then identified as those for which the nearest neighbor is in a different supergroup. Because some supergroups only have a single representative, the method is most effective at finding candidate recombination to or from A–D supergroups. Next, an IR score was used to quantify the degree of divergence of the gene from its strains’ supergroup. The distance from the candidate gene to its nearest neighbor (Nn) is compared with the average interclade distance (IC) distance of the recombination candidate gene to other members of its strain’s supergroup (based on the concatenated phylogeny) using the IR metric below.
An IR score can range from 0 to 100, with a larger score indicating a recombination between supergroups.
We further manually compared gene trees in both nucleotide and protein level as follows: 1) the nucleotide ML trees were compared with the concatenated ML tree to manually confirm the recombination events; 2) nucleotide sequence alignments were further inspected for informative SNPs that separate different supergroups; and 3) the single-gene protein trees were also compared with the concatenated protein tree to check for consistency of supergroup classification. Inference of intragenic recombination events and the breakpoints was conducted on nucleotide sequence alignments using the GARD algorithm (Kosakovsky Pond et al. 2006) with default parameters using the datamonkey web server (http://www.datamonkey.org/, last accessed June 2020). The individual trees with potential recombination events were defined as trees with IR score larger than 65 for A/B recombination. Three additional candidate genes are all <60. We inspected the trees and found they are not IR events. The A/E recombination was identified by manual evaluations as only one species is available in E supergroup.
Phylogenetic Analysis of Wolbachia in Nasonia Using MLST Genes
The five multilocus sequence typing (MLST) genes (Baldo, Dunning Hotopp, et al. 2006; Jolley and Maiden 2010) were examined to further characterize the phylogenetic relationships of Wolbachia strains in Nasonia. These genes include gatB (aspartyl/glutamyl-tRNA [Gln] amidotransferase, subunit B), coxA (cytochrome c oxidase subunit I), hcpA (conserved hypothetical protein), ftsZ (cell division protein), and fbpA (fructose-bisphosphate aldolase). The pairwise evolutionary divergence distances between 33 Wolbachia species were estimated with both the core gene set identified in this study, five MLST genes, and the concatenated sequence of these five MLST genes in 33 Wolbachia species by using the Maximum Composite Likelihood model (Tamura et al. 2004) in MEGA7 (Kumar et al. 2016). Estimates of evolutionary divergence using the ftsZ gene were only conducted among 31 Wolbachia species, excluding wBm and wWb, because of the inability to correctly annotate ftsZ in these species. The Pearson correlation coefficient of estimated evolutionary divergences with the core gene set and the MLST gene set (each MLST gene and the concatenated sequence of five MLST genes) was calculated with Hmisc package (Harrell FE and Harrell MFE 2019) in R.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This project is supported by an Auburn University Intramural Grant Program Award (AUIGP-180271 to Xu Wang) and an USDA National Institute of Food and Agriculture Hatch project 1018100. Xu Wang is supported by National Science Foundation EPSCoR RII Track-4 Research Fellowship (NSF OIA 1928770), an Alabama Agricultural Experiment Station (AAES) ARES Agriculture Research Enhancement, Exploration and Development (AgR-SEED) award, and a generous laboratory start-up fund from Auburn University College of Veterinary Medicine. X.X. and W.C. are supported by Auburn University Presidential Graduate Research Fellowship and Auburn University College of Veterinary Medicine Dean’s Fellowship. Contributions of J.H.W. were supported by US National Science Foundation awards 1456233 and 1950078, and the Nathaniel and Helen Wisch Professorship. S Cheng is thanked for conducting tests for directional and purifying selection on recombined gene lineages. The authors thank the Auburn Hopper supercomputer clusters for computational support and two anonymous reviewers for their valuable suggestions.
Data Availability
The data underlying this article are available in the Dryad Digital Repository, at https://doi.org/10.5061/dryad.kg87554.
Literature Cited
- Bailly-Bechet M, et al. 2017. How long does Wolbachia remain on board? Mol Biol Evol. 34(5):1183–1193. [DOI] [PubMed] [Google Scholar]
- Baldo L, Bordenstein S, Wernegreen JJ, Werren JH. 2006. Widespread recombination throughout Wolbachia genomes. Mol Biol Evol. 23(2):437–449. [DOI] [PubMed] [Google Scholar]
- Baldo L, Dunning Hotopp JC, et al. 2006. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 72(11):7098–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Lo N, Werren JH. 2005. Mosaic nature of the Wolbachia surface protein. J Bacteriol. 187(15):5406–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, Werren JH. 2007. Revisiting Wolbachia supergroup typing based on WSP: spurious lineages and discordance with MLST. Curr Microbiol. 55(1):81–87. [DOI] [PubMed] [Google Scholar]
- Bleidorn C, Gerth M. 2018. A critical re-evaluation of multilocus sequence typing (MLST) efforts in Wolbachia. FEMS Microbiol Ecol. 94(1). [DOI] [PubMed] [Google Scholar]
- Bordenstein SR, Bordenstein SR. 2016. Eukaryotic association module in phage WO genomes from Wolbachia. Nat Commun. 7(1):13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chafee ME, Funk DJ, Harrison RG, Bordenstein SR. 2010. Lateral phage transfer in obligate intracellular bacteria (Wolbachia): verification from natural populations. Mol Biol Evol. 27(3):501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnetzki AB, Tebbe CC. 2004. Detection and phylogenetic analysis of Wolbachia in Collembola. Environ Microbiol. 6(1):35–44. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27(8):1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplouy A, et al. 2013. Draft genome sequence of the male-killing Wolbachia strain wBol1 reveals recent horizontal gene transfers from diverse sources. BMC Genomics. 14(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duron O, et al. 2005. Transposable element polymorphism of Wolbachia in the mosquito Culex pipiens: evidence of genetic diversity, superinfection and recombination. Mol Ecol. 14(5):1561–1573. [DOI] [PubMed] [Google Scholar]
- Ellegaard KM, Klasson L, Näslund K, Bourtzis K, Andersson SGE. 2013. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 9(4):e1003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16(1):157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn K, Blaxter M. 2006. Wolbachia genomes: revealing the biology of parasitism and mutualism. Trends Parasitol. 22(2):60–65. [DOI] [PubMed] [Google Scholar]
- Foster J, Slatko B, Bandi C, Kumar S. 2011. Recombination in Wolbachia endosymbionts of filarial nematodes? Appl Environ Microbiol. 77(5):1921–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster J, et al. 2005. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 3(4):e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountain MT, Hopkin SP. 2005. Folsomia candida (Collembola): a “standard” soil arthropod. Annu Rev Entomol. 50(1):201–222. [DOI] [PubMed] [Google Scholar]
- Harrell FE, Jr, Harrell MFE., Jr 2019. Package ‘Hmisc’. CRAN 2018:235–236. [Google Scholar]
- Heath BD, Butcher RD, Whitfield WG, Hubbard SF. 1999. Horizontal transfer of Wolbachia between phylogenetically distant insect species by a naturally occurring mechanism. Curr Biol. 9(6):313–316. [DOI] [PubMed] [Google Scholar]
- Hedges LM, Brownlie JC, O’Neill SL, Johnson KN. 2008. Wolbachia and virus protection in insects. Science 322(5902):702. [DOI] [PubMed] [Google Scholar]
- Hilgenboecker K, Hammerstein P, Schlattmann P, Telschow A, Werren JH. 2008. How many species are infected with Wolbachia?—a statistical analysis of current data. FEMS Microbiol Lett. 281(2):215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa T, Koga R, Kikuchi Y, Meng XY, Fukatsu T. 2010. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc Natl Acad Sci U S A. 107(2):769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilinsky Y, Kosterin OE. 2017. Molecular diversity of Wolbachia in Lepidoptera: prevalent allelic content and high recombination of MLST genes. Mol Phylogenet Evol. 109:164–179. [DOI] [PubMed] [Google Scholar]
- Jiggins FM. 2002. The rate of recombination in Wolbachia bacteria. Mol Biol Evol. 19(9):1640–1643. [DOI] [PubMed] [Google Scholar]
- Jiggins FM, von Der Schulenburg JH, Hurst GD, Majerus ME. 2001. Recombination confounds interpretations of Wolbachia evolution. Proc Biol Sci. 268(1474):1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinf. 11(1):595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2014. MAFFT: iterative refinement and additional methods. Methods Mol Biol. 1079:131–146. [DOI] [PubMed] [Google Scholar]
- Kent BN, et al. 2011. Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture. Genome Biol Evol. 3:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L, et al. 2009. The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc Natl Acad Sci U S A. 106(14):5725–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfstein S, van Der Schyff G, Tierney S, Austin AD. 2018. Wolbachia infections in Australian ichneumonid parasitoid wasps (Hymenoptera: Ichneumonidae): evidence for adherence to the global equilibrium hypothesis. Biol J Linn Soc. 123(3):518–534. [Google Scholar]
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD. 2006. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 23(10):1891–1901. [DOI] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey AR, Werren JH, Richards S, Stouthamer R. 2016. Comparative genomics of a parthenogenesis-inducing Wolbachia symbiont. G3 (Bethesda) 6:2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, et al. 2017. Revisiting the phylogeny of Wolbachia in Collembola. Ecol Evol. 7(7):2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguiza JA, et al. 1993. A gene encoding arginyl-tRNA synthetase is located in the upstream region of the lysA gene in Brevibacterium lactofermentum: regulation of argS-lysA cluster expression by arginine. J Bacteriol. 175(22):7356–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. 2018. FigTree v1.4.4, A Graphical Viewer of Phylogenetic Trees [Internet]. [cited 2020 May 4]. Available from: https://github.com/rambaut/figtree/
- Raychoudhury R, Baldo L, Oliveira DCSG, Werren JH. 2009. Modes of acquisition of Wolbachia: horizontal transfer, hybrid introgression, and codivergence in the Nasonia species complex. Evolution 63(1):165–183. [DOI] [PubMed] [Google Scholar]
- Reuter M, Keller L. 2003. High levels of multiple Wolbachia infection and recombination in the ant Formica exsecta. Mol Biol Evol. 20(5):748–753. [DOI] [PubMed] [Google Scholar]
- Ros VI, Fleming VM, Feil EJ, Breeuwer JA. 2012. Diversity and recombination in Wolbachia and Cardinium from Bryobia spider mites. BMC Microbiol. 12(Suppl 1):S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stouthamer R, Breeuwer JAJ, Hurst GDD. 1999. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol. 53(1):71–102. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34(Web Server):W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Nei M, Kumar S. 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci U S A. 101(30):11030–11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove TTM, et al. 1999. Phylogenetic analysis of the 16S rDNA of the cytoplasmic bacterium Wolbachia from the novel host Folsomia candida (Hexapoda, Collembola) and its implications for wolbachial taxonomy. FEMS Microbiol Lett. 180(2):279–286. [DOI] [PubMed] [Google Scholar]
- Verne S, Johnson M, Bouchon D, Grandjean F. 2007. Evidence for recombination between feminizing Wolbachia in the isopod genus Armadillidium. Gene 397(1–2):58–66. [DOI] [PubMed] [Google Scholar]
- Vladimirov SN, Druzina Z, Wang R, Cooperman BS. 2000. Identification of 50S components neighboring 23S rRNA nucleotides A2448 and U2604 within the peptidyl transferase center of Escherichia coli ribosomes. Biochemistry 39(1):183–193. [DOI] [PubMed] [Google Scholar]
- Wang GH, et al. 2016. Bacteriophage WO can mediate horizontal gene transfer in endosymbiotic Wolbachia genomes. Front Microbiol. 7:1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. 2019. Genome assembly of the A-group Wolbachia in Nasonia oneida using linked-reads technology. Genome Biol Evol. 11(10):3008–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH. 1997. Biology of Wolbachia. Annu Rev Entomol. 42(1):587–609. [DOI] [PubMed] [Google Scholar]
- Werren JH, Baldo L, Clark ME. 2008. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 6(10):741–751. [DOI] [PubMed] [Google Scholar]
- Werren JH, Bartos JD. 2001. Recombination in Wolbachia. Curr Biol. 11(6):431–435. [DOI] [PubMed] [Google Scholar]
- Werren JH, Windsor DM. 2000. Wolbachia infection frequencies in insects: evidence of a global equilibrium? Proc Biol Sci. 267(1450):1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, Zhang W, Guo LR. 1995. Evolution and phylogeny of Wolbachia: reproductive parasites of arthropods. Proc Biol Sci. 261(1360):55–63. [DOI] [PubMed] [Google Scholar]
- Wolfgang S. 1999. FtsH—a single-chain charonin? FEMS Microbiol Rev. 23(1):1–11. [DOI] [PubMed] [Google Scholar]
- Wu M, et al. 2004. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2(3):e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zug R, Hammerstein P. 2012. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One 7(6):e38544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the Dryad Digital Repository, at https://doi.org/10.5061/dryad.kg87554.