

Abstract
Interleukin-15 (IL-15) is an inflammatory cytokine whose role in autoimmune diseases has not been fully elucidated. Th17 cells have been shown to play critical roles in experimental autoimmune encephalomyelitis (EAE) models. In this study, we demonstrate that blockade of IL-15 signaling by TMβ-1 mAb treatment aggravated EAE severity. The key mechanism was not NK-cell depletion but depletion of CD8+CD122+ T cells. Adoptive transfer of exogenous CD8+CD122+ T cells to TMβ-1-treated mice rescued animals from severe disease. Moreover, transfer of preactivated CD8+CD122+ T cells prevented EAE development and significantly reduced IL-17 secretion. Naïve effector CD4+CD25− T cells cultured with either CD8+CD122+ T cells from wild-type mice or IL-15 transgenic mice displayed lower frequencies of IL-17A production with lower amounts of IL-17 in the supernatants when compared with production by effector CD4+CD25− T cells cultured alone. Addition of a neutralizing antibody to IL-10 led to recovery of IL-17A production in Th17 cultures. Furthermore, coculture of CD8+CD122+ T cells with effector CD4+ T cells inhibited their proliferation significantly, suggesting a regulatory function for IL-15 dependent CD8+CD122+ T cells. Taken together, these observations suggest that IL-15, acting through CD8+CD122+ T cells, has a negative regulatory role in reducing IL-17 production and Th17-mediated EAE inflammation.
Keywords: CD8+CD122+ T cells, Experimental autoimmune encephalomyelitis, Interleukin-15, Interleukin-17, Multiple sclerosis
Introduction
IL-15 is a proinflammatory cytokine that stimulates NK-cell and memory CD8+ T-cell activity [1–4]. IL-15 signals through heterotrimeric receptors that include the IL-15 specific receptor subunit, IL-15Rα and IL-2/IL-15Rβ(CD122) that is shared with IL-2, as well as the common γ-chain (γc) that is shared with IL-2, IL-4, IL-7, IL-9, and IL-21[5, 6]. IL-15 is not secreted but acts as part of an immunological synapse. Activated dendritic cells and monocytes coexpress IL-15Rα and IL-15, which is transpresented to NK cells and CD8+ T cells which express IL-2/IL-15Rβ and γc [6]. Dysregulated IL-15 expression has been reported in patients with a range of autoimmune diseases including multiple sclerosis [7], rheumatoid arthritis [8], Crohn’s and ulcerative colitis [9, 10] refractory celiac disease [11–13], and type 1-diabetes [14, 15]. In order to treat these diseases, agents that inhibit IL-15 action have been developed, including soluble IL-15Rα, a dominant-negative IL-15 molecule [16], and neutralizing antibodies specific for IL-15 or IL-2/IL-15Rβ [17, 18]. These data showed that blockade of IL-15 signaling ameliorated symptoms and led to the proposal that IL-15 and its receptors as therapeutic targets for certain IL-15 mediated autoimmune diseases.
Multiple sclerosis (MS) is a demyelinating disease of the central nervous system (CNS). It is thought to have an etiology involving autoimmunity and cytokine dysregulation [19]. Patients with MS were found to have increased numbers of IL-15 mRNA–expressing mononuclear cells in blood and cerebrospinal fluid compared with non-MS patients [20]. Another study confirmed a significant increase of IL-15 in the sera of patients with MS when compared with those of a control group [21]. These studies suggested that IL-15 plays an important role in MS and provided the theoretical rational for a focus on inhibiting IL-15 action in MS.
Experimental autoimmune encephalomyelitis (EAE) is an inflammatory demyelinating disease of the CNS that shares clinical and pathological features with MS, and that has been used as a model for MS. Studies in EAE have provided convincing evidence that both Th1 and Th17 are the main effector cells responsible for the autoimmune inflammation [22–24]. Compared with CD4+ T cells, the role of CD8+ T cells in EAE is ambiguous. Earlier studies focused on the ability of myelin Ag-specific CD8+ T cells to function as potent effectors of CNS autoimmunity [25, 26]. More recently, suppressive functions of CD8+ T cells in EAE models have been reported [27, 28].
To study the function of IL-15 in EAE and the inter-regulation between IL-15 dependent CD8+ T cells and Th17 cells, we took advantage of IL-15−/− mice and the administration of antimurine CD122(TMβ-1), which blocks IL-15 transpresentation, to wild-type animals. We demonstrated that the absence of IL-15 or TMβ-1 mediated depletion of IL-15 dependent CD8+CD122+ T cells was associated with increased severity of EAE symptoms in mice. Furthermore, transfer of pre-activated CD8+CD122+ T cells into mice prevented EAE development, thereby supporting the view that CD8+CD122+ T cells have important regulatory activity in the initiation and acute phases of EAE. Additionally, in this study we identified a mechanism by which IL-15 through its action on CD8+CD122+ T cells inhibits Th17-cell differentiation and modulates the secretion of IL-17 in the EAE model.
Results
Blockade of IL-15 transpresentation by TMβ-1 treatment aggravates EAE severity
It has been reported that humanized Mikβ-1(anti-IL-2/IL-15Rβ, CD122) blocks IL-15 transpresentation in humans [18, 29–31]. TMβ-1 is a murine antibody equivalent to Mikβ-1 [32]. In the present study one day after TMβ-1 treatment, animals were induced to have EAE. Mice that received rat IgG showed the typical course of EAE symptoms that appeared 11–12 days after MOG immunization. The disease reached its peak around days 18–20, and then gradually declined. In contrast, TMβ-1-treated EAE mice showed earlier initiation of EAE symptoms with high clinical scores from day 9 until day 40, with the maximum score significantly higher than that observed in the control group (Fig. 1A). Flow cytometric analysis detected significantly reduced numbers of NK and CD8+CD122+ T cells 4 days after TMβ-1 administration (Fig. 1B). To detect CD122 expression on CD8+ T cells, we did not use the TMβ-1 but rather another antibody 5H4 that targets an alternative noncompeting epitope. The percentage of CD122+ cells in the CD8+ population was significantly decreased in mice treated with TMβ-1. In order to avoid the possibility of ADCC mediated by TMβ-1, mutated TMβ-1 which does not participate in ADCC was also administered to the mice, with similar affects being observed. This observation suggested that blockade of transpresentation of IL-15 signaling rather than ADCC was responsible for the diminished numbers of NK and CD8+CD122+ T cells upon TMβ-1 administration. In contrast, CD4+CD25+FoxP3+ cells were not affected by TMβ-1 treatment (Fig. 1B). Further study with IL-15 transgenic mice and IL-15−/− mice confirmed that both NK and CD8+CD122+ T cells are dependent on IL-15 (Fig. 1C). Summary of flow cytometry data are available in Supporting Information Figure 1. More severe EAE was observed in IL-15−/− mice compared to that in wild-type animals (Fig. 1D) which provides further evidence that cells dependent on IL-15 play critical ameliorating roles in EAE disease.
Figure 1.
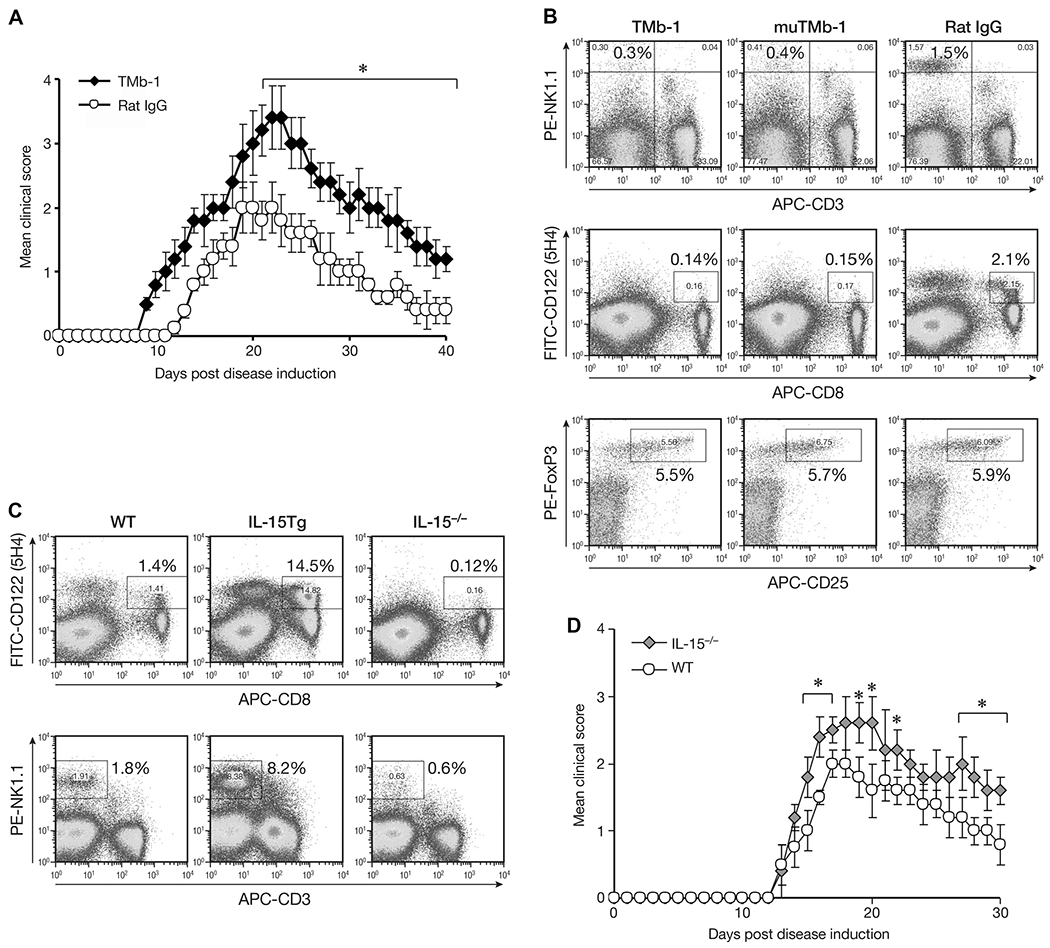
Effect of TMβ-1 treatment on EAE. (A) MOG EAE was induced in Ly5.1 C57BL/6 mice one day after i.p injection of 50 μg TMβ-1 Abs (TMb-1, filled diamonds) or rat IgG (Rat IgG, open circles). The clinical scores were assessed as described in the Materials and methods and are shown as mean ± SD of N = 10/group. (B) Flow cytometric analysis of NK cells, CD8+CD122+ T cells and CD4+CD25+FoxP3+ Treg cells upon administration of TMβ-1. Ly5.1 C57BL/6 mice were sacrificed on day 4 after i.p. injection of 50 μg TMβ-1, mutated TMβ-1 or rat IgG. The percentages of CD3−NK1.1+ cells and CD8+CD122+ T cells in total splenic lymphocytes are shown. After gating on CD4+ T cells, the percentages of CD25+FoxP3+ T cells in the entire CD4+ population were evaluated. (C) Flow cytometric analysis of CD8+CD122+ T cells and NK cells in wild type (WT), IL-15 transgeneic (IL-15 Tg), and IL-15−/− mice. (D) MOG EAE in IL-15−/− mice. MOG EAE was induced in IL-15−/− mice (IL-15−/−, gray diamonds) and age-matched Ly5.1 C57BL/6 mice (WT, open circles). The clinical scores are shown as mean ± SD of N = 10/group. (A–D) Data shown are from single experiments representative of four experiments performed. *p < 0.05 compared to control group by Student’s t-test.
Adoptive transfer of NK cells does not rescue animals from severe EAE symptoms upon TMβ-1 treatment
In adoptive transfer studies to avoid continued depletion of transferred cells by the remaining antibody following TMβ-1 administration, instead of using TMβ-1, we administered TMβ-1 Fab fragments to mice. The study outlined in Figure 2A demonstrates comparable EAE disease severity and development between TMβ-1 Fab and TMβ-1 mAb treatments. On days 5 and 10 after EAE induction, NK cells separated from Ly5.2 RAG-1−/− mice were transferred into Ly5.1C57BL/6 mice. The persistent presence of Ly5.2+NK1.1+ confirmed the survival of transferred cells in the subsequent days (Fig. 2B). It has been reported that NK cells originating in the thymus differ from bone marrow-derived NK cells and have functional properties closely resembling those of the human subset CD56hiCD16− NK cells with specific immunoregulatory properties of these human NK cells [33]. Meanwhile, it has been recognized that CD56hiCD16− NK cells play a critical role in the amelioration of MS achieved with daclizumab treatment [34]. Currently, we transferred thymic or splenic NK cells into recipient mice simultaneously with TMβ-1 Fab administration. No protection was observed compared with TMβ-1 Fab treatment alone (Fig. 2C and D).
Figure 2.
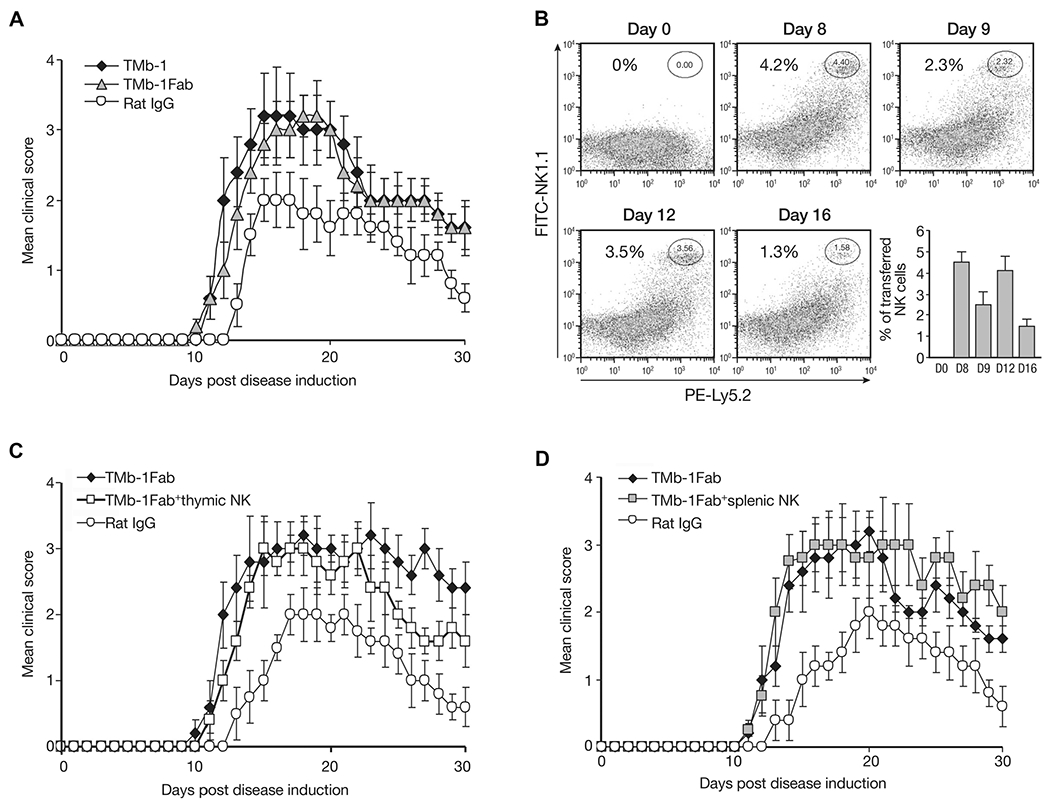
Effect of transferred NK cells on EAE disease. Ly5.1C57BL/6 mice were induced to develop EAE on day 0, along with 4 days of TMβ-1 Fab treatments (day −1 to day 3, 500 μg/injection). (A) The effect of TMβ-1 Fab administration compared to TMβ-1 mAb treatment on the mean clinical scores was evaluated. A group of mice received TMβ-1 Fab (TMβ-1 Fab, gray triangles) and mice that received a single injection (i.p.) of 50 μg TMβ-1 mAb (TMb-1, filled diamonds) or rat IgG (Rat IgG, open circles) on day −1 were set up as controls. Data are shown as mean ± SD of N = 10/group. (B) The presence of transferred NK cells in the peripheral blood of mice after TMβ-1 Fab treatment was evaluated by flow cytometry. On day 5 and 10, NK cells separated from RAG-1−Ly5.2C57BL/6 mice were transferred into Ly5.1 C57BL/6 mice. At different time points, PBMCs from the recipient mice were collected to detect transferred Ly5.2 NK cells by flow cytometry. A summary of data pooled from three experiments is shown (bottom right). (C and D) The effect of (C) thymic-derived NK cells or (D) splenic-derived NK cells on EAE after TMβ-1 Fab treatment was evaluated. On day 5 and 10, 1 × 106/mouse thymic NK cells (C) or splenic NK cells (D) separated from Ly5.2 RAG1−/− mice were adoptively transferred into EAE mice (TMb-1 Fab + thymic NK, open squares, TMb-1 Fab + splenic NK, gray squares). Groups of mice received TMβ-1 Fab alone (TMb-1 Fab, filled diamonds) or received rat IgG (Rat IgG, open circles) were set up as controls. The clinical scores are shown as mean ± SD of N = 10/group. (A–D) Data shown are from single experiments representative of three experiments performed, except for (B), in which the data from 15 mice from three experiments was pooled.
Transfer of CD8+CD122+ T cells effectively suppresses EAE symptoms
In order to examine the effect of CD8+CD122+ T cells on EAE, along with 4 day TMβ-1 Fab treatment and induction of EAE, on days 5 and 10, we transferred CD8+CD122+ T cells separated from IL-15-transgenic (IL-15-Tg) mice into the recipient animals. The transfer of cells was rapidly associated with a decrease in the severity of EAE symptoms compared with TMβ-1 Fab treated alone group (Fig. 3A). Furthermore when we provided anti-IL-10 on day 20 after disease induction, the decreased symptoms due to transferred CD8+CD122+ T cells were not observed, and the mean clinical score reached a comparable level with that of TMβ-1 Fab treated group (Fig. 3A). These observations suggest that the protection derived from CD8+CD122+ T cells is at least partially dependent on the action of IL-10 in vivo. Meanwhile, mice that received either CD8+CD122− T cells or bulk CD8+ T cells showed continued EAE symptoms with as high a clinical score as did TMβ-1 Fab treated group (Fig. 3B). In summary, reintroduction of IL-15-dependent-CD8+CD122+ T cells partially protected the animals from severe EAE that was associated with TMβ-1 treatment.
Figure 3.
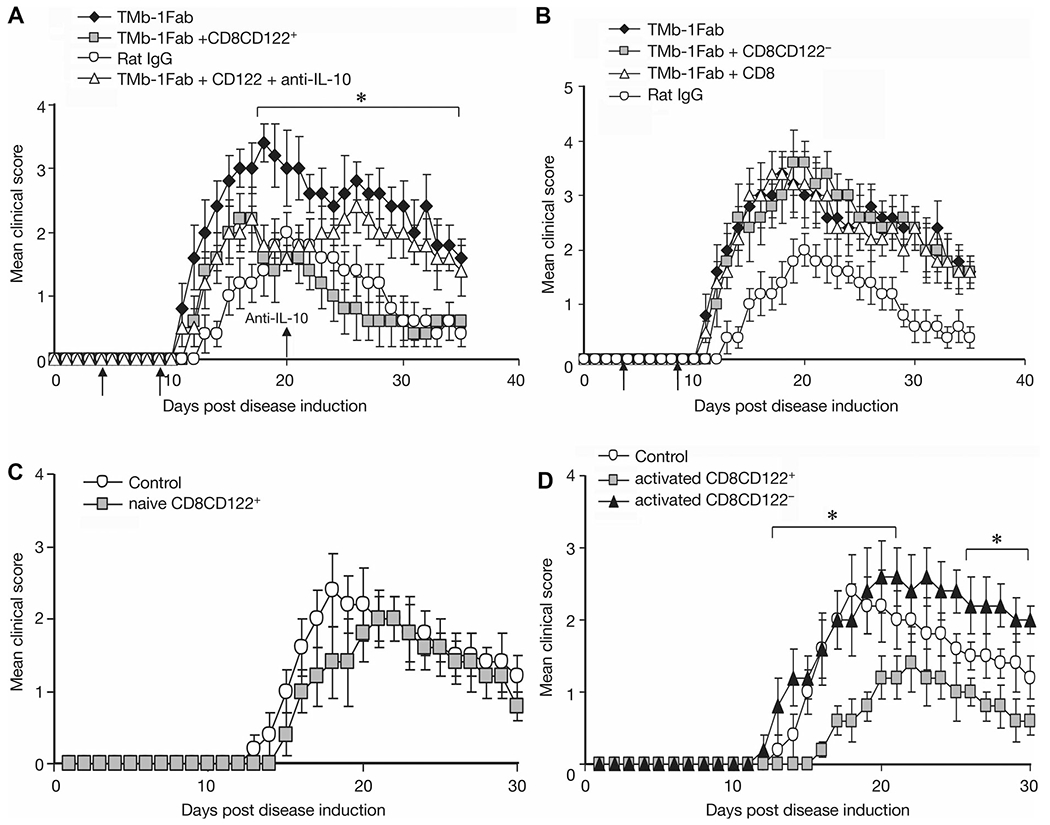
Effect of transferred CD8+CD122+ T cells on EAE. (A) Effect of transferred CD8+CD122+ T cells on EAE after TMβ-1 Fab treatment and the contribution of IL-10 in vivo. Ly5.1 C57BL/6 mice were induced to develop MOG EAE on day 0, along with 4 days of TMβ-1 Fab treatment (day −1 to day 3). On days 5 and 10, 1 × 106/mouse CD8+CD122+ T cells separated from IL-15-Tg mice were transferred into EAE mice (TMb-1 Fab+CD8CD122+, gray squares). A 100 μg/mL of anti-IL-10 was administered to selected animals on day 20, and then twice a week for 2 weeks (TMb-1 Fab+CD122++anti-IL-10, open triangles). EAE mice that received only rat IgG (Rat IgG, open circles) and that only received TMβ-1 Fab (TMb-1 Fab, filled diamonds) were set up as controls. *p < 0.05 TMb-1 Fab+CD8CD122+ group compared to TMb-1 Fab group analyzed by Student’s t-test. (B) Effect of transferred CD8+ T cells or CD8+CD122− T cells on EAE. Similar procedures were performed and with same control groups as in (A). 1 × 106/mouse CD8 (TMb-1 Fab + CD8, open triangles) or CD8+CD122− T cells (TMb-1 Fab + CD8CD122−, gray squares) from IL-15-Tg mice were transferred into TMβ-1 Fab treated MOG EAE mice. (C) Effect of naïve CD8+CD122+ T cells on EAE. On day −1, 1 × 106/mouse CD8+CD122+ T cells from IL-15 transgenic mice were transferred into Ly5.1 C57BL/6 mice, on day 0, MOG EAE was induced. Repeated adoptive cell transfer was performed on day 5 (naïve CD8CD122+, gray squares). (D) Effect of transferred preactivated CD8+CD122+/CD8+CD122− T cells on EAE. On day —2, 1 × 106/mouse CD8+CD122+/CD8+CD122− T cells from IL-15-Tg mice were stimulated with anti-CD3 plus anti-CD28. Cells were washed and transferred into Ly5.1 C57BL/6 mice 24 h later, followed by MOG EAE induction on day 0. Repeat transfers were performed on day 5 (activated CD8+CD122+, gray squares; activated CD8CD122−, filled triangles). *p < 0.05 activated CD8CD122+ group versus control group by Student’s t-test. (A–D) The clinical scores are shown as mean ± SD. N = 10/group, from single experiments representative of three experiments performed.
Transfer of preactivated CD8+CD122+ T cells delays the initiation of EAE
We determined whether CD8+CD122+ T cells could prevent or delay EAE initiation. CD8+CD122+ T cells derived from IL-15-Tg mice were transferred to recipient mice one day before EAE induction. On day 5, repeated transfers were performed. No significant benefit was observed compared to nontransferred group (Fig. 3C). In subsequent studies, instead of transferring naïve T cells, selected animals received CD8+CD122+ or CD8+CD122− T cells preactivated with anti-CD3 plus anti-CD28 one day before EAE induction. These activated CD8+CD122+ T-cell-transferred EAE mice showed a delay of disease initiation and a meaningful decrease in the clinical score at the peak of the EAE course as compared to CD8+CD122− T-cell-transferred EAE mice or simple EAE mice (Fig. 3D). This result provides evidence that preactivated CD8+CD122+ T cells can inhibit EAE symptoms.
TMβ-1 administration and CD8+CD122+ cellular transfer to EAE mice affects IL-17 production in vivo
To investigate the mechanism underlying CD8+CD122+ T-cell suppression and prevention of EAE, splenocytes were separated from EAE mice that had received rat IgG or TMβ-1 (Fig. 4A), IL-15−/− mice along with WT mice (Fig. 4B), naive CD8+CD122+-transferred (Fig. 4C), or preactivated CD8+CD122+/CD8+CD122− T cells transferred groups and their controls (Fig. 4D), then stimulated with MOG35–55 peptide. Cytokine levels in culture supernatants were examined by ELISA.
Figure 4.
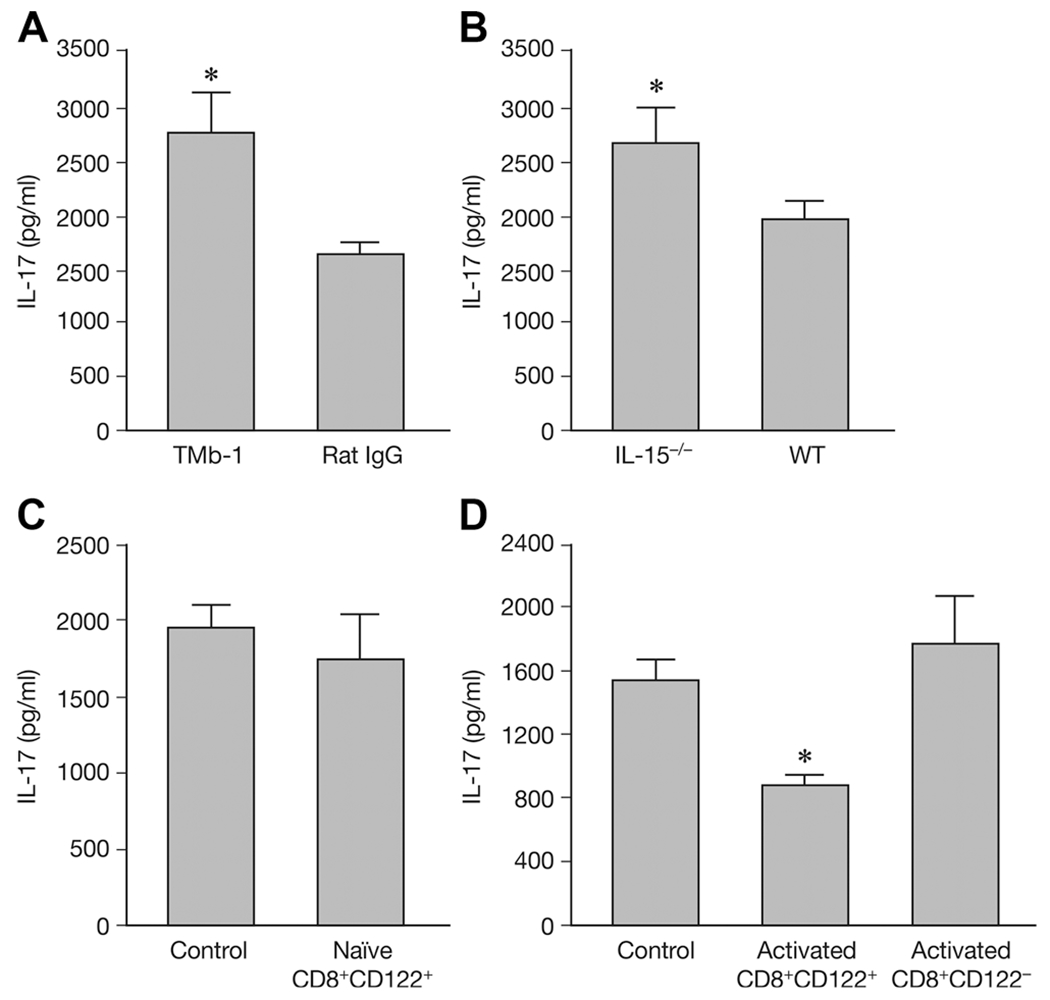
Secretion of IL-17 with different treatments. On day 19 after EAE induction splenocytes from EAE mouse were stimulated with MOG peptide and IL-17 secretion was detected by ELISA. (A) TMβ-1 (TMb-1) or control rat IgG (Rat IgG) treated groups were examined. (B) IL-15−/− mice and wild type (WT) animals were examined. (C) IL-17 secretion by transfer of naïve CD8+CD122+ T cells (naïve CD8+CD122+) and nontransferred control group (Control) are shown. (D) IL-17 secretion by preactivated CD8+CD122+/CD8+CD122− T cells transferred groups (Activated CD8+CD122+/Activated CD8+CD122−) are shown. Nontransferred control group (Control) were provided as control. Data are shown as mean ± SD of three to five mice from a single experiment representative of three experiments performed. *p < 0.05 compared to control group (Student’s t-test).
Nineteen days after MOG immunization, production of IL-17 was increased in splenocytes from TMβ-1 treated EAE mice, as well as in IL-15−/− mice when compared with those of control mice (Fig. 4A and B), p < 0.05. However, no difference in IL-17 secretion was detected between naïve CD8+CD122+ T-cell transferred mice and the control group (Fig. 4C). Intriguingly, mice transferred with preactivated CD8+CD122+ T cells produced significantly lower amounts of IL-17 when compared with the control group and with mice transferred with preactivated CD8+CD122− T cells, p = 0.031 and 0.036 (Fig. 4D).
Phenotypic study of the regulatory subset of CD8+CD122+ T cells
A number of markers has been associated with regulatory/suppressor CD8+ T cells, such as the presence of PD-1, CD25, Qa-1 Ly49 Class I MHC receptor or the absence of CD28 [35–39]. Our studies also showed heterogeneity of the CD8+CD122+ T-cell population. Splenocytes of naïve IL-15-Tg mice were used to phenotype CD8+CD122+ T cells. Analysis of the Ly49 gene family including the inhibitory receptors Ly49A and Ly49G2 expressed by CD8+CD122+ T cells revealed that this subset had increased mean fluorescence intensity (MFI) compared to those with isotype control staining. These cells did not express activating Ly49 receptors (Ly49D and H) at detectable levels (Fig. 5). Meanwhile, the markers of CD4 regulatory T cells-CD25 and FoxP3 were not detectable on CD8+CD122+ T cells. No CD69 expression was found which suggested that the expression of CD122 and CD44 were not due to the activation. The MFI with anti-IL-15Rα staining was also increased in CD8+CD122+ T cells along with PD-1, CTLA-4, and CD28 expression on the cell surface (Fig. 5A). A summary of multiple experiments are shown in Figure 5B.
Figure 5.
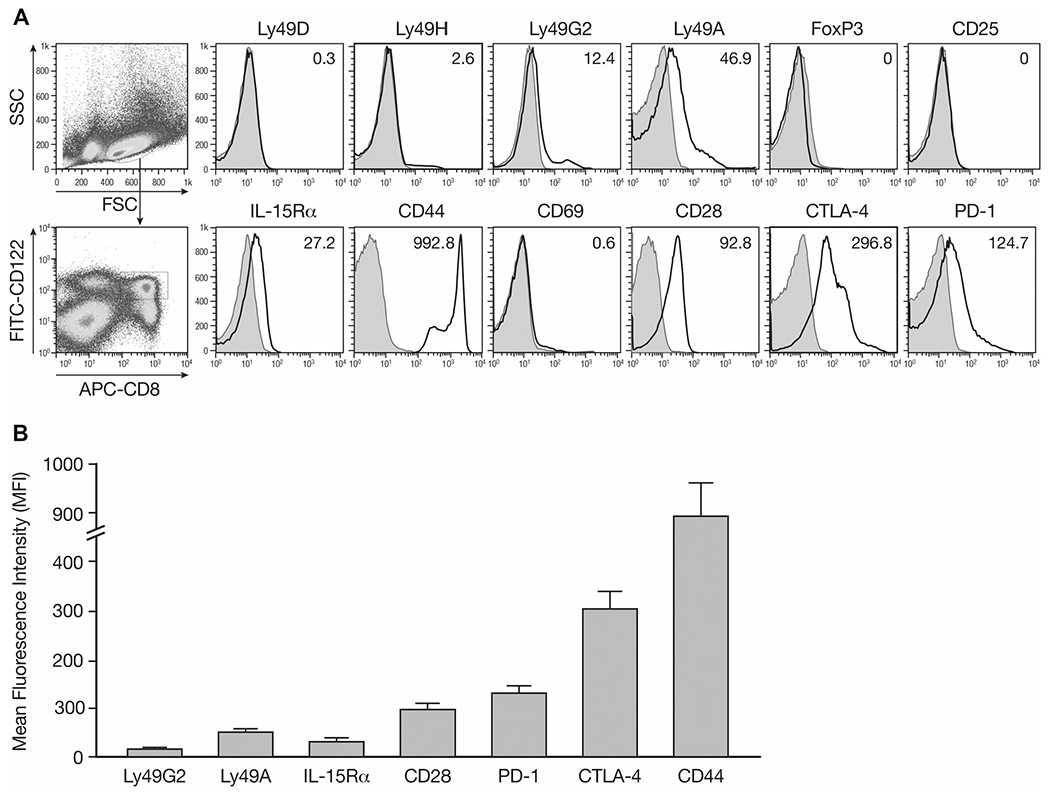
Phenotypic study of surface markers of CD8+CD122+ T cells. Splenocytes were isolated from naïve IL-15-Tg mice and stained for surface expression of the indicated molecules and analyzed by flow cytometry. The analysis was performed with FlowJo software. (A) Mean fluorescence intensity (MFI) was shown for each molecule after gating on CD8+CD122+ T cells. Filled histograms, cells stained with isotype controls; open histograms, cells stained with the indicated mAbs. (B) A summary of three independent experiments is shown as mean ± SD of 15 samples pooled from the three experiments.
Activated CD8+CD122+ T cells reduce Th17 generation and inhibit effector T-cell proliferation
To clarify the interaction between CD8+CD122+ T cells with Th17 cells, we took advantage of an in vitro assay to detect the effect of CD8+CD122+ T cells on Th17 generation. Naive CD4+CD25− T cells were stimulated under Th17 conditions. Different ratios of CD8+CD122+ T cells either from naïve Ly5.2 C57BL/6 or from Ly5.2 IL-15-Tg mice were added to the assay and cocultured for 4 days. When CD8+CD122+ T cells were added at the beginning of the stimulation period under Th17 polarizing conditions, IL-17A produced by CD4+ cells was reduced in a dose-dependent manner, as assessed by intracellular staining (Fig. 6A). However, if CD8+CD122+ T cells from IL-15-Tg mice were provided on day 3 and followed with the procedures to generate and detect Th17 cells on day 4, no significant effect could be observed on the IL-17A secretion (Fig. 6B). Summary of flow cytometry data are available in Supporting Information Figure 2. Meanwhile, the presence of anti-IL-10 dramatically reduced IL-17 expression by CD4+CD25− T cells even when cocultured with activated CD8+CD122+ T cells in the assay (Fig. 6B and C). ELISA data confirmed this inhibition of IL-17 secretion by activated CD8+CD122+ T cells. Both CD8+CD122+ T cells derived from IL-15-Tg mice and Ly5.2 WT mice effectively reduced IL-17 in the supernatant, while addition of CD8+CD122− T cells did not affect IL-17 production. Along with the inhibitory activities on IL-17 secretion, the proliferation of naïve CD4+CD25− T cells was tested. CD4+CD25+ regulatory T cells were used as a positive control. As in Figure 6D, CD8+CD122+ T cells suppressed naïve CD4+CD25− T cells in a dose-dependent manner, while CD8+CD122− T cells had no effect on the proliferation of effector CD4+ T cells.
Figure 6.
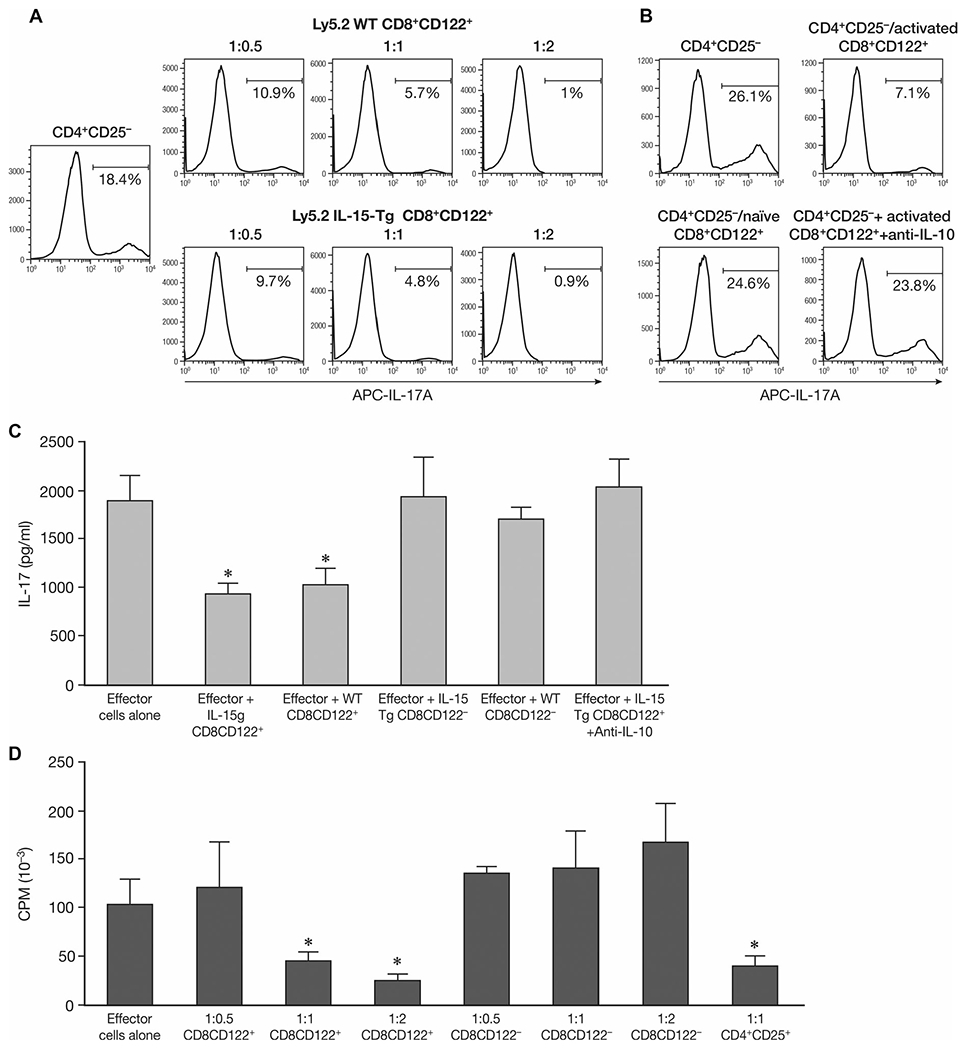
Effect on Th17-cell generation and effector T-cell proliferation by CD8+CD122+ T cells in vitro. (A) Effect on Th17-cell generation by coculturing CD8+CD122+ T cells with effector CD4+CD25− T cells. Naïve CD4+CD25− T cells from Ly5.1 C57BL/6 mice were stimulated under Th17 conditions. Different ratios of effector cells to CD8+CD122+ T cells either from naïve Ly5.2 wild type C57BL/6 (Ly5.2 WT) or from Ly5.2 IL-15 transgenic C57BL/6 mice (Ly5.2 IL-15-Tg) were cocultured with CD4+CD25− T cells for 4 days. The percentage of TM7 cells was detected by intracelluar staining for IL-17A by flow cytometry. (B) Effect on Th17-cell generation by naive CD8+CD122+ T cells in vitro. Instead of coculture with CD4+CD25− T cells for 4 days, the CD8+CD122+ T cells from Ly5.2 IL-15-Tg mice were provided on day 3 and continued with the following procedures to generate and detect Th17 cells (CD4+CD25−/naïve CD8+CD122+). Selected groups were cultured with CD8+CD122+ T cells for 4 days along with 10 μg/mL anti-IL-10 (CD4+CD25−+activated CD8+CD122++anti-IL-10). The percentage of Th17 cells was detected by intracellular staining for IL-17A by flow cytometery. Data shown are from single experiments representative of three experiments performed. (C) Effect of CD8+CD122+ T cells on IL-17 secretion by CD4+CD25− T cells. Naïve CD4+CD25− T cells from Ly5.1 C57BL/6 mice were stimulated under Th17 conditions, CD8+CD122+/CD8+CD122− T cells either from naïve Ly5.2 wild type C57BL/6 (WT) or from Ly5.2 IL-15 transgenic mice (IL-15 Tg) were cocultured with CD4+CD25− T cells for 4 days at a 1:1 ratio. Supernatants were collected and IL-17 was detected by ELISA. In selected groups, 10 μg/mL of anti-IL-10 was added at the initiation of the incubation. *p < 0.05 compared to effector cells alone group analyzed by Student’s t-test. (D) Effect of CD8+CD122+ T cells on the proliferation of CD4+CD25− T-cell proliferation upon anti-CD3 stimulation. Different ratios of effector cells to potential regulatory cells (CD8+ CD122+ or CD8+ CD122− T cells) were cocultured with naïve CD4+CD25− T cells upon plate bound anti-CD3 stimulation for 72 h, cell proliferation was measured in the final 6 h by 3H-TdR incorporation. (C, D) Data are shown as mean ± SD of three samples from a single experiment representative of three independent experiments performed. *p < 0.05 compared to effector cells alone group (Student’s t-test).
Taken together, these results suggest that CD8+CD122+ T cells play an important role in limiting IL-17 production, in differentiating Th17 cells, and that IL-10 is one of the effectors produced by CD8+CD122+ T cells that suppress EAE.
Discussion
IL-15 and IL-2 are common γ chain cytokines that play pivotal roles in the regulation of the immune response. Their heterotrimeric receptors share the γc with IL-4, IL-7, IL-9, and IL-21 [4, 5]. Furthermore IL-2 and IL-15 also share the IL-2/IL-15β receptor subunit (IL-2/IL-15Rβ, CD122) [1]. The high-affinity forms of IL-2 and IL-15 receptors also include the private receptor subunits IL-2Rα(CD25) and IL-15Rα(CD215). IL-2 and IL-15 have a number of related functions due to their sharing of receptor subunits and signaling elements, but also have distinct and often competing roles in select adaptive immune responses. Both IL-2 and IL-15 have roles in the generation, maintenance, and activation of multiple elements of the immune system including stimulation of T-cell proliferation, induction of cytotoxic T cells, generation, and maintenance of NK cells. However, both cytokines are also responsible for maintenance of distinct negative regulatory checkpoints on the immune system that act to prevent the development of autoimmune disorders. In particular, IL-2 is involved in the process of activation-induced cell death (AICD) and in the maintenance of CD4+CD25+ regulatory T (Treg) cells [4, 40]. In the case of IL-15, its administration is associated with the production of inhibitory elements including IL-10 and with PD-1 expression on CD8+ T cells, and as shown in this and previous reports the generation and maintenance of CD8+CD122+ negative regulatory T cells [41, 42]. Both IL-2 and IL-15 have been administered clinically to activate the immune response in the treatment of patients with malignant melanoma and metastatic renal cell cancer, and as an element of molecular vaccines. Conversely, antibody-mediated blockade of both IL-2 and IL-15 have been proposed to treat certain autoimmune disorders. In light of checkpoint actions, one must take into consideration induction of IL-15 dependent-CD8+CD122+ regulatory cells and inhibitory cytokines in design of clinical trials that involve IL-15 or alternatively agents that inhibit its action. For example, we demonstrated that although IL-15 showed efficacy in CT-26 and TRAMP-C2 tumor models in mice, it was not optimal because of its induction of immunological checkpoints [41, 42]. We addressed this issue by simultaneous coadministration with IL-15, of anti-PD-L1 and anti-CTLA-4, thereby markedly increasing the efficacy of IL-15 in the treatment of these murine tumor models [41, 42].
In parallel in treatment of patients with autoimmune disorders, one must take into account induction by IL-15 of immunoregulatory CD8+CD122+ T cells when contemplating use of an antibody such as Mikβ1(anti-CD122, IL-2/IL-15β) that blocks IL-15 transpresentation to human cells expressing CD122. Such a blockade would appear to be rational in situations where IL-15 plays a central role in the pathogenesis of the disease but should be avoided in situations where its role in facilitating the actions of immunoregulatory CD8+CD122+ T cells predominates. In particular, antibody mediated blockade of IL-15 transpresentation to CD122 expressing T cells would appear to be rational in treatment of patients with refractory celiac disease where evidence has implicated a pivotal pathogenic role for IL-15 [11, 12]. In contrast to the situation in refractory celiac disease, IL-15 blockade would appear to be contraindicated in certain other autoimmune disorders. For example, IL-15 has been shown to exacerbate collagen-induced arthritis in mice [43]. Furthermore, as shown in the present and previous studies antibody-mediated blockade of transpresentation of IL-15 to CD122 expressing CD8+ T cells with TMβ-1 exacerbated EAE [27]. Moreover, the exaggerated EAE disease in IL-15−/− mice confirmed the positive contribution of IL-15 to ameliorate this disease development in mice. Flow cytometeric analysis suggested that NK cells and CD8+CD122+ T cells were missing due to TMβ-1 treatment. Furthermore, studies in IL-15-Tg mice and those in IL-15−/− mice also supported the view that the subset of the CD8+CD122+ T-cell is an IL-15 dependent population.
No protective effects were observed after adoptive transfer of thymic and splenic derived NK cells to the TMβ-1 Fab treated animals (Fig. 2). This appears to provide some negative information for a role for NK cells in this model. However, it remains quite controversial to select particular markers of murine NK cells to resemble human CD56brightNK cells. Studies by others had suggested murine CD27 and CD11b [44], or CXCR3+CD27bright NK-cell [45] as the murine equivalent. Other investigators reported that CD11b and CD27 also reflect distinct populations and functional specializations in human NK cells [46]. Moreover, currently we used nonprimed 6 week-old RAG1−/− mice as the donor of NK cells. This choice may not reflect the full spectrum of NK function upon adoptive transfer. Further studies with additional selection markers and variable donors of NK cells to evaluate the role of NK cells would be of value.
Next we focused on CD8+CD122+ T cells. Compared with the conclusion that CD4+CD25+ regulatory T cells are involved in the prevention of select autoimmune disorders, the role of CD8+ regulatory T cells in EAE has still not been widely studied. A number of markers have been described for regulatory CD8 T-cell subsets. Some groups have shown certain CD8 regulatory T cells that act on follicular helper T cells express Qa-1 antigen [47]. Others have reported CD8 regulatory T cells include CD8+CD28− T cells [48], as well as human CD8+CD25+ regulatory T cells that have been shown to suppress Th1-cells[37]. Recently CD122 expressing CD8 regulatory T cells have been reported [27, 28, 35, 49]. In the experimental EAE model we observed that a subset of IL-15 dependent CD8+CD122+ T cells manifested regulatory/suppressive function. Adoptive transfer of these CD8+CD122+ T cells to TMβ-1 treated mice was associated with a diminution of the severe EAE features compared to transfer of CD8+CD122− or un-separated CD8+ T cells to TMβ-1 treated mice. Moreover, transfer of preactivated CD8+CD122+ T cells to naive recipient mice was associated with protection of animals from EAE. These observations confirmed the previous report and supported the hypothesis that CD8+CD122+ T cells manifest a regulatory action on EAE [27].
A phenotypic analysis demonstrated that a subset of CD8+CD122+ T cells express IL-15Rα, along with high expression of PD-1, CTLA-4, inhibitory MHC class I Ly49A, but did not express CD25, FoxP3 or CD69. It has been reported that the heterogeneous population of CD8+CD122+ T cells contain PD-1− memory cells and PD-1 expressing cells with suppressive action, while CD8+CD122+PD-1+cells produce IL-10 that regulates other T cells [49]. When we administered anti-IL-10 to the CD8+CD122+ T-cell recipient mice that had been treated with TMβ-1, the protective effect was diminished (Fig. 2A). This suggested that IL-10 was one of the factors produced by CD8+CD122+ T cells that ameliorated EAE symptoms.
As an effector cytokine, IL-17 has been associated with a broad variety of chronic disorders, suggesting a role in these diseases [50]. A crucial role for IL-17 has been reported in multiple sclerosis and its animal disease model EAE [22, 23]. In the present study, we utilized an in vitro system involving two cytokines, TGF-β and IL-6 to drive naïve CD4+ T cells to differentiate into IL-17-producing CD4+ T cells (Th17 cells). The addition of activated CD8+CD122+ T cells to this system was associated with a significant inhibition of the differentiation process (Fig. 6A). The suppression of CD4 T-cell proliferation and reduction of the secretion of IL-17 in vitro also reflected the regulatory function by CD8+CD122+ T cells on EAE disease (Fig. 5 and 6). Moreover, the addition of anti-IL-10 ameliorated the inhibition. This observation suggests that at least part of the regulatory function mediated by CD8+CD122+ T cells on Th17 differentiation was dependent on IL-10 (Fig. 6B). However, when we added the CD8+CD122+ T cells to the in vitro assay on day 3 instead of on day 0, no obvious suppression could be observed on Th17 differentiation. This echoes the in vivo EAE model results where as shown in Figure 5C, transferred naïve CD8+CD122+ T cells did not delay the disease initiation or ameliorate disease severity. The preactivation of CD8+CD122+ T cells was required for the amelioration of EAE at the initialization phase.
Intriguingly, concerning the IL-17 secretion in MOG EAE, there was a report by Lee and colleagues who collected the splenic cells from mice 14 days after MOG EAE induction and stimulated them with either anti-CD3 or MOG peptide, no significant difference was found between TMβ-1 treated and the control mice [27]. In contrast to Lee, currently we examined IL-17 secretion at the peak stage of disease development (day 19 after EAE induction), and detected significantly increased IL-17 secretion in the supernatant upon MOG peptide stimulation from mice that received TMβ-1 when compared to the control IgG treated group, p < 0.05 (Fig. 4A). In our case, the secretion level of IL-17 paralleled the severity of EAE. This conclusion was also confirmed in IL-15−/− mice that had more severe disease along with a higher level of MOG specific IL-17 secretion compared to wild-type animals (Fig. 4B). Thus our observations suggest that IL-15 acting through CD8+CD122+ T cells has a negative regulatory role reducing IL-17 production and Th17 mediated EAE inflammation.
In this study, we demonstrated an action of IL-15 dependent CD8+CD122+ T cells in reducing the symptoms of EAE in the acute phase. In particular, the administration of preactivated CD8+CD122+ T cells prevented EAE in the initialization phase.
Materials and methods
Mice
Ly5.1 C57BL/6, I115−/− mice, RAG1−/− Ly5.2 C57BL/6, Ly5.2 C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Human IL-15 transgenic C57BL/6 mice were generated by our group [3]. All animal experiments were performed at the National Cancer Institute (NCI) under a protocol approved by the NCI Animal Care and Use Committee.
Reagents
Anti-CD122 (TMβ-1) mAb was purchased from BioXcell (West Lebanon, NH). Mutated TMβ-1 mAb which does not mediate ADCC was a gift from the UCB Company (Smyrna, GA). All other antibodies were purchased from eBioscience (San Diego, CA). Mouse CD4+CD25+ regulatory T-cell isolation kit, Mouse NK Cell Isolation Kit, Mouse CD8 isolation kit and antibiotin microbeads were purchased from Miltenyi Biotec (Auburn, CA). Mouse IL-17 Quantikine ELISA kits, recombinant mouse IL-6 and human TGF-β were purchased from R&D systems (Minneapolis, MN). The myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (MEVGWYR-SPFSRVVHLYRNGK) was synthesized by EZBiolab (Westfield, IN).
In vivo blockade of IL-15 signaling by administration of the TMβ-1 mAb
To block IL-15 signaling in vivo, 50 μg of anti-CD122 mAb(TMβ-1) or control rat IgG was administered on day −1 by i.p. injection (MOG immunization: day 0). Where indicated, 500 μg of Fab fragments of TMβ-1 or control Ab (rat IgG) was injected intraperitoneally twice daily from day −1 to day 3. To obtain Fab fragments, Thermo Scientific Pierce Fab Preparation Kits were used to digest, separate Fab and Fc fragments and subsequently purify the Fab fragment (Thermo Scientific).
EAE induction in mice
Female mice (6–8 week old) were induced to develop EAE by s.c. injection at the base of the limbs with 200 μg of MOG35–55 peptide emulsified in CFA containing 4 mg/mL heat-killed Mycobacterium tuberculosis H37RA (Difco, Lawrence, KS) on day 0. 6 and 48 h later, the mice were administered i.v. with 500 ng of purified Bordetella Pertussis toxin (List Biological Laboratories, Campbell, CA). Disease severity was monitored according to the following scale: 0, no clinical score; 1, loss of tail tone; 2, hind limb weakness; 3, hind limb paralysis; 4, forelimb paralysis; and 5, moribund or dead. The score recordings were performed by different reviewers one blinded and the other unblinded to the experimental groups. These evaluations were associated with comparable clinical scores.
Isolation and adoptive transfer of NK cells
Thymocytes or splenocytes separated from 6 week old RAG1−/− Ly5.2 C57BL/6 mice were prepared. NK cells were isolated by a mouse NK Cell Isolation kit according to manufacturer’s instructions. 1 × 106 cells/ mouse thymic or splenic NK cells were intravenously injected into the recipients.
Isolation and adoptive transfer of CD8+ T cells or CD8+CD122+/ CD8+CD122− T cells
Spelnic CD8+ T cells were purified by a Mouse CD8 Isolation Kit (Miltenyi Biotec). CD8+ T cells were incubated with biotin-anti-CD122 antibody (eBioscience) and selected by Strepavidin-conjugated magnetic beads (Miltenyi Biotec). For the adoptive transfer study, 1 × 106 cells/ mouse cells were intravenously injected into the recipients. Where indicated, CD8+CD122+ T cells were activated by plate-bound anti-CD3 (1 μg/mL) plus soluble anti-CD28 (1 μg/mL) for 24 h, then washed with PBS and transferred into mice.
Th17 differentiation
Naïve splenic CD4+CD25− T cells from naive Ly5.1 C57BL/6 mice were separated using a mouse CD4+CD25+ Regulatory T-Cell Isolation Kit (Miltenyi Biotec). Cells were cultured in the presence of plate bound anti-CD3 (1 μg/mL) and soluble anti-CD28 (1 μg/mL) Abs under Th17 conditions for 4 days. Th17 cells were polarized using mIL-6 (10 ng/mL), hTGF-β (1 ng/mL), anti–IFN-γ (5 μg/mL) and anti–IL-4 (5 μg/mL). Where indicated, CD8+CD122+ T cells were added at an effector cell/ regulatory cell ratio of 1:0.5, 1:1 or 1:2 either at the beginning of the culture, or on day 3 of the assay.
Intracellular staining of cytokines and flow cytometry
Cells were cultured under Th17 conditions and fixed with a Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA). Before fixation, cells were restimulated with PMA (50 ng/mL) and ionomycin (500 ng/mL) for 4 h, and Brefeldin A (10 μg/mL) was added during the last 2 h. Data were acquired using a BD FACSCalibur cytometer (BD Biosciences) and were analyzed using FlowJo software (Tree Star).
Cell culture and cytokine detection
Splenocytes taken from EAE mice on day 19 were cultured with MOG35–55 for 72 h. Supernatants from cell cultures were collected and ELISA was performed according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN).
In vitro proliferation assay
Naïve CD4+CD25− T cells were cultured with plate bound anti-CD3 (1 μg/mL) with or without CD8+CD122+/CD8+CD122− T cells at various effector to regulatory T-cell ratios (1:0.5, 1:1 or 1:2). After culture for 72 h, 3H-TdR was added to the assay to quantitate proliferation of the cells.
Statistical analyses
The p values were calculated by Student’s t-test using Microsoft Excel software exploring unpaired, two-tailed distribution and two-sample equal variance parameters using JMP statistical software (SAS Institute, Cary, NC). A value of p < 0.05 was considered significant.
Supplementary Material
Acknowledgments:
This work was supported by the Intramural Research Program of the Center for Cancer Research, NCI, NIH.
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
Additional supporting information may be found in the online version of this article at the publisher’s web-site
References
- 1.Bamford RN, Grant AJ, Burton JD, Peters C, Kurys G, Goldman GK, Brennan J et al. , The IL-2 receptor beta chain is shared by IL-2 and a cytokine, provisionally designated IL-T, that stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc. Natl. Acad. Sci. USA 1994. 91: 4940–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldmann TA and Tagaya Y, The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu. Rev. Immunol 1999. 17: 19–49. [DOI] [PubMed] [Google Scholar]
- 3.Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, Waldmann TA et al. , IL-2 induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc. Natl. Acad. Sci. USA 2000. 97: 11445–11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waldmann TA, The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat. Rev. Immunol 2006. 6: 595–601. [DOI] [PubMed] [Google Scholar]
- 5.Fehniger TA and Galigiuri MA, Interleukin 15: biology and relevance to human disease. Blood 2001. 97:14–32. [DOI] [PubMed] [Google Scholar]
- 6.Dubois S, Mariner J, Waldmann TA and Tagaya Y, IL-15R alpha recycles and presents IL-15 in trans to neighboring cells. Immunity 2002. 17: 537–547. [DOI] [PubMed] [Google Scholar]
- 7.Blanco-Jerez C, Plaza JF, Masjuan J, Orensanz LM and Alvarez-Cermeno JC, Increased levels of IL-15 mRNA in relapsing-remitting multiple sclerosis attacks. J. Neuroimmunol 2002. 128: 90–94. [DOI] [PubMed] [Google Scholar]
- 8.McInnes IB, Leung BP, Sturrock RD, Field M and Liew FY, Interleukin-15 mediates T-cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat. Med 1997. 3: 189–195. [DOI] [PubMed] [Google Scholar]
- 9.Kirman I and Nielsen OH, Increased numbers of interleukin-15-expressing cells in active ulcerative colitis. Am. J. Gastroenterol 1996. 91: 1789–1794. [PubMed] [Google Scholar]
- 10.Liu Z, Geboes K, Colpaert S, D’Haens GR, Rutgeerts P and Ceuppens JL, IL-15 is highly expressed in inflammatory bowel disease and regulates local T-cell-dependent cytokine production. J. Immunol 2000. 164: 3608–3615. [DOI] [PubMed] [Google Scholar]
- 11.Benhmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, Allez M et al. , Inhibition of TGF-beta signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 2007. 132: 994–1008. [DOI] [PubMed] [Google Scholar]
- 12.Yokoyama S, Watanabe N, Sato N, Perera PY, Filkoski L, Tanaka T, Miyasaka M et al. , Antibody-mediated blockade of IL-15 reverses the autoimmune intestinal damage in transgenic mice that overexpress IL-15 in enterocytes. Proc. Natl. Acad. Sci. USA 2009. 106:15849–15854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meresse B, Curran SA, Ciszewski C, Orbelyan G, Setty M, Bhagat G, Lee L et al. , Reprogramming of CTLs into natural killer-like cells in celiac disease. J. Exp. Med 2006. 203:1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen J, Feigenbaum L, Awasthi P, Butcher DO, Anver MR, Golubeva YG, Bamford R et al. , Insulin-dependent diabetes induced by pancreatic beta cell expression of IL-15 and IL-15Rα. Proc. Natl. Acad. Sci. USA 2013. 110:13534–13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuczyński S, Winiarska H, Abramczyk M, Szczawinska K, Wierusz-Wysocka B and Dworacka M, IL-15 is elevated in serum patients with type 1-diabetes mellitus. Diabetes Res. Clin. Pract 2005. 69: 231–236. [DOI] [PubMed] [Google Scholar]
- 16.Kim YS, Maslinski W, Zheng XX, Stevens AC, Li XC, Tesch GH, Kelley VR et al. , Targeting the IL-15 receptor with an antagonist IL-15 mutant/Fcγ2a protein blocks delayed-type hypersensitivity. J. Immunol 1998. 160: 5742–5748. [PMC free article] [PubMed] [Google Scholar]
- 17.Baslund B, Tvede N, Danneskiold-Samsoe B, Larsson P, Panayi G, Petersen J, Petersen LJ et al. , Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis. Rheum 2005. 52:2686–2692. [DOI] [PubMed] [Google Scholar]
- 18.Morris JC, Janik JE, White JD, Fleisher TA, Brown M, Tsudo M, Goldman CK et al. , Preclinical and phase I clinical trial of blockade of IL-15 using Mikbeta1 monoclonal antibody in T cell large granular lymphocyte leukemia. Proc. Natl. Acad. Sci. USA 2006. 103: 401–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinman L, Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 1996. 85:299–302. [DOI] [PubMed] [Google Scholar]
- 20.Kivisakk P, Matusevicius D, He B, Soderstrom M, Fredrikson S and Link H, IL-15 mRNA expression is up-regulated in blood and cerebrospinal fluid mononuclear cells in multiple sclerosis (MS). Clin. Exp. Immunol 1998. 111: 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Losy J, Niezgoda A and Zaremba J, IL-15 is elevated in sera of patients with relapsing-remitting multiple sclerosis. Folia Neuropathol. 2002. 40: 151–153. [PubMed] [Google Scholar]
- 22.Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S and Link H, Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult. Scler 1999. 5:101–104. [DOI] [PubMed] [Google Scholar]
- 23.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K et al. , IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol 2006. 177:566–573. [DOI] [PubMed] [Google Scholar]
- 24.Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N and Mills KH, T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol 2010. 162:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun D, Whitaker JN, Huang Z, Liu D, Coleclough C, Wekerle H and Raine CS, Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J. Immunol 2001. 166: 7579–7587. [DOI] [PubMed] [Google Scholar]
- 26.Huseby ES, Liggitt D, Brabb T, Schnabel B, Ohlén G and Goverman J, A pathogenic role for myelin-specific CD8+ T cells in a model for multiple sclerosis. J. Exp. Med 2001. 194:669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YH, Ishida Y, Rifa’i M, Shi Z, Isobe K and Suzuki H, Essential role of CD8+CD122+ regulatory T cells in the recovery from experimental autoimmune encephalomyelitis. J. Immunol 2008. 180:825–832. [DOI] [PubMed] [Google Scholar]
- 28.Mangalam AK, Luckey D, Giri S, Smart M, Pease LR, Rodriguez M and David CS, Two discreet subsets of CD8 T cells modulate PLP(91–110) induced EAE in HLA-DR3 transgenic mice. J. Autoimmun 2012. 38:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsudo M, Kitamura F and Miyasaka M, Characterization of the IL-2R1538; chain using 3 distinct monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1989. 86: 1982–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waldmann TA, Conlon KC, Stewart DM, Worthy TA, Janik JE, Fleisher TA, Albert PS et al. , Phase 1 trial of IL-15 trans-presentation blockade using humanized Mikβ1 mAb in patients with T-cell large granular lymphocytic leukemia. Blood 2013. 121: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hakimi J, Ha VC, Lin P, Campbell E, Gately MK, Tsudo M, Payne PW et al. , Humanized Mikβ1, a humanized antibody to the IL-2Rβ chain that acts synergistically with humanized anti-TAC. J. Immunol 1993. 151: 1075–1085. [PubMed] [Google Scholar]
- 32.Tanaka T, Tsudo M, Karasuyama H, Kitamura F, Kono T, Hatakeyama M, Taniguchi T et al. , A novel monoclonal antibody against murine IL-2Rβ chain. Characterization of receptor expression in normal lymphoid cells and EL-4 cells. J. Immunol 1991. 147: 2222–2228. [PubMed] [Google Scholar]
- 33.Vosshenrich CA, García-Ojeda ME, Samson-Villíger SI, Pasqualetto V, Enault L, Richard- Le, Goff O et al. , A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol 2006. 7: 1217–1224. [DOI] [PubMed] [Google Scholar]
- 34.Bielekova B, Gatalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H et al. , Regulatory CD56bright natural killer-cells mediate immunomodulatory effects of IL-2R1537;-targeted therapy (daclizumab) in multiple sclerosis. Proc. Natl Acad. Sci. USA 2006. 103: 5941–5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rifa’i M, Kawamoto Y, Nakashima I and Suzuki H, Essential roles of CD8+CD122+ regulatory T cells in the maintenance of T-cell homeostasis. J. Exp. Med 2004. 200: 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai H, Wan N, Zhang S, Moore Y, Wan F and Dai Z, Programmed death-1 defines CD8+CD122+ T cells as regulatory versus memory T cells. J. Immunol 2010. 185: 803–807. [DOI] [PubMed] [Google Scholar]
- 37.Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, Santarlasci V et al. , Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood 2003. 102: 4107–4114. [DOI] [PubMed] [Google Scholar]
- 38.Najafian N, Chitnis T, Salama AD, Zhu B, Benou C, Yuan X, Clarkson MR et al. , Regulatory functions of CD8+CD28− T cells in an autoimmune disease model. J. Clin. Invest 2003. 112: 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kima H-J, Wang X, Radfara S, Sproule T-J, Oopenian DJ and Cantor H, CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc. Natl Acad. Sci. USA 2011. 108: 2010–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakaguchi S, Yamaguchi T, Nomura T and Ono M, Regulatory T cells and immune tolerance. Cell 2008. 133: 775–787. [DOI] [PubMed] [Google Scholar]
- 41.Yu P, Steel JC, Zhang M, Morris JC and Waldmann TA, Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin. Cancer. Res 2010. 16: 6019–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu P, Steel JC, Zhang M, Morris JC, Waitz R,Fasso M, Allison JP et al. , Simultaneous inhibition of two regulatory T-cell subsets enhanced Interleukin-15 efficacy in a prostate tumor model. Proc. Natl. Acad. Sci. USA 2012. 109: 6187–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshihara K, Yamada H, Hori A, Yajima T, Kubo C and Yoshikai Y, IL-15 exacerbates collagen-induced arthritis with an enhanced CD4+ T-cell response to produce IL-17. Eur. J. Immunol 2007. 37: 2744–2752. [DOI] [PubMed] [Google Scholar]
- 44.Hayakawa Y, Huntington ND, Nutt SL and Smyth MJ, Functional subsets of mouse natural killer cells. Immunol. Rev 2006. 214: 47–55. [DOI] [PubMed] [Google Scholar]
- 45.Marquardt N, Wilk E,Pokoyski C, Schmidt RE and Jacobs R, Murine CXCR3+CD27bright NK cells resemble the human CD56bright NK-cell population. Eur. J. Immunol 2010. 40:1428–1439. [DOI] [PubMed] [Google Scholar]
- 46.Fu B, Wang F, Sun R, Ling B, Tian Z and Wei H, CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology 2011. 133: 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang H, Ware R, Stall A, Flaherty L, Chess L and Pernis B, Murine CD8+ T cells that specifically delete autologous CD4+ T cells expressing V1538;8 TCR: a role of the Qa-1 molecule. Immunity 1995. 2:185–194. [DOI] [PubMed] [Google Scholar]
- 48.Cortesini R, LeMaoult J, Ciubotariu R and Cortesini NS, CD8+CD28− T-suppressor cells and the induction of antigen-specific, antigen-presenting cell-mediated suppression of Th reactivity. Immunol. Rev 2001. 182: 201–206. [DOI] [PubMed] [Google Scholar]
- 49.Endharti AT, Rifa’I M, Shi Z, Fukuoka Y, Nakahara Y, Kawamoto Y, Takeda K et al. , CD8+CD122+ regulatory T cells produce IL-10 to suppress IFN-γ production and proliferation of CD8+ T cells. J. Immunol 2005. 175: 7093–7097. [DOI] [PubMed] [Google Scholar]
- 50.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y et al. , A distinct lineage of CD4 T cells regulates tissue inflammation by producing IL-17. Nat. Immunol 2005. 6:1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.