

Abstract
Pathological changes resulting from myocardial infarction (MI) include extracellular matrix alterations of the left ventricle, which can lead to cardiac stiffness and impair systolic and diastolic function. The signals released from necrotic tissue initiate the immune cascade, triggering an extensive inflammatory response followed by reparative fibrosis of the infarct area. Immune cells such as neutrophils, monocytes, macrophages, mast cells, T-cells, and dendritic cells play distinct roles in orchestrating this complex pathological condition, and regulate the balance between pro-fibrotic and anti-fibrotic responses. This review discusses how molecular signals between fibroblasts and immune cells mutually regulate fibrosis post-MI, and outlines the emerging pharmacological targets and therapies for modulating inflammation and cardiac fibrosis associated with MI.
Keywords: fibrosis, myocardial infarction, leukocytes, T-cells, inflammation, heart failure
1. Introduction
Heart failure (HF) is a consequence of various cardiovascular diseases (CVD) [1]. Myocardial infarction (MI) remains the most common cause of HF worldwide [2]. Following an MI, necrosis of cardiomyocytes within the ischemic area elicits intense inflammation, and ultimately leads to replacement of dead cells with an extracellular matrix (ECM)-rich scar. Optimal healing in the infarct zone is critical for restoration of left ventricle (LV) integrity and function. Dysregulated healing causes reactive interstitial fibrosis in the non-infarct and remote zones due to impaired suppression of inflammation and excessive accumulation of ECM proteins such as collagen. These changes results in LV stiffness and thereby contributes to the pathogenesis of HF [3, 4]. Preservation of homeostasis requires an ongoing balance between ECM synthesis and degradation, which is regulated by matrix metalloproteinase (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) [4, 5].
This review is devoted to the cellular and molecular cascades mediating the pathogenesis of cardiac fibrosis post-MI, focusing mainly on the pro-fibrotic and anti-fibrotic aspects of the immune system. We also provide insight into the therapeutic opportunities for targeting different cell types and molecules involved in the pathogenesis of cardiac fibrosis post-MI, along with summarizing therapies currently available, and those in development.
2. Immune cells orchestrate the complex and dynamic response post-MI
Numerous immune cells, such as neutrophils (PMNs), monocyte/macrophages, mast cells, T-cells, dendritic cells (DCs) and their associated molecular signals are involved in regulating the balance between pro-fibrotic and anti-fibrotic responses (Figure 1) [5, 6]. The interaction between inflammatory cells and fibroblasts regulates cardiac fibrosis post-MI. Immune cells coordinate cardiac scar formation directly by producing proteases and other mediators that break down and inhibit ECM deposition or indirectly by secreting mediators that initiate the differentiation of fibroblast into activated ECM producing myofibroblast [7, 8]. Furthermore, timely clearance of leukocytes is crucial for optimal healing in infarct. Despite recent significant advancements in the field, there is a need to accumulate more in-depth knowledge of the mechanisms that contribute to the process of cardiac fibrosis and development of HF post-MI, so that effective translatable therapies can be developed.
Figure 1: Pro-fibrotic and anti-fibrotic signaling factors in immune regulation of cardiac fibrosis post-MI.
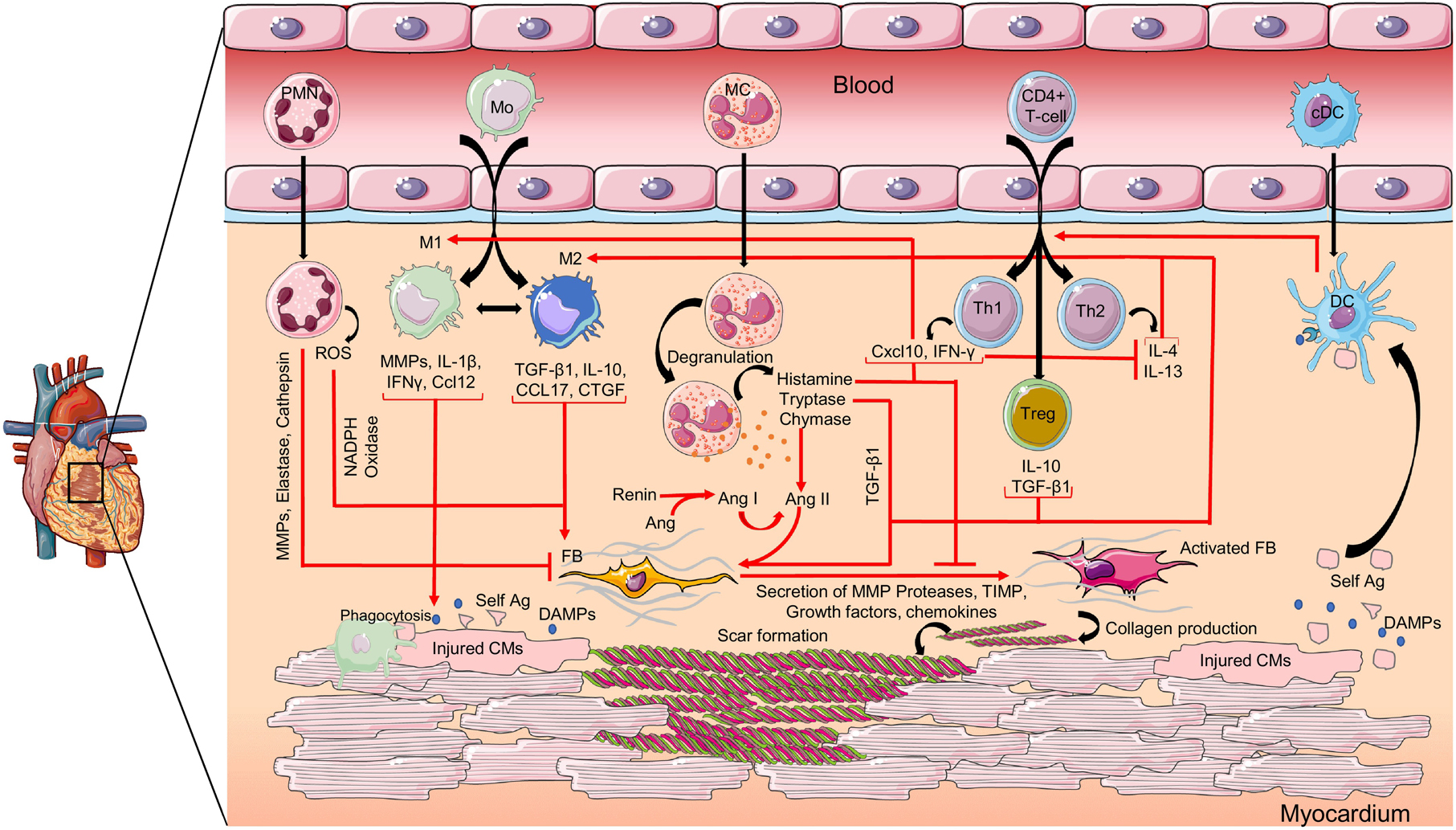
Following cardiac injury, the necrotic cardiomyocytes release inflammatory mediators like DAMPs, which recruit immune cells to the infarcted heart. Cytokines, chemokines, growth factors and enzymes released by activated immune cells directly or indirectly regulate fibroblast activation and collagen synthesis. PMN: neutrophil; Mo: monocyte; M1: monocyte-derived inflammatory macrophage; M2: monocyte-derived anti-inflammatory/pro-fibrotic macrophage; MC: mast cell; Th1, Th2: T helper type 1, −2 cells; Treg: regulatory T-cell; cDC: conventional dendritic cell; DC: mature dendritic cell; ROS: reactive oxygen species; MMP: matrix metalloproteinase; IL: interleukin; IFN-γ: interferon gamma; CCL: chemokine ligand; TGF-β: transforming growth factor beta; CTGF: connective tissue growth factor; Ang: angiotensin; IP-10: IFN-γ induced protein-10; FB: fibroblast; self-Ag: self antigen; CM: cardiomyocyte; DAMP: damage-associated molecular pattern; TIMP: tissue inhibitor of metalloproteinase. Graphics were created using Servier Medical Art templates; https://smart.servier.com
2.1. Neutrophils (PMNs)
PMNs are the first responders at the ischemic injury site. During acute inflammation, PMNs are not only vital for the clearance of necrotic tissue, but also for the resolution of inflammation and maintenance of tissue homeostasis. Upon arrival to the infarct, PMNs secrete granules which contain various factors including neutrophil gelatinase associated lipocalin (NGAL), reactive oxygen species (ROS), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, MMPs, elastase, and cathepsins, all of which have direct or indirect roles in the regulation of cardiac fibrosis post-MI [9, 10]. PMNs indirectly contribute to the tissue repair, in part by modulating the phenotype of macrophages. A recent study provided evidence that PMNs are required for resolving post-MI inflammation and promoting cardiac healing, as PMN depletion aggravated cardiac function and increased fibrosis [11]. Mice with PMN depletion showed a significant down-regulation of M1 markers (interleukin (IL)-12, tumor necrosis factor (TNF)α, interferon (IFN)γ, IL1β) and up-regulation of M2 signature markers (CX3CR1, arginase, chitinase-3-like protein 3 or YM1, IL4) in infarcted hearts at day 7 post-MI, illustrating that the effect on fibrosis was likely due to alterations in macrophage population. In addition, PMN-depleted mice had decreased macrophage expression of phagocytosis receptor, Mer-tyrosine protein kinase (MertK), indicating a worsened capacity to clear necrotic cardiomyocytes [11]. PMNs also release large amount of ROS via NADPH oxidase [12, 13]. Inhibition of NADPH oxidase resulted in attenuation of cardiac fibrosis post-MI while reducing cardiac Nox4 expression, ROS production, NFκB activation, and plasma MMP2 activity in rats [13].
In reparative response, apoptotic PMNs helped in wound healing by directly secreting cytokines and growth factors. Contrasting to what was previously thought; Dasake et al. demonstrated PMNs isolated from the infarct of mice at days 3–7 post-MI had a reparative signature that included expression of fibronectin and fibrinogen [14]. Interestingly, stimulation of PMNs in vitro with fibronectin induced MMP9 and NGAL secretion, indicating a possible negative feedback loop. MMP9 facilitates the removal of necrotic myocytes by degrading collagen, fibronectin, and other ECM components. However, MMP9 also prevents neutrophil apoptosis leading to prolonged inflammation, which results in enhanced degradation of collagen and ECM components. This may compromise tissue integrity and leads to infarct expansion propagating adverse LV remodeling [15]. Understanding the detrimental and beneficial roles of PMNs in the post-MI remodeling process will provide mechanistic insight and possible therapeutic targets that would tip the balance towards favorable cardiovascular remodeling.
2.2. Monocytes/Macrophages
Monocytes and monocyte-derived macrophages play an important role in the initiation and progression of the fibrotic responses. As a result of their remarkable plasticity and heterogeneity, macrophages exert a wide range of pro-fibrotic and anti-fibrotic effects in the myocardium [16]. Yano and colleagues have shown that the level of collagen deposited post-MI directly correlates with macrophage numbers [17]. Typically, monocyte-derived macrophages are classified into either pro-inflammatory M1 cells or anti-inflammatory/pro-fibrotic M2 cells; however, this nomenclature is simplistic and does not truly capture the complexity of macrophage biology. Resident and monocyte-derived (infiltrating) macrophages likely lie between (and outside of) the classical M1 and M2 definitions [16]. Resident macrophages maintain tissue-specific homeostasis by clearing cellular debris, recruiting other inflammatory leukocytes and resolving inflammation. After ischemic injury, resident macrophages drive the influx of monocytes into the infarct zone [18]. Upon infiltration, monocytes begin to differentiate into inflammatory macrophages [19]. During acute phase post-MI (day 1–3 after ischemia in mice), the MI environment is highly inflammatory [8, 16]. Ly-6Chigh monocytes and inflammatory macrophages secrete multiple proteases including MMP2 and −9 [20, 21]. Together they form a ‘demolition crew’ that clears the debris and digests ECM in order to replace the injured tissue with granulation tissue, which includes newly laid connective tissue and developing blood vessels and the evolving scar [22].
In addition to the degradation of the ECM via proteases, pro-inflammatory macrophages also secrete inflammatory markers that regulate fibroblast function. Previously, we demonstrated that secretion of CCL12 by macrophages inhibits fibroblast activation, leading to decreased collagen deposition [7]. In mice, macrophages at days 1 and 3 post-MI display a unique signaling profile composed of genes associated with pro-inflammatory, phagocytic, proliferative, and metabolic reprogramming [16]. Macrophage-derived cytokines, IL1β and IFNγ are the major promoters of the pro-inflammatory environment early post-MI. In vitro stimulation of human cardiac myofibroblasts with IFNγ arrested proliferation and reduced the expression of α-smooth muscle actin (αSMA), implying that IFNγ is a negative regulator of cardiac fibrosis [23]. Similarly, blocking IL1β signaling reduced cardiac fibrosis and MMP expression by decreasing leukocyte recruitment and attenuating inflammation [24].
The reparative phase post-MI is characterized by a phenotypic transition of pro-inflammatory macrophages to anti-inflammatory macrophages [16, 25]. Macrophages during this phase influence the encompassing cells, and activate fibroblasts by producing an abundance of inflammatory mediators, such as tissue growth factor (TGF)-β, IL10, and CCL17. Together, these mediators activate fibroblasts and enhance ECM protein synthesis, while suppressing the degradation of ECM, thus promoting tissue remodeling and repair post-MI [26–28]. Increasing the number of anti-inflammatory macrophages by constant infusion of recombinant IL10 has been shown to decrease MMP expression and improve LV function post-MI [27, 29]. Jung et al. demonstrated that macrophages from IL10-infused mice increased cardiac fibrosis compared to wild-type (WT) mice by stimulating proliferation and migration of cardiac fibroblasts [27]. Despite the increase in fibroblast activation, IL10 also decreased the collagen I to collagen III ratio post-MI, improving myocardial wall compliance and cardiac function.
Among all pro-fibrotic mediators, TGFβ is the main player in fibrotic remodeling. It stimulates production of pro-fibrotic factors such as connective tissue growth factor (CTGF) and type I collagen, via SMAD3-dependent pathway [26]. Interestingly, in addition to decreased Tgfβ and IL10 expression, Smad3 deletion in macrophages has been shown to reduce expression of genes associated with phagocytosis, such as milk fat globule-EGF factor 8 (Mfge8), indicating that the transition to the anti-inflammatory phenotype may be regulated by phagocytosis [30]. Similarly, human monocyte-derived macrophages increased TGFβ and TGFβ-induced (TGFβi) expression following ingestion of apoptotic debris [31]. In vitro stimulation of fibroblasts with TGFβi decreased MMP14 levels and subsequent collagen accumulation.
New evidence suggests that macrophages may directly mediate maintenance of the fibrotic scar through secretion of ECM proteins, including collagen [16, 32]. These studies have shown that macrophages can convert themselves into fibroblast-like cells, and then upregulate fibroblast-specific ECM organization genes (e.g. Col1a1 and Postn) in mice at day 7 post-MI [16, 32, 33]. The newly converted fibroblasts, however, expressed very low levels of fibroblast marker discoidin domain receptor 2 (DDR2), which is thought to play a role in monitoring the state of the cardiac ECM and directing collagen synthesis/turnover [32]. In a mouse model of chronic renal allograft injury, fibroblast-like macrophages (CD68+/F4/80+ and α-SMA+) contributed to interstitial fibrosis and correlated with allograft rejection [34]. Therefore, ECM-producing macrophages may represent a subgroup of pathogenic fibroblasts. Future studies to focus on determining whether these cells are friend or foe are needed. Defining the spatio-temporal proteomic profile by combining techniques such as MALDI-imaging and histology would provide insight into whether immune cells-mediated alterations in ECM can alter the pliability of the infarct scar. Gaining a better understanding of the stimulus needed to activate ECM production by macrophages would also provide possible targets that may facilitate in our understanding of cardiac fibrosis post-MI.
2.3. Mast cells
Mast cells are characterized by their abundant and diverse granules, which produce a variety of fibrogenic mediators, including proteases (i.e. tryptase, chymase) and growth factors. Not much is known about the role of mast cells post-MI but recent studies have begun to utilize the mast cell deficient mice in ischemia reperfusion (I/R) injury and MI [35–37]. Frangogiannis et al. first showed that mast cell density increased during the healing phase in a canine model of I/R, with maximum accumulation in the areas of collagen deposition [37]. Cimini et al. reported that decreased fibroblast activation and accumulation around the infarct could be associated with diminished mast cell numbers [35].
Histamine, a biogenic amine known to be stored in and secreted from mast cells, was found to decrease cardiac fibrogenesis by inhibiting TGF-β through STAT6-dependent signaling pathway [38]. In vitro human mast cells have been shown to induce αSMA expression in dermal fibroblasts through the release of histamine and tryptase [39]. Histamine, however, did not stimulate fibroblasts’ contraction in the collagen contraction assay. In contrast, inhibition of tryptase eliminated mast cells’ ability to stimulate fibroblast contraction, suggesting that tryptase, but not histamine, is a key mediator for fibroblast physiology [39]. Studies have shown that both tryptase and chymase activate TGFβ1, which in turn stimulates fibroblast activation, myofibroblast differentiation, and collagen synthesis [40, 41]. Interestingly, chymase released by mast cells can also generate angiotensin (Ang) II (a profibrotic regulator), which leads to an increase in collagen synthesis, and development of diastolic dysfunction [42, 43]. Achieving a better understanding of how the multifaceted mast cells mediate post-MI healing could increase the potential to harness their activities and provide opportunities for the treatment of MI-induced HF.
2.4. T-cells
After cardiac injury, T-cells are recruited to the site of injury as a result of cytokine production, and release a multitier of pro- and anti-inflammatory molecules (e.g. TNFα, IL1β, TGFβ) [11, 44]. T-cells are categorized into CD4+ helper T-cells (Th1, Th2), CD4+ regulatory T-cells (Treg), and CD8+ cytotoxic T-cells. During the first 7 days after MI, Th1 and CD8+ are the predominant T-cells. In the chronic post-MI environment (8 weeks after MI in mice), Th2 and Treg become the predominant phenotype within the infarcted tissue [45, 46]. These changes in the temporal T-cell phenotype facilitate in cardiac fibrosis post-MI.
2.4.1. Th1 cells
Th1 cells are associated with imbalanced ECM turnover and decreased myofibroblast differentiation, thus promoting cardiac rupture [47]. Th1 cells release IFN-γ, which acts as an anti-fibrotic mediator by blocking the pro-fibrotic activity of TGF-β, thereby inhibiting fibroblast proliferation and subsequently the expression of collagen I and III mRNA [26]. In addition, IFNγ impedes Th2-mediated fibroblast activation by reducing IL4 and IL13 secretion [48, 49]. Another potent anti-fibrotic mediator secreted by Th1 cells after MI is an IFNγ induced protein-10 (i.e. CXCL10) [50]. The anti-fibrotic role of CXCL10 is believed to be due to inhibition of basic fibroblast growth factor (bFGF)-induced cellular migration and enhancement of growth factor-mediated wound contraction in fibroblast-populated collagen lattices [51]. However, the role of Th1 cells is complex, as IFNγ has been shown to promote differentiation of a distinct Treg population that limit Th1 and CD8+ T-cell-mediated pathology and could ultimately promote cardiac healing [52, 53].
2.4.2. Th2 cells
Th2 cells secrete various pro-fibrotic mediators including IL4 and IL13, which are potent stimulators of collagen synthesis post-MI [48, 54, 55]. In a mouse model of MI, IL-13 deficiency aggravated cardiac wound healing and increased LV dilation likely due to decreased macrophage recruitment and M2 polarization in the infarct and border area [55]. Even still, there was no effect of IL13 deficiency on de novo expression of collagen in mice at day 3 post-MI. IL4 is known to promote cardiac fibrosis by stimulating the recruitment of monocyte-derived M2 macrophages, thus indirectly regulating cardiac fibroblasts [54]. In mice, constant in vivo infusion of IL4 starting 24 hours after occlusion upregulated T-cell mediated inflammation and was linked to an overall environment of pro-resolution while stimulating anti-inflammatory macrophages at day 3 post-MI [56]. Although these observations demonstrate a putative pro-fibrotic role for Th2 in the heart, more studies are needed to further clarify the role of Th2 cells in post-MI remodeling.
2.4.3. Treg
Tregs are thought to have a dual role in contributing to cardiac fibrosis by releasing the pro-fibrotic molecule, TGF-β and by inhibiting secretion of IL-10 [57, 58]. Co-culturing of Tregs with cardiac fibroblasts led to decreased αSMA and Mmp3 expression and attenuated fibroblast mediated-contraction of collagen pads, suggesting that direct contact may be necessary for Tregs to stimulate fibroblasts and preserve the matrix [59]. Similarly, Treg expansion by CD28 antibodies significantly decreased MMP-mediated degradation of collagen, and led to improved survival and attenuation of cardiac ruptures during the first 7 days after MI [60]. Within the healing myocardium, Tregs activation has also been linked to increased expression of macrophage-derived proteins via M2 macrophage polarization, thus implicating Tregs as indirect regulators of fibrosis via activation of macrophages [59, 60]. Co-culturing Tregs with macrophages increased the expression of genes associated with healing, such as osteopontin and arginase-1, supporting the notion that Tregs facilitate cardiac fibrosis indirectly via macrophages [60].
2.4.4. CD8+ T-cells
Despite the recent advancements in our understanding of the role that CD4+ T-helpers and CD4+ Tregs play in cardiac fibrosis post MI, little is known about the role of CD8+ T-cells in this context. Our recent study showed that CD8+ T-cell deficient mice had increased cardiac rupture due to poor collagen cross-linking [61]. The data suggested that this was due to indirect effects on fibroblast activation through macrophage and Th1 cell activation. Furthermore, we revealed that CD8+ T-cells are needed for activation of macrophage-mediated removal of necrotic debris, which is essential to keep the inflammatory response in check, and proper collagen scar formation [61]. A delay in the removal of necrotic debris resulted in an exacerbation of inflammation and poor collagen scar formation. Focusing on T-cell mediated responses may be a way to selectively target the antagonistic inflammatory responses while preserving other protective processes like host-defense and wound healing.
2.5. Dendritic cells (DCs)
After necrotic injury, antigen-presenting cells internalize damage-associated molecular patterns (DAMPs) and migrate into the lymph nodes where they present these molecules to other inflammatory cells, and initiate their recruitment to the infarcted myocardium [62]. DCs play a crucial role in initiation of adaptive response through antigen presentation and stimulation of T-cells and natural killer cells [63]. Several subtypes of DCs have been described with myeloid DC (mDC) and plasmacytoid DC (pDC) being the most predominant. DCs that are CD1c+CD11chi, secrete IL12 upon stimulation, and drive a Th1-polarized immune response are classified as mDCs [64, 65]. On the other hand, pDC are CD123+CD11c−/lo, produce IFNα upon viral activation, and induce a Th2 polarization of naive T-cells [64, 65]. The most prominent DC in the infarcted heart is the mDC which peaks at day 7 in rat/mouse models whereas as pDCs are found mainly in circulation and peripheral lymphoid organs [25, 66]. In a study by Kretzschmar et al., CD209-positive mDCs were found to be increased in the infarct tissue collected post-mortem in MI patients, while circulating DCs (DC precursors) were significantly reduced, likely due to enhanced recruitment to the infarcted myocardium [67]. Similarly, Nagai et al. demonstrated that the number of infiltrated CD209 and mDCs directly correlates with the extent of reparative fibrosis in the infarcted myocardium [68].
The pro-fibrotic role of DCs was also demonstrated in a study by Lee et al., where ablation of conventional but not precursor DCs resulted in decreased cardiac fibrosis in mice at 3 weeks post-MI [69]. This decrease in cardiac fibrosis corresponds to reduced numbers of cardiac macrophages, neutrophils, and T-cells, indicating a blunted inflammatory response. Similarly, DC depletion reduced post-MI survival rates, increased MMP9 activity, and disorganized collagen fibers within the infarct [70]. Additional insight is needed to assess the influence of DCs on cardiac fibrosis post-MI. In particular, attention needs to be devoted to temporal changes in the DC population, and how these changes will influence the cardiac remodeling process.
3. Balance in immunity and fibrosis
Taken together, the processes involved in resolution of inflammation and initiation of fibrosis are tightly regulated to achieve optimal healing. Slight deviations in timing and magnitude of the inflammatory response can tip the balance from fibrosis necessary for wound healing post-MI, towards reactive fibrosis [6, 71]. Excessive collagen crosslinking and progressive fibrous tissue contraction results in a stiff non-compliant scar and thereby leads to diastolic and systolic dysfunction [72]. In contrast, attenuated collagen deposition and cross-linking is associated with a weak scar that ultimately leads to increased LV dilation and cardiac rupture [7, 61, 73]. Elevated Ly-6Chi monocytosis in both lipopolysaccharide exposed and in atherosclerotic (apoE−/−) mice has been shown to interrupt resolution of inflammation and consequently enhance adverse remodeling [74]. Interestingly, Ly-6Chi monocytes have also been shown to orchestrate the reparative phase post-MI, by giving rise to Ly-6Clow F4/80hi macrophages that proliferate locally and contribute to cardiac firbosis [75]. Chronically activated Ly-6Chi monocytes due to the absence of orphan nuclear hormone receptor NR4A1 became abnormally differentiated pro-inflammatory macrophages that contributed to defective cardiac healing and compromised heart function [75]. Previously, we demonstrated that under LPS-induced chronic inflammation, macrophage recruitment is accelerated, leading to reduced fibroblast activation and increased cardiac rupture [7]. These studies highlight that fibrosis is not always a negative consequence, and that some fibrosis is needed for cardiac wound healing.
The exact mechanisms behind reactive fibrosis are not clear, however preliminary evidence suggests that continual release of cytokines and other inflammatory mediators may lead to increased fibroblast proliferation and matrix deposition in the infarct border zone, which may expand fibrosis into the viable tissue [76]. Immune cells including macrophages and T-cells accumulate in the remote zone weeks after the MI, both in patients and animal models [77, 78]. Comparing Balb/c and C57Bl/J mice, Toor et al. demonstrated that a more pronounced and prolonged inflammatory response in the infarct and remote zone of C57 mice may contribute to increased scar size and impaired survival compared to Balb/c mice [79].
Persistent fibroblast activation as well as Ang II and TGF-β synthesized and secreted at the infarction site have also been suggested to play a role in the development of reactive fibrosis in the non-infarcted myocardium [72, 80]. In vitro stimulation of fibroblasts with Ang II has been shown to promote myofibroblast transdifferentiation, collagen production, and secretion of growth factors and ROS [81, 82]. Stimulation of neonatal fibroblast with Ang II increases cellular proliferation however, the data in adult cardiac fibroblasts isn’t as convincing [83]. This maybe in part due to the fact that Ang II can effectively increase the synthesis of pro-fibrotic substances, like TGF-β, through activation of the AT1 receptor [84–87]. Simm and Diez observed that confluent cultures (above 1×105 cells/cm2) of rat adult cardiac fibroblasts did not respond at all to Ang II (10−7 mol/L), whereas lower density cultures (below 2×104 cells/cm2) showed evidence of cell proliferation [88]. This positive response however, was only observed after 40 hours of incubation. Interestingly, the addition of a platelet-derived growth factor (PDGF) blocking antibody inhibited this growth suggesting Ang II indirectly regulated cell proliferation in adult cardiac fibroblasts by cell density-dependent expression of growth factors including PDGF.
In all, these studies highlight the importance of maintaining a precise balance between pro-inflammatory and pro-healing processes for optimal healing of damaged tissue. Improving our understanding of the drivers that are maintaining this precise balance may identify novel regulators that are key in orchestrating beneficial tissue repair. Development of more specific, effective, and clinically translatable therapeutic targets will emerge if we learn to influence the fundamental regulatory proteins and enhance healing to improve clinical outcomes.
4. Drugs and therapies under pre-clinical and clinical trials targeting inflammation and cardiac fibrosis associated with MI
There are multiple preclinical and clinical trials that are targeting the inflammatory response with the overall goal of inhibiting cardiac fibrosis and progression of ischemic HF with reduced ejection fraction (HFrEF; Table 1). Angiotensin receptor-neprilysin inhibition (ARNI) has received much attention as the newest addition to the established therapies that are used as first-line treatments. This therapy combines an Ang II receptor blocker (ARB, Valsartan), with a compound that inhibits neprilysin (Sacubitril). This particular Valsartan-Sacubitril combination, more commonly known as Entresto, was previously observed to have cardioprotective and desirable anti-inflammatory effects in mice during the first 7 days post-MI [89]. This was further supported by the PARADIGM-HF trial (Prospective Comparison of ARNI With an ACE-Inhibitor to Determine Impact on Global Mortality and Morbidity in Heart Failure), which reported that combination of neprilysin with an ARB further improved clinical outcomes in HFrEF [90]. Due to the beneficial effects on HFrEF, Entresto has been more recently evaluated in the pathological setting of HF with preserved ejection fraction (HFpEF) [91–93]. Total hospitalizations for HFpEF and death from cardiovascular causes were not statistically significant compared to valsartan alone. This study highlights that the etiology of non-ischemic and ischemic heart failure are likely different and not all treatments will work for both. This therapy continues to be studied, as the current clinical trials programmed to terminate in 2022 relate to examining its pharmacodynamics.
Table 1.
Current therapies in pre-clinical and clinical trials targeting cardiac fibrosis
| Target molecule/Pathway | Drug or therapy | Trial Phase | Current findings | Ref |
|---|---|---|---|---|
| Angiotensin receptor/Neprilysin | Entresto | Clinical phase IV (Oct 2018–July 2022) | Anti-inflammatory effects in animal models; improved composite end point of death from cardiovascular cause or hospitalization for HFrEF patients | [89, 90] |
| Baroreceptors | Baroreflex activation therapy | NA (April 2016–December 2021) | Improve outcomes including quality of life, exercise capability, and NT-proBNP levels | [94] |
| Relaxin/Relaxin receptor | Serelaxin | Clinical phase III (completed) | Anti-inflammatory effects (|NOS); reduced worsening heart failure and all-cause and cardiovascular mortality in heart failure patients | [97, 99, 100] |
| Interleukin-1 receptor | Anakinra | Clinical phase II (Jan 2019-July 2024) | Decreased CRP; improved exercise capacity and quality of life measures after 12 weeks of treatment | [101] |
| Interleukin-1 | Canakinumab | Clinical phase III (completed) | Decreased CRP; decreased risk for non-fatal MI, non-fatal stroke, or cardiovascular related death. | [102] |
| Engineered T-cells | NA | Preclinical | Reduction in cardiac fibrosis; restoration of function after continuous Ang II infusion | [105] |
Heart failure with reduced ejection fraction (HFrEF); N-terminal pro-B-type natriuretic peptide (NT-proBNP); Nitric oxide synthase (NOS); C-reactive protein (CRP); Angiotensin (Ang)
Baroreflex activation therapy in patients with HFrEF has also been shown to improve outcomes including quality of life, exercise capability, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels compared to the established optimal medical management therapies [94]. NT-proBNP release is associated with increased levels of IL-6 and C-reactive protein (CRP), suggesting baroreflex treatment would likely attenuate cardiac inflammation [95, 96]. How baroreflex activation therapy affected morbidity and mortality or change in cardiovascular structure or function endpoints however, was not evaluated. The PARADIGM-HF and GUIDE-IT (Guiding Evidence Based Therapy Using Biomarker Intensified Treatment) studies demonstrated that a reduction in NT-proBNP was associated with significant improvements in left ventricular systolic function and left ventricular remodeling, suggesting that baroreflex activation therapy will likely improve morbidity and cardiovascular structure.
Recent clinical trials of serelaxin (Pre-RELAX-AHF; RELAX-AHF) have provided a new hope in HF. Studies have shown that relaxin, the hormone serelaxin is modeled after, has protective effects on end organs via alterations in inflammatory mediators such as nitric oxide, endothelial endothelin type B receptor, vascular endothelial growth factor, and cAMP [97, 98]. Clinical studies have demonstrated significant serelaxin-related improvement in HF symptoms, length of hospital stay as well as mortality reduction in HF patients [97, 99, 100]. Seralaxin treatment reduced worsening of HF by 47% through day 5 and all-cause and cardiovascular mortality by 37% through day 180 [99, 100].
In a pilot study from REDHART (REcently Decompensated Heart failure Anakinra Response Trial), inhibition of IL-1 receptor decreased CRP levels and improved exercise capacity and quality of life measures but only in patients given 12 weeks of treatment [101]. In the CANTOS trial (Canakinumab Anti-inflammatory Thrombosis Outcome Study), IL1β blockade via Canakinumab was effective at preventing adverse cardiac events over a median of 3.7 years [102]. However, neutropenia and sepsis were more common in the canakinumab groups than placebo group. Neutropenia was dose-dependent supporting the notion that IL1 signaling regulates both detrimental and beneficial processes. These studies highlight that we still have a lot to learn about the inflammatory system. Future studies must focus on strategies that inhibit the detrimental effects of inflammation while retaining and promoting the beneficial processes.
The possibility of using immunotherapy to treat cardiac fibrosis and the subsequent development of HF is supported by the enormous progress in the treatment of certain cancers through the use of engineered T-cells [103]. More recently, chimeric antigen receptor (CAR) T-cell therapy has been approved by the FDA (US Food and Drug Administration) for use in patients with some forms of leukemia and lymphoma [103, 104]. Aghajanian et al. was the first to use engineered T-cells that target a chimeric antigen receptor expressed on activated fibroblast to treat cardiac fibrosis [105]. Adoptive transfer of engineered CD8+ T-cells reduced cardiac fibrosis and restored cardiac function in mice with Ang II induced cardiac dysfunction. To date, there have been no studies that use the same techniques to treat post-MI hearts. Continued investigation to identify unique antigens that are expressed by activated cardiac fibroblasts may identify alternative antigens, or combinations of antigens, that could be effective as targets for immunotherapy in patients with heart disease. A major limitation of CAR T-cell therapies is that they can have serious and not readily predictable off-target and organ-specific toxicities including severe myocardial damage [106]. This highlights the need for improved methods to identify novel antigens and minimize any off-target side effects.
5. Conclusion and Perspectives
This review highlights the importance and diversity of the inflammatory processes in the development of ischemic HF. We have attempted to target the immune system as a means for treatment and while we have made great strides, there is much that still needs to be understood. This is especially important since anti-inflammatory treatments have been associated with adverse effects. Studies like the CANTOS trial highlighted that IL-1 signaling and inflammation are critical including for infection and wound healing. Pharmaceutical inhibition of IL-1 therefore, led to increased incidence of sepsis in patients on these drugs. We must first learn how to favorably tilt the balance to find a way to inhibit the adverse effects of inflammation without affecting the beneficial modulators.
Highlights.
Crosstalk among fibroblasts and immune cells regulate fibrosis post-MI.
Cells secrete pro- and anti-fibrotic signals to regulate scar quality post-MI.
Therapies that target inflammation may be beneficial for ischemic heart failure.
Funding
This study was supported by the National Institute of Health [R00DK105160, R01HL148114, and U54DA016511], Polycystic Kidney Disease Foundation [221G18a], American Heart Association [20IPA35260039], American Physiological Society [Research Career Enhancement and Lazaro J Mandel Young Investigator and S&R Foundation Ryuji Ueno Award] and the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development [IK2BX003922].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB, Tsao CW, E. American Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association, Circulation 141(9) (2020) e139–e596. [DOI] [PubMed] [Google Scholar]
- [2].Roger VL, Epidemiology of heart failure, Circ Res 113(6) (2013) 646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu Y, Xu J, Wu M, Kang L, Xu B, The effector cells and cellular mediators of immune system involved in cardiac inflammation and fibrosis after myocardial infarction, J Cell Physiol (2020) 1–9. [DOI] [PubMed] [Google Scholar]
- [4].Nielsen SH, Mouton AJ, DeLeon-Pennell KY, Genovese F, Karsdal M, Lindsey ML, Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes, Matrix biology : journal of the International Society for Matrix Biology 75–76 (2019) 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].DeLeon KY, Yabluchanskiy A, Winniford MD, Lange RA, Chilton RJ, Lindsey ML, Modifying Matrix Remodeling To Prevent Heart Failure, in: Li R, Weisel RD (Eds.), Cardiac Regeneration And Repair Volume I: Pathology And Therapies, Woodhead; 2013. [Google Scholar]
- [6].Voorhees AP, DeLeon-Pennell KY, Ma Y, Halade GV, Yabluchanskiy A, Iyer RP, Flynn E, Cates CA, Lindsey ML, Han HC, Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction, J Mol Cell Cardiol 85 (2015) 229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, Jung M, Shannon D, Garrett MR, Buchanan W, Hall ME, Ma Y, Lindsey ML, Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing, JCI insight 2(18) (2017) e94207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jin YF, Han HC, Berger J, Dai Q, Lindsey ML, Combining experimental and mathematical modeling to reveal mechanisms of macrophage-dependent left ventricular remodeling, BMC systems biology 5 (2011) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A, Neutrophil function: from mechanisms to disease, Annu Rev Immunol 30 (2012) 459–89. [DOI] [PubMed] [Google Scholar]
- [10].Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML, Temporal neutrophil polarization following myocardial infarction, Cardiovascular research 110(1) (2016) 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S, Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype, European heart journal 38(3) (2017) 187–197. [DOI] [PubMed] [Google Scholar]
- [12].Mohazzab HK, Kaminski PM, Wolin MS, Lactate and PO2 modulate superoxide anion production in bovine cardiac myocytes: potential role of NADH oxidase, Circulation 96(2) (1997) 614–20. [DOI] [PubMed] [Google Scholar]
- [13].Liu XH, Pan LL, Deng HY, Xiong QH, Wu D, Huang GY, Gong QH, Zhu YZ, Leonurine (SCM-198) attenuates myocardial fibrotic response via inhibition of NADPH oxidase 4, Free Radic Biol Med 54 (2013) 93–104. [DOI] [PubMed] [Google Scholar]
- [14].Daseke MJ, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML, Neutrophil proteome shifts over the myocardial infarction time continuum, Basic research in cardiology 114(5) (2019) 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML, CD36 Is a Matrix Metalloproteinase-9 Substrate That Stimulates Neutrophil Apoptosis and Removal During Cardiac Remodeling, Circulation. Cardiovascular genetics 9(1) (2016) 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML, Mapping macrophage polarization over the myocardial infarction time continuum, Basic research in cardiology 113(4) (2018) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yano T, Miura T, Whittaker P, Miki T, Sakamoto J, Nakamura Y, Ichikawa Y, Ikeda Y, Kobayashi H, Ohori K, Macrophage colony-stimulating factor treatment after myocardial infarction attenuates left ventricular dysfunction by accelerating infarct repair, Journal of the American College of Cardiology 47(3) (2006) 626–634. [DOI] [PubMed] [Google Scholar]
- [18].Davies LC, Jenkins SJ, Allen JE, Taylor PR, Tissue-resident macrophages, Nat Immunol 14(10) (2013) 986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Weerasinghe D, McHugh KP, Ross FP, Brown EJ, Gisler RH, Imhof BA, A role for the alphavbeta3 integrin in the transmigration of monocytes, J Cell Biol 142(2) (1998) 595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT, Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction, J Clin Invest 106(1) (2000) 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zamilpa R, Ibarra J, de Castro Bras LE, Ramirez TA, Nguyen N, Halade GV, Zhang J, Dai Q, Dayah T, Chiao YA, Lowell W, Ahuja SS, D’Armiento J, Jin YF, Lindsey ML, Transgenic overexpression of matrix metalloproteinase-9 in macrophages attenuates the inflammatory response and improves left ventricular function post-myocardial infarction, J Mol Cell Cardiol 53(5) (2012) 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nahrendorf M, Swirski FK, Monocyte and macrophage heterogeneity in the heart, Circ Res 112(12) (2013) 1624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee JW, Oh JE, Rhee KJ, Yoo BS, Eom YW, Park SW, Lee JH, Son JW, Youn YJ, Ahn MS, Ahn SG, Kim JY, Lee SH, Yoon J, Co-treatment with interferon-gamma and 1-methyl tryptophan ameliorates cardiac fibrosis through cardiac myofibroblasts apoptosis, Mol Cell Biochem 458(1–2) (2019) 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, Weissleder R, Libby P, Swirski FK, Nahrendorf M, Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction, Circulation 132(20) (2015) 1880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M, Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction, J Mol Cell Cardiol 62 (2013) 24–35. [DOI] [PubMed] [Google Scholar]
- [26].Ulloa L, Doody J, Massague J, Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway, Nature 397(6721) (1999) 710–3. [DOI] [PubMed] [Google Scholar]
- [27].Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML, IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation, Basic research in cardiology 112(3) (2017) 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS, Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts, Cell Immunol 204(1) (2000) 19–28. [DOI] [PubMed] [Google Scholar]
- [29].Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R, IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR, Circ Res 104(2) (2009) e9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen B, Huang S, Su Y, Wu YJ, Hanna A, Brickshawana A, Graff J, Frangogiannis NG, Macrophage Smad3 Protects the Infarcted Heart, Stimulating Phagocytosis and Regulating Inflammation, Circ Res 125(1) (2019) 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nacu N, Luzina IG, Highsmith K, Lockatell V, Pochetuhen K, Cooper ZA, Gillmeister MP, Todd NW, Atamas SP, Macrophages produce TGF-beta-induced (beta-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts, J Immunol 180(7) (2008) 5036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Haider N, Bosca L, Zandbergen HR, Kovacic JC, Narula N, Gonzalez-Ramos S, Fernandez-Velasco M, Agrawal S, Paz-Garcia M, Gupta S, DeLeon-Pennell K, Fuster V, Ibanez B, Narula J, Transition of Macrophages to Fibroblast-Like Cells in Healing Myocardial Infarction, J Am Coll Cardiol 74(25) (2019) 3124–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chang MY, Chan CK, Braun KR, Green PS, O’Brien KD, Chait A, Day AJ, Wight TN, Monocyte-to-Macrophage differentiation synthesis and secretion of a complex extracellular matrix, J Biol Chem 287(17) (2012) 14122–14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang YY, Jiang H, Pan J, Huang XR, Wang YC, Huang HF, To KF, Nikolic-Paterson DJ, Lan HY, Chen JH, Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury, J Am Soc Nephrol 28(7) (2017) 2053–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD, Li RK, c-kit dysfunction impairs myocardial healing after infarction, Circulation 116(11 Suppl) (2007) I77–82. [DOI] [PubMed] [Google Scholar]
- [36].Bhattacharya K, Farwell K, Huang M, Kempuraj D, Donelan J, Papaliodis D, Vasiadi M, Theoharides TC, Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion, Int J Immunopathol Pharmacol 20(1) (2007) 69–74. [DOI] [PubMed] [Google Scholar]
- [37].Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML, Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion, Circulation 98(7) (1998) 687–98. [DOI] [PubMed] [Google Scholar]
- [38].Chen J, Hong T, Ding S, Deng L, Abudupataer M, Zhang W, Tong M, Jia J, Gong H, Zou Y, Wang TC, Ge J, Yang X, Aggravated myocardial infarction-induced cardiac remodeling and heart failure in histamine-deficient mice, Sci Rep 7 (2017) 44007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gailit J, Marchese MJ, Kew RR, Gruber BL, The differentiation and function of myofibroblasts is regulated by mast cell mediators, J Invest Dermatol 117(5) (2001) 1113–9. [DOI] [PubMed] [Google Scholar]
- [40].Tatler AL, Porte J, Knox A, Jenkins G, Pang L, Tryptase activates TGFbeta in human airway smooth muscle cells via direct proteolysis, Biochemical and biophysical research communications 370(2) (2008) 239–42. [DOI] [PubMed] [Google Scholar]
- [41].Zhao XY, Zhao LY, Zheng QS, Su JL, Guan H, Shang FJ, Niu XL, He YP, Lu XL, Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts, Mol Cell Biochem 310(1–2) (2008) 159–66. [DOI] [PubMed] [Google Scholar]
- [42].Chester AH, Borland JA, Chymase-dependent angiotensin II formation in human blood vessels, J Hum Hypertens 14(6) (2000) 373–6. [DOI] [PubMed] [Google Scholar]
- [43].Matsumoto T, Wada A, Tsutamoto T, Ohnishi M, Isono T, Kinoshita M, Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure, Circulation 107(20) (2003) 2555–8. [DOI] [PubMed] [Google Scholar]
- [44].Barbul A, Breslin RJ, Woodyard JP, Wasserkrug HL, Efron G, The effect of in vivo T helper and T suppressor lymphocyte depletion on wound healing, Ann Surg 209(4) (1989) 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD, Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure, Circ Heart Fail 10(3) (2017) e003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD, Dysfunctional and Proinflammatory Regulatory T-Lymphocytes Are Essential for Adverse Cardiac Remodeling in Ischemic Cardiomyopathy, Circulation 139(2) (2019) 206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yan X, Zhang H, Fan Q, Hu J, Tao R, Chen Q, Iwakura Y, Shen W, Lu L, Zhang Q, Zhang R, Dectin-2 Deficiency Modulates Th1 Differentiation and Improves Wound Healing After Myocardial Infarction, Circ Res 120(7) (2017) 1116–1129. [DOI] [PubMed] [Google Scholar]
- [48].Shao DD, Suresh R, Vakil V, Gomer RH, Pilling D, Pivotal Advance: Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation, Journal of leukocyte biology 83(6) (2008) 1323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gurujeyalakshmi G, Giri SN, Molecular mechanisms of antifibrotic effect of interferon gamma in bleomycin-mouse model of lung fibrosis: downregulation of TGF-beta and procollagen I and III gene expression, Exp Lung Res 21(5) (1995) 791–808. [DOI] [PubMed] [Google Scholar]
- [50].Saxena A, Bujak M, Frunza O, Dobaczewski M, Gonzalez-Quesada C, Lu B, Gerard C, Frangogiannis NG, CXCR3-independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans, Cardiovascular research 103(2) (2014) 217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, Frangogiannis NG, Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction, Circ Res 105(10) (2009) 973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hall AO, Beiting DP, Tato C, John B, Oldenhove G, Lombana CG, Pritchard GH, Silver JS, Bouladoux N, Stumhofer JS, Harris TH, Grainger J, Wojno ED, Wagage S, Roos DS, Scott P, Turka LA, Cherry S, Reiner SL, Cua D, Belkaid Y, Elloso MM, Hunter CA, The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology, Immunity 37(3) (2012) 511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, Fujisawa S, Chudakov DM, Treuting PM, Rudensky AY, Stability and function of regulatory T cells expressing the transcription factor T-bet, Nature 546(7658) (2017) 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N, Podaru M, Kainuma S, Tanaka H, Suzuki K, IL-4 as a Repurposed Biological Drug for Myocardial Infarction through Augmentation of Reparative Cardiac Macrophages: Proof-of-Concept Data in Mice, Scientific reports 7(1) (2017) 6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S, Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction, Circ Heart Fail 7(5) (2014) 822–30. [DOI] [PubMed] [Google Scholar]
- [56].Daseke MJ, Tenkorang-Impraim MAA, Ma Y, Chalise U, Konfrst SR, Garrett MR, DeLeon-Pennell KY, Lindsey ML, Exogenous IL-4 shuts off pro-inflammation in neutrophils while stimulating anti-inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction, J Mol Cell Cardiol 145 (2020) 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Van Linthout S, Miteva K, Tschope C, Crosstalk between fibroblasts and inflammatory cells, Cardiovascular research 102(2) (2014) 258–69. [DOI] [PubMed] [Google Scholar]
- [58].Crome SQ, Clive B, Wang AY, Kang CY, Chow V, Yu J, Lai A, Ghahary A, Broady R, Levings MK, Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells, Journal of immunology 185(6) (2010) 3199–208. [DOI] [PubMed] [Google Scholar]
- [59].Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG, Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function, Am J Physiol Heart Circ Physiol 307(8) (2014) H1233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S, Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation, Circ Res 115(1) (2014) 55–67. [DOI] [PubMed] [Google Scholar]
- [61].Ilatovskaya DV, Pitts C, Clayton J, Domondon M, Troncoso M, Pippin S, DeLeon-Pennell KY, CD8(+) T-cells negatively regulate inflammation post-myocardial infarction, Am J Physiol Heart Circ Physiol 317(3) (2019) H581–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Van der Borght K, Lambrecht BN, Heart macrophages and dendritic cells in sickness and in health: A tale of a complicated marriage, Cell Immunol 330 (2018) 105–113. [DOI] [PubMed] [Google Scholar]
- [63].Durai V, Murphy KM, Functions of Murine Dendritic Cells, Immunity 45(4) (2016) 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, Matsuki Y, Mount AM, Belz GT, O’Keeffe M, Ohmura-Hoshino M, Ishido S, Stoorvogel W, Heath WR, Shortman K, Villadangos JA, Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells, Nat Immunol 9(11) (2008) 1244–52. [DOI] [PubMed] [Google Scholar]
- [65].Masten BJ, Olson GK, Tarleton CA, Rund C, Schuyler M, Mehran R, Archibeque T, Lipscomb MF, Characterization of myeloid and plasmacytoid dendritic cells in human lung, Journal of immunology 177(11) (2006) 7784–93. [DOI] [PubMed] [Google Scholar]
- [66].Naito K, Anzai T, Sugano Y, Maekawa Y, Kohno T, Yoshikawa T, Matsuno K, Ogawa S, Differential effects of GM-CSF and G-CSF on infiltration of dendritic cells during early left ventricular remodeling after myocardial infarction, Journal of immunology 181(8) (2008) 5691–701. [DOI] [PubMed] [Google Scholar]
- [67].Kretzschmar D, Betge S, Windisch A, Pistulli R, Rohm I, Fritzenwanger M, Jung C, Schubert K, Theis B, Petersen I, Drobnik S, Mall G, Figulla HR, Yilmaz A, Recruitment of circulating dendritic cell precursors into the infarcted myocardium and pro-inflammatory response in acute myocardial infarction, Clin Sci (Lond) 123(6) (2012) 387–98. [DOI] [PubMed] [Google Scholar]
- [68].Nagai T, Honda S, Sugano Y, Matsuyama TA, Ohta-Ogo K, Asaumi Y, Ikeda Y, Kusano K, Ishihara M, Yasuda S, Ogawa H, Ishibashi-Ueda H, Anzai T, Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans, J Am Heart Assoc 3(3) (2014) e000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee JS, Jeong SJ, Kim S, Chalifour L, Yun TJ, Miah MA, Li B, Majdoubi A, Sabourin A, Keler T, Guimond JV, Haddad E, Choi EY, Epelman S, Choi JH, Thibodeau J, Oh GT, Cheong C, Conventional Dendritic Cells Impair Recovery after Myocardial Infarction, J Immunol 201(6) (2018) 1784–1798. [DOI] [PubMed] [Google Scholar]
- [70].Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K, Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling, Circulation 125(10) (2012) 1234–45. [DOI] [PubMed] [Google Scholar]
- [71].Tian Y, Koganti T, Yao Z, Cannon P, Shah P, Pietrovito L, Modesti A, Aiyetan P, DeLeon-Pennell K, Ma Y, Halade GV, Hicks C, Zhang H, Lindsey ML, Cardiac extracellular proteome profiling and membrane topology analysis using glycoproteomics, Proteomics Clin Appl 8(7–8) (2014) 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Myofibroblast-mediated mechanisms of pathological remodelling of the heart, Nat Rev Cardiol 10(1) (2013) 15–26. [DOI] [PubMed] [Google Scholar]
- [73].DeLeon-Pennell KY, de Castro Bras LE, Iyer RP, Bratton DR, Jin YF, Ripplinger CM, Lindsey ML, gingivalis P lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9, J Mol Cell Cardiol 76C (2014) 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R, Nahrendorf M, Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis, J Am Coll Cardiol 55(15) (2010) 1629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, Hedrick CC, Libby P, Nahrendorf M, Weissleder R, Swirski FK, Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium, Circ Res 114(10) (2014) 1611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG, Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling, Am J Pathol 173(1) (2008) 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lee WW, Marinelli B, van der Laan AM, Sena BF, Gorbatov R, Leuschner F, Dutta P, Iwamoto Y, Ueno T, Begieneman MP, Niessen HW, Piek JJ, Vinegoni C, Pittet MJ, Swirski FK, Tawakol A, Di Carli M, Weissleder R, Nahrendorf M, PET/MRI of inflammation in myocardial infarction, J Am Coll Cardiol 59(2) (2012) 153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Abbate A, Bonanno E, Mauriello A, Bussani R, Biondi-Zoccai GG, Liuzzo G, Leone AM, Silvestri F, Dobrina A, Baldi F, Pandolfi F, Biasucci LM, Baldi A, Spagnoli LG, Crea F, Widespread myocardial inflammation and infarct-related artery patency, Circulation 110(1) (2004) 46–50. [DOI] [PubMed] [Google Scholar]
- [79].Toor IS, Ruckerl D, Mair I, Thomson A, Rossi AG, Newby DE, Allen JE, Gray GA, Enhanced monocyte recruitment and delayed alternative macrophage polarization accompanies impaired repair following myocardial infarction in C57BL/6 compared to BALB/c mice, Clin Exp Immunol 198(1) (2019) 83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Talman V, Ruskoaho H, Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration, Cell Tissue Res 365(3) (2016) 563–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R, Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species, J Hypertens 24(4) (2006) 757–66. [DOI] [PubMed] [Google Scholar]
- [82].Singh VP, Baker KM, Kumar R, Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production, Am J Physiol Heart Circ Physiol 294(4) (2008) H1675–84. [DOI] [PubMed] [Google Scholar]
- [83].Bouzegrhane F, Thibault G, Is angiotensin II a proliferative factor of cardiac fibroblasts?, Cardiovascular research 53(2) (2002) 304–12. [DOI] [PubMed] [Google Scholar]
- [84].Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T, TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II, J Clin Invest 109(6) (2002) 787–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Campbell SE, Katwa LC, Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts, J Mol Cell Cardiol 29(7) (1997) 1947–58. [DOI] [PubMed] [Google Scholar]
- [86].Fisher SA, Absher M, Norepinephrine and ANG II stimulate secretion of TGF-beta by neonatal rat cardiac fibroblasts in vitro, Am J Physiol 268(4 Pt 1) (1995) C910–7. [DOI] [PubMed] [Google Scholar]
- [87].Lee AA, Dillmann WH, McCulloch AD, Villarreal FJ, Angiotensin II stimulates the autocrine production of transforming growth factor-beta 1 in adult rat cardiac fibroblasts, J Mol Cell Cardiol 27(10) (1995) 2347–57. [DOI] [PubMed] [Google Scholar]
- [88].Simm A, Diez C, Density dependent expression of PDGF-A modulates the angiotensin II dependent proliferation of rat cardiac fibroblasts, Basic research in cardiology 94(6) (1999) 464–71. [DOI] [PubMed] [Google Scholar]
- [89].Ishii M, Kaikita K, Sato K, Sueta D, Fujisue K, Arima Y, Oimatsu Y, Mitsuse T, Onoue Y, Araki S, Yamamuro M, Nakamura T, Izumiya Y, Yamamoto E, Kojima S, Kim-Mitsuyama S, Ogawa H, Tsujita K, Cardioprotective Effects of LCZ696 (Sacubitril/Valsartan) After Experimental Acute Myocardial Infarction, JACC Basic Transl Sci 2(6) (2017) 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Oatmen KE, Zile MR, Burnett JC Jr., Spinale FG, Bioactive Signaling in Next-Generation Pharmacotherapies for Heart Failure: A Review, JAMA Cardiol 3(12) (2018) 1232–1243. [DOI] [PubMed] [Google Scholar]
- [91].Shah AM, Cikes M, Prasad N, Li G, Getchevski S, Claggett B, Rizkala A, Lukashevich I, O’Meara E, Ryan JJ, Shah SJ, Mullens W, Zile MR, Lam CSP, McMurray JJV, Solomon SD, Investigators P-H, Echocardiographic Features of Patients With Heart Failure and Preserved Left Ventricular Ejection Fraction, J Am Coll Cardiol 74(23) (2019) 2858–2873. [DOI] [PubMed] [Google Scholar]
- [92].Solomon SD, McMurray JJV, Committee P-HS, Investigators, Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. Reply, N Engl J Med 382(12) (2020) 1182–1183. [DOI] [PubMed] [Google Scholar]
- [93].Cunningham JW, Claggett BL, O’Meara E, Prescott MF, Pfeffer MA, Shah SJ, Redfield MM, Zannad F, Chiang LM, Rizkala AR, Shi VC, Lefkowitz MP, Rouleau J, McMurray JJV, Solomon SD, Zile MR, Effect of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFpEF, J Am Coll Cardiol 76(5) (2020) 503–514. [DOI] [PubMed] [Google Scholar]
- [94].Zile MR, Lindenfeld J, Weaver FA, Zannad F, Galle E, Rogers T, Abraham WT, Baroreflex Activation Therapy in Patients With Heart Failure With Reduced Ejection Fraction, J Am Coll Cardiol 76(1) (2020) 1–13. [DOI] [PubMed] [Google Scholar]
- [95].Fish-Trotter H, Ferguson JF, Patel N, Arora P, Allen NB, Bachmann KN, Daniels LB, Reilly MP, Lima JAC, Wang TJ, Gupta DK, Inflammation and Circulating Natriuretic Peptide Levels, Circ Heart Fail 13(7) (2020) e006570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jensen J, Ma LP, Fu ML, Svaninger D, Lundberg PA, Hammarsten O, Inflammation increases NT-proBNP and the NT-proBNP/BNP ratio, Clin Res Cardiol 99(7) (2010) 445–52. [DOI] [PubMed] [Google Scholar]
- [97].Metra M, Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Pang PS, Ponikowski P, Voors AA, Adams KF, Anker SD, Arias-Mendoza A, Avendano P, Bacal F, Bohm M, Bortman G, Cleland JGF, Cohen-Solal A, Crespo-Leiro MG, Dorobantu M, Echeverria LE, Ferrari R, Goland S, Goncalvesova E, Goudev A, Kober L, Lema-Osores J, Levy PD, McDonald K, Manga P, Merkely B, Mueller C, Pieske B, Silva-Cardoso J, Spinar J, Squire I, Stepinska J, Van Mieghem W, von Lewinski D, Wikstrom G, Yilmaz MB, Hagner N, Holbro T, Hua TA, Sabarwal SV, Severin T, Szecsody P, Gimpelewicz C, Investigators R-A-C, Effects of Serelaxin in Patients with Acute Heart Failure, N Engl J Med 381(8) (2019) 716–726. [DOI] [PubMed] [Google Scholar]
- [98].Du XJ, Bathgate RA, Samuel CS, Dart AM, Summers RJ, Cardiovascular effects of relaxin: from basic science to clinical therapy, Nat Rev Cardiol 7(1) (2010) 48–58. [DOI] [PubMed] [Google Scholar]
- [99].Teerlink JR, Voors AA, Ponikowski P, Pang PS, Greenberg BH, Filippatos G, Felker GM, Davison BA, Cotter G, Gimpelewicz C, Boer-Martins L, Wernsing M, Hua TA, Severin T, Metra M, Serelaxin in addition to standard therapy in acute heart failure: rationale and design of the RELAX-AHF-2 study, Eur J Heart Fail 19(6) (2017) 800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Teerlink JR, Davison BA, Cotter G, Maggioni AP, Sato N, Chioncel O, Ertl G, Felker GM, Filippatos G, Greenberg BH, Pang PS, Ponikowski P, Edwards C, Senger S, Teichman SL, Nielsen OW, Voors AA, Metra M, Effects of serelaxin in patients admitted for acute heart failure: a meta-analysis, Eur J Heart Fail 22(2) (2020) 315–329. [DOI] [PubMed] [Google Scholar]
- [101].Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, Abouzaki NA, Dixon D, Kadariya D, Christopher S, Schatz A, Regan J, Viscusi M, Del Buono M, Melchior R, Mankad P, Lu J, Sculthorpe R, Biondi-Zoccai G, Lesnefsky E, Arena R, Abbate A, Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial), Circ Heart Fail 10(11) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT, Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease, N Engl J Med 377(12) (2017) 1119–1131. [DOI] [PubMed] [Google Scholar]
- [103].Ghobadi A, Chimeric antigen receptor T cell therapy for non-Hodgkin lymphoma, Curr Res Transl Med 66(2) (2018) 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Mullard A, FDA approves first CAR T therapy, Nat Rev Drug Discov 16(10) (2017) 669. [DOI] [PubMed] [Google Scholar]
- [105].Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM, Epstein JA, Targeting cardiac fibrosis with engineered T cells, Nature 573(7774) (2019) 430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH, Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma, Blood 122(6) (2013) 863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]