

Abstract
This open‐label, multicenter, phase I therapeutic protein‐drug interaction study was designed to evaluate the potential effect of guselkumab, a fully human anti‐interleukin‐23 immunoglobulin G1 lambda monoclonal antibody, on the pharmacokinetics of a cocktail of representative cytochrome P450 (CYP) probe substrates (midazolam (CYP3A4), S‐warfarin (CYP2C9), omeprazole (CYP2C19), dextromethorphan (CYP2D6), and caffeine (CYP1A2)). Fourteen participants with psoriasis received a single subcutaneous dose of guselkumab 200 mg on day 8 and an oral probe cocktail on days 1, 15, and 36. Blood samples were collected for measuring plasma concentrations of these probe substrates on days 1, 15, and 36. No consistent trends in observed maximum plasma concentration and area under the curve from time 0 to infinity values of each probe CYP‐substrate before (day 1) and after guselkumab treatment (days 15 and 36) could be identified in each individual patient, suggesting that the use of guselkumab in patients with psoriasis is unlikely to influence the systemic exposure of drugs metabolized by CYP isozymes (CYP3A4, CYP2C9, CYP2C19, CYP2D6, and CYP1A2). The probe cocktail was generally well‐tolerated when administered in combination with guselkumab in patients with psoriasis.
Clinicaltrials.gov Identifiers: NCT02397382.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Therapeutic proteins (TPs) that modulate cytokine concentrations and activity can indirectly influence expression of cytochrome P450 (CYP) isoenzymes and may alter CYP‐mediated metabolism of concomitantly administrated small molecule drugs. An in vitro study 1 and two phase I studies 2 , 3 were previously conducted to assess if interleukin (IL)‐23 modulates the expression or activity of multiple CYP isoenzymes (including CYP1A2, 2C9, 2C19, 2D6, and 3A4). These results suggest that potential TP‐drug interactions between guselkumab and drugs metabolized by CYP450 could be low.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This phase I study evaluated whether treatment with guselkumab, which selectively binds and inhibits IL‐23, affects CYP450 isoenzyme activity in patients with moderate‐to‐severe psoriasis.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Subcutaneous administration of guselkumab to patients with psoriasis has no effect on the pharmacokinetics (PK) of the evaluated CYP substrates.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ These results suggest that guselkumab can be used for the treatment of psoriasis without significant PK interactions with drugs metabolized by CYP3A4, CYP2C9, CYP2C19, CYP2D6, or CYP1A2.
Psoriasis is a chronic inflammatory disease affecting 1–3% of the world’s population. 4 Traditional systemic therapies for psoriasis have not fully met patients’ needs. 5 Highly effective antibody‐based or fusion protein‐based biologics targeting key inflammatory mediators have been developed for psoriasis treatment. 6 Based on their mechanisms of action, biological psoriasis therapies can be classified as: (i) T‐cell modulating agents, (ii) tumor necrosis factor (TNF)‐α antagonists, (iii) interleukin (IL)‐12/23 and/or IL‐23 inhibitors, and (iv) IL‐17 inhibitors. 4 , 7
Guselkumab (Tremfya, Janssen Research & Development, Spring House, PA) is a fully human immunoglobulin G1 lambda (IgG1λ) monoclonal antibody (mAb) that selectively binds and inhibits IL‐23, a critical driver of pathogenic T cells in chronic plaque psoriasis. Clinical trials have demonstrated that guselkumab had favorable efficacy and safety profiles for the treatment of moderate‐to‐severe plaque psoriasis. 8 , 9 , 10
As a fully human IgG1λ mAb, guselkumab is expected to be metabolized in the same manner as any other endogenous IgG antibody (degraded into small peptides and amino acids via catabolic pathways) and subject to similar routes for elimination. 11 Therefore, the likelihood of direct therapeutic protein (TP)‐drug interaction occurring during co‐administration of guselkumab and other concomitant small molecule medications is assumed to be low. In line with this, clinically relevant information has been published about potential TP‐drug interactions, 12 , 13 , 14 , 15 , 16 and supports that mAbs do not elicit a direct effect on the metabolic/clearance pathways of small molecular therapeutics. However, the immunomodulatory properties of mAbs may indirectly alter the clearance of certain small molecules through noncatabolic hepatic metabolism pathways. 14 , 15
An in vitro study 1 using cryopreserved human hepatocytes to assess whether IL‐12 and/or IL‐23 modulate the expression or activity of multiple cytochrome P450 (CYP) enzymes (i.e., CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) suggested that TP‐drug interactions between guselkumab and CYP450 substrates are unlikely. However, in vitro studies may have limitations in predicting clinical interactions between TPs and small molecule drugs. 17
To confirm these findings, we conducted a phase I study in patients with moderate‐to‐severe plaque psoriasis to determine if blocking IL‐23 with guselkumab for treatment of psoriasis would clinically alter the metabolism of probe substrates metabolized by CYP isozymes (CYP3A4, CYP2C9, CYP2C19, CYP2D6, or CYP1A2).
METHODS
Study design
This was an open‐label, multicenter, phase I drug interaction study (ClinicalTrials.gov identifier: NCT02397382) designed to evaluate the potential effect of a single subcutaneous (s.c.) dose of guselkumab 200 mg on the pharmacokinetics (PK) of a cocktail of representative probe substrates of CYP isozymes (midazolam (CYP3A4), S‐warfarin (CYP2C9), omeprazole (CYP2C19), dextromethorphan (CYP2D6), and caffeine (CYP1A2). All participants were to receive a single s.c. dose of guselkumab 200 mg on day 8 and an oral probe cocktail on days 1, 15, and 36 (Figure 1 ). The 200‐mg guselkumab dose was selected because it was twofold higher than the 100‐mg dose selected for the guselkumab phase III psoriasis studies, 8 , 9 and serum guselkumab concentrations were expected to be maintained at a high level through week 4. All components of the probe cocktail included in this study were reported to be systemically cleared within 7 days after a single oral administration. Therefore, the likelihood of systemic probe CYP‐substrate exposures carried over from previous probe cocktail administrations (i.e., on day 15 following administrations on day 1, and on day 36 following administrations on day 15) is low. The day‐15 visit (i.e., 1 week after guselkumab administration) was chosen because this time point was expected to reflect the approximate time to reach the maximum observed concentration (Tmax) following guselkumab s.c. administration on day 8, which would allow time for occurrence of any potential CYP isozyme induction or inhibition. The day‐36 visit (i.e., 4 weeks after guselkumab administration) was chosen because, based on the terminal half‐life (t 1/2) of ~ 18 days for guselkumab, serum guselkumab concentrations would still be high enough to provide additional time to detect any potential indirect effect of guselkumab on CYP isozyme induction or inhibition.
Figure 1.

Study design.
There was no formal hypothesis to be tested for this trial. Therefore, no formal sample‐size power calculation was performed. Approximately 18 patients were planned to be enrolled in this study such that at least 12 patients would be anticipated to complete the day‐40 assessments for the PK of probe CYP‐substrates. The total duration of study participation was ~ 17 weeks, including a screening visit up to 4 weeks prior to first probe cocktail administration, 4 inpatient visits during the study (1 each on days 1, 8, 15, and 36), and study visits on days 64 and 92. The study was conducted at seven investigational sites in the United States from June 2015 to August 2016. It was conducted in accordance with applicable laws and regulations, the current International Council for Harmonization guidelines for Good Clinical Practices, and the Declaration of Helsinki. The study protocol and amendments were reviewed and approved by institutional review board/governing ethical bodies. Written informed consent was obtained from all participants prior to enrollment.
Patients
Eligible participants were men or women age ≥ 18 years with a diagnosis of moderate‐to‐severe plaque‐type psoriasis (with or without psoriatic arthritis) for at least 6 months before day 1. Other key inclusion criteria were Psoriasis Area and Severity Index (PASI) ≥ 12, Investigator’s Global Assessment (IGA) ≥ 3, involved body surface area ≥ 10% at screening, and patients had to be candidates for phototherapy or systemic treatment for psoriasis (either naïve or history of previous treatment).
Patients were excluded if they had a history of or current signs or symptoms of severe, progressive, or uncontrolled renal, hepatic, cardiac, vascular, pulmonary, gastrointestinal, endocrine, neurologic, hematologic, bleeding disorder, rheumatologic, psychiatric, or metabolic disturbances; had previously received guselkumab; had received any anti‐TNF therapy within the longer of 3 months or 5 half‐lives; had received any therapeutic agent directly targeted to IL‐17 or IL‐23 within the last 6 months; had received natalizumab, belimumab, or agents that modulate B cells or T cells (e.g., rituximab, alemtuzumab, abatacept, or visilizumab) within the last 12 months; had received any systemic immunosuppressants (e.g., methotrexate, azathioprine, cyclosporine, 6‐thioguanine, mercaptopurine, mycophenolate mofetil, or tacrolimus) or anakinra within the last 4 weeks; had received phototherapy or any systemic medications/treatments within the last 4 weeks or topical medications/treatments within the last 2 weeks that could affect psoriasis or IGA evaluations; had received an experimental antibody or biologic therapy within the last 6 months or any other experimental therapy or new investigational agent within the last 30 days or 5 half‐lives; had received omeprazole, esomeprazole, warfarin, dextromethorphan, or midazolam within the last 2 weeks; used medications that are known inhibitors or inducers of CYP3A4, CYP2C9, CYP2C19, CYP2D6, or CYP1A2 within 1 month prior to baseline (except for oral corticosteroids); had consumed any foods or beverages known to be inducers or inhibitors of CYP3A4, CYP2C9, CYP2C19, CYP2D6, or CYP1A2 (such as grapefruit/grapefruit juice, Seville oranges (e.g., marmalade), vegetables from the mustard green family (e.g., kale, broccoli, watercress, collard greens, kohlrabi, Brussel sprouts, mustard greens), charcoal grilled meats, star fruit, and apple juice) within 96 hours prior to baseline; consumed caffeine containing products within 48 hours prior to baseline; or consumed alcohol within 72 hours prior to baseline. Of note, smokers (≤ 20 cigarettes/day) were permitted in the study to be more reflective of the population.
Cytochrome P450 genotyping
A single blood sample was obtained at screening from each patient to determine eligibility for the study. DNA samples were genotyped for common polymorphisms in the CYP2C9, CYP2C19, and CYP2D6 genes to identify poor metabolizers to CYP2C9, CYP2C19, and CYP2D6 (Covance Laboratory, Indianapolis, IN): for CYP2C9, patients were screened for the presence of *2 and *3 alleles; for CYP2C19, patients were screened for presence of *2, *3, and *4 alleles; and for CYP2D6, patients were screened for the presence of *9, *10, *17, *29, and *41 alleles. Genetically determined poor metabolizers of CYP2C9, CYP2C19, or CYP2D6 substrates (i.e., patients who did not have at least one functional allele for CYP2C9, CYP2C19, and CYP2D6) were excluded from participation because these patients had little or no catalytic activity to metabolize probe substrates.
Interventions
Guselkumab was supplied as a sterile, preservative‐free, clear, colorless‐to‐light yellow solution assembled in a 1‐mL single‐use prefilled syringe assembled with a passive needle‐guard. The formulation comprises 100 mg/mL guselkumab, containing L‐histidine, L‐histidine monohydrochloride monohydrate, polysorbate‐80, sucrose, and water for injection at pH 5.8. 18
The probe cocktail consisted of oral doses of 0.03 mg/kg midazolam, 10 mg warfarin (+ 10 mg vitamin K), 20 mg omeprazole, 30 mg dextromethorphan, and 100 mg caffeine. The probe cocktail used in this study was expected to have no mutual interactions among the individual medications. 19
Drug administrations
Prior to each probe cocktail administration on days 1, 15, and 36, patients were to have fasted overnight for at least 8 hours; however, water was permitted until 2 hours prior to dosing. Each probe cocktail was to be administered orally with a total of 240 mL water, which was used to rinse the probe cocktail container prior to administration. Two hours after probe cocktail administration, consumption of nonprohibited food and beverages could resume. All patients had to remain in a semi‐supine position (~ 30° upper body elevation) for ~ 1 hour after ingesting the probe cocktail. Patients had to remain seated during and for a minimum of 4 hours after administration of the probe cocktail. On Day 8, guselkumab was administered as a single s.c. dose of 200 mg (2 injections, 100 mg each) at a recommended injection‐site (lower abdomen, upper thigh, or upper arm). 18
PK sample collection
Blood samples were collected for analysis of plasma concentrations of midazolam, S‐warfarin, omeprazole, dextromethorphan, and caffeine prior to administration of the probe cocktail and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours following the administration of the probe cocktail on days 1, 15, and 36. Additional blood samples were collected for analysis of plasma concentrations of S‐warfarin at 48, 72, and 96 hours following each probe cocktail administration. Blood samples for analysis of serum guselkumab concentrations were collected prior to guselkumab administration on day 8, prior to probe cocktail administrations on days 1, 15, and 36, and on days 64 and 92.
Bioanalytical methods
Plasma samples were analyzed to determine concentrations of midazolam, S‐warfarin, omeprazole, dextromethorphan, and caffeine using validated, specific, and sensitive liquid chromatography‐mass spectrometry/mass spectrometry methods. The lower limits of quantitation (LLOQ) were 5, 20, 0.1, 0.05, and 1 ng/mL, respectively, for S‐warfarin, caffeine, midazolam, dextromethorphan, and omeprazole; the accuracy (%Bias) values across the assays were within ± 12%; and the precision (percentage coefficients of variation (%CV)) values across the assays were ≤ 7.3% (Frontage Laboratories, Exton, PA). Serum guselkumab concentrations were quantified using a validated electrochemiluminescence immunoassay method. The lowest quantifiable concentration for a sample was 0.01 µg/mL (LLOQ multiplied by the minimum required dilution of 1:10). 20
PK analyses
The PK parameters of each probe CYP‐substrate following each probe cocktail administration were calculated from the plasma drug concentration over time data using noncompartmental analyses 21 implemented in Phoenix WinNonlin (version 6.3; Pharsight; a Certara Company, St. Louis, MO). All calculations were based on actual sampling times. For estimation of PK parameters, plasma concentrations below the LLOQ were assigned a value of 0 if they preceded the first quantifiable sample in the profile. Any other values below the LLOQ were set to “missing.” The area under the curve (AUC) values were calculated using a combination of linear and logarithmic trapezoidal methods: the linear trapezoidal method was used before Tmax and the logarithmic trapezoidal rule was used after Tmax. Linear regression of the log‐linear portion of the terminal phase was used to estimate the terminal rate constant (λz). A minimum of three data points at the terminal phase, not including the maximum concentration (Cmax), were used in calculating λz. Any measurable plasma concentration values occurring after consecutive samples that were below the LLOQ at the terminal portion of the profile, were not used for the estimation of λz. The calculated PK parameters for probe CYP‐substrates included, but were not limited to, Cmax, Tmax, area under the curve from time 0 to infinity (AUCinf), and t1/2.
Statistical analysis
Descriptive statistics, including arithmetic mean and standard deviation (SD), were used to summarize plasma concentration‐time data and derived PK parameters of each probe CYP‐substrate. Concentrations below LLOQ were to be treated as zero in the summary statistics. For the descriptive statistics, PK parameters, including Cmax, Tmax, AUCinf, and t1/2, were to be excluded if: (i) the dose amount of the probe CYP‐substrate could not be verified; (ii) the baseline (predose) concentration was > 10% of the Cmax value; (iii) the concentration‐time profiles were abnormal; (iv) the concentration‐time data points were insufficient for noncompartmental analysis; and/or (v) the probe CYP‐substrate concentration values were identified as outliers using the Dixon test 22 at a significance level of 0.01. In addition, PK parameters of AUCinf and t1/2 were to be excluded from descriptive statistics if: (i) the adjusted coefficient of determination (R 2) values of the terminal data points were < 0.80; or (ii) the percentage of extrapolated AUC after the last quantifiable plasma concentration (%AUCinf,ex) exceeded 25% of the AUCinf value. The geometric mean ratios (GMRs) of Cmax and AUCinf were calculated and the 90% confidence intervals (CIs) were estimated for each probe CYP‐substrate: day 15 (1 week after guselkumab treatment) vs. day 1 (before guselkumab treatment) and day 36 (4 weeks after guselkumab treatment) vs. day 1. Only patients with available paired data (both before and after guselkumab administration) were included in the comparisons. The P values for exact median tests 23 were provided to evaluate the difference in median Tmax values between day‐15/day‐36 and day 1. Of note, this was not a statistically powered study; therefore, the results from the statistical analysis should be interpreted with caution.
Clinical efficacy and safety evaluations
Efficacy assessments, such as IGA and PASI, were conducted from the screening period through day 64 for all patients who received at least one dose of guselkumab. Safety assessments, such as type, incidence, and severity of treatment‐emergent adverse events (TEAEs), injection‐site reactions, vital sign measurements, clinical laboratory test results, pulse oximetry, electrocardiogram, and physical examinations, were performed from the screening period through day 92 (~ 12 weeks after guselkumab administration) for all patients who received at least 1 dose of probe cocktail or guselkumab.
RESULTS
Patient disposition and demographics
A total of 16 patients with moderate‐to‐severe psoriasis, genotyped to exclude poor metabolizers of CYP2C9, CYP2C19, and CYP2D6 were enrolled and received probe cocktail on day 1. Overall, patients had a mean age of 43 years (range 18–68 years), body weight of 96 kg (range 58–150 kg), and body mass index of 35 kg/m2 (range 21–51 kg/m2). Among these patients, 14 received guselkumab and 12 completed the study. Four patients discontinued for various reasons (Table 1 ).
Table 1.
Patient disposition
| Patients enrolled and treated, n | |
| Patients received probe cocktail on day 1 | 16 |
| Patients received guselkumab on day 8 | 14 |
| Patients received probe cocktail on day 15 | 13 |
| Patients received probe cocktail on day 36 | 12 |
| Patients completed study | 12 |
| Reason for discontinuation, n (%) | |
| Adverse event | 1 (6.3) a , b |
| Death | 0 (0.0) |
| Lost to follow‐up | 1 (6.3) a , c |
| Noncompliance with study drug | 0 (0.0) |
| Physician decision (difficulty in blood draw) | 1 (6.3) a , d |
| Pregnancy | 1 (6.3) a , e |
| Protocol violation | 0 (0.0) |
| Study terminated by sponsor | 0 (0.0) |
| Technical problems | 0 (0.0) |
| Withdrawal by patients | 0 (0.0) |
| Product quality complaint | 0 (0.0) |
| Other | 0 (0.0) |
The percentage is based on the total number of patients who were enrolled and received probe cocktail administration on day 1.
Discontinued on day 17.
Discontinued on day 87.
Discontinued on day 7.
Discontinued on day 20.
Exposure to guselkumab
A total of 14 patients received a single s.c. administration of guselkumab 200 mg on day 8. The mean serum guselkumab concentrations were 15.47 μg/mL (range 7.47−23.77 μg/mL; N = 14) on day 15 and 5.69 μg/mL (range 0.01−11.03 μg/mL; N = 12) on day 36, indicating that patients were exposed to guselkumab when receiving the CYP probe cocktail. Serum guselkumab concentrations were still quantifiable (mean: 0.53 μg/mL (range 0.01−1.34 μg/mL) on day 92. Of note, the t 1/2 of guselkumab is ~ 18 days in patients with psoriasis. 24
PKs of probe CYP450 substrates
Mean plasma concentration‐time profiles for each probe CYP‐substrate following probe cocktail administrations on day 1 (1 week prior to guselkumab treatment), and on days 15 and 36 (1 and 4 weeks, respectively, after guselkumab treatment) are plotted on a semilogarithmic scale in Figure 2. Descriptive statistics for the derived PK parameters of each probe CYP‐substrate before and after probe cocktail administrations are summarized in Table 2 . Of note, among the 16 patients who received probe cocktail on day 1, 3 patients received an unverified dose of midazolam on day 1; 2 patients received unverified doses for midazolam on day 15; and 1 patient received an unverified dose of midazolam on day 36. PK parameters of midazolam from these patients were, therefore, excluded from the analyses. In addition, PK parameters for any probe CYP‐substrate were excluded from the analyses if the parameters met any of the exclusion criteria described in the Methods section.
Table 2.
PK parameters of probe CYP450 substrates before and after treatment with guselkumab
| Substrate/parameter | Day 1 (1 week before initiating guselkumab) | Day 15 (1 week after initiating guselkumab | Day 36 (4 weeks after initiating guselkumab) |
|---|---|---|---|
| Midazolam | |||
| N | 13 | 11 | 11 |
| Cmax, ng/mL | 13.2 (7.0) | 14.6 (6.8) | 15.2 (8.0) |
| Tmax, hour | 1.0 (0.5; 3.0) | 0.5 (0.5; 1.1) | 1.0 (0.5; 1.6) |
| AUCinf, ng·hour/mL | 49.8 (24.0) | 51.2 (22.9) | 51.5 (23.1) |
| t1/2, hour | 7.3 (1.9) | 7.4 (2.7) | 7.0 (2.0) |
| S‐Warfarin | |||
| N | 16 | 13 | 12 |
| Cmax, ng/mL | 582.9 (159.7) | 618.7 (132.7) | 540.0 (142.5) |
| Tmax, hour | 1.8 (0.5; 3.0) | 1.5 (0.5; 4.0) | 1.6 (0.5; 3.1) |
| AUCinf, ng·hour/mL | 18398.2 (6037.8) a | 20774.2 (5871.5) | 19522.5 (5726.0)b |
| t1/2, hour | 34.1 (7.1) a | 36.1 (6.7) | 36.4 (6.7)b |
| Omeprazole | |||
| N | 15 | 12 | 11 |
| Cmax, ng/mL | 350.6 (132.6) | 331.3 (130.8) | 330.9 (175.5) |
| Tmax, hour | 2.8 (1.5; 4.1) | 3.0 (1.5; 4.0) | 3.0 (2.0; 7.7) |
| AUCinf, ng·hour/mL | 1029.9 (686.6)c | 952.8 (646.8)b | 795.6 (369.7)d |
| t1/2, hour | 1.4 (0.6)c | 1.3 (0.5)b | 1.2 (0.3)d |
| Dextromethorphan | |||
| N | 15 | 12 | 11 |
| Cmax, ng/mL | 1.8 (2.0) | 2.1 (2.7) | 2.5 (3.3) |
| Tmax, hour | 3.0 (1.0; 4.1) | 3.2 (1.5; 6.3) | 3.1 (1.5; 4.0) |
| AUCinf, ng·hour/mL | 23.0 (29.6)e | 17.2 (21.7)f | 26.4 (33.8)g |
| t1/2, hour | 6.5 (1.1)e | 6.6 (1.0)f | 6.9 (1.2)g |
| Caffeine | |||
| N | 16 | 13 | 11 |
| Cmax, ng/mL | 2096.3 (533.5) | 2166.2 (358.9) | 2183.6 (499.9) |
| Tmax, hour | 1.5 (0.5; 4.0) | 1.5 (0.5; 4.0) | 1.0 (0.5; 3.0) |
| AUCinf, ng·hour/mL | 22766.7 (12312.0) | 21019.2 (8215.7)e | 20856.9 (7874.5) |
| t1/2, hour | 6.4 (1.9) | 6.2 (1.9)e | 6.5 (2.5) |
AUCinf, area under the plasma concentration versus time curve from time 0 to infinity with extrapolation of the terminal phase; Cmax, maximum observed plasma concentration; PK, pharmacokinetic; SD, standard deviation; t1/2, terminal half‐life; Tmax, time to reach the maximum observed plasma concentration.
Median (minimum, maximum) is reported for Tmax; arithmetic mean (SD) is reported for other PK parameters.
n = 14; b n = 11; c n = 13; d n = 7; e n = 12; f n = 9; g n = 10. Patients were excluded from midazolam analysis because of unverified midazolam dose; patients were excluded from s‐warfarin analysis because of the percentage of extrapolated AUC after the last quantifiable plasma concentration (%AUCinf,ex) exceeded 25% of the AUCinf value; patients were excluded from omeprazole analysis because of (i) insufficient data points; (ii) R 2 < 0.80; and/or (iii) concentration values were outliers identified using Dixon test; patients were excluded from dextromethorphan analysis because of (i) insufficient data points; (ii) abnormal PK profile; and/or R 2 < 0.80; patients were excluded from caffeine analysis because of (i) %AUCinf,ex > 25% of the AUCinf value, and (ii) predose concentration (632 ng/mL) is > 10% of Cmax.
Overall, the mean plasma concentration‐time profiles before and after guselkumab treatment were superimposed for each individual probe CYP‐substrate, and the estimated PK parameters for each individual probe CYP‐substrate were also generally comparable before and after guselkumab treatment. Of note, the interpatient variability in PK parameters of omeprazole was moderate‐to‐large (%CV up to 53% for Cmax and 68% for AUCinf). The interpatient variability in PK parameters of dextromethorphan were even larger (%CV for Cmax and AUCinf were > 100%). Nevertheless, considering interpatient variability in PK, the plasma concentration‐time profiles and estimated PK parameters for omeprazole and dextromethorphan were generally comparable before and after guselkumab treatment.
The GMRs (day‐15/day‐1 and day‐36/day‐1) with 90% CIs for exposure parameters (Cmax and AUCinf) of each probe CYP‐substrate are summarized in Table 3 . Of note, only patients with available paired data (i.e., both days 1 and 15, or days 1 and 36) were included in the comparisons. The GMRs for Cmax and AUCinf of midazolam, S‐warfarin, omeprazole, and caffeine ranged from 0.90−1.14 and 0.96−1.19, respectively. These ratios indicate that changes in Cmax and AUCinf values of these probe CYP‐substrates were within ± 20% before and after guselkumab treatment. The GMRs for Cmax and AUCinf of dextromethorphan for day‐36/day‐1 were 1.33 (90% CI 0.55−3.18) and 1.24 (90% CI 0.46 − 3.31), respectively. The numerically higher Cmax and AUCinf values of dextromethorphan on day 36 vs. day 1 were likely attributed to the large interpatient variability in PK of dextromethorphan (i.e., %CV for Cmax and AUCinf > 100%). The mean Tmax values of midazolam, S‐warfarin, omeprazole, dextromethorphan, and caffeine was not affected by guselkumab treatment (all P values of exact median test 23 for the difference in median Tmax between day 15 and day 1, or day 36 and day 1 were > 0.05; data on file).
Table 3.
GMRs with 90% CIs of exposure parameters of probe CYP450 substrates after treatment with guselkumab
| Substrate | Parameter | Day 15/day 1 | Day 36/day 1 | ||
|---|---|---|---|---|---|
| n a | GMR (90% CI) | n a | GMR (90% CI) | ||
| Midazolam | Cmax (ng/mL) | 11 | 1.11 (0.75–1.65) | 11 | 1.14 (0.77–1.69) |
| AUCinf (ng·hour/mL) | 11 | 1.01 (0.70–1.45) | 11 | 1.04 (0.75–1.44) | |
| S‐warfarin | Cmax (ng/mL) | 13 | 1.07 (0.90–1.27) | 12 | 0.90 (0.74–1.11) |
| AUCinf (ng·hour/mL) | 13 | 1.12 (0.90–1.40) | 11 | 1.05 (0.82–1.36) | |
| Omeprazole | Cmax (ng/mL) | 12 | 0.96 (0.72–1.28) | 11 | 0.96 (0.67–1.36) |
| AUCinf (ng·hour/mL) | 10 | 0.96 (0.61–1.52) | 6 | 1.19 (0.75–1.90) | |
| Dextromethorphan | Cmax (ng/mL) | 12 | 1.06 (0.46–2.43) | 11 | 1.33 (0.55–3.18) |
| AUCinf (ng·hour/mL) | 8 | 1.13 (0.56–2.28) | 8 | 1.24 (0.46–3.31) | |
| Caffeine | Cmax (ng/mL) | 13 | 1.07 (0.94–1.22) | 11 | 1.06 (0.89–1.26) |
| AUCinf (ng·hour/mL) | 12 | 1.00 (0.77–1.31) | 11 | 1.02 (0.77–1.35) | |
AUCinf, area under the plasma concentration versus time curve from time 0 to infinity with extrapolation of the terminal phase; CI, confidence interval; Cmax, maximum observed plasma concentration; GMR, geometric mean ratio.
Only patients with paired data were included in the comparison (i.e., patients who had both day 1 and day 15 pharmacokinetic (PK) parameters were included in comparison of day 15/day 1 and patients who had both day 1 and day 36 PK parameters were included in comparison of day 36/day 1).
No consistent trends could be identified for each individual AUCinf values of each probe CYP‐substrate, including dextromethorphan, before (day 1) and after guselkumab treatment (days 15 and 36; Figure 3). Similarly, no consistent trend could be identified for individual Cmax values of each probe CYP‐substrate, including dextromethorphan, before and after guselkumab treatment (data on file).
Clinical efficacy
The PASI and IGA data from this study indicated that the majority of patients experienced substantial clinical improvement in their psoriasis after a single s.c. administration of guselkumab 200 mg (Table 4 ). On day 64 (i.e., 8 weeks following guselkumab administration), 9 of 12 patients (75.0%) achieved at least 75% improvement in PASI score from baseline (PASI 75) and 8 of 11 patients (72.7%) achieved an IGA score of clear (0) or minimal (1).
Table 4.
Overview of efficacy results
| Day 19 (1 week + 4 days after initiating guselkumab) | Day 40 (4 weeks + 4 days after initiating guselkumab) | Day 64 (8 weeks after initiating guselkumab) | |
|---|---|---|---|
| Patients with PASI data available, n | 13 | 12 | 12 |
| PASI 100 responders | 0 (0.0) | 1 (8.3) | 2 (16.7) |
| PASI 90 responders | 1 (7.7) | 1 (8.3) | 5 (41.7) |
| PASI 75 responders | 1 (7.7) | 2 (16.7) | 9 (75.0) |
| PASI 50 responders | 5 (38.5) | 9 (75.0) | 11 (91.7) |
| Patients with IGA data available, n | 13 | 11 | 11 |
| IGA 0 responders | 0 (0.0) | 1 (9.1) | 1 (9.1) |
| IGA 0/1 responders | 1 (7.7) | 4 (36.4) | 8 (72.7) |
| IGA ≤ 2 responders | 7 (53.8) | 9 (81.8) | 11 (100.0) |
Values are n (%) unless otherwise indicated.
IGA, Investigator’s Global Assessment; IGA 0 responders, patients achieving an IGA score of cleared (0); IGA0/1 responders, patients achieving an IGA score of cleared (0) or minimal (1); PASI, Psoriasis Area and Severity Index; PASI 50/75/90/100 responders, patients achieving ≥ 50%/75%/90%/100% improvement in PASI score from baseline.
Safety and tolerability
The probe cocktail was generally well‐tolerated when administered alone or in combination with guselkumab to patients with psoriasis. A total of 10 patients reported at least 1 TEAE: 4 patients who received probe cocktail only (after day 1 but before receiving guselkumab on day 8), 1 patient who received guselkumab only (received guselkumab on day 8 but before receiving probe cocktail on day 15), and 5 patients who received probe cocktail after receiving guselkumab (after receiving probe cocktail on day 15; data on file). The rate of TEAEs reported during the period of treatment with probe cocktail in combination with guselkumab (38.5%) was slightly higher than that for treatment with probe cocktail alone (25.0%); overall, there was no pattern suggestive of a meaningful difference in TEAEs between treatment with probe cocktail alone and in combination with guselkumab. The majority of TEAEs were mild or moderate in intensity. No clinically significant changes were observed for vital signs, electrocardiogram, physical examinations, and/or laboratory parameters.
DISCUSSION
The formation of CYP450 enzymes can be altered by increased levels of certain cytokines (e.g., IL‐1, IL‐6, IL‐10, TNFα, and interferon) during chronic inflammation. 18 For example, sirukumab, which targets IL‐6 directly, and tocilizumab, which targets the IL‐6 cell surface receptor, could reverse suppression of CYP enzyme activity in vitro or in vivo. In patients with rheumatoid arthritis, effects of sirukumab on midazolam (CYP3A4‐substrate), omeprazole (CYP2C19‐substrate), and S‐warfarin (CYP2C9‐substrate) were observed, 16 and effects of tocilizumab on simvastatin (CYP3A4‐substrate) and omeprazole (CYP2C19‐substrate) were also reported. 13 Consequently, TP‐drug interaction studies are important for determining the effects of therapeutic monoclonal antibodies that target or modulate cytokines on CYP enzymes. 12 , 25 Although recent studies reported that tildrakizumab and risankizumab (other IL‐23p19 inhibitors) did not have any clinically meaningful effects on CYP metabolism, 2 , 3 the implication of these study results to guselkumab is questionable because guselkumab has different binding affinity to the p19 subunit of human IL‐23, distinct PK profile, and different dosing regimen when compared with tildrakizumab and risankizumab. 26 , 27 , 28
This study was conducted in patients with psoriasis to evaluate whether blocking IL‐23p19 with guselkumab would alter the metabolism of probe substrates for CYP isozymes. Although a single‐dose of guselkumab was used, the dose selected (200 mg) was twofold higher than the therapeutic dose level (100 mg) approved in patients with psoriasis. 18 A sufficiently high level of serum concentration was maintained through week 4. Therefore, the activity of IL‐23 ligand would be fully blocked during the 4‐week study period, which is deemed to be adequate to restore the activities of CYP‐enzymes. Effects of cytokines on CYP‐enzymes were evident following 24–48 hours of incubation of human hepatocytes with known influential cytokines. 29 Overall, results from this exploratory TP‐drug interaction study demonstrate that there were no clinically relevant changes in systemic exposure (Cmax and AUCinf) of midazolam, S‐warfarin, omeprazole, dextromethorphan, and caffeine (probe substrates of CYP3A4, CYP2C9, CYP2C19, CYP2D6, and CYP1A2, respectively) after a single s.c. dose of guselkumab. These results confirm in vitro findings that IL‐23 inhibition does not modulate the expression or activity of multiple CYP450 enzymes, 1 and indicate that drug interactions between guselkumab and substrates of various CYP enzymes are unlikely in patients with psoriasis. Our findings also suggest that the metabolic activity of CYP3A4, CYP2C9, CYP2C19, CYP2D6, and CYP1A2 was not affected by the decreased inflammation associated with improvement in disease achieved by blocking IL‐23 in patients with psoriasis.
Dextromethorphan, a substrate of CYP2D6 (and also CYP3A4), has been widely used as a probe substrate for the measurement of CYP2D6 activity, as assessed by dextromethorphan exposure in plasma, or metabolic ratios of dextromethorphan to dextrorphan (its major metabolite) in urine or plasma. 30 Due to the large sample size required and the low correlation of urinary metabolic ratio of dextromethorphan to dextrorphan with oral clearance of dextromethorphan, 31 urine samples were not collected in this study. Large interpatient variability in PK parameters of dextromethorphan has been reported, with CV% of plasma AUC values over 100% in extensive metabolizers, 32 which might be related to the high interpatient variability in CYP2D6 hepatic intrinsic clearance (~ 60–70% CV% among extensive metabolizers). 33 Dextromethorphan has also been reported to exhibit moderate‐to‐large intrapatient variability (37–56%) in metabolic ratios, 34 which would further complicate the assessment of CYP2D6‐related drug interactions using this probe substrate. The large interpatient variability in dextromethorphan PK observed in this study (CV% ~ 115–130% for Cmax and AUCinf) was consistent with literature reports cited above.
Because increased cytokine levels may downregulate CYPs, blockade of cytokines may enhance CYP activity and consequently lead to reduction in systemic exposure of drugs metabolized by CYPs. In this study, however, the systemic exposure of dextromethorphan following guselkumab treatment seemed to be higher rather than lower. This cannot be explained by inhibition of the potentially downregulating effect of IL‐23 on CYPs by guselkumab. The numerically higher mean Cmax and AUCinf values of dextromethorphan at 4 weeks following guselkumab treatment were more likely attributable to the large interpatient and intrapatient variability in the PK of dextromethorphan. Regardless of the numeric differences in the geometric mean values of Cmax and AUCinf for dextromethorphan before and after treatment with guselkumab, no consistent trend could be identified for the individual Cmax and AUCinf values of dextromethorphan before and after guselkumab treatment, further demonstrating that systemic exposure of dextromethorphan was not affected by treatment with guselkumab.
Based on the prespecified criteria, some PK parameters were excluded from statistical analyses (Table S1 ). Additional sensitivity analyses were conducted to include all paired PK parameters, including the outliers for omeprazole and dextromethorphan, in the statistical comparison. The results from the sensitivity analysis (data on file) were similar to those presented from the original analysis. With the inclusion of the outliers, the GMRs were slightly closer to 1, but the 90% CIs were slightly larger.
The efficacy of guselkumab for treatment of psoriasis observed in this study was expected and comparable to published results. 8 , 9 The probe cocktail of CYP substrates was generally well‐tolerated when administered alone or in combination with guselkumab. In two large‐scale phase III studies (N = 1,721) with up to 156 weeks of follow‐up, the rates of serious infections, infections requiring treatment, malignancies, and major adverse cardiovascular events were low and stable with increased duration of exposure to guselkumab over time. 10
Compared with the in vitro study, 1 this phase I study is more informative in predicting clinically relevant interactions between guselkumab and small molecule drugs. However, this exploratory study had the limitation of small sample size, which may not be sufficient to definitively assess a potentially downregulating effect of IL‐23 on dextromethorphan (CYP2D6 substrate) by guselkumab. Drug‐drug interaction studies with a cocktail of substrates are generally not powered to use the strict bioequivalence criteria to determine the presence or absence of drug‐drug interactions for all evaluated exposure parameters of each component of the cocktail. 3
Based on the findings from this study, drug interactions between guselkumab and substrates of various CYP enzymes are unlikely. Consequently, dose adjustment for concomitant CYP substrates in patients treated with guselkumab does not seem to be necessary.
Funding
This study was funded by Janssen Research & Development, LLC.
Conflicts of Interest
All authors are employed by Janssen Research & Development, LLC, and own stocks in Johnson & Johnson, of which Janssen is a subsidiary.
Author Contributions
All authors wrote the manuscript, designed the research, performed the research, and analyzed the data.
Figure 2.
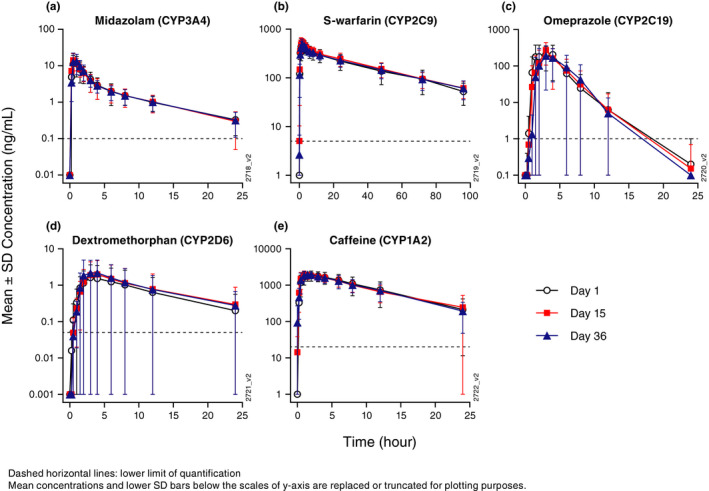
Arithmetic mean (± SD) plasma concentration‐time profiles of: (a) midazolam, (b) S‐warfarin, (c) omeprazole, (d) dextromethorphan, and (e) caffeine before and after treatment with guselkumab. SD, standard deviation
Figure 3.
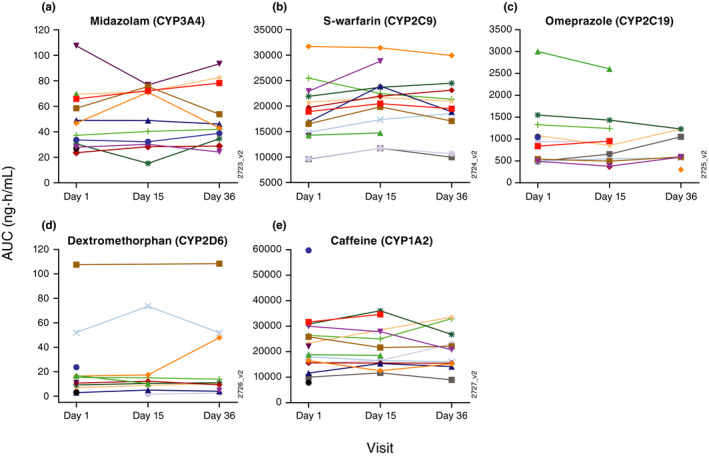
Individual AUCinf of: (a) midazolam, (b) S‐warfarin, (c) omeprazole, (d) dextromethorphan, and (e) caffeine before and after treatment with guselkumab. AUC, area under the curve; AUCinf, AUC from time 0 to infinity with extrapolation of the terminal phase.
Supporting information
Table S1
Acknowledgments
The authors thank the patients, investigators, and study personnel who made this phase I study successful. We also thank Kristin Ruley Sharples, PhD, of Janssen Scientific Affairs, LLC, for assistance with manuscript preparation and submission.
References
- 1. Dallas, S. , Chattopadhyay, S. , Sensenhauser, C. , Batheja, A. , Singer, M. & Silva, J. Interleukins‐12 and ‐23 do not alter expression or activity of multiple cytochrome P450 enzymes in cryopreserved human hepatocytes. Drug Metab. Dispos. 41, 689–693 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Khalilieh, S. et al Effect of tildrakizumab (MK‐3222), a high affinity, selective anti‐IL23p19 monoclonal antibody, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br. J. Clin. Pharmacol. 84, 2292–2302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khatri, A. , Cheng, L. , Camez, A. , Ignatenko, S. , Pang, Y. & Othman, A.A. Lack of effect of 12‐Week treatment with risankizumab on the pharmacokinetics of cytochrome P450 probe substrates in patients with moderate to severe chronic plaque psoriasis. Clin. Pharmacokinet. 58, 805–814 (2019). [DOI] [PubMed] [Google Scholar]
- 4. Weger, W. Current status and new developments in the treatment of psoriasis and psoriatic arthritis with biological agents. Br. J. Pharmacol. 160, 810–820 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nijsten, T. , Margolis, D.J. , Feldman, S.R. , Rolstad, T. & Stern, R.S. Traditional systemic treatments have not fully met the needs of psoriasis patients: results from a national survey. J. Am. Acad. Dermatol. 52, 434–444 (2005). [DOI] [PubMed] [Google Scholar]
- 6. Nestle, F.O. , Kaplan, D.H. & Barker, J. Psoriasis. N. Engl. J. Med. 361, 496–509 (2009). [DOI] [PubMed] [Google Scholar]
- 7. Lee, E.B. , Amin, M. , Bhutani, T. & Wu, J.J. Emerging therapies in psoriasis: a systematic review. Cutis 101, 5–9 (2018). [PubMed] [Google Scholar]
- 8. Blauvelt, A. et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double‐blinded, placebo‐ and active comparator‐controlled VOYAGE 1 trial. J. Am. Acad. Dermatol. 76, 405–417 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Reich, K. et al Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 76, 418–431 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Reich, K. et al Maintenance of clinical response and consistent safety profile with up to three years of continuous treatment with guselkumab: results from the VOYAGE 1 and VOYAGE 2 trials. J. Am. Acad. 82, 936–945 (2020). [DOI] [PubMed] [Google Scholar]
- 11. Tabrizi, M.A. , Tseng, C.M. & Roskos, L.K. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov. Today 11, 81–88 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Girish, S. et al AAPS workshop report: strategies to address therapeutic protein‐drug interactions during clinical development. AAPS J. 13, 405–416 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim, S. , Östör, A.J. & Nisar, M.K. Interleukin‐6 and cytochrome‐P450, reason for concern? Rheumatol. Int. 32, 2601–2604 (2012). [DOI] [PubMed] [Google Scholar]
- 14. Morgan, E.T. Impact of infectious and inflammatory disease on cytochrome P450‐mediated drug metabolism and pharmacokinetics. Clin. Pharmacol. Ther. 85, 434–438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou, H. & Mascelli, M.A. Mechanisms of monoclonal antibody‐drug interactions. Annu. Rev. Pharmacol. Toxicol. 51, 359–372 (2011). [DOI] [PubMed] [Google Scholar]
- 16. Zhuang, Y. et al Evaluation of disease mediated therapeutic protein‐drug interactions between an anti‐interleukin‐6 monoclonal antibody (sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J. Clin. Pharmacol. 55, 1386–1394 (2015). [DOI] [PubMed] [Google Scholar]
- 17. Evers, R. et al Critical review of preclinical approaches to investigate cytochrome p450‐mediated therapeutic protein drug‐drug interactions and recommendations for best practices: a white paper. Drug Metab. Dispos. 41, 1598–1609 (2013). [DOI] [PubMed] [Google Scholar]
- 18. TREMFYA® (Guselkumab) [package insert]. Janssen Biotech Inc, Horsham, PA, USA; November 2019. [Google Scholar]
- 19. Chainuvati, S. et al Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the "Cooperstown 5+1 cocktail". Clin. Pharmacol. Ther. 74, 437–447 (2003). [DOI] [PubMed] [Google Scholar]
- 20. Zhu, Y. et al Use of phase 2 guselkumab exposure response relationship in psoriasis to inform phase 3 dose selection. 26th European Academy of Dermatology and Venereology Congress, September 13–17, 2017, Geneva, Switzerland (Poster 1827).
- 21. Gabrielsson, J. & Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis: Concepts and Applications, 4th edn. (Swedish Pharmaceutical Press, Sweden, 2007). [Google Scholar]
- 22. Dixon, W.J. Processing data for outliers. Biometrics 9, 74–89 (1953). [Google Scholar]
- 23. Conover, W.J. Practical Nonparametric Statistics, 3rd edn. (John Wiley & Sons, Hoboken, NJ, 1999). [Google Scholar]
- 24. Yao, Z. et al Population pharmacokinetic modeling of guselkumab, a human IgG1λ monoclonal antibody targeting IL‐23, in patients with moderate to severe plaque psoriasis. J. Clin. Pharmacol. 58, 613–627 (2018). [DOI] [PubMed] [Google Scholar]
- 25. US Food and Drug Administration (FDA) Guidance for Industry . Clinical drug interaction studies — study design, data analysis, and clinical implications. 2017. <https://www.fda.gov/media/82734/download>. Accessed September 3, 2019.
- 26. US Food and Drug Administration . Clinical pharmacology review. 761061Orig1s000. [BLA#761021] 2016. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761061Orig1s000MultidisciplineR.pdf>. Accessed February 25, 2020.
- 27. US Food and Drug Administration . Clinical pharmacology review. 761067Orig1s000. [BLA#761027] 2017. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761067Orig1s000MultdisciplineR.pdf>. Accessed February 25, 2020.
- 28. US Food and Drug Administration . Clinical pharmacology review. 761105Orig1s000. [BLA#761105] 2019. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761105Orig1s000MultidisciplineR.pdf>. Accessed February 25, 2020.
- 29. Guillén, M.I. et al Oncostatin M down‐regulates basal and induced cytochromes P450 in human hepatocytes. J. Pharmacol. Exp. Ther. 285, 127–134 (1998). [PubMed] [Google Scholar]
- 30. Capon, D.A. , Bochner, F. , Kerry, N. , Mikus, G. , Danz, C. & Somogyi, A.A. The influence of CYP2D6 polymorphism and quinidine on the disposition and antitussive effect of dextromethorphan in humans. Clin. Pharmacol. Ther. 60, 295–307 (1996). [DOI] [PubMed] [Google Scholar]
- 31. Borges, S. , Li, L. , Hamman, M.A. , Jones, D.R. , Hall, S.D. & Gorski, J.C. Dextromethorphan to dextrorphan urinary metabolic ratio does not reflect dextromethorphan oral clearance. Drug Metab. Dispos. 33, 1052–1055 (2005). [DOI] [PubMed] [Google Scholar]
- 32. Vetticaden, S.J. et al Phenotypic differences in dextromethorphan metabolism. Pharm. Res. 6, 13–19 (1989). [DOI] [PubMed] [Google Scholar]
- 33. Chiba, K. , Kato, M. , Ito, T. , Suwa, T. & Sugiyama, Y. Inter‐individual variability of in vivo CYP2D6 activity in different genotypes. Drug Metab. Pharmacokinet. 27, 405–413 (2012). [DOI] [PubMed] [Google Scholar]
- 34. Zhou, H. , Tong, Z. & McLeod, J.F. "Cocktail" approaches and strategies in drug development: valuable tool or flawed science? J. Clin. Pharmacol. 44, 120–134 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1