

Abstract
Xanthine oxidase inhibitors febuxostat and allopurinol are commonly used in the treatment of gout. Febuxostat inhibits the breast cancer resistance protein (BCRP) in vitro. Rosuvastatin is a BCRP substrate and genetic variability in BCRP markedly affects rosuvastatin pharmacokinetics. In this study, we investigated possible effects of febuxostat and allopurinol on rosuvastatin pharmacokinetics. In a randomized crossover study with 3 phases, 10 healthy volunteers ingested once daily placebo for 7 days, 300 mg allopurinol for 7 days, or placebo for 3 days, followed by 120 mg febuxostat for 4 days, and a single 10 mg dose of rosuvastatin on day 6. Febuxostat increased the peak plasma concentration and area under the plasma concentration‐time curve of rosuvastatin 2.1‐fold (90% confidence interval 1.8–2.6; P = 5 × 10−5) and 1.9‐fold (1.5–2.5; P = 0.001), but had no effect on rosuvastatin half‐life or renal clearance. Allopurinol, on the other hand, did not affect rosuvastatin pharmacokinetics. In vitro, febuxostat inhibited the ATP‐dependent uptake of rosuvastatin into BCRP‐overexpressing membrane vesicles with a half‐maximal inhibitory concentration of 0.35 µM, whereas allopurinol showed no inhibition with concentrations up to 200 µM. Taken together, the results suggest that febuxostat increases rosuvastatin exposure by inhibiting its BCRP‐mediated efflux in the small intestine. Febuxostat may, therefore, serve as a useful index inhibitor of BCRP in drug‐drug interaction studies in humans. Moreover, concomitant use of febuxostat may increase the exposure to BCRP substrate drugs and, thus, the risk of dose‐dependent adverse effects.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Rosuvastatin is a known substrate of breast cancer resistance protein (BCRP). Recent in vitro findings indicate that the urate‐lowering drug febuxostat inhibits BCRP at clinically relevant concentrations. Febuxostat also markedly increased the exposure to the BCRP substrate sulfasalazine in mice. Allopurinol is not known to inhibit BCRP in vitro.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study investigated possible effects of febuxostat and allopurinol on rosuvastatin pharmacokinetics in healthy volunteers.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE?
☑ Febuxostat almost doubles the exposure to rosuvastatin in healthy volunteers, whereas allopurinol has no effect on the rosuvastatin pharmacokinetics. The mechanism underlying the febuxostat‐rosuvastatin interaction is likely inhibition of the BCRP‐mediated efflux of rosuvastatin in the small intestine.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ Potential BCRP‐mediated interactions should be taken into consideration when planning urate‐lowering therapy for patients with multiple medications. Febuxostat may be a useful probe inhibitor for BCRP in humans.
Gout is an increasingly common condition, and the most common form of inflammatory arthritis in developed countries. The main cause of gout is chronic hyperuricemia. Elevated serum uric acid concentration leads to accumulation of monosodium urate in joints, where urate crystals cause an extremely painful inflammatory arthritis and recurrent gout flares. Risk factors for gout include obesity, renal failure, genetic variability, diet, medications, such as diuretics, and excessive consumption of alcohol. 1 , 2 , 3 , 4 The increasing prevalence rates of gout worldwide are concordant with the role of lifestyle in its development.
Allopurinol is the first‐line treatment of chronic hyperuricemia and gout. It is used as a long‐term medication to prevent attacks of gout and avoid permanent damage to the joints. Being a nonselective inhibitor of the xanthine oxidase enzyme, allopurinol prevents biotransformation of hypoxanthine to xanthine and xanthine further to uric acid. Many patients on allopurinol, however, fail to achieve the target serum uric acid levels. 5 In addition, some patients on allopurinol experience cutaneous adverse effects, ranging from mild rash to severe, but rare, cutaneous adverse reactions associated with the HLA‐B*58:01‐allele, 6 , 7 necessitating discontinuation of allopurinol treatment.
Febuxostat is a newer xanthine oxidase inhibitor, which can be used as an alternative to allopurinol in the treatment of gout. Febuxostat was recently found to strongly inhibit the breast cancer resistance protein (BCRP; encoded by ABCG2) mediated transport of urate in vitro and to markedly increase the exposure to the BCRP substrate sulfasalazine in mice. 8 BCRP is an efflux transporter expressed on the apical membrane of epithelial cells in the gastrointestinal wall, liver canalicular membranes, blood‐brain barrier, placental syncytiotrophoblasts, testis, and the apical membrane of proximal tubular cells in the kidneys. 9 BCRP can limit the oral bioavailability of its substrates and enhance their excretion into the bile and urine. The most convincing evidence for a role of BCRP in drug pharmacokinetics comes from pharmacogenetic studies demonstrating increased exposure to BCRP substrate drugs in carriers of reduced function ABCG2 variants. 10 For example, the area under the plasma concentration‐time curve (AUC) of the cholesterol‐lowering drug rosuvastatin is increased 2.4‐fold in individuals homozygous for the ABCG2 c.421C>A (p.Gln141Lys, rs2231142) reduced function single nucleotide variation (SNV), as compared with noncarriers. 11
Patients with gout often receive medications for multiple comorbid conditions. Therefore, we found it important to investigate the potential pharmacokinetic interactions of the xanthine oxidase inhibitors febuxostat and allopurinol with the BCRP substrate rosuvastatin. To this end, we carried out a randomized, placebo‐controlled, crossover study to investigate possible effects of febuxostat and allopurinol on rosuvastatin pharmacokinetics in healthy subjects.
Methods
Subjects and study design
Following a written informed consent, 10 healthy volunteers participated in the study (Table 1 ). The participants’ health was confirmed by medical history, physical examination, and routine laboratory tests before entering the study. The participants were genotyped for the HLA‐B*58:01 allele and carriers of the allele were excluded from the study. Genotyping for the HLA‐B*58:01 allele was carried out in the Finnish Red Cross Blood Service (Helsinki, Finland) using the Luminex bead array technology (One Lambda, Los Angeles, CA) together with sequence‐specific primers (Olerup, Stockholm, Sweden). None of the participants used any continuous medication, including hormonal contraception, and all were nonsmokers.
Table 1.
Subject characteristics
| Sex | |
| Women | 3 |
| Men | 7 |
| Age, years | 26 ± 4 (21–35) |
| Weight, kg | 76 ± 14 (58–98) |
| BMI, kg/m2 | 24 ± 4 (19–31) |
| ABCG2 c.421C>A genotype | |
| C/C | 7 |
| C/A | 2 |
| A/A | 1 |
| SLCO1B1 c.521T>C genotype | |
| T/T | 6 |
| T/C | 1 |
| C/C | 3 |
Data are given as mean ± SD (range), except for sex and the ABCG2 and SLCO1B1 genotypes, which are given as number of participants.
BMI, body mass index.
The study protocol was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District (record number HUS/2942/2017) and the Finnish Medicines Agency Fimea (EudraCT number 2016‐002297‐13). In a randomized, crossover study with three phases, the volunteers ingested as pretreatment either placebo (placebo tablets; University Pharmacy, Helsinki, Finland) once daily for 7 days; or 300 mg allopurinol (Apurin Sandoz; Lek Pharmaceuticals d.d., Ljubljana, Slovenia) once daily for 7 days; or placebo for 3 days (days 1–3) followed by 120 mg febuxostat (Adenuric, Menarini—Von Heyden GmbH, Dresden, Germany) once daily for 4 days (days 4–7; Figure 1 ). The participants were randomized to all six possible pretreatment sequences. A single 10 mg dose of rosuvastatin (Crestor; AstraZeneca UK Ltd., Cheshire, UK) was administered at 9 am on day 6, 1 hour after the intake of placebo, allopurinol, or febuxostat. Between the days of rosuvastatin administration, there was a washout period of 3 weeks (2 weeks washout between the last day of pretreatment of the previous phase and the first day of pretreatment in the next phase).
Figure 1.
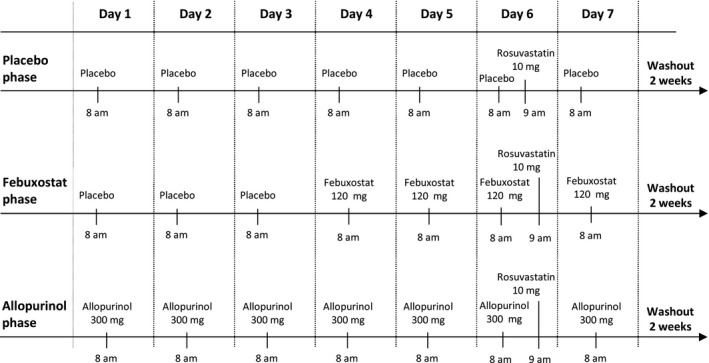
Study design. In a randomized crossover study, 10 healthy individuals ingested as pretreatment either placebo, 120 mg febuxostat, or 300 mg allopurinol once daily at 8 am. On day 6 of each phase, the participants ingested 10 mg of rosuvastatin at 9 am. There was a washout period of 2 weeks between the last day of pretreatment and the start of the pretreatment in the next phase.
The volunteers fasted overnight before the days of rosuvastatin administration. A standard warm meal was served 4 hours, and light meals 7 and 10 hours after the ingestion of rosuvastatin. The participants were not allowed to use any grapefruit products during the study. No other drugs were allowed from 1 week before to 1 week after the days of rosuvastatin administration. Use of alcohol was prohibited during the pretreatment and the days of rosuvastatin administration in each phase.
On the days of rosuvastatin administration, timed 4 mL EDTA venous blood samples were drawn before the administration of pretreatment, and 5 minutes prior to and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 23, and 47 hours after rosuvastatin ingestion for the determination of drug concentrations. The sample tubes were placed on ice immediately after sampling, and plasma was separated within 30 minutes. Urine was collected up to 10 hours after rosuvastatin ingestion. Plasma and urine aliquots were stored at –70°C until analysis.
Analysis of drug concentrations in plasma and urine samples
Rosuvastatin, allopurinol, febuxostat, and the corresponding stable isotope‐labeled internal standards were purchased from Toronto Research Chemicals (North York, Ontario, Canada). Oxypurinol and its isotope analog were purchased from Santa Cruz Biotechnology (Dallas, TX). Other reagents and organic solvents were commercially available analytical grade. Deionized water was obtained from an in‐house PRIMA water purification system (ELGA LabWater, High Wycombe, UK).
For the determination of rosuvastatin, plasma samples (150 µL) were mixed with acetonitrile (450 µL) containing the internal standard, and the mixture was filtered using a Phree Phospholipid Removal 96‐well extraction plate (Phenomenex, Torrance, CA). The supernatants were then evaporated using a centrifugal evaporator (GeneVac, Thermo Fisher Scientific, Waltham, MA) and reconstituted in 100 µL of 0.1% formic acid:acetonitrile (80:20, v:v).
The sample pretreatment for urine samples was performed with Strata‐X solid‐phase extraction cartridges (10 mg/cartridge; Phenomenex), which were preconditioned with methanol and water according to the manufacturer's instructions. Internal standard and 100 µL of phosphate buffer (0.1 M, pH 7.4) were added to 100 µL urine sample, and the sample was drawn through the cartridge followed by 200 µL of 20% methanol. Rosuvastatin was eluted with 2 × 100 µL of 1% formic acid in acetonitrile and reconstituted in 0.1% formic acid:acetonitrile (80:20, v:v).
Rosuvastatin concentrations were determined by using a Shimadzu Nexera liquid chromatography system (Shimadzu, Kyoto, Japan) coupled to an API 3000 tandem mass spectrometer (AB Sciex, Toronto, Ontario, Canada). The chromatographic separation was performed with a Kinetex C18 column (2.6 µm particle size, 2.1 mm × 50 mm internal diameter; Phenomenex) equipped with a Kinetex C18 pre‐column. The mobile phase consisted of 0.1% formic acid in water (A) and acetonitrile (B), and the mobile phase gradient was from 15% B to 95% B. The mobile phase flow rate was set at 300 µL/minute and the injection volume was 10 µL. The mass spectrometric detection was performed using electro‐spray ionization in positive multiple reaction monitoring (MRM) mode, and the targeted ion transition was a mass‐to‐charge ratio (m/z) 482 to m/z 258. The lower and upper limits of quantification in plasma were 0.1 ng/mL and 50 ng/mL. The intra‐day coefficient of variation of the method was 3.2% (n = 5) and day‐to‐day coefficient of variation was 6.9% (n = 30) at relevant concentrations.
For the determination of allopurinol and febuxostat, the plasma samples were pretreated using simple protein precipitation with acetonitrile containing 1% formic acid and isotope‐labeled internal standards. The supernatants were then diluted with water prior to liquid chromatography mass spectrometry analysis. All measurements were carried out using a Nexera X2 UHPLC system (Shimadzu, Kyoto, Japan) coupled to a 5500 Qtrap or a 6500 Qtrap mass spectrometer interfaced with an electrospray ion source (AB Sciex, Toronto, Ontario, Canada).
Plasma allopurinol and oxypurinol were separated on a Luna Omega Polar C18 column using a mobile phase consisting of 0.2% acetic acid (A) and acetonitrile (B). The mobile phase gradient was a linear ramp of B from 4% to 23% over 2 minutes, followed by a washing step (90% B) and an equilibration back to the starting composition. The flow rate and the column temperature were maintained at 200 µL/minute and 30°C. The mass spectrometer (5500 Qtrap) was operated in a negative polarity mode and the targeted MRM m/z ion transitions were 135 to 92 for allopurinol and 151 to 42 for oxypurinol.
The chromatographic separation of plasma febuxostat was performed on a Kinetex C18 column (2.6 µm particle size, 2.1 × 100 mm internal diameter; Phenomenex). The mobile phase gradient was a mixture of 0.05% formic acid (A) and acetonitrile (B) as follows: 0.5 minutes at 25% B on hold, a linear ramp from 25% B to 95% B over 2.5 minutes, and 2 minutes at 95% B on hold followed by equilibration back to 25% B. The mobile phase was delivered at 250 µL/minute and the oven temperature was set at 30°C. The mass spectrometer (6500 Qtrap) was operated in positive MRM mode using the characteristic m/z transition of 317 to 261 for febuxostat. The lower limits of quantification were 5.0 ng/mL, 100 ng/mL, and 10 ng/mL for allopurinol, oxypurinol, and febuxostat, respectively. The day‐to‐day coefficients of variation were below 15% for all analytes in relevant drug concentrations.
Pharmacokinetics
The pharmacokinetic variables (peak plasma concentration (Cmax); time to Cmax, time of peak plasma concentration (Tmax); AUC from zero to infinity (AUC0–∞) for rosuvastatin and dose‐interval AUC from zero to 24 hours (AUC0–24) for allopurinol and febuxostat; elimination half‐life (t ½); amount excreted into urine; and renal clearance (Clrenal)) of rosuvastatin, allopurinol, and febuxostat were calculated by standard noncompartmental methods using Phoenix WinNonlin, version 8.1 (Certara, Princeton, NJ).
Genotyping
Genomic DNA was extracted from buffy coats using the Maxwell 16 LEV Blood DNA Kit on a Maxwell 16 Research automated nucleic acid extraction system (Promega, Madison, WI). The samples were genotyped for the ABCG2 c.421C>A (rs2231142) and SLCO1B1 c.521T>C (rs4149056) SNVs using TaqMan genotyping assays with OpenArray technology on a QuantStudio 12K Flex Real‐Time polymerase chain reaction system (Life Technologies, Carlsbad, CA).
Statistical analysis
The number of subjects was estimated to be sufficient to detect a potentially clinically meaningful effect size of 30% difference in the AUC0–∞ of rosuvastatin among the 3 phases, with a power of 80% (α‐level 5%). The data were analyzed using the IBM SPSS Statistics for Windows, version 25 (Armonk, NY). Before analysis, the pharmacokinetic parameters, apart from Tmax, were logarithmically transformed. Statistical comparisons between the phases were made using repeated‐measures analysis of variance with treatment phase as a within‐subjects factor. The results are given as geometric means with geometric coefficients of variation and geometric mean ratios with 90% confidence intervals (CIs), except for Tmax, which is given as median with range. Any P values < 0.05 were considered statistically significant.
In vitro transport assays with BCRP‐expressing plasma membrane vesicles
The vesicular transport assays were carried out with membrane vesicles produced from BCRP‐overexpressing human embryonic kidney (HEK293) cells (PharmTox; Radboud UMC, Nijmegen, The Netherlands). The assays were performed essentially as described previously, 12 with rosuvastatin as a probe substrate at a final concentration of 5 µM. In short, membrane vesicles (7.5 µg) were incubated in transport assay buffer (PharmTox; Radboud UMC) supplemented with 10 mM MgCl2, to which various concentrations of febuxostat or allopurinol, or vehicle was added. The plate was preincubated at 37°C for 10 minutes. After the preincubation, the transport was started by adding prewarmed Mg‐ATP solution (final concentration 4 mM) or distilled water to the wells. The samples were incubated at 37°C for 5 minutes, after which transport was terminated with 200 µL ice‐cold stop buffer (PharmTox; Radboud UMC), and the samples were immediately transferred on a MultiScreenHTS FB Filter Plate 1.0 μm/0.65 μm (Merck KGaA, Darmstadt, Germany). The vesicles were washed twice with 200 μl stop buffer and twice with ice‐cold washing buffer (40 mM MOPS‐Tris pH 7.0 and 70 mM KCl), and dried. Rosuvastatin was subsequently eluted from the wells with 50% methanol containing stable isotope‐labeled rosuvastatin as internal standard and quantified with the 5500 Qtrap mass spectrometer using the method parameters described above for the plasma samples. All assays were performed in triplicates on 96‐well plates. Ko143 (10 µM) served as a positive inhibitor control in the allopurinol experiment and completely inhibited BCRP‐mediated rosuvastatin transport.
ATP‐dependent transport was calculated as the difference between rosuvastatin uptake in the presence and absence of ATP. Relative transport values were then calculated by comparing the ATP‐dependent rosuvastatin uptake in the presence of febuxostat or allopurinol with that in the presence of vehicle. To determine the half‐maximal inhibitory concentration of febuxostat and allopurinol, the relative transport values were fitted to the four‐parameter logistic regression model as shown in Eq. 1, using GraphPad Prism version 8 (GraphPad Software, San Diego, CA):
| (1) |
where minimum and maximum correspond to the plateaus of minimum and maximum relative transport (%), [I] is the concentration of inhibitor, and h is the Hill slope. In the case of febuxostat, the minimum value was constraint to 0.
Results
Effect of febuxostat on rosuvastatin pharmacokinetics
Febuxostat increased the Cmax of rosuvastatin on average 2.1‐fold (90% CI 1.8–2.6; P = 5 × 10−5) and the AUC0–∞ of rosuvastatin 1.9‐fold (90% CI 1.5–2.5; P = 0.001; Figure 2 , Table 2 ). In addition, the amount of rosuvastatin excreted into urine was increased 1.8‐fold (90% CI 1.2–2.7; P = 0.03) by febuxostat. The t ½ and Clrenal of rosuvastatin remained unaffected by febuxostat.
Figure 2.
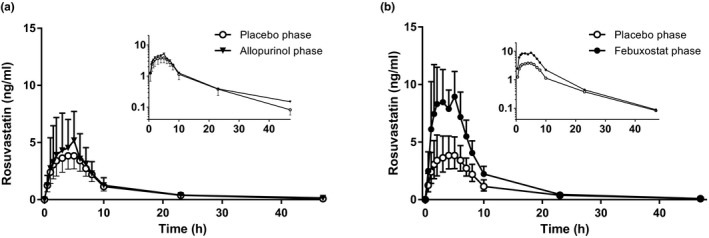
The effect of allopurinol and febuxostat on the plasma concentrations of rosuvastatin. Ten healthy volunteers ingested as pretreatment either 300 mg allopurinol on days 1–7 (a), or 120 mg febuxostat on days 4–7 (b), or placebo. Rosuvastatin 10 mg was administered 1 hour after the administration of pretreatment on day 6 of each of the three phases. Data are geometric means with 90% confidence interval. For clarity, some error bars have been omitted. Insets depict the same data on a semilogarithmic scale.
Table 2.
Effects of allopurinol and febuxostat on the pharmacokinetics of rosuvastatin
| Variable | Placebo phase | Allopurinol phase | Allopurinol phase to placebo phase ratio (90% CI); P value | Febuxostat phase | Febuxostat phase to placebo phase ratio (90% CI); P value |
|---|---|---|---|---|---|
| Rosuvastatin | |||||
| Cmax, ng/mL | 5.0 (75%) | 6.1 (100%) | 1.22 (1.02–1.47); P = 0.08 | 10.7 (62%) | 2.13 (1.76–2.58); P = 5 × 10−5 |
| Tmax, hour | 4.5 (0.5–6.0) | 5.0 (1.5–8.0) | P = 0.15 | 3.5 (1.0–5.0) | P = 0.15 |
| t ½, hour | 6.4 (63%) | 10.9 (62%) | 1.72 (1.03–2.85); P = 0.08 | 6.3 (29%) | 1.00 (0.75–1.33); P = 0.99 |
| AUC0–∞, ng·hour/mL | 42.6 (76%) | 53.5 (77%) | 1.26 (0.98–1.61); P = 0.13 | 82.5 (44%) | 1.93 (1.51–2.48); P = 0.001 |
| Ae, mg | 0.45 (78%) | 0.45 (101%) | 0.99 (0.69–1.43); P = 0.98 | 0.79 (55%) | 1.76 (1.16–2.66); P = 0.03 |
| Clrenal, mL/minute | 261 (43%) | 225 (36%) | 0.86 (0.65–1.14); P = 0.36 | 213 (36%) | 0.82 (0.58–1.14); P = 0.30 |
The pretreatments in the three phases were as follows: placebo (days 1–7), allopurinol 300 mg once daily (days 1–7), and febuxostat (placebo on days 1–3 and 120 mg febuxostat on days 4–7). Data are given as geometric mean with geometric coefficient of variation, except for Tmax, which is given as median with range. The geometric mean ratios between the phases are given with 90% CIs.
Ae, amount excreted into urine; AUC, area under the plasma concentration‐time curve from zero to infinity; Clrenal, renal clearance; Cmax, peak plasma concentration; t ½, elimination half‐life; Tmax, time to Cmax.
Marked interindividual variability was observed in the interaction between febuxostat and rosuvastatin (Figure 3 ). The effect of febuxostat on rosuvastatin AUC0–∞ ranged among the individual participants from no increase to 5.3‐fold increase. Three of the participants were homozygous and one was heterozygous for the SLCO1B1 c.521T>C reduced function variant. One participant was homozygous and two heterozygous for the ABCG2 c.421C>A variant. The SLCO1B1 or ABCG2 genotypes did not seem to be related to the extent of interaction (Figure 3 ).
Figure 3.
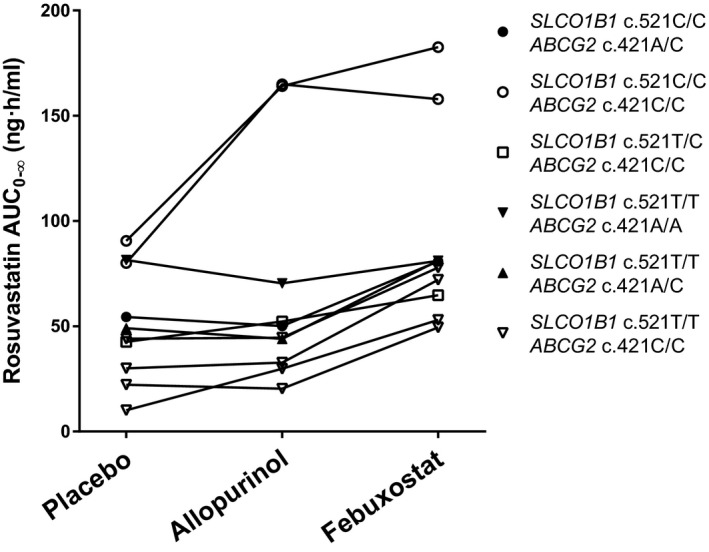
The individual rosuvastatin area under the plasma concentration‐time curve from zero to infinity (AUC0–∞) values in healthy volunteers after the administration of 10 mg rosuvastatin in a crossover study during placebo, allopurinol, and febuxostat phases.
Effect of allopurinol on rosuvastatin pharmacokinetics
In contrast to febuxostat, allopurinol had no statistically significant effect on the pharmacokinetics of rosuvastatin ( Figure 2 ; Table 2 ). However, there was a tendency for a prolonged t ½ of rosuvastatin during allopurinol treatment (10.9 hours vs. 6.4 hours during placebo), but the effect was not statistically significant (P = 0.08). In two of three individuals with the SLCO1B1 c.521C/C genotype, allopurinol seemed to increase the AUC0–∞ of rosuvastatin to the same extent as febuxostat (Figure 3 ).
Effects of febuxostat and allopurinol on BCRP‐mediated rosuvastatin transport in vitro
In membrane vesicles prepared from BCRP‐overexpressing human embryonic kidney (HEK293) cells, febuxostat inhibited the ATP‐dependent uptake of rosuvastatin with an half‐maximal inhibitory concentration of 0.35 µM (Figure 4 ). Allopurinol did not inhibit rosuvastatin uptake with concentrations up to 200 µM.
Figure 4.
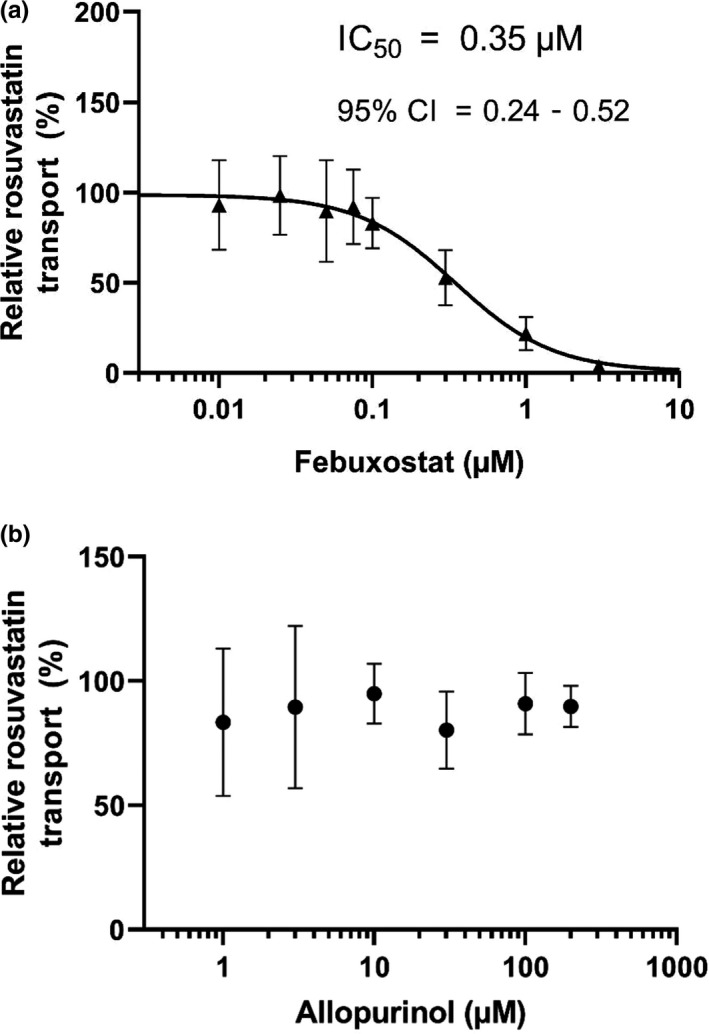
The effect of febuxostat and allopurinol on the BCRP‐mediated transport of rosuvastatin in the vesicular transport assay. BCRP‐expressing membrane vesicles were incubated in the presence of rosuvastatin and febuxostat (a) or allopurinol (b). The ATP‐dependent transport of rosuvastatin in the absence of test compounds was set as 100%. Data are means ± SD (n = 6).
Febuxostat and allopurinol pharmacokinetics
The plasma concentrations of febuxostat and allopurinol varied moderately between individual participants (Table 3 ). The Cmax and AUC0–24 of febuxostat varied 5.0‐fold and 3.0‐fold and those of allopurinol 1.8‐fold and 4.7‐fold.
Table 3.
Pharmacokinetic variables of allopurinol and febuxostat on day 6 of 300 mg allopurinol once daily, or on day 3 of 120 mg febuxostat once daily in 10 healthy volunteers
| Variable | Geometric mean (geometric CV) |
|---|---|
| Allopurinol | |
| Cmax, µg/mL | 2.0 (21%) |
| t ½, hour | 1.3 (50%) |
| AUC0–24, µg·hour/mL | 5.7 (46%) |
| Febuxostat | |
| Cmax, µg/mL | 4.3 (49%) |
| t ½, hour | 6.0 (21%) |
| AUC0–24, µg·hour/mL | 12.3 (39%) |
AUC, area under the plasma concentration‐time curve from zero to 24 hours; Cmax, peak plasma concentration; CV, coefficient of variation; t ½, elimination half‐life.
Discussion
Previous in vitro data have suggested that febuxostat inhibits BCRP at clinically relevant concentrations. 8 By contrast, allopurinol and its metabolite oxypurinol have not inhibited BCRP in vitro. 13 To study the effects of the two urate‐lowering drugs on BCRP activity in humans, we investigated their effects on the pharmacokinetics of the BCRP‐substrate rosuvastatin in healthy volunteers. Our results indicate a difference in the interaction potentials of the two xanthine oxidase inhibitors. In this study, febuxostat markedly increased the plasma concentrations of rosuvastatin. After the administration of febuxostat, the Cmax of rosuvastatin was more than doubled and the AUC0–∞ almost doubled when compared with those measured after the administration of placebo. The Clrenal of rosuvastatin, however, was not altered by administration of febuxostat. In contrast to febuxostat, allopurinol had no significant effect on the pharmacokinetic variables of rosuvastatin. The t ½ of rosuvastatin, however, tended to be prolonged by allopurinol. In vitro, febuxostat, but not allopurinol, strongly inhibited the BCRP‐mediated transport of rosuvastatin, consistent with the observations from previous in vitro studies using urate or pitavastatin as a substrate. 8 , 13
The findings that febuxostat raised the Cmax and AUC0–∞ of rosuvastatin with no effect on its t ½ or renal clearance suggest that febuxostat increased the oral bioavailability of rosuvastatin. Given that febuxostat is a potent inhibitor of BCRP in vitro, the most likely mechanism underlying the interaction is inhibition of the BCRP‐mediated efflux of rosuvastatin in the small intestinal wall. If the transporter inhibition is restricted to the small intestinal wall, it should not affect the t ½ of rosuvastatin. The intestinal concentrations of febuxostat are likely much higher than its systemic concentrations, which may result in a strong inhibition of the BCRP in the gut, but not necessarily in other tissues. Of note is that homozygosity for the ABCG2 c.421C>A SNV has a remarkably similar impact on rosuvastatin pharmacokinetics, having no effect on its t ½. 11 However, rosuvastatin is also a substrate of the organic anion transporting polypeptide (OATP) 1B1, 1B3, and 2B1 influx transporters expressed on the basolateral membrane of human hepatocytes. 14 , 15 , 16 , 17 As yet, there are no data on febuxostat's potential to inhibit OATPs. In vivo in humans, homozygosity for the reduced function variant c.521T>C in the SLCO1B1 gene encoding OATP1B1 has raised rosuvastatin Cmax about 1.8–2.7‐fold and AUC about 1.7–2.2‐fold. 18 , 19 , 20 In addition, a single intravenous dose of rifampin (INN, rifampicin), a strong inhibitor of OATP1B1 and OATP1B3, has raised the Cmax and AUC of rosuvastatin about 8‐fold to 9‐fold and 3‐fold to 3.5‐fold, respectively. 21 However, unlike the ABCG2 c.421C>A variant or febuxostat, both the SLCO1B1 c.521T>C variant and rifampin tended to shorten the t ½ of rosuvastatin. The shortened t ½ could indicate a reduced volume of distribution due to impaired hepatic uptake. Taken together, these findings indicate that the febuxostat‐rosuvastatin interaction is indeed most likely caused by inhibition of the BCRP in the small intestine.
Three individuals recruited for the study were homozygous for the SLCO1B1 c.521T>C variant and one for the ABCG2 c.421C>A variant. These variants did not seem to be related to the extent of interaction between febuxostat and rosuvastatin, but due to the limited sample size, no conclusions can be drawn. Interestingly, the AUC0–24 of febuxostat was increased 1.8‐fold in individuals homozygous for SLCO1B1 c.521T>C as compared with the noncarriers and one heterozygote (data not shown), suggesting that OATP1B1 is important for the hepatic uptake of febuxostat. Further studies are required to verify this preliminary finding.
The finding that febuxostat inhibits BCRP in vivo in humans may make it a useful index inhibitor for drug‐drug interaction studies in humans. Moreover, it can be safely administered to healthy volunteers and its pharmacokinetic steady‐state is reached rapidly, which are desirable characteristics of index inhibitors. 22 In vitro, febuxostat is a weak inhibitor of CYP2C8 and CYP2D6, but it does not inhibit CYP1A2, CYP2C9, CYP2C19, or CYP3A4. 23 , 24 In vivo in humans, febuxostat had no effect on the pharmacokinetics of the CYP2C8 probe substrate rosiglitazone and only slightly increased the AUC of the sensitive CYP2D6 substrate desipramine (by 12%). 24 , 25 These data indicate relative selectivity for BCRP compared with the CYP enzymes. However, more studies are needed to determine whether febuxostat inhibits OATPs or other drug transporters. Moreover, as febuxostat is a strong inhibitor of xanthine oxidase, it can interact with drugs metabolized by this enzyme, such as the thiopurines.
Cyclosporine and eltrombopag are among the best documented examples of clinical BCRP inhibitors, but both of them are nonselective. 22 Cyclosporine has raised the AUC of rosuvastatin 7.1‐fold. 26 However, cyclosporine is also a potent inhibitor of OATP1B1, OATP1B3, and OATP2B1, which likely explains the extent of the cyclosporine‐rosuvastatin interaction. 16 , 27 In addition, cyclosporine inhibits, for example, the P‐glycoprotein and CYP3A4. 28 , 29 On the other hand, eltrombopag has raised rosuvastatin AUC only 1.6‐fold. Eltrombopag also inhibits OATP1B1, OATP1B3, and OATP2B1 in vitro. 30 , 31 , 32
Patients on urate‐lowering pharmacotherapy are at high risk for comorbidities and polypharmacotherapy. Gout is strongly associated with metabolic syndrome and cardiovascular diseases. 4 , 33 Given that febuxostat presumably increased the intestinal absorption of rosuvastatin, the concomitant use of febuxostat may enhance the cholesterol‐lowering efficacy of rosuvastatin and the risk of rosuvastatin‐induced muscle toxicity. 34 , 35 , 36 In addition to rosuvastatin, several other statins are BCRP substrates. 37 In previous pharmacokinetic studies in humans, the AUCs of atorvastatin and fluvastatin were increased 1.7‐fold in individuals homozygous for the ABCG2 c.421C>A variant, as compared with noncarriers. 11 , 38 Thus, febuxostat may increase the exposure to atorvastatin and fluvastatin. On the other hand, the ABCG2 c.421C>A variant has had no significant effect on the AUC of simvastatin acid, pitavastatin, or pravastatin. 38 , 39 , 40 However, in patients with established cardiovascular disease, febuxostat increases all‐cause and cardiovascular mortality compared with allopurinol. 41 Febuxostat use is, therefore, restricted to exceptional cases only and its concomitant use with statins is unlikely.
The present study was carried out with a single low dose of rosuvastatin in healthy volunteers. Because rosuvastatin shows linear pharmacokinetics and its pharmacokinetics do not change after multiple dosing, the present results can be extrapolated to higher doses and continuous treatment. 36 Marked interindividual variability existed in the interaction between febuxostat and rosuvastatin within the homogenous group of healthy volunteers of the present study. Patients using febuxostat often have comorbid diseases and may use other drugs as well, which may further increase interindividual variability in the extent of its interactions with BCRP substrates.
To conclude, febuxostat but not allopurinol markedly increases the plasma concentrations of rosuvastatin in healthy volunteers. It is plausible that the mechanism underlying the febuxostat‐rosuvastatin interaction is inhibition of the BCRP‐mediated efflux of rosuvastatin in the gut wall. Potential BCRP‐mediated interactions should be taken into consideration when planning urate‐lowering therapy for patients with multiple medications. Furthermore, febuxostat could be a useful BCRP index inhibitor in drug‐drug interaction studies.
Funding
This study was supported by grants from the Sigrid Jusélius Foundation and State funding for university‐level health research (Helsinki, Finland).
Conflicts of Interest
The authors declared no competing interests for this work.
Author Contributions
M.L. and M.Ni. wrote the manuscript. M.L., J.E.K., A.T., O.L.‐R., J.V., M.Ne., J.T.B., and M.Ni. designed the research. M.L., J.E.K., A.T., O.L.‐R., F.D., T.J., J.V., M.Ne., J.T.B., and M.Ni. performed the research. M.L. and M.Ni. analyzed the data.
Acknowledgments
The authors thank Eija Mäkinen‐Pulli and Lisbet Partanen for their skillful technical assistance.
References
- 1. Choi, H.K. , Atkinson, K. , Karlson, E.W. , Willett, W. & Curhan, G. Purine‐rich foods, dairy and protein intake, and the risk of gout in men. N. Engl. J. Med. 350, 1093–1103 (2004). [DOI] [PubMed] [Google Scholar]
- 2. Choi, H.K. , Atkinson, K. , Karlson, E.W. , Willett, W. & Curhan, G. Alcohol intake and risk of incident gout in men: a prospective study. Lancet 363, 1277–1281 (2004). [DOI] [PubMed] [Google Scholar]
- 3. Choi, H.K. , Atkinson, K. , Karlson, E.W. & Curhan, G. Obesity, weight change, hypertension, diuretic use, and risk of gout in men: the health professionals follow‐up study. Arch. Intern. Med. 165, 742–748 (2005). [DOI] [PubMed] [Google Scholar]
- 4. Roddy, E. & Doherty, M. Epidemiology of gout. Arthritis Res. Ther. 12, 223 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Becker, M.A. et al. The urate‐lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res. Ther. 12, R63 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hung, S.I. et al. HLA‐B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc. Natl. Acad. Sci. USA 102, 4134–4139 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim, S.C. , Newcomb, C. , Margolis, D. , Roy, J. & Hennessy, S. Severe cutaneous reactions requiring hospitalization in allopurinol initiators: a population‐based cohort study. Arthritis Care Res. 65, 578–584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyata, H. , Takada, T. , Toyoda, Y. , Matsuo, H. , Ichida, K. & Suzuki, H. Identification of febuxostat as a new strong ABCG2 inhibitor: potential applications and risks in clinical situations. Front. Pharmacol. 7, 518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horsey, A.J. , Cox, M.H. , Sarwat, S. & Kerr, I.D. The multidrug transporter ABCG2: still more questions than answers. Biochem. Soc. Trans. 44, 824–830 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giacomini, K.M. et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin. Pharmacol. Ther. 94, 23–26 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Keskitalo, J.E. , Zolk, O. , Fromm, M.F. , Kurkinen, K.J. , Neuvonen, P.J. & Niemi, M. ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203 (2009). [DOI] [PubMed] [Google Scholar]
- 12. Sjöstedt, N. , Deng, F. , Rauvala, O. , Tepponen, T. & Kidron, H. Interaction of food additives with intestinal efflux transporters. Mol. Pharm. 14, 3824–3833 (2017). [DOI] [PubMed] [Google Scholar]
- 13. Wen, C.C. et al. Genome‐wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin. Pharmacol. Ther. 97, 518–525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schneck, D.W. et al. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin. Pharmacol. Ther. 75, 455–463 (2004). [DOI] [PubMed] [Google Scholar]
- 15. Ho, R.H. & Kim, R.B. Transporters and drug therapy: implications for drug disposition and disease. Clin. Pharmacol. Ther. 78, 260–277 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Ho, R.H. et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology 130, 1793–1806 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181 (2011). [DOI] [PubMed] [Google Scholar]
- 18. Lee, E. et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin. Pharmacol. Ther. 78, 330–341 (2005). [DOI] [PubMed] [Google Scholar]
- 19. Pasanen, M.K. , Fredrikson, H. , Neuvonen, P.J. & Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 82, 726–733 (2007). [DOI] [PubMed] [Google Scholar]
- 20. Choi, J.H. , Lee, M.G. , Cho, J.Y. , Lee, J.E. , Kim, K.H. & Park, K. Influence of OATP1B1 genotype on the pharmacokinetics of rosuvastatin in Koreans. Clin. Pharmacol. Ther. 83, 251–257 (2008). [DOI] [PubMed] [Google Scholar]
- 21. Wu, H.F. et al. Rosuvastatin pharmacokinetics in Asian and white subjects wild type for both OATP1B1 and BCRP under control and inhibited conditions. J. Pharm. Sci. 106, 2751–2757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tornio, A. , Filppula, A.M. , Niemi, M. & Backman, J.T. Clinical studies on drug‐drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin. Pharmacol. Ther. 105, 1345–1361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mukoyoshi, M. et al. In vitro drug‐drug interaction studies with febuxostat, a novel non‐purine selective inhibitor of xanthine oxidase: plasma protein binding, identification of metabolic enzymes and cytochrome P450 inhibition. Xenobiotica 38, 496–510 (2008). [DOI] [PubMed] [Google Scholar]
- 24. Naik, H. , Wu, J.T. , Palmer, R. & McLean, L. The effects of febuxostat on the pharmacokinetic parameters of rosiglitazone, a CYP2C8 substrate. Br. J. Clin. Pharmacol. 74, 327–335 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khosravan, R. , Erdman, K. , Vernillet, L. , Wu, J. , Josephridge, N. , Umeda, S. & Mulford, D. Effect of febuxostat on pharmacokinetics of desipramine, a CYP2D6 substrate, in healthy subjects. Clin. Pharmacol. Ther. 77, P43 (2005). [Google Scholar]
- 26. Simonson, S.G. et al. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 76, 167–177 (2004). [DOI] [PubMed] [Google Scholar]
- 27. Shitara, Y. , Itoh, T. , Sato, H. , Li, A.P. & Sugiyama, Y. Inhibition of transporter‐mediated hepatic uptake as a mechanism for drug‐drug interaction between cerivastatin and cyclosporin A. J. Pharmacol. Exp. Ther. 304, 610–616 (2003). [DOI] [PubMed] [Google Scholar]
- 28. Kajosaari, L.I. , Niemi, M. , Neuvonen, M. , Laitila, J. , Neuvonen, P.J. & Backman, J.T. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin. Pharmacol. Ther. 78, 388–399 (2005). [DOI] [PubMed] [Google Scholar]
- 29. Tamai, I. & Safa, A.R. Competitive interaction of cyclosporins with the Vinca alkaloid‐binding site of P‐glycoprotein in multidrug‐resistant cells. J. Biol. Chem. 265, 16509–16513 (1990). [PubMed] [Google Scholar]
- 30. Takeuchi, K. et al. Pharmacokinetics and hepatic uptake of eltrombopag, a novel platelet‐increasing agent. Drug Metab. Dispos. 39, 1088–1096 (2011). [DOI] [PubMed] [Google Scholar]
- 31. Allred, A.J. et al. Eltrombopag increases plasma rosuvastatin exposure in healthy volunteers. Br. J. Clin. Pharmacol. 72, 321–329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takeuchi, K. et al. Interaction of novel platelet‐increasing agent eltrombopag with rosuvastatin via breast cancer resistance protein in humans. Drug Metab. Dispos. 42, 726–734 (2014). [DOI] [PubMed] [Google Scholar]
- 33. Stamp, L.K. & Chapman, P.T. Gout and its comorbidities: implications for therapy. Rheumatology 52, 34–44 (2013). [DOI] [PubMed] [Google Scholar]
- 34. Neuvonen, P.J. , Niemi, M. & Backman, J.T. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin. Pharmacol. Ther. 80, 565–581 (2006). [DOI] [PubMed] [Google Scholar]
- 35. Thompson, P.D. , Clarkson, P. & Karas, R.H. Statin‐associated myopathy. JAMA 289, 1681–1690 (2003). [DOI] [PubMed] [Google Scholar]
- 36. US Food and Drug Administration . Crestor clinical pharmacology and biopharmaceutics review (2003). <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21-366_Crestor_BioPharmr.pdf>.
- 37. Niemi, M. Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 87, 130–133 (2010). [DOI] [PubMed] [Google Scholar]
- 38. Keskitalo, J.E. , Pasanen, M.K. , Neuvonen, P.J. & Niemi, M. Different effects of the ABCG2 c.421C>A single nucleotide polymorphism on the pharmacokinetics of fluvastatin, pravastatin, and simvastatin. Pharmacogenomics 10, 1617–1624 (2009). [DOI] [PubMed] [Google Scholar]
- 39. Ho, R.H. et al. Effect of drug transporter genotypes on pravastatin disposition in European‐ and African‐American participants. Pharmacogenet. Genomics 17, 647–656 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ieiri, I. et al. SLCO1B1 (OATP1B1, an uptake transporter) and ABCG2 (BCRP, an efflux transporter) variant alleles and pharmacokinetics of pitavastatin in healthy volunteers. Clin. Pharmacol. Ther. 82, 541–547 (2007). [DOI] [PubMed] [Google Scholar]
- 41. White, W.B. et al. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N. Engl. J. Med. 378, 1200–1210 (2018). [DOI] [PubMed] [Google Scholar]