

Abstract
MycG is a cytochrome P450 that performs two sequential oxidation reactions on the 16-membered ring macrolide M-IV. The enzyme evolved to oxidize M-IV preferentially over M-III and M-VI, which differ structurally by the presence of methoxy vs free hydroxyl groups on one of the macrolide sugar moieties. We utilized a two-pronged computational approach to study both the chemoselective reactivity and substrate specificity of MycG. Density functional theory computations determined that epoxidation of the substrate hampers its ability to undergo C-H abstraction primarily due to a loss of hyperconjugation in the transition state. Metadynamics and molecular dynamics simulations revealed a hydrophobic sugar-binding pocket deep within the enzyme that is responsible for substrate recognition/specificity and was not apparent in crystal structures of the enzyme/substrate complex. The residues in the sugar-binding pocket are found to be conserved in the cytochrome P450 superfamily, with mutation of these residues leading to low protein expression. Computational results also led to the identification of other interactions between the enzyme and its substrates that had not previously been observed in the experimental cocrystal structures. Site-directed mutagenesis was then employed to test the effects of mutations hypothesized to broaden the substrate scope and alter the product profile of MycG. The results of these experiments validated this complementary effort to engineer MycG variants with improved catalytic activity toward earlier stage mycinamicin substrates.
Keywords: P450, quantum mechanics, DFT, molecular dynamics, enzyme engineering
Graphical Abstract
INTRODUCTION
Cytochrome P450 enzymes catalyze a wide array of reactions, including, but not limited to, hydroxylation, epoxidation, C-C coupling, N-dealkylation, sulfoxidation, and arene oxidation.1–6 While P450S have evolved to catalyze reactions with excellent selectivity and efficiency, it is rare to find members of the P450 superfamily that are capable of catalyzing multiple oxidative steps involving separate reaction mechanisms. Initially described as a key player in the biosynthesis of mycinamicin macrolide antibiotics, MycG catalyzes the sequential hydroxylation and epoxidation of mycinamicin IV (M-IV) to generate mycinamicin II (M-II).7–9 Notably, the order in which these oxidative steps are carried out is critical: direct epoxidation of M-IV leads to the formation of mycinamicin I (M-I), a shunt metabolite that cannot undergo MycG-catalyzed hydroxylation to produce M-II (Scheme 1).
Scheme 1.
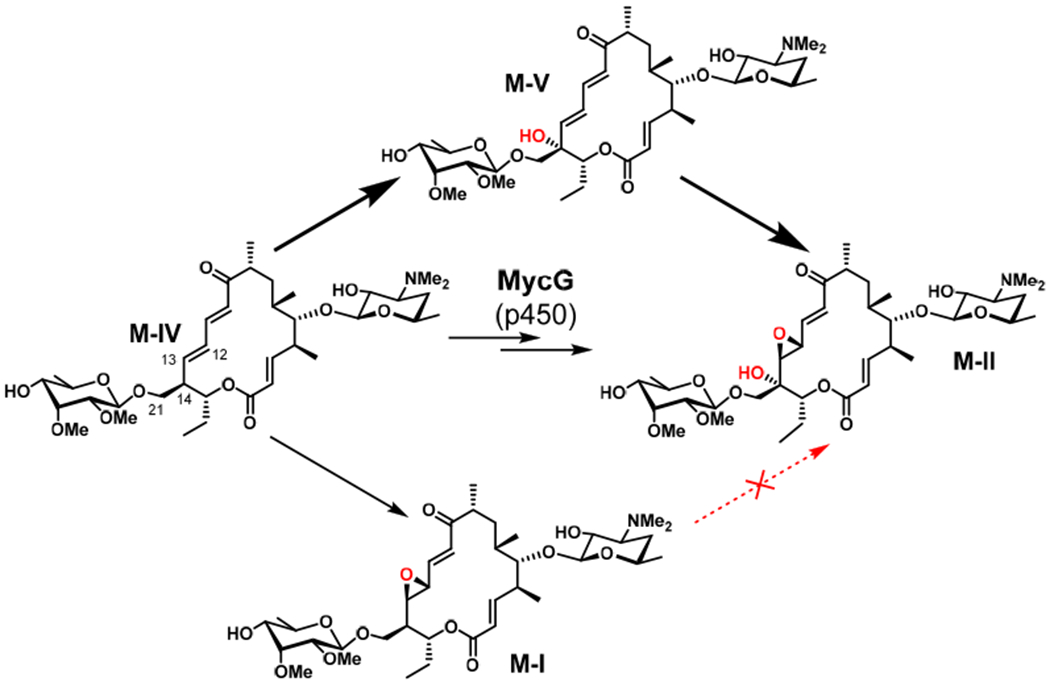
MycG-catalyzed oxidation of M-IV. Hydroxylation to form M-V followed by epoxidation leads to M-II, whereas direct epoxidation of M-IV leads to shunt metabolite M-I.
In addition to selectively performing two sequential and distinct oxidative steps, MycG is highly substrate specific. M-IV (bearing a C-21 mycinose), M-III (bearing a C-21 javose), and M-VI (bearing a C-21 6-deoxyallose) are identical in macrocyclic structure, differing only in the extent of O-methylation of the sugar moiety linked to C-21. Despite having ≥110 atoms in common (>94% structural identity to M-IV), substrates M-III and M-VI undergo low levels of oxidation by MycG. The number of methoxy groups present on the C-21-linked deoxysugar directly correlates with the reactivity of the substrate, as the yield of oxidation products decreases in the following order: M-IV (two methoxy groups) > M-III (one methoxy group) > M-VI (no methoxy groups).
Due to the unusual substrate selectivity and reactivity preferences of MycG, we pursued a two-pronged computational study of this P450. First, we used density functional theory (DFT) calculations to understand the specific oxidation sequence of M-IV, which involves initial hydroxylation (C-14) followed by epoxidation (C-12/C-13 double bond). Then, molecular dynamics (MD) simulations and metadynamics were employed to determine how minor differences in the sugar linked to C-21, remote from the sites of oxidation, impact substrate selectivity. Collectively, these complementary approaches provided a new understanding of this complex, multifunctional P450. Based on computation-derived prediction, subsequent mutagenesis experiments helped to improve reactivity of MycG towards M-VI and M-III.
RESULTS
Using A as a model for M-IV and B as a model for M-I (Figure 2), we performed DFT calculations to determine the barriers to C-H abstraction for each of these substrates. With respect to separated reactants, the free energy barrier to C-H abstraction from A (A-TS) was 14.6 kcal/mol while that from B (B-TS) was 20.1 kcal/mol (Figure 2). This 5.5 kcal/mol increase in energy for B-TS means that C-H abstraction from the epoxidized substrate is ~10,000 times slower than that from the non-epoxidized substrate. This drastic difference in reactivity is in accord with the lack of activity of MycG toward M-I. Thus, the presence of the epoxide in M-I decreases the inherent reactivity of the substrate such that MycG is unable to effectively catalyze hydroxylation of this biosynthetic intermediate.
Figure 2.
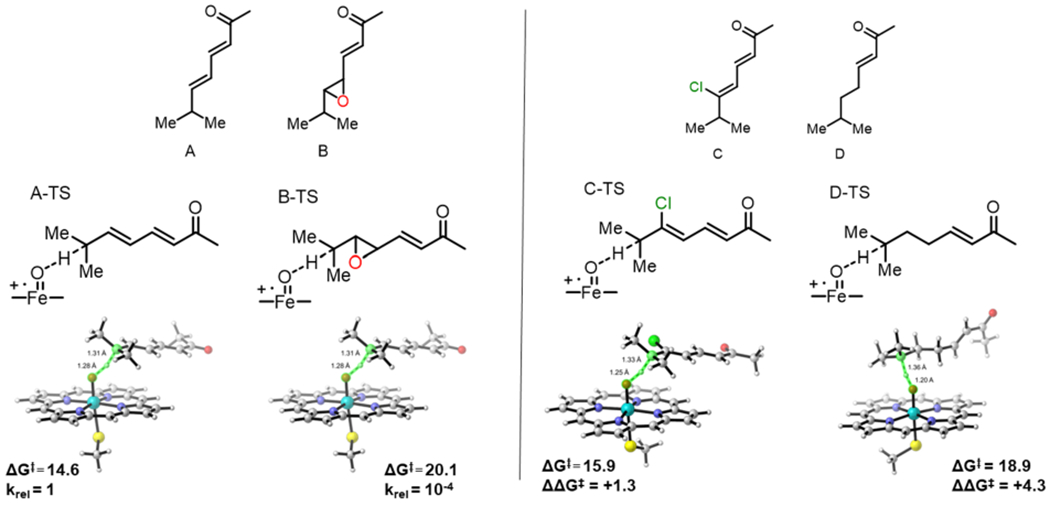
Left panel: free energies (in kcal/mol) and transition structures of C-H abstraction from substrate models A and B. Right panel: substrate models C and D (top) and the respective transition structures with transition state free energy barriers (ΔG‡) and difference from A-TS (ΔΔG‡) for C-TS and D-TS (bottom).
We reasoned that there are two attributes of the M-I epoxide that could destabilize the developing radical in the transition state and could therefore be the origin of the barrier increase: 1) the lack of allylic stabilization or 2) the electron-withdrawing nature of the oxygen atom. To probe these two properties of the epoxide independently, we computed the barriers to C-H abstraction of two additional substrate models. Model C mimics the electron-withdrawing nature of the oxygen in M-I but maintains the conjugation present in M-IV. Model D mimics the loss of conjugation due to the epoxide, but no electron withdrawing groups are present. The barriers to C-H abstraction from C and D were 15.9 (C-TS) and 18.9 (D-TS) kcal/mol, respectively (Figure 2). Compared with A-TS, the barrier increases by +1.3 kcal/mol for C-TS and by +4.3 kcal/mol for D-TS. Therefore, while both factors contribute to the barrier increase in B-TS relative to A-TS, the lack of allylic stabilization plays a more significant role as demonstrated by the more significant barrier increase for D-TS. While the finding that removal of allylic stabilization is not surprising or novel, our findings do highlight that though P450S catalyze many C-H hydroxylations of unactivated C-H bonds, the disfavoring of some substrates is too significant to overcome. In this case, the inherent reactivity of the substrate prevails over the activity of the catalyst enzyme.
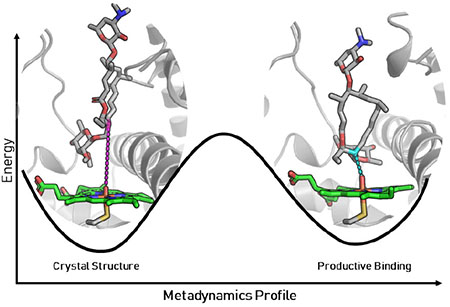
With the reactivity and chemoselectivity of MycG explored and elucidated with DFT calculations, we sought to understand the substrate specificity using molecular dynamics. The crystal structure of MycG in complex with M-IV shows that the substrate binds in a “mycinose-in desosamine-out” binding mode.7 However, the mycinose sugar of M-IV is located directly above the heme cofactor, hindering access of the macrolactone to the heme iron. The site of hydroxylation of M-IV is about 8.9 Å away from the heme iron, a distance long enough to preclude productive substrate oxidation. This observation indicates that the crystal structure does not capture an on-cycle, reactive pre-complex. A novel binding mode was revealed by roo ns restrained MD simulations with harmonic restraints added along the distance from C-14 to the oxygen of the iron-oxo species. The restrained complex led to geometries more consistent with productive C-H-abstraction (Figure 3). The simulations illuminated a different binding mode for M-IV characterized by a much shorter C-14 to O=Fe distance of 3.5 Å (Figure 3). In this binding mode, the macrolactone ring is rotated by ~90 degrees, with the deoxysugar mycinose pushed into a hydrophobic cavity composed of residues L227, L83, and L94. The desosamine sugar on the opposite end of the macrocycle is also pulled into the binding cavity, and the BC and FG loops form a “closed gate” mediated by a salt bridge interaction between R75 and E173 (Figure 3).
Figure 3.
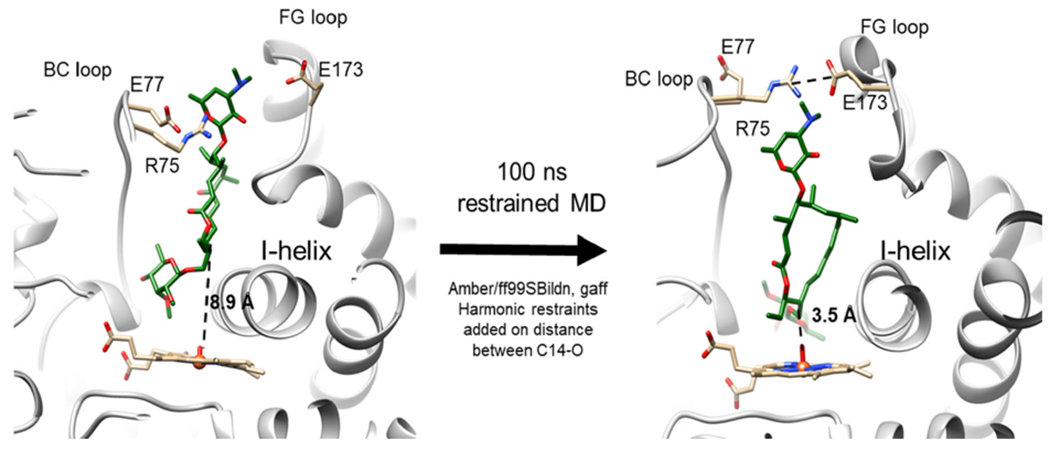
Binding configuration of M-IV in MycG as observed in the cocrystal structure (PDB 2Y98; left), and binding configuration of the same substrate with C-14 constrained to be proximal to the heme iron of MycG (right). The FG (containing E173) and BC (containing R75 and E77) loops are shown and labeled accordingly. The I-helix (containing L227) adjacent to the heme group is also shown and labeled accordingly.
To compare the free energies of the two different binding modes, metadynamics were carried out on M-IV, M-III, and M-VI bound to MycG. 250 ns metadynamics calculations were conducted using the Amber/ff99SBildn force field, and the free energy profiles were plotted along the HC14—OFe distance from 1.1Å to 7.2 Å (Figure 4). The free energy profiles show that there are two minima for each substrate within MycG. One minimum is located at a HC14—OFe distance of ~7.1 Å, representing a non-reactive binding mode (similar to that shown in the crystal structure). The other minimum is located at a distance of ~2.7 Å, corresponding to a reactive binding configuration. A small energy barrier of only ~5-7 kcal/mol must be overcome for C-14 to approach the iron-oxo species by rearrangement of both the enzyme and bound substrate (Figure 4). While these two minima are essentially isoenergetic for M-IV, the reactive binding pose is significantly less stable than the non-reactive binding conformation for both M-III and M-VI. This observation is consistent with the reduced activity of MycG toward M-III and M-VI since the simulations indicate that a stable, reactive complex cannot be readily attained. The reason for the destabilization of the reactive binding pose for both M-III and M-VI relative to that for M-IV can be understood upon further investigation of the deoxysugar binding pocket.
Figure 4.
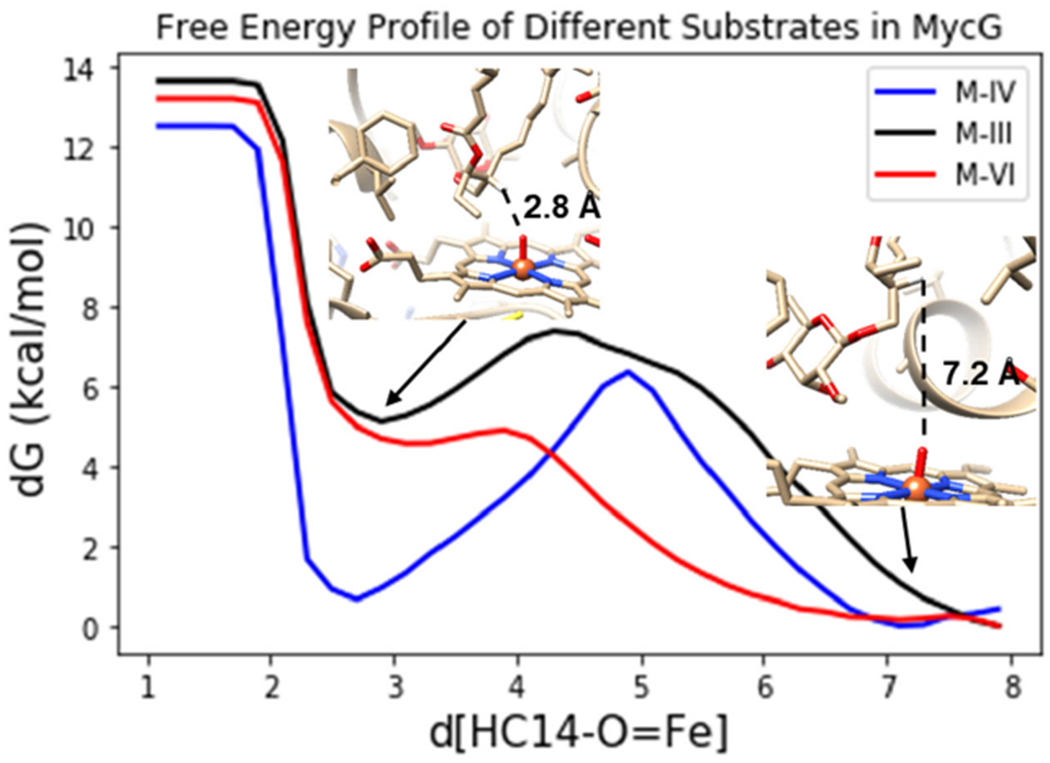
Free energy profiles of M-IV (blue), M-III (black), M-VI (red) binding in MycG along the HC14—OFe bond.
As previously noted, the only difference between these three substrates is the identity of the deoxysugar attached to C-21 (mycinose in M-IV, javose in M-III, and 6-deoxyallose in M-VI). As elucidated by the restrained MD, the mycinose of M-IV resides in a hydrophobic pocket composed of L227, L94, and L83 in the reactive binding pose. The metadynamics show that in this active pose, the MycG-M-IV complex is just as stable as that seen in the non-reactive crystal structure pose. In contrast, M-III and M-VI are destabilized in the active pose relative to the inactive pose (Figure 4). This destabilization is most likely due to the decreased hydrophobicity of the deoxysugars of M-III and M-VI when they are forced into the hydrophobic sugar-binding pocket (Figure 5). Additional 500 ns MD simulations were performed starting from each the reactive binding poses of M-IV, M-III, and M-VI. Over the course of the additional MD simulations, the average distance of the M-IV mycinose to the center of the hydrophobic residues (L227, L94, and L83) is only 3.4 Å, a reasonable van der Waals interaction distance. This distance increases to 5.4 Å for javose (M-III) and 7.6 Å for 6-deoxyallose (M-VI).
Figure 5.
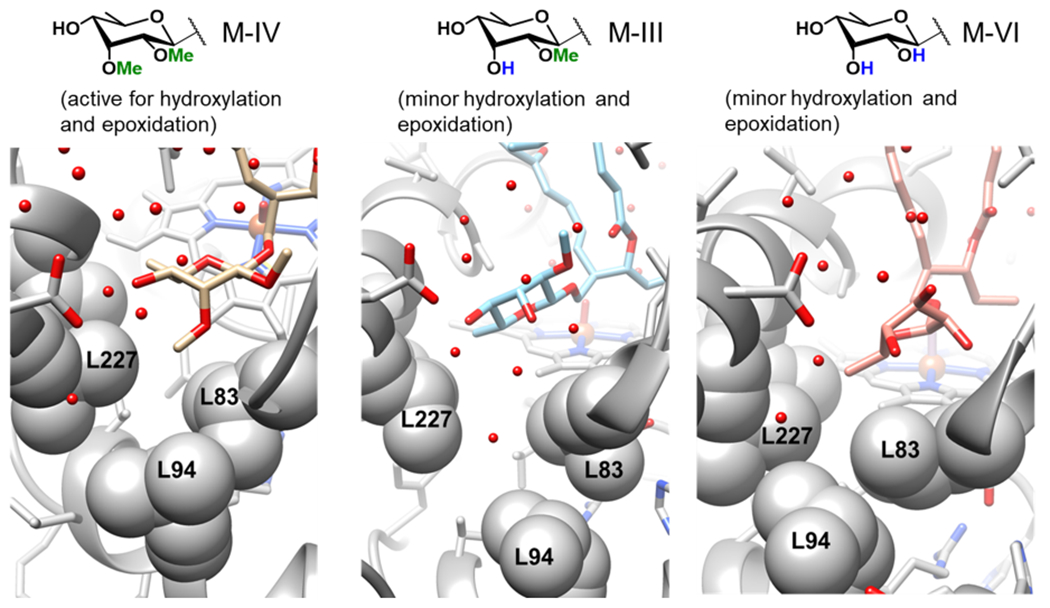
Deoxysugars of M-IV (left, tan), M-III (middle, blue), M-VI (right, pink) bound in the hydrophobic pocket composed of L227, L94, and L83.
The FG and BC loops of MycG at the exterior of the binding pocket contain residues E173 and E77, respectively, which form salt bridges with the desosamine sugar of M-IV in the cocrystal structure. These loops also play a key role in the interactions between MycG and its substrates in their productive binding poses. The two loops form a “closed” conformation for a reasonable portion of the simulation with M-IV in the productive binding mode. This closed conformation is maintained by a new salt bridge interaction between R75 of the BC loop and E173 of the FG loop for about 100 ns during the MD simulation with M-IV. However, in the simulations with M-III and M-VI, this salt bridge interaction is not present or stable, with the distances between the two residues averaging 12 Å and 20 Å for M-III and M-VI, respectively (Figure 6 and Figure S1). For M-IV, the two salt bridges between desosamine and either E173 or E77 are still observed in the closed conformation, but the desosamine-E173 interaction breaks after 100 ns. The simulation with M-III reveals that, while the desosamine-E77 interaction persists throughout the entire MD simulation, no salt bridge interaction involving E173 is established. Neither salt bridge is present in the MD simulation with M-VI (see the Supporting Information).
Figure 6.
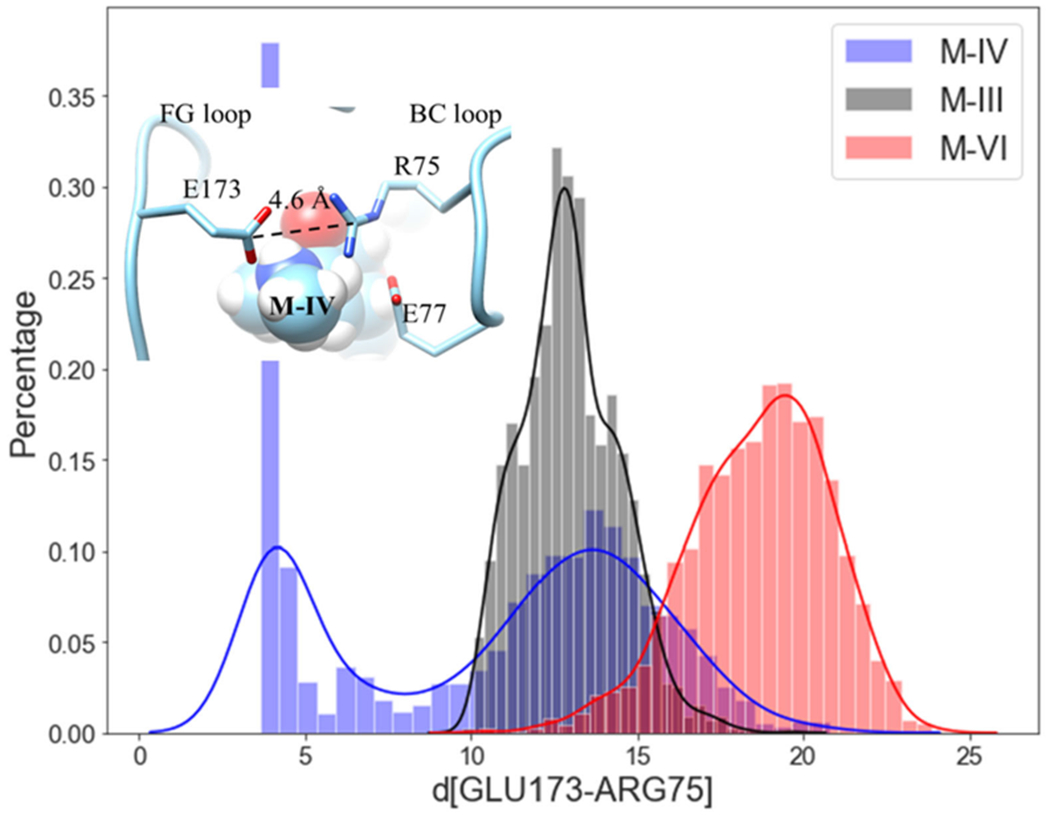
The distribution of the distances between E173 and R75 for M-IV (blue), M-III (black), and M-VI (red).
While the hydrophobic pocket and the FG/BC loop salt bridges are far from one another, they both appear to be linked to the substrate specificity of MycG. Taken together, our data reveal how the enzyme has evolved to exhibit optimal activity toward M-IV. In the reactive pose revealed by constrained MD and metadynamics, the mycinose sugar of M-IV binds in the hydrophobic pocket comprising L227, L94, and L83. This binding shifts L227 and L83 apart to create space for the sugar moiety, resulting in the movement of the I-helix proximal to the heme (Figure 3). This movement creates a more spacious pocket for the macrolactone core of M-IV and enables C-14 to more closely approach the iron-oxo species. In turn, movement of the macrolactone deeper into the binding pocket enables the FG and BC loops to move closer together. Closure of the FG/BC gate further stabilizes the salt bridges between desosamine and E173/E77. This cascade of motion and binding of M-IV relies upon initial effective and tight binding of the sugar and the subsequent establishment of FG/BC loop salt bridge interactions (Figure S2). When the hydrophobicity of the sugar at C-21 is decreased as in M-III and M-VI, this binding cascade fails at the start. Binding of the more hydrophilic sugars in the hydrophobic pocket is destabilized, which partially pushes the substrate out of the binding pocket. Consequently, the substrate is more distal from the iron-oxo species, tending to force the FG/BC gate open. From these results, we conclude that the specificity of MycG toward M-IV originates from the binding of a more hydrophobic deoxysugar in the hydrophobic pocket, but it is also a result of interactions in three different binding regions (Figure S2).
On the basis of the results of our computational studies, a selection of amino acid residues located in these three regions was targeted for mutagenesis with the goal of expanding the substrate tolerance of MycG. Mutations at L227, L94, and L83 were designed to modify the hydrophobicity of the active site cavity where the deoxysugar binds, and mutations adjacent to E173, R75, and E77 were designed to induce a tighter connection between the FG and BC loops. S170D was proposed to form a new salt bridge interaction with desosamine (Figure S3). Additionally, L280, located in the central region of the binding pocket, was targeted in an effort to alter the binding pocket so as to favor the “desosamine-out” binding mode for improved catalytic activity toward M-III and M-VI (Figure S4).
Due to its ease of expression and purification as well as demonstrated functional activity in vitro,20 we employed the catalytically self-sufficient MycG-RhFRED fusion protein as a template for mutagenesis in order to test our hypotheses for improving MycG activity toward M-III and M-VI. The wild-type enzyme exhibited the highest level of activity on its physiologically preferred substrate M-IV, converting nearly all of it to a mixture of singly (32% M-V + 39% M-I) and doubly (22% M-II) oxidized products. Consistent with prior results,7,8 MycG-RhFRED was also capable of oxidizing, to a lesser extent, M-III (21% conversion) and M-VI (9% conversion), producing a mixture of hydroxylated, epoxidized, and N-demethylated products. However, virtually no products bearing any combination of these modifications were detected. While oxidized products result from productive “desosamine-out” binding as discussed above, N-demethylation must arise via the non-productive “desosamine-in” binding pose in which the desosamine sugar binds proximal to the heme (Figure S4). Notably, while 14-hydroxy-M-III (M-IX), 14-hydroxy-M-VI (M-XV), and 12,13-epoxy-M-III have all previously been isolated from the fermentation broth of Micromonospora griseorubida,8,21 12,13-epoxy-M-VI remains uncharacterized.
Various residues and combinations thereof were targeted for mutagenesis, and all MycG-RhFRED mutants were tested with each of the three substrates (for results with M-IV, see Figure 7; for results with M-III and M-VI, see Figure 8). Mutating the putative mycinose binding pocket of MycG (L83/L94/L227) resulted in essentially complete abrogation of activity across all substrates tested (Figures 7 and 8). These triple mutants expressed at very low levels relative to the wild-type enzyme. Given that the hydrophobic nature of these residues is conserved among functional homologs of MycG and other macrolide biosynthetic P450S, these residues are likely critical to maintaining overall protein stability. A E237A/G281IK double mutant also expressed very poorly and exhibited essentially no activity toward any of the substrates. Multiple sequence alignment indicated that E237 likely plays the role of the catalytic acid required for O2 activation in this and related P450S.
Figure 7.
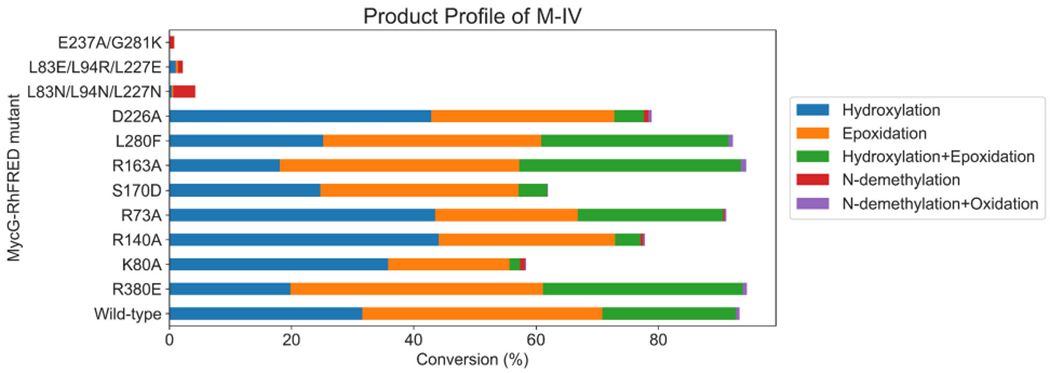
Product profiles of MycG-RhFRED mutants with M-IV as substrate.
Figure 8.
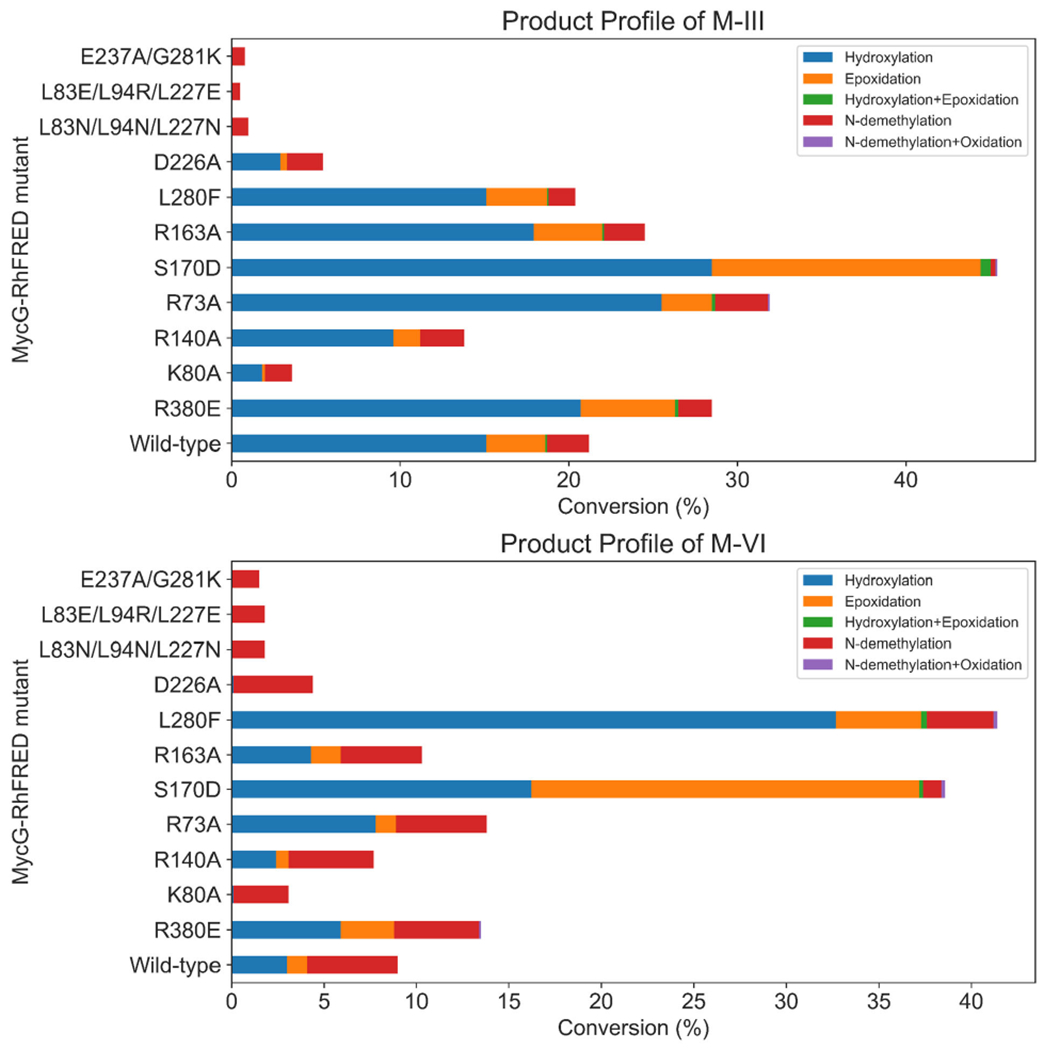
Product profiles of MycG-RhFRED mutants with M-III (top) and M-VI (bottom) as substrates.
Two notable MycG mutants that had the most substantial impact on activity toward M-III and M-VI were variants S170D and L280F. The S170D mutant was designed to improve the FG/BC loop salt bridge interactions with the goal of improving the “desosamine-out” binding mode for all substrates, particularly M-III and M-VI. The overall activity of the S170D mutant on M-III (45% conversion) was more than double that of the wild-type enzyme acting on the same substrate. Moreover, the S170D mutant converted nearly 40% of M-VI to a mixture of singly oxidized products. Similar to M-IV, only trace amounts of doubly oxidized product were observed with the S170D mutant. Concomitant with these increased levels of oxidation products was a significant decrease in N-demethylation activity, indicating that the enzyme was more likely to bind these substrates in the catalytically competent “desosamine-out” binding mode, consistent with the hypothesis.20,22 Thus, with the introduction of the S170D mutation, MycG gained tolerance for accepting alternative substrates beyond its wild-type preference for M-IV. We reason that D170, which is located in the FG loop region of MycG, could form a stable salt-bridge interaction with R75 in the BC loop and/or the desosamine group of the substrate, thereby promoting the enzyme to adopt a closed conformation after substrate binding, leading in turn to higher turnover.
While very low levels of N-demethylated products were observed with wild-type MycG acting on M-IV, such products comprised a more significant portion of the total reaction mixture when M-III and M-VI were employed as substrates. L280 is located in a loop proximal to the heme cofactor that forms the inner surface of the MycG substrate binding pocket. Taking note of the fact that L280 is located next to the desosamine group when the substrate is in the “desosamine-in” binding mode, we hypothesized that the L280F mutation would add steric bulk and sufficiently alter the binding pocket so as to favor the “desosamine-out” binding mode for the different mycinamicin substrates. While the activity of the L280F mutant toward M-IV and M-III was similar to that of the wild-type enzyme, this mutant demonstrated a significantly higher overall conversion of M-VI (41%). Since wild-type MycG produces the N-demethylated product in greater proportion relative to the hydroxylation/epoxidation products when acting on M-VI, the latter appears to prefer adopting the “desosamine-in” binding mode in the wild-type enzyme. We posit that the L280F mutant disfavors the “desosamine-in” binding mode for M-VI, thus indirectly promoting the binding of this substrate in the “desosamine-out” configuration, leading to higher levels of the hydroxylation/epoxidation products. Given the relatively low levels of JV-demethylated products when M-IV and M-III were used as substrates, the “desosamine-in” binding mode is likely minor for these substrates; thus, the L280F mutation has little to no effect on the product profiles for these substrates. The S170D and L280F mutants achieved comparable turnover of M-VI, but the product profile observed with S170D showed accumulation of both hydroxylated and epoxidized products in approximately equal amounts. In contrast, the L280F mutant exhibited a strong preference for hydroxylation rather than epoxidation of M-VI (~7:1 hydroxy:epoxy). Therefore, these mutants could be used as effective starting points toward the generation of highly regioselective biocatalysts for oxidation of M-VI to produce both rare (M-XV) and non-naturally occurring (12,13-epoxy-M-VI) mycinamicin analogs.
Additional studies with MycG variants D226A, S170D, R140A, and K80A showed altered product profiles relative to the wild-type enzyme when acting on M-IV. While the yields of epoxidation (M-I) and hydroxylation (M-V) products for these mutants were similar to or slightly lower than those for the wild-type enzyme, we observed a much smaller amount of the sequential oxidation product M-II (hydroxylation + epoxidation). However, the product profiles of the R380E, L280F, R163A, and R73A mutants were largely the same compared to wild-type. Similar yields of monooxidized products M-I and M-V compared to the wild-type enzyme suggest that the binding of M-IV is largely unaffected by these mutations. We reasoned that a change in M-II yield is most likely related to altered binding of the monohydroxylated intermediate M-V in MycG, and thus hypothesize that the D226A, S170D, R140A, and K80A mutations lead to disfavored M-V binding and faster release of this intermediate from the enzyme active site upon formation. While this finding is unexpected, these mutants could serve as reasonable starting points for production and isolation of monorather than dioxidized mycinamicin products.
CONCLUSION
We have applied several computational methods to investigate the substrate scope and product profile of the multifunctional P450 MycG. Using DFT, we found that the lack of activity toward M-I, the epoxidation product of M-IV, is primarily due to the lack of allylic stabilization of the developing radical during the C-H abstraction transition state. Therefore, the sequence of oxidation is primarily controlled by the inherent reactivity of the substrate and intermediates rather than by the enzyme itself. Moreover, MycG favors its native substrate M-IV over earlier biosynthetic intermediates M-III and M-VI, which are identical to M-IV aside from one of the deoxysugar moieties. MD simulations revealed that the hydrophobicity of the remote deoxysugar group in the substrate is crucial for stabilizing the binding conformation of the substrate, which is distinct from that observed in the crystal structure. M-IV is the preferred substrate due to the hydrophobic nature of the two methoxy groups present in the mycinose deoxysugar. Subsequent mutagenesis experiments enabled us to alter both the substrate scope and product profile of MycG acting on all three mycinamicin substrates. Two mutations (L280F and SryoD) rendered MycG more reactive toward M-III and M-VI, while several additional mutations simplified the product profile of M-IV by diminishing formation of the doubly-oxidized hydroxylation/epoxidation product M-II. The S170D and L280F mutants could serve as promising starting points for future engineering efforts aimed toward generating robust biocatalysts for site-selective oxidation of M-VI.
Supplementary Material
Figure 1.
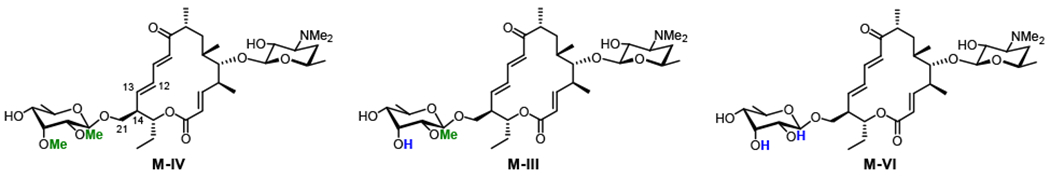
Macrocycles in the biosynthesis of M-II that differ in the extent of methylation of the deoxysugar appended to C-21. Only M-IV is effectively oxidized by MycG.
ACKNOWLEDGEMENT
This work was supported jointly by grants from the National Institute of Health, National Institute for General Medical Sciences (124480), NSF Center for Selective CH Functionalization (CHE-1700982). We are also grateful to the National Institutes of Health (R35 GM118101), and the Hans W. Vahlteich Professorship (to D.H.S.) for financial support.
Footnotes
SUPPORTING INFORMATION
Experimental methods, protein expression and purification, supplementary graphics, computational methods, coordinates of QM calculations are available in the Supporting Information.
REFERENCES
- (1).Meesters RJW; Duisken M; Hollender J Cytochrome P450-Catalysed Arene-Epoxidation of the Bioactive Tea Tree Oil Ingredient p-Cymene: Indication for the Formation of a Reactive Allergenic Intermediate? Xenobiotica 2009,39 (9), 663–671. [DOI] [PubMed] [Google Scholar]
- (2).Vaz ADN; McGinnity DF; Coon MJ Epoxidation of Olefins by Cytochrome P450: Evidence from Site-Specific Mutagenesis for Hydroperoxo-Iron as an Electrophilic Oxidant. PNAS 1998, 95 (7), 3555–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem Rev 2010, 110 (2), 932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lin H-C; McMahon TC; Patel A; Corsello M; Simon A; Xu W; Zhao M; Houk KN; Garg NK; Tang Y P450-Mediated Coupling of Indole Fragments To Forge Communesin and Unnatural Isomers. J. Am. Chem. Soc 2016,138 (12), 4002–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Usmani KA; Karoly ED; Hodgson E; Rose RL In Vitro Sulfoxidation of Thioether Compounds by Human Cytochrome P450 and Flavin-Containing Monooxygenase Isoforms with Particular Reference to the CYP2C Subfamily. Drug Metab. Dispos 2004, 32 (3), 333–339. [DOI] [PubMed] [Google Scholar]
- (6).Lee HS; Park EJ; Ji HY; Kim SY; Im G-J; Lee SM; Jang IJ Identification of Cytochrome P450 Enzymes Responsible for N -Dealkylation of a New Oral Erectogenic, Mirodenafil. Xertobiodca 2008, 38 (1), 21–33. [DOI] [PubMed] [Google Scholar]
- (7).Anzai Y; Li S; Chaulagain MR; Kinoshita K; Kato F; Montgomery J; Sherman DH Functional Analysis of MycCI and MycG, Cytochrome P450 Enzymes Involved in Biosynthesis of Mycinamicin Macrolide Antibiotics. Chem Biol 2008, 15 (9). 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Anzai Y; Tsukada S; Sakai A; Masuda R; Harada C; Domeki A; Li S; Kinoshita K; Sherman DH; Kato F Function of Cytochrome P450 Enzymes MycCI and MycG in Micromonospora Griseorubida, a Producer of the Macrolide Antibiotic Mycinamicin. Antimicrob. Agents Chemother 2012,56 (7), 3648–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tietz DR; Podust LM; Sherman DH; Pochapsky TC Solution Conformations and Dynamics of Substrate-Bound Cytochrome P450 MycG. Biochemistry 2017, 56 (21), 2701–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford CT, 2009. [Google Scholar]
- (11).Case DA; Babin V; Berryman JT; Betz RM; Cai Q; Cerutti DS; Cheatham TE; Darden TA; Duke RE; Gohlke H; et al. Amber 14; University of California, San Francisco, 2014. [Google Scholar]
- (12).Salomon-Ferrer R; Götz AW; Poole D; Le Grand S; Walker RC Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 2013, 9 (9), 3878–3888. [DOI] [PubMed] [Google Scholar]
- (13).Le Grand S; Götz AW; Walker RC SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Computer Physics Communications 2013, 184 (2), 374–380. [Google Scholar]
- (14).Shahrokh K; Orendt A; Yost GS; Cheatham TE Quantum Mechanically Derived AMBER-Compatible Heme Parameters for Various States of the Cytochrome P450 Catalytic Cycle. J. Comput. Chem 2012, 33 (2), 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang J; Wang W; Kollman PA; Case DA Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. Journal of Molecular Graphics and Modelling 2006, 25 (2), 247–260. [DOI] [PubMed] [Google Scholar]
- (16).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25 (9), 1157–1174. [DOI] [PubMed] [Google Scholar]
- (17).Bayly CI; Cieplak P; Cornell W; Kollman PA A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem 1993, 97 (40), 10269–10280. [Google Scholar]
- (18).Laio A; Parrinello M Escaping Free-Energy Minima. Proc Natl Acad Sci U S A 2002, 99 (20), 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26 (16), 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang W; Liu Y; Yan J; Cao S; Bai F; Yang Y; Huang S; Yao L; Anzai Y; Kato F; Podust LM; Sherman DH; Li S New Reactions and Products Resulting from Alternative Interactions between the P450 Enzyme and Redox Partners. J. Am. Chem. Soc 2014, 136, 3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kinoshita K; Takenaka S; Suzuki H; Morohoshi T; Hayashi M Mycinamicins, New Macrolide Antibiotics. J. Antibiot 1992. 451. [DOI] [PubMed] [Google Scholar]
- (22).Li S; Tietz DR; Rutaganira FU; Kells PM; Anzai Y; Kato F; Pochapsky TC; Sherman DH; Podust LM Substrate recognition by the multifunctional cytochrome P450 MycG in mycinamicin hydroxylation and epoxidation reactions. J. Biol. Chem 2012, 287, 37880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.