

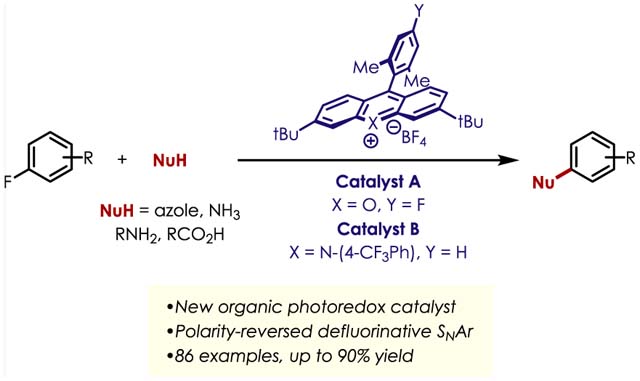
Abstract
Nucleophilic aromatic substitution (SNAr) is a classical reaction with well-known reactivity toward electron-poor fluoroarenes. However, electron-neutral and electron-rich fluoro(hetero)arenes are considerably underrepresented. Herein, we present a method for the nucleophilic defluorination of unactivated fluoroarenes enabled by cation radical-accelerated nucleophilic aromatic substitution. The use of organic photoredox catalysis renders this method operationally simple under mild conditions and is amenable to various nucleophile classes, including azoles, amines, and carboxylic acids. Select fluorinated heterocycles can be functionalized using this method. In addition, the late-stage functionalization of pharmaceuticals is also presented. Computational studies demonstrate that the site selectivity of the reaction is dictated by arene electronics.
Graphical Abstract
INTRODUCTION
The (hetero)aromatic ring is a common structural unit found in countless agrochemicals, natural products, and pharmaceuticals. A 2011 study of three major pharmaceutical companies found that 99% of their medicinal chemistry targets possessed at least one aromatic ring.1 Substitution reactions from common precursors are a desirable strategy that can allow for the rapid synthesis, derivatization, and screening of potential therapeutics.2
To this end, nucleophilic aromatic substitution (SNAr) has been employed by chemists to build up molecular complexity in (hetero)aromatic systems. Mechanistically, SNAr chemistry can take various forms (Figure 1A). Aryne intermediates have been employed in formal SNAr chemistry, but generation of arynes and difficulties associated with regiocontrol have limited their use in synthesis.3 Concerted addition–elimination SNAr via nucleophilic attack on the π-system is also possible.4–7 This concerted process tolerates a wide array of arene electronics by avoiding a buildup of formal charge in the arene. The most well-known mechanistic path is perhaps stepwise addition–elimination, which is commonly restricted to arenes possessing electron-withdrawing groups ortho and/or para to the nucleofuge (commonly fluoride, chloride, or nitro groups). This electronic requirement greatly restricts the aromatic scaffolds toward which such reactivity is amenable. The prevalence of electron-neutral and -rich fluoroarenes makes these compounds attractive targets for further derivatization, but the electronic limitations of addition–elimination SNAr must be overcome in cases where concerted SNAr is inoperative.
Figure 1.
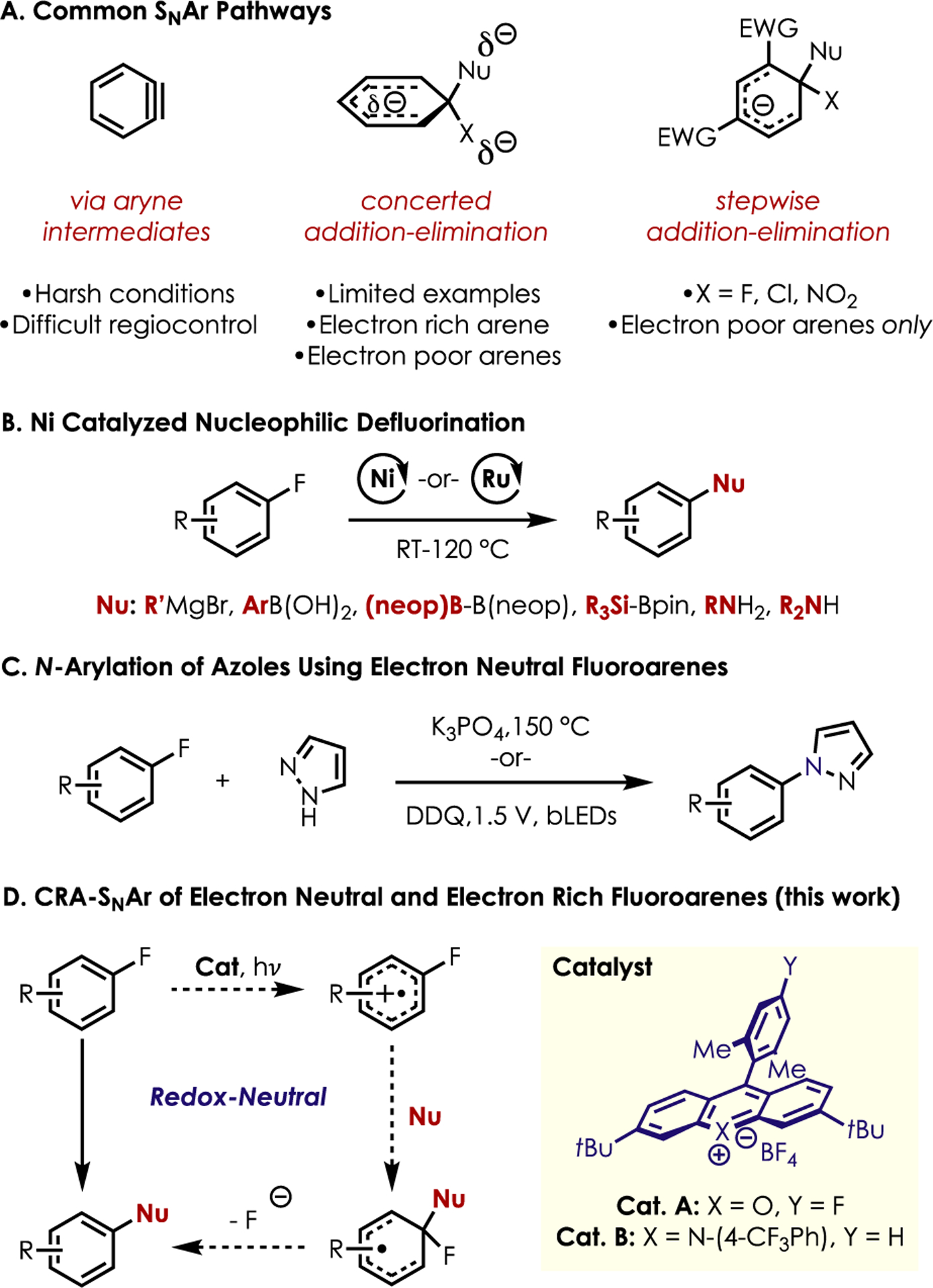
Methods for fluoroarene functionalization.
Transition-metal-catalyzed couplings with fluoroarenes are underrepresented due to the strength of Csp2–F bonds (126 kcal/mol) as compared to Csp2–Br or Csp2–I bonds (80 and 65 kcal/mol, respectively).8 Kumada9–11 and Suzuki12 couplings, defluoroborylation,13,14 and defluorosilylation15 have all been reported via nickel catalysis (Figure 1B). However, specialized ligands and/or high temperatures can limit these methods. Amination of aryl fluorides with primary16 and secondary17 amines have also been documented using nickel and ruthenium, respectively.
The metal-free functionalization of unactivated fluoroarenes has even fewer precedents. Azole N-arylation of such electron-neutral substrates has been disclosed by the Diness18 and Lambert groups19 (Figure 1C). Both methods are selective for fluorine in the presence of other halogens, compatible with various azole nucleophiles, and represent a significant step forward in the development of metal-free functionalization of electron-neutral fluoroarenes. However, both require an excess of the typically more valuable fluoroarene (2–5 equivalents), and electron-rich arenes cannot be functionalized. In addition, the Diness method requires very high temperatures (150–190 °C) and microwave heating in select cases. Lambert and coworkers developed an interesting electrophotocatalytic method that operates at room temperature. This enables a broader substrate scope and greater functional group tolerance, yet a specialized electrode operating at a constant voltage of +1.5 V vs SCE precludes the use of easily oxidized functionality.
Previously, our laboratory has demonstrated that acridinium salts can facilitate cation radical accelerated nucleophilic aromatic substitution (CRA-SNAr) using alkoxy and phenoxy nucleofuges with a variety of nucleophiles.20–23 The limitations outlined above present an opportunity to use CRA-SNAr with fluoride as a nucleofuge to achieve nucleophilic defluorination using both electron-neutral and electron-rich fluoroarenes in a redox-neutral manifold (Figure 1D).
RESULTS AND DISCUSSION
Development of Xanthylium Salts as Potent Excited State Oxidants.
Nucleophilic defluorination of electron-neutral fluoroarenes was examined first in conjunction with acridinium salts. Little to no reaction was observed using 4-fluorotoluene (Ep/2 = +2.24 V vs SCE) in the presence of blue light irradiation with pyrazole as a nucleophile; this is due to an endergonic electron transfer (Table 1, entries 1 and 2). A pyrylium salt photooxidant (E*1/2red = +2.32 V vs SCE)24 could also be used with moderate success (37%, Table S1) but showed visible decomposition over the course of the reaction, demonstrating a need for further catalyst development.
Table 1.
Catalyst Development for Fluorotoluene SNAr
| entry | R | R1 | X | E*1/2red (V)a | yieldb |
|---|---|---|---|---|---|
| 1 | Me | Me | NPh | +2.10 | 0% |
| 2 | Cl | H | NPh | +2.21 | 8% |
| 3 | Me | Me | O | +2.51 | <5% |
| 4 | Cl | H | O | +2.66 | 35% |
| 5 | Me | F | O | +2.57 | 55% |
Saturated calomel electrode (SCE) as reference.
Yield determined by 1H NMR using HMDSO as an internal standard.
A recent publication from our group detailed a modular synthesis of xanthylium salts en route to acridinium salts.25 We were interested in probing the photophysical properties of xanthylium salts considering their structural similarity to pyrylium salts (see the SI for detailed characterization) in order to develop a stronger excited state photooxidant. The xanthylium salts were found to be highly oxidizing (E*1/2red > +2.5 V vs SCE), rendering the electron transfer exergonic and giving yields of the desired product in poor to modest amounts (Table 1, entries 3–5). This scaffold possesses significant steric bulk around the catalyst core (tBu groups in the 3 and 6 positions, orthogonal arene ring in the 9 position) to prevent catalyst decomposition via known nucleophilic decomposition pathways.26
Further optimization revealed that a fluorinated alcohol solvent is a crucial component of the reaction (Table 2, entries 1–4). This may be due to the stabilization of high-energy cation radical intermediates via hydrogen-bond interactions with the solvent.27,28 Nucleophile loadings could be lowered to three equivalents without a noticeable decline in yield (entries 5–7). Ultimately, higher catalyst loadings in conjunction with 1,1,1,3,3,3-hexfluoro-2-propanol (HFIP) as a solvent proved to be ideal across a range of substrates (entries 8 and 9). Finally, the method of irradiation proved to be more important than the central wavelength of light employed (entries 10–12). Kessil lamps in combination with a foil barrier gave the best yields. This is likely due to an increase in photon flux as well as thermal activation (ca. 45–50 °C).
Table 2.
Optimization of Fluorotoluene SNAr using Pyrazole
| entry | deviations from the above conditions | yielda |
|---|---|---|
| 1 | none | 55% |
| 2 | DCM | 4% |
| 3 | MeCN | 10% |
| 4 | HFIP as solvent | 72% |
| 5 | 3 equiv of pyrazole | 68% |
| 6 | 2 equiv of pyrazole | 51% |
| 7 | 1 equiv of pyrazole | 45% |
| 8 | 0.01 equiv of catalyst | 48% |
| 9 | 0.075 equiv of catalyst; HFIP | 72% |
| 10 | 456 nm Kessils | 63% |
| 11 | 427 nm Kessils | 62% |
| 12 | 427 nm Kessils with foil barrier | 82% |
Yield determined by 1H NMR using HMDSO as an internal standard
SNAr of Electron-Neutral Fluoroarenes with Azoles.
A variety of mono- and dimethyl fluorobenzene derivatives served as competent electrophiles in modest to poor yields (Scheme 1, 1–5). Bromine was well tolerated with no substitution observed at the C–Br bond (6), thus offering a complementary functional handle for downstream transformations. The pharmaceutically relevant trifluoromethyl moiety29,30 was tolerated in modest yield (7) as well. Various azoles were also competent nucleophiles for this reaction, providing the desired C–N coupled product in poor to excellent yields. Particularly noteworthy was Fomepizole (9), an FDA-approved treatment for ethylene glycol poisoning,31 and ethyl-4-pyrazole carboxylate (11), a precursor to several pesticides and antivirals.32,33 The various biological activities observed with aryl-substituted pyrazole derivatives make this approach an attractive method to construct this valuable motif in a mild, straightforward manner.34 Three different triazoles, including benzotriazole 14, gave the desired substitution products in modest to excellent yields. Imidazoles were largely unreactive in this system due to their poor nucleophilicities. In addition to azole nucleophiles, tethered carboxylic acids were competent in an intramolecular defluorination (15 and 16). It should also be noted that intermolecular SNAr using carboxylic acid nucleophiles was unsuccessful.
Scheme 1.

Scope of the Nucleophilic Defluorination of Electron-Neutral Fluoroarenesa
aAverage isolated yields are reported (0.300 mmol, n = 2); 45–50 °C represents the ambient temperature of the light setup using external fan cooling. b4 equiv of azole nucleophile was used. cIsolated yield (0.150 mmol, n = 1), 0.075 equiv of Catalyst A.
SNAr of Electron-Rich Fluoroarenes Using Ammonia, Primary Amines, and Carboxylic Acids.
To the best of our knowledge, a metal-free defluoroamination and defluorooxygenation of electron-rich fluoroarenes has yet to be developed.35 In doing so, regioselective C–F bond functionalization was a significant obstacle that needed to be addressed.20–23 Moreover, xanthylium salt catalysts are incompatible with primary amine nucleophiles.25 Consequently, acridinium salt catalysts were employed as their excited states are sufficiently strong to oxidize electron-rich fluoroarenes.
Construction of anilines and N-aryl secondary amines was pursued first. For the former, ammonium carbamate served as an ammonia source considering our previous success with this nucleophile.20,26 After extensive optimization (Table S3), a 3:1 solvent mixture of 1,2-dichloroethane (DCE) and 2,2,2-trifluoroethanol (TFE) afforded the desired aniline product in the highest yield. This solvent system allows for the controlled release of ammonia, as ammonium carbamate is sparingly soluble in DCE. For the latter, minor alterations to the previously optimized conditions proved to be ideal (Table S4). Optimization using carboxylic acid nucleophiles revealed that using TFE as the sole solvent increased reaction yields (Table S5). Interestingly, use of Catalyst B enabled moderate to good yields of the desired substitution products across a variety of arenes, potentially due to its longer excited state lifetime (τ = 20.7 ns) as compared to other acridinium catalysts.25
A collection of electron-rich fluoroarenes was screened with three standard nucleophiles: ammonium carbamate, 2-picolylamine, and benzoic acid (Scheme 2). It should be noted that in all cases, except one, the nucleophilic defluorination product was isolated as a single regioisomer (vide infra) with unreacted starting material accounting for the remainder of the mass balance. Both 4- and 2-methoxy-1-fluorobenzene were aminated in good yields, while oxygenation was observed in fair yield (17 and 18). Other (pseudo)halides were well tolerated in this methodology with yields ranging from poor to excellent across the three nucleophiles screened (19–24). Substrates possessing extended π-systems (25–27) were also competent electrophiles with amine nucleophiles in modest to very good yields, while benzoic acid afforded lower yields of the desired product. An acetophenone derivative was also compatible with the developed reaction conditions, albeit in low yield (28). 2,4-Difluoroanisole gave interesting results with moderate yields for all three nucleophiles. Largely unselective reactivity was observed using ammonia (29A); however, amination occurs ortho- to the methoxy group preferentially (29B). This is surprising given the greatest degree of positive charge is located para- to the methoxy group (see the SI for computational data). We believe the difference in reactivity observed in these cases can be rationalized by hydrogen bonding. The methoxy group acts as a directing group by serving as a hydrogen-bond acceptor from the amine nucleophile; conversely, ammonia is a very weak hydrogen-bond donor, and its reactivity is dictated primarily by arene cation radical electronics.36,37 The inability for benzoate to act as a hydrogen-bond donor results in the expected para selectivity (29C) with disubstitution occurring as a minor product (not observed for amine nucleophiles).
Scheme 2.
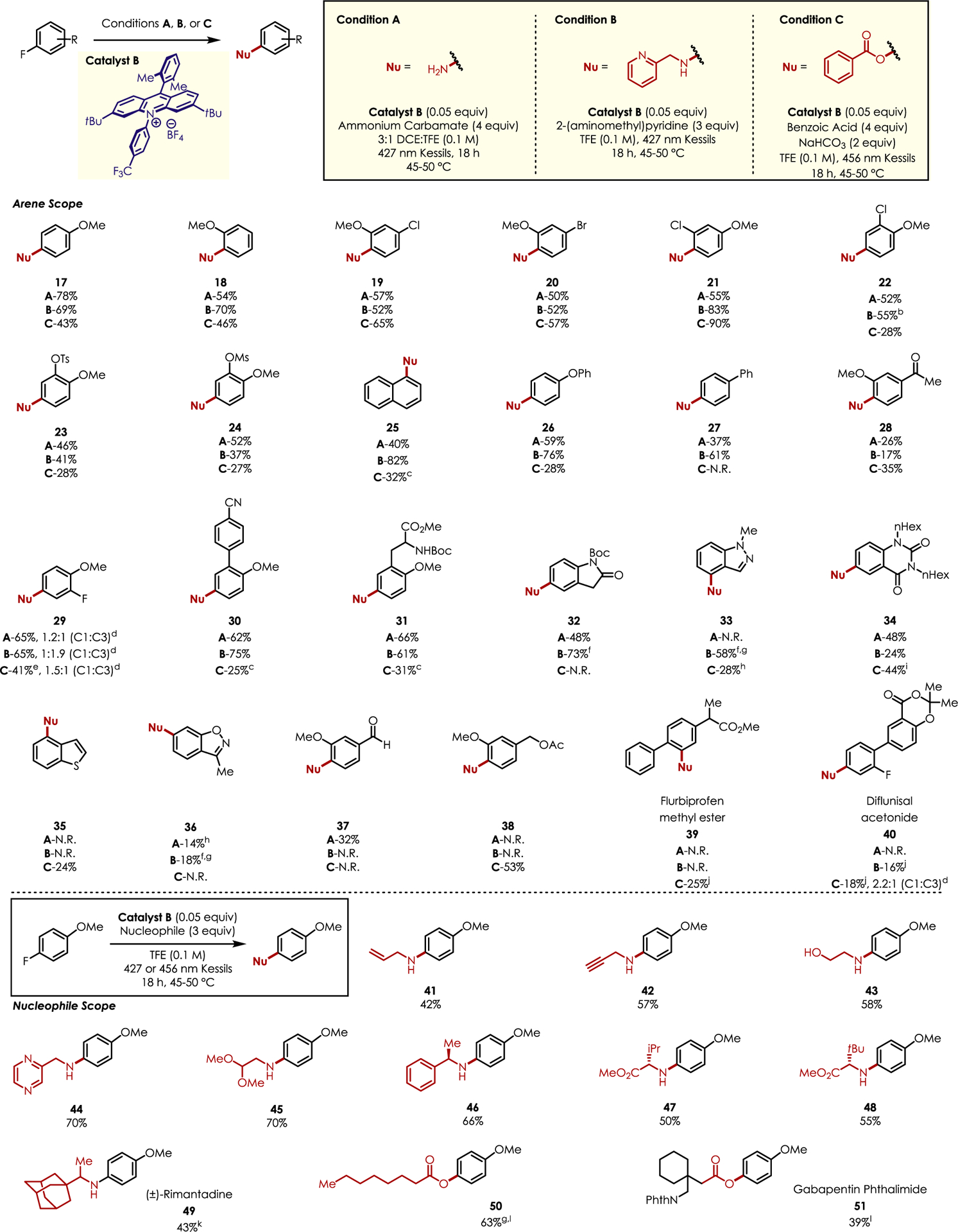
Scope of the Nucleophilic Defluorination of Electron-Rich Fluoroarenesa
aAverage isolated yields are reported (0.3–0.5 mmol, n = 2); 45–50 °C represents the ambient temperature of the light setup using external fan cooling. bEleven percent C–O substitution product. c0.10 equiv of Catalyst B used. dRatio determined by 1H NMR. eNine percent disbustitution observed. f3 equiv of benzyl amine as nucleophile gIsolated yield (0.150 mmol, n = 1). hYield determined by 1H NMR using HMDSO as an internal standard. iIsolated yield (0.050 mmol, n = 1). j0.075 equiv of Catalyst B used. kDCE used as in place of TFE. l4 equiv of carboxylic acid and 2 equiv of NaHCO3 employed.
Substrates with applications toward liquid crystals (30) or biology (31) gave good yields of the desired amination products while demonstrating limited oxygenation. Heterocyclic substrates were also tolerated, including protected indolinone, indazole, quinazolinone, and benzothiophene motifs in modest yields (32–35). Benzisoxazole 36 is uniquely interesting as this aromatic core is found in a family of antipsychotics, including risperidone and iloperidone.38 Some benzenoids did not demonstrate reactivity across all nucleophile classes. For example, benzaldehyde derivative 37 was only reactive toward ammonia. Similarly, only oxygenation was observed with a protected benzylic alcohol (38). The inability of amine nucleophiles to tolerate a benzylic functionality in this case is likely the result of an increased acidity of benzylic protons upon arene oxidation.39 The presence of these sites leads to deprotonation instead of nucleophilic addition in the presence of amines. Finally, late-stage functionalization of pharmaceuticals was demonstrated with flurbiprofen methyl ester 39 (oxygenation) as well as diflunisal acetonide 40 (amination and oxygenation), albeit in low yields.
Primary amine nucleophiles possessing a variety of functional groups (41–45) were amenable to the reaction conditions in fair to good yields. Enantiomerically pure phenylethylamine (46) did not racemize under the reaction conditions nor did amino esters (47 and 48), thus preserving the configuration of the starting nucleophile. The free base of rimantadine, an antiviral drug, also gave the desired product 49 in moderate yield. Alkyl carboxylic acid 50 and gabapentin phthalimide 51 were also compatible with the reaction conditions, demonstrating the method is not restricted to benzoic acid derivatives. Although detailed mechanistic work has not been undertaken, we envision our defluorination reaction proceeding in an analogous manner to our previously reported CRA-SNAr transformations.20,21
Product Inhibition.
It is worth noting the products of these reactions can also be oxidized by the excited photocatlyst. Product inhibition is likely responsible for the poor yields obtained in some cases. A case study was performed using fluoroarene 20 and the corresponding SNAr products. Cyclic voltammetry demonstrated that the products 20A–C are preferentially oxidized over the starting fluoroarene 20 on a thermodynamic basis (Figure 2). As a result, competitive oxidation of the substitution products by the excited state photoredox catalyst occurs more at higher levels of conversion, rendering the desired substitution process less efficient as the reaction proceeds. Experimentally, we tested this hypothesis via competition experiments performed with equimolar amounts of starting material and product. In this case, a 50% yield represents no further formation of product (complete product inhibition), whereas a yield of 100% demonstrates full conversion of the starting fluoroarene to the desired substitution product (no product inhibition). The reactions using aniline products 20A or 20B demonstrate little to no product formation under otherwise normal reaction conditions, highlighting the severity of product inhibition present when synthesizing anilines using this methodology. Interestingly, product inhibition was less significant using benzoic acid as a nucleophile (20C). This is somewhat surprising given that these reactions tend to have lower yields than those of the amination products. While oxidation of these oxygenation products occurs, it is possible that the lower yields observed are the result of reduced nucleophilicity. The negative charge of a carboxylate is delocalized across two oxygen atoms, meaning it is less nucleophilic than amines. This is likely a significant factor in the lower yields obtained using carboxylic acids as nucleophiles. Taken together, competitive oxidation of the desired substitution products (especially anilines) and the poor nucleophilicity of carboxylate anions account for the poor yields obtained in some cases.
Figure 2.
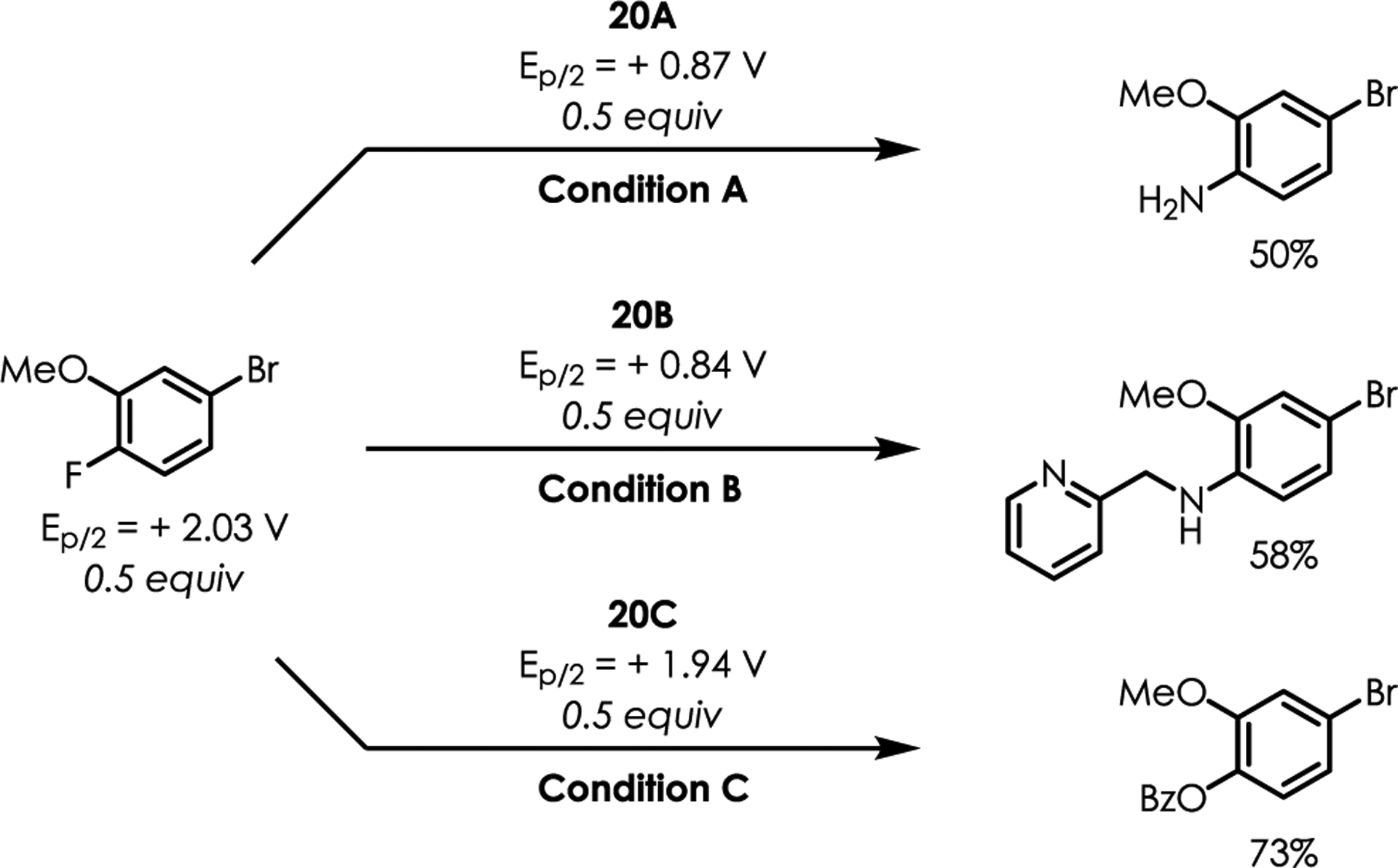
Competition experiments using equimolar amounts of fluoroarene and products. All potentials are reported vs SCE in MeCN.
Rationale for Regioselectivity.
A major concern in this methodology was selective functionalization of C–F bonds in alkoxy-substituted fluoroarenes. In all cases, save for one example, the desired C–F substitution product was isolated as a single regioisomer without additives. . This triggered a brief computational study (see Supporting Information for details) of the alkoxy-substituted fluoroarenes and their corresponding cation radicals using natural population analysis (NPA). In all cases, the greatest degree of positive charge was found to reside on the fluorine-bearing carbon of both the ground state and the cation radicals of the fluoroarenes studied. The decreased electron density at the C–F carbon in arene cation radicals allows for highly selective nucleophilic substitution to occur with a range of substrates (Figure 3). Previous computational models demonstrate functionalization to occur at the site which undergoes the largest change in NPA values upon oxidation;21,40,41 this marked change we observe is the result of the superior leaving group ability of fluoride as well as being less sterically encumbered than methoxide. The notable exception is 22B, where C–O substitution is isolated as a minor product. The difference in NPA values at both C–F and C–O sites is nearly identical, which leads to lower chemoselectivity in this example. It should be noted though that C–F substitution is still isolated as the major product (~5:1).
Figure 3.

Computational model for selective C–F regiocontrol.
User Guide for SNAr of Unactivated Fluoroarenes.
We thought it would be instructive to construct a user guide to inform the decision-making process involved in using this chemistry (Figure 4). The first point one should consider is the oxidation potential of a given substrate, which can be obtained via cyclic voltammetry using a standard three-electrode setup. If this potential falls between +2.14 and +2.57 V vs SCE, functionalization using Catalyst A is possible. Azoles and intramolecular carboxylic acids can participate as nucleophiles, while mono- and dialkyl fluoroarenes are representative electrophiles that fall in this category. Primary amines are incompatible with Catalyst A and should not be used. If the oxidation potential of the fluoroarene is less than +2.14 V vs SCE, a variety of nucleophiles, such as ammonia, amines, and carboxylic acids, can be used in conjunction with Catalyst B. Typical substrates that fall in this category include ortho- and/or para-alkoxy-substituted fluoroarenes, biaryls, and various heterocycles. Meta-substituted alkoxy fluoroarenes and fluoropyridines are unreactive under these conditions, though fluoropyridines can undergo thermal SNAr under acidic conditions.
Figure 4.
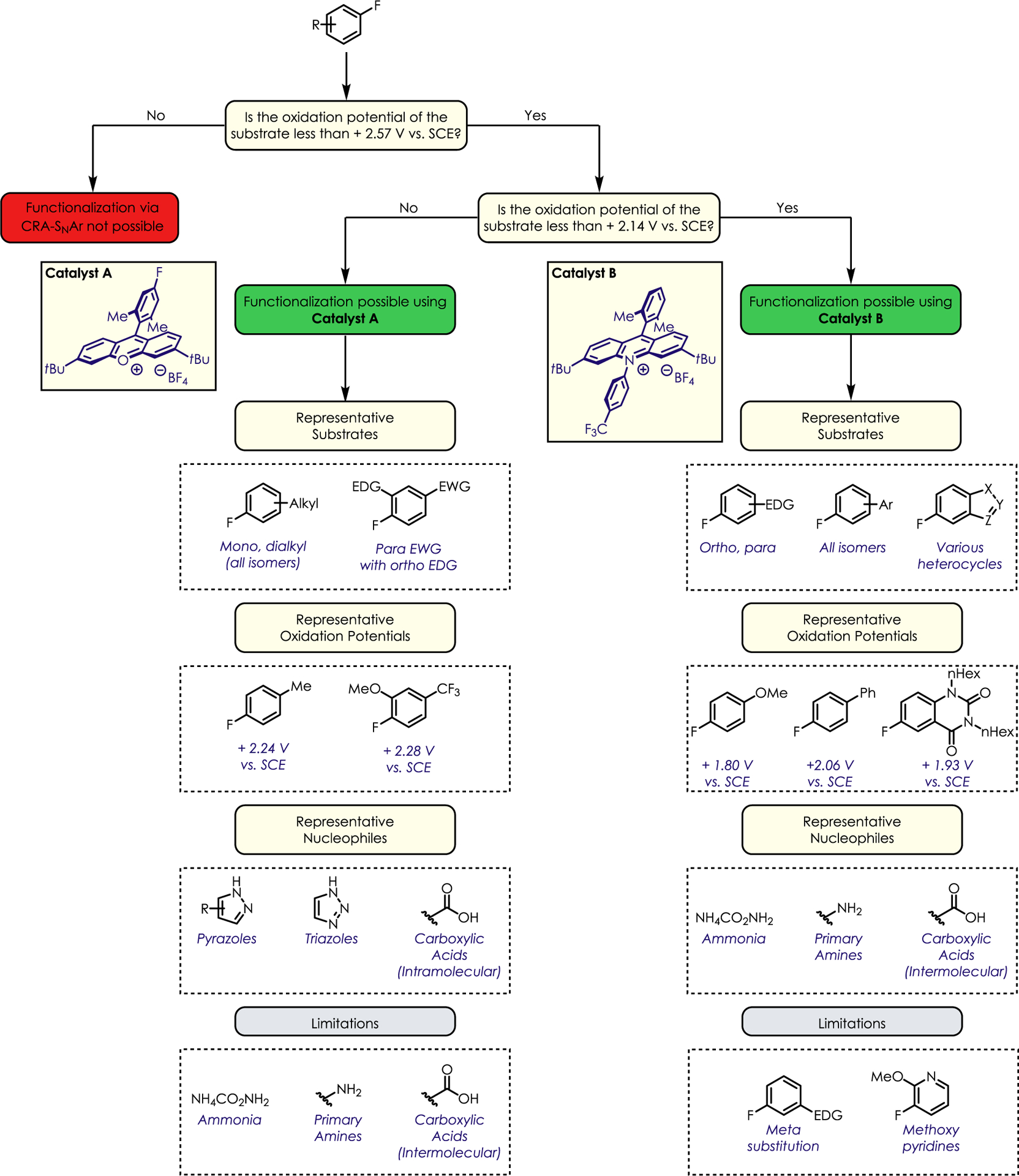
User guide to nucleophilic defluorination of unactivated fluoroarenes.
CONCLUSION
In summary, we developed a method to achieve nucleophilic defluorination of unactivated fluoroarenes using organic photoredox catalysis. A novel xanthylium catalyst mediates such reactivity with electron-neutral substrates and azole nucleophiles to give substitution products in good yields. A variety of nucleophiles can be used to achieve both defluoroamination and defluorooxygenation of functionally diverse electron-rich arenes, including the late-stage functionalization of pharmaceutical derivatives. Computational data support a model wherein the electronics of arene cation radicals dictate the chemoselectivity in substrates bearing both C–O and C–F bonds. This work demonstrates polarity-reversed defluorinative SNAr reactivity, offering another approach to utilize fluoroarenes in chemical syntheses.
Supplementary Material
ACKNOWLEDGMENTS
Financial support was provided in part by the National Institutes of Health (NIGMS) Award No. R01 GM120186. V.A.P. and M.E.S.H. recognize the NSF Graduate Research Fellowship Program for support. Photophysical measurements were performed in the AMPED EFRC Instrumentation Facility established by the Alliance for Molecular PhotoElectrode Design for Solar Fuels (AMPED), an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001011. We also acknowledge Dr. Alexander R. White for his assistance in obtaining photophysical measurements and useful discussion.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c09296
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09296.
Experimental procedures and supporting 1H and 13C NMR spectra (PDF)
REFERENCES
- (1).Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54 (10), 3451–3479. [DOI] [PubMed] [Google Scholar]
- (2).Shahlaei M Descriptor Selection Methods in Quantitative Structure–Activity Relationship Studies: A Review Study. Chem. Rev 2013, 113 (10), 8093–8103. [DOI] [PubMed] [Google Scholar]
- (3).Roberts JD; Simmons HE; Carlsmith LA; Vaughan CW REARRANGEMENT IN THE REACTION OF CHLOROBENZENE-1-C14 WITH POTASSIUM AMIDE1. J. Am. Chem. Soc 1953, 75 (13), 3290–3291. [Google Scholar]
- (4).Neumann CN; Hooker JM; Ritter T Concerted Nucleophilic Aromatic Substitution with 19 F− and 18 F−. Nature 2016, 534 (7607), 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Neumann CN; Ritter T Facile C–F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent. Acc. Chem. Res 2017, 50 (11), 2822–2833. [DOI] [PubMed] [Google Scholar]
- (6).Kwan EE; Zeng Y; Besser HA; Jacobsen EN Concerted Nucleophilic Aromatic Substitutions. Nat. Chem 2018, 10 (9), 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rohrbach S; Smith AJ; Pang JH; Poole DL; Tuttle T; Chiba S; Murphy JA Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem., Int. Ed 2019, 58 (46), 16368–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Luo Y-R Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press LLC: St. Petersburg, FL, 2003. [Google Scholar]
- (9).Kiso Y; Tamao K; Kumada M Effects of the Nature of Halides on the Alkyl Group Isomerization in the Nickel-Catalyzed Cross-Coupling of Secondary Alkyl Grignard Reagents with Organic Halides. J. Organomet. Chem 1973, 50 (1), C12–C14. [Google Scholar]
- (10).Böhm VPW; Gstöttmayr CWK; Weskamp T; Herrmann WA Catalytic C–C Bond Formation through Selective Activation of C–F Bonds. Angew. Chem., Int. Ed 2001, 40 (18), 3387–3389. [DOI] [PubMed] [Google Scholar]
- (11).Ackermann L; Born R; Spatz JH; Meyer D Efficient Aryl–(Hetero)Aryl Coupling by Activation of C–Cl and C–F Bonds Using Nickel Complexes of Air-Stable Phosphine Oxides. Angew. Chem., Int. Ed 2005, 44 (44), 7216–7219. [DOI] [PubMed] [Google Scholar]
- (12).Tobisu M; Xu T; Shimasaki T; Chatani N Nickel-Catalyzed Suzuki–Miyaura Reaction of Aryl Fluorides. J. Am. Chem. Soc 2011, 133 (48), 19505–19511. [DOI] [PubMed] [Google Scholar]
- (13).Liu X-W; Echavarren J; Zarate C; Martin R Ni-Catalyzed Borylation of Aryl Fluorides via C–F Cleavage. J. Am. Chem. Soc 2015, 137 (39), 12470–12473. [DOI] [PubMed] [Google Scholar]
- (14).Niwa T; Ochiai H; Watanabe Y; Hosoya T Ni/Cu-Catalyzed Defluoroborylation of Fluoroarenes for Diverse C–F Bond Functionalizations. J. Am. Chem. Soc 2015, 137 (45), 14313–14318. [DOI] [PubMed] [Google Scholar]
- (15).Cui B; Jia S; Tokunaga E; Shibata N Defluorosilylation of Fluoroarenes and Fluoroalkanes. Nat. Commun 2018, 9 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Harada T; Ueda Y; Iwai T; Sawamura M Nickel-Catalyzed Amination of Aryl Fluorides with Primary Amines. Chem. Commun 2018, 54 (14), 1718–1721. [DOI] [PubMed] [Google Scholar]
- (17).Kang Q-K; Lin Y; Li Y; Shi H Ru(II)-Catalyzed Amination of Aryl Fluorides via H6-Coordination. J. Am. Chem. Soc 2020, 142 (8), 3706–3711. [DOI] [PubMed] [Google Scholar]
- (18).Diness F; Fairlie DP Catalyst-Free N-Arylation Using Unactivated Fluorobenzenes. Angew. Chem., Int. Ed 2012, 51 (32), 8012–8016. [DOI] [PubMed] [Google Scholar]
- (19).Huang H; Lambert TH Electrophotocatalytic SNAr Reactions of Unactivated Aryl Fluorides at Ambient Temperature and Without Base. Angew. Chem., Int. Ed 2020, 59 (2), 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tay NES; Nicewicz DA Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139 (45), 16100–16104. [DOI] [PubMed] [Google Scholar]
- (21).Holmberg-Douglas N; Nicewicz DA Arene Cyanation via Cation-Radical Accelerated-Nucleophilic Aromatic Substitution. Org. Lett 2019, 21 (17), 7114–7118. [DOI] [PubMed] [Google Scholar]
- (22).Venditto NJ; Nicewicz DA Cation Radical-Accelerated Nucleophilic Aromatic Substitution for Amination of Alkoxyarenes. Org. Lett 2020, 22 (12), 4817–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tay NES; Chen W; Levens A; Pistritto VA; Huang Z; Wu Z; Li Z; Nicewicz DA 19 F- and 18 F-Arene Deoxyfluorination via Organic Photoredox-Catalysed Polarity-Reversed Nucleophilic Aromatic Substitution. Nature Catalysis 2020, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang Y; Haze O; Dinnocenzo JP; Farid S; Farid RS; Gould IR Bonded Exciplexes. A New Concept in Photochemical Reactions. J. Org. Chem 2007, 72 (18), 6970–6981. [DOI] [PubMed] [Google Scholar]
- (25).White AR; Wang L; Nicewicz DA Synthesis and Characterization of Acridinium Dyes for Photoredox Catalysis. Synlett 2019, 30 (07), 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Romero NA; Margrey KA; Tay NE; Nicewicz DA Site-Selective Arene C-H Amination via Photoredox Catalysis. Science 2015, 349 (6254), 1326–1330. [DOI] [PubMed] [Google Scholar]
- (27).Colomer I; Chamberlain AER; Haughey MB; Donohoe TJ Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Rev. Chem 2017, 1 (11), 1–12. [Google Scholar]
- (28).Shida N; Imada Y; Nagahara S; Okada Y; Chiba K Interplay of Arene Radical Cations with Anions and Fluorinated Alcohols in Hole Catalysis. Commun. Chem 2019, 2 (1), 1–8. [Google Scholar]
- (29).Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev 2014, 114 (4), 2432–2506. [DOI] [PubMed] [Google Scholar]
- (30).Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceña JL; Soloshonok VA; Izawa K; Liu H Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev 2016, 116 (2), 422–518. [DOI] [PubMed] [Google Scholar]
- (31).Casavant MJ Fomepizole in the Treatment of Poisoning. Pediatrics 2001, 107 (1), 170–170. [DOI] [PubMed] [Google Scholar]
- (32).Bretschneider T; Koehler A; Fischer R; Fueslein M; Jeschke P; Kluth J; Muehlthau FA; Voerste A; Malsam O; Goergens U; Sato Y Novel Heterocyclic Compounds as Pesticides. US2012095023 (A1). [Google Scholar]
- (33).Lançois DFA; Guillemont JÉG; Raboisson PJ-MB; Roymans DAE; Rogovoy B; Bichko V; Lardeau DYR; Michaut AB Rsv Antiviral Pyrazolo- and Triazolo-Pyrimidine Compounds. WO2016174079 (A1), 2016. [Google Scholar]
- (34).Naim MJ; Alam O; Nawaz F; Alam MJ; Alam P Current Status of Pyrazole and Its Biological Activities. J. Pharm. BioAllied Sci 2016, 8 (1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zhou S; Shi W; Zhang J; Zhao F; Wei W; Liang F; Zhang Y Nucleophilic Aromatic Substitution of Unactivated Aryl Fluorides with Primary Aliphatic Amines via Organic Photoredox Catalysis. Chem. - Eur. J 2020. DOI: 10.1002/chem.202002315. [DOI] [PubMed] [Google Scholar]
- (36).Nelson DD; Fraser GT; Klemperer W Does Ammonia Hydrogen Bond? Science 1987, 238 (4834), 1670–1674. [DOI] [PubMed] [Google Scholar]
- (37).Rodham DA; Suzuki S; Suenram RD; Lovas FJ; Dasgupta S; Goddard WA; Blake GA Hydrogen Bonding in the Benzene–Ammonia Dimer. Nature 1993, 362 (6422), 735–737. [Google Scholar]
- (38).Caccia S; Pasina L; Nobili A New Atypical Antipsychotics for Schizophrenia: Iloperidone. Drug Des., Dev. Ther 2010, 4, 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schmittel M; Burghart A Understanding Reactivity Patterns of Radical Cations. Angew. Chem., Int. Ed. Engl 1997, 36 (23), 2550–2589. [Google Scholar]
- (40).Margrey KA; McManus JB; Bonazzi S; Zecri F; Nicewicz DA Predictive Model for Site-Selective Aryl and Heteroaryl C–H Functionalization via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139 (32), 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).McManus JB; Onuska NPR; Jeffreys MS; Goodwin NC; Nicewicz DA Site-Selective C–H Alkylation of Piperazine Substrates via Organic Photoredox Catalysis. Org. Lett 2020, 22 (2), 679–683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.