

Abstract
This study aimed to predict long‐term progression‐free survival (PFS) using early M‐protein dynamic measurements in patients with relapsed/refractory multiple myeloma (MM). The PFS was modeled based on dynamic M‐protein data from two phase III studies, POLLUX and CASTOR, which included 569 and 498 patients with relapsed/refractory MM, respectively. Both studies compared active controls (lenalidomide and dexamethasone, and bortezomib and dexamethasone, respectively) alone vs. in combination with daratumumab. Three M‐protein dynamic features from the longitudinal M‐protein data were evaluated up to different time cutoffs (1, 2, 3, and 6 months). The abilities of early M‐protein dynamic measurements to predict the PFS were evaluated using Cox proportional hazards survival models. Both univariate and multivariable analyses suggest that maximum reduction of M‐protein (i.e., depth of response) was the most predictive of PFS. Despite the statistical significance, the baseline covariates provided very limited predictive value regarding the treatment effect of daratumumab. However, M‐protein dynamic features obtained within the first 2 months reasonably predicted PFS and the associated treatment effect of daratumumab. Specifically, the areas under the time‐varying receiver operating characteristic curves for the model with the first 2 months of M‐protein dynamic data were ~ 0.8 and 0.85 for POLLUX and CASTOR, respectively. Early M‐protein data within the first 2 months can provide a prospective and reasonable prediction of future long‐term clinical benefit for patients with MM.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ M‐protein is a biomarker for tumor burden and its levels in the serum and urine have been used to assess treatment responses for patients with multiple myeloma (MM).
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Whether early M‐protein data in the first several months can provide a prospective prediction for long‐term benefit and treatment effect.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ By using clinical data collected from two pivotal phase III trials of daratumumab in relapsed or refractory MM, we demonstrated that M‐protein dynamic data collected during the first 2 months of therapy can provide a prospective and reasonable prediction for not only long‐term progression‐free survival (PFS) but also the treatment effects of daratumumab on PFS compared with other active controls.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Early M‐protein dynamic data within the first 2 months of therapy may enable the designers of clinical trials to predict the probability of treatment success, and thus facilitate decision making in developing new drugs for MM.
Daratumumab is a human immunoglobulin G (IgGκ) monoclonal antibody that targets CD38 and kills malignant plasma cells via direct antitumor and immunomodulatory mechanisms of action. 1 , 2 , 3 , 4 , 5 , 6 , 7 Daratumumab induces antitumor activity through several CD38 immune‐mediated actions, including complement‐dependent cytotoxicity, antibody‐dependent cellular cytotoxicity, antibody‐dependent cellular phagocytosis, apoptosis, and modulation of CD38 enzymatic activity. 1 , 2 , 3 , 4 , 5 Daratumumab also induces an immunomodulatory effect by minimizing the immune‐suppressive functions of CD38+ myeloid‐derived suppressor cells, regulatory T cells, and regulatory B cells, and increasing T‐cell clonality. 6
In the phase III clinical studies POLLUX (MMY3003) and CASTOR (MMY3004), daratumumab in combination with standards of care regimens lenalidomide and dexamethasone (Rd), and bortezomib and dexamethasone (Vd), respectively, reduced the risk of disease progression or death by ≥ 50%, doubled the rates of complete response or better, and more than tripled the rates of minimal residual disease negativity based on next‐generation sequencing at the 10–5 sensitivity threshold vs. standard of care alone in patients with relapsed/refractory multiple myeloma (MM). 8 , 9 , 10 , 11 These findings led to the approval of daratumumab (16 mg/kg) in combination with Rd, and Vd in patients with relapsed and refractory MM in many countries worldwide. Daratumumab has also been approved as monotherapy in many countries and in combination with pomalidomide and dexamethasone in the United States.
In MM, tumor plasma cell produces a large amount of monoclonal IgG or IgG free light chain (FLC), known as M‐protein. M‐protein is a biomarker for tumor burden and its levels in the serum and urine have been used to assess treatment responses for patients with MM. 12 Dispenzieri et al. 13 demonstrated the utility of FLC after 2 months of therapy to predict the overall response. Furthermore, several other studies have shown retrospective association between FLC/M‐protein reduction and the long‐term benefit, such as progression‐free survival (PFS) or overall survival, 13 , 14 , 15 , 16 , 17 , 18 which is critical for predicting antitumor activities for new agents and personalizing therapy for patients with myeloma.
To date, the retrospective studies of the association between M‐protein dynamics and PFS or overall survival have been mainly based on complete M‐protein dynamic data collected up to the time of disease progression. 13 , 14 , 15 , 16 , 17 , 18 Therefore, the prospective ability of M‐protein, particularly early measurements within 2–3 months of therapy, to predict long‐term survivals remains unknown. Moreover, because most of existing association studies were performed based on nonrandomized, single‐arm phase II studies, 13 , 14 , 15 , 17 , 19 translation of the significant association between M‐protein and survival in these studies to treatment effects compared with control arms in the randomized phase III studies is still challenging. Furthermore, existing association studies have mainly focused on reduction in the M‐protein level at a static single time point (e.g., end of 8 weeks) without considering dynamic information or features of M‐protein (e.g., variation in M‐protein levels over time, and rate of M‐protein changes). 13 , 14 , 18
Here, we investigated the usefulness of early M‐protein dynamic data collected during the first several months after treatment initiation as a predictor of PFS and the effects of treatment on PFS. This analysis is based on the clinical data of two, large‐scale phase III studies, POLLUX and CASTOR for daratumumab. We evaluated different features of M‐protein dynamics in addition to M‐protein reduction and compared the predictive performance of M‐protein dynamic data within different periods after treatment. With this analysis, we hypothesized that an early interim analysis based on M‐protein dynamics within a couple of months after treatment could prospectively predict the future long‐term treatment effect on PFS. Such predictions would contribute greatly to predictions of the probability of success of future clinical trials, decision making in drug development, and the prevision of individualized guidance for treatment and patient care.
METHODS
Study design and data collection
POLLUX (ClinicalTrials.gov identifier, NCT02076009) and CASTOR (ClinicalTrials.gov identifier, NCT02136134) were phase III, multicenter, randomized (1:1), active‐controlled studies evaluating the efficacy and safety of daratumumab plus active control vs. active control alone in patients with relapsed and refractory MM. In POLLUX, 569 patients were randomly assigned to the daratumumab group (n = 286) or control group (n = 283). Daratumumab 16 mg/kg was administered once weekly for 8 weeks (cycles 1–2), followed by every 2 weeks for 16 weeks (cycles 3–6), and then every 4 weeks thereafter. 8 Both groups received lenalidomide and dexamethasone throughout the treatment period. Combination of daratumumab with lenalidomide and dexamethasone were compared with lenalidomide and dexamethasone (Rd) alone in patients with relapsed or refractory MM. In CASTOR, 498 patients were randomly assigned to the daratumumab group (n = 251) or control group (n = 247). Patients in the former group received daratumumab 16 mg/kg once weekly for 9 weeks (cycles 1–3), followed by every 3 weeks for 15 weeks (cycles 4–8), and every 4 weeks thereafter. 9 In contrast to POLLUX, all patients in CASTOR only received up to eight cycles (3 weeks per cycle) of bortezomib and dexamethasone. Daratumumab plus bortezomib and dexamethasone were compared with bortezomib and dexamethasone (Vd) alone in patients with relapsed and refractory disease treated with a median of two lines of therapy. Table S1 presented a comparison of POLLUX and CASTOR in terms of study design.
For POLLUX, M‐protein measurements were collected at screening, and on day 1 of each treatment cycle for 18 months. Subsequently, M‐protein measurements were collected every two cycles until the end of treatment. For CASTOR, M‐protein measurements were collected at screening, then day 1 of each treatment cycle for the first eight cycles. After cycle 8 (i.e., beginning of cycle 9), M‐protein measurements were collected every cycle for the first 18 months of the study and every other month thereafter until the end of the treatment.
The clinical data cutoff dates were March 7, 2016, for POLLUX and January 11, 2016, for CASTOR. 8 , 9 The dataset of POLLUX and CASTOR contain 169 and 189 events of disease progression or death, respectively. Details of the study designs have been described previously. 8 , 9 Both studies were approved by the institutional review boards of the participating institutions and were conducted in accordance with the ethical principles of the World Medical Association Declaration of Helsinki. All patients provided written informed consent.
Statistical analysis
Cox proportional hazards (PHs) analyses were performed on PFS using the survival package in R 3.5.3. 20 M‐protein dynamic data from each patient were extracted. For each patient, if serum M‐protein was measurable, dynamic data for serum M‐protein was used. However, if serum M‐protein was not measurable, whereas urine M‐protein was measurable, dynamic data for urine M‐protein was used. Finally, if both serum and urine M‐protein were unmeasurable, serum FLC data were used. Because the units of M‐protein are different among different measurements, the M‐protein dynamic data were normalized as the observed percent change of M‐protein from the baseline vs. time curve as shown in Figure S1 . The normalization is based on the following equation:
where MPt represents the M‐protein measurements over time, and MP0 represents the baseline M‐protein measurements. Three M‐protein dynamic features were extracted, namely the maximum percent reduction (representing maximal tumor reduction), last observed percent change of M‐protein (representing tumor burden change at the last observation), and rate of M‐protein change (representing the rate of tumor progression; i.e., the slope of the regression line connecting the measurements between the last and previous observations, as illustrated in Figure S1 ). To study the predictive ability of early M‐protein dynamics, we extracted three dynamic features within different time cutoffs for M‐protein measurements (1, 2, 3, and 6 months as well as complete M‐protein data up to the clinical cutoff dates). That is, a set of dynamic features was extracted for each cutoff of the M‐protein data, without changing the number of patients in the datasets. Univariate Cox analysis was conducted for individual M‐protein features. In the subsequent multivariable analysis, a stepwise screening was conducted to identify the most significant baseline covariates using Bayesian information criterion. The three M‐protein dynamic features at different time cutoffs were then added to the model without further variable selection. Predictive abilities of three dynamic features were assessed in this multivariable setting. Furthermore, the areas under the curve (AUC) corresponding to the time‐varying receiver operating characteristic (ROC) curve of the different models were calculated and compared to evaluate the predictive performance of the final Cox PH model. 21 The models were then validated further using external cross‐validation methods; specifically, the models constructed based on POLLUX data were used to predict PFS from the CASTOR data and vice versa. A landmark analysis was also conducted at different time points corresponding to the M‐protein cutoff dates. 22
We explored whether predictions based on dynamic M‐protein data would be superior to those based on static M‐protein data. The M‐protein measurements at static time points (i.e., 1 and 2 months) were used to construct models that were then compared with the models based on dynamic features. The AUCs of the ROC curves based on these models were calculated and compared.
M‐protein dynamic modeling
Longitudinal M‐protein dynamic modeling was performed using a tumor growth inhibition (TGI) model to evaluate the treatment effect of daratumumab. 23 , 24 , 25 The details of this model are described in the Supplementary Materials .
Figure 1.
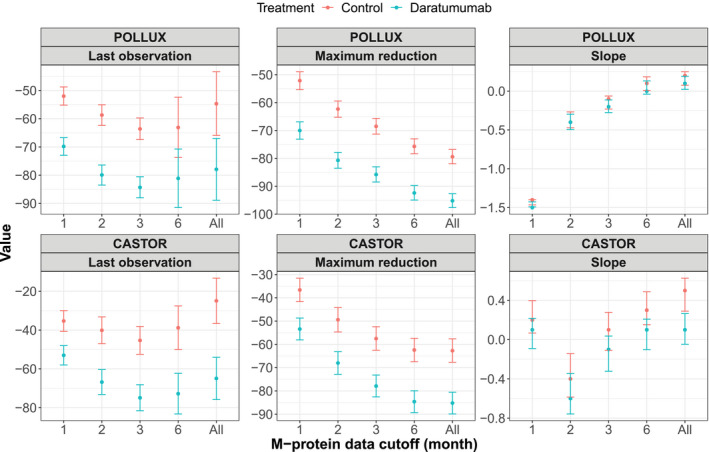
Observed means and 95% confidence intervals of the dynamic features of baseline‐normalized M‐protein data up to different time cutoffs from the POLLUX and CASTOR studies.
RESULTS
Treatment effect of daratumumab as demonstrated by univariate analysis and M‐protein dynamic modeling
Univariate analysis showed a strong treatment effect of daratumumab. In the univariate Cox analysis provided in Tables S2 and S3, all M‐protein dynamic features were significantly associated with PFS (P < 0.0001) except for the rate of M‐protein change within the first month, indicating that M‐protein dynamics are highly correlated with PFS. Figure 1 presents a comparison of the mean values of (with 95% confidence interval) of M‐protein dynamic features observed in the daratumumab and control arm of both POLLUX and CASTOR studies. This comparison revealed an apparent difference in these features between daratumumab and control groups. Specifically, for all the investigated time cutoffs, the last observation and maximum reduction in the daratumumab‐treated group were significantly lower than that of the control group, thus demonstrating a strong treatment effect of daratumumab and potential predictive abilities of these two M‐protein dynamic features. However, the difference in the rate of change in M‐protein between the two groups was less apparent.
We also evaluate the treatment effect of daratumumab using a TGI model for M‐protein dynamics. The model diagnostic plots of the TGI modeling are provided in the Figure S2 , which showed that the TGI model adequately describes the M‐protein dynamics data from the POLLUX and CASTOR studies. The parameter estimates are provided in Table S4 . The precisions of parameter estimates are expressed as relative standard error and are all within 30%. Interestingly, the estimated treatment effect of daratumumab in the combination therapy compared with standard of cares are 0.546 for POLLUX and 0.526 for CASTOR, indicating an increase of over 50% in the tumor decay rate induced by daratumumab. The model‐based simulation in Figure 2 showed daratumumab combination therapies in CASTOR and POLLUX provided a greater reduction in M‐protein and more persistent inhibition of tumor growth compared with the active control arm (Rd in CASTOR and Vd in POLLUX).
Figure 2.
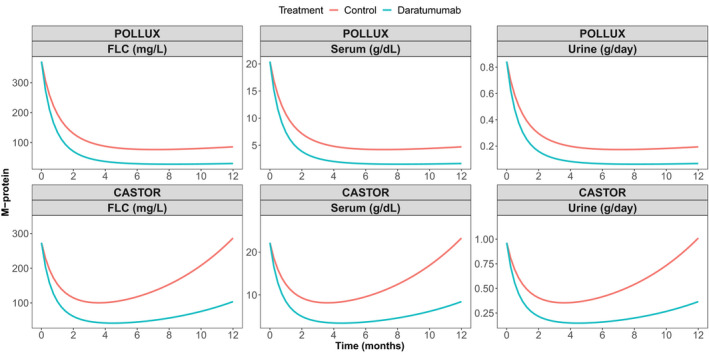
Predicted M‐protein dynamics based on a tumor growth inhibition model of the POLLUX and CASTOR studies. FLC, free light chain.
Association between PFS and M‐protein dynamic features after adjusting for baseline variables as demonstrated by multivariable analysis
Figure 3 shows the statistical significance of all covariates included in the model using the M‐protein up to 1 month, 2 months, and the last observation (i.e., all data). Slope of M‐protein change was only significant in PULLUX using M‐protein data collected up to 1 month. Except for the 1 month data cutoff in PULLUX, the maximum reduction of M‐protein was significant in both studies for different data cutoffs. The last observation of M‐protein was significant when using the M‐protein up to the last observation (i.e., all data) in POLLUX, and when using 2 months cutoff and all data in CASTOR.
Figure 3.
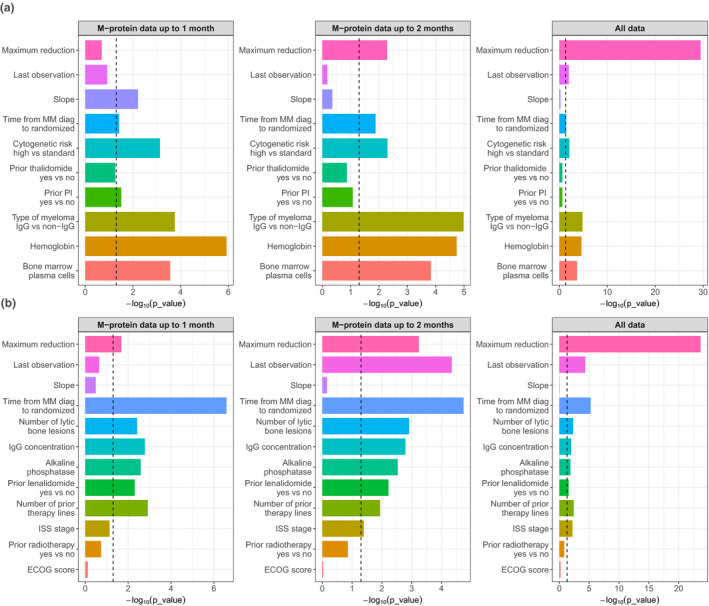
Statistical significance (‐log10 P) of each variable in a multivariable analysis from the (a) POLLUX and (b) CASTOR studies. The vertical dashed line indicates a P value of 0.05. Maximum reduction = maximum M‐protein reduction (% change from baseline); Last observation = last M‐protein observation (% change from baseline); slope = rate of M‐protein change (%/week). ECOG, Eastern Cooperative Oncology Group; IgG, interleukin G; ISS, International Staging System; MM, multiple myeloma; PI, prior lenalidomide.
Tables 1 and 2 present the hazard ratio for each covariate included in the final multivariable Cox PH models for POLLUX and CASTOR, along with the M‐protein dynamic features derived from the data up to different time cutoffs. When only 1 month of M‐protein data were included, it was very challenging to obtain reasonable estimates of the effect of M‐protein dynamics on PFS (Tables 1 and 2 ). This is demonstrated by that the estimates of hazard ratios associated with the selected dynamic features using 1 month data differed markedly from those obtained from all data (Tables 1 and 2 ). However, with inclusion of 2 months or more M‐protein data, it appears that the effect of M‐protein could be much better estimated. Consistent from the results shown in Figure 3 , all three M‐protein features barely showed any statistical significance with only 1 month of data. The M‐protein dynamic features (maximum reduction or/and last observed value) started to show significance once 2 months of data were included, and become highly significant when all data were used to construct the models (Figure 3 ).
Table 1.
Multivariable analysis of M‐protein dynamic features and identified baseline variables for PFS in the POLLUX (n = 569) study
| Variable, unit | HRs (95% CIs) based on M‐protein data up to the indicated time cutoffs | |||||
|---|---|---|---|---|---|---|
| Baseline | 1 month | 2 months | 3 months | 6 months | All data | |
| Maximum reduction of M‐protein, % | ‐ | 0.715 (0.426–1.2) | 1.19 (1.05–1.35) | 1.17 (1.06–1.3) | 1.34 (1.27–1.41) | 1.39 (1.32–1.48) |
| Last observed M‐protein change from baseline, % | ‐ | 10.2 (1.94–54) | 1.1 (0.86–1.41) | 1.01 (0.705–1.44) | 1.23 (0.831–1.82) | 0.903 (0.63–1.29) |
| Rate of M‐protein change, % per week | ‐ | 1.5 (0.899–2.51) | 1.02 (0.921–1.14) | 1.07 (0.986–1.16) | 1.01 (0.984–1.03) | 1.03 (1.01–1.05) |
| Time from MM diagnosis to randomization, years | 1.1 (1.04–1.16) | 0.947 (0.9–0.997) | 0.936 (0.888–0.986) | 0.943 (0.896–0.993) | 0.947 (0.9–0.997) | 0.945 (0.897–0.997) |
| Cytogenetic risk high vs. standard | 1.92 (1.30–2.83) | 1.96 (1.33–2.89) | 1.77 (1.19–2.64) | 1.76 (1.17–2.65) | 1.77 (1.18–2.66) | 1.72 (1.15–2.58) |
| Prior thalidomide yes vs. no | 1.45(1.06–1.98) | 1.38 (0.993–1.91) | 1.28 (0.926–1.76) | 1.14 (0.83–1.58) | 1.22 (0.886–1.68) | 1.22 (0.883–1.68) |
| Prior PI yes vs. no | 1.78 (1.07–2.96) | 1.76 (1.05–2.94) | 1.57 (0.942–2.61) | 1.43 (0.86–2.38) | 1.37 (0.82–2.27) | 1.37 (0.82–2.28) |
| Type of myeloma IgG vs. non‐IgG | 0.565 (0.415–0.77) | 0.55 (0.402–0.751) | 0.489 (0.356–0.672) | 0.506 (0.367–0.698) | 0.51 (0.371–0.701) | 0.493 (0.358–0.679) |
| Hemoglobin, g/L | 0.808 (0.736– 0.887) | 0.789 (0.717–0.868) | 0.807 (0.732–0.89) | 0.802 (0.728–0.884) | 0.803 (0.73–0.884) | 0.814 (0.74–0.896) |
| Bone marrow plasma cells, % | 0.94 (0.892–0.991) | 1.12 (1.05–1.18) | 1.12 (1.06–1.19) | 1.12 (1.06–1.19) | 1.11 (1.05–1.18) | 1.12 (1.05–1.18) |
For hemoglobin (g/L), bone marrow plasma cells (%), maximum reduction of M‐protein (%), and last observed M‐protein change from baseline (%), the hazard ratio is associated with each 10‐unit increase.
Table 2.
Multivariable analysis of M‐protein dynamic features and identified baseline variables for PFS in the CASTOR (n = 498) study
| Variable | HRs (95% CIs) based on M‐protein data up to the indicated time cutoffs | |||||
|---|---|---|---|---|---|---|
| Baseline | 1 month | 2 months | 3 months | 6 months | All Data | |
| Maximum % reduction of M‐protein, % | ‐ | 1.42 (1.06–1.92) | 1.09 (1.04–1.15) | 1.08 (1.03–1.13) | 1.21 (1.16–1.25) | 1.21 (1.17–1.25) |
| Last observed M‐protein change from baseline, % | ‐ | 0.838 (0.63–1.11) | 1.08 (1.04–1.12) | 1.11 (1.08–1.15) | 1.02 (1.01–1.04) | 1.02 (1.01–1.04) |
| Rate of M‐protein change, % per week | ‐ | 1.09 (0.92–1.28) | 0.987 (0.927–1.05) | 0.934 (0.874–0.999) | 0.998 (0.945–1.05) | 0.996 (0.943–1.05) |
| Time from MM diagnosis to randomized, years | 0.819 (0.764–0.879) | 0.839 (0.785–0.897) | 0.868 (0.813–0.926) | 0.873 (0.817–0.932) | 0.857 (0.801–0.917) | 0.855 (0.799–0.915) |
| Number of lytic bone lesions | 1.3 (1.16–1.47) | 1.83 (1.22–2.77) | 2 (1.31–3.05) | 1.87 (1.23–2.85) | 1.8 (1.18–2.74) | 1.82 (1.2–2.77) |
| IgG concentration, g/dL | 1.01 (1.01–1.02) | 1.01 (1–1.02) | 1.01 (1–1.02) | 1.01 (1–1.02) | 1.01 (1–1.02) | 1.01 (1–1.02) |
| Alkaline phosphatase, IU/L | 1.01 (1–1.01) | 1.04 (1.02–1.08) | 1.04 (1.02–1.08) | 1.04 (1.01–1.07) | 1.04 (1.01–1.07) | 1.04 (1.01–1.07) |
| Prior lenalidomide yes vs. no | 2.260 (1.62–1.47) | 1.65 (1.16–2.33) | 1.62 (1.15–2.29) | 1.5 (1.05–2.12) | 1.55 (1.1–2.18) | 1.47 (1.04–2.07) |
| Number of prior therapy lines | 1.880 (1.25–2.82) | 1.24 (1.09–1.42) | 1.18 (1.04–1.35) | 1.18 (1.04–1.35) | 1.2 (1.06–1.37) | 1.21 (1.06–1.37) |
| ISS, II or III vs. I | 1.600 (1.14–2.40) | 1.36 (0.971–1.92) | 1.43 (1.02–2) | 1.26 (0.893–1.77) | 1.63 (1.15–2.3) | 1.62 (1.15–2.3) |
| Prior radiotherapy yes vs. no | 1.510 (1.09–2.1) | 1.25 (0.9–1.74) | 1.29 (0.921–1.8) | 1.13 (0.802–1.58) | 1.3 (0.929–1.83) | 1.28 (0.914–1.8) |
| ECOG score, 1 or 2 vs. 0 | 0.950 (0.697–1.29) | 0.954 (0.704–1.29) | 0.985 (0.726–1.34) | 1.1 (0.81–1.5) | 0.927 (0.679–1.27) | 0.923 (0.676–1.26) |
For maximum reduction of M‐protein (%) and last observed M‐protein change from baseline (%), the hazard ratio is associated with each 10‐unit increase.
Based on the analysis with M‐protein data up to the last observation (i.e., all data), time from MM diagnosis to randomization was significantly associated with PFS for both studies (Figure 3 ), suggesting that patients who survived a longer time tend to have better PFS. Furthermore, based on analysis of all the data in POLLUX (Table 1 and Figure 3 ), the cytogenetic risk, history of treatment with thalidomide or proteasome inhibitors, and higher bone marrow plasma cell levels were associated with a shorter PFS, whereas a higher baseline hemoglobin level was significantly associated with a longer PFS (e.g., lower risk). Moreover, patients with IgG type of M‐protein tend to have better PFS. Consistent with the POLLUX data, our previously reported findings also suggested that compared with patients with non‐IgG MM, patients with IgG MM appear to be more sensitive to daratumumab treatment. 26 However, this result might be confounded with the baseline level of disease severity, as the non‐IgG group included a larger portion of patients with International Staging System stage III (24.4%) relative to the IgG group (16.6%). Based on the analysis of all data in CASTOR (Table 2 and Figure 3 ), a higher number of lytic bone lesions, higher IgG concentration, elevated alkaline phosphatase levels, history of lenalidomide therapy, higher number of previous lines of therapy lines, and International Staging System stage (II or III), were significantly associated with a higher risk of progression or death, suggesting that more severe disease might lead to a shorter PFS.
M‐protein dynamics as early as the first 2 months can provide reasonable predictions for PFS and treatment effect
We further overlaid the Cox‐PH prediction based on the multivariable model with observed survival curve to evaluate whether M‐protein dynamic features are predictive of the long‐term benefit of treatment effect for patients with MM (Figure 4 ). Despite the statistical significance, the baseline covariates have very limited predictive value in terms of the treatment effect as there was no separation of the predicted PFS for the daratumumab arm and active control arm in POLLUX. For CASTOR, there is a slight separation for the predicted PFS curves. This is not surprising as for each study, patients in the daratumumab arm should have similar baseline characteristics compared with those in the control arm due to randomization. However, the predicted PFS for daratumumab and control arms started to show separation when the M‐protein dynamic features from the first month of M‐protein data were included in the models. With the addition of further M‐protein data from the first 2 months, a reasonable prediction of treatment effect on PFS can be observed for both CASTOR and POLLUX. Boxplots of AUCs from the time‐varying ROC curves of each model (Figure 5 ) confirmed that, with the inclusion of M‐protein dynamic features from the first 2 months of data, the AUC increased to > 0.7 for POLLUX and > 0.8 for CASTOR. For POLLUX, the model continued to improve with the inclusion of more M‐protein data (Figure 5 ). For CASTOR, the performance of the model using M‐protein data obtained within the first 2 months was almost equal to that of the model that included all available M‐protein data (Figure 5 ). Moreover, our sensitivity landmark analysis demonstrated that the predictive performance for both CASTOR and POLLUX models was robust after accounting for the lead bias (Figures S3 and S4 ).
Figure 4.
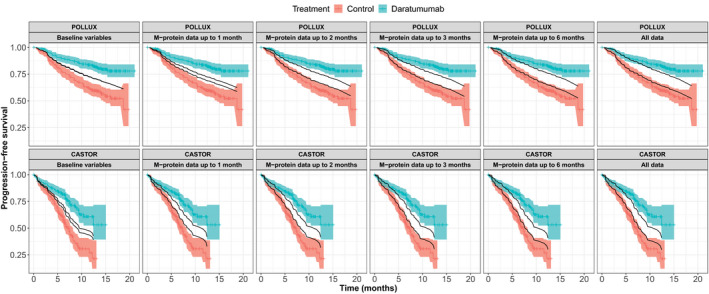
Comparison of the predicted probability of progression‐free survival based on the final multivariable survival models, with the observed probability over time (Kaplan–Meier survival curves) in the POLLUX (n = 569) and CASTOR (n = 498) studies. For POLLUX and CASTOR, the hazard ratio for progression‐free survival was in favor of daratumumab treated group with P < 0.001. 8 , 9 The predict ability of M‐protein data was evaluated together with the baseline variables.
Figure 5.
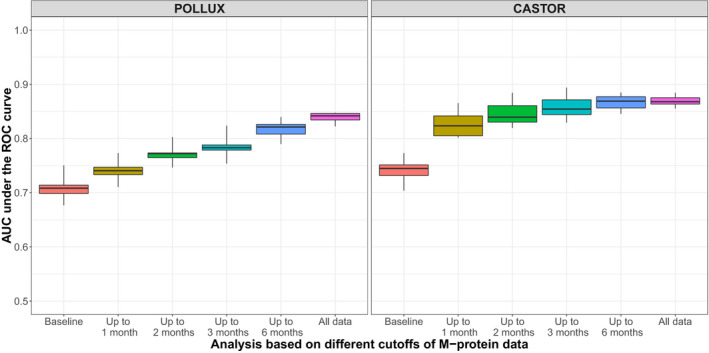
AUCs under the time‐varying ROC curves from the multivariable survival models based on M‐protein data from the POLLUX (n = 569) and CASTOR (n = 498) studies collected up to the indicated cutoff time points. AUC, area under the curve; ROC, receiver operating characteristic.
To evaluate whether the dynamic M‐protein data can provide a better prediction for PFS compared with the static M‐protein data at fixed time points, boxplots comparing the AUC values of time‐varying ROC curves from different approaches are presented in Figure S5 . For both the POLLUX and CASTOR studies, the AUCs of the ROC curves based on dynamic M‐protein data were consistently higher than those based on static M‐protein data. Interestingly, for CASTOR, the static M‐protein measurement at the end of 1 month provided better predictions than the static measurement at the end of 2 months, indicating the inconsistency of static M‐protein data for PFS prediction. These results highlight the superiority of dynamic data compared with static measurements to predict long‐term benefit.
Cross (external) validation further confirmed the predictive capacity of early M‐protein dynamics (Figure S6 ). The model based on early M‐protein dynamics (first 2 months) from POLLUX yielded a similar prediction of the treatment effect on PFS for the CASTOR data compared with that using the original CASTOR model. Similarly, the CASTOR model provided an adequate prediction for the treatment effect on PFS for POLLUX data.
DISCUSSION
The change in tumor size has been demonstrated to correlate with the survival for various cancers, including non‐small cell lung cancer, 27 colon cancer, 28 breast cancer, 29 and MM. 13 , 14 , 15 However, most previous correlative analyses were based either on complete dynamic data collected during the studies or on the tumor size at specific static time points (e.g., 8 weeks). Therefore, these analyses generally provided information only regarding the retrospective association between long‐term survival and the full tumor profile or partial static data. Early dynamic measurements may help to predict the impact of drug activity on survival and guide early decision making or treatment individualization. Unfortunately, to date, very little research has been performed to evaluate the predictive performance of early dynamic measurements in terms of long‐term survival benefits.
In this paper, we compared the predictive ability of M‐protein dynamic features based on data collected up to 1, 2, 3, and 6 months, and all available data following daratumumab administration using clinical data collected from POLLUX and CASTOR (sample size 569 and 498, respectively). We demonstrated that M‐protein dynamic data collected during the first 2 months of therapy can provide a prospective and reasonable prediction for not only long‐term PFS but also the treatment effects of daratumumab on PFS compared with other active controls. Although M‐protein has different subclasses, such as IgA, IgD, IgE, IgG, IgM, and FLC, the IgG M‐protein has the longest half‐life (about 20 days on average). 30 Therefore, active agents would be expected to exert sizeable treatment effects on M‐protein dynamics in the majority of patients after 2 months (60 days, 3 half‐lives) of treatment.
The analysis showed that the reduction of M‐protein is the most important predictor for PFS. This is consistent with previous studies for other cancers where the tumor shrinkage is a significant predictor for survival. 15 , 29 Furthermore, we demonstrated the superiority of dynamic data for predicting PFS compared with static measurements at fixed time points. Given the heterogeneous nature of MM, 31 the M‐protein dynamic data are more informative than static measurements collected at a specific time point.
We also investigated the predictive performance of two other M‐protein dynamic features, namely last observation of M‐protein within certain time windows and slope of change in M‐protein trajectory. The simple univariate analysis indicated that all features were important, supporting the importance of M‐protein dynamics for prediction of long‐term PFS. However, multivariable analysis results revealed that the maximum reduction appears to be the most statistically significant predictors for PFS, confirming that the depth of response is the most important predictor for the long‐term benefit. 32 In contrast, the last observation of M‐protein was a statistically significant predictor only for the CASTOR dataset. In POLLUX, the continuous deepening of the M‐protein response during the first year of therapy might have led to minimum differentiation between maximum reduction and last observation, and, thus, confounded these two dynamic features. In CASTOR, however, the M‐protein levels, particularly in the control arm, appeared to progress a few months after the initiation of the therapy (Figure 2 ), and differentiated between the maximum reduction and last observation (Figure 2 ). Accordingly, both features provided useful information for PFS prediction in the CASTOR study. Regarding the slope, this factor was no longer a significant predictor in the multivariable analysis after adjusting for the reduction and the last observation of M‐protein, indicating that the other two features completely explained its information. In CASTOR, the background treatment of bortezomib and dexamethasone were only administered up to eight cycles (~ 4 months). The beginning of progress of M‐protein at 4 months in the control arm is reflective of this cessation of treatment. In contrast, the M‐protein response in the daratumumab arm demonstrates the continued benefit of the experimental treatment.
The survival prediction based on M‐protein dynamic features differed slightly between POLLUX and CASTOR. The predictive ability of M‐protein dynamics in terms of PFS of patients in POLLUX continuously improve with more longitudinal M‐protein data, whereas the prediction performance of the M‐protein data from CASTOR started to plateau after 2 months (Figure 5 ). This observation is consistent with the clinical responses to daratumumab observed in these two studies. In POLLUX, the median time to complete remission or better was 13.2 months in the daratumumab arm, indicating a continuous deepening of response over the first year of treatment. In CASTOR, the corresponding time was 4.21 months, suggesting a much more rapid onset of maximum drug effect. It is well known that deep response in MM is associated with long‐term survival benefit. 32 Therefore, due to the rapid onset of maximum drug effect in CASTOR, it is not surprising that the maximum predictive performance can be attained with early M‐protein dynamic measurements within 2 months of therapy in this study. In POLLUX, however, 2 months of data could also yield a reasonable prediction of the treatment effect (i.e., separation of the PFS curves), although the addition of further dynamic data yielded slightly better prediction. The deepening of response over the long time observed in POLLUX is consistent with the observation that under Rd, the response can continue to deepen after years. 33 Therefore, the lack of a rapid very good partial response to lenalidomide‐based therapy should not discourage treatment continuation to a deeper response or encourage a too‐early switch in treatment. Nevertheless, the M‐protein dynamic data clearly shows that unlike other diseases where combination therapy may trigger more resistance, 34 combination therapies containing daratumumab could significantly slow down disease progression in patients with MM.
Our analysis had some limitations of note. First, although the M‐protein dynamics could provide a reasonable prediction of the treatment effect on PFS, it still tended to underpredict the treatment effect as the predicted separation in PFS curves was slightly smaller than the observed separation (Figure 4 ). To further improve the prediction, a more sensitive biomarker than M‐protein is required. Recent research suggested that minimal residual disease, which is measured using flow cytometry, multiparameter flow cytometry, polymerase chain reaction, next‐generation sequencing, and positron emission tomography/computed tomography methods, may be used as a surrogate biomarker to evaluate the efficacy. 32 Future research is warranted to evaluate the predictive capability of minimal residual disease dynamics, particularly early measurements, for survival outcomes. Second, this analysis was conducted using clinical data from two phase III studies of daratumumab in patients with relapsed and refractory MM. The generalizability of the model was not externally validated with data involving other agents. Clinical data from studies of other agents would be helpful to further confirm our findings. Nevertheless, strong associations between M‐protein and clinical outcomes (based on complete data across entire study duration) have been reported in multiple publications and for different treatments. 14 , 15 , 19 , 35 , 36 In addition, the percent reduction of M‐protein has been used to define international response criteria for MM, which has been widely used in clinical trials. 12 Therefore, our findings in the current study regarding the good predict ability of early M‐protein dynamics is very likely generalizable. Third, as the objective of the present research was to provide a practical modeling tool for clinical researchers and scientists to predict long‐term clinical efficacy endpoints (e.g., PFS) and associated treatment effect using early (e.g., first 2 months) M‐protein dynamics, we did not investigate the disease biology, particularly mechanism of drug resistance, using the M‐protein data. Recently, Tang et al. constructed sophisticated biological models using M‐protein dynamics data from three randomized controlled trials of bortezomib, and showed that bortezomib‐based therapy exerts a selection pressure on the tumor cells, leading to interpatient variability. 37 Future research using Tang et al.’s approach may be warranted to explore if daratumumab has a similar effect on myeloma cells.
In conclusion, our analyses based on the clinical data from POLLUX (n = 569) and CASTOR (n = 498) demonstrated that early M‐protein dynamic features obtained from data within the first 2 months following treatment could provide prospective and reasonable predictions of future long‐term clinical benefits in patients with MM. The models developed in this paper allow prediction of the probability of success for clinical trials to facilitate the decision making in drug development (e.g., early predictions of phase III clinical study outcomes at early interim analyses or when only early phase I/II data are available). Early M‐protein dynamic readouts may also enable individualized guidance for treatment and patient care.
Funding
This work was supported by the start‐up grant from School of Pharmacy, The Chinese University of Hong Kong.
Conflict of Interest
X.S.X., T.P., J.U., K.B., Q.M., S.S., and H.Z. are employees of Janssen Research & Development, LLC. X.S.X., T.P., J.U., K.B., Q.M., S.S., and H.Z. own stock in Johnson & Johnson. K.W. reports research funding from Janssen, Amgen, Sanofi, and Celgene Corporation; honoraria from Amgen, Bristol‐Myers Squibb, Celgene Corporation, Janssen, Novartis, Onyx, and Takeda; and advisory board membership for Adaptive Biotech, Amgen, Bristol‐Myers Squibb, Celgene Corporation, Janssen, Novartis, Onyx, and Takeda. M.V.M. received honoraria from and consulted for Janssen, Celgene, Takeda, and Amgen. A.S. received research funding and personal fees from Janssen Cilag. P.S. reports research funding from Janssen, Celgene, Amgen, and Takeda; received honoraria from Janssen, Celgene, Amgen, and Takeda; and provided expert testimony for Pharma Mar. M.A.D. consulted for Amgen, Janssen‐Cilag, and Takeda; received travel expenses from Janssen‐Ortho and Genesis Pharmaceuticals; received honoraria from Amgen, Novartis, Celgene, Takeda, Genesis Pharmaceuticals, Janssen‐Cilag, and Bristol‐Myers Squibb; and received research funding from Janssen‐Cilag and Amgen. S.Z.U. consulted for Abbvie, GlaxoSmithKline, Celgene, Amgen/Onyx, Takeda/Millennium, Sanofi, Seattle Genetics, Skyline, Merck, and Janssen; received research funding from Celgene, Amgen/Onyx, Takeda/Millennium, Sanofi, Seattle Genetics, Skyline, Merck, Janssen, Array BioPharma, and Pharmacyclics; served on speakers bureaus for Celgene, Amgen, Janssen, Sanofi, and Takeda; and received travel expenses from Janssen, Celgene, Amgen, and Takeda. N.J.B. received honoraria and travel expenses from Celgene, Takeda, Janssen, and Amgen; served on advisory boards for Celgene, Takeda, Janssen, and Amgen; served on speaker’s bureaus for Celgene, Janssen, and Amgen; and received research funding from and provided expert testimony to Celgene and Janssen. All other authors declared no competing interests for this work.
Author Contributions
X.Y., X.S.X., K.C.W., M.V.M., P.S., M.A.D., S.Z.U., N.J.B., T.P., J.U., K.B., Q.M., S.S., and H.Z. wrote the manuscript. X.Y., X.S.X., K.C.W., M.V.M., P.S., M.A.D., S.Z.U., N.J.B., N.J.B., T.P., J.U., K.B., Q.M., S.S., and H.Z. designed the research. X.Y., X.S.X., K.C.W., M.V.M., P.S., M.A.D., S.Z.U., N.J.B., N.J.B., T.P., J.U., K.B., Q.M., S.S., and H.Z. performed the research. X.Y. and X.S.X analyzed the data.
Supporting information
Supplementary Material
Acknowledgments
The authors thank the patients participating in this study and their families, as well as the global network of investigators, research nurses, study coordinators, and operations staff.
Contributor Information
Xiaoyu Yan, Email: xiaoyuyan@cuhk.edu.hk.
Xu Steven Xu, Email: xuxu93@gmail.com.
References
- 1. de Weers, M. et al Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 186, 1840–1848 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Overdijk, M. B. et al Antibody‐mediated phagocytosis contributes to the anti‐tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. 7, 311–321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Overdijk, M. B. et al The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcgamma receptor‐mediated cross‐linking. J. Immunol. 197, 807–813 (2016). [DOI] [PubMed] [Google Scholar]
- 4. Lammerts van Bueren, J. et al Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650984 and Ab79. Blood 124, 3474 (2014). [Google Scholar]
- 5. van de Donk, N. W. C. J. et al Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 270, 95–112 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krejcik, J. et al Daratumumab depletes CD38+ immune‐regulatory cells, promotes T‐cell expansion, and skews T‐cell repertoire in multiple myeloma. Blood 128, 384–394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DARZALEX™®(daratumumab) injection [packet insert]. (Janssen Biotech, Inc, Horsham, PA, 2019). [Google Scholar]
- 8. Palumbo, A. et al Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med. 375, 754–766 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Dimopoulos, M. A. et al Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 375, 1319–1331 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Mateos, M. V. et al Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N. Engl. J. Med. 378, 518–528 (2018). [DOI] [PubMed] [Google Scholar]
- 11. Spencer, A. et al Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: updated analysis of CASTOR. Haematologica 103, 2079–2087 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Durie, B. G. et al International uniform response criteria for multiple myeloma. Leukemia 20, 1467–1473 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Dispenzieri, A. et al Appraisal of immunoglobulin free light chain as a marker of response. Blood 111, 4908–4915 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dytfeld, D. et al Superior overall survival of patients with myeloma achieving very good partial response or better to initial treatment with bortezomib, pegylated liposomal doxorubicin, and dexamethasone, predicted after two cycles by a free light chain‐ and M‐protein‐based model: extended follow‐up of a phase II trial. Leuk. Lymphoma. 52, 1271–1280 (2011). [DOI] [PubMed] [Google Scholar]
- 15. Vij, R. et al Serum free light chain reduction correlates with response and progression‐free survival following carfilzomib therapy in relapsed/refractory multiple myeloma. Leuk. Lymphoma 56, 2959–2961 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Shimizu, K. et al Posttreatment M‐protein nadir level is a significant prognostic factor associated with survival in multiple myeloma. Nagoya Myeloma Cooperative Study Group. Jpn. J. Cancer Res. 90, 355–360 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oivanen, T. M. Prognostic value of serum M‐protein doubling time at escape from plateau of multiple myeloma. The Finnish Leukaemia Group. Eur. J. Haematol. 57, 247–253 (1996). [DOI] [PubMed] [Google Scholar]
- 18. Shah, J. et al Rapid early monoclonal protein reduction after therapy with bortezomib or bortezomib and pegylated liposomal doxorubicin in relapsed/refractory myeloma is associated with a longer time to progression. Cancer 117, 3758–3762 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michallet, M. et al Heavy+light chain monitoring correlates with clinical outcome in multiple myeloma patients. Leukemia 32, 376–382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. R Core Team . R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2018). [Google Scholar]
- 21. Blanche, P. , Dartigues, J.F. & Jacqmin‐Gadda, H. Estimating and comparing time‐dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat Med. 32, 5381–5397 (2013). [DOI] [PubMed] [Google Scholar]
- 22. Dafni, U. Landmark analysis at the 25‐year landmark point. Circ. Cardiovasc. Qual. Outcomes 4, 363–371 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Jonsson, F. et al A tumor growth inhibition model based on M‐protein levels in subjects with relapsed/refractory multiple myeloma following single‐agent carfilzomib use. CPT Pharmacometrics Syst. Pharmacol. 4, 711–719 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ribba, B. et al A review of mixed‐effects models of tumor growth and effects of anticancer drug treatment used in population analysis. CPT Pharmacometrics Syst. Pharmacol. 3, e113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu, X.S. et al Correlation between prostate‐specific antigen kinetics and overall survival in abiraterone acetate‐treated castration‐resistant prostate cancer patients. Clin. Cancer Res. 21, 3170–3177 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Yan, X. et al Influence of disease and patient characteristics on daratumumab exposure and clinical outcomes in relapsed or refractory multiple myeloma. Clin. Pharmacokinet. 57, 529–538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Claret, L. et al A model of overall survival predicts treatment outcomes with atezolizumab versus chemotherapy in non‐small cell lung cancer based on early tumor kinetics. Clin. Cancer Res. 24, 3292–3298 (2018). [DOI] [PubMed] [Google Scholar]
- 28. Claret, L. et al Evaluation of tumor‐size response metrics to predict overall survival in Western and Chinese patients with first‐line metastatic colorectal cancer. J. Clin. Oncol. 31, 2110–2114 (2013). [DOI] [PubMed] [Google Scholar]
- 29. Wang, Y. et al Elucidation of relationship between tumor size and survival in non‐small‐cell lung cancer patients can aid early decision making in clinical drug development. Clin. Pharmacol. Ther. 86, 167–174 (2009). [DOI] [PubMed] [Google Scholar]
- 30. Lobo, E.D. , Hansen, R.J. & Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 93, 2645–2668 (2004). [DOI] [PubMed] [Google Scholar]
- 31. Manier, S. et al Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 14, 100–113 (2017). [DOI] [PubMed] [Google Scholar]
- 32. Paiva, B. , van Dongen, J.J. & Orfao, A. New criteria for response assessment: role of minimal residual disease in multiple myeloma. Blood 125, 3059–3068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Facon, T. et al Final analysis of survival outcomes in the phase 3 FIRST trial of up‐front treatment for multiple myeloma. Blood 131, 301–310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leary, M. , Heerboth, S. , Lapinska, K. & Sarkar, S. Sensitization of drug resistant cancer cells: a matter of combination therapy. Cancers (Basel) 10, 483 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harutyunyan, N.M. et al Levels of uninvolved immunoglobulins predict clinical status and progression‐free survival for multiple myeloma patients. Br. J. Haematol. 174, 81–87 (2016). [DOI] [PubMed] [Google Scholar]
- 36. Schaefer, E.W. et al Residual serum monoclonal protein predicts progression‐free survival in patients with previously untreated multiple myeloma. Cancer 116, 640–646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tang, M. et al Myeloma cell dynamics in response to treatment supports a model of hierarchical differentiation and clonal evolution. Clin. Cancer Res. 22, 4206–4214 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material