

Abstract
Heart failure (HF) is a complex disease in which cardiomyocyte injury leads to a cascade of inflammatory and fibrosis pathway activation, thereby causing decrease in cardiac function. As a result, several biomolecules are released which can be identified easily in circulating body fluids. The complex biological processes involved in the development and worsening of HF require an early treatment strategy to stop deterioration of cardiac function. Circulating biomarkers provide not only an ideal platform to detect subclinical changes, their clinical application also offers the opportunity to monitor disease treatment. Many of these biomarkers can be quantified with high sensitivity; allowing their clinical application to be evaluated beyond diagnostic purposes as potential tools for HF prognosis. Though the field of biomarkers is dominated by protein molecules, non-coding RNAs (microRNAs, long non-coding RNAs, and circular RNAs) are novel and promising biomarker candidates that encompass several ideal characteristics required in the biomarker field. The application of genetic biomarkers as genetic risk scores in disease prognosis, albeit in its infancy, holds promise to improve disease risk estimation. Despite the multitude of biomarkers that have been available and identified, the majority of novel biomarker candidates are not cardiac-specific, and instead may simply be a readout of systemic inflammation or other pathological processes. Thus, the true value of novel biomarker candidates in HF prognostication remains unclear. In this article, we discuss the current state of application of protein, genetic as well as non-coding RNA biomarkers in HF risk prognosis.
Keywords: biomarker, heart failure, prognosis, protein biomarker, NT-proBNP, non-coding RNA, genetic risk score
Introduction
Heart failure (HF) is a complex cardiovascular disease (CVD) in which the heart's functional capacity is reduced, leading to failure in meeting the body's blood and oxygen demand (1). The most common risk factors are age, sex, environmental risk factors, genetic disposition, and diseases such as diabetes, hypertension, coronary artery disease, and atrial fibrillation. HF is described as a global pandemic as it affects ~26 million people worldwide (2). In North America and Europe, >80% of HF cases comprise of people who are ≥65 years old (2–5). Survival rate of HF patients is poor with 2–17% of HF patients dying while in hospital, 17–45% patients die within 1-year of admission and the majority dies within 5-years of admission (6). Due to the high mortality rates associated with HF, early diagnosis of the first subclinical signs is essential to prevent severe outcomes.
HF is a multi-system disorder which is characterized by a decrease in functional capacity of the heart. Reduced cardiac output due to impairment in left ventricular function leads to an activation of the neuro-hormonal system. This, in turn, stimulates the renin-angiotensin-aldosterone system leading to increased concentrations of renin, angiotensin II and aldosterone, each of which causes salt and water retention, vasoconstriction, and enhanced sympathetic activity. Prolonged exposure to neuro-hormonal activation leads to dilation and structural changes in the myocardium and fibrosis, thereby further worsening cardiac output (7). The severity of HF is graded in accordance to the New York Heart Association (NYHA) classification I, II, III, and IV. This gradation is based on patient clinical symptoms and effect of HF on their physical mobility. This definition also takes into account the decrease in the left ventricular ejection fraction (LVEF), classifying HF as either HF with preserved ejection fraction (HFpEF; LVEF ≥50%) or HF with reduced ejection fraction (HFrEF, LVEF <50%). HF involves micro- and macro-structural changes, each involving activation of inflammatory and neuro-hormonal system that release several biomolecules to compensate for the failing heart. Consequently, a storm of cytokines and regulatory molecules are released. The abundance of dysregulated molecules make it difficult to identify biomarkers that can specifically aid in HF prognosis.
A biomarker is defined as a biological compound that is easily accessible and measurable in the body. The biomarker can be classified as molecular, cellular, or imaging as long as they help in identifying the disease or provide therapeutic guidance. Diagnosis of HF through different diagnostic tools such as chest X-ray (8, 9), echocardiography (ECG) (10–12), and cardiac magnetic resonance (CMR) (13–15), have been highly reliable in guiding treatment (16). Imaging biomarkers provide great insight into the structural and functional abnormalities of the heart, however, the imaging biomarker readout fails to detect early and subclinical states of HF. Moreover, the reliability of imaging biomarker readout is biased depending on image quality, imaging modality, the observer, and center experience, as there exist differences based on age, sex, and imaging modality (16). Natriuretic Peptides (NP), i.e., brain-type natriuretic peptide (BNP) and N-terminal prohormone of BNP, and cardiac troponin measurements have been included in the guidelines for HF diagnosis and treatment of the European Society of Cardiology (ESC) (16) and the American Heart Association (AHA) (17). Addition of other diagnostic biomarkers such as markers of inflammation (e.g., soluble ST2 receptor), oxidative stress (e.g., growth differentiation factor-15) and cardiac remodeling (e.g., galectin-3) can be beneficial in guiding HF therapy (17). Though there are several well-established diagnostic biomarkers for HF, the prognostic biomarkers for such a complex disease still remain poorly evolved. Therefore, in addition to existing imaging diagnosis techniques, it is important to identify biological markers which focus on HF pathogenesis and molecular function that can aid in risk stratification and patient care (Figure 1).
Figure 1.

Advantage of having a prognostic biomarker for personalized treatment strategy. Presence of prognosis biomarkers allows for early identification of individuals at-risk of developing HF. For individuals with HF, measurement of prognosis biomarkers guide the treatment strategy leading to reduced risk of mortality and improved quality of life (green color), when compared to absence of prognosis biomarkers and standard of care management (red color). Depiction of human figures: pink, humans at-risk of HF; red, humans having HF; gray, healthy humans.
In this review article, we elucidate the role of the most prominent circulating protein biomarkers which show promising results in HF prognosis. In addition, we provide details on genetic biomarkers and polygenic risk scores that are currently being developed along with details about emerging evidence on circulating non-coding RNA biomarkers.
Protein-Based Biomarkers
Protein biomarkers are released into the circulation and can be detected using various assays. Protein biomarkers that have entered the prognostic field for HF are either released from the heart exhibiting its value of tissue-specific damage, or are released from other cells as a systemic response to HF (Figure 2). In addition to tissue specificity, the half-life of protein biomarkers is often the crucial factor for its potential use as a biomarker (Table 1). The ease of measurement of circulating protein biomarkers and the speed of assay results make them invaluable to HF diagnosis and prognosis.
Figure 2.
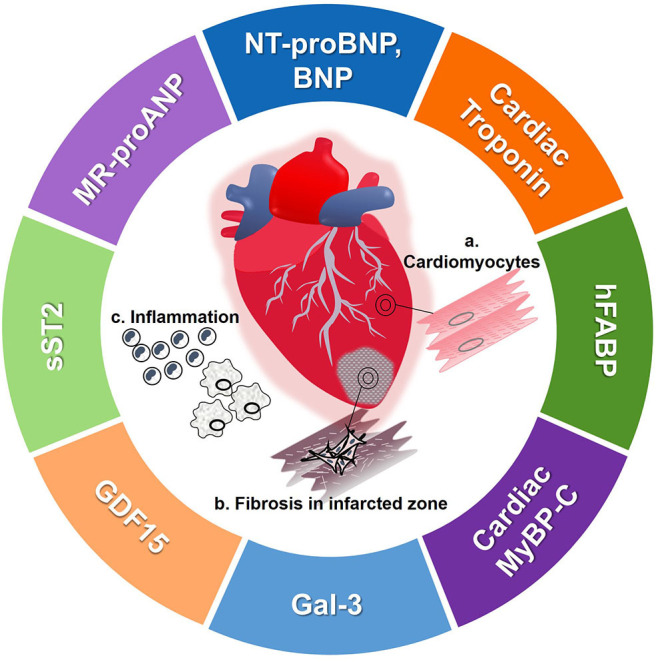
Pictorial representation of protein biomarkers detailed in this paper. (a) During HF, cardiomyocyte damage and restructuring leads to active and passive release of cardiomyocyte specific biomarkers. (b) After an event of myocardial infarction, the cardiomyocytes are destroyed and fibrosis occurs. (c) HF activates inflammatory pathways which also release other biomarkers, representative of systemic inflammation. BNP, brain-type natriuretic peptide; Gal-3, galectin 3; GDF15, growth differentiation factor 15; hFABP, heart-type fatty acid binding protein; MR-proANP, mid-region of N-terminal prohormone of atrial-type natriuretic peptide; MyBP-C, myosin binding protein-C; NT-proBNP, N-terminal prohormone of BNP; sST2, soluble suppression of tumorigenicity 2.
Table 1.
Basic information on the prognostic/diagnostic capability, organ/cell of origin of protein biomarkers that are currently used for HF prognosis or diagnosis.
| Biomarker | Prognostic/diagnostic | Organ/cell of origin | Mode of release | Half-life |
|---|---|---|---|---|
| NT-proBNP | Prognostic and diagnostic | Cardiac ventricles | Actively upon cardiomyocyte injury | 120 min |
| MR-proANP | Prognostic and diagnostic | Cardiac atrial | Actively upon cardiomyocyte injury | 60–120 min |
| Troponin I, T | Diagnostic and maybe prognostic | Cardiac thin filament | Passively upon myofibrillar injury | 120 min |
| cMyBP-C | Diagnostic | Cardiac thick filament | Passively upon myofibrillar injury | Not known |
| hFABP | Prognostic | Cardiomyocyte cytoplasmic protein | Passively upon cardiomyocyte membrane injury | 27 min |
| Gal-3 | Diagnostic | Multiple cells | Upon inflammation and cardiac remodeling | Not known |
| GDF-15 | Diagnostic | Multiple cells | Not clear | Not known |
| sST2 | Diagnostic | Cardiomyocytes, cardiac fibroblasts, and vascular endothelial cells | Upon fibrosis and cardiac remodeling | Not known |
Natriuretic Peptides
Natriuretic peptides (NPs) are hormones that play an important role in regulating volume and pressure overload, thereby maintaining homeostasis within the cardiovascular system (18). NPs are initially produced as pre-prohormones which are then cleaved to form prohormones and later the final hormones. Atrial NP (ANP) released from cardiac atria, brain NP (BNP) released from cardiac ventricles and C-type NP (CNP) are the three different types of NP hormones. This NP system includes three NP receptors (NPRs) namely NPR-A (or guanylyl cyclase A), NPR-B (or guanylyl cyclase B), and NPR-C (or clearance receptor) (19). Both ANP and BNP have similar physiological roles; they act by binding to NPR-A (19) inducing diuresis, natriuresis, and vasodilation in an attempt to reduce cardiac preload. Furthermore, they inhibit aldosterone synthesis and renin secretion, which reduces cardiac afterload (19–21). ANP, BNP and CNP bind to NPR-C which plays a role in their clearing from the system by receptor internalization and hormone degradation (18, 19).
BNP and N-Terminal Prohormone of BNP
BNP, encoded by the NPPB gene, is secreted in response to increased ventricular stretching (22). BNP acts by binding to NPR-A on target tissues, activating the cGMP signaling cascade to reduce vascular stress, diuresis, natriuresis, and inhibition of renin-angiotensin-aldosterone system (23, 24). BNPs are present in a healthy individual at ~3.5 pg/mL in plasma, which increases by 100-fold in HF patients (25). However, BNP has a short half-life of 20 min when compared to the prohormone form of BNP known as N-terminal prohormone of BNP (NT-proBNP) with a longer circulation half-life of 60–120 min (26) (Table 1). Therefore, NT-proBNP is widely used in clinical assessment of HF.
There are numerous studies that have evaluated the prognostic ability of NT-proBNP and BNP biomarkers in various HF settings such as acute or chronic HF, providing robust evidence of their incremental value (27–30). BNP was measured in a study consisting of 122 acute decompensated HF patients along with worsening renal function. A significant reduction in BNP value of ≥40% over the period of hospitalization i.e., from baseline to discharge had a positive prognostic value in reduced rehospitalization (31). The PARADIGM-HF trial (2016) quantified NT-proBNP levels in 2,080 HF patients. Patients with reduction in NT-proBNP levels had a subsequent lower rate of HF hospitalization and CV death (32). Later in 2019, the PARADIGM-HF trial tested the prognostic value of BNP and NT-proBNP before and during treatment with neprilysin inhibitors (sacubitril/valsartan) in HF. From a patho-mechanistic point of view, the authors reported that the treatment caused a direct inhibition of neprilysin leading to a treatment-associated increase in circulating BNP levels early after treatment initiation. This report indicates that inhibiting neprilysin causes a direct increase of BNP in the circulation, while NT-proBNP was not affected. Therefore, the group concluded that the use of NT-proBNP as a biomarker (which is not a substrate for neprilysin inhibition) rather than BNP avoids clinical confusion (33). The combination of biological pathways and the reported clinical findings strengthen NT-proBNP's position as an excellent circulating biomarker for HF. Additionally, serial measurements of NT-proBNP at the time of hospital presentation and over the course of treatment has the potential to provide better prognostic information on disease outcome. In the TRANSITION study (2020), NT-proBNP was studied at the time of randomization, after 4 weeks and after 10 weeks in 982 HFrEF patients with acute decompensated HF. Reduced NT-proBNP levels at 4 weeks was indicative of a lower risk of HF re-hospitalization and CV death (hazard ratio: 0.57; 95% confidence interval (CI): 0.38–0.86; p = 0.007). In another prospective multi-center study of 171 patients with acute decompensated HF, BNP, and NT-proBNP measurements were taken at the time of hospital presentation, after 24 h, after 48 h and at the time of discharge. The primary end point (1-year all-cause mortality) was reliably predicted by BNP and NT-proBNP, with the prognostic accuracy of both biomarkers increasing over the course of hospitalization. The area under the receiver operating curves (ROC) curve (AUC) increased during the course of hospitalization for BNP (AUC presentation: 0.67; AUC 24 h: 0.77; AUC 48 h: 0.78; AUC discharge: 0.78) and NT-proBNP (AUC presentation: 0.67; AUC 24 h: 0.73; AUC 48 h: 0.75; AUC discharge: 0.77) (34). This observation seems comprehensible given inter-individual differences in the severity of the disease upon presentation to hospital, with these differences being reduced upon HF treatment. It is to be noted that the reported AUC values with a maximum of <80% don't seem to support BNP or NT-proBNP as clinically applicable biomarkers—at least not when applied as single markers.
NT-proBNP has further been evaluated as a means of guiding HF therapy in various studies with mixed results: The PROTECT study (2014) investigated whether lowering of NT-proBNP concentration (NT-proBNP guided therapy) led to an improvement in echocardiographic parameters when compared to standard of care management in 151 chronic HF patients with LV systolic dysfunction. The authors reported that guiding therapy through NT-proBNP levels demonstrated prognostically meaningful improvement in echocardiographic parameters such as LV volumes and function (35). By contrast, when treatment of n = 405 acute decompensated HF patients was guided by NT-proBNP, 6-month outcome was not improved in the PRIMA II trial (2018) (36).
BNP and especially NT-proBNP are reliable gold standard diagnostic biomarkers in heart failure, likely driven by their well-understood and cardiac-specific biological function. Their prognostic potential seems promising, while at the same time, when used as single markers, their accuracy doesn't yet allow for clinical applicability and their use in HF treatment guidance still needs to be further evaluated.
Mid-Region of N-Terminal Prohormone ANP (MR-proANP)
ANP, encoded by the NPPA gene, is secreted in response to atrial volume stretch receptors (37). The physiologic activity of ANP is similar to BNP, ANP acts by binding to NPR-A in cardiac atria, kidney, adrenal glands, and vascular smooth muscle cells, causing an increase in renal sodium excretion. This results in decreased extracellular fluid volume and blood volume, thereby improving cardiac ejection fraction and reduction of blood pressure (38). In healthy individuals, plasma levels of ANP are ~20 pg/mL and it rises 10–100 times in HF patients (39). It is difficult to measure the bioactive form of ANP due to its short half-life (2 min) (40–43) and although the N-terminal prohormone form of ANP (NT-proANP) is more chemically stable, it is easily degraded (40). Therefore, mid region of NT-proANP (MR-proANP) which is less susceptible to proteolytic degradation is used in clinical assessment (40, 44).
MR-proANP retains its prognostic incremental value in regards to the study end-points and has mostly been studied in combination with NT-proBNP and/or other biomarkers. In acute HF patients, higher MR-proANP levels provided long-term (5-year follow-up) prognostic value [MR-proANP (AUC = 0.668) vs. BNP (AUC = 0.604), p = 0.042 and vs. NT-proBNP (AUC = 0.564), p = 0.004] (45). A secondary analysis from the MOLITOR trial (2019) investigated serial measurements of MR-proANP and NT-proBNP biomarkers in 104 acute decompensated HF patients for betterment in quality of life (QoL). The authors concluded that MR-proANP predicted the reduced physical and mental QoL whereas NT-proBNP was predictive of reduced physical QoL (46). In the GISSI-HF trial (2010) investigators quantified MR-proANP along with three other circulating biomarkers in 1,237 chronic and stable HF patients at baseline and 3-months follow-up. Serial measurements of MR-proANP had the best prognostic accuracy with AUC of 0.74 (95% CI = 0.71–0.77) independent of NT-proBNP, which had an AUC 0.73 (95% CI = 0.70–0.76) (47). Similar results were reported in other clinical trials (48, 49). Thus, while not as clinically established as NT-proBNP in the diagnostic field, there is some evidence that MR-proANP may serve as an equally precise prognostic biomarker in HF patients.
NPs function as cardiac-specific hormones, released in pathologic states of increased cardiac strain. Therefore, their biomarker function is directly derived from a cardiac-specific pathology and comprises a strong link to a natural therapeutic attempt. This may well be an important reason why NPs remain unparalleled in the field of biomarkers for heart failure diagnosis, prognosis, and now also in guided therapy (18).
Troponin I and T
Troponin protein is a component of skeletal and cardiac muscle thin filaments and has three isoforms, namely troponin C (Tn-C), troponin I (Tn-I encoded by TNNI3 gene) and troponin T (Tn-T encoded by TNNT2 gene), known as the troponin complex. Cardiac isoforms of Tn-I (cTn-I) and Tn-T (cTn-T) are exclusively expressed in cardiac muscle, whereas Tn-C is expressed as one isoform in both cardiac (encoded by TNNC1 gene) and skeletal muscle (encoded by TNNC2 gene). The troponin complex plays a key role in cardiac and skeletal muscular contraction (50), regulated by calcium concentration in the myocyte cytoplasm. This is achieved by allowing calcium binding (by Tn-C), inhibition of ATPase activity of actomyosin complex (by Tn-I) and interaction with tropomyosin which is wrapped around the actin (by Tn-T). cTns are the primary biomarker for MI and acute coronary syndrome (ACS) and can also be elevated during HF. Therefore, cTns present a picture of myocardial damage and not HF itself, and are known as cardiac injury biomarkers. Guidelines recommend measuring cTn along with NPs at the time of hospitalization to identify acute coronary syndrome as the underlying cause of acute HF patients as well as for prognosis of HF disease (16, 17).
The ASCEND-HF study examined the prognostic importance of cardiac troponin I (cTn-I) in patients with acute decompensated HF. Elevated levels of cTn-I (above 99% upper reference limit) was observed in 50% of acute decompensated HF patients and helped in predicting in-hospital outcome (p=0.01), but was not an independent predictor of post discharge outcomes (51). The CORONA trial examined high-sensitivity cardiac troponin T (hs-cTn-T) in a subgroup of elderly patients (≥60 years, n = 1,245) with systolic HF of ischemic origin. The authors reported that elevated hs-cTn-T levels (above 99% upper reference limit) provided a strong and independent prognosis of CV death, non-fatal MI, non-fatal stroke and HF hospitalization in older patients with chronic ischemic HF (52). The RELAX-AHF study investigated the association between serial measurements (baseline, 2, 5, and 14 days) of hs-cTn-T and outcomes [CV death (180 days), HF/renal hospitalization (60 days), and dyspnea relief]. The authors concluded that hs-cTn-T was elevated above 99% upper reference limit in 90% of patients. Higher levels of hs-cTn-T was associated with worse outcomes and CV death (180 days) (hazard ratio adjusted hs-cTnT = 1.36, 95% CI 1.15–1.60, p = 0.0004) (53).
When measured with other biomarkers, Tn-T retained its prognostic value. The RED-HF trial tested the prognostic values of various cardiac inflammatory and renal biomarkers in HFrEF patients. The authors concluded that NT-proBNP and hs-Tn improved risk stratification in HFrEF subjects, outperforming other emerging biomarkers tested by the group (54). A biomarker sub-study of the PARADIGM-HF trial investigated the prognostic importance of NT-proBNP and Tn-T measurements in HF patients with and without diabetes. NT-proBNP levels were not influenced by the presence of diabetes, whereas Tn-T levels were elevated in HF patients with diabetes. Diabetes, high NT-proBNP and high Tn-T were highly predictive for CV death or HF hospitalization (55).
The above-mentioned studies show a combined prognostic value of using cTn with BNP or NT-proBNP for predicting HF morbidity and mortality, as is also mentioned in the ESC and the AHA guidelines (16, 17). Comparable to NT-proBNP, cTns are cardiac-specific, which provides them with high tissue specificity. At the same time, latest advances in improving high and ultra-high sensitive assays has largely improved sensitivity of cTn detection. This has lately opened new approaches to evaluate cTn as a prognostic biomarker even in the general population, with detectable values in nearly 100% of healthy individuals (56). These new opportunities to evaluate cTn as a marker for subclinical cardiac pathologies seems promising. On the other hand, such discrete elevations of circulating cTn levels may merely represent higher protein turnover. Patho-mechanistically, cTns are passively released into the blood flow upon myocyte injury as opposed to NT-proBNP which is actively secreted upon pathological triggers in early cardiac disease states. This could make NT-proBNP the better, more sensitive, cardiac-specific HF prognosis biomarker—independent of the sensitivity of the used assay. Nevertheless, hs-cTn assays have been proven beneficial since they allow detection of Tn in patients with stable chronic HF in the absence of acute myocardial damage (57) and could aid in improving HF prognosis, when used additionally to NT-proBNP in a multi-biomarker approach.
Cardiac Myosin-Binding Protein C
Cardiac myosin-binding protein C (cMyBP-C), encoded by the MYBPC3 gene, is a component of the sarcomere thick filament complex along with titin and myosin. cMyBP-C plays an important role in sarcomere structure and maintenance, and regulation of muscle contraction through modulation of actin-myosin cross-bridges (58, 59). cMyBP-C is a large modular protein of 140.8 kDa with 11 globular domains and belongs to the intracellular immunoglobulin (Ig) superfamily (60, 61). cMyBP-C differs from the skeletal isoform sMyBP-C, in that cMyBP-C contains 3 additional domains: C0 (at the N-terminus), C1 and C2. The region between C1 and C2 domains contain functionally important phosphorylation sites, which confer cardio-protection and reduce cMyBP-C association to actin and myosin (60, 62–64). After an event of MI, phosphorylation of cMyBP-C results in the release of a cleaved ~40 kDa N-terminal fragment of cMyBP-C into the circulation (65–67). This phosphorylation protects cMyBP-C from proteolysis during ischemic injury and can therefore serve as a diagnosis biomarker for cardiac injury upon hospital admission (65, 68). Phosphorylation by phosphatase kinase A (PKA) is important, because reduction in phosphorylation of sarcomeric target protein c-MyBP-C has been reported in end-stage failing myocardium irrespective of the cause of HF (69–73).
As a circulating biomarker, cMyBP-C has first been assessed as a highly sensitive marker for myocardial injury and recent studies provide evidence that cMyBP-C could outperform cTn in the early detection of MI (74–77). Reasons for cMyBP-C's earlier detectability in the circulation could be its higher abundance in cardiomyocytes compared to cTn (74) and, more importantly, an ischemia-induced shedding of cleaved N-terminal fragments of cMyBP-C (78). These cardiac-specific and highly sensitive characteristics make circulating cMyBP-C an ideal target to be evaluated as a biomarker for subclinical CVD states and also a candidate biomarker for HF prognostication. In a prospective case-control study involving 50 children with acute HF, cMyBP-C levels were measured at the time of hospital admission and 1 month after treatment with a follow-up at 3 months. The authors reported that higher levels of cMyBP-C at the time of hospital admission were associated with worse prognosis and higher rate of readmission and mortality (79). Surprisingly, these promising initial results in HF outcome prognostication have so far not been re-assessed. Only one further study assessed cMyBP-C in early subclinical CVD disease states; Anand et al. reported cMyBP-C to be associated with myocardial hypertrophy and fibrosis, as potential causes for HF, in aortic stenosis patients (80). The authors furthermore report an association of cMyBP-C with mortality in the assessed cohort.
cMyBP-C is an important regulatory protein of the cardiac contractile complex. As a biomarker, there is convincing data on its promising potential to be clinically used to improve early rule-in and rule-out of MI. With respect to HF prognostication further trials are warranted to validate the prognostic and diagnostic ability of cMyBP-C.
Heart Type Fatty Acid Binding Protein
Heart type fatty acid binding protein (hFABP), encoded by the fatty acid-binding proteins 3 (FABP3) gene, is a member of cytoplasmic protein group and is classified as a cardiac cell death marker (81). In general, FABPs play a role in cellular fatty acid metabolism by binding to and transporting long-chain polyunsaturated fatty acids from the cell membrane to mitochondria (82). hFABP is a 15 kDa protein, expressed abundantly in the cytoplasm of striated muscle cells. Unlike Tn or cMyBP-C, hFABP is not a component of muscle structure (83–86). Therefore, it is likely that hFABP is released immediately upon cardiomyocyte injury, as in case of MI, and may be detectable earlier than cTn and cMyBP-C. Importantly, the cytosolic localization of hFABP could make an important patho-mechanistic difference in terms of active secretion as opposed to passive release after cardiomyocyte injury. This may provide a different quality of information on cardiac pathology in terms of early detection of subclinical disease or HF prognostication. hFABP starts a negative cycle of cardiac damage because increased extracellular hFABP levels affect cardiac contraction by decreasing intracellular calcium levels, therein causing further damage and more extracellular hFABP levels (82). hFABP levels in serum rise immediately after cardiomyocyte injury, making it a promising molecule to investigate with respect to cardiac function and HF, although the concentration variability of hFABP in HF patients is less well known.
Kazimierczyk et al. investigated the prognostic ability of hFABP in 77 patients with acute decompensated HF at hospital admission and discharge. The authors reported that constantly higher levels of hFABP might reflect ongoing myocardial damage and might be a valuable biomarker to predict poor outcome in acute decompensated HF patients (87). A study by Niizeki et al. investigated serial measurements of hFABP levels in 113 chronic HF patients at the time of hospital admission and at the time of hospital discharge. The patients with consistently high levels of hFABP had subsequent higher cardiac events in the follow-up period (624 ± 299 days) when compared to patients with normal hFABP levels or those, whose levels decreased between admission and discharge. The authors concluded that such serial measurement of hFABP can be informative for guiding therapy and management of chronic HF patients (88). Another study by Niizeki et al. investigated whether the combination of hFABP and BNP would provide information on risk stratification in 186 chronic HF patients. High hFABP and BNP levels at the time of hospital admission were associated with increased number of cardiac events and mortality, therefore were helpful in risk stratifying chronic HF patients upon hospitalization (89). A post-hoc analysis of the MANPRO study (2015) by Hoffmann et al. investigated the prognostic ability of hFABP when compared to Tn-I in patients suspected of acute HF with a 5-year follow-up period. Higher hFABP levels were associated with all-cause mortality and acute HF related hospitalization at 5-years, and hFABP levels could predict the acute HF related hospitalization better than Tn-I (90). In a study by Kutsuzawa et al. (91), hFABP and Tn-T levels were measured in 151 prospectively enrolled HFpEF patients. Higher levels of hFABP was observed more frequently in patients when compared to circulating Tn-T levels, indicating that cardiomyocyte membrane injury occurred frequently compared to myofibrillar damage in HFpEF patients. Circulating levels of hFABP increased with advancing NYHA functional class and was an independent predictor of future CV events (91).
The above reports suggest hFABP as a biomarker for CVD, not primarily focusing on MI like cTn and cMyBP-C. Instead, circulating hFABP may well be a marker of cardiomyocyte-specific metabolic disorders as they occur not only during but also before the onset of HF, thus making hFABP a promising candidate biomarker for very early stages of subclinical HF, and for HF prognostication. On the other hand, currently available data is limited to a low number of trials and large-scale validation is needed in order to gain more information on its potential for clinical implementation.
Galectin 3
Galectin 3 (Gal-3), encoded by LGALS3, is a member of the galectin family and is a beta-galactosidase-binding lectin with an atypical N-terminal domain (92). Gal-3 is ubiquitously expressed and plays an important function in several biological processes such as in cell-cell adhesion, cell-matrix interactions, macrophage activation, angiogenesis, metastasis, and apoptosis (92). Hence, Gal-3 is not cardiac-specific. Basal Gal-3 expression varies depending on tissue type and tissue maturity (93). Within hematopoietic tissue, macrophages express Gal-3 more than monocytes and it is undetected in human peripheral blood lymphocytes (94–97). This increased expression of Gal-3 in macrophages is assumed to induce inflammation, fibroblast proliferation, and collagen deposition in the heart, thereby promoting ventricular restructuring, which is a central patho-mechanistic process in HF (98–100).
In the CORONA study, the prognostic value of Gal-3 was tested in 1,462 patients aged >60 years with systolic, ischemic HF. Using an unadjusted analysis, Gal-3 was not associated with HF hospitalization but with CV death. When adjusted for NT-proBNP, Gal-3 showed no significant association with any end-points. The authors concluded that prognosis of HF in elderly patients with systolic HF is limited using Gal-3 (101). In the HF-ACTION study, the association between Gal-3 and long-term clinical outcome in ambulatory HF patients was evaluated. In a univariate analysis, Gal-3 was significantly predictive of long-term outcomes [unadjusted hazard ratio, 1.14 (per 3-ng/mL increase in Gal-3), 95% CI 1.09–1.19; p < 0.0001], however this association did not withstand multiple testing or adjustment for NT-proBNP (adjusted hazard ratio: 1.03, 95% CI 0.98–1.08, p = 0.27) (102). Conversely, there are studies or analyses which have shown that Gal-3 can be helpful in predicting adverse outcomes when measured serially (baseline and follow-up). In the DEAL-HF study (2010), Gal-3 was measured in 232 patients with chronic HF (NYHA class III or IV) at baseline and 6.5-year follow-up. Plasma levels of Gal-3 proved to be a significant predictor of mortality (103). In two large cohorts of chronic HF and acute decompensated HF patients, the prognostic value of Gal-3 serial measurements in HF patients at baseline vs. 3-months follow-up (CORONA study) and baseline vs. 6-month follow-up (COACH study) revealed that an increase of Gal-3 at follow-up was associated with an increased rate of re-hospitalization and mortality (hazard ratio in CORONA, 1.60; 95% CI, 1.13–2.25; p = 0.007; hazard ratio in COACH, 2.38; 95% CI, 1.02–5.55; p = 0.046) (104).
The above information suggests that Gal-3 may well be a biomarker in the setting of HF. Its expression is upregulated in fibrosis, a common phenomenon associated with HF. The tissue of origin remains a matter of debate and thus its lack of cardiac specificity seems to be reflected by a limited prognostic value when compared with more established biomarkers such as NT-proBNP.
Growth Differentiation Factor 15
Growth Differentiation Factor 15 (GDF15), also known as macrophage inhibitory cytokine-1 is a distant member of the transforming growth factor-β cytokine superfamily (105–107). The secreted form of GDF15 is a ~28 kDa disulphide-linked dimer that is expressed in low levels in all organs except the placenta (105, 108). More specifically, GDF15 expression arises from macrophages, vascular smooth muscle cells, endothelial cells and adipocytes (109–114). An increase in GDF15 levels is observed under cardiac inflammation, injury, and restructuring (115–117). GDF15 was detected in atherosclerotic plaques in coronary arteries (109, 118) although the origin tissue of GDF15 expression in HF remains unclear.
GDF15 biomarker measurements in HF patients has provided evidence of its capability in HF prognosis, either through serial measurements or when measured along with other prominent biomarkers. In the Valsartan Heart Failure Trial (Val-HeFT), the investigators evaluated serial measurements of GDF15 at baseline (n = 1,734) and 12-months follow-up (n = 1,517) in HF patients. GDF15 levels were independently associated with risk of death, even after adjustment for BNP, hs-Tn-T, and hs-C reactive protein. Serially increasing GDF15 levels at follow-up were associated with worsening renal function and increase in cardiac strain biomarkers (BNP, Tn-T) (119). In another study, elevated levels of GDF15 were associated with increased risk of death in 455 chronic HF patients. Even after adjusting to NT-proBNP and other markers, GDF15 retained the prognostic ability in predicting HF mortality (120).
HF causes release of GDF15, whereas the specific tissue of origin is not completely determined. Instead, the utility of GDF15 must be taken as marker for systemic causes or effects of HF. Whether GDF15 allows for specific HF-related prognostication remains matter of debate. We can speculate that in patients with other causes of systemic inflammatory processes, GDF15 may lose its HF-specific prognostic value. Nevertheless, results from clinical trials suggest a promising role of GDF15 as a biomarker in HF prognostication. In this respect, further investigations and validations remain to be undertaken.
Soluble Suppression of Tumorigenicity 2
Soluble suppression of tumorigenicity 2 (sST2), is a member of interleukin (IL)-1 receptor family and is a ligand for IL-33 (121). IL-33 is an IL-1 like cytokine that can be secreted by most cells in response to damage (122). Whereas, sST2 is produced by vascular endothelial cells, cardiomyocytes, and cardiac fibroblasts in response to stress or injury providing a certain degree of cardiovascular specificity. sST2 is the circulating isoform of the cellular membrane receptor ST2L, lacking the cytoplasmic, and transmembrane domains. IL-33/ST2L is beneficial to the heart because it inhibits cardiac hypertrophy and fibrosis, thereby mitigating adverse cardiac remodeling (123). This beneficial effect is blocked by increased levels of sST2 (acting as decoy receptor) (123), since sST2 binds to IL-33 and interrupts the IL-33/ST2L downstream signaling.
Serial measurement of sST2 at baseline and at follow-up provides evidence of the prognostic impact of sST2 in predicting HF hospitalization outcomes. In the ASCEND-HF trial, serial measurement of sST2 levels at baseline and follow-up at 48, 72 h, and 30 days were investigated in 858 acute HF patients for adverse HF outcomes. Continuously higher levels of sST2 was associated with increased risk of adverse HF events and higher risk of death at 180 days [hazard ratio at baseline: 2.21 (p < 0.001); at follow-up: 2.64; (p < 0.001)]. However, the prognostic value of sST2 decreased after adjustment with NT-proBNP (124). In the MADIT-CRT trial, sST2 levels were serially measured at baseline and 1-year follow up in 684 patients with mild symptoms of HF and reduced LV function. Elevated sST2 levels at baseline was associated with increased risk of death, HF or ventricular arrhythmia (VA) events, and serially increasing levels of sST2 was associated with higher risk of VA and death (125). In the Valsartan Heart Failure Trial (Val-HeFT), sST2 was serially measured at baseline, 4 and 12-months follow-up in patients with HF. Increased sST2 levels were independently associated with morbidity, mortality and HF hospitalization. However, it failed to provide prognostic information when adjusted with NT-proBNP (126).
The IL-33/ST2L system exerts cardio-protective function, which is interrupted by sST2's decoy receptor capabilities. Circulating sST2 can provide information about cardiac stress and can be targeted pharmaceutically to maintain the IL-33/ST2L cardio-protective role. Clinical evaluation of circulating sST2 provide initial data on its use as a prognostic biomarker in HF, albeit its additional values compared to more cardiac-specific markers remains limited and subject to further large-scale investigation.
Genetic Biomarkers
Currently, the clinical standard for prognosis for HF depend on protein-based biomarkers as they bear many advantages such as easy accessibility, comparably low costs, and easy handling. Nevertheless, protein-based biomarkers might not be specific for HF and therein definite prognostic markers for HF remain scarce (127). This scarcity is partly caused by the complex and diverse etiology and pathophysiology of HF (1, 128). Recently, new opportunities for genetic analyses have risen as a novel approach to understanding the pathophysiology of CVD, paving the way for the development of gene-based biomarkers. Omics technology that identifies genome-wide (GW) and transciptome-wide (TW) gene variation is an innovative approach to identify DNA/RNA-based biomarkers. Omics analyses not only allow to identify genetic variations that can contribute to at-risk HF identification but also for understanding the molecular mechanisms behind the diseases. By identifying a common single nucleotide polymorphism (SNP) (through GW) or gene expression variation (through TW), it is possible to stratify at-risk populations for a disease. This brings us one step closer to personalized medicine (Figure 3). It has to be noted, however, that given the origin and novelty of these analyses and the complexity of CVD, most analyses broadly consider CVD and only few limited studies are available specifically for HF. Although it was well-established that there is a genetic component for the development of CVD (129), the causal role of genetics in the development of CVD remained largely elusive until genome-wide association studies (GWAS) became available. In 2007, four groups discovered the 9p21 risk locus simultaneously by using GWAS analyses (130–133). The locus encodes different transcripts of the long non-coding RNA ANRIL (134). Current studies suggest that the ratio of circular to linear ANRIL, which affects basic cellular mechanisms, is associated with the risk of coronary artery disease (CAD) and could potentially serve as a biomarker (135). Subsequently after discovery of the 9p21 locus, multiple genetic loci were identified that, all together, account for ~25% of the estimated CVD heritability (135). These results changed the understanding of the genetic architecture of CVD where instead of rare variants of SNPs having large effects on CVD risk in most patients, the genetic risk for CVD derives from the cumulative effect of many common risk alleles, each of them with small effect sizes (135).
Figure 3.
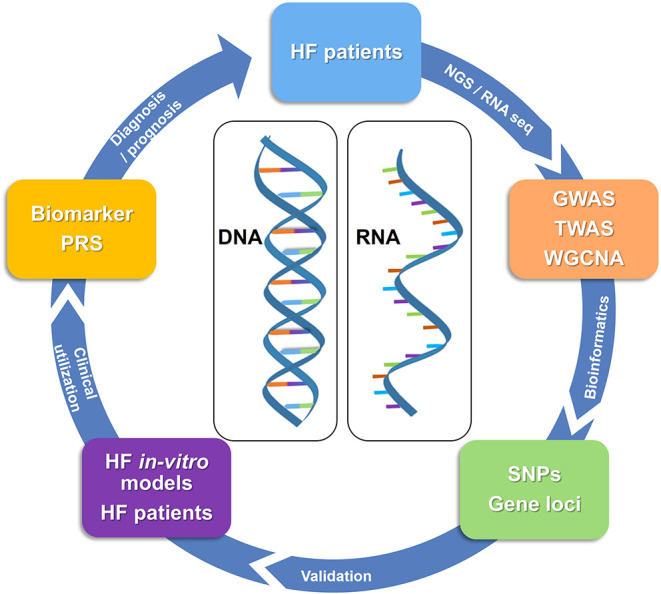
Identification and validation of genetic biomarkers in HF. In order to identify genetic biomarkers, samples taken from HF patients are sequenced and analyzed via GWAS, TWAS as well as WGCNA. Results are then further processed using bioinformatics providing information on SNPs or gene loci associated with HF. Validation of these SNPs by in-vitro models for HF and HF patients provides concrete evidence on the clinical utilization of genetic biomarkers or PRS. Ultimately, genetic biomarkers can be used for diagnosis/prognosis of HF patients in the future. DNA, deoxyribonucleic acid; GWAS, genome-wide association studies; NGS, next generation sequencing; PRS, polygenic risk score; RNA, ribonucleic acid; RNA seq, RNA sequencing; SNPs, single nucleotide polymorphisms; TWAS, transcriptome-wide association studies; WGCNA, weighted gene co-expression network analyses.
Genetic Biomarkers Combined With Lifestyle Risk Factors
As HF is a multi-causal disease, risk scores integrating clinical diagnostics, protein, and genetic biomarkers as well as lifestyle factors bear a huge potential for personalized risk stratification and prevention strategies for HF. In this regard, easily accessible biomarkers for lifestyle risk factors like obesity and smoking can also be an important approach for objective risk assessment and the identification of molecular mechanism for the development of HF. This in turn might lead to the identification of novel HF biomarkers as well. Adipokines like leptin and chemerin are examples of protein-based markers derived from the adipose tissue. They are not only obesity-associated but also dysregulated in cardiovascular diseases (136, 137). For smoking, a number of affected loci have been identified by genetic and epigenetic analyses, including the AHRR, GPR15, GFI1, and RARA genes, which in turn might be involved in cardiovascular diseases (138, 139). The bioinformatics integration of these data and validation in large cohorts can result in a biomarker panel for risk stratification. For HF prognosis, genetic analyses bear the potential for future identification of genetic and protein biomarkers for early as well as late HF prognosis, thereby improving personalized treatment. As a future scenario, a panel of protein-based and genetic-based biomarkers for inheritable and lifestyle factors might be measurable from just one single blood draw, which altogether integrate into a highly accurate HF risk score.
Genome Wide Association Studies (GWAS) and Weighted Gene Co-expression Network Analyses (WGCNA)
Currently, more than 160 chromosomal loci associated with CAD risk have been identified by GWAS consortia. These loci affect pathways such as lipid metabolism, blood pressure, inflammation, blood coagulation, cell cycle and proliferation, signal transduction, apoptosis, and transcription splicing regulation (135, 140). Fewer gene loci have been identified specifically for HF in contrast to the large number of GWAS loci associated with CAD risk. An overview on HF-specific genomic loci identified by GWAS analyses is given by van der Ende et al. (141). The likely affected genes comprise KIAA1598, USP3, LRIG3, HSPB7, BAG3, and HCG22 (141). Interestingly, USP3, LRIG3, and HSPB7 have also been linked to HF in other studies. USP3 codes for the ubiquitin specific peptidase 3, which might be involved in the development of HF (141). LRIG3 has previously been connected to several types of cancers, but Lrig3 knockout in mice also impaired cardiac function (142), HSPB7 encodes the heat shock protein B7, which is mostly expressed in cardiac and skeletal muscle and preserves contractile integrity. It is also referred to as the cardiovascular heat shock protein (cvHSP) and has been associated with advanced HF (141, 143). A recent study by Shah et al. combining a GWAS meta-analysis with Mendelian randomization analysis identified eleven HF-associated loci, among them another variant within the above mentioned BAG3 locus (128). BAG3 encodes the B cell lymphoma 2-associated anthanogene protein, which is an anti-apoptotic co-chaperone protein (144). It plays an essential protective role in dilated cardiomyopathy and is associated with LVEF (144–146). These genes and proteins are interesting candidates for novel prognostic markers for HF and their role and mechanisms should further be experimentally elucidated. An example for a potential GWAS-derived HF biomarker is the SNP rs12564445 within the TNNT2 gene which encodes the cardiac troponin T protein. The SNP rs12564445 was associated with incident HF in European Americans (147). As described in this review, cardiac troponin is clinically used as a protein-based biomarker for the diagnosis of HF. Furthermore, cytokine gene polymorphisms, e.g., in the Interleukin 10 (IL-10) gene might act as biomarkers to identify individuals more susceptible to HF (148). IL-10 is an anti-inflammatory cytokine with pleiotropic effects in immune regulation. Experimentally, IL-10 has been shown to act in a cardio-protective way and, amongst other mechanisms, antagonizes TNF-α, which is an important cytokine for heart failure progression (149). Given its manifold effects, it has to be further evaluated whether IL-10 can be used as a heart failure-specific biomarker in the future. Hypertrophic cardiomyopathy (HCM), a disease of the sarcomere, is identified by mutations in MYBPC3 gene encoding cMyBP-C. More than 350 mutations have been identified in the MYBPC3 gene, representing 40–50% of all HCM mutations, making it the most frequently mutated gene in HCM disease (150, 151). More than 60% of MYBPC3 mutations are truncating, slicing, or branch point mutations leading to COOH-terminally truncated cMyBP-C protein that lacks major myosin- and/or titin- binding sites (151).
As a step further than GWAS, weighted gene co-expression network analyses (WGCNA) allow functional interpretations of gene network modules (152). In a recent WGCNA analyses, the six hub genes BCL3, HCK, PPIF, S100A9, SERPINA1, and TBC1D9B were identified in HF patients after acute MI and could potentially serve as early prognostic biomarkers for HF (152). These hub genes might be involved in the development of HF by regulating local and systemic inflammatory pathways (152). For instance, BCL3 encodes the proto-oncogene B-cell lymphoma 3-encoded protein (153). Amongst others, BCL3 is involved in the transition from compensated cardiac hypertrophy to HF (154). Even though these hub genes and many other genetic risk loci have been described in relation to HF and CVD, their translation as biomarkers into the clinic remains difficult as described below. The longest known genetic CVD biomarkers that were translated into the clinic include mutations in the LDLR, PCSK9, and APOB genes, which can lead to hypercholesterolaemia (135). Screening for these mutations gives the opportunity for early diagnosis and personalized treatment with lipid-lowering medication (135). Although novel genomic analyses have not led to genomic biomarkers yet, transcriptome analyses were the means for identification of protein-based biomarkers like GDF15 and ST2. GDF15 has been identified by gene expression array in cardiomyocytes under nitrosative stress and ST2 was identified by microarray technology to be upregulated in cardiac myocytes after mechanical strain (115, 155).
Polygenic Risk Scores
The identification of CVD loci has not revolutionized diagnostics or treatment of CVD yet because CVD is a complex multi-genic disease and SNP variants have mainly small effect sizes. As strong genetic biomarkers have not been established for HF, genetic risk scores were a new approach to consolidate the small effect sizes of SNP variants. Initial studies creating genetic risk scores had only limited success due to small size of the initial GWAS, limited computational methods and lack of large datasets for validation and testing (156). Presently, polygenic (multiple genes) risk scores (PRS) include a large number of genetic variants that are able to identify more people at risk for CVD, as it has been shown to be possible with rare monogenic mutations that appear to be the main cause for positive CVD family history (156, 157) In 2018, Khera et al. published a PRS, wherein 8% of the population were identified to have a genetic predisposition, resulting in a more than 3-fold increase in CVD risk (156). Participants with a high genetic risk score would benefit from a 50% lower risk of CVD by adhering to a healthy lifestyle (158). Furthermore, another current clinical study combined a PRS for CAD with lifetime exposure of LDL-C and systolic blood pressure (159). This study concluded that although the PRS categorizes the lifetime risk for CAD, modifiable risk factors are the main influence for the development of the disease (159).
Presently, few gene panel assays are commercially available but have not been applied in clinical settings (160, 161). Even though PRS were able to identify individuals at risk for CAD, it did not add significant predictive value compared to traditional risk factors and the clinical relevance of PRS might be less than expected (162, 163).
Challenges and Prospects of Using Genetic Biomarkers
Though many genetic loci for increased CVD risk have been identified, the heritability of CVD is still largely unknown. One reason is that the functions of the identified loci remain largely elusive. Only 30% of the SNPs were found to modify classic risk factors for CVD and very few affect the protein structure (140). Most variants are located within non-coding regions, which impedes the identification of their function or targets. Furthermore, most of the loci affect multiple genes and phenotypes, making it difficult to identify the causal variant (135). Additionally, CVD is a complex heterogeneous disease and genetic analyses mostly did not consider environmental factors, the various triggers for HF and were limited to CAD/CVD patients of European ancestry (156, 164, 165). Therefore, performance of PRS and the sensitivity and specificity of genomic loci as HF prognosis biomarkers needs to be validated in different large-scale studies as well as in different ethnicities (166, 167). Large-scale population-based cohort studies are required to allow for the evaluation of a broad range of phenotypes together with high-density genetic mapping.
Presently, the laboratory methodology for genetic biomarkers is time-consuming, complex, and expensive for clinical routine compared to protein-based biomarkers and currently these genetic biomarkers do not add significant additive value to classic cardiovascular risk factors. However, in a mid- to long-term perspective, genetic biomarkers may provide a unique opportunity for treatment and early risk prediction through lifestyle changes or medication. If genetic biomarkers are combined with classic and clinical risk factors, they can add significant value by helping clinicians to practice personalized medicine at an early point of the disease process (159).
Transcriptomics Based Biomarkers—Non-coding RNAs
Transcripts of non-coding regions in the genome have been widely implicated in biology. Several subcategories of non-coding RNAs have been described (Figure 4). Of these, most attention has been focused on microRNAs (miRNAs) since their discovery in the early 1990's (168). Over the last decade, other classes have been identified, such as long non-coding RNAs (lncRNAs) and circular RNA (circRNAs). Several of these have been studied in the context of cardiovascular disease (CVD). Most strikingly, several subclasses of ncRNA are readily detected in the circulation (169)—freely circulating as well as associated with and derived from circulating cells such as leukocytes and platelets. This has opened up new avenues of biomarker research, which has traditionally relied on protein measurements (78).
Figure 4.

Synthesis, length, cellular location and molecular function of non-coding RNAs. (A) miRNAs are expressed from miRNA genes. Located in the nucleus, pri-miRNAs (a) are transcribed from the DNA and are processed by Drosha-Pasha processing complexes giving rise to pre-miRNAs (b). Pre-miRNAs are translocated from the nucleus into the cytoplasm via Exportin-5, where they are further processed by Dicer enzyme to form miRNA duplex (c) and later the final miRNA (d). (B) lncRNAs arise from sense and antisense strands of introns (e), exons (f), and intergenic regions (g) of the DNA and are located within the nucleus or mitochondria. (C) circRNAs are generated from the pre-mRNA in a process called back-splicing' resulting in circRNAs made up of either exon-exon junctions (h), intron-intron junctions (i) or exon-intron junctions (j). Red indicates the back-splice junction. Black arrows indicate direction of back-splicing in circRNAs. circRNA = circular RNA, lncRNA = long non-coding RNA, miRNA = microRNA, mRNA = messenger RNA.
miRNAs
miRNAs are an abundant class of non-coding RNAs of 19–25 nucleotides in length, recognized as evolutionarily conserved RNA molecules that fine-tune protein synthesis by regulating gene expression at the post-transcriptional level (Figure 4). By binding to the 3′-untranslated region (UTR) of messenger RNAs (mRNAs) through sequence complementarity, miRNAs either initiate translational repression or mRNA degradation. For some miRNAs, expression is confined to a specific organ or cell type (170–172) and thus, these miRNAs are expressed in a tissue- and cell-type specific fashion (173, 174). Intracellular miRNAs have been proven to be important modulators of cell function under pathological conditions (175). A single miRNA can suppress more than one gene. Conversely, single genes can be targeted by multiple miRNAs in parallel. Targets of individual miRNAs are often found to be functionally related. Gene suppression is usually partial rather than total, indicating that miRNAs fine-tune protein levels (169).
miRNAs are not just confined to the intracellular space of organ tissues, but also present in the circulation (176) within circulating cells such as platelets and as a pool of extracellular miRNAs. The stable detection of these miRNAs was surprising, given the high levels of RNase activity in blood plasma. In contrast to mRNA, miRNAs are protected from instant degradation through several mechanisms (177–179). Circulating miRNAs were initially thought to be a passive spill-over from various tissues. However, there is emerging evidence for their function as extracellular messengers in cell-to-cell communication. This might provide an alternative pathway of gene regulation in physiological and disease conditions (180), although the biological relevance of this phenomenon remains to be matter of debate (181). Independent of a biological function of circulating miRNAs, their potential use as circulating biomarkers for CVD has been increasingly recognized over the past years (182, 183). In vivo findings suggest that specific subsets of miRNAs are dysregulated with different features of CVD (184) and several studies have explored their potential clinical utility.
Most miRNAs are ubiquitously expressed across tissues. The earliest reports in the cardiovascular field were published in 2005, identifying miR-1 as a regulator of cardiac differentiation (185, 186). miR-1 plays a key role in the differentiation process of mesodermal precursors to cardiomyocytes and is involved in reprogramming cells into cardiomyocytes (187). Furthermore, miR-1 restoration in failing hearts improved cardiac function (188), suggesting not only its essential biological role but pointing out miR-1′s potential as a therapeutic target in heart failure treatment. Patho-mechanistically, cardiac remodeling and fibrosis are essential and causal pathological processes in the development of HF and there is proof that miRNAs are key regulators in developing and sustaining remodeling and fibrosis (172). In fact, miR-1 was identified as downregulated in an in-vivo mouse model of induced cardiac hypertrophy via regulation of calmodulin and Mef2a—both known to be hypertrophy-associated (189). In cardiac tissue of n=34 HF patients mir-1, among other miRNAs, was upregulated compared to healthy controls (190). When assessed as a circulating biomarker, in a highly controlled setting of doxorubicin-induced heart failure in n = 56 female patients with breast cancer, miR-1 was identified as a potential prognostic biomarker to identify individuals who later on develop a reduction in EF (191). Although derived from a small cohort, these results are particularly interesting when comparing the AUC of 0.851 for miR-1 with that of cTn (0.544). These results were validated in n=49 MI patients in which miR-1 was reported to negatively correlate with ejection fraction, suggesting miR-1 as a predictive biomarker for HF onset after MI (192). Other reports were not able to validate these findings (193), which may be caused by a number of different potential reasons: (1) the detectability of cardiac miRNAs in the circulation is generally low unless measured in samples of patients suffering from severe MI (74), introducing a potential detectability bias in quantification efforts; (2) up to date there are no harmonized methods with respect to miRNA quantification, which is particularly problematic for miRNA quantification in low-RNA-yield samples such as plasma and serum in terms of comparability of the results (78); (3) Only few large-scale studies have been conducted assessing the prognostic properties of miRNAs in CVD and their common interpretation of the results is, that not single miRNAs, but instead miRNA combinations comprise prognostic potential as biomarker (194, 195).
Detailed reviews of specific cardiac-enriched (187) and other miRNAs (196) involved in HFrEF and HFpEF (172) are beyond the scope of this article and can be found elsewhere. Using miR-1 as an example, it can be seen that miRNAs are important biological players in the development of HF and are suggested as promising circulating biomarker in HF prognostication, while on the other hand validation of initial findings is pending and methodological issues remain to be solved.
lncRNAs
Further to miRNAs, long non-coding RNAs (lncRNAs) were recently investigated as regulators of protein function (197). lncRNAs are a broad group of RNA, >200 nucleotides in length (198). There is thus far no agreement on sub-classification for these RNAs. A recent review summarized different characteristic features that could be used for this purpose, comprising of their length, their relation to protein-coding genes or their relation to DNA/promoter elements (199). Unlike miRNAs, lncRNAs are mainly located within the nucleus or in mitochondria (200, 201) (Figure 4) and their biosynthesis seems to largely overlap with that of mRNAs with regards to their transcription, polyadenylation, capping, and splicing (202). For the majority of identified lncRNAs, the function remains unclear. Nuclear lncRNAs are involved in regulation of neighboring loci through transcriptional regulation or by inhibiting expression of a gene through sequestration of transcription factors (203). Conversely, other lncRNAs were shown to enhance transcription of genes. lncRNAs are more tissue-specific than protein coding genes (200) and compared with miRNAs, many more transcripts have been identified (204). They have emerged as mediators of protein translation (205). Data is available suggesting key regulatory roles of lncRNAs in cardiac and vascular tissue with respect to CVD (205). Both, miRNAs and lncRNAs are potent regulators of translation and their expression influences protein levels, while at the same time these two ncRNA species influence each other's expression (206).
Several lncRNAs are also readily detected in the circulation. This indicates the presence of protective mechanisms against RNase-mediated degradation, for which the mechanisms show overlap with those of miRNAs (198). The plasma level of Long Intergenic ncRNA Predicting CArdiac Remodeling (LIPCAR) was found to predict adverse cardiac remodeling and death in the aftermath of MI, imposing an increased risk for ischemic cardiomyopathy and HF (201). Thus, LIPCAR has potential as a circulating biomarker for HF prognostication, but has not yet been evaluated in a clinical trial. Myosin Heavy Chain Associated RNA Transcripts (MHRT) levels were found dysregulated in plasma depending on the SNP alleles (rs7140721, rs3729829, and rs3729825) in chronic HF patients. Significant difference in risk of mortality was observed based on these SNP genotypes (p < 0.001) indicating an association of these SNPs with chronic HF risk and prognosis (207). The latter might provide a way to link a single circulating molecule/biomarker with genetic risk prediction, while additional evaluation of this interesting link remains to be further explored. lncRNA H19 was discovered to be down-regulated in failing hearts from mice and was validated in pig and human hearts (208). The authors were further able to prove H19's essential HF-reversing effect. While these findings are backed-up by similar results on cardiac tissue level (209), validation of H19 as a circulating biomarker for HF prognostication is still pending. These findings indicate the potential use of lncRNAs as prognostic circulating biomarkers for CVD—similar to some miRNAs.
lncRNAs are promising RNA molecules with good characteristics as circulating biomarkers for CVD such as detectability in the circulation and distinct biological function in the heart. Their general exploration as circulating biomarkers is still in its infancy and more interesting results can be expected in the near future.
circRNAs
The first single-stranded DNA product (replicating form of DNA) that was shown to have a circular shape was described by Chandler et al. in 1964 (210), whereas the first circular RNA was described a decade later in plant viroids (211). Before circular RNAs were first described in humans in 1993, RNA species were identified, where “exons were joined accurately at consensus splice sites, but in an order different from that present in genomic DNA”(212). These “scrambled exons” were described as stable and situated in the cellular cytoplasm (213). Only during the past decade however, novel RNA analysis tools such as biochemical enrichment strategies and high-throughput deep sequencing methods have allowed for large numbers of circRNAs to be detected (214). circRNAs are a stable RNA species, endogenous to mammalian cells and proven to be expressed in a tissue- and developmental-specific context (214, 215). They can either emerge from exons or introns of primary gene transcripts (pre-mRNA) (215, 216) and are products of alternative splicing in a head-to-tail fashion known as “back-splicing” (214) (Figure 4). circRNAs are resistant to degradation by the exonuclease RNase R—a type of RNase that cleaves linear RNA. RNase R treatment can therefore be used to enrich circRNAs over their linear counterparts (217, 218). In combination with the use of divergent primers in polymerase chain reaction (PCR) amplification, this approach yields high specificity for the detection of circular transcripts.
Functionally, circRNAs appear to influence gene expression in different ways. They act as potent miRNA sponges—decreasing the inhibitory effect of miRNAs on protein synthesis (219). More recently, circRNAs were reported to be translated into proteins (220). At the same time, their expression is regulated by proteins such as RNA-binding proteins. circRNAs appear to influence gene expression by competing with splicing of their linear counterparts (173, 218, 221). circRNA expression has been mapped in different tissue types and it is now clear that they can be reliably detected in a tissue- and cell-specific manner, whilst also showing a certain degree of conservation across species (173, 222).
Bearing in mind the vast opportunities for disease detection and possibly treatment offered by miRNAs, efforts have been undertaken to evaluate circRNAs for their applicability as biomarkers and disease modifiers. A growing number of studies have reported the involvement of circRNAs across features of CVD, indicating diagnostic potential as well as potential relevance as regulators of biology (223). Sequencing data revealed more than 15,000 circRNAs present in the human heart, some in high abundance (224). A number of studies have described cardiac circRNAs to be involved in MI-related apoptosis in the myocardium (225, 226) and circRNA MICRA was identified to predict left ventricular dysfunction in MI patients (227). The results were validated in a different study where circRNA MICRA was reported to improve risk stratification of post-MI patients (228). Recently, cardiac circRNAs were assessed for their detectability in the circulation after MI in a controlled stepwise approach (74). Interestingly, none of the screened and validated circRNAs were identified as well-enough detectable in neither plasma nor serum to be used as circulating cell-free biomarkers. The findings question the validity of quantifying circulating circRNAs in cell-free body liquids using currently available technology. In fact, when studying literature regarding circulating circRNAs including the abovementioned studies regarding circRNA MICRA, an interesting fact can be observed: all circulating circRNA biomarker studies report their findings in whole blood samples—containing a large number of circulating cells instead of cell-free serum or plasma. The use of whole blood samples in the assessment of disease biomarkers yields a risk of confounding by cells such as platelets and leukocytes. Thus, the assessment of circRNAs as circulating biomarkers in CVD currently suffers from detectability problems and efforts to improve detectability are needed to further evaluate this issue.
Discussion
The identification and further exploration of biomolecules suitable as biomarkers for specific disease is a complex process, which requires numerous prerequisites to be met such as detectability in the circulation, reliable quantification methods, pathophysiologic relation to the suspected disease, and many more (Table 2). Proteins have been evaluated as circulating biomarkers for quite a long time and their quantification methods are established. They have also been analyzed for their applicability in heart failure prognosis with promising results already available. Nevertheless, validation of existing results is crucial and has only just begun in this respect. Interestingly, the added value of promising protein biomarkers on top of classic risk factors still remains limited. Therefore, the exploration of alternative biomarkers is a focus of current biomarker research. Alternatives such as genetic risk scores and also ncRNAs have caught scientists' attention for a few years. Genetic biomarkers provide a promising platform to improve mid- and long-term prognostication of HF, in particular with regards to improving individualized approaches of risk evaluation. On the other hand, the current laboratory methodology for their determination is complex, expensive, and time-consuming, limiting their implementation into clinical routine at the current stage. ncRNAs can be stably detected in the circulation and their potential as circulating biomarkers has been recognized. Several ncRNAs have been studied in the context of CVD. miRNAs, lncRNAs, and circRNAs are the most promising ncRNA species being evaluated for their biomarker potential. Several of them are expressed in a cell type- and tissue-specific manner and are involved in distinct physiological and pathological processes, raising hopes for them to evolve as helpful in diagnosis and prognosis of CVD and HF. Currently, application of ncRNAs in clinical settings is hampered by methodological issues such as lack of harmonized quantification methods and suboptimal detectability in the circulation of some transcripts.
Table 2.
Characteristics of a biomarker.
| Characteristics of a biomarker | Protein biomarkers | Genetic biomarkers | Non-coding RNA biomarkers |
|---|---|---|---|
| Pathophysiological reliability |  |
|
|
| Stability - of biomolecule in body fluids |
|
|
|
| Accessability - through routine clinical procedures |
|
|
|
| Added value - does the biomarker improve standard clinical evaluation/risk stratification? |
|
|
|
| Detectability - is the biomarker stably detectable in target phenotype? |
|
|
|
| Diagnostic and/or prognostic validation - can the biomarker differentiate affected vs. non-affected individuals? |
|
|
|
| Consensual agreement - are the quantification techniques standardized? |
|
 |
|
| Reference values - are they available and reliable? |
|
|
|
| Comparability - of results across centers |
|
|
|
| Gender specificity | |
|
|
| Tested in various ethnicities | |
|
|
Protein biomarkers are the most widely used, largely due to methodological advances in their quantification methods and consensual agreement on their detection techniques. Genetic risk scores have the advantage of gender specificity tested across various ethnicities. ncRNAs specifically lack consensual agreement on the quantification methods, reference values and thus comparability of results.
With respect to HF prognostication, currently the best data is available for NT-proBNP, which has been used in the diagnosis of HF for decades. Its value in HF prognostication has recently been recognized and validation of currently available results seems to be only a matter of time. NT-proBNP has been included into the ESC and AHA guidelines not only because it provides insights into the severity of cardiac damage, but also because current assays allow its detection even at small amounts. But importantly, there is still need for validation of sensitivity and specificity. Large-scale population-based cohort studies applying state-of-the-art laboratory methodology will give the opportunity to identify additional prognostic biomarkers such as genetic biomarkers and validate existing biomarkers for the prognosis of HF. Tissue-specificity seems to play a major role in the application of biomolecules as biomarkers when assessing single markers and it is not surprising that NT-proBNP, as one of only few heart-specific markers, ranks high in the list of biomarker candidates for HF prognostication. On the other hand, given the complex etiology of HF, up until now trials failed to identify single biomarkers in the prognostic assessment of patients with HF. This stretches the importance of the idea to identify complementary biomarkers in order to define biomarker panels as a promising way of improving prognostication of multifactorial disease entities such as HF and argues to include non-organ-specific molecules, which may provide a readout of systemic disease, such as i.e., inflammation, in the seek for biomarkers in HF.
Author Contributions
AS, TH, and CS wrote the manuscript. TZ and CS edited the manuscript, figures and tables. All authors were responsible for overall editing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. TZ is funded by the German Center for Cardiovascular Research (DZHK). CS was funded by a research fellowship by the Deutsche Forschungsgemeinschaft (DFG) (SCHU 2983/1-1 and SCHU 2983/2-1). TH is funded by Grant No. 8/18 from the Ernst und Berta Grimmke-Stiftung and the Research Promotion Fund of the Faculty of Medicine (FFM) of the University Medical Center Eppendorf, Hamburg (UKE).
References
- 1.National Heart Lung and Blood Institute What Is Heart Failure? Maryland, MD: Nhlbi; (2015). Available online at: https://www.nhlbi.nih.gov/health-topics/heart-failure. [Google Scholar]
- 2.Ambrosy AP, Fonarow GC, Butler J, Chioncel O, Greene SJ, Vaduganathan M, et al. The global health and economic burden of hospitalizations for heart failure: lessons learned from hospitalized heart failure registries. J Am Coll Cardiol. (2014) 63:1123–33. 10.1016/j.jacc.2013.11.053 [DOI] [PubMed] [Google Scholar]
- 3.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics−2014 update: a report from the American heart association. Circulation. (2014) 129:e28–292. 10.1161/01.cir.0000441139.02102.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ceia F, Fonseca C, Mota T, Morais H, Matias F, de Sousa A, et al. Prevalence of chronic heart failure in Southwestern Europe: the EPICA study. Eur Heart J. (2002) 4:531–9. 10.1016/S1388-9842(02)00034-X [DOI] [PubMed] [Google Scholar]
- 5.Townsend N, Wickramasinghe K, Bhatnagar P, Nichols MS, Leal J, et al. Coronary Heart Disease Statistics (2012 Edition). London: British Heart Foundation; (2012). Available online at: https://www.bhf.org.uk/informationsupport/publications/statistics/coronary-heart-disease-statistics-2012 (accessed July 26, 2020). [Google Scholar]
- 6.Ponikowski P, Anker SD, Alhabib KF, Cowie MR, Force TL, Hu S, et al. Heart failure: preventing disease and death worldwide. ESC Heart Fail. (2014) 1:4–25. 10.1002/ehf2.12005 [DOI] [PubMed] [Google Scholar]
- 7.Jackson G, Gibbs CR, Davies MK, Lip GY. ABC of heart failure Pathophysiology. BMJ. (2000) 320:167–70. 10.1136/bmj.320.7228.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawkins NM, Petrie MC, Jhund PS, Chalmers GW, Dunn FG, Mcmurray JJ. Heart failure and chronic obstructive pulmonary disease: diagnostic pitfalls and epidemiology. Eur J Heart Fail. (2009) 11:130–9. 10.1093/eurjhf/hfn013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas JT, Kelly RF, Thomas SJ, Stamos TD, Albasha K, Parrillo JE, et al. Utility of history, physical examination, electrocardiogram, and chest radiograph for differentiating normal from decreased systolic function in patients with heart failure. Am J Med. (2002) 112:437–45. 10.1016/S0002-9343(02)01048-3 [DOI] [PubMed] [Google Scholar]
- 10.Kirkpatrick JN, Vannan MA, Narula J, Lang RM. Echocardiography in heart failure: applications, utility, and new horizons. J Am Coll Cardiol. (2007) 50:381–96. 10.1016/j.jacc.2007.03.048 [DOI] [PubMed] [Google Scholar]
- 11.Nagueh SF, Bhatt R, Vivo RP, Krim SR, Sarvari SI, Russell K, et al. Echocardiographic evaluation of hemodynamics in patients with decompensated systolic heart failure. Circ Cardiovasc Imaging. (2011) 4:220–7. 10.1161/CIRCIMAGING.111.963496 [DOI] [PubMed] [Google Scholar]
- 12.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American society of echocardiography and the European Association of cardiovascular imaging. J Am Soc Echocardiogr. (2015) 28:1–39.e14. 10.1016/j.echo.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez JA, Kramer CM. Role of imaging techniques for diagnosis, prognosis and management of heart failure patients: cardiac magnetic resonance. Curr Heart Fail Rep. (2015) 12:276–83. 10.1007/s11897-015-0261-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hundley WG, Bluemke DA, Finn JP, Flamm SD, Fogel MA, Friedrich MG, et al. ACCF/ACR/AHA/NASCI/SCMR 2010 expert consensus document on cardiovascular magnetic resonance: a report of the American College of cardiology foundation task force on expert consensus documents. J Am Coll Cardiol. (2010) 55:2614–62. 10.1016/j.jacc.2009.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kilner PJ, Geva T, Kaemmerer H, Trindade PT, Schwitter J, Webb GD. Recommendations for cardiovascular magnetic resonance in adults with congenital heart disease from the respective working groups of the European society of cardiology. Eur Heart J. (2010) 31:794–805. 10.1093/eurheartj/ehp586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur J Heart Fail. (2016) 18:891–975. 10.1002/ejhf.592 [DOI] [PubMed] [Google Scholar]
- 17.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Colvin MM, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the management of heart failure: a report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. Circulation. (2017) 136:e137–e161. 10.1161/CIR.0000000000000509 [DOI] [PubMed] [Google Scholar]
- 18.Maisel AS, Duran JM, Wettersten N. Natriuretic peptides in heart failure: atrial and B-type natriuretic peptides. Heart Fail Clin. (2018) 14:13–25. 10.1016/j.hfc.2017.08.002 [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa Y, Nishikimi T, Kuwahara K. Atrial and brain natriuretic peptides: hormones secreted from the heart. Peptides. (2019) 111:18–25. 10.1016/j.peptides.2018.05.012 [DOI] [PubMed] [Google Scholar]
- 20.Nishikimi T, Maeda N, Matsuoka H. The role of natriuretic peptides in cardioprotection. Cardiovasc Res. (2006) 69:318–28. 10.1016/j.cardiores.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 21.Nishikimi T, Kuwahara K, Nakao K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J Cardiol. (2011) 57:131–40. 10.1016/j.jjcc.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 22.Tokola H, Hautala N, Marttila M, Magga J, Pikkarainen S, Kerkelä R, et al. Mechanical load-induced alterations in B-type natriuretic peptide gene expression. Canad J Physiol Pharmacol. (2001) 79:646–53. 10.1139/y01-031 [DOI] [PubMed] [Google Scholar]
- 23.Daniels LB, Maisel AS. Natriuretic peptides. J Am College Cardiol. (2007) 50:2357–68. 10.1016/j.jacc.2007.09.021 [DOI] [PubMed] [Google Scholar]
- 24.Potter LR. Natriuretic peptide metabolism, clearance and degradation. FEBS J. (2011) 278:1808–17. 10.1111/j.1742-4658.2011.08082.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abassi Z, Karram T, Ellaham S, Winaver J, Hoffman A. Implications of the natriuretic peptide system in the pathogenesis of heart failure: diagnostic and therapeutic importance. Pharmacol Ther. (2004) 102:223–41. 10.1016/j.pharmthera.2004.04.004 [DOI] [PubMed] [Google Scholar]
- 26.Baba M, Yoshida K, Ieda M. Clinical applications of natriuretic peptides in heart failure and atrial fibrillation. Int J Mol Sci. (2019) 20:2824 10.3390/ijms20112824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masson S, Latini R, Anand IS, Barlera S, Angelici L, Vago T. Prognostic value of changes in N-terminal pro-brain natriuretic peptide in Val-HeFT (valsartan heart failure trial). J Am College Cardiol. (2008) 52:997–1003. 10.1016/j.jacc.2008.04.069 [DOI] [PubMed] [Google Scholar]
- 28.Bettencourt P, Azevedo A, Pimenta J, Friões F, Ferreira S, Ferreira A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation. (2004) 110:2168–74. 10.1161/01.CIR.0000144310.04433.BE [DOI] [PubMed] [Google Scholar]
- 29.Hartmann F, Packer M, Coats AJS, Fowler MB, Krum H, Mohacsi P, et al. Prognostic impact of plasma N-terminal pro-brain natriuretic peptide in severe chronic congestive heart failure. Circulation. (2004) 110:1780–6. 10.1161/01.CIR.0000143059.68996.A7 [DOI] [PubMed] [Google Scholar]
- 30.Logeart D, Thabut G, Jourdain P, Chavelas C, Beyne P, Beauvais F, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol. (2004) 43:635–41. 10.1016/j.jacc.2003.09.044 [DOI] [PubMed] [Google Scholar]
- 31.Stolfo D, Stenner E, Merlo M, Porto AG, Moras C, Barbati G, et al. Prognostic impact of BNP variations in patients admitted for acute decompensated heart failure with in-hospital worsening renal function. Heart Lung Circ. (2017) 26:226–34. 10.1016/j.hlc.2016.06.1205 [DOI] [PubMed] [Google Scholar]
- 32.Zile MR, Claggett BL, Prescott MF, Mcmurray JJV, Packer M, Rouleau JL, et al. Prognostic implications of changes in N-terminal pro-B-type natriuretic peptide in patients with heart failure. J Am Coll Cardiol. (2016) 68:2425–36. 10.1016/j.jacc.2016.09.931 [DOI] [PubMed] [Google Scholar]
- 33.Myhre PL, Vaduganathan M, Claggett B, Packer M, Desai AS, Rouleau JL, et al. B-type natriuretic peptide during treatment with sacubitril/valsartan. the PARADIGM-HF trial. J Am Coll Cardiol. (2019) 73:1264–72. 10.1016/j.jacc.2019.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noveanu M, Breidthardt T, Potocki M, Reichlin T, Twerenbold R, Uthoff H, et al. Direct comparison of serial B-type natriuretic peptide and NT-proBNP levels for prediction of short- and long-term outcome in acute decompensated heart failure. Critical Care. (2011) 15:R1 10.1186/cc9398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiner RB, Baggish AL, Chen-Tournoux A, Marshall JE, Gaggin HK, Bhardwaj A, et al. Improvement in structural and functional echocardiographic parameters during chronic heart failure therapy guided by natriuretic peptides: mechanistic insights from the ProBNP Outpatient Tailored Chronic Heart Failure (PROTECT) study. Eur J Heart Fail. (2013) 15:342–51. 10.1093/eurjhf/hfs180 [DOI] [PubMed] [Google Scholar]
- 36.Stienen S, Salah K, Moons AH, Bakx AL, Van Pol P, Kortz R, et al. NT-proBNP (N-terminal pro-B-type natriuretic peptide)-guided therapy in acute decompensated heart failure: PRIMA II randomized controlled trial (Can NT-ProBNP-guided therapy during hospital admission for acute decompensated heart failure reduce mortality and readmissions?). Circulation. (2018) 137:1671–83. 10.1161/CIRCULATIONAHA.117.029882 [DOI] [PubMed] [Google Scholar]
- 37.Edwards BS, Zimmerman RS, Schwab TR, Heublein DM, Burnett JC., Jr Atrial stretch, not pressure, is the principal determinant controlling the acute release of atrial natriuretic factor. Circ Res. (1988) 62:191–5. [DOI] [PubMed] [Google Scholar]
- 38.Marin-Grez M, Fleming JT, Steinhausen M. Atrial natriuretic peptide causes pre-glomerular vasodilatation and post-glomerular vasoconstriction in rat kidney. Nature. (1986) 324:473–6. [DOI] [PubMed] [Google Scholar]
- 39.Cody RJ, Atlas SA, Laragh JH, Kubo SH, Covit AB, Ryman KS, et al. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J Clin Invest. (1986) 78:1362–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goetze JP, Hansen LH, Terzic D, Zois NE, Albrethsen J, Timm A, et al. Atrial natriuretic peptides in plasma. Clin Chim Acta. (2015) 443:25–8. 10.1016/j.cca.2014.08.017 [DOI] [PubMed] [Google Scholar]
- 41.Lee CY, Burnett JC., Jr Natriuretic peptides and therapeutic applications. Heart Fail Rev. (2007) 12:131–42. 10.1007/s10741-007-9016-3 [DOI] [PubMed] [Google Scholar]
- 42.Yandle TG, Richards AM, Nicholls MG, Cuneo R, Espiner EA, Livesey JH. Metabolic clearance rate and plasma half life of alpha-human atrial natriuretic peptide in man. Life Sci. (1986) 38:1827–33. [DOI] [PubMed] [Google Scholar]
- 43.Nakao K, Sugawara A, Morii N, Sakamoto M, Yamada T, Itoh H, et al. The pharmacokinetics of α-human atrial natriuretic polypeptide in healthy subjects. Euro J of Clin. (1986) Pharmacol. 31:101–3. [DOI] [PubMed] [Google Scholar]
- 44.Morgenthaler NG, Struck J, Thomas B, Bergmann A. Immunoluminometric assay for the midregion of pro-atrial natriuretic peptide in human plasma. Clin Chem. (2004) 50:234–6. 10.1373/clinchem.2003.021204 [DOI] [PubMed] [Google Scholar]
- 45.Seronde M-F, Gayat E, Logeart D, Lassus J, Laribi S, Boukef R, et al. Comparison of the diagnostic and prognostic values of B-type and atrial-type natriuretic peptides in acute heart failure. Int J Cardiol. (2013) 168:3404–11. 10.1016/j.ijcard.2013.04.164 [DOI] [PubMed] [Google Scholar]
- 46.Zelenak C, Chavanon M-L, Tahirovic E, Trippel TD, Tscholl V, Stroux A, et al. Early NT-proBNP and MR-proANP associated with QoL 1 year after acutely decompensated heart failure: secondary analysis from the MOLITOR trial. Biomark Med. (2019) 13:1493–507. 10.2217/bmm-2019-0083 [DOI] [PubMed] [Google Scholar]
- 47.Masson S, Latini R, Carbonieri E, Moretti L, Rossi MG, Ciricugno S, et al. The predictive value of stable precursor fragments of vasoactive peptides in patients with chronic heart failure: data from the GISSI-heart failure (GISSI-HF) trial. Euro J Heart Failure. (2010) 12, 338–47. 10.1093/eurjhf/hfp206 [DOI] [PubMed] [Google Scholar]
- 48.Tzikas S, Keller T, Ojeda FM, Zeller T, Wild PS, Lubos E, et al. MR-proANP and MR-proADM for risk stratification of patients with acute chest pain. Heart. (2013) 99:388–95. 10.1136/heartjnl-2012-302956 [DOI] [PubMed] [Google Scholar]
- 49.Arrigo M, Truong QA, Szymonifka J, Rivas-Lasarte M, Tolppanen H, Sadoune M, et al. Mid-regional pro-atrial natriuretic peptide to predict clinical course in heart failure patients undergoing cardiac resynchronization therapy. Europace. (2017) 19:1848–54. 10.1093/europace/euw305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parmacek MS, Solaro RJ. Biology of the troponin complex in cardiac myocytes. Prog Cardiovasc Dis. (2004) 47:159–76. 10.1016/j.pcad.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 51.Felker GM, Hasselblad V, Tang WHW, Hernandez AF, Armstrong PW, Fonarow GC, et al. Troponin I in acute decompensated heart failure: insights from the ASCEND-HF study. Eur J Heart Fail. (2012) 14:1257–64. 10.1093/eurjhf/hfs110 [DOI] [PubMed] [Google Scholar]
- 52.Gravning J, Askevold ET, Nymo SH, Ueland T, Wikstrand J, Mcmurray JJV, et al. Prognostic effect of high-sensitive troponin t assessment in elderly patients with chronic heart failure. Circulation. (2014) 7:96–103. 10.1161/CIRCHEARTFAILURE.113.000450 [DOI] [PubMed] [Google Scholar]
- 53.Felker GM, Mentz RJ, Teerlink JR, Voors AA, Pang PS, Ponikowski P, et al. Serial high sensitivity cardiac troponin T measurement in acute heart failure: insights from the RELAX-AHF study. Eur J Heart Fail. (2015) 17:1262–70. 10.1002/ejhf.341 [DOI] [PubMed] [Google Scholar]
- 54.Welsh P, Kou L, Yu C, Anand I, Van Veldhuisen DJ, Maggioni AP, et al. Prognostic importance of emerging cardiac, inflammatory, and renal biomarkers in chronic heart failure patients with reduced ejection fraction and anaemia: RED-HF study. Eur J Heart Fail. (2018) 20:268–77. 10.1002/ejhf.988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rørth R, Jhund PS, Kristensen SL, Desai AS, Køber L, Rouleau JL, et al. The prognostic value of troponin T and N-terminal pro B-type natriuretic peptide, alone and in combination, in heart failure patients with and without diabetes. Eur J Heart Fail. (2019) 21:40–9. 10.1002/ejhf.1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ford I, Shah AS, Zhang R, Mcallister DA, Strachan FE, Caslake M, et al. High-sensitivity cardiac troponin, statin therapy, and risk of coronary heart disease. J Am Coll Cardiol. (2016) 68:2719–28. 10.1016/j.jacc.2016.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Omland T, Røsj, ø H, Giannitsis E, Agewall S. Troponins in heart failure. Clin Chim Acta. (2015) 443:78–84. 10.1016/j.cca.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 58.Ackermann MA, Kontrogianni-Konstantopoulos A. Myosin binding protein-C: a regulator of actomyosin interaction in striated muscle. J Biomed Biotechnol. (2011) 2011:636403 10.1155/2011/636403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finley NL, Cuperman TI. Cardiac myosin binding protein-C: a structurally dynamic regulator of myocardial contractility. Pflügers Archiv. (2014) 466:433–8. 10.1007/s00424-014-1451-0 [DOI] [PubMed] [Google Scholar]
- 60.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, et al. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci USA. (2006) 103:16918–23. 10.1073/pnas.0607069103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin B, Govindan S, Lee K, Zhao P, Han R, Runte KE, et al. Cardiac myosin binding protein-c plays no regulatory role in skeletal muscle structure and function. PLoS ONE. (2013) 8:e69671 10.1371/journal.pone.0069671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ababou A, Gautel M, Pfuhl M. Dissecting the N-terminal myosin binding site of human cardiac myosin-binding protein C. Structure and myosin binding of domain C2. J Biol Chem. (2007) 282:9204–15. 10.1074/jbc.M610899200 [DOI] [PubMed] [Google Scholar]
- 63.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, et al. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ Res. (2011) 109:141–50. 10.1161/CIRCRESAHA.111.242560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shaffer JF, Kensler RW, Harris SP. (2009). The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J Biol Chem. (2011) 284:12318–27. 10.1074/jbc.M808850200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Govindan S, Sarkey J, Ji X, Sundaresan NR, Gupta MP, De Tombe PP, et al. Pathogenic properties of the N-terminal region of cardiac myosin binding protein-C in vitro. J Muscle Res Cell Motil. (2012) 33:17–30. 10.1007/s10974-012-9292-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Govindan S, Mcelligott A, Muthusamy S, Nair N, Barefield D, Martin JL, et al. Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction. J Mol Cell Cardiol. (2012) 52:154–64. 10.1016/j.yjmcc.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Razzaque MA, Gupta M, Osinska H, Gulick J, Blaxall BC, Robbins J. An endogenously produced fragment of cardiac myosin-binding protein C is pathogenic and can lead to heart failure. Circ Res. (2013) 113:553–61. 10.1161/CIRCRESAHA.113.301225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Govindan S, Kuster DW, Lin B, Kahn DJ, Jeske WP, Walenga JM, et al. Increase in cardiac myosin binding protein-C plasma levels is a sensitive and cardiac-specific biomarker of myocardial infarction. Am J Cardiovasc Dis. (2013) 3:60–70. [PMC free article] [PubMed] [Google Scholar]
- 69.El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, et al. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J Mol Cell Cardiol. (2007) 43:223–9. 10.1016/j.yjmcc.2007.05.003 [DOI] [PubMed] [Google Scholar]
- 70.Jacques AM, Copeland O, Messer AE, Gallon CE, King K, Mckenna WJ, et al. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol. (2008) 45:209–16. 10.1016/j.yjmcc.2008.05.020 [DOI] [PubMed] [Google Scholar]
- 71.Zaremba R, Merkus D, Hamdani N, Lamers JM, Paulus WJ, Dos Remedios C, et al. Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteomics Clin Appl. (2007) 1:1285–90. 10.1002/prca.200600891 [DOI] [PubMed] [Google Scholar]
- 72.Copeland O, Sadayappan S, Messer AE, Steinen GJ, Van Der Velden J, Marston SB. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J Mol Cell Cardiol. (2010) 9:1003–11. 10.1016/j.yjmcc.2010.09.007 [DOI] [PubMed] [Google Scholar]
- 73.Hamdani N, Borbély A, Veenstra SP, Kooij V, Vrydag W, Zaremba R, et al. More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared to ischemic heart disease. J Muscle Res Cell Motil. (2010) 31:289–301. 10.1007/s10974-010-9231-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schulte C, Barwari T, Joshi A, Theofilatos K, Zampetaki A, Barallobre-Barreiro J, et al. Comparative analysis of circulating noncoding RNAs versus protein biomarkers in the detection of myocardial injury. Circ Res. (2019) 125:328–40. 10.1161/CIRCRESAHA.119.314937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacquet S, Yin X, Sicard P, Clark J, Kanaganayagam GS, Mayr M, et al. Identification of cardiac myosin-binding protein C as a candidate biomarker of myocardial infarction by proteomics analysis. Mol Cell Proteomics. (2009) 8:2687–99. 10.1074/mcp.M900176-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaier TE, Twerenbold R, Puelacher C, Marjot J, Imambaccus N, Boeddinghaus J, et al. Direct comparison of cardiac myosin-binding protein C with cardiac troponins for the early diagnosis of acute myocardial infarction. Circulation. (2017) 136:1495–508. 10.1161/CIRCULATIONAHA.117.028084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baker JO, Tyther R, Liebetrau C, Clark J, Howarth R, Patterson T, et al. Cardiac myosin-binding protein C: a potential early biomarker of myocardial injury. Basic Res Cardiol. (2015) 110:23 10.1007/s00395-015-0478-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schulte C, Barwari T, Joshi A, Zeller T, Mayr M. Noncoding RNAs versus protein biomarkers in cardiovascular disease. Trends Mol Med. (2020) 26:583–96. 10.1016/j.molmed.2020.02.001 [DOI] [PubMed] [Google Scholar]
- 79.El Amrousy D, Hodeib H, Suliman G, Hablas N, Salama ER, Esam A. Diagnostic and prognostic value of plasma levels of cardiac myosin binding protein-C as a novel biomarker in heart failure. Pediatric Cardiol. 38:418–24. 10.1007/s00246-016-1532-2 [DOI] [PubMed] [Google Scholar]
- 80.Anand A, Chin C, Shah ASV, Kwiecinski J, Vesey A, Cowell J, et al. Cardiac myosin-binding protein C is a novel marker of myocardial injury and fibrosis in aortic stenosis. Heart. (2018) 104:1101–8. 10.1136/heartjnl-2017-312257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Piek A, Du W, De Boer RA, Silljé HHW. Novel heart failure biomarkers: why do we fail to exploit their potential? Crit Rev Clin Lab Sci. (2018) 55:246–63. 10.1080/10408363.2018.1460576 [DOI] [PubMed] [Google Scholar]
- 82.Otaki Y, Watanabe T, Kubota I. Heart-type fatty acid-binding protein in cardiovascular disease: a systemic review. Clin Chim Acta. (2017) 474:44–53. 10.1016/j.cca.2017.09.007 [DOI] [PubMed] [Google Scholar]
- 83.Okamoto F, Sohmiya K, Ohkaru Y, Kawamura K, Asayama K, Kimura H, et al. Human heart-type cytoplasmic fatty acid-binding protein (H-FABP) for the diagnosis of acute myocardial infarction. clinical evaluation of H-FABP in comparison with myoglobin and creatine kinase isoenzyme MB. Clin Chem Lab Med. (2000) 38:231–8. 10.1515/CCLM.2000.034 [DOI] [PubMed] [Google Scholar]
- 84.Aartsen WM, Pelsers MM, Hermens WT, Glatz JF, Daemen MJ, Smits JF. Heart fatty acid binding protein and cardiac troponin T plasma concentrations as markers for myocardial infarction after coronary artery ligation in mice. Pflugers Arch. (2000) 439:416–22. 10.1007/s004249900180 [DOI] [PubMed] [Google Scholar]
- 85.Kleine AH, Glatz JF, Van Nieuwenhoven FA, Van Der Vusse GJ. Release of heart fatty acid-binding protein into plasma after acute myocardial infarction in man. Mol Cell Biochem. (1992) 116:155–62. 10.1007/BF01270583 [DOI] [PubMed] [Google Scholar]
- 86.Ishii J, Ozaki Y, Lu J, Kitagawa F, Kuno T, Nakano T, et al. Prognostic value of serum concentration of heart-type fatty acid-binding protein relative to cardiac troponin T on admission in the early hours of acute coronary syndrome. Clin Chem. (2005) 51:1397–404. 10.1373/clinchem.2004.047662 [DOI] [PubMed] [Google Scholar]
- 87.Kazimierczyk E, Kazimierczyk R, Harasim-Symbor E, Kaminski K, Sobkowicz B, Chabowski A, et al. Persistently elevated plasma heart-type fatty acid binding protein concentration is related with poor outcome in acute decompensated heart failure patients. Clin Chim Acta. (2018) 487:48–53. 10.1016/j.cca.2018.09.004 [DOI] [PubMed] [Google Scholar]
- 88.Niizeki T, Takeishi Y, Arimoto T, Nozaki N, Hirono O, Watanabe T, et al. Persistently increased serum concentration of heart-type fatty acid-binding protein predicts adverse clinical outcomes in patients with chronic heart failure. Circ J. (2008) 72:109–14. 10.1253/circj.72.109 [DOI] [PubMed] [Google Scholar]
- 89.Niizeki T, Takeishi Y, Arimoto T, Takahashi T, Okuyama H, Takabatake N, et al. Combination of heart-type fatty acid binding protein and brain natriuretic peptide can reliably risk stratify patients hospitalized for chronic heart failure. Circ J. (2005) 69:922–7. 10.1253/circj.69.922 [DOI] [PubMed] [Google Scholar]
- 90.Hoffmann U, Espeter F, Weiß C, Ahmad-Nejad P, Lang S, Brueckmann M, et al. Ischemic biomarker heart-type fatty acid binding protein (hFABP) in acute heart failure - diagnostic and prognostic insights compared to NT-proBNP and troponin I. BMC Cardiovasc Disord. (2015) 15:50 10.1186/s12872-015-0026-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kutsuzawa D, Arimoto T, Watanabe T, Shishido T, Miyamoto T, Miyashita T, et al. Ongoing myocardial damage in patients with heart failure and preserved ejection fraction. J Cardiol. (2012) 60:454–61. 10.1016/j.jjcc.2012.06.006 [DOI] [PubMed] [Google Scholar]
- 92.Dumic J, Dabelic S, Flögel M. Galectin-3: an open-ended story. Biochim Biophys Acta. (2006) 1760:616–35. 10.1016/j.bbagen.2005.12.020 [DOI] [PubMed] [Google Scholar]
- 93.Kim H, Lee J, Hyun JW, Park JW, Joo HG, Shin T. Expression and immunohistochemical localization of galectin-3 in various mouse tissues. Cell Biol Int. (2007) 31:655–62. 10.1016/j.cellbi.2006.11.036 [DOI] [PubMed] [Google Scholar]
- 94.Yang RY, Hsu DK, Liu FT. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci USA. (1996) 93:6737–42. 10.1073/pnas.93.13.6737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hsu DK, Hammes SR, Kuwabara I, Greene WC, Liu FT. Human T lymphotropic virus-I infection of human T lymphocytes induces expression of the beta-galactoside-binding lectin, galectin-3. Am J Pathol. (1996) 148:1661–70. [PMC free article] [PubMed] [Google Scholar]
- 96.Sundblad V, Croci DO, Rabinovich GA. Regulated expression of galectin-3, a multifunctional glycan-binding protein, in haematopoietic and non-haematopoietic tissues. Histol Histopathol. (2011) 26:247–65. 10.14670/HH-26.247 [DOI] [PubMed] [Google Scholar]
- 97.Kim K, Mayer EP, Nachtigal M. Galectin-3 expression in macrophages is signaled by Ras/MAP kinase pathway and up-regulated by modified lipoproteins. Biochim Biophys Acta. (2003) 1641:13–23. 10.1016/S0167-4889(03)00045-4 [DOI] [PubMed] [Google Scholar]
- 98.Sharma UC, Pokharel S, Van Brakel TJ, Van Berlo JH, Cleutjens JP, Schroen B, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. (2004) 110:3121–8. 10.1161/01.CIR.0000147181.65298.4D [DOI] [PubMed] [Google Scholar]
- 99.Liu YH, D'ambrosio M, Liao TD, Peng H, Rhaleb NE, Sharma U, et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol. (2009) 296:H404–12. 10.1152/ajpheart.00747.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lin YH, Lin LY, Wu YW, Chien KL, Lee CM, Hsu RB, et al. The relationship between serum galectin-3 and serum markers of cardiac extracellular matrix turnover in heart failure patients. Clin Chim Acta. (2009) 409:96–9. 10.1016/j.cca.2009.09.001 [DOI] [PubMed] [Google Scholar]
- 101.Gullestad L, Ueland T, Kjekshus J, Nymo SH, Hulthe J, Muntendam P, et al. The predictive value of galectin-3 for mortality and cardiovascular events in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Am Heart J. (2012) 164:878–83. 10.1016/j.ahj.2012.08.021 [DOI] [PubMed] [Google Scholar]
- 102.Felker GM, Fiuzat M, Shaw LK, Clare R, Whellan DJ, Bettari L, et al. Galectin-3 in ambulatory patients with heart failure. Circulation. (2012) 5:72–8. 10.1161/CIRCHEARTFAILURE.111.963637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lok DJA, Van Der Meer P, De La Porte PWB-A, Lipsic E, Van Wijngaarden J, Hillege HL, et al. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: data from the DEAL-HF study. Clin Res Cardiol. (2010) 99:323–8. 10.1007/s00392-010-0125-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Velde ARVD, Gullestad L, Ueland T, Aukrust P, Guo Y, Adourian A, et al. Prognostic value of changes in galectin-3 levels over time in patients with heart failure. Circulation. (2013) 6:219–26. 10.1161/CIRCHEARTFAILURE.112.000129 [DOI] [PubMed] [Google Scholar]
- 105.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-β superfamily. Proc Natl Acad Sci USA. (1997) 94:11514–9. 10.1073/pnas.94.21.11514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hromas R, Hufford M, Sutton J, Xu D, Li Y, Lu L. PLAB, a novel placental bone morphogenetic protein. Biochim Biophys Acta. (1997) 1354:40–4. 10.1016/S0167-4781(97)00122-X [DOI] [PubMed] [Google Scholar]
- 107.Lawton LN, Bonaldo MDF, Jelenc PC, Qiu L, Baumes SA, Marcelino RA, et al. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene. (1997) 203:17–26. 10.1016/S0378-1119(97)00485-X [DOI] [PubMed] [Google Scholar]
- 108.Bauskin AR, Zhang HP, Fairlie WD, He XY, Russell PK, Moore AG, et al. The propeptide of macrophage inhibitory cytokine (MIC-1), a TGF-beta superfamily member, acts as a quality control determinant for correctly folded MIC-1. EMBO J. (2000) 19:2212–20. 10.1093/emboj/19.10.2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schlittenhardt D, Schober A, Strelau J, Bonaterra GA, Schmiedt W, Unsicker K, et al. Involvement of growth differentiation factor-15/macrophage inhibitory cytokine-1 (GDF-15/MIC-1) in oxLDL-induced apoptosis of human macrophages in vitro and in arteriosclerotic lesions. Cell Tissue Res. (2004) 318:325–33. 10.1007/s00441-004-0986-3 [DOI] [PubMed] [Google Scholar]
- 110.Bermúdez B, López S, Pacheco YM, Villar J, Muriana FJ, Hoheisel JD, et al. Influence of postprandial triglyceride-rich lipoproteins on lipid-mediated gene expression in smooth muscle cells of the human coronary artery. Cardiovasc Res. (2008) 79:294–303. 10.1093/cvr/cvn082 [DOI] [PubMed] [Google Scholar]
- 111.Ferrari N, Pfeffer U, Dell'eva R, Ambrosini C, Noonan DM, Albini A. The transforming growth factor-beta family members bone morphogenetic protein-2 and macrophage inhibitory cytokine-1 as mediators of the antiangiogenic activity of N-(4-hydroxyphenyl)retinamide. Clin Cancer Res. (2005) 11:4610–9. 10.1158/1078-0432.CCR-04-2210 [DOI] [PubMed] [Google Scholar]
- 112.Secchiero P, Corallini F, Gonelli A, Dell'eva R, Vitale M, Capitani S, et al. Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res. (2007) 100:61–9. 10.1161/01.RES.0000253975.76198.ff [DOI] [PubMed] [Google Scholar]
- 113.Nickel N, Jonigk D, Kempf T, Bockmeyer CL, Maegel L, Rische J, et al. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir Res. (2011) 12:62 10.1186/1465-9921-12-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ding Q, Mracek T, Gonzalez-Muniesa P, Kos K, Wilding J, Trayhurn P, et al. Identification of macrophage inhibitory cytokine-1 in adipose tissue and its secretion as an adipokine by human adipocytes. Endocrinology. (2009) 150:1688–96. 10.1210/en.2008-0952 [DOI] [PubMed] [Google Scholar]
- 115.Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. (2006) 98:351–60. 10.1161/01.RES.0000202805.73038.48 [DOI] [PubMed] [Google Scholar]
- 116.Clerk A, Kemp TJ, Zoumpoulidou G, Sugden PH. Cardiac myocyte gene expression profiling during H2O2-induced apoptosis. Physiol Genom. (2007) 29:118–27. 10.1152/physiolgenomics.00168.2006 [DOI] [PubMed] [Google Scholar]
- 117.Frank D, Kuhn C, Brors B, Hanselmann C, Lüdde M, Katus HA, et al. Gene expression pattern in biomechanically stretched cardiomyocytes: evidence for a stretch-specific gene program. Hypertension. (2008) 51:309–18. 10.1161/HYPERTENSIONAHA.107.098046 [DOI] [PubMed] [Google Scholar]
- 118.De Jager SC, Bermúdez B, Bot I, Koenen RR, Bot M, Kavelaars A, et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med. (2011) 208:217–25. 10.1084/jem.20100370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Anand IS, Kempf T, Rector TS, Tapken H, Allhoff T, Jantzen F. Serial measurement of growth-differentiation factor-15 in heart failure: relation to disease severity and prognosis in the valsartan heart failure trial. Circulation. (2010) 122:1387–95. 10.1161/CIRCULATIONAHA.109.928846 [DOI] [PubMed] [Google Scholar]
- 120.Kempf T, Von Haehling S, Peter T, Allhoff T, Cicoira M, Doehner W, et al. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J Am Coll Cardiol. (2007) 50:1054–60. 10.1016/j.jacc.2007.04.091 [DOI] [PubMed] [Google Scholar]
- 121.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, Mcclanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. 10.1016/j.immuni.2005.09.015 [DOI] [PubMed] [Google Scholar]
- 122.Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. (2012) 287:6941–8. 10.1074/jbc.M111.298703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pascual-Figal DA, Januzzi JL. The biology of ST2: the international ST2 consensus panel. Am J Cardiol. (2015) 115:3B−7. 10.1016/j.amjcard.2015.01.034 [DOI] [PubMed] [Google Scholar]
- 124.Tang WHW, Wu Y, Grodin JL, Hsu AP, Hernandez AF, Butler J, et al. Prognostic value of baseline and changes in circulating soluble ST2 levels and the effects of nesiritide in acute decompensated heart failure. JACC Heart Fail. (2016) 4:68–77. 10.1016/j.jchf.2015.07.015 [DOI] [PubMed] [Google Scholar]
- 125.Skali H, Gerwien R, Meyer TE, Snider JV, Solomon SD, Stolen CM. Soluble ST2 and risk of arrhythmias, heart failure, or death in patients with mildly symptomatic heart failure: results from MADIT-CRT. J Cardiovasc Transl Res. (2016) 9:421–8. 10.1007/s12265-016-9713-1 [DOI] [PubMed] [Google Scholar]
- 126.Anand IS, Rector TS, Kuskowski M, Snider J, Cohn JN. Prognostic value of soluble ST2 in the valsartan heart failure trial. Circ Heart Fail. (2014) 7:418–26. 10.1161/CIRCHEARTFAILURE.113.001036 [DOI] [PubMed] [Google Scholar]
- 127.Liu Z, Ma C, Gu J, Yu M. Potential biomarkers of acute myocardial infarction based on weighted gene co-expression network analysis. Biomed Eng Online. (2019) 18:9 10.1186/s12938-019-0625-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. (2020) 11:163 10.1038/s41467-019-13690-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Myers RH, Kiely DK, Cupples LA, Kannel WB. Parental history is an independent risk factor for coronary artery disease: the Framingham study. Am Heart J. (1990) 120:963–9. 10.1016/0002-8703(90)90216-K [DOI] [PubMed] [Google Scholar]
- 130.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. (2007) 357:443–53. 10.1056/NEJMoa072366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mcpherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. (2007) 316:1488–91. 10.1126/science.1142447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. (2007) 316:1491–3. 10.1126/science.1142842 [DOI] [PubMed] [Google Scholar]
- 133.Consortium WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. 10.1038/nature05911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. (2016) 7:12429 10.1038/ncomms12429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. (2018) 114:1241–57. 10.1093/cvr/cvy084 [DOI] [PubMed] [Google Scholar]
- 136.Poetsch MS, Strano A, Guan K. Role of leptin in cardiovascular diseases. Front Endocrinol. (2020) 11:354 10.3389/fendo.2020.00354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kaur J, Mattu HS, Chatha K, Randeva HS. Chemerin in human cardiovascular disease. Vascul Pharmacol. (2018) 110:1–6. 10.1016/j.vph.2018.06.018 [DOI] [PubMed] [Google Scholar]
- 138.Silva CP, Kamens HM. Cigarette smoke-induced alterations in blood: a review of research on DNA methylation and gene expression. Exp Clin Psychopharmacol. (2020). 10.1037/pha0000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Haase T, Müller C, Krause J, Röthemeier C, Stenzig J, Kunze S, et al. Novel DNA methylation sites influence GPR15 expression in relation to smoking. Biomolecules. (2018) 8:74 10.3390/biom8030074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schunkert H, Von Scheidt M, Kessler T, Stiller B, Zeng L, Vilne B. Genetics of coronary artery disease in the light of genome-wide association studies. Clin Res Cardiol. (2018) 107:2–9. 10.1007/s00392-018-1324-1 [DOI] [PubMed] [Google Scholar]
- 141.Van Der Ende MY, Said MA, Van Veldhuisen DJ, Verweij N, Van Der Harst P. Genome-wide studies of heart failure and endophenotypes: lessons learned and future directions. Cardiovasc Res. (2018) 114:1209–25. 10.1093/cvr/cvy083 [DOI] [PubMed] [Google Scholar]
- 142.Hellström M, Ericsson M, Johansson B, Faraz M, Anderson F, Henriksson R, et al. Cardiac hypertrophy and decreased high-density lipoprotein cholesterol in Lrig3-deficient mice. Am J Physiol Regul Integr Comp Physiol. (2016) 310:R1045–52. 10.1152/ajpregu.00309.2015 [DOI] [PubMed] [Google Scholar]
- 143.Cappola TP, Li M, He J, Ky B, Gilmore J, Qu L, et al. Common variants in HSPB7 and FRMD4B associated with advanced heart failure. Circ Cardiovasc Genet. (2010) 3:147–54. 10.1161/CIRCGENETICS.109.898395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu L, Sun K, Zhang X, Tang Y, Xu D. Advances in the role and mechanism of BAG3 in dilated cardiomyopathy. Heart Fail Rev. (2019). 10.1007/s10741-019-09899-7. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 145.Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. (2020) 141:387–98. 10.1161/CIRCULATIONAHA.119.037661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Choquet H, Thai KK, Jiang C, Ranatunga DK, Hoffmann TJ, Go AS, et al. Meta-analysis of 26,638 individuals identifies two genetic loci associated with left ventricular ejection fraction. Circ Genom Precis Med. (2020) 13:e002804 10.1161/CIRCGEN.119.002804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yu B, Barbalic M, Brautbar A, Nambi V, Hoogeveen RC, Tang W, et al. Association of genome-wide variation with highly sensitive cardiac troponin-T levels in European Americans and blacks: a meta-analysis from atherosclerosis risk in communities and cardiovascular health studies. Circ Cardiovasc Genet. (2013) 6:82–8. 10.1161/CIRCGENETICS.112.963058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mahmoudi MJ, Hedayat M, Taghvaei M, Harsini S, Nematipour E, Rezaei N, et al. Interleukin-10 and transforming growth factor beta1 gene polymorphisms in chronic heart failure. Acta Biomed. (2019) 90:221–7. 10.23750/abm.v90i2.6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bartekova M, Radosinska J, Jelemensky M, Dhalla NS. Role of cytokines and inflammation in heart function during health and disease. Heart Fail Rev. (2018) 23:733–58. 10.1007/s10741-018-9716-x [DOI] [PubMed] [Google Scholar]
- 150.Schlossarek S, Mearini G, Carrier L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol. (2011) 50:613–20. 10.1016/j.yjmcc.2011.01.014 [DOI] [PubMed] [Google Scholar]
- 151.Behrens-Gawlik V, Mearini G, Gedicke-Hornung C, Richard P, Carrier L. MYBPC3 in hypertrophic cardiomyopathy: from mutation identification to RNA-based correction. Pflugers Arch. (2014) 466:215–23. 10.1007/s00424-013-1409-7 [DOI] [PubMed] [Google Scholar]
- 152.Niu X, Zhang J, Zhang L, Hou Y, Pu S, Chu A, et al. Weighted gene co-expression network analysis identifies critical genes in the development of heart failure after acute myocardial infarction. Front Genet. (2019) 10:1214 10.3389/fgene.2019.01214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kreisel D, Sugimoto S, Tietjens J, Zhu J, Yamamoto S, Krupnick AS, et al. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest. (2011) 121:265–76. 10.1172/JCI42596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schilling J, Kelly DP. The PGC-1 cascade as a therapeutic target for heart failure. J Mol Cell Cardiol. (2011) 51:578–83. 10.1016/j.yjmcc.2010.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Weinberg EO, Shimpo M, De Keulenaer GW, Macgillivray C, Tominaga S, Solomon SD, et al. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation. (2002) 106:2961–6. 10.1161/01.CIR.0000038705.69871.D9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. (2018) 50:1219–24. 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, De Wit C, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. (2013) 504:432–6. 10.1038/nature12722 [DOI] [PubMed] [Google Scholar]
- 158.Khera AV, Emdin CA, Kathiresan S. Genetic risk, lifestyle, and coronary artery disease. N Engl J Med. (2017) 376:1194–5. 10.1056/NEJMc1700362 [DOI] [PubMed] [Google Scholar]
- 159.Jia X, Al Rifai M, Liu J, Agarwala A, Gulati M, Virani SS. Highlights of studies in cardiovascular disease prevention presented at the 2020 american college of cardiology annual scientific session. Curr Atheroscler Rep. (2020) 22:32 10.1007/s11883-020-00856-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Siemelink MA, Zeller T. Biomarkers of coronary artery disease: the promise of the transcriptome. Curr Cardiol Rep. (2014) 16:513 10.1007/s11886-014-0513-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pua CJ, Bhalshankar J, Miao K, Walsh R, John S, Lim SQ, et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J Cardiovasc Transl Res. (2016) 9:3–11. 10.1007/s12265-016-9673-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rao AS, Knowles JW. Polygenic risk scores in coronary artery disease. Curr Opin Cardiol. (2019) 34:435–40. 10.1097/HCO.0000000000000629 [DOI] [PubMed] [Google Scholar]
- 163.Curtis D. Clinical relevance of genome-wide polygenic score may be less than claimed. Ann Hum Genet. (2019) 83:274–7. 10.1111/ahg.12302 [DOI] [PubMed] [Google Scholar]
- 164.Soler-Botija C, Gálvez-Montón C, Bayés-Genís A. Epigenetic biomarkers in cardiovascular diseases. Front Genet. (2019) 10:950 10.3389/fgene.2019.00950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sweet ME, Cocciolo A, Slavov D, Jones KL, Sweet JR, Graw SL, et al. Transcriptome analysis of human heart failure reveals dysregulated cell adhesion in dilated cardiomyopathy and activated immune pathways in ischemic heart failure. BMC Genomics. (2018) 19:812 10.1186/s12864-018-5213-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schulte C, Zeller T. Biomarkers in primary prevention: meaningful diagnosis based on biomarker scores? Herz. (2020) 45:10–6. 10.1007/s00059-019-04874-2 [DOI] [PubMed] [Google Scholar]
- 167.Osman J, Tan SC, Lee PY, Low TY, Jamal R. Sudden Cardiac Death (SCD) – risk stratification and prediction with molecular biomarkers. J Biomed Sci. (2019) 26:39 10.1186/s12929-019-0535-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. 10.1016/0092-8674(93)90529-Y [DOI] [PubMed] [Google Scholar]
- 169.Barwari T, Joshi A, Mayr M. MicroRNAs in cardiovascular disease. J Am Coll Cardiol. (2016) 68:2577–84. 10.1016/j.jacc.2016.09.945 [DOI] [PubMed] [Google Scholar]
- 170.Bauersachs J, Thum T. Biogenesis and regulation of cardiovascular microRNAs. Circ Res. (2011) 109:334–47. 10.1161/CIRCRESAHA.110.228676 [DOI] [PubMed] [Google Scholar]
- 171.Schulte C, Zeller T. microRNA-based diagnostics and therapy in cardiovascular disease-Summing up the facts. Cardiovasc Diagn Ther. (2015) 5:17–36. 10.3978/j.issn.2223-3652.2014.12.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schulte C, Westermann D, Blankenberg S, Zeller T. Diagnostic and prognostic value of circulating microRNAs in heart failure with preserved and reduced ejection fraction. World J Cardiol. (2015) 7:843–60. 10.4330/wjc.v7.i12.843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rybak-Wolf A, Stottmeister C, GlaŽar P, Jens M, Pino N, Giusti S, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Molecular Cell. (2015) 58:870–85. 10.1016/j.molcel.2015.03.027 [DOI] [PubMed] [Google Scholar]
- 174.Kaudewitz D, Zampetaki A, Mayr M. MicroRNA biomarkers for coronary artery disease? Curr Atheroscler Rep. (2015) 17:70 10.1007/s11883-015-0548-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. (2012) 148:1172–87. 10.1016/j.cell.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. (2008) 105:10513–8. 10.1073/pnas.0804549105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. (2011) 108:5003–8. 10.1073/pnas.1019055108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. (2011) 13:423–33. 10.1038/ncb2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Turchinovich A, Weiz L, Langheinz A, Burwinkel B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. (2011) 39:7223–33. 10.1093/nar/gkr254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Boon RA, Vickers KC. Intercellular transport of microRNAs. Arterioscler Thromb Vasc Biol. (2013) 33:186–92. 10.1161/ATVBAHA.112.300139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sunderland N, Skroblin P, Barwari T, Huntley RP, Lu R, Joshi A, et al. MicroRNA biomarkers and platelet reactivity: the clot thickens. Circ Res. (2017) 120:418–35. 10.1161/CIRCRESAHA.116.309303 [DOI] [PubMed] [Google Scholar]
- 182.Reid G, Kirschner MB, Van Zandwijk N. Circulating microRNAs: association with disease and potential use as biomarkers. Crit Rev Oncol Hematol. (2011) 80:193–208. 10.1016/j.critrevonc.2010.11.004 [DOI] [PubMed] [Google Scholar]
- 183.Simons M, Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol. (2009) 21:575–81. 10.1016/j.ceb.2009.03.007 [DOI] [PubMed] [Google Scholar]
- 184.Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. (2012) 110:483–95. 10.1161/CIRCRESAHA.111.247452 [DOI] [PubMed] [Google Scholar]
- 185.Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in drosophila and regulates Notch signaling. Proc Natl Acad Sci USA. (2005) 102:18986–91. 10.1073/pnas.0509535102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. (2005) 436:214–20. 10.1038/nature03817 [DOI] [PubMed] [Google Scholar]
- 187.Chistiakov DA, Orekhov AN, Bobryshev YV. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J Mol Cell Cardiol. (2016) 94:107–21. 10.1016/j.yjmcc.2016.03.015 [DOI] [PubMed] [Google Scholar]
- 188.Kumarswamy R, Lyon AR, Volkmann I, Mills AM, Bretthauer J, Pahuja A, et al. SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur Heart J. (2012) 33:1067–75. 10.1093/eurheartj/ehs043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. (2009) 29:2193–204. 10.1128/MCB.01222-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lai KB, Sanderson JE, Izzat MB, Yu CM. Micro-RNA and mRNA myocardial tissue expression in biopsy specimen from patients with heart failure. Int J Cardiol. (2015) 199:79–83. 10.1016/j.ijcard.2015.07.043 [DOI] [PubMed] [Google Scholar]
- 191.Rigaud VO, Ferreira LR, Ayub-Ferreira SM, Ávila MS, Brandão SM, Cruz FD, et al. Circulating miR-1 as a potential biomarker of doxorubicin-induced cardiotoxicity in breast cancer patients. Oncotarget. (2017) 8:6994–7002. 10.18632/oncotarget.14355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang R, Niu H, Ban T, Xu L, Li Y, Wang N, et al. Elevated plasma microRNA-1 predicts heart failure after acute myocardial infarction. Int J Cardiol. (2013) 166:259–60. 10.1016/j.ijcard.2012.09.108 [DOI] [PubMed] [Google Scholar]
- 193.Seronde MF, Vausort M, Gayat E, Goretti E, Ng LL, Squire IB, et al. Circulating microRNAs and outcome in patients with acute heart failure. PLoS ONE. (2015) 10:e0142237 10.1371/journal.pone.0142237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schulte C, Molz S, Appelbaum S, Karakas M, Ojeda F, Lau DM, et al. miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS ONE. (2015) 10:e0145930 10.1371/journal.pone.0145930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zampetaki A, Willeit P, Tilling L, Drozdov I, Prokopi M, Renard JM, et al. Prospective study on circulating MicroRNAs and risk of myocardial infarction. J Am Coll Cardiol. (2012) 60:290–9. 10.1016/j.jacc.2012.03.056 [DOI] [PubMed] [Google Scholar]
- 196.Cruz MS, Da Silva AMG, De Souza KSC, Luchessi AD, Silbiger VN. miRNAs emerge as circulating biomarkers of post-myocardial infarction heart failure. Heart Fail Rev. (2020) 25:321–9. 10.1007/s10741-019-09821-1 [DOI] [PubMed] [Google Scholar]
- 197.Li DY, Busch A, Jin H, Chernogubova E, Pelisek J, Karlsson J, et al. H19 induces abdominal aortic aneurysm development and progression. Circulation. (2018) 138:1551–68. 10.1161/CIRCULATIONAHA.117.032184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Viereck J, Thum T. Circulating noncoding RNAs as biomarkers of cardiovascular disease and injury. Circ Res. (2017) 120:381–99. 10.1161/CIRCRESAHA.116.308434 [DOI] [PubMed] [Google Scholar]
- 199.Sallam T, Sandhu J, Tontonoz P. Long noncoding RNA discovery in cardiovascular disease: decoding form to function. Circ Res. (2018) 122:155–66. 10.1161/CIRCRESAHA.117.311802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. 10.1101/gr.132159.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res. (2014) 114:1569–75. 10.1161/CIRCRESAHA.114.303915 [DOI] [PubMed] [Google Scholar]
- 202.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. (2009) 23:1494–504. 10.1101/gad.1800909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gangwar RS, Rajagopalan S, Natarajan R, Deiuliis JA. Noncoding RNAs in cardiovascular disease: pathological relevance and emerging role as biomarkers and therapeutics. Am J Hypertens. (2018) 31:150–65. 10.1093/ajh/hpx197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. (2015) 116:737–50. 10.1161/CIRCRESAHA.116.302521 [DOI] [PubMed] [Google Scholar]
- 205.Tan P, Guo YH, Zhan JK, Long LM, Xu ML, Ye L, et al. LncRNA-ANRIL inhibits cell senescence of vascular smooth muscle cells by regulating miR-181a/Sirt1. Biochem Cell Biol. (2019) 97:571–80. 10.1139/bcb-2018-0126 [DOI] [PubMed] [Google Scholar]
- 206.Natarelli L, Geißler C, Csaba G, Wei Y, Zhu M, Di Francesco A, et al. miR-103 promotes endothelial maladaptation by targeting lncWDR59. Nat Commun. (2018) 9:2645 10.1038/s41467-018-05065-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhang G, Dou L, Chen Y. Association of long-chain non-coding RNA MHRT gene single nucleotide polymorphism with risk and prognosis of chronic heart failure. Medicine. (2020) 99:e19703 10.1097/MD.0000000000019703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Viereck J, Bührke A, Foinquinos A, Chatterjee S, Kleeberger JA, Xiao K, et al. (2020). Targeting muscle-enriched long non-coding RNA H19 reverses pathological cardiac hypertrophy. Eur Heart J. 41:3462–74. 10.1093/eurheartj/ehaa519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Greco S, Zaccagnini G, Perfetti A, Fuschi P, Valaperta R, Voellenkle C, et al. Long noncoding RNA dysregulation in ischemic heart failure. J Transl Med. (2016) 14:183 10.1186/s12967-016-0926-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chandler B, Hayashi M, Hayashi MN, Spiegelman S. Circularity of the replicating form of a single-stranded DNA virus. Science. (1964) 143:47–9. 10.1126/science.143.3601.47 [DOI] [PubMed] [Google Scholar]
- 211.Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci USA. (1976) 73:3852–6. 10.1073/pnas.73.11.3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, et al. Scrambled exons. Cell. (1991) 64:607–13. 10.1016/0092-8674(91)90244-S [DOI] [PubMed] [Google Scholar]
- 213.Cocquerelle C, Mascrez B, Hétuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. (1993) 7:155–60. 10.1096/fasebj.7.1.7678559 [DOI] [PubMed] [Google Scholar]
- 214.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. (2014) 32:453–61. 10.1038/nbt.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. (2013) 495:333–8. 10.1038/nature11928 [DOI] [PubMed] [Google Scholar]
- 216.Zhang Y, Zhang X-O, Chen T, Xiang J-F, Yin Q-F, Xing Y-H, et al. Circular intronic long noncoding RNAs. Mol Cell. (2013) 51:792–806. 10.1016/j.molcel.2013.08.017 [DOI] [PubMed] [Google Scholar]
- 217.Suzuki H, Zuo Y, Wang J, Zhang MQ, Malhotra A, Mayeda A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. (2006) 34:e63–e63. 10.1093/nar/gkl151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Khan MA, Reckman YJ, Aufiero S, van den Hoogenhof MM, van der Made I, Beqqali A, et al. (2016). RBM20 regulates circular RNA production from the titin gene. Circ Res. 119, 996–1003. 10.1161/CIRCRESAHA.116.309568 [DOI] [PubMed] [Google Scholar]
- 219.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. (2013) 495:384–8. 10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- 220.Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, et al. Circ-ZNF609 Is a circular RNA that can be translated and functions in myogenesis. Mol Cell. (2017) 66:22–37.e9. 10.1016/j.molcel.2017.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. (2014) 56:55–66. 10.1016/j.molcel.2014.08.019 [DOI] [PubMed] [Google Scholar]
- 222.Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development. (2016) 143:1838–47. 10.1242/dev.128074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Han B, Chao J, Yao H. Circular RNA and its mechanisms in disease: from the bench to the clinic. Pharmacol Ther. (2018) 187:31–44. 10.1016/j.pharmthera.2018.01.010 [DOI] [PubMed] [Google Scholar]
- 224.Tan WL, Lim BT, Anene-Nzelu CG, Ackers-Johnson M, Dashi A, See K, et al. A landscape of circular RNA expression in the human heart. Cardiovasc Res. (2017) 113:298–309. 10.1093/cvr/cvw250 [DOI] [PubMed] [Google Scholar]
- 225.Wang K, Gan TY, Li N, Liu CY, Zhou LY, Gao JN, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. (2017) 24:1111–20. 10.1038/cdd.2017.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Geng HH, Li R, Su YM, Xiao J, Pan M, Cai XX, et al. The Circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of mir-7a on its target genes expression. PLoS ONE. (2016) 11:e0151753 10.1371/journal.pone.0151753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Vausort M, Salgado-Somoza A, Zhang L, Leszek P, Scholz M, Teren A, et al. Myocardial infarction-associated circular RNA predicting left ventricular dysfunction. J Am Coll Cardiol. (2016) 68:1247–8. 10.1016/j.jacc.2016.06.040 [DOI] [PubMed] [Google Scholar]
- 228.Salgado-Somoza A, Zhang L, Vausort M, Devaux Y. The circular RNA MICRA for risk stratification after myocardial infarction. Int J Cardiol Heart Vasc. (2017) 17:33–6. 10.1016/j.ijcha.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]