

Abstract
Spinal cord injury (SCI) results in the disruption of supraspinal control of spinal networks and an increase in the relative influence of afferent feedback to sublesional neural networks, both of which contribute to enhancing spinal reflex excitability. Hyperreflexia occurs in ~75% of individuals with chronic SCI and critically hinders functional recovery and quality of life. It is suggested to result from an increase in motoneuronal excitability and a decrease in presynaptic and postsynaptic inhibitory mechanisms. In contrast, locomotor training decreases hyperreflexia by restoring presynaptic inhibition.
Primary afferent depolarization (PAD) is a powerful presynaptic inhibitory mechanism that selectively gates primary afferent transmission to spinal neurons to adjust reflex excitability and ensure smooth movement. However, the effect of chronic SCI and step-training on the reorganization of presynaptic inhibition evoked by hindlimb afferents, and the contribution of PAD has never been demonstrated. The objective of this study is to directly measure changes in presynaptic inhibition through dorsal root potentials (DRPs) and its association to plantar H-reflex inhibition. We provide direct evidence that H-reflex hyperexcitability is associated with a decrease in transmission of PAD pathways activated by PBSt afferents after chronic SCI. More precisely, we illustrate that the pattern of inhibition evoked by PBSt group I muscle afferents onto both L4-DRPs and plantar H-reflexes evoked by the distal tibial nerve is impaired after chronic SCI. These changes are not observed in step-trained animals, suggesting a role for activity-dependent plasticity to regulate PAD pathways activated by flexor muscle group I afferents.
Keywords: Spinal cord injury, Step-training, Presynaptic inhibition, H-reflex, Dorsal root potential
INTRODUCTION
Following chronic spinal cord injury (SCI), pathological adaptations lead to the development of spasticity and hyperreflexia in ~75% of individuals (Maynard et al., 1990; Skold et al., 1999; Holtz et al., 2017). The symptoms range from hyperactive stretch reflex to increased muscle tone and from involuntary flexor withdrawal to extensor spasms (Nielsen et al., 2007). These symptoms are the consequence of multiple mechanisms including the disengagement of supraspinal control below the injury site and subsequent increase in the relative influence of afferent feedback, hyperexcitability of motoneurons and decrease in spinal inhibitory mechanisms (Hultborn & Malmsten, 1983; Malmsten, 1983; Bennett et al., 1999; 2001; Edgerton et al., 2001; Boulenguez et al., 2010; Brocard et al., 2016). Increased activation of motoneurons by Ia afferents is thought to contribute to hyperactive stretch reflexes after SCI, which is a hallmark of spasticity. Historically, the hyperexcitability of the monosynaptic reflex (or H-reflex) after SCI has been suggested to rely on presynaptic inhibitory mechanisms (Thompson et al., 1992; Calancie et al., 1993; Faist et al., 1994; Nielsen et al., 1995; Schindler-Ivens & Shields, 2000; Morita et al., 2001; Grey et al., 2008). Presynaptic inhibition is a powerful mechanism that is under the control of supraspinal centers and sensory afferents. It selectively gates primary afferent transmission to spinal neurons by decreasing the probability of transmitter release from presynaptic terminals (reviewed in Rudomin & Schmidt, 1999; Rudomin, 2009). Classical presynaptic inhibition was described by Eccles and colleagues more than half a century ago (Eccles, 1964) and shown to rely on GABAergic axo-axonic synapse onto sensory afferents. Presynaptic inhibition of the monosynaptic reflex is paralleled by primary afferent depolarization (PAD) and its antidromic spread to dorsal roots recorded as a dorsal root potential (DRP) which is mostly mediated by GABAergic receptors on sensory afferents and can be used as an estimate of its strength (Barron & Matthews, 1938; Eccles et al., 1963a-b; Curtis et al., 1971).
After acute SCI, tonic supraspinal inhibition of spinal reflex pathways is impaired, leading to disinhibition of spinal reflexes (Eccles & Lundberg, 1959; Carpenter et al., 1963; Quevedo et al., 1993). Presynaptic inhibition of the monosynaptic reflex evoked by posterior biceps – semitendinosus (PBSt) muscles group I afferents remains after an acute SCI in cats, when tonic descending inhibition is removed and supraspinal control disrupted but is reduced as the injury becomes chronic (Hancock et al., 1973; Naftchi et al., 1979). The presence of presynaptic inhibition after acute injury suggests that PAD pathways involved in presynaptic inhibition of the monosynaptic reflex are mostly located below the injury site, likely in the lumbar cord spinal cord. This further suggests that the decrease in presynaptic inhibition of the monosynaptic reflex after chronic SCI is unlikely due to the acute loss of descending control, but rather to plastic changes in the strength of transmission in PAD pathways activated by hindlimb afferents in local spinal networks, but this remains to be determined. Therefore, the first objective of this study is to directly investigate the changes in transmission in PAD pathways originating from hindlimb afferents through L4-DRP recordings comparing acute SCI to chronic SCI to assess if a reduction in the compound PAD amplitude is associated with a decrease in presynaptic inhibition of the H-reflex.
After chronic SCI and other conditions that lead to long-lasting immobilization, spinal reflex modulation is improved by locomotor training (Côté et al., 2003; Côté & Gossard, 2004; Martin Ginis & Latimer, 2007; Knikou & Mummidisetty, 2014; Caron et al., 2016). Activity-dependent recovery relies on sensory feedback during stepping as descending modulation is disrupted (Edgerton et al., 2001). Increased phase-dependent modulation of sensory input with locomotor training after chronic SCI has been hypothesized to rely on the recovery of presynaptic inhibition for years (Bouyer & Rossignol, 2003; Côté et al., 2003). In agreement with this putative return of presynaptic inhibition, the hyperexcitability of the monosynaptic reflex (or H-reflex) is decreased in both SCI humans and animals that followed a step-training regimen (Côté et al., 2003; Knikou & Mummidisetty, 2014). The second objective of this study is to directly investigate if step-training improves primary afferent control on presynaptic inhibition of the H-reflex after chronic SCI through an increase in transmission in PAD pathways.
We hypothesized that 1) a decrease in PAD evoked by primary afferents originating from the hindlimbs contributes to decrease presynaptic inhibition of the H-reflex after chronic SCI and 2) the beneficial effect of step-training on presynaptic inhibition of the H-reflex is associated with a concomitant increase of the PAD after chronic SCI. Using a decerebrated rat preparation to preserve spinal neurons excitability, we first evaluated PAD pathways activated by PBSt muscle afferents as it has been shown to be an efficient source of presynaptic inhibition of the monosynaptic reflex (Eccles, 1964; Hancock et al., 1973; Naftchi et al., 1979). The regulation of presynaptic inhibition is highly specific with primary afferents of different modalities receiving specific PAD patterns. In addition, different terminals can be selectively regulated (Lomeli et al., 1998) so that PAD originating from muscles and cutaneous afferents may be differentially regulated (Rudomin & Schmidt, 1999). Therefore, we further evaluated transmission in PAD pathways activated by low threshold distal tibial afferents, which is a mixed muscle and cutaneous nerve, but mostly contains cutaneous afferents evoking PAD on other cutaneous and Ib-II afferents (Janig et al., 1968a-b; Harrison & Jankowska, 1989; Quevedo et al., 1995; Enriquez et al., 1996a-b). We limited our investigation to stimulation intensities up to 5T, as the activation of higher threshold cutaneous afferents (~20T) evokes delayed presynaptic inhibition of the monosynaptic reflex after an acute SCI, that was shown to rely on extrasynaptic mechanisms in addition to the classical PAD in Ia afferents (Bergmans et al., 1974).We found that after chronic SCI, the compound PAD recorded as a DRP from a cut L4 dorsal rootlet is decreased when evoked by PBSt but not tibial afferents.
We also simultaneously measured the L4-DRP and the plantar H-reflex evoked by the distal tibial nerve and tested the effect of a PBSt conditioning stimulation. We show that there is a decrease in transmission in PAD pathways activated by PBSt group I afferents, which is associated with a reduction in presynaptic inhibition of the plantar H-reflex, and contributes to hyperreflexia after chronic SCI. The increase in presynaptic inhibition of the plantar H-reflex evoked by PBSt group I afferents with step-training further suggests a better modulation of tibial Ia afferents by PBSt. Because flexor muscle proprioceptors affect the activity of hindlimb muscles and exert a powerful control on locomotor rhythm (Grillner & Rossignol, 1978; Rossignol et al., 2006), this suggests a contribution of activity-dependent plasticity in decreasing hyperreflexia and improving walking ability through restoration of presynaptic inhibition.
METHODS
Ethical approval.
All procedures complied with ARRIVE guidelines, were performed in accordance with protocols approved by Drexel University Institutional Animal Care and Use Committee (IACUC, project No 1044959) and followed the National Institutes of Health guidelines for the care and use of laboratory animals. Rats were housed in facilities accredited by AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) on a 12h light-dark cycle, with controlled room temperature, and ad libitum access to food and water. Rats were singly housed for 3 days following injury, then in pairs for the remainder of the study.
Surgical procedures and postoperative care.
Adult female Sprague Dawley rats (240-300g, Charles Rivers Laboratories) underwent a complete spinal transection at the low thoracic level (T12) as described previously (Côté et al., 2011; Côté et al., 2014). Briefly, rats were anesthetized with isoflurane (~2.5%) in O2. The depth of anesthesia was monitored through a lack of response to toe pinch and reaction to incision and the body temperature maintained to ~37°C by a thermal barrier to limit heat loss during and after operating procedures. A laminectomy was performed at the T10–T11 vertebral level under aseptic conditions. The dura was carefully slit open, the spinal cord completely transected with small scissors, and saline-soaked gel foam inserted in the cavity to achieve hemostasis. The completeness of the spinal cord lesion was confirmed by the retraction of the rostral and caudal portions of the cord and by examining the ventral floor of the spinal canal. Paravertebral muscles were sutured back together, and the skin closed with wound clips. Upon completion of the surgery, animals received a single injection of slow release buprenorphine (0.05 mg/kg, s.c.) and daily saline (5 ml, s.c.) and Baytril (100 mg/kg, s.c.) for 7 days to prevent dehydration and infection, respectively. In the immediate period following surgery, food pellets were provided on the cage floor. Although mobility is impaired following SCI, animals were able to reach food pellets in the standard feeding tray within hours of surgery. Post-operative care also included monitoring animals for pain and discomfort twice a day. Positive signs included, but were not limited to, piloerection, naso-ocular discharge, excess vocalization, labor breathing, unresponsiveness to external stimuli, autophagia, loss of body weight, infection, dehydration. Veterinarian assistance was requested, and the appropriate treatment administered if any of those signs were present. Bladders were also expressed manually at least twice daily until voiding reflex returned.
Rats were randomly assigned to one of the following groups: chronic spinal cord injury (Chronic SCI; n=7) or chronic spinal cord injury + step-training (SCI + Step-training; n=6) with the terminal experiment taking place ~8 weeks post-injury. Another group of animals used as “Controls” (Acute SCI; n=8; 260-460g) were acutely transected at the beginning of the terminal experiment at the same spinal level at least 3h before starting terminal experiment recordings (see Terminal experiment section).
Locomotor training.
Beginning on day 5-7 post-injury, the SCI + Step-training group received 10 min of daily locomotor training, 5 days per week until the terminal experiment. Animals were placed on a treadmill belt (11-15cm/s) with the forelimbs resting on an acrylic glass platform. Perineal stimulation was provided to induce and maintain locomotor movements in the hindlimbs (video in Côté et al., 2011, see also Alluin et al., 2015), likely through the activation of the Central Pattern Generator (CPG) by sacro-caudal afferents (Cherniak et al., 2014; Etlin et al., 2010). The pressure applied to the perineal region was adjusted on a case by case basis (light/strong, tonic/rhythmic) to achieve the best rhythmic behavior and maximal support during stance. Additional weight support was provided by the experimenter and adjusted at a level sufficient to prevent collapse of the locomotion until the animals recovered weight-bearing stepping (Côté et al., 2011). Animals are very tolerant, with no apparent stress or pain associated with this procedure. It should be emphasized that the animals subjected to this form of training are paralyzed and experience no cognitive pain below the level of spinal cord injury.
Terminal experiments.
Isoflurane anesthesia (~3.5%) in O2 was induced by mask and was maintained through tracheotomy during surgery. One common carotid artery was cannulated to monitor blood pressure while the other one was ligated. The depth of anesthesia was continuously monitored through blood pressure, heart rate and respiratory rate. A cannula was inserted in a jugular vein for administration of Ringer-Locke solution (2.5% dextrose) to maintain the mean arterial blood pressure (>80 mmHg). The posterior biceps and semitendinosus nerves (PBSt) were dissected free from surrounding tissue and cut distally, while the distal portion of the tibial nerve (proximal to the medial and lateral plantar nerve bifurcation) was dissected free from surrounding tissue and left intact. A laminectomy was performed from T13 to L3 to expose the lumbar enlargement of the spinal cord and to allow appropriate identification of dorsal roots. At this point, the Acute SCI group was transected at the same spinal level as Chronic SCIs (T12), with a minimum of 3 hours before starting electrophysiological recordings. Anesthesia was maintained during this recovery period. Skin flaps surrounding the spinal cord and peripheral nerves were then used to construct mineral oil pools. A precollicular decerebration was performed, the rostral tissue was removed by suction and residual bleeding was prevented by packing the cranial fossae with small pieces of gelfoam soaked in cold thrombin solution. Anesthesia was then slowly discontinued, and rats artificially ventilated to maintain the expired CO2 near 4%. Animal rectal temperature was controlled and kept at 37°C via a DC heating pad and heating lamp. Recordings started at least 60 min after isoflurane was discontinued to ensure complete anesthesia removal and animal stabilization (Marchenko et al., 2002). After completion of the terminal experiment, rats were overdosed with Euthasol (390mg/kg sodium pentobarbital and 50mg/kg phenytoin, i.p.), followed by a thoracotomy and the section of the right atrium to facilitate exsanguination.
Stimulation and recordings.
The experimental setup is illustrated in Figure 1A. PBSt and tibial nerves were mounted on bipolar hook electrodes (FHC, Bowdoin, ME, USA) for stimulation. The cord dorsum potential (CDP), elicited by the stimulation of peripheral nerves, is used to record the incoming volley with a monopolar silver ball electrode placed at the L4-L5 dorsal root entry. The stimulation threshold for the most excitable afferent was defined as the minimum intensity capable of producing an incoming volley as recorded from the CDP. The stimulation strength is thereafter expressed as multiples of this threshold (xCDP). Dorsal root potentials (DRPs) were recorded from cut L4 dorsal rootlet (~1/3 of L4) using a bipolar tungsten hook electrode, placed 1mm from the cord, in response to a stimulation (200 μs duration) of various intensities to the tibial or PBSt nerves. CDP and DRP recordings were amplified (x1000) and band-pass filtered (0.1 Hz-5 kHz; A-M System, Carlsborg, WA, USA). H-reflexes were evoked by a stimulation (200 μs duration) to the tibial nerve and recorded using bipolar wire electrodes (Cooner Wire, Chatsworth, CA, USA) inserted in the interosseous muscles. EMG recordings were amplified (x1000) and band-pass filtered (10 Hz-5 kHz; A-M Systems). All signals were digitized (10 kHz) and fed to Signal 5 software (Cambridge Electronic Design, UK).
Figure 1. Experimental set-up.
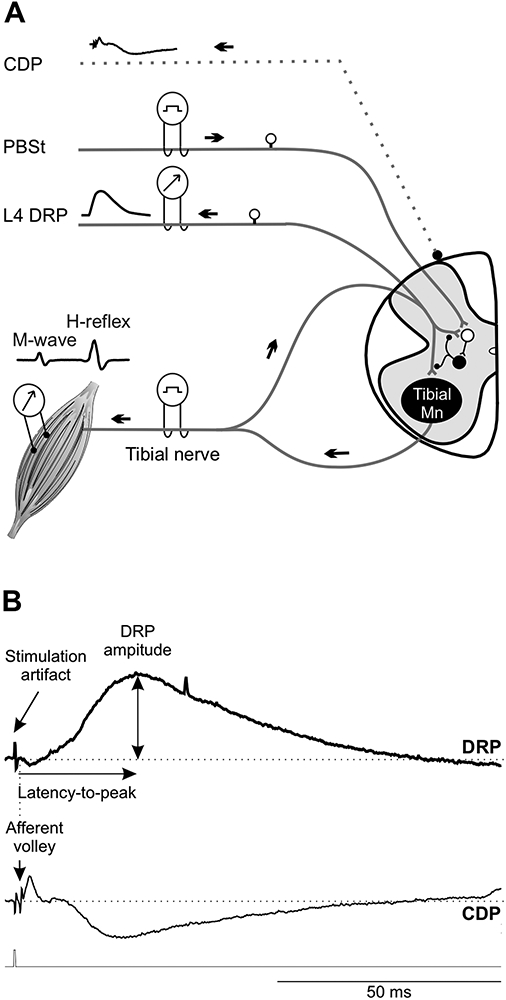
A, The PBSt nerve, tibial nerve and cut dorsal rootlet were mounted on bipolar hook electrodes. The cord dorsum potential (CDP) was recorded using a monopolar silver ball electrode placed at the L4-L5 dorsal root entry to determine the activation threshold for the most excitable afferent fiber. Dorsal root potentials (DRPs) were recorded in response to a stimulation to the tibial or PBSt nerves. H-reflexes were evoked by a stimulation to the tibial nerve and obtained using bipolar wire electrodes inserted under the plantar surface of the foot. In the conditioning protocol, PBSt stimulation was used as the conditioning stimulus and preceded the tibial stimulation. B, Typical DRP recording displaying a long-lasting upward deflection. By convention, DRP negativity is represented upwards and CDP negativity downwards. The latency-to-peak was determined as the time between the onset of the afferent volley (CDP) and the maximal DRP amplitude from baseline to peak.
To determine the input-output relationship, PBSt- and Tib-evoked DRPs were recorded in response to single stimuli of increasing intensity until reaching DRPmax to built an input-output curve. M-waves and H-reflexes were also recorded in response to single stimuli of increasing intensity to build a recruitment curve. Presynaptic inhibition evoked by group I afferents from PBSt on DRPs and H-reflex evoked by a stimulation to the tibial nerve were estimated using a conditioning-test protocol. Tibial nerve stimulations (1 pulse, 1.2-1.8T) were alternatively preceded (conditioned) or not (test) by stimulation of PBSt (4 pulses, 1.5T, 250 Hz) with conditioning-test (C-T) intervals ranging from 0 to 150ms. Each C-T combination was delivered at 0.2Hz and a total of 8-10 frames were averaged.
Data analysis.
A typical DRP trace is shown in Figure 1B. The stimulation artifact is followed by a short downward deflection. The subsequent long-lasting upward response, representing the DRP, is clearly distinguishable. We measured the latency-to-peak as determined by the time between the onset of the afferent volley (cord dorsum potential, CDP) and the maximal amplitude of a given DRP. The DRP amplitude was measured and plotted as a function of stimulus intensity to build an input-output curve for each animal. The stimulation intensity required to reach the maximal DRP amplitude (DRPmax) was then determined. A Boltzmann sigmoid function was then fitted to the data (Klimstra & Zehr, 2008; Lundbye-Jensen & Nielsen, 2008a; Smith et al., 2015), and the goodness of the fit (R2) determined.
The estimated parameters of the function include the maximal amplitude (amax), the slope of the function (m) which reflects the gain independently from the absolute amplitude (Carroll et al., 2001), and the stimulation intensity required to elicit a DRP equivalent to 50% of DRPmax (s50) at a given stimulation intensity (s). Group averages were calculated from the individual values for amax, m and s50, and used to establish a sigmoid function for each group. Similarly, we also performed measurements of the input-output curve for the M-wave and H-reflex for each animal. The motor threshold, H-reflex threshold, maximal H-reflex and M-wave amplitude and latency were also determined.
To determine the strength of inhibition evoked by PBSt group I afferents on the H-reflex, the amplitude of the conditioned H-reflex was expressed as a percentage of the amplitude of the unconditioned test reflex and was plotted against the C-T interval. To determine the strength of inhibition, the amplitude of the PBSt-evoked DRP was subtracted from the conditioned Tib-DRP to isolate the remaining Tib-DRP (Eccles et al., 1963b; Wall & Lidierth, 1997). The amplitude of the conditioned Tib-DRP response expressed as a percentage of the unconditioned response was then averaged (n=8-10 traces) and plotted against the C-T interval.
STATISTICS
One-way ANOVA followed by Holm-Sidak post hoc test (or Kruskal-Wallis followed by Dunn’s post hoc test if normality or equal variance test failed) was used to determine significant differences between groups unless stated below. To address the effect of the conditioning PBSt stimulation on Tib-DRP or H-reflex amplitude, a two-way repeated measures ANOVA followed by a Holm-Sidak post hoc test was performed to determine significant differences between groups, the C-T interval and their interaction. Results are presented as mean values ± standard deviation (SD). A linear regression analysis was used to correlate the conditioned H-reflex and DRP amplitude evoked by the same stimulation to the tibial nerve. Statistical analysis was performed using Sigma Plot software version 14.0 (SYSTAT software). For all statistical tests, the effects were considered significant when P < 0.05.
RESULTS
Chronic SCI decreases transmission in PAD pathways activated by PBSt afferents
To investigate the effect of chronic SCI and potential benefits of step-training on the strength of presynaptic inhibition, we first recorded dorsal root potentials (DRPs) evoked by the stimulation of 1) the posterior biceps-semitendinosus (PBSt) nerve, a knee flexor and hip extensor, known to be an efficient source of presynaptic inhibition (Eccles et al., 1962c; Rudomin & Schmidt, 1999; Côté & Gossard, 2003), and 2) the distal portion of the tibial nerve, a mixed nerve containing both cutaneous and muscle afferents innervating the plantar surface of the foot. DRPs were recorded from a cut L4 dorsal rootlet in response to a stimulation of the tibial nerve (Tib-DRP) or of the PBSt nerve (PBSt-DRP) (Fig. 1A).
The properties of the recorded DRPs are depicted in Table 1. Chronic SCI did not significantly affect the latency to reach peak DRP (Tib-DRP: F2,18 = 1.750; P = 0.202; PBSt-DRP: F2,18 = 2.145; P = 0.146, one-way ANOVA). However, the stimulation intensity required to evoke a DRP of maximal amplitude (DRPmax) was different between groups, but only when induced by a stimulation to PBSt (H2,18 = 13.986; P < 0.001, Kruskal-Wallis) and not the tibial nerve (F2,18 = 0.106; P = 0.9, one-way ANOVA). Overall, the stimulation intensity required to evoke PBSt-DRPmax almost doubled after chronic SCI as compared to acute SCI from 2.32T ± 0.49 to 4.30T ± 0.55 (P < 0.05, Dunn’s). This increase was independent from the stimulation intensity required to evoke an incoming afferent volley, as the CDP was not different between groups whether evoked by the tibial (H2,18 = 3.262; P = 0.196, one-way ANOVA on Ranks) or the PBSt nerve (F2,18 = 0.686; P = 0.516, one-way ANOVA).
Table 1.
Properties of DRPs evoked by PBSt or tibial nerve stimulation
| PBST DRPmax | Tibial DRPmax | ||||||
|---|---|---|---|---|---|---|---|
| n= | Threshold for CDP (uA) |
Stimulation intensity (xCDP) |
Latency-to- peak (ms) |
CDP threshold (uA) |
Stimulation intensity (xCDP) |
Latency-to- peak (ms) |
|
| Acute SCI | 8 | 16.24 ± 8.72 | 2.32 ± 0.49 | 26.21 ± 2.16 | 7.13 ± 5.47 | 2.39 ± 0.41 | 20.86 ± 2.32 |
| Chronic SCI | 7 | 11.01 ± 6.17 | 4.30 ± 0.55* | 23.89 ± 2.77 | 9.81 ± 2.48 | 2.34 ± 0.41 | 19.59 ± 1.91 |
| SCI + Step-Training | 6 | 15.00 ± 11.46 | 2.65 ± 0.55 | 23.61 ± 2.51 | 7.72 ± 4.10 | 2.29 ± 0.27 | 18.94 ± 1.43 |
The threshold for CDP (Tib-DRP: H2,18 = 3.262; P = 0.196, Kruskal-Wallis; PBSt-DRP: F2,18 = 0.686; P = 0.516, one-way ANOVA) and the latency to reach peak DRP amplitude (Tib-DRP: F2,18 = 1.750; P = 0.202; PBSt-DRP: F2,18 = 2.145; P = 0.146, one-way ANOVA) were similar across groups. However, the stimulation intensity required to evoke a DRP of maximal amplitude (DRPmax) was significantly different between groups, but only when induced by a stimulation to PBSt (H2,18 = 13.986; P < 0.001, Kruskal-Wallis) and not the tibial nerve (F2,18 = 0.106; P = 0.9, one-way ANOVA). The stimulation intensity required to evoke PBSt-DRPmax was increased after chronic SCI as compared to acute SCI and SCI + Step-training (P < 0.05, Dunn’s), while SCI + Step-training animals were no different to acute SCI (P > 0.05, Dunn’s). Values are mean ± SD.
P<0.05 (vs Acute SCI and SCI + Step-training). DRP, Dorsal root potential; CDP, cord dorsum potential.
The DRP represents the compound potential from activated afferents in the recorded rootlet at a given stimulation intensity. As the amplitude of the DRP reflects the amount of PAD evoked by the stimulated primary afferents (Lucas-Osma et al., 2018), we therefore sought to investigate whether chronic SCI affects the recruitment gain of DRPs evoked by tibial and PBSt afferents. Figure 2A illustrates examples of DRPs recorded in response to stimulations of increasing intensity in an animal from the Acute SCI, Chronic SCI and SCI + Step-training group. The DRP amplitude augmented with stimulation intensity, whether evoked by PBSt or the tibial nerve, and the resulting input-output curve was fitted to a sigmoid function (Fig. 2B). All animals displayed input-output relationships that tightly fit a sigmoid function (P < 0.001) with individual R2 ranging from 0.93-0.98 after acute SCIs, 0.94-0.99 after chronic SCI and 0.90-0.99 after SCI + Step-training. Function parameters were then averaged by group (Fig. 2C-E), and the sigmoid functions for each group plotted with a 95% confidence interval (Fig. 2F-G). Overall, chronic SCI did not significantly affect DRPmax evoked by either the tibial (F2,18 = 0.642; P = 0.538) or the PBSt nerve (F2,18 = 1.049; P = 0.371, one-way ANOVA) whether the animals were step-trained or not (Fig. 2C). However, the stimulation intensity required to reach 50% of DRPmax (s50) was significantly different between groups (Fig. 2D) both when evoked by the tibial (F2,18 = 7.457; P = 0.004) or PBSt (H2,18 = 15.068; P < 0.001, Kruskal-Wallis). Chronic SCI increased the s50 from 1.26T ± 0.08 to 1.39T ± 0.13 as compared to acute SCI when evoked by the tibial (P = 0.020, Holm-Sidak) and from 1.35T ± 0.11 to 2.10T ± 0.38 when evoked by PBSt (P < 0.05, Dunn’s). These results are further supported by a significant difference in the slope of the PBSt-DRP evoked input-output curve (F2,18 = 12.617; P < 0.001, one-way ANOVA) between groups (Fig. 2E), with chronic SCI significantly decreasing the steepness of the slope (P < 0.001, Holm-Sidak). In contrast, the slope of Tib-DRPs input-output curve was not different between groups (Fig. 2E; F2,18 = 3.162; P = 0.067, one-way ANOVA). Overall, our results suggest that although chronic SCI does not affect the amplitude of the maximal DRP that can be evoked, it decreases the amplitude of DRPs generated at submaximal stimulation intensity, specifically when evoked by PBSt (but not the tibial).
Figure 2. Chronic SCI and step-training specifically modulates DRPs evoked by PBSt group I muscle afferents.

A, Example of DRPs evoked by the tibial (top) or PBSt (bottom) nerves at different stimulation intensities (1.2, 1.5, 2, or 5T) in an Acute SCI, Chronic SCI and SCI + Step-training animal. B, Example of the individual sigmoid function that was fitted to input-output relationships for each animal. Overall, all animals displayed curves that tightly fitted a sigmoid function (P < 0.001) with R2 ranging from 0.93-0.98 after acute SCI, 0.94-0.99 after chronic SCI and 0.90-0.99 after SCI + Step-training. C-E, Chronic SCI did not significantly affect the amplitude of Tib-DRPmax (P = 0.538) and PBst-DRPmax (P = 0.371) whether the animals were step-trained or not (C). The stimulation intensity to reach 50% of DRPmax (s50) when evoked by the tibial (P = 0.004) or PBSt nerve (P < 0.001) was different across groups (D). Chronic SCI significantly increased s50 as compared to acute SCI (tibial, P = 0.020; PBSt, P < 0.001). Step-trained animals had similar s50 than Chronic SCIs when DRPs were evoked by the tibial nerve (P = 0.225 vs. Chronic SCI) but lower s50 when evoked by PBSt (p<0.001) with values similar to acute SCI (P = 0.182). The slope of PBST-DRPs input-output relationship was different across groups only when evoked by PBSt (P < 0.001), not tibial nerve (P = 0.067) (E). Chronic SCI decreased the slope (P < 0.001), while step-training had no further effect (P = 0.100 vs. Chronic SCI). Results are expressed as mean ± SD, one-way ANOVA followed by Holm-Sidak post hoc test. *P < 0.05; ** P < 0.01; ***P < 0.001. Acute SCI, n = 8; Chronic SCI, n = 7; SCI + Step-training, n = 6. CDP, cord dorsum potential; F-G, Sigmoid function resulting from group averages is illustrated with 95% confidence interval for Tib- and PBSt-DRPs.
Step-training increases the transmission in PAD pathways activated by PBSt afferents after chronic SCI
Among the beneficial effect of step-training after SCI is a decrease in hyperreflexia and spasticity and improvement in spinal reflex modulation that is believed to rely, at least in part, on a restoration in presynaptic inhibition (Bouyer & Rossignol, 2003; Côté & Gossard, 2004). We sought to investigate whether step-training more specifically affected the recruitment gain of Tib- and PBSt-DRPs after chronic SCI. As reported in the previous paragraph, no group difference was observed in latency to reach peak DRP amplitude, nor in the stimulation intensity required to reach DRPmax, suggesting that step-training did not affect those properties (Table 1). Also, step-training did not affect any of the parameters of the input-output relationship of evoked Tib-DRPs (Fig. 2C-E). However, the stimulation intensity required to evoke PBSt-DRPmax was decreased after step-training as compared to untrained chronic SCI animals from 4.30T ± 0.55 to 2.65T ± 0.55 (P < 0.05, Dunn’s), and was no different from the acute SCI Controls (P > 0.05, Dunn’s) (Table 1). Step-training restored the stimulation intensity required to reach 50% of DRPmax (Fig. 2D, P < 0.001) with values similar to after acute SCI (P = 0.182, Dunn’s). These results indicate that step-training is associated with larger DRP amplitudes originating from PBSt afferents after chronic SCI.
PAD pathways activated by PBSt group I muscle afferents are disrupted by chronic SCI
The decrease in s50 after chronic SCI in the absence of a decrease in the maximal DRP amplitude evoked by PBSt suggests a decrease in DRP amplitude at submaximal intensity, in the range of large PBSt group I muscle afferent activation. Previous studies have suggested that stimulation intensities < 2T and frequencies >200Hz specifically increase the activation of PAD pathways by group I muscle afferents (Tasaki, 1953; Eccles et al., 1963b). To specifically address the role of PAD pathways activated by group I muscle afferents, without engaging group II afferents, we measured the amplitude of the DRP evoked in L4 dorsal rootlet by a tetanic stimulation to PBSt nerve at submaximal group I afferents strength (4p, 250Hz, 1.5T). Figure 3A shows an example of DRPs evoked by PBSt group I afferents in an animal form the Acute SCI, Chronic SCI and SCI + Step-training group, with the associated DRPmax illustrated for reference. We found a significant difference in the relative DRP amplitude between groups (F2,18 = 14.336; P < 0.001, one-way ANOVA). Chronic SCI decreased the amplitude of DRPs evoked by PBSt group I afferents (P = 0.002, Holm-Sidak). In addition, step-trained animals displayed significantly larger PBSt-DRPs amplitude than chronic SCI (P < 0.001, Holm-Sidak), and were similar to acute SCIs (P = 0.058, Holm-Sidak), suggesting that step-training is associated with increased presynaptic inhibition originating from PBSt group I afferents after chronic SCI (Fig. 3B). Because the size of the DRP is highly sensitive to the number of afferents in the recorded rootlet and the distance between the recording electrode and the cord, we chose to express the amplitude of the DRP evoked by PBSt group I afferents as a function of the DRPmax recorded in the same experimental conditions. It is important to note that similar differences in DRP absolute amplitude (in mV, not shown) were also observed between groups (F2,18 = 16.315; P < 0.001, one-way ANOVA) with a decrease in DRP amplitude in chronic SCI animals as compared to acute SCI (P = 0.026, Holm-Sidak), unless the animal was step-trained , with DRP amplitude significantly larger in SCI + Step-training than in chronic SCI (P < 0.001, Holm-Sidak) and acute SCI animals (P = 0.002, Holm-Sidak).
Figure 3. Disruption in the transmission of group I PBSt afferents in PAD pathways after chronic SCI is improved by step-training.

A, Representative traces of DRPs evoked by a stimulation to PBSt nerve at group I afferent strength (4p, 1.5T, 250Hz) in an Acute SCI, a Chronic SCI and a SCI + Step-training animal. For reference purpose, the dotted line illustrates DRPmax in the same animal. B, The amplitude of DRPs evoked by PBSt group I afferents was different across groups (P < 0.001). Chronic SCI decreased DRP amplitude (P = 0.002, vs. Acute SCI). This decrease was not observed after step-training with values similar to acute SCIs (P = 0.058) but significantly larger than chronic SCI animals (P < 0.001). Results are expressed as mean ± SD, one-way ANOVA followed by Holm-Sidak post hoc test ** P < 0.01; *** P < 0.001. Acute SCI, n = 8; Chronic SCI, n = 7; SCI + Step-training, n = 6.
Step-training promotes the inhibition of Tib-DRPs induced by PBSt group I afferents after chronic SCI
To assess the strength and latency of presynaptic inhibition originating from PBSt group I afferents, we used a condition-test protocol and recorded DRPs evoked by a stimulation to the tibial nerve when preceded or not by a conditioning stimulation of PBSt group I afferents (Eccles et al., 1962b; 1963b) (Fig 4A). Figure 4B illustrates that after chronic SCI, there is minimal effect of a conditioning stimulation to PBSt, unless the animal was step-trained. Overall, there was a significant difference between groups (F2,171 = 9.985; P = 0.001), C-T intervals (F9,171 = 58.299; P < 0.001), and an interaction between groups and C-T intervals (F18,171 = 1.847; P = 0.023, two-way RM ANOVA). The inhibition of the Tib-DRP exerted by PBSt group I afferents was significantly decreased after chronic SCI at C-T intervals ranging from 10 to 60ms as compared to acute SCI (Fig. 4C, P < 0.05, Holm-Sidak). Step-training was associated with an increase in inhibition at intervals ranging from 10 to 50ms (P < 0.05, Holm-Sidak) and was similar to acute SCIs (P > 0.05, Holm-Sidak). These results suggest that presynaptic inhibition induced by PBSt group I afferents is decreased after chronic SCI unless the animals are step-trained indicating that activity-based therapies may contribute to either increase inhibition in this pathway or prevent the chronic SCI induced decrease in inhibition.
Figure 4. Step-training improves the inhibition induced by PBSt group I afferents on L4-DRPs evoked by the tibial nerve after chronic SCI.

A, Conditioning-test protocol used to estimate the level of inhibition evoked by a conditioning stimulation to PBSt group I afferents (4p, 250Hz, 1.5T) on Tib-DRPs (1p, 0.2 Hz, 1.2-1.8T). B, DRPs evoked by the tibial nerve when preceded (black) or not (gray) by a conditioning stimulation to PBSt (C-T: 50 ms) in an Acute SCI, a Chronic SCI and a SCI + Step-training animal. The inhibitory effect of the conditioning stimulation is not present after chronic SCI. C, Overall, chronic SCI significantly decreases the strength of inhibition at C-T intervals ranging from 10 to 60ms as compared to acute SCI (P < 0.05, gray stars). This decrease was not observed in step-trained animals with values similar to acute SCIs and inhibition significantly larger than chronic SCI animals at intervals ranging from 10 to 50ms (P < 0.05, green stars). Two-way RM ANOVA followed by Holm-Sidak post hoc test, *p<0.05; **p<0.01; ***p<0.001. Acute SCI, n = 8; Chronic SCI, n = 7; SCI + Step-training, n = 6. C-T: Conditioning-Test.
Presynaptic inhibition exerted by PBSt group I muscle afferents contributes to plantar H-reflex hyperexcitability after chronic SCI.
In order to investigate if the decrease in presynaptic inhibition contributes to hyperreflexia, we assessed the effect of a conditioning stimulation to PBSt on DRPs and H-reflexes evoked by a stimulation to the tibial nerve simultaneously. We first examined the excitability of the H-reflex by analyzing the general features of the M-waves and H-reflexes. Chronic SCI did not alter the latency, the threshold for activation, nor the maximal amplitude of the M-wave and H-reflex (Table 2). Figure 5A illustrates examples of M-wave and H-reflex recruitment curves in an animal from the Acute SCI, Chronic SCI and SCI + Step-training group. The recruitment curves of the M-wave and H-reflex were individually fitted to a sigmoid function for each animal. All animals displayed recruitment curves that tightly fitted a sigmoid function (P < 0.001), with R2 ranging from 0.93-0.99 after acute SCI, 0.90-0.99 after chronic SCI and 0.91-0.99 after SCI + step-training. Function parameters were then averaged by group and the resulting sigmoid functions for each group plotted with a 95% confidence interval (Fig. 5B-C). Further analysis of parameters of the sigmoid function illustrate that chronic SCI did not change the maximal H-reflex amplitude (Fig. 5D; F2,18 = 0.826; P = 0.454, one-way ANOVA) nor the stimulation intensity to obtain 50% of Hmax (Fig. 5E; F2,18 = 0.510; P = 0.609, one-way ANOVA) whether the animals were step-trained or not. However, the slope of the H-reflex sigmoid function was significantly different between groups (Fig. 5F; F2,18 = 7.682; P = 0.004, one-way ANOVA). The slope of the H-reflex recruitment curve was dramatically steeper after chronic SCI as compared to acute SCIs (P = 0.002) and SCI + Step-training (P = 0.015, Holm-Sidak). This observation is unlikely due to changes at the neuromuscular junction as we found no significant difference in Mmax (Fig. 5D; F2,18 = 0.534; P = 0.595, one-way ANOVA), s50 (Fig. 5E; F2,18 = 0.951; P = 0.405, one-way ANOVA) or the slope of the M-wave recruitment curve (Fig. 5F; F2,18 = 2.463; P = 0.113, one-way ANOVA). Altogether, these results suggest an increase in the recruitment gain of the motor pool by tibial afferents after chronic SCI that is normalized by step-training.
Table 2.
Properties of the M-Wave and H-Reflex
| M-wave | H-reflex | ||||||
|---|---|---|---|---|---|---|---|
| n= | Motor threshold (uA) |
Latency (ms) |
Mmax (xMT) |
Threshold (x MT) |
Latency (ms) |
Hmax (xMT) |
|
| Acute SCI | 8 | 10.49 ± 9.23 | 1.88 ± 0.33 | 1.46 ± 0.26 | 0.96 ± 0.12 | 8.20 ± 0.79 | 1.45 ± 0.24 |
| Chronic SCI | 7 | 12.21 ± 2.87 | 2.11 ± 0.16 | 1.48 ± 0.21 | 1.03 ± 0.13 | 8.00 ± 0.38 | 1.21 ± 0.14 |
| SCI + Step-Training | 6 | 10.43 ± 5.84 | 2.02 ± 0.49 | 1.52 ± 0.27 | 0.94 ± 0.17 | 7.93 ± 0.59 | 1.34 ± 0.22 |
The motor threshold (MT, H2,18 = 2.175; P = 0.337, Kruskal-Wallis), the stimulation intensity to evoke a M-wave of maximal amplitude (Mmax; H2,18 = 0.440; P = 0.803, Kruskal-Wallis) and the M-wave latency (H2,18 = 1.521; P = 0.468, Kruskal-Wallis) were similar across groups. Similarly, the H-reflex threshold (F2,18 = 0.828; P = 0.453, One-way ANOVA), the stimulation intensity to evoke a H-reflex of maximal amplitude (Hmax; F2,18 = 2.437; P = 0.116, One-way ANOVA), and the H-reflex latency (F2,18 = 0.337; P = 0.718, One-way ANOVA) were not different between groups. Values are mean ± SD.
Figure 5. H-reflex excitability is impaired following chronic SCI and normalized by step-training.

A, Example of M-wave and H-reflex recruitment curves in an Acute SCI, a Chronic SCI and a SCI + Step-training animal. Sigmoid functions were individually fitted to the recruitment curves. Overall, all animals displayed curves that tightly fitted a sigmoid function (P < 0.001) with R2 ranging from 0.93-0.99 in acute SCIs, 0.90-0.99 in chronic SCIs and 0.91-0.99 in SCI + Step-training animals. B-C, The sigmoid function resulting from group averages is illustrated with 95% confidence interval for both the M-wave (B) and the H-reflex (C). D-F, Chronic SCI did not alter the Hmax and Mmax amplitude (D) (P = 0.454 and P = 0.595 respectively) and the stimulation intensity to reach 50% of the maximal amplitude (E) (P = 0.609 and P = 0.405 respectively) whether the animals were step-trained or not. However, the slope of the H-reflex sigmoid function was significantly different across groups (F) (P = 0.004). Chronic SCI increased the steepness of the slope of the recruitment curve for the H-reflex as compared to acute SCI (P = 0.002) and step-trained animals (P = 0.015), while step-training shifted the slope toward values similar to acute SCIs (P = 0.465). The slope of the M-wave recruitment curve was similar across groups (P = 0.113). One-way ANOVA followed by Holm-Sidak post hoc test. * P < 0.05; ** P < 0.01. Acute SCI, n = 8; Chronic SCI, n = 7; SCI + Step-training, n = 6. MT, motor threshold.
Figure 6A illustrate an example of conditioned and unconditioned H-reflexes from an animal from the Acute SCI, Chronic SCI and SCI + Step-training groups at 10 and 50ms C-T intervals. The conditioning stimulation to PBSt dramatically decreases the H-reflex amplitude at both C-T intervals in the acute SCI and SCI + Step-training animals, but not in chronic SCIs. Overall, there was a significant difference in the inhibition of the H-reflex between groups (F2,171 = 32.056; P < 0.001), C-T interval (F9,171 = 29.865; P < 0.001), and an interaction between groups and C-T intervals (F18,171 = 2.506; P = 0.001, two-way RM ANOVA). For clarity, these significant differences across C-T intervals within a given group are not illustrated in Figure 6B. The lack of modulation after chronic SCI is further supported by a decrease in H-reflex inhibition between chronic SCI as compared to acute SCI (P < 0.01, grey stars) and step-trained animals (P < 0.05, green stars) at intervals ranging from 10-150ms. Hence, step-training was associated with improved H-reflex inhibition after chronic SCI for intervals ranging from 10-150ms to values similar to acute SCIs, except for the 20-30ms intervals where step-trained animals displayed more inhibition than chronic SCI, but less than acute SCIs (P < 0.05). Because the quantification of H-reflex inhibition by the conditioning stimulation is critically dependent on the amplitude of the test H-reflex response (Crone et al., 1990), we further ensured that the amplitude of the unconditioned H-reflex (expressed as a percent of Mmax) was similar across groups (Fig. 6C, F2,18 = 0.614; P = 0.552, one-way ANOVA).
Figure 6. Step-training improves H-reflex inhibition after chronic SCI.

A, Representative recordings of H-reflexes evoked by a stimulation to the tibial nerve with or without a conditioning stimulation to PBSt in an Acute SCI, a Chronic SCI and SCI + Step-training animal at C-T intervals of 10ms and 50ms. B, There was a significant difference in the strength of inhibition of the H-reflex between groups (F2,171=32.056; p<0.001), C-T interval (F9,171=29.865; P < 0.001), and an interaction between groups and C-T intervals (F18,171=2.506; P = 0.001). For clarity purpose, only significance between groups, but not intragroup is illustrated. Chronic SCI significantly decreased the depression of the H-reflex as compared to acute SCI at intervals ranging from 10-150ms (P < 0.01). In addition, the strength of inhibition in step-trained animals was larger than in non-trained animals after chronic SCI (P < 0.05, green stars) and similar to acute SCIs at similar C-T intervals. Two-way RM ANOVA followed by Holm-Sidak post hoc test. C-T, Conditioning-Test. C, The amplitude of the unconditioned H-reflex (% of Mma) was similar between experimental groups (P = 0.552). One-way ANOVA. D, There was a significant decrease in postsynaptic (C-T intervals 10-20ms; F2,18 = 15.567, P < 0.001) and presynaptic inhibition (C-T intervals 50-150ms; F2,18 = 16.597, P < 0.001). Chronic SCI decreased the strength of postsynaptic (P < 0.001, gray stars) and presynaptic inhibition (P < 0.001, gray stars), unless the animals were step-trained (respectively P = 0.002 and P < 0.001, green stars). One-way ANOVA followed by Holm-Sidak post hoc test. *P < 0.05; ** P < 0.01; *** P < 0.001. Acute SCI, n = 8; Chronic SCI, n = 7; SCI + Step-training, n = 6.
PBSt afferents simultaneously activate GABAergic pathways involved in PAD generation, but also contribute to postsynaptic inhibitory pathways leading to IPSPs in motoneurons (Hultborn et al., 1987a; Stuart & Redman, 1992; Pierce & Mendell, 1993; Hughes et al., 2005), both of which contribute to modulating the H-reflex amplitude. We therefore sought to isolate presynaptic inhibitory effects from postsynaptic inhibition. To do so, we included the longest C-T intervals (50-150ms) to isolate presynaptic inhibition, while postsynaptic inhibition was estimated at shorter intervals (10-20ms) (Eccles et al., 1961a; 1962c). There was a significant difference in postsynaptic (F2,18 = 15.567; P < 0.001, one-way ANOVA) and presynaptic inhibition (F2,18 = 16.597; P < 0.001, one-way ANOVA). Chronic SCI decreased postsynaptic (P < 0.001) and presynaptic inhibition (P < 0.001) as compared to acute SCI, while step-training was associated with greater inhibition (respectively P = 0.002 and P < 0.001, Holm-Sidak) as compared to untrained chronic SCIs (Fig.6D). These results suggest that chronic SCI reduces inhibition originating from group I PBSt afferents onto tibial Ia afferents projecting to motoneurons (presynaptic inhibition) and onto interosseous motoneurons (postsynaptic inhibition). Step-training counteracted these effects by promoting both the postsynaptic and presynaptic inhibition.
Chronic SCI disrupts the association between DRP and H-reflex inhibition
The inhibition of the monosynaptic reflex and Tib-DRP evoked by PBSt group I muscle afferents are believed to rely on similar mechanisms due to the similarity in the strength and duration of their inhibition (Eccles et al., 1962b-c; 1963b). We further performed a regression analysis to assess if the strength of inhibition evoked by PBSt group I afferents on Tib-DRPs is predictive of the strength of inhibition incurred on the plantar H-reflex (Fig. 7). The inhibition of the conditioned Tib-DRP at a given C-T interval significantly co-varied with the inhibition of the conditioned H-reflex after acute SCI (R2: 0.244, P < 0.001) and in SCI + Step-training animals (R2: 0.264, P < 0.001, linear regression), but not in chronic SCI animals (R2: 0.0150, P = 0.301). The lack of correlation in chronic SCI animals is further supported by the clustering of data in the top right corner with little inhibition of both the DRP and H-reflex amplitude (Fig. 7B) contrary to acute SCIs (Fig. 7A) and SCI + Step-training animals (Fig. 7C). Together, these results suggest that the inhibition evoked by PBSt group I afferents on Tib-DRP is predictive of the inhibition incurred on the plantar H-reflex under acute conditions, even in the absence of supraspinal control, but is lost after chronic SCI unless the animals are step-trained.
Figure 7. Chronic SCI disrupts the relationship between DRP amplitude and H-reflex inhibition unless the animal is step-trained.

The linear relationship between the amplitude of the H-reflex and DRP conditioned by a stimulation to PBSt after acute SCI (R2: 0.244, P < 0.001) is not present after chronic SCI (R2: 0.0150, P = 0.301), unless the animals were step-trained (R2: 0.264, P < 0.001). Regression Analysis.
DISCUSSION
In rodents, most studies investigating presynaptic inhibition derive from reduced preparations of adult spinal cords ex vivo (Lucas-Osma et al., 2018), neonates (Hochman et al., 2010; Garcia-Ramirez et al., 2014) in which the state of the spinal cord is different from adulthood or adult anaesthetized preparations in which the excitatory/inhibitory state of the spinal cord is altered (Wall & Lidierth, 1997). Here, we show a decrease in the activation of lumbar PAD pathways associated with a reduction in presynaptic inhibition of the H-reflex in vivo in the adult decerebrated rat after a chronic low thoracic transection that is not observed acutely after injury. More precisely, we have demonstrated that the activation of flexor muscle group I PAD pathways activated by PBSt group afferents is significantly disrupted after chronic SCI and contributes to decrease presynaptic inhibition of the plantar H-reflex in association with an increased recruitment of motor pools. We further show that step-training reestablishes presynaptic inhibition originating from PBSt group I afferents on the H-reflex through an increase in transmission in PAD pathways after chronic SCI.
Afferent control of presynaptic inhibition from PBSt group I afferents is selectively impaired after chronic SCI
Pathways mediating presynaptic inhibition provide a powerful mechanism by which the incoming sensory information flooding the spinal cord is regulated before it even reaches the first central synapse (reviewed in Rudomin & Schmidt, 1999; Rudomin, 2009). Presynaptic inhibition is mainly produced via primary afferent depolarization (PAD) of intraspinal terminals, which can be measured experimentally following the antidromic electrotonic spread to the dorsal roots and recorded as a dorsal root potential (DRP) (Barron & Matthews, 1938; Eccles et al., 1963b). Presynaptic inhibition is under the control of both supraspinal centers and primary afferents in order to gate afferent feedback and to adjust reflex excitability to meet task requirements (Rudomin & Schmidt, 1999). While the supraspinal control of PAD pathways has been well described, little is known about maladaptive plasticity triggered by chronic SCI when supraspinal control is disrupted, and how it impacts sensory-evoked presynaptic inhibitory mechanisms.
In our experiment, chronic SCI decreased the amplitude of DRPs evoked by tibial afferents to a much lesser extent than DRPs evoked by PBSt as compared to Acute SCI. Since tonic descending inhibition of PAD pathways is impaired in both groups (Carpenter et al., 1963; Quevedo et al., 1993), the modulation in DRP amplitude observed after chronic SCI likely reflects plasticity occurring in local spinal networks associated to the transmission in PBSt- and Tib-PAD pathways.
The distal tibial nerve is a mixed nerve, innervating multiple muscles of the foot and the plantar surface of the foot, and contains mostly cutaneous afferents. Although the specific contribution of cutaneous and low threshold muscle afferents to Tib-DRP amplitude cannot be determined with certainty, the location of our electrode on the distal part of the tibial nerve suggests a contribution of mostly cutaneous origin (Eccles et al., 1962b; Loeb, 1993; Frigon et al., 2012). PAD pathways activated by cutaneous afferents primarily generate PAD on other cutaneous and Ib-II afferents (Janig et al., 1968a-b; Harrison & Jankowska, 1989; Quevedo et al., 1995; Enriquez et al., 1996a-b) and receive tonic inhibition from supraspinal centers (Carpenter et al., 1963; Quevedo et al., 1993). Our results indicate that the DRPs evoked by tibial afferents are barely altered by chronic SCI and suggest that the increased gain of sensory afferents vs. supraspinal input on spinal networks after chronic SCI (Edgerton et al., 2001) does not significantly alter the activation of tibial PAD pathways. In contrast, DRPs evoked by PBSt afferents were significantly smaller, suggesting that PAD pathways activated by hip extensor and/or knee flexor afferents are critically disrupted after chronic SCI. PBSt is a purely muscle nerve known to be an efficient source of presynaptic inhibition (Frank & Fuortes, 1957; Eccles et al., 1962c; Côté & Gossard, 2003). While chronic SCI did not decrease the amplitude of PBSt-DRPmax, the input-output curve was significantly shifted toward higher stimulation intensities, i.e. higher stimulation intensities were required to evoke PBSt-DRPmax and to reach s50 (Fig. 2 and Table 1). In decerebrated acutely transected cats, the recruitment of group Ia PBSt afferents is initiated at 1T and reaches a maximum ~1.5T. Higher intensity of stimulation is required to activate group Ib afferents (~1.4T), reaching maximal activation ~2T, which overlaps with the lowest threshold for group II afferents activation (~1.6T) (Eccles & Lundberg, 1959; Carpenter et al., 1963). While we have not directly investigated the type of muscle afferents recruited, the stimulation intensity required to reach PBSt-DRPmax was consistent with group I strength acutely after SCI and with group II afferents following chronic SCI (Fig. 2G).
Because the alteration in the amplitude of PBSt-evoked DRP is mainly observed at group I strength, but not seen when group II afferents were engaged, we stimulated PBSt at submaximal group I strength (1.5T) to minimize the contribution of group II afferents. Hence, stimulation at maximal group I strength (2T) or higher was shown to not only maximally activate group I PAD pathways, but also engage low threshold group II PAD pathways, which brings DRP amplitude closer to DRPmax values (Eccles & Lundberg, 1959; Naftchi et al., 1979). Consistent with this, the additional recruitment of PBSt group II afferents barely increases the DRP amplitude, as group I afferents already evoked a large DRP that is close to maximal amplitude. In contrast, when DRPs evoked by group I afferents are small, the additional recruitment of group II afferents produces a large increase in DRP amplitude (Eccles et al., 1961a). This is likely due to the activation of separate pathways by PBSt group I and II afferents that respectively evoke PAD on Ia, Ib and cutaneous afferents or on group II afferents (Riddell et al., 1993). Accordingly, our results suggest that PAD evoked by PBSt group I afferents on Ia, Ib and cutaneous afferents is specifically decreased after chronic SCI and that engaging PBSt group II afferents does not reveal any further change. Together, our results indicate that presynaptic inhibition evoked by PBSt group I muscle afferents is specifically altered after chronic SCI, while presynaptic inhibition evoked by PBSt group II or tibial cutaneous afferents (historically termed flexor reflex afferents) is unlikely to contribute. Whether decreased transmission in group I PAD pathway is the consequence of a change in interneuronal excitability, primary afferents or other factors remains to be determined.
Both the PBSt (Nicolopoulos-Stournaras & Iles, 1983) and distal tibial nerve afferents innervating the plantar surface (Crockett et al., 1987) enters the spinal cord ~L5, it is therefore unlikely that the distance between afferents entry in the spinal cord and the injury site plays a role in the difference observed in PAD evoked by PBSt and tibial afferents after chronic SCI. However, large proprioceptive and cutaneous afferents projections areas in the spinal cord are anatomically different (Brown, 1981). Proprioceptive group Ia-Ib afferents both project to laminae V, VI and dorsal part of lamina VII, which contains last-order interneurons from group I PAD pathway (Rudomin and Schmidt, 1999; Jankowska, 1992). In contrast, proprioceptive group II afferents project to laminae IV-V and VI-VIII, with interneurons from laminae IV-V being activated by cutaneous afferents, but not group I afferents (Edgley & Jankowska, 1987). Interestingly, dorsal horn interneurons involved in evoking PAD on group II afferent are activated by cutaneous afferents but not group I afferents (Jankowska & Riddell, 1995). In our experiment, there was a decrease in transmission in PAD pathways activated by PBSt group I afferents that was not observed when group II afferents were engaged and when PAD pathways were activated by distal tibial nerve afferents. Together, this suggest that chronic SCI may induce plasticity in specific interneuronal populations (group I vs group II, Tib-In vs PBSt-In, etc), but this remains to be determined.
In addition, there is an increased proportion of primary afferents (VGlut1+) projecting to motoneurons (the majority of type Ia and some type II afferents) that do not receive presynaptic GABAergic boutons after chronic SCI (Khalki et al., 2018). This suggests the presence of an anatomical correlate that could contribute to the decrease of both presynaptic inhibition of the H-reflex and the PAD evoked by PBSt group I afferents after chronic SCI. Alternatively, proprioceptive afferents projecting to interneurons rather than motoneurons also receive presynaptic inhibition (Eccles et al., 1961b) and likely contribute to our observations. In contrast to proprioceptive afferents, cutaneous afferents significanlty sprout after chronic SCI, which expands the projection area of large cutaneous afferents (VGlut1+) in laminae III-IV but also in lamina II (Lee et al., 2019). While anatomical differences between the location and density of the terminations of proprioceptive and cutaneous afferents could contribute to the difference in PAD evoked by PBSt vs. distal tibial afferents after chronic SCI, it remains to be determined.
H-reflex hyperexcitability is associated with a concurrent increase in the recruitment of motor pools and impairment in presynaptic and postsynaptic inhibition after chronic SCI
The DRP represents the sum of PADs from afferents present in the recorded dorsal rootlet. However, unlike the PAD, DRPs do not yield information on the specific type of afferents receiving presynaptic inhibition. A convenient method of testing this is to study the effect of a conditioning stimulation known to evoke PAD on primary afferents in a specific pathway. The H-reflex, the electrical analogue of the stretch reflex, is commonly used to assess the excitability of the monosynaptic reflex. Because of its monosynaptic nature, the H-reflex prevents the possible involvement of a change in excitability of interneurons present in the reflex pathway under different conditions. The modulation of the H-reflex has been extensively investigated in the rat using repeated homonymous sensory activation (Thompson et al., 1992; Côté et al., 2014; Caron et al., 2016), which includes presynaptic, homosynaptic and postsynaptic inhibitory mechanisms (Hultborn et al., 1996; Zucker & Regehr, 2002).
Presynaptic inhibition of hindlimb muscle group Ia afferents is mainly under the influence of flexor muscle group I afferents (Eccles et al., 1961a; 1962a, c; 1963a-b). Last-order inhibitory interneurons involved in presynaptic inhibition of the monosynaptic reflex synapse onto terminals of Ia muscle spindle afferents (Hughes et al., 2005) and are mostly activated by flexor muscle group I afferents, inhibited by flexor reflex afferents, and controlled by descending tracts (Jankowska, 2001). Presynaptic inhibition exerted by PBSt group I afferents on the triceps surae monosynaptic reflex in the cat includes a short-lasting inhibition with an onset around 5ms and reaching maximal amplitude around 30ms, followed by long-lasting inhibition which persists for over 100ms (Eccles et al., 1962a). Intracellular motoneuron recordings, following the same stimulation, similarly showed that PBSt group I afferents induce 40ms IPSPs in triceps surae motoneurons (postsynaptic inhibition) and decreased monosynaptic EPSPs (presynaptic inhibition) (Frank & Fuortes, 1957; Hultborn et al., 1987a; Stuart & Redman, 1992). In this study, the heteronymous inhibition of the plantar H-reflex by PBSt eliminates the possibility for post-activation depression, so that long C-T intervals (50-150ms) reflect presynaptic inhibition exerted on tibial Ia afferents and short C-T intervals (10-20ms) mainly reflect postsynaptic inhibition exerted on motoneurons, with a minimal contribution of presynaptic inhibition onto tibial Ia afferents. Our data show that chronic SCI did not modify the latency, but noticeably decreased the strength of both postsynaptic and presynaptic inhibition. Chronic SCI noticeably decreased the strength of both postsynaptic and presynaptic inhibition of the H-reflex. However, changes in motoneuronal properties can also influence the strength of H-reflex, which would not be reflected by measures of either pre- or postsynaptic inhibition (Kernell & Hultborn, 1990). An increase in the slope of the H-reflex recruitment curve is associated with hyperreflexia and routinely used as a clinical evaluation of spasticity (Biering-Sorensen et al., 2006). Accordingly, our data show that chronic SCI increased the slope of the plantar H-reflex recruitment curve, suggesting an increase in the recruitment gain of motor pools by Ia afferents (Capaday, 1997). Under normal conditions, Henneman’s size principle suggest that Ia afferents first activate slow motor units, such that motor units producing the smallest force are recruited the earliest (Henneman, 1957; Burke & Rymer, 1976; Harrison & Taylor, 1981). However, increasing the recruitment gain of the motor pool induces a fast and synchronous activation of the entire motoneuronal pool (Nielsen et al., 2019), which contributes to further increase the excitability of the H-reflex (Kernell & Hultborn, 1990). This effect can be the consequence of several factors, including, 1) homogenization of motor unit properties, 2) increased inhibition biased towards slow motor units, 3) increased excitation biased towards large faster motor units, 4) a combination of aforementioned factors (Kernell & Hultborn, 1990).
After SCI and other conditions inducing immobilization, the intrinsic properties of slow motor units change towards faster motor unit properties (Burnham et al., 1997; Talmadge, 2000; Beaumont et al., 2004; Cormery et al., 2005), which would likely contribute to increase the recruitment gain of the motor pool and influence the strength of H-reflex inhibition (Kernell & Hultborn, 1990). To account for this possibility, we have simultaneously recorded the inhibition exerted by PBSt group I afferent onto the plantar H-reflex, which includes the motoneuronal response, and onto the Tib-DRP, which does not involve the motoneuronal response. We show that the inhibition exerted by PBSt group I afferents on both the H-reflex and Tib-DRP is decreased after chronic SCI, confirming that chronic SCI also decreases inhibition independently of motoneuron activation. In addition, the correlation between the strength of inhibition exerted by PBSt on Tib-DRP and on the H-reflex observed after acute injury, was not present in chronic SCI animals. This lack of correlation further supports a disruption in presynaptic inhibition after SCI.
Together, our results suggest that flexor muscle group I afferents PAD pathways are specifically undergoing plastic changes after chronic SCI and contribute to the loss of presynaptic inhibition associated with hyperreflexia. While we can confirm with certainty that presynaptic inhibition initiated by PBSt on the plantar H-reflex is disrupted by chronic SCI, whether the decreased transmission in PBSt PAD pathway is the consequence of a change in interneuronal excitability, failure to activate said interneurons by sensory afferents and/or a decrease in PAD activation remains to be determined.
Step-training increases transmission in PBSt group I PAD pathways after chronic SCI.
Our results indicate that step-training did not affect input-output curve of L4-DRPs evoked by the tibial nerve suggesting that this pathway is not undergoing activity-dependent plasticity after SCI. In contrast, we found that step-training restored the amplitude of L4-DRPs evoked by PBSt group I muscle afferents, which unlike untrained chronic SCI animals was not associated with a shift toward a requirement for higher intensity of stimulation to evoke a DRPmax. Tib-DRPs were barely affected by chronic SCI, while PBSt-DRPs were significantly decreased, suggesting that step-training may specifically target pathways that are disrupted by chronic SCI. Together, these results suggest that the SCI-induced decrease in transmission in PAD pathways activated by PBSt group I afferents is altered in an activity-dependent manner, whereas PAD pathways activated by PBSt group II or tibial cutaneous afferents (historically termed flexor reflex afferents) are not. However, cutaneous afferents can evoke different PAD patterns in similarly identified afferents, including a decrease in PAD evoked by PBSt group I afferents (Quevedo et al., 1993; 1995), the occurrence of which can be shifted after a neuronal insult such as a crush injury (Enriquez et al., 1996a). Whether chronic SCI or step-training altered the PAD pattern evoked by tibial cutaneous afferents remains to be determined as it would not have been reflected by Tib-DRPs input-output properties.
Presynaptic and postsynaptic inhibition of the H-reflex are regulated in an activity-dependent manner.
Our data show that step-training did not modify the latency, but noticeably increased the strength of presynaptic inhibition. After chronic SCI and other conditions inducing long-lasting immobilization, motor training decreases the hyperexcitability of the monosynaptic reflex and improves reflex modulation (Côté et al., 2003; Côté & Gossard, 2004; Martin Ginis & Latimer, 2007; Lundbye-Jensen & Nielsen, 2008b; Knikou & Mummidisetty, 2014; Caron et al., 2016). Accordingly, our results show that the slope of the plantar H-reflex recruitment curve is increased after chronic SCI, suggesting an increase in the recruitment grain of motor pools by tibial Ia afferents (Capaday, 1997). This shift was not observed in step-trained animals, in agreement with previous studies suggesting that step-training decreases the slope of the H-reflex recruitment curve in SCI patients (Smith et al., 2015) and more generally that the slope of the H-reflex recruitment curve is altered in an activity-dependent manner. (Lundbye-Jensen & Nielsen, 2008a). A decreased recruitment of motor pools suggests a less homogenous activation of motor units.
SCI and immobilization induce changes in the intrinsic properties of slow motor units towards faster motor unit properties (Burnham et al., 1997; Talmadge, 2000; Beaumont et al., 2004; Cormery et al., 2005). Motor activity can counteract these effects by shifting back fast motor unit properties towards slower properties (Yarar-Fisher et al., 2018). Although not directly measured in this study, our results suggest a possible contribution of an activity-dependent alteration in motor unit properties to the normalization of the motor pool recruitment and to the excitability of the H-reflex. In addition to a decrease in the recruitment gain of the plantar H-reflex with step-training, our results show that both short- (10-20ms C-T interval) and long-lasting inhibition (50-150ms C-T intervals) induced by PBSt group I afferents are increased with step-training, suggesting an increase in presynaptic inhibition of the plantar H-reflex, and a likely augmentation of postsynaptic inhibition. An increase in presynaptic inhibition of the H-reflex with locomotor training has been suggested in extensor muscles both in animals (Côté et al., 2003) and humans (Knikou & Mummidisetty, 2014). The increase in DRP amplitude evoked by PBSt group I afferents in step-trained animals suggests that the increase in presynaptic inhibition of the plantar H-reflex relies on a larger PAD evoked by PBSt on tibial Ia afferents. While our results suggest an increase in postsynaptic inhibition, most likely through increased IPSPs in motoneurons also evoked by PBSt group I afferents (Hultborn et al., 1987a; Stuart & Redman, 1992), it remains to be demonstrated. Together, our results suggest that step-training decreases hyperreflexia through a simultaneous normalization in the recruitment of motor pools and a an augmentation of both presynaptic and postsynaptic inhibition induced by PBSt group I afferents. Whether step-training prevented the decrease in pre/post synaptic inhibition induced by chronic SCI or increased inhibition after it was disrupted remains to be determined.
Possible mechanisms contributing to the reorganization of afferent control of spinal networks after SCI and step-training
In this study, we stimulated PBSt, which is an efficient sensory source of presynaptic inhibition frequently used to inhibit the monosynaptic reflex (Frank & Fuortes, 1957; Eccles et al., 1962c; Côté & Gossard, 2003). Presynaptic inhibition is largely mediated by the shunting action of GABAA receptors at the afferent terminal, which decreases the size of the action potential that invades the terminal and reduces the activation of calcium current and associated transmitter release (Curtis et al., 1971; Stuart & Redman, 1992; Curtis, 1998; Cattaert & El Manira, 1999). This inhibition is described as the phasic PAD, in opposition to the tonic PAD, which regulates afferents transmission through extrasynaptic GABAA receptors (Lucas-Osma et al., 2018). While a change in tonic PAD has the potential to be of particular importance for the propagation of action potentials at branching points and for steady state inhibition (Decandia et al., 1968; Hultborn et al., 1987a), our recording configuration for DRPs (6mm interelectrode distance and 0.1Hz high pass) prevented its estimation in this experiment. Overall, whether the decreased transmission in PBSt PAD pathway is the consequence of a change in interneuronal excitability, failure to activate said interneurons by sensory afferents and/or a decrease in phasic vs. tonic PAD activation on primary afferents remains to be determined.
More recently, last order GABAergic interneurons specifically involved in presynaptic inhibition have been genetically identified in rodent models (Fink et al., 2014; Koch et al., 2017) and presynaptic inhibitory mechanisms were shown to rely not only on GABAergic signaling, but also on other neurotransmitters, neuromodulators and receptors that also could contribute to our results (Kremer & Lev-Tov, 1998; Hochman et al., 2010; Lucas-Osma et al., 2018). Finally, mechanisms other than PAD are also likely to contribute to presynaptic inhibition of Ia afferents and possibly contribute to the PAD-independent presynaptic inhibition of the H-reflex in our experiments (Price et al., 1984; Stuart & Redman, 1992; Hultborn et al., 1996; Curtis et al., 1997; Garcia-Ramirez et al., 2014).
Supplementary Material
{kind=link}
Key point summary.
Presynaptic inhibition is modulated by supraspinal centers and primary afferents in order to filter sensory information, adjust spinal reflex excitability, and ensure smooth movements.
After SCI, the supraspinal control of primary afferent depolarization (PAD) interneurons is disengaged, suggesting an increased role for sensory afferents. While increased H-reflex excitability in spastic individuals indicates a possible decrease in presynaptic inhibition, it remains unclear whether a decrease in sensory-evoked PAD contributes to this effect.
We investigated whether the PAD evoked by hindlimb afferents contributes to the change in presynaptic inhibition of the H-reflex in a decerebrated rat preparation. We found that chronic SCI decreases presynaptic inhibition of the plantar H-reflex through a reduction in PAD evoked by PBSt muscle group I afferents.
We further found that step-training restored presynaptic inhibition of the plantar H-reflex evoked by PBSt, suggesting the presence of activity-dependent plasticity of PAD pathways activated by flexor muscle group I afferents.
Clinical significance.
Pathological changes in presynaptic inhibition are crucial to several diseases including peripheral inflammation, abnormal pain processing, hyperreflexia and spasticity (Wall & Devor, 1981; Calancie et al., 1993; Enriquez-Denton et al., 2004; Witschi et al., 2011). Sensory feedback plays a critical role in determining and refining spatiotemporal features of motor output (Rossignol et al., 2006). The spinal cord is continuously flooded by sensory input via primary afferents, and the integrity of presynaptic inhibitory mechanisms to effectively select the relevant information and filter sensory noise is critical.
The CNS is believed to adjust the level of presynaptic inhibition of extensor group Ia afferents to modulate the central gain of the stretch reflex (Capaday & Stein, 1986; Hultborn et al., 1987b), and also the locomotor rhythm in animals and humans (Simonsen & Dyhre-Poulsen, 1999; Morita et al., 2001; Knikou, 2006). For example, it was shown that maintaining the Achilles’ tendon stretched maintains extensor muscles activity, which during locomotion prevents the switch from stance to swing phase (Duysens & Pearson, 1980). Thus, presynaptic inhibition evoked by PBSt group I afferents could play an important role in the initiation of the swing phase when the Achilles’ tendon is loaded during stance. In contrast, plantar cutaneous afferents inhibit presynaptic inhibition evoked by PBSt group I afferents on Ia afferents (Quevedo et al., 1993; 1995), which during locomotion could contribute to increase extensor muscles activity and decrease the inhibition of the extensor H-reflex with ground contact (Duysens & Pearson, 1976; Duysens, 1977; Guertin et al., 1995; Iles, 1996). Thus, the decrease in presynaptic inhibition observed after chronic SCI impairs this ability to tailor motor output to environmental constraints and may contribute to the presence of co-contraction during walking preventing the transition from stance to swing and to the development of tremors or clonus, which are frequently observed in spastic patients (Nielsen et al., 2007).
The recovery of presynaptic inhibition with step-training illustrates the importance of activity-based therapies after chronic SCI. Understanding the physiological effects of rehabilitation programs is critical to the design of optimized therapies in the clinic. For example, step-training changes the pattern of postsynaptic potentials evoked by cutaneous afferents in specific motor pools. It more specifically leads to an increase in the excitation of ankle extensors by cutaneous afferents from the plantar surface of the foot, presumably to improve weight-bearing during ground contact (Côté & Gossard, 2004; Knikou, 2010). The concomitant increase in presynaptic inhibition evoked by PBSt group I afferents would not only counteract the aforementioned debilitative effects of decreased presynaptic inhibition after SCI, but would also allow for the modulation of sensory inputs resulting from plastic changes after SCI. As presynaptic inhibition is regulated in a task-dependent manner (Côté & Gossard, 2003), more studies are needed to investigate if the restoration of presynaptic inhibition is specific to certain movements and how it responds to a comprehensive rehabilitation program that includes an array of motor training and pharmacological tools to manage spastic symptoms.
Acknowledgements:
We thank Dr. Jean-Pierre Gossard for helpful comments on an earlier version of this manuscript and Kyle Yeakle for assistance in data collection.
Funding: This work was supported by grants from the National Institute of Neurological Disorders and Stroke (RO1 NS083666) and the Craig H. Neilsen Foundation (189758).
Biography
Guillaume Caron received his PhD in exercise physiology from Aix-Marseille University, France and is currently a postdoctoral fellow at Drexel University, PA, USA. His research interest is to study mechanisms involved in the plasticity of the sensorimotor system and to implement therapies for individuals with sensory and/or motor dysfunction. His current work aims to use in vivo electrophysiology and pharmacological strategies in adult mammals to delineate mechanisms underlying the regulation of spinal reflexes excitability involved in the development of spasticity after SCI.
Footnotes
Competing interests
The authors declare no competing financial interests.
Data availability statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alluin O, Delivet-Mongrain H & Rossignol S. (2015). Inducing hindlimb locomotor recovery in adult rat after complete thoracic spinal cord section using repeated treadmill training with perineal stimulation only. J Neurophysiol 114, 1931–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron DH & Matthews BH. (1938). The interpretation of potential changes in the spinal cord. J Physiol 92, 276–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont E, Houle JD, Peterson CA & Gardiner PF. (2004). Passive exercise and fetal spinal cord transplant both help to restore motoneuronal properties after spinal cord transection in rats. Muscle Nerve 29, 234–242. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Gorassini M, Fouad K, Sanelli L, Han Y & Cheng J. (1999). Spasticity in rats with sacral spinal cord injury. JNeurotrauma 16, 69–84. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Li Y & Siu M. (2001). Plateau potentials in sacrocaudal motoneurons of chronic spinal rats, recorded in vitro. Journal of Neurophysiology 86, 1955–1971. [DOI] [PubMed] [Google Scholar]
- Bergmans J, Burke R, Fedina L & Lundberg A. (1974). The effect of dopa on the spinal cord. 8. Presynaptic and "remote" inhibition of transmission from Ia afferents to alpha motoneurones. Acta Physiol Scand 90, 618–639. [DOI] [PubMed] [Google Scholar]
- Biering-Sorensen F, Nielsen JB & Klinge K. (2006). Spasticity-assessment: a review. Spinal Cord 44, 708–722. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M & Vinay L. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. NatMed 16, 302–307. [DOI] [PubMed] [Google Scholar]
- Bouyer LJ & Rossignol S. (2003). Contribution of cutaneous inputs from the hindpaw to the control of locomotion. II. Spinal cats. Journal of Neurophysiology 90, 3640–3653. [DOI] [PubMed] [Google Scholar]
- Brocard C, Plantier V, Boulenguez P, Liabeuf S, Bouhadfane M, Viallat-Lieutaud A, Vinay L & Brocard F. (2016). Cleavage of Na(+) channels by calpain increases persistent Na(+) current and promotes spasticity after spinal cord injury. Nat Med 22, 404–411. [DOI] [PubMed] [Google Scholar]
- Burke RE & Rymer WZ. (1976). Relative strength of synaptic input from short-latency pathways to motor units of defined type in cat medial gastrocnemius. J Neurophysiol 39, 447–458. [DOI] [PubMed] [Google Scholar]
- Burnham R, Martin T, Stein R, Bell G, MacLean I & Steadward R. (1997). Skeletal muscle fibre type transformation following spinal cord injury. Spinal Cord 35, 86–91. [DOI] [PubMed] [Google Scholar]
- Calancie B, Broton JG, Klose KJ, Traad M, Difini J & Ayyar DR. (1993). Evidence that alterations in presynaptic inhibition contribute to segmental hypo- and hyperexcitability after spinal cord injury in man. ElectroencephalogrClinNeurophysiol 89, 177–186. [DOI] [PubMed] [Google Scholar]
- Capaday C (1997). Neurophysiological methods for studies of the motor system in freely moving human subjects. J Neurosci Methods 74, 201–218. [DOI] [PubMed] [Google Scholar]
- Capaday C & Stein RB. (1986). Amplitude modulation of the soleus H-reflex in the human during walking and standing. Journal of Neuroscience 6, 1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron G, Marqueste T & Decherchi P. (2016). Restoration of post-activation depression of the H-reflex by treadmill exercise in aged rats. Neurobiol Aging 42, 61–68. [DOI] [PubMed] [Google Scholar]
- Carpenter D, Engberg I, Funkenstein H & Lundberg A. (1963). Decerebrate Control of Reflexes to Primary Afferents. Acta Physiol Scand 59, 424–437. [DOI] [PubMed] [Google Scholar]
- Carroll TJ, Riek S & Carson RG. (2001). Reliability of the input-output properties of the corticospinal pathway obtained from transcranial magnetic and electrical stimulation. J Neurosci Methods 112, 193–202. [DOI] [PubMed] [Google Scholar]
- Cattaert D & El Manira A. (1999). Shunting versus inactivation: analysis of presynaptic inhibitory mechanisms in primary afferents of the crayfish. J Neurosci 19, 6079–6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherniak M, Etlin A, Strauss I, Anglister L & Lev-Tov A. (2014). The sacral networks and neural pathways used to elicit lumbar motor rhythm in the rodent spinal cord. Front Neural Circuits 8, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormery B, Beaumont E, Csukly K & Gardiner P. (2005). Hindlimb unweighting for 2 weeks alters physiological properties of rat hindlimb motoneurones. J Physiol 568, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M-P, Azzam GA, Lemay MA, Zhukareva V & Houle JD. (2011). Activity-dependent increase in neurotrophic factors is associated with an enhanced modulation of spinal reflexes after spinal cord injury. J Neurotrauma 28, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M-P, Gandhi S, Zambrotta M & Houle JD. (2014). Exercise modulates chloride homeostasis after spinal cord injury. J Neurosci 34, 8976–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M-P & Gossard JP. (2003). Task-dependent presynaptic inhibition. J Neurosci 23, 1886–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M-P & Gossard JP. (2004). Step training-dependent plasticity in spinal cutaneous pathways. J Neurosci 24, 11317–11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M-P, Menard A & Gossard JP. (2003). Spinal cats on the treadmill: changes in load pathways. J Neurosci 23, 2789–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockett DP, Harris SL & Egger MD. (1987). Plantar motoneuron columns in the rat. J Comp Neurol 265, 109–118. [DOI] [PubMed] [Google Scholar]
- Crone C, Hultborn H, Mazieres L, Morin C, Nielsen J & Pierrot-Deseilligny E. (1990). Sensitivity of monosynaptic test reflexes to facilitation and inhibition as a function of the test reflex size: a study in man and the cat. Exp Brain Res 81, 35–45. [DOI] [PubMed] [Google Scholar]
- Curtis DR. (1998). Two types of inhibition in the spinal cord In Preynaptic inhibition and neuron control, ed. Rudomin P, Romo R & Mendell LM, pp. 150–161. Oxford University Press, New York. [Google Scholar]
- Curtis DR, Duggan AW, Felix D & Johnston GA. (1971). Bicuculline, an antagonist of GABA and synaptic inhibition in the spinal cord of the cat. Brain Res 32, 69–96. [DOI] [PubMed] [Google Scholar]
- Curtis DR, Gynther BD, Lacey G & Beattie DT. (1997). Baclofen: reduction of presynaptic calcium influx in the cat spinal cord in vivo. Exp Brain Res 113, 520–533. [DOI] [PubMed] [Google Scholar]
- Decandia M, Gasteiger EL & Mann MD. (1968). Escape of the extensor monosynaptic reflex from presynaptic inhibition. Brain Res 7, 317–319. [DOI] [PubMed] [Google Scholar]
- Duysens J. (1977). Reflex control of locomotion as revealed by stimulation of cutaneous afferents in spontaneously walking premammillary cats. Journal of Neurophysiology 40, 737–751. [DOI] [PubMed] [Google Scholar]
- Duysens J & Pearson KG. (1976). The role of cutaneous afferents from the distal hindlimb in the regulation of the step cycle of thalamic cats. Exp Brain Res 24, 245–255. [DOI] [PubMed] [Google Scholar]
- Duysens J & Pearson KG. (1980). Inhibition of flexor burst generation by loading ankle extensor muscles in walking cats. Brain Research 187, 321–332. [DOI] [PubMed] [Google Scholar]
- Eccles JC. (1964). Presynaptic Inhibition in the Spinal Cord. Prog Brain Res 12, 65–91. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Eccles RM & Magni F. (1961a). Central inhibitory action attributable to presynaptic depolarization produced by muscle afferent volleys. J Physiol 159, 147–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Oscarsson O & Willis WD. (1961b). Synaptic action of group I and II afferent fibres of muscle on the cells of the dorsal spinocerebellar tract. J Physiol 158, 517–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Kostyuk PG & Schmidt RF. (1962a). Central pathways responsible for depolarization of primary afferent fibres. J Physiol 161, 237–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Kostyuk PG & Schmidt RF. (1962b). Presynaptic inhibition of the central actions of flexor reflex afferents. J Physiol 161, 258–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Schmidt R & Willis WD. (1963a). Pharmacological Studies on Presynaptic Inhibition. J Physiol 168, 500–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Schmidt RF & Willis WD. (1962c). Presynaptic inhibition of the spinal monosynaptic reflex pathway. J Physiol 161, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Schmidt RF & Willis WD. (1963b). The location and the mode of action of the presynaptic inhibitory pathways on to group I afferent fibers from muscle. J Neurophysiol 26, 506–522. [Google Scholar]
- Eccles RM & Lundberg A. (1959). Supraspinal control of interneurones mediating spinal reflexes. J Physiol 147, 565–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton VR, Leon RD, Harkema SJ, Hodgson JA, London N, Reinkensmeyer DJ, Roy RR, Talmadge RJ, Tillakaratne NJ, Timoszyk W & Tobin A. (2001). Retraining the injured spinal cord. JPhysiol 533, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgley SA & Jankowska E. (1987). An interneuronal relay for group I and II muscle afferents in the midlumbar segments of the cat spinal cord. J Physiol 389, 647–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez M, Jimenez I & Rudomin P. (1996a). Changes in PAD patterns of group I muscle afferents after a peripheral nerve crush. Exp Brain Res 107, 405–420. [DOI] [PubMed] [Google Scholar]
- Enriquez M, Jimenez I & Rudomin P. (1996b). Segmental and supraspinal control of synaptic effectiveness of functionally identified muscle afferents in the cat. Exp Brain Res 107, 391–404. [DOI] [PubMed] [Google Scholar]
- Enriquez-Denton M, Manjarrez E & Rudomin P. (2004). Persistence of PAD and presynaptic inhibition of muscle spindle afferents after peripheral nerve crush. Brain Res 1027, 179–187. [DOI] [PubMed] [Google Scholar]
- Etlin A, Blivis D, Ben-Zwi M & Lev-Tov A. (2010). Long and short multifunicular projections of sacral neurons are activated by sensory input to produce locomotor activity in the absence of supraspinal control. Journal of Neuroscience 30, 10324–10336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faist M, Mazevet D, Dietz V & Pierrot-Deseilligny E. (1994). A quantitative assessment of presynaptic inhibition of Ia afferents in spastics. Differences in hemiplegics and paraplegics. Brain 117 ( Pt 6), 1449–1455. [DOI] [PubMed] [Google Scholar]
- Fink AJ, Croce KR, Huang ZJ, Abbott LF, Jessell TM & Azim E. (2014). Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature 509, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank K & Fuortes MGF. (1957). Presynaptic and postsynaptic inhibition of monosynaptic reflex. Fed Proc 16, 39–40. [Google Scholar]
- Frigon A, Hurteau MF, Johnson MD, Heckman CJ, Telonio A & Thibaudier Y. (2012). Synchronous and asynchronous electrically evoked motor activities during wind-up stimulation are differentially modulated following an acute spinal transection. J Neurophysiol 108, 3322–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ramirez DL, Calvo JR, Hochman S & Quevedo JN. (2014). Serotonin, dopamine and noradrenaline adjust actions of myelinated afferents via modulation of presynaptic inhibition in the mouse spinal cord. PLoS One 9, e89999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey MJ, Klinge K, Crone C, Lorentzen J, Biering-Sorensen F, Ravnborg M & Nielsen JB. (2008). Post-activation depression of soleus stretch reflexes in healthy and spastic humans. Experimental Brain Research 185, 189–197. [DOI] [PubMed] [Google Scholar]
- Grillner S & Rossignol S. (1978). On the initiation of the swing phase of locomotion in chronic spinal cats. Brain Research 146, 269–277. [DOI] [PubMed] [Google Scholar]
- Guertin P, Angel MJ, Perreault MC & McCrea DA. (1995). Ankle extensor group I afferents excite extensors throughout the hindlimb during fictive locomotion in the cat. JPhysiol 487 ( Pt 1), 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock J, Knowles L & Gillies JD. (1973). Segmental reflex changes in acute and chronic spinal cats. Proc Aust Assoc Neurol 10, 139–143. [PubMed] [Google Scholar]
- Harrison PJ & Jankowska E. (1989). Primary afferent depolarization of central terminals of group II muscle afferents in the cat spinal cord. J Physiol 411, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ & Taylor A. (1981). Individual excitatory post-synaptic potentials due to muscle spindle Ia afferents in cat triceps surae motoneurones. J Physiol 312, 455–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman E (1957). Relation between size of neurons and their susceptibility to discharge. Science 126, 1345–1347. [DOI] [PubMed] [Google Scholar]
- Hochman S, Shreckengost J, Kimura H & Quevedo J. (2010). Presynaptic inhibition of primary afferents by depolarization: observations supporting nontraditional mechanisms. Ann N Y Acad Sci 1198, 140–152. [DOI] [PubMed] [Google Scholar]
- Holtz KA, Lipson R, Noonan VK, Kwon BK & Mills PB. (2017). Prevalence and Effect of Problematic Spasticity After Traumatic Spinal Cord Injury. Arch Phys Med Rehabil 98, 1132–1138. [DOI] [PubMed] [Google Scholar]
- Hughes DI, Mackie M, Nagy GG, Riddell JS, Maxwell DJ, Szabo G, Erdelyi F, Veress G, Szucs P, Antal M & Todd AJ. (2005). P boutons in lamina IX of the rodent spinal cord express high levels of glutamic acid decarboxylase-65 and originate from cells in deep medial dorsal horn. ProcNatlAcadSciUSA 102, 9038–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultborn H, Illert M, Nielsen J, Paul A, Ballegaard M & Wiese H. (1996). On the mechanism of the post-activation depression of the H-reflex in human subjects. Experimental Brain Research 108, 450–462. [DOI] [PubMed] [Google Scholar]
- Hultborn H & Malmsten J. (1983). Changes in segmental reflexes following chronic spinal cord hemisection in the cat. I. Increased monosynaptic and polysynaptic ventral root discharges. Acta Physiol Scand 119, 405–422. [DOI] [PubMed] [Google Scholar]
- Hultborn H, Meunier S, Morin C & Pierrot-Deseilligny E. (1987a). Assessing changes in presynaptic inhibition of I a fibres: a study in man and the cat. J Physiol 389, 729–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultborn H, Meunier S, Pierrot-Deseilligny E & Shindo M. (1987b). Changes in presynaptic inhibition of Ia fibres at the onset of voluntary contraction in man. J Physiol 389, 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iles JF. (1996). Evidence for cutaneous and corticospinal modulation of presynaptic inhibition of Ia afferents from the human lower limb. J Physiol 491 ( Pt 1), 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janig W, Schmidt RF & Zimmermann M. (1968a). Single unit responses and the total afferent outflow from the cat's foot pad upon mechanical stimulation. Exp Brain Res 6, 100–115. [DOI] [PubMed] [Google Scholar]
- Janig W, Schmidt RF & Zimmermann M. (1968b). Two specific feedback pathways to the central afferent terminals of phasic and tonic mechanoreceptors. Exp Brain Res 6, 116–129. [DOI] [PubMed] [Google Scholar]
- Jankowska E & Riddell JS. (1995). Interneurones mediating presynaptic inhibition of group II muscle afferents in the cat spinal cord. J Physiol 483 ( Pt 2), 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska E (2001). Spinal interneuronal systems: identification, multifunctional character and reconfigurations in mammals. J Physiol 533, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska E (1992). Interneuronal relay in spinal pathways from proprioceptors. ProgNeurobiol 38, 335–378. [DOI] [PubMed] [Google Scholar]
- Kernell D & Hultborn H. (1990). Synaptic effects on recruitment gain: a mechanism of importance for the input-output relations of motoneurone pools? Brain Research 507, 176–179. [DOI] [PubMed] [Google Scholar]
- Khalki L, Sadlaoud K, Lerond J, Coq JO, Brezun JM, Vinay L, Coulon P & Bras H. (2018). Changes in innervation of lumbar motoneurons and organization of premotor network following training of transected adult rats. Exp Neurol 299, 1–14. [DOI] [PubMed] [Google Scholar]
- Klimstra M & Zehr EP. (2008). A sigmoid function is the best fit for the ascending limb of the Hoffmann reflex recruitment curve. Exp Brain Res 186, 93–105. [DOI] [PubMed] [Google Scholar]
- Knikou M (2006). Plantar cutaneous input modulates differently spinal reflexes in subjects with intact and injured spinal cord. Spinal Cord. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knikou M (2010). Plantar cutaneous afferents normalize the reflex modulation patterns during stepping in chronic human spinal cord injury. J Neurophysiol 103, 1304–1314. [DOI] [PubMed] [Google Scholar]
- Knikou M & Mummidisetty CK. (2014). Locomotor training improves premotoneuronal control after chronic spinal cord injury. J Neurophysiol 111, 2264–2275. [DOI] [PubMed] [Google Scholar]
- Koch SC, Del Barrio MG, Dalet A, Gatto G, Gunther T, Zhang J, Seidler B, Saur D, Schule R & Goulding M. (2017). RORbeta Spinal Interneurons Gate Sensory Transmission during Locomotion to Secure a Fluid Walking Gait. Neuron 96, 1419–1431 e1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer E & Lev-Tov A. (1998). GABA-receptor-independent dorsal root afferents depolarization in the neonatal rat spinal cord. Journal of Neurophysiology 79, 2581–2592. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Malone PS, Chung J, White JM, Wilson N, Tidwell J & Tansey KE. (2019). Central Plasticity of Cutaneous Afferents Is Associated with Nociceptive Hyperreflexia after Spinal Cord Injury in Rats. Neural Plast 2019, 6147878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb GE. (1993). The distal hindlimb musculature of the cat: interanimal variability of locomotor activity and cutaneous reflexes. Exp Brain Res 96, 125–140. [DOI] [PubMed] [Google Scholar]
- Lomeli J, Quevedo J, Linares P & Rudomin P. (1998). Local control of information flow in segmental and ascending collaterals of single afferents. Nature 395, 600–604. [DOI] [PubMed] [Google Scholar]
- Lucas-Osma AM, Li Y, Lin S, Black S, Singla R, Fouad K, Fenrich KK & Bennett DJ. (2018). Extrasynaptic alpha5GABAA receptors on proprioceptive afferents produce a tonic depolarization that modulates sodium channel function in the rat spinal cord. J Neurophysiol 120, 2953–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbye-Jensen J & Nielsen JB. (2008a). Central nervous adaptations following 1 wk of wrist and hand immobilization. J Appl Physiol (1985) 105, 139–151. [DOI] [PubMed] [Google Scholar]
- Lundbye-Jensen J & Nielsen JB. (2008b). Immobilization induces changes in presynaptic control of group Ia afferents in healthy humans. J Physiol 586, 4121–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmsten J. (1983). Time course of segmental reflex changes after chronic spinal cord hemisection in the rat. Acta Physiol Scand 119, 435–443. [DOI] [PubMed] [Google Scholar]
- Marchenko V, Granata AR & Cohen MI. (2002). Respiratory cycle timing and fast inspiratory discharge rhythms in the adult decerebrate rat. Am J Physiol Regul Integr Comp Physiol 283, R931–940. [DOI] [PubMed] [Google Scholar]
- Martin Ginis KA & Latimer AE. (2007). The effects of single bouts of body-weight supported treadmill training on the feeling states of people with spinal cord injury. Spinal Cord 45, 112–115. [DOI] [PubMed] [Google Scholar]
- Maynard FM, Karunas RS & Waring WP 3rd. (1990). Epidemiology of spasticity following traumatic spinal cord injury. Archives of physical medicine and rehabilitation 71, 566–569. [PubMed] [Google Scholar]
- Morita H, Crone C, Christenhuis D, Petersen NT & Nielsen JB. (2001). Modulation of presynaptic inhibition and disynaptic reciprocal Ia inhibition during voluntary movement in spasticity. Brain 124, 826–837. [DOI] [PubMed] [Google Scholar]
- Naftchi NE, Schlosser W & Horst WD. (1979). Correlation of changes in the GABA-ergic system with the development of spasticity in paraplegic cats. Adv Exp Med Biol 123, 431–450. [DOI] [PubMed] [Google Scholar]
- Nicolopoulos-Stournaras S & Iles JF. (1983). Motor neuron columns in the lumbar spinal cord of the rat. J Comp Neurol 217, 75–85. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Petersen N & Crone C. (1995). Changes in transmission across synapses of Ia afferents in spastic patients. Brain 118 ( Pt 4), 995–1004. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Crone C & Hultborn H. (2007). The spinal pathophysiology of spasticity--from a basic science point of view. Acta Physiol (Oxf) 189, 171–180. [DOI] [PubMed] [Google Scholar]
- Nielsen JB, Morita H, Wenzelburger R, Deuschl G, Gossard JP & Hultborn H. (2019). Recruitment gain of spinal motor neuron pools in cat and human. Exp Brain Res 237, 2897–2909. [DOI] [PubMed] [Google Scholar]
- Pierce JP & Mendell LM. (1993). Quantitative ultrastructure of Ia boutons in the ventral horn: scaling and positional relationships. J Neurosci 13, 4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price GW, Wilkin GP, Turnbull MJ & Bowery NG. (1984). Are baclofen-sensitive GABAB receptors present on primary afferent terminals of the spinal cord? Nature 307, 71–74. [DOI] [PubMed] [Google Scholar]
- Quevedo J, Eguibar JR, Jimenez I & Rudomin P. (1995). Raphe magnus and reticulospinal actions on primary afferent depolarization of group I muscle afferents in the cat. J Physiol 482 ( Pt 3), 623–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo J, Eguibar JR, Jimenez I, Schmidt RF & Rudomin P. (1993). Primary afferent depolarization of muscle afferents elicited by stimulation of joint afferents in cats with intact neuraxis and during reversible spinalization. J Neurophysiol 70, 1899–1910. [DOI] [PubMed] [Google Scholar]
- Riddell JS, Jankowska E & Eide E. (1993). Depolarization of group II muscle afferents by stimuli applied in the locus coeruleus and raphe nuclei of the cat. J Physiol 461, 723–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol S, Dubuc R & Gossard JP. (2006). Dynamic sensorimotor interactions in locomotion. Physiol Rev 86, 89–154. [DOI] [PubMed] [Google Scholar]
- Rudomin P. (1990). Presynaptic inhibition of muscle spindle and tendon organ afferents in the mammalian spinal cord. Trends Neurosci 13, 499–505. [DOI] [PubMed] [Google Scholar]
- Rudomin P. (2009). In search of lost presynaptic inhibition. Exp Brain Res 196, 139–151. [DOI] [PubMed] [Google Scholar]
- Rudomin P & Schmidt RF. (1999). Presynaptic inhibition in the vertebrate spinal cord revisited. Exp Brain Res 129, 1–37. [DOI] [PubMed] [Google Scholar]
- Schindler-Ivens S & Shields RK. (2000). Low frequency depression of H-reflexes in humans with acute and chronic spinal-cord injury. Experimental Brain Research 133, 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen EB & Dyhre-Poulsen P. (1999). Amplitude of the human soleus H reflex during walking and running. JPhysiol 515 ( Pt 3), 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skold C, Levi R & Seiger A. (1999). Spasticity after traumatic spinal cord injury: nature, severity, and location. Archives of physical medicine and rehabilitation 80, 1548–1557. [DOI] [PubMed] [Google Scholar]
- Smith AC, Rymer WZ & Knikou M. (2015). Locomotor training modifies soleus monosynaptic motoneuron responses in human spinal cord injury. Exp Brain Res 233, 89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ & Redman SJ. (1992). The role of GABAA and GABAB receptors in presynaptic inhibition of Ia EPSPs in cat spinal motoneurones. J Physiol 447, 675–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmadge RJ. (2000). Myosin heavy chain isoform expression following reduced neuromuscular activity: potential regulatory mechanisms. Muscle Nerve 23, 661–679. [DOI] [PubMed] [Google Scholar]
- Tasaki I. (1953). Nervous transmission. Thomas Charles C., Springfield, Illinois. [Google Scholar]
- Thompson FJ, Reier PJ, Lucas CC & Parmer R. (1992). Altered patterns of reflex excitability subsequent to contusion injury of the rat spinal cord. Journal of Neurophysiology 68, 1473–1486. [DOI] [PubMed] [Google Scholar]
- Wall PD & Devor M. (1981). The effect of peripheral nerve injury on dorsal root potentials and on transmission of afferent signals into the spinal cord. Brain Res 209, 95–111. [DOI] [PubMed] [Google Scholar]
- Wall PD & Lidierth M. (1997). Five sources of a dorsal root potential: their interactions and origins in the superficial dorsal horn. J Neurophysiol 78, 860–871. [DOI] [PubMed] [Google Scholar]
- Witschi R, Punnakkal P, Paul J, Walczak JS, Cervero F, Fritschy JM, Kuner R, Keist R, Rudolph U & Zeilhofer HU. (2011). Presynaptic alpha2-GABAA receptors in primary afferent depolarization and spinal pain control. J Neurosci 31, 8134–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar-Fisher C, Polston KFL, Eraslan M, Henley KY, Kinikli GI, Bickel CS, Windham ST, McLain AB, Oster RA & Bamman MM. (2018). Paralytic and nonparalytic muscle adaptations to exercise training versus high-protein diet in individuals with long-standing spinal cord injury. Journal of applied physiology 125, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS & Regehr WG. (2002). Short-term synaptic plasticity. Annu Rev Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.