

Abstract
A low lymphocyte count puts immune‐compromised patients at risk of mortality. hIL‐7‐hyFc is a homodimeric interleukin‐7 (IL‐7), a potent T‐cell amplifier, fused to the hybridizing IgD/IgG4 immunoglobulin domain. We performed a randomized, double‐blind, placebo‐controlled, dose‐escalation, phase I study to assess the pharmacokinetic, pharmacodynamic, safety, tolerability, and immunogenicity profiles of hIL‐7‐hyFc administered s.c. and i.m. to healthy volunteers. Thirty subjects randomly received hIL‐7‐hyFc or its matching placebo in an 8:2 ratio at 20, 60 μg/kg s.c., or 60 μg/kg i.m. The hIL‐7‐hyFc was slowly absorbed and its terminal half‐life was 63.26 hours after i.m. administration. The hIL‐7‐hyFc increased absolute lymphocyte count, mostly in T‐cells, which peaked 3 weeks after administration and then lasted for several additional weeks. The hIL‐7‐hyFc was well‐tolerated after a single s.c. and i.m. administration. Injection site reaction was the most common treatment‐emergent adverse event, which resolved spontaneously without treatment. The hIL‐7‐hyFc can be developed into a beneficial treatment option for patients with compromised T‐cell immunity. This trial was registered at www.clinicaltrials.gov as #NCT02860715.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
☑ Exogenously administered interleukin (IL)‐7 has shown to increase T cells in various immune‐deficient patients. However, exogenously administered IL‐7, particularly nonglycosylated IL‐7, are known to have a relatively short half‐life.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We performed this study to assess the pharmacokinetics, pharmacodynamics, safety, and tolerability profiles of hIL‐7‐hyFc, a novel long‐acting IL‐7, in humans.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Our results clearly showed that hIL‐7‐hyFc stays longer in the body, is well‐tolerated, and increases T cells in a dose‐dependent manner.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Our results provide evidence that a single dose of hIL‐7‐hyFc can be further developed into a beneficial treatment option for a variety of patients with a low T‐cell count or compromised T‐cell immunity. Several phase II studies in immune‐compromised patients or patients with a low T‐cell count are currently ongoing (ClinicalTrials.gov NCT03478995, NCT03619239, NCT03733587, and NCT03752723).
Decrease in lymphocyte counts caused by anticancer therapy is associated with frequent relapse, severe toxicity, and mortality. 1 , 2 , 3 Likewise, increased neutrophil‐to‐lymphocyte ratio was a poor prognostic predictor in patients with cancer. 4 Furthermore, patients with a low lymphocyte count were significantly less benefited from immune checkpoint inhibitors. 5 Additionally, low lymphocyte count increased the risk for serious opportunistic infections in patients with AIDS, primary immunodeficiency, and idiopathic CD4 lymphopenia or patients receiving chronic immunosuppressive therapy. 6 , 7 , 8 , 9 , 10 , 11
Interleukin‐7 (IL‐7) plays a critical role in the development, proliferation, maintenance, and reinvigoration of T‐cells. 12 The effects of IL‐7 are mediated via a heterodimeric receptor consisting of the IL‐7 receptor α‐chain (IL‐7Rα) and the common cytokine receptor γ‐chain. IL‐7Rα, expressed on naïve T‐cells, is downregulated upon activation of IL‐7‐mediated signaling and re‐expressed in memory T‐cells. 13 , 14
Exogenously administered IL‐7 to humans and mice proliferated T‐cells in the peripheral blood, increased tumor infiltrating lymphocytes, enhanced the survival of T‐cells, and generated memory T‐cells. 15 , 16 , 17 Furthermore, exogenously administered IL‐7 was well‐tolerated and increased the number of T‐cells in a dose‐dependent manner in patients with viral infections (e.g., HIV and hepatitis B and C), cancer, and rare disease, such as progressive multifocal leukoencephalopathy and idiopathic CD4 lymphopenia. 18 , 19 , 20 , 21 , 22 However, exogenously administered IL‐7 had a relatively short half‐life in humans, particularly for non‐glycosylated IL‐7, which ranged only 6.46–9.80 hours, 15 similar to other cytokines. 23 , 24 Therefore, a novel long‐standing formulation of IL‐7 has been an unmet medical need.
The hIL‐7‐hyFc is a homodimeric IL‐7 fused to the hybridizing IgD/IgG4 immunoglobulin domain (hyFc). The hyFc region in hIL‐7‐hyFc is composed of the hinge and N‐terminal portion of the heavy chain constant region 2 (hinge‐CH2) of human IgD, which is fused to the C‐terminal region of CH2 and the entire CH3 region of human IgG4. The hyFc region helps IL‐7 stay longer in the body through neonatal Fc receptor‐mediated recycling. 25 A single s.c. administration of hIL‐7‐hyFc at 0.1, 0.3, or 1.0 mg/kg in rats increased the peripheral lymphocyte count by ~ 1.5‐fold. Additionally, the terminal half‐life of hIL‐7‐hyFc after a single s.c. administration in rats ranged from 19–25 hours.
Based on the preclinical findings that support the notion that hIL‐7‐hyFc can be developed as a beneficial therapeutic option for patients with low lymphocyte count, we performed a phase I study to assess the pharmacokinetic (PK), pharmacodynamic (PD), safety, tolerability, and immunogenicity profiles of hIL‐7‐hyFc in humans.
Methods
Study design and subjects
This was a randomized, double‐blind, placebo‐controlled, dose‐escalation, phase I study with hIL‐7‐hyFc after a single s.c. and i.m. administration in healthy subjects. Men and women 19–45 years of age were eligible if they were healthy at screening, which was performed 4 weeks before study drug administration, on the basis of medical history, physical examination, vital signs (i.e., blood pressure, pulse rate, body temperature, and respiratory rate), 12‐lead electrocardiography, serology (i.e., hepatitis B, hepatitis C, and human immunodeficiency virus), clinical laboratory tests (i.e., hematology, chemistry, coagulation, and urinalysis), breath alcohol test, and urinary drug screening.
Eligible subjects were admitted to the clinical trial center of Seoul National University Hospital the night before dosing. After an overnight fasting of > 12 hours, subjects randomly received hIL‐7‐hyFc or its matching placebo in a ratio of 8:2 at one of the following doses and routes of administration: 20 μg/kg s.c., 60 μg/kg s.c., or 60 μg/kg i.m. The no observed adverse effect level of hIL‐7‐hyFc determined from the repeated dose toxicity study was 6 mg/kg s.c. in cynomolgus monkeys, which is translated into a human equivalent dose of 1.9 mg/kg s.c. To ensure the safety of hIL‐7‐hyFc, we applied a safety factor of 95. Therefore, the starting dose in this study (i.e., 20 μg/kg s.c.) was considered sufficiently safe. After completing the protocol‐specified procedures up to 72 hours postdose, subjects were discharged. On day 28 postdose, the investigators decided whether to escalate dose (i.e., 20–60 μg/kg s.c.) and to switch the route of administration (i.e., s.c. to i.m. at 60 μg/kg) based on the safety data collected by that day.
This study was approved by the Institutional Review Board of Seoul National University Hospital, and conducted in full accordance with the principles of the Declaration of Helsinki and Korean Good Clinical Practice (www.clinicaltrials.gov registration number: NCT02860715). Written informed consent was obtained from each subject before any study‐related procedure was performed.
Determination of serum concentrations of IL‐7 and PK analysis
For PK analysis, serial blood samples were collected on day −1, and at 0 (i.e., predose), 1, 2, 4, 6, 8, 11, 14, 24, 36, 48, 60, 72, 96, 168, 240, 336, 504, and 672 hours postdose. Serum IL‐7 concentrations were determined using Quantikine HS ELISA Human IL‐7 immunoassay kit (R&D Systems, Minneapolis, MN). Briefly, the calibration standards, quality control samples, and blood samples were added to a precoated plate with the capture antibody, specific for human IL‐7. After the first incubation, the plate was washed to remove any unbound material. Then, a human IL‐7 antibody conjugate was added to the wells, and this antibody bound to the immobilized IL‐7 captured during the first incubation. Following the second incubation and washing to remove the excess conjugate, the detection solution for the human IL‐7 bound complex was added and was converted to a color signal. The intensity of this colored product was directly proportional to the concentration of human IL‐7. The lower limit of quantitation was 0.031 ng/mL, and the calibration curve was linear over the concentration range of 0.031–2.00 ng/mL. The accuracy ranged 92.48–97.53% and the precision was ≤ 7.24% (coefficient of variation). The baseline‐adjusted IL‐7 concentrations were obtained by subtracting the mean value of concentrations determined before dose (i.e., at screening, day‐1, and 0 hour (predose)).
PK parameters were derived using a noncompartmental approach in the Phoenix WinNonlin software (version 7.0, Certara, St. Louis, MO). The maximum concentration (Cmax) and the time to reach Cmax (Tmax) were obtained directly from the observed values. The area under the serum concentration vs. time curve (AUC) from time zero to the last measurable time‐point (AUClast) was determined using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. On the other hand, the AUC from time zero to infinity (AUCinf) was calculated as AUClast + C t/λz, where Ct is the last measurable concentration and λz is the elimination rate constant estimated from linear regression of the log‐linear portion of the serum concentration–time curve. The terminal half‐life (t 1/2) was the natural logarithm of 2 divided by λz. The apparent clearance was the dose divided by AUCinf.
PD analysis
The PD variables were absolute lymphocyte count (ALC), absolute T‐cell count, and absolute B‐cell count, assessed at screening, on day −1, and at 0, 72, 168, 240, 336, 504, 672, and 1,344 hours postdose. Absolute T‐cell and B‐cell counts were ALCs multiplied by the proportions of CD3+ and CD19+ cells against total lymphocytes, respectively, determined using multicolor flow cytometry. Cryopreserved peripheral blood mononuclear cells were thawed and stained using the LIVE/DEAD Fixable Red Dead Cell Stain Kit (Life Technologies, Waltham, MA). The cells were then washed once and stained with anti‐CD3 (UCTH1; BD Biosciences, San Jose, CA) and anti‐CD19 (HIB19; BD Biosciences) in the dark at 4°C for 30 minutes. Multicolor flow cytometry was performed on an LSR II instrument (BD Biosciences) and data were analyzed using FlowJo software (Treestar, Ashland, OR).
For the PD variables, the percent change from baselines was calculated, where the baseline value was the average of those measured at screening, on day –1, and at 0 hour postdose on day 1. Furthermore, the maximum ALC and the area under the ALC‐time curve until the last sampling time were calculated using the Phoenix WinNonlin software after correcting the baseline values.
Safety, tolerability, and immunogenicity assessments
The safety and tolerability of hIL‐7‐hyFc was assessed for 56 days postdose through adverse event (AE) monitoring, vital signs, and physical examination, including evaluation of the injection sites, clinical laboratory tests, 12‐lead electrocardiography, upper abdomen ultrasound, and measurement of auto‐antibodies (i.e., antinuclear antibody and anti‐dsDNA antibody). Antidrug antibody (ADA) and neutralizing antibody (nAb), if ADA was positive, were determined prior to study drug administration (day 0), and 7, 10, 14, 28, and 56 days after study drug administration. If subjects were positive on ADA, they were further followed up to 20 months after study drug administration with a 1‐month to 6‐month interval.
ADA and nAb against hIL‐7‐hyFc were determined using a validated enzyme‐linked immunosorbent assay with a sensitivity of 0.01 µg/mL. Briefly, the samples were incubated with hIL‐7‐hyFc coated plates. After incubation, the plates were washed and the conjugate (biotin‐labeled hIL‐7‐hyFc) was added. After another incubation and wash, the bound complex was detected using streptavidin‐horseradish peroxidase, which was visualized with tetramethylbenzidine substrate. All of the ADA‐positive samples were analyzed for nAbs.
Statistical analysis
All of the demographic characteristics, PK, and PD parameters are summarized using descriptive statistics. Comparability of the demographic characteristics between treatment groups were assessed using the analysis of variance test. On the other hand, the Kruskal–Wallis test was used to compare the PK and PD parameters between treatment groups. Additionally, linear regression was used to assess the PK‐PD relationship. A nonparametric Spearman correlation coefficient was derived between the PK parameters (V d/F, CL/F, Cmax, and AUClast) and body weight in each treatment group to assess the correlation. A P value of < 0.05 was considered statistically significant. All analyses were conducted using the SAS software (version 9.4; SAS Institute, Cary, NC).
Results
Subjects
We enrolled a total of 31 subjects, one of whom withdrew consent and was dropped out before the study drug was administered. As a result, 30 subjects (20 men and 10 women) completed the study, and their demographic characteristics were comparable between the treatment groups (P value > 0.05, analysis of variance). The mean ± SD of age, height, body weight, and body mass index of the subjects were 27.1 ± 6.0 years, 1.70 ± 0.08 m, 65.3 ± 11.1 kg, and 22.4 ± 2.4 kg/m2, respectively.
Pharmacokinetics
The hIL‐7‐hyFc was more rapidly absorbed after i.m. administration than after s.c. administration (median T max: 4 vs. 36 hours postdose for i.m. vs. s.c., respectively; Table 1 ), and the exposure to hIL‐7‐hyFc after i.m. administration was also ~ 2 times greater than that after s.c. administration at the same dose of 60 μg/kg (mean AUCinf: 83.4 vs. 33.3 h∙μg/L for i.m. vs. s.c., respectively; Table 1 ). The hIL‐7‐hyFc was slowly removed from the body such that it was still detectable 168 hours postdose (Figure 1 ). The mean t 1/2 ranged from 26.83–52.70 hours after s.c. administration and was 63.26 hours after i.m. administration, which showed considerable variability among subjects. After s.c. administration, the exposure to IL‐7 increased in a dose‐proportional manner (Figure 1 , Table 1 ). None of the PK parameters, including V d/F, CL/F, Cmax, and AUClast, showed a statistically significant correlation with body weight.
Table 1.
Pharmacokinetic parameters of hIL‐7‐hyFc after a single subcutaneous and intramuscular administration
| Pharmacokinetic parameters | 20 μg/kg s.c. (N = 8) | 60 μg/kg s.c. (N = 8) | 60 μg/kg i.m. (N = 8) |
|---|---|---|---|
| Tmax, hour | 42.0 (6.0–72.0) | 36.0 (4.0–72.0) | 4.0 (2.0–35.8) |
| Cmax, μg/L | 0.1 ± 0.0 | 0.5 ± 0.3 | 1.2 ± 0.5 |
| AUClast, h∙μg/L | 10.2 ± 2.0 | 31.0 ± 8.4 | 64.9 ± 25.1 |
| AUCinf, h∙μg/L | 12.4 ± 2.8 | 33.3 ± 8.8 | 83.4 ± 57.9 |
| t 1/2, hour | 52.7 ± 32.1 | 26.8 ± 15.6 | 63.3 ± 49.4 |
| CL/F, L/hour | 101.3 ± 25.2 | 128.4 ± 31.4 | 55.5 ± 20.8 |
| V d/F, L | 7243.3 ± 3500.7 | 5097.8 ± 3707.5 | 4577.9 ± 3666.4 |
AUCinf, area under the serum concentration vs. time curve from time zero to infinity; AUClast, area under the serum concentration vs. time curve from time zero to the last measurable time‐point; CL/F, apparent clearance; Cmax, maximum concentration; t 1/2, terminal half‐life; Tmax, time to reach maximum concentration; V d/F, apparent volume of distribution.
All data are presented as mean ± SD, except for Tmax, which are presented as median (minimum–maximum).
Figure 1.
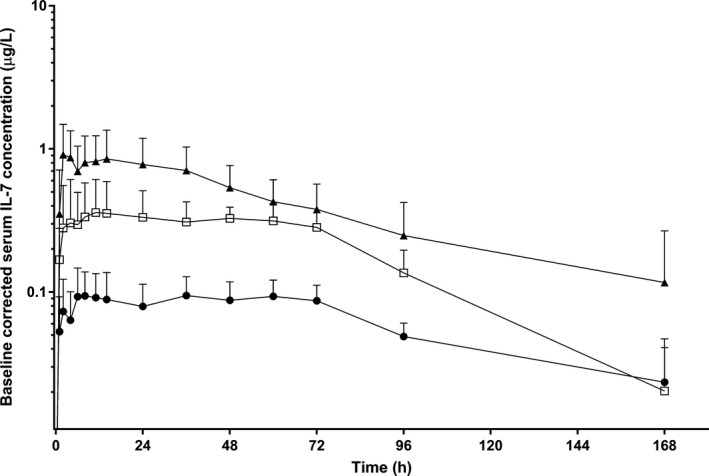
Mean serum concentration‐time profiles of interleukin (IL)‐7 after a single subcutaneous or intramuscular administration of hIL‐7‐hyFc. The error bars denote the SDs (● = 20 μg/kg s.c., N = 8; □ = 60 μg/kg s.c., N = 8; ▲ = 60 μg/kg i.m., N = 8).
Pharmacodynamics
The hIL‐7‐hyFc markedly increased ALC in a dose‐dependent manner after an initial decrease for 4–5 days, whereas ALC remained stable after placebo (Figure 2a,b ). Furthermore, the increase in ALC was greater after i.m. administration than after s.c. administration at the same dose of 60 μg/kg. The peak ALC was reached in ~ 3 weeks postdose, after which the increase in ALC lasted for several additional weeks. Furthermore, the increase in ALC was mostly due to the increase in T‐cells (Figure 2c,d ), whereas the effect of hIL‐7‐hyFc on B‐cells was minimal and variable (Figure 2e,f ). Compared with placebo, hIL‐7‐hyFc significantly more increased the baseline‐corrected maximum ALC (ΔEmax) and the area under the ALC‐time curve until the last sampling time (ΔAUEC) in a dose‐dependent manner (ΔEmax 316, 638, 1,589, and 1,763/µL for placebo, 20 μg/kg s.c., 60 μg/kg s.c., 60 μg/kg i.m., respectively; ΔAUEC 147708, 416752, 1119131, and 1439902 h/µL for placebo, 20 μg/kg s.c., 60 μg/kg s.c., and 60 μg/kg i.m., respectively; Figure 3 ).
Figure 2.
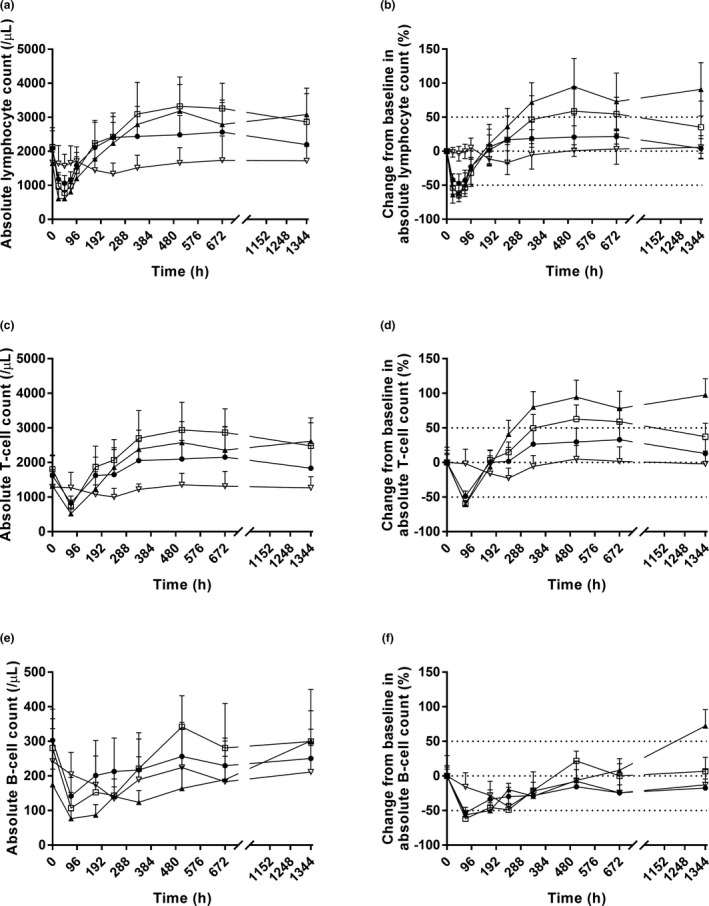
Time course of mean pharmacodynamic parameters after a single subcutaneous or intramuscular administration of hIL‐7‐hyFc or placebo. (a) Absolute lymphocyte count. (b) Percent change from baseline in absolute lymphocyte count. (c) Absolute T‐cell count. (d) Percent change from baseline in absolute T‐cell count. (e) Absolute B‐cell count. (f) Percent change from baseline in absolute B‐cell count. The error bars denote the SDs. Upper dotted line: 50% change from baseline; middle dotted line: 0% change from baseline; lower dotted line: −50% change from baseline (● = 20 μg/kg s.c., N = 8; □ = 60 μg/kg s.c., N = 8; ▲ = 60 μg/kg i.m., N = 8; ▽ = placebo, N = 6).
Figure 3.
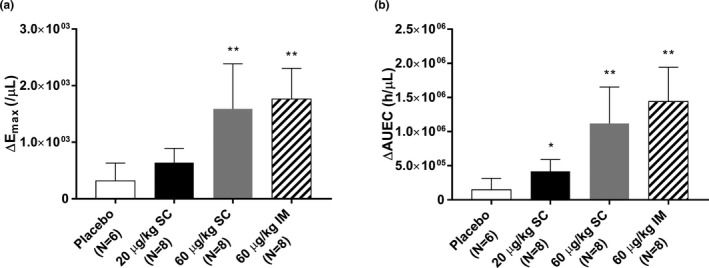
Baseline‐corrected pharmacodynamic parameters after a single subcutaneous or intramuscular administration of hIL‐7‐hyFc or placebo. (a) maximum absolute lymphocyte count (ΔEmax), (b) area under the absolute lymphocyte count‐time curve until the last sampling time (ΔAUEC). The error bars represent the standard deviations (*P value < 0.05 vs. placebo; **P value < 0.01 vs. placebo).
PK‐PD relationship
There was a time delay between the PK and PD of hIL‐7‐hyFc (i.e., the maximum effect on the increase in ALC was seen much later than when the peak serum concentration of hIL‐7‐hyFc occurred; Figure S1 ). The increase in ALC, as assessed by ΔEmax and ΔAUEC, was positively correlated with the systemic exposure to IL‐7 (i.e., Cmax and AUClast), particularly between ΔAUEC and Cmax (R 2 = 0.6039; Figure 4 ).
Figure 4.
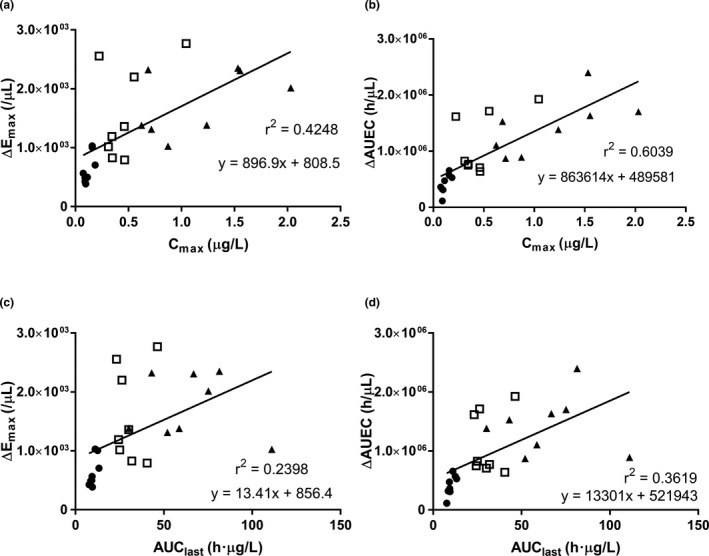
Relationships between individual pharmacokinetic parameters vs. pharmacodynamic parameters. (a) maximum concentration (Cmax) of interleukin (IL)‐7 vs. maximum absolute lymphocyte count (ΔEmax), (b) Cmax of IL‐7 vs. absolute lymphocyte count‐time curve until the last sampling time (ΔAUEC), (c) area under the serum concentration vs. time curve (AUC) from time zero to the last measurable time‐point (AUClast) of IL‐7 vs. ΔEmax, (d) AUClast of IL‐7 vs. ΔAUEC of absolute lymphocyte count after a single subcutaneous or intramuscular administration of IL‐7 (● = 20 μg/kg s.c., N = 8; □ = 60 μg/kg s.c., N = 8; ▲ = 60 μg/kg i.m., N = 8).
Safety, tolerability, and immunogenicity
Overall, hIL‐7‐hyFc was safe and well‐tolerated after a single s.c. and i.m. administration at 20–60 μg/kg (Table 2 ). A total of 58 AEs occurred in 26 subjects (86.7%), all of which resolved spontaneously. The majority of AEs were mild in intensity, and no death or serious AE was seen during the entire study. The most frequent AE was injection site reactions (n = 25), the intensity of which was dose‐dependent such that they were all grade 1 in the 20 μg/kg s.c. group, whereas all subjects in the 60 μg/kg s.c. group experienced grade 2. Additionally, injection site reactions were much milder after i.m. administration than after s.c. administration; only one subject in the 60 μg/kg i.m. group reported a grade 2 injection site reaction with the rest grade 1 (Figure S2 ). Two subjects reported an enlargement of the inguinal lymph node on days 5 and 10, respectively, after SC hIL‐7‐hyFc at 60 µg/kg, which resolved subsequently. One subject in the 60 μg/kg s.c. group experienced a grade 3 elevation in alanine transaminase (ALT) on day 1 (one day after dosing) without any other clinical signs and symptoms. In this subject, the ALT returned to normal within 7 months without any treatment. Nonalcoholic steatohepatitis was strongly suggested for the subject, who gained 6 kg in body weight rapidly over a month after enrollment. The increase in ALT in this subject was judged “unlikely related” to the study drug.
Table 2.
Summary of adverse events by treatment
|
System organ class preferred term |
Placebo (N = 6) | hIL‐7‐hyFc | |||
|---|---|---|---|---|---|
| 20 μg/kg s.c. (N = 8) | 60 μg/kg s.c. (N = 8) | 60 μg/kg i.m. (N = 8) | Total (N = 24) | ||
| Number of subjects with AEs | 2 (33.3) [10] | 8 (100.0) [11] | 8 (100.0) [23] | 8 (100.0) [14] | 24 (100) [48] |
| Number of subjects with AEs that were reported by more than one subject | |||||
| Blood and lymphatic system disorders | |||||
| Lymphadenopathy | 2 (25.0) [2] | 2 (8.3) [2] | |||
| General disorders and injection site conditions | |||||
| Injection site reaction | 1 (16.7) [1] | 8 (100.0) [8] | 8 (100.0) [8] | 8 (100.0) [8] | 24 (100) [24] |
| Pyrexia | 1 (16.7) [1] | 1 (12.5) [1] | 1 (4.2) [1] | ||
| Nervous system disorders | |||||
| Headache | 2 (33.3) [3] | 1 (12.5) [1] | 1 (12.5) [1] | 2 (8.3) [2] | |
| Dizziness | 1 (12.5) [1] | 1 (12.5) [1] | 2 (8.3) [2] | ||
| Respiratory, thoracic, and mediastinal disorders | |||||
| Oropharyngeal pain | 1 (16.7) [1] | 1 (12.5) [1] | 1 (4.2) [1] | ||
| Productive cough | 1 (33.3) [2] | 1 (12.5) [1] | 1 (4.2) [1] | ||
AEs, adverse events.
The values are displayed as number of subjects (percentage of subjects) [number of events].
Denominator of percentage is the number of subjects in the column.
ADA was positive in 75%, 100%, and 100% of subjects 56 days after hIL‐7‐hyFc at 20 μg/kg s.c., 60 μg/kg s.c., and 60 μg/kg i.m., respectively, whereas no subject receiving placebo became ADA‐positive (Figure 5a ). A subject receiving i.m. hIL‐7‐hyFc at 60 μg/kg was ADA‐positive before treatment, who was also ADA‐positive as early as 10 days after administration. Further follow‐up of these subjects up to 600 days after administration, however, showed many of them became ADA‐negative. Approximately, 30–50% of ADA was neutralizing, which eventually became undetected 330 days after s.c. or i.m. administration at 60 μg/kg (Figure 5b ). No abnormality in clinical laboratory tests or physical examination was seen in those nAb‐positive subjects.
Figure 5.
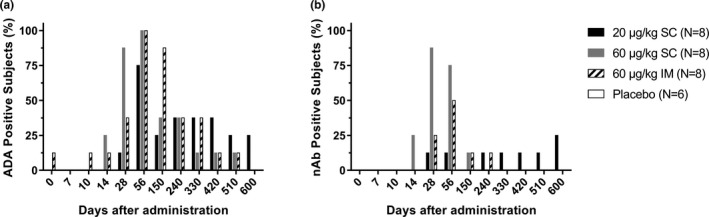
Percentages of subjects positive on anti‐drug antibody against hIL‐7‐hyFc over time after administration by treatment group. (a) anti‐drug antibody (ADA), (b) neutralizing antibody (nAb).
Discussion
We successfully assessed the PK, PD, safety, and tolerability profiles of hIL‐7‐hyFc in humans. The mean t 1/2 of hIL‐7‐hyFc (s.c.: 26.8–52.7 hours, i.m.: 63.3 hours; Table 1 ) was much longer than those seen with non‐hyFc human recombinant IL‐7s (< 10 hours for nonglycosylated IL‐7). 15 , 26 Two mechanisms can explain the longer half‐life of hIL‐7‐hyFc. First, IgG, a part of the hyFc region in hIL‐7‐hyFc, is recycled by interacting with the neonatal Fc receptor. 25 This recycling process could have protected hIL‐7‐hyFc from lysosomal catabolism, thereby prolonging its half‐life. Second, the large molecular size of hIL‐7‐hyFc (~ 104 kDa) makes it less subject to being cleared through the kidneys, contributing also to its longer half‐life. 27
It was interesting to note that hIL‐7‐hyFc was much quickly absorbed after i.m. than s.c. administration (Table 1 , Figure 1 ). This observation may be related to the greater vascular supply in the muscle tissue than in the subcutaneous tissue. Greater blood supply in the muscle tissue might have led to a more rapid absorption and greater systemic exposure to hIL‐7‐hyFc, thereby a larger effect (see the next paragraph) as we showed in this study.
The hIL‐7‐hyFc markedly increased ALC, which was not only dose‐dependent, but also more prominent when given via the i.m. route than via the s.c. route (Figures 2 and 3) probably because the systemic exposure to hIL‐7‐hyFc was ~ 2 times greater after i.m. than after s.c. administration at the same dose of 60 µg/kg (Table 1 , Figure 1 ). The initial decrease in ALC for a short duration of ~ 4 days after hIL‐7‐hyFc may have been caused by early cell trafficking out of the circulation into the peripheral lymphoid organs and tissues, such as lymph nodes, spleen, and bone marrow, which was previously reported. 15 , 28 Transient decrease in ALC has also been described in patients treated with IL‐2. 29 Two cases of lymph node enlargement seen in the present study may also explain the mechanism behind the early cell trafficking by hIL‐7‐hyFc. However, no subject experienced an ultrasonographic confirmed splenomegaly (Table 2 ). Furthermore, the lymph node enlargement may signify increased metabolic activities by hIL‐7‐hyFc in lymphoid organs. 30
It is both revealing and promising that hIL‐7‐hyFc had a minimal effect on circulating B‐cells. Although hIL‐7‐hyFc dramatically increased ALC in the peripheral blood, the effect was mostly through the increase in T‐cells (Figure 2 ). The selective effect on T cells by IL‐7 was reported previously in patients with refractory malignancy. 15 These findings are also in agreement with previous in vitro results that, whereas IL‐7 induced proliferation and differentiation of pro‐B cells into pre‐B cells, other stromal cell‐derived signals are essential for the survival and growth of pre‐B cells. 31 , 32 , 33
PD effects are frequently delayed in many biologics, including monoclonal antibodies. 34 For example, the peak concentration of EPO‐hyFc was seen 30.8 hours postdose, whereas the maximum increase in hemoglobin by EPO‐hyFc occurred much later (i.e., ~ 10 days after administration). 35 The present study also showed that there was a marked time delay between the PK and PD of hIL‐7‐hyFc (Figure S1 ).
We showed that hIL‐7‐hyFc was relatively well‐tolerated after a single s.c. and i.m. administration at all doses. Most AEs were mild, all of the AEs spontaneously resolved without sequelae, and none of the AEs that were regarded as “likely related” to the study drug were grade 3 or higher. Injection site reaction, the most common AE in the present study, showed dose‐dependent nature in intensity, particularly after s.c. administration, which was associated with the dose‐dependent increase in ALC of hIL‐7‐hyFc. Furthermore, injection site reactions were much milder after i.m. than s.c. administration, which can be understood in that the skin is one of the most active immune organs. Although many subjects were positive on ADA, particularly after s.c. administration, only a few of them were nAb‐positive (Figure 5 ). Both ADA and nAb have become undetectable in many subjects as they were followed. Similarly, neither ADA nor nAb was associated with any clinical or laboratory test abnormality. Furthermore, when hIL‐7‐hyFc 1 mg/kg was co‐administered to mice with plasma obtained from ADA‐positive subjects in this study, no discernible negative effect was noted in increasing ALC (Figure S2 ). However, because this study was performed with a single‐dose design, further multiple‐dose studies are warranted to adequately evaluate the effect of repeated dosing with hIL‐7‐hyFc on the development of ADA, which are currently on‐going.
This study had two major limitations. First, because this study was conducted in heathy subjects, the findings may not translate to immune‐compromised patients or patients with a low T‐cell count of any cause. However, the proposed mechanisms for the long half‐life of hIL‐7‐hyFc and the ALC‐increasing effect of IL‐7 are likely to be reproduced in patients, regardless of the disease status. In fact, exogenous IL‐7 has been repeatedly shown to increase ALC in a variety of immune‐deficient patients. 15 , 19 , 36 , 37 Additionally, the dose range investigated in the present study was not wide enough for the evaluation of a full dose‐response relationship. However, we decided not to go beyond 60 μg/kg in healthy volunteers because of the ample effect in increasing ALC seen with this dose and potential safety concern at > 60 μg/kg, such as antidrug antibody.
In conclusion, hIL‐7‐hyFc was well‐tolerated in healthy subjects after a single s.c. and i.m. administration. The hIL‐7‐hyFc has a long half‐life and its ALC‐increasing effect, mostly selective on T cells, strongly supports the notion that hIL‐7‐hyFc could be developed into a beneficial treatment option for patients with compromised T‐cell immunity. Further studies in patients are warranted to test this hypothesis.
Funding
This study was funded by Genexine, Inc., Seongnam‐si, Gyeonggi‐do 13488, Republic of Korea.
Conflict of Interest
D.C., B.H.L., and S.H.Y. are employees of NeoImmuneTech, Inc. M.H., Y.‐K.O., and Y.C. Sung are employees of Genexine, Inc. All other authors declared no competing interests for this work.
Author Contributions
S.W.L., D.C., and H.L. wrote the manuscript. All authors designed the research. All authors performed the research. S.W.L. and H.L. analyzed the data.
Supporting information
Figure S1
Figure S2
Figure S3
[Correction added on 12th August 2020, after first online publication: the term ‘hhIL’ has been corrected to ‘hIL’ in the article title.]
References
- 1. Péron, J. et al. CD4 lymphopenia to identify end‐of‐life metastatic cancer patients. Eur. J. Cancer 49, 1080–1089 (2013). [DOI] [PubMed] [Google Scholar]
- 2. Grossman, S.A. et al. Survival in patients with severe lymphopenia following treatment with radiation and chemotherapy for newly diagnosed solid tumors. J. Natl. Compr. Canc. Netw. 13, 1225–1231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ray‐Coquard, I. et al. Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 69, 5383–5391 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. He, J.R. et al. Pretreatment levels of peripheral neutrophils and lymphocytes as independent prognostic factors in patients with nasopharyngeal carcinoma. Head Neck 34, 1769–1776 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Gong, J. , Chehrazi‐Raffle, A. , Reddi, S. & Salgia, R. Development of PD‐1 and PD‐L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J. Immunotherapy Cancer 6, 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith, D.K. , Neal, J.J. & Holmberg, S.D. Unexplained opportunistic infections and CD4+ T‐lymphocytopenia without HIV infection–an investigation of cases in the United States. N. Engl. J. Med. 328, 373–379 (1993). [DOI] [PubMed] [Google Scholar]
- 7. Walker, U.A. & Warnatz, K. Idiopathic CD4 lymphocytopenia. Curr. Opin. Rheumatol. 18, 389–395 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Hequet, O. et al. Multifocal progressive leukoencephalopathy occurring after refractory anemia and multiple infectious disorders consecutive to severe lymphopenia. Ann. Hematol. 81, 340–342 (2002). [DOI] [PubMed] [Google Scholar]
- 9. Glück, T. , Kiefmann, B. , Grohmann, M. , Falk, W. , Straub, R.H. & Schölmerich, J. Immune status and risk for infection in patients receiving chronic immunosuppressive therapy. J. Rheumatol. 32, 1473–1480 (2005). [PubMed] [Google Scholar]
- 10. Duncan, R.A. , von Reyn, C.F. , Alliegro, G.M. , Toossi, Z. , Sugar, A.M. & Levitz, S.M. Idiopathic CD4+ T‐lymphocytopenia–four patients with opportunistic infections and no evidence of HIV infection. N. Engl. J. Med. 328, 393–398 (1993). [DOI] [PubMed] [Google Scholar]
- 11. Cunningham‐Rundles, C. & Bodian, C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin. Immunol. 92, 34–48 (1999). [DOI] [PubMed] [Google Scholar]
- 12. Valiathan, R. , Deeb, K. , Diamante, M. , Ashman, M. , Sachdeva, N. & Asthana, D. Reference ranges of lymphocyte subsets in healthy adults and adolescents with special mention of T cell maturation subsets in adults of South Florida. Immunobiology 219, 487–496 (2014). [DOI] [PubMed] [Google Scholar]
- 13. Park, J.‐H. et al. Suppression of IL7Rα transcription by IL‐7 and other prosurvival cytokines: a novel mechanism for maximizing IL‐7‐dependent T cell survival. Immunity 21, 289–302 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Fry, T.J. et al. IL‐7 therapy dramatically alters peripheral T‐cell homeostasis in normal and SIV‐infected nonhuman primates. Blood 101, 2294–2299 (2003). [DOI] [PubMed] [Google Scholar]
- 15. Sportès, C. et al. Phase I study of recombinant human interleukin‐7 administration in subjects with refractory malignancy. Clin. Cancer Res. 16, 727–735 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andersson, Å. et al. IL‐7 promotes CXCR3 ligand‐dependent T cell antitumor reactivity in lung cancer. J. Immunol. 182, 6951–6958 (2009). [DOI] [PubMed] [Google Scholar]
- 17. Tan, J.T. et al. IL‐7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl. Acad. Sci. 98, 8732–8737 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sheikh, V. et al. Administration of interleukin‐7 increases CD4 T cells in idiopathic CD4 lymphocytopenia. Blood 127, 977–988 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levy, Y. et al. Enhanced T cell recovery in HIV‐1–infected adults through IL‐7 treatment. J. Clin. Investig. 119, 997–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alstadhaug, K.B. et al. Treatment of progressive multifocal leukoencephalopathy with interleukin 7. JAMA Neurol. 71, 1030–1035 (2014). [DOI] [PubMed] [Google Scholar]
- 21. Zhong, H. et al. Interleukin‐7 in patients with chronic hepatitis B may have effect on T follicular helper cells and specific cellular immunity. Hepatitis Monthly 16, e36068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou, H. , Kang, Y. , Zeng, Y. , Li, Y. & Shang, J. Interleukin‐7 augments CD8+ T cells function and promotes viral clearance in chronic hepatitis C virus infection. Cytokine 102, 26–33 (2018). [DOI] [PubMed] [Google Scholar]
- 23. Bocci, V. Interleukins. Clin. Pharmacokinet. 21, 274–284 (1991). [DOI] [PubMed] [Google Scholar]
- 24. Huhn, R.D.C. Clinical pharmacology and development of recombinant human biological agents In: Biotechnology and Safety Assessment (eds. Thomas J.A. & Myers L.A.) 59–78 (Raven Press, New York, 1993). [Google Scholar]
- 25. Sockolosky, J.T. & Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 91, 109–124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sereti, I. et al. IL‐7 administration drives T cell–cycle entry and expansion in HIV‐1 infection. Blood 113, 6304–6314 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Knauf, M.J. , Bell, D.P. , Hirtzer, P. , Luo, Z.‐P. , Young, J.D. & Katre, N.V. Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin‐2 chemically modified with water‐soluble polymers. J. Biol. Chem. 263, 15064–15070 (1988). [PubMed] [Google Scholar]
- 28. Beq, S. et al. Injection of glycosylated recombinant simian IL‐7 provokes rapid and massive T‐cell homing in rhesus macaques. Blood 114, 816–825 (2009). [DOI] [PubMed] [Google Scholar]
- 29. Kovacs, J.A. et al. Interleukin‐2 induced immune effects in human immunodeficiency virus‐infected patients receiving intermittent interleukin‐2 immunotherapy. Eur. J. Immunol. 31, 1351–1360 (2001). [DOI] [PubMed] [Google Scholar]
- 30. Sportès, C. et al. Administration of rhIL‐7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 205, 1701–1714 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LeBien, T.W. Fates of human B‐cell precursors. Blood 96, 9–23 (2000). [PubMed] [Google Scholar]
- 32. Dittel, B.N. & LeBien, T.W. The growth response to IL‐7 during normal human B cell ontogeny is restricted to B‐lineage cells expressing CD34. J. Immunol. 154, 58–67 (1995). [PubMed] [Google Scholar]
- 33. Johnson, S.E. , Shah, N. , Panoskaltsis‐Mortari, A. & LeBien, T.W. Murine and human IL‐7 activate STAT5 and induce proliferation of normal human pro‐B cells. J. Immunol. 175, 7325–7331 (2005). [DOI] [PubMed] [Google Scholar]
- 34. Mould, D.R. & Sweeney, K.R. The pharmacokinetics and pharmacodynamics of monoclonal antibodies‐mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Devel. 10, 84 (2007). [PubMed] [Google Scholar]
- 35. Im, S.J. et al. Natural form of noncytolytic flexible human Fc as a long‐acting carrier of agonistic ligand, erythropoietin. PLoS One 6, e24574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levy, Y. et al. Effects of recombinant human interleukin 7 on T‐cell recovery and thymic output in HIV‐infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo‐controlled, multicenter study. Clin. Infect. Dis. 55, 291–300 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perales, M.‐A. et al. Recombinant human interleukin‐7 (CYT107) promotes T‐cell recovery after allogeneic stem cell transplantation. Blood 120, 4882–4891 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3