

Abstract
Background
The IGF2 (insulin-like growth factor 2) and H19 gene cluster plays an important role during pregnancy as it promotes both foetal and placental growth. We investigated the association between cord blood DNA methylation status of the IGF2/H19 gene cluster and maternal fine particulate matter exposure during fetal life. To the best of our knowledge, this is the first study investigating the association between prenatal PM2.5 exposure and newborn DNA methylation of the IGF2/H19.
Methods
Cord blood DNA methylation status of IGF2/H19 cluster was measured in 189 mother-newborn pairs from the ENVIRONAGE birth cohort (Flanders, Belgium). We assessed the sex-specific association between residential PM2.5 exposure during pregnancy and the methylation level of CpG loci mapping to the IGF2/H19 cluster, and identified prenatal vulnerability by investigating susceptible time windows of exposure. We also addressed the biological functionality of DNA methylation level in the gene cluster.
Results
Prenatal PM2.5 exposure was found to have genetic region-specific significant association with IGF2 and H19 during specific gestational weeks. The association was found to be sex-specific in both gene regions. Functionality of the DNA methylation was annotated by the association to fetal growth and cellular pathways.
Conclusions
The results of our study provided evidence that prenatal PM2.5 exposure is associated with DNA methylation in newborns’ IGF2/H19. The consequences within the context of fetal development of future phenotyping should be addressed.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12940-020-00677-9.
Keywords: Imprinted genes, IGF2, H19, Methylation, Factor analysis, DLNM, Air pollution, PM2.5, Gestation, Newborn, Fetal growth
Background
Genomic imprinting is an epigenetic process leading to monoallelic gene expression and one of the major regulators of genomic imprinting is DNA methylation. Imprinted genes are involved in cellular pathways crucial for growth and development [1]. Among over a hundred imprinted genes in humans, a pair of widely investigated is the insulin-like growth factor 2 (IGF2) gene, clustered with the reciprocally imprinted neighboring H19 gene. With one of the two alleles silenced, the paternal allele of IGF2 gene is expressed and the maternally active H19 gene downstream to IGF2 is transcribed into a non-coding RNA. Studies have shown IGF2 as a contributor to maternal nutrient supply to the fetus [2] and that loss of imprinting in IGF2 resulted in fetal overgrowth [3]. H19 partly serves as a regulator of IGF2 expression [4]. The DNA methylation levels in differentially methylated regions (DMR) of these two imprinted genes in placenta and cord blood have been reported to associate with birth size [5–8].
Ambient airborne fine particulate matter with diameter smaller than 2.5 μm, PM2.5, is one of the air pollution components with the strongest adverse effects on health and mortality as it can penetrate the respiratory system, circulate via bloodstream to other organs [9, 10] and cross the maternal–fetal placental barrier [11, 12]. Epidemiological studies have shown that maternal exposure to PM2.5 is associated with preterm birth and low birth weight [13, 14], elevated blood pressure [15, 16], changes in heart rate variability [17], respiratory disease [18] and central nervous system diseases [19, 20]. The imprinting status of genes is susceptible to environmental changes especially during fetal development, when DNA synthesis and cell division are extremely active. Prenatal exposure to PM2.5 may have even life-long consequences as, according to the Developmental Origins of Health and Diseases (DOHaD) theory, perturbations in the intrauterine environment are involved in the development of disease in later life [21]. Alterations in methylation of the imprinted IGF2/H19 cluster might be a potential mechanism underlying the association between in utero PM2.5 exposure and fetal growth, as maternal residential PM2.5 has been reported to alter their expression [22]. In this study, we assessed the association between maternal PM2.5 exposure during pregnancy and the DNA methylation level specific to IGF2/H19 gene cluster in cord blood collected at birth.
Methods
Study population
From the ongoing population-based prospective ENVIRONmental influence ON AGEing in early life (ENVIRONAGE) birth cohort [23], 199 mother-newborn (all singleton) pairs that were recruited between July 2014 and June 2015 were included. The study has been approved by the ethical committees of Hasselt University and East-Limburg Hospital and was conducted according to the ethical principles in Helsinki declaration [24]. Owing to missing exposure measurements (n = 2) and unavailable covariate information (n = 8), the final sample size in model fitting was 189.
The ENVIRONAGE birth cohort recruits mothers with a singleton newborn at arrival for delivery at the East-Limburg Hospital. At the first antennal visit (weeks 7–9 of pregnancy), maternal body mass index (BMI) was determined and the date of conception was estimated on the basis of the first day of the mother’s last menstrual period combined with the first ultrasonographic examination. We collected detailed information as newborn’s sex, gestational age, birth date, maternal age and parity from medical records. Ethnicity of a newborn was classified as European if at least 2 grandparents were Europeans and classified as non-European otherwise. Educational level of the mothers was coded “low” if they did not obtain a high school diploma, as “middle” if they obtained a high school diploma, and as “high” if they obtained a college or university degree. Maternal smoking status was categorized as “never smoked”, “former smoker” (when having quit smoking before pregnancy), or “smoker” (in case of continuing smoking during pregnancy). The presence of pregnancy complications was obtained from medical records on gestational diabetes, hypertension, hyperthyroidism or hypothyroidism, infectious disease, preclampsia, vaginal bleeding, phenylketonuria and allergy or asthma. Birth weight and length of newborns were also collected, based on which the Ponderal index (PI), an indicator of fetal growth status, was calculated according to Rohrer’s formulas: PI = 100 × Birth weight (in grams) / [Birth length (in centimeter)]3 [25].
PM2.5 exposure assessment
Daily PM2.5 concentration (μg/m3) measurements were obtained from the Belgian Interregional Environment Agency. Residential PM2.5 concentrations of the mothers during pregnancy was estimated for each mother’s address by the combination of land-cover data from satellite images and pollutant data from official fixed monitoring stations, processed by a model chain of spatial-temporal interpolation [23] and a dispersion model [26], providing high-resolution daily exposure values (on 100 m grids). Address changes during pregnancy were considered. In the Flemish region, this interpolation model predicts 80% of the spatial and temporal variance (based on R2) [27], and was validated with measurements of internal exposure in urine [10] and for gestational exposure with placental carbon load [11]. Based on the daily residential PM2.5 concentrations, we calculated the weekly mean PM2.5 concentrations of gestational week 1 to week 40 for each mother, with week 1 starting from the estimated date of conception. In case that gestational age was less than 40 weeks, the exposure values after delivery until week 40 were set to zero.
DNA methylation measurement
Details on cord blood sample collection are provided in Method S1, Additional file 1. DNA methylation was measured at the International Agency for Research on Cancer of Lyon in France. DNA extracted from buffy coat of cord blood samples collected at birth were used to determine the epigenome-wide DNA methylation profile through hybridization to the Illumina Infinium HumanMethylation450K BeadChip arrays [28]. The methylated (M) and unmethylated (U) signal intensities were detected and processed in R using the minfi package [29] to calculate the beta-value indicating the methylation level at each CpG site: β = M/(M + U + α), with α = 100 an offset value used to stabilize the calculation when both M and U are small [30]. The resulting beta-values, ranging from 0 (unmethylated) to 1 (fully methylated), were exported for quality control and pre-processing. The methylation data was filtered from cross-reactive probes and low-quality probes (probes with bead counts lower than 3 in at least 5% samples). Data quality was further evaluated by checking the distribution of the methylated and unmethylated signals. Sample outliers and gender mismatches were removed based on multidimensional scaling plots and the results of unsupervised clustering. Samples which had more than 1% of the probes with detection p-value > 0.05 were removed. The remaining DNA methylation data (485,577 probes) underwent functional normalization [31] using the minfi package. Each CpG locus was trimmed for potential outliers based on the range [Q1–3 × IQR, Q3 + 3 × IQR], with Q1 and Q3 the first and the third quartiles respectively, and IQR the inter-quartile range. Beta-values identified as outliers were replaced by missing values. Additionally, 40,590 probes targeting non-specific CpGs, 15,702 probes with missing values in over 20% of the samples, and 11,648 probes located on X or Y chromosomes were excluded. The remaining probes were examined if they contained single nucleotide polymorphisms located within 10 bps (SNP < 10 bps) of the target CpG, as suggested by Illumina [32], and the probe filtering for SNP < 10 bps was restricted to those found in newborns [33]. The CpG loci with their UCSC reference gene name referring to IGF2 or H19 were selected for the present study. In total, 145 CpGs mapping to IGF2 gene or its adjacent region including IGF2-INS and 62 CpG loci mapping to H19 were selected. Imputation methods were not applied because they tend to modify the correlation structure [34] and the amount of missing values was not substantial (Method S2, Additional file 1). We therefore excluded the CpG loci with incomplete records. 109 CpG loci of IGF2 and 53 of H19 remained for further analysis.
Statistical modelling
The beta-values and correlation structure of the CpGs mapping to each one gene was visualized using R package ComplexHeatmap [35]. In order to address the inter-correlation of the CpG loci [36], reduce the number of independent hypothesis tests and remove less relevant CpG loci, we performed factor analysis on the CpG loci mapping to IGF2 and H19, respectively. Factor analysis was performed based on the correlation matrices using iterative principal component factoring and varimax rotation, maximizing the sum of the squared loadings. Parallel analysis [37, 38] was used to decide the number of common factors to extract. The extracted factors were orthogonal to each other and the standardized factor scores were used as integrated measures for the actual methylation beta-values of the CpG loci and each entered a regression model as the response variable. CpG loci were selected as relevant to a factor if the absolute value of their factor loadings were larger than 0.45, which was visualized in chord diagrams using R package circlize [39]. Since the methylation array was performed in batches with 2 × 6 samples per each array chip, batch effects existed due to sample plate and sentrix positions (row- and column-coordinates). At least one of these grouping categories were introduced into the analysis as random effects, based on test results from the exact likelihood ratio test (LRT) for the presence of random effects for each of the IGF2 and H19 factors (Method S3, Additional file 1).
The models were adjusted for covariates chosen a priori based on previous studies [40, 41]. Those were parameters characterizing the mother and newborns: maternal age, pre-pregnancy BMI, educational level, smoking status, parity and presence of any pregnancy complication; newborn’s sex, gestational age, birth date, birth season and ethnicity. All categorical variables were contrast-coded, and all continuous variables except birth date were centered around the mean. The date of birth was calculated as the time difference in days between the actual birth date of each newborn and the first birth date in this data set. An interaction between newborn’s sex and PM2.5 exposure was included in the models based on previous evidence of sex-specific differences on molecular level in response to prenatal environmental exposures [42, 43]. Whether newborn’s sex was an effect modifier was assessed by performing a likelihood ratio test (LRT) on the interaction term. Afterwards, for each sex-group the change of factor scores for a 5 μg/m3 increment in PM2.5 concentration was estimated at each gestational week using distributed lag nonlinear models (DLNMs) proposed by Gasparrini et al. [44]. The DLNMs provide a flexible method to model the level of exposures while adjusting for lagged exposure values and thereby allows the identification of vulnerable exposure windows, which in turn provides hints on mechanisms through which exposure acts on fetal health [41, 45]. The exposure-response relationship and lag-response relationship are simultaneously involved in one model, via the construction of a cross-basis combining two basis-functions corresponding to exposure structure and lag structure, respectively. We assumed the exposure-response relationship to be linear and specified for the lag structure a natural cubic spline with 3 inner knots equally spaced along the original lag scale (week 1 to week 40) based on a previous study [41]. The total degree of freedom (DF) of the cross-basis was 5. The association between the factors and prenatal PM2.5 exposure was estimated for each gestational week. Based on the same DLNM models, the cumulative association over the entire pregnancy as well as for each trimester was calculated as the incremental cumulative predicted associations from gestational week 1 to week 40, from gestational week 1 to week 13, from gestational week 14 to 26, and from gestational week 27 to 40, respectively.
Since the maximum likelihood (ML) or restricted maximum likelihood (REML) estimators used in mixed model estimation are not robust to outliers, we applied a robust estimator using a smoothed Huber ψ-function and squared robustness weights. This method allows controlling the robustness on single observation level as well as on the group level. For the main analysis we had the estimator’s efficiency fixed at 95% relative to REML. This was done by setting the tuning parameter k for both fixed and random effects at 1.345 [46]. When likelihood needed to be calculated for model selection or LRT, the ML- or REML-estimator was used instead of the robust estimator.
In sensitivity analyses, (1) we excluded all pre-term birth observations to lower the potential influence of the missing PM2.5 measurements entering the model as zeros; (2) the flexibility of the natural cubic spline function for modelling lag structure was varied so that the total DF of cross-basis was compared between DF = 5 and DF = 7 or DF = 9; in parallel, an unconstrained DLM was fitted where all 40 weekly mean exposures entered the model separately; (3) the choice of robustness and estimation efficiency of the estimator was assessed, by comparing the main results (k = 1.345) to a non-robust REML estimator (k = ∞) and to a more robust but less efficient estimator (k = 1.69).
In order to address the functionality of DNA methylation in these two genes, we correlated the factor scores to cord blood DNA transcriptome. The detailed procedure of profiling the transcriptome, quality control, normalization and preprocessing is provided in Method S4, Additional file 1. In total, pairwise Pearson correlation was calculated between each factor and 29,164 transcripts. Based on the ascendingly ordered p-values of the correlation tests which were smaller than 0.05, the first 100 transcripts were used to perform the overrepresentation analysis (ORA) using the R package ReactomePA [47]. A pathway was considered significantly enriched if the p-value was smaller than 0.01 and q-value was smaller than 0.05 with at least 3 genes included in the functional set of a pathway. In addition, we assessed the association between the methylation level and fetal growth by regressing newborn’s birth weight or PI on the factor scores. The birth weight as well as PI were surrogates of fetal growth which have been reported to associate with changes in expression level of growth- and development-related genes [48]. The regression models were adjusted for maternal age, pre-pregnancy BMI, educational level, smoking status, parity, presence of pregnancy complications, newborn’s sex, gestational age, birth date, birth season and ethnicity. We assumed causality for the association among PM2.5 exposure, DNA methylation and fetal growth, and performed mediation analysis [49] to assess whether the change in IGF2/H19 DNA methylation mediates the association between prenatal PM2.5 exposure and fetal growth.
Data analyses were conducted in R (version 3.6.0) and SAS 9.4 (SAS Institute, Cary NC). Based on the number of extracted factors per gene, the family wise error rate (FWER) was controlled below 0.05 using the Sidak correction.
Results
Descriptive statistics
Details of the characteristics of the mother-newborn pairs are summarized in Table 1. 91 of the 189 newborns were girls (48.1%). PI of all newborns was 2.69 ± 0.22 g/cm3. 14 newborns (7.4%) was born preterm (10 boys and 4 girls). The weekly average residential PM2.5 concentration of 40 weeks for all mothers is summarized in Table 2, with a global mean of 12.97 and high variability (SD: 8.25 μg/m3). The correlation structure of the weekly average PM2.5 is shown in a heatmap of pairwise Pearson correlations in Figure S1, Additional file 1. PM2.5 levels of adjacent weeks were in general positively correlated. Some of the correlations were relatively higher than others, such as those near the end of gestation. Correlation between non-neighboring gestational weeks mainly appeared in the middle part of gestation. The beta-values as well as the correlation structures of the CpG loci mapping to IGF2 and H19 are illustrated in Figure 1a. Using a cutoff value of 0.50 for the beta-value, most of the CpGs resided in IGF2 gene body and close to CpG island showed hypomethylation, while those neighboring the transcription starting site (TSS) and distant to CpG island were hypermethylated. All 53 H19 CpGs were inside or close to CpG island and the majority of these CpGs displayed hypermethylation, especially those within gene body. The correlation heatmaps suggested that the hypermethylated loci tended to be highly positively correlated in both sets of CpGs and intercorrelation was also present within the hypomethylated IGF2 CpGs. This intercorrelation was consistent with the Kaiser-Meyer-Olkin (KMO) measure of sampling adequacy, which was 0.79 for CpG loci of IGF2 and 0.92 for CpG loci of H19, indicating factorability of each of the two sets of CpGs (KMO > 0.50 is considered suitable [50]). Both the heatmaps and KMO also suggested that correlations among all the H19 CpGs are higher than that among all the IGF2 CpGs.
Table 1.
Characteristics of the study population (189 mother-newborn pairs)
| Characteristic | N (%) | Mean ± SD |
|---|---|---|
| Newborns | ||
| Sex | ||
| Male | 98 (51.9) | |
| Female | 91 (48.1) | |
| Ethnicity | ||
| European | 170 (89.9) | |
| Non-European | 19 (10.1) | |
| Season of birth | ||
| Winter | 42 (22.2) | |
| Spring | 82 (43.4) | |
| Summer | 26 (13.8) | |
| Autumn | 39 (20.6) | |
| Birth weight (kg) | 3.40 ± 0.48 | |
| Ponderal index (g/cm3) | 2.69 ± 0.22 | |
| Gestational age (weeks) | 39.1 ± 1.6 | |
| < 37 | 14 (7.4) | |
| 37 | 6 (3.2) | |
| 38 | 26 (13.8) | |
| 39 | 46 (24.3) | |
| 40 | 72 (38.1) | |
| 41 | 25 (13.2) | |
| Date of birth (days) | 207 ± 102 | |
| Mothers | ||
| Educational level | ||
| Low | 27 (14.3) | |
| Middle | 65 (34.4) | |
| High | 97 (51.3) | |
| Smoking status | ||
| Never smoker | 122 (64.6) | |
| Former smoker | 41 (21.7) | |
| Smoker | 26 (13.8) | |
| Parity | ||
| Primiparous | 104 (55.0) | |
| Secundiparous | 59 (31.2) | |
| Multiparous | 26 (13.8) | |
| With pregnancy complications | 40 (21.2) | |
| Age (years) | 29.3 ± 4.3 | |
| Pre-pregnancy BMI (kg/m2) | 24.2 ± 4.2 | |
Table 2.
Summary statistics of weekly average PM2.5 concentration in μg/m3
| Mean ± SD | Minimum | 1st quartile | Median | 3rd quartile | Maximum |
|---|---|---|---|---|---|
| 12.97 ± 8.25 | 1.89 | 7.29 | 10.74 | 16.34 | 91.30 |
The missing weekly average PM2.5 values that were imputed as zero were excluded
Fig. 1.

Heatmaps of the CpGs and cord diagrams of the CpGs and factors. (a). Heatmaps of the 109 IGF2-related CpGs (upper rows) and 53 H19-related CpGs (lower rows). From left to right panels: heatmap of the beta-values, gene region annotation of the CpGs, CpG island annotation of the CpGs and correlation heatmap of the pairwise Pearson correlations. b. IGF2 CpGs and c H19 CpGs shown with beta-value averaged over all observations and their genomic context (obtained via UCSC Genome Browser). Factors were shown in different colors and the width represents the relative amount of variance explained by each factor. "F." is the abbreviation of "Factor". Factors and CpGs were connected with colored ribbons representing the factor loadings, where only loadings with absolute value larger than 0.45 were shown and higher color saturation indicates higher value of the loading. Except the loading of cg26913576 (the second CpG from left in (B)) on IGF2 Factor 1 was − 0.62, all loadings shown in these two diagrams were positive
Constructing integrated cord blood methylation measures of IGF2 and H19
An 8-factor solution and a 6-factor solution were used to construct the factor models for IGF2 and H19, respectively. When assessing the factor solution, we referred to the amount of total variance explained by the factors (higher than 60% as acceptable) and the square root of mean squared off-diagonal residuals (RMSR) (smaller than 0.042 as acceptable according to Kelly’s criterion). The 8 factors of IGF2 accounted for 51.2% of the total variance of the 109 CpGs and the corresponding RMSR was 0.04. Within the explained variance, the proportions of variance explained by Factor1–8 were 23.0, 21.1, 12.4, 12.3, 11.8, 7.7, 6.2 and 5.5%, respectively. For H19 gene, the 6 factors explained 64.0% of the total variance of the 53 CpGs and the model RMSR was 0.03. Factor1–6 covered 34.3, 22.5, 15.2, 12.6, 8.7 and 6.6% of the explained variance, respectively. After varimax rotation of the factor axes, the standardized factor scores were obtained, which ranged from − 3.79 to 4.54 in the 8 IGF2 factors and from − 3.76 to 3.02 in the 6 H19 factors. Factor loadings were shown in Table S1 and S2, Additional file 2. For each factor, the CpG loci with the absolute value of factor loading higher than 0.45 were grouped to that factor and are shown in the chord diagrams in Fig. 1b and c. All these factor loadings shown in the chord diagrams had a positive value, except that the loading of cg26913576 on IGF2 Factor1 was − 0.62. IGF2 Factor1, Factor4, Factor7 and Factor8 mostly pointed to a hypomethylated region near the IGF2 promoter and Factor 2 covered several genomic regions which were hypermethylated. Factor3 was mostly linked to CpGs inside IGF2 gene body and Factor5 was mostly related to the INS gene. H19 Factor1, Factor2 and Factor 5 mainly related to the CpGs outside H19 gene body and Factor5 also pointed to the promoter region. In the meanwhile, H19 Factor3, Factor4 and Factor6 were linked with CpGs within H19 gene body, which showed a relatively higher methylation level.
The standardized factor scores of the factors were used as response variables in the models, hence 8 independent hypotheses were tested for IGF2 and 6 independent hypotheses were tested for H19. By controlling the FWER at 0.05 for each gene using Sidak correction, the individual confidence level for IGF2 was 99.36% and for H19 was 99.15%. In sex-stratified analysis, the corresponding confidence levels were 99.68 and 99.57% due to the doubling of number of hypothesis tests.
Integrated cord blood DNA methylation levels of IGF2 and H19 in association with prenatal PM2.5 exposure
The interaction between newborn’s sex and PM2.5 cross-basis was significant in H19 Factor2 (LRT p-value = 0.036) indicating the effect of PM2.5 on methylation being sex-specific. Although the effect modification was not detected in other H19-factors and IGF2-factors (Table S3 and S4, Additional file 1), we report our results for all observations as well as in a sex-stratified way. Only the results for IGF2 Factor1 and Factor5 and H19 Factor2 and Factor5 were displayed because in other factors no significant associations were found. Figure 2 shows the week-specific estimates of the association between maternal exposure to PM2.5 during pregnancy and the standardized factor scores with confidence intervals. Table 3 shows the cumulative associations over the entire gestation as well as the three trimesters. All of the IGF2-factors were not found to be associated with PM2.5 exposures when all observations were considered together. However, specific to sexes, IGF2 Factor1 was significantly inversely associated with PM2.5 exposure in only boys during gestational week 38–40 and IGF2 Factor5 was significantly inversely associated with PM2.5 exposure in only girls during gestational week 38–40. For H19 gene, Factor2 was inversely associated with PM2.5 exposure during weeks 28–36 in all observations and during week 33–38 only in girls. In addition, the estimated cumulative change per 5-μg/m3 increment of PM2.5 in H19 Factor2 over trimester 3 was − 0.46 (99.15% CI: [− 0.79, − 0.13]) in all observations and − 0.73 (99.57% CI: [− 1.20, − 0.26]) in girls. H19 Factor5 showed a significant positive association with PM2.5 exposure during week 5–12 with a trimester-specific cumulative effect over trimester 1: 0.70 (99.15% CI: [0.05, 1.35]) per 5-μg/m3 increment of PM2.5 in all observations.
Fig. 2.
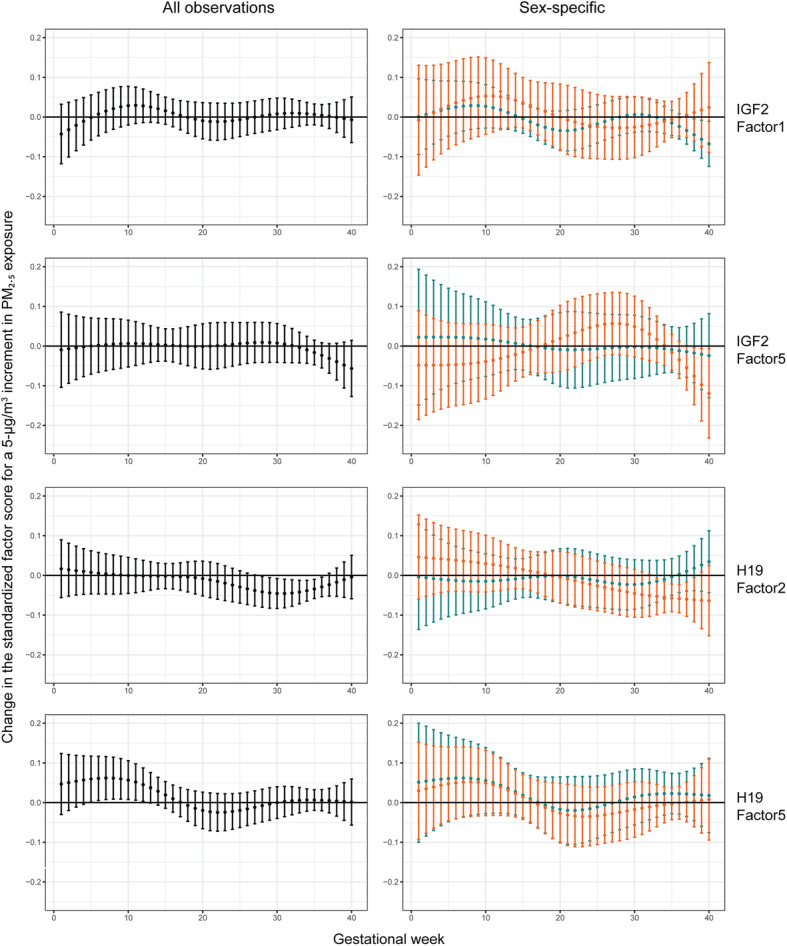
DLNM estimates of the week-specific associations. The estimates were in all observation (left column, n = 189), specific to newborn boys (n = 98, green) or girls (n = 91, orange) (right column), for IGF2 Factor1 (row 1) and Factor5 (row 2), and H19 Factor2 (row 3) and Factor5 (row 4), respectively. Estimates are presented as the change in factor scores for a 5-μg/m3 increase in PM2.5 concentration. Whiskers around the point estimates represent the confidence intervals of the week-specific estimates. For IGF2 factors, confidence level is 99.36% for all-observation and 99.68% for sex-specific analysis. For H19 factors, confidence level is 99.15% for all-observation and 99.57% for sex-specific analysis
Table 3.
The DLNM estimates of the cumulative association
| Exposure window | All newborns | Boys | Girls | |
|---|---|---|---|---|
|
IGF2 Factor1 |
Overall | 0.11 (− 0.71, 0.94) | − 0.25 (− 1.15, 0.64) | 0.35 (− 1.11, 1.81) |
| Trimester 1 | 0.07 (− 0.57, 0.71) | 0.25 (− 0.52, 1.01) | 0.44 (− 0.79, 1.67) | |
| Trimester 2 | − 0.02 (− 0.49, 0.45) | − 0.27 (− 0.79, 0.24) | 0.04 (− 0.81, 0.89) | |
| Trimester 3 | 0.06 (− 0.30, 0.41) | − 0.23 (− 0.68, 0.21) | − 0.13 (− 0.73, 0.48) | |
|
IGF2 Factor5 |
Overall | − 0.14 (− 1.11, 0.83) | 0.06 (− 1.52, 1.64) | − 0.40 (− 1.83, 1.02) |
| Trimester 1 | 0.02 (− 0.77, 0.81) | 0.25 (− 1.10, 1.60) | − 0.55 (− 1.75, 0.65) | |
| Trimester 2 | 0.03 (− 0.55, 0.61) | − 0.06 (− 1.02, 0.90) | 0.24 (− 0.60, 1.09) | |
| Trimester 3 | − 0.19 (− 0.61, 0.24) | −0.13 (− 0.93, 0.67) | −0.09 (− 0.69, 0.51) | |
|
H19 Factor2 |
Overall | −0.54 (− 1.29, 0.20) | −0.29 (− 1.47, 0.89) | −0.34 (− 1.42, 0.74) |
| Trimester 1 | 0.07 (−0.54, 0.68) | − 0.15 (− 1.15, 0.86) | 0.46 (− 0.45, 1.37) | |
| Trimester 2 | −0.16 (− 0.60, 0.29) | −0.08 (− 0.80, 0.64) | −0.07 (− 0.73, 0.59) | |
| Trimester 3 | −0.46 (− 0.79, − 0.13) | −0.06 (− 0.71, 0.58) | −0.73 (− 1.20, − 0.26) | |
|
H19 Factor5 |
Overall | 0.61 (−1.19, 1.42) | 0.87 (− 0.53, 2.27) | 0.23 (− 1.02, 1.48) |
| Trimester 1 | 0.70 (0.05, 1.35) | 0.71 (−0.48, 1.90) | 0.56 (− 0.49, 1.61) | |
| Trimester 2 | −0.11 (− 0.58, 0.36) | −0.07 (− 0.92, 0.78) | −0.22 (− 0.98, 0.54) | |
| Trimester 3 | 0.02 (−0.33, 0.37) | 0.23 (− 0.47, 0.94) | −0.11 (− 0.66, 0.43) |
The cumulative effect of a 5 μg/m3 increase in PM2.5 concentration on IGF2 Factor1 and Factor5 and H19 Factor2 and Factor5 over different exposure windows. Estimated in all observations (n = 189), in boys (n = 98) and in girls (n = 91), respectively. Confidence intervals are shown with lower and upper bounds. Confidence level for IGF2 factors in all-observation analysis is 99.36% and that of sex-specific analysis is 99.68%. Confidence level for H19 factors in all-observation analysis is 99.15% and that of sex-specific analysis is 99.57%
In sensitivity analysis, we repeated the sex-specific associations with exclusion of 14 preterm births (10 boys and 4 girls), which validated our main results since the estimated associations were only slightly more evident if preterm birth observations were excluded (Table S5, Additional file 1). Therefore, there was not much estimation bias induced by setting the missing values to zero at the end of gestation. Sensitivity analyses on the DF of the cross-basis suggested DF = 5 as the best based on model information criterion (Table S6, Additional file 1). In addition, the existence of vulnerable exposure window was confirmed by performing LRT to the unconstrained model where all 40 weekly average exposures entered the model equally. The application of the robust estimator facilitated finding the association while the classical REML estimator failed for H19 Factor2 (Table S7, Additional file 1).
Biological indications of the integrated DNA methylation
The pathways that the selected transcripts were mapping to are shown in Figure S2, Additional file 1. The majority of the listed pathways of IGF2 Factor1 were related hemoglobin metabolism and detoxification of reactive oxygen species. H19 Factor2 showed relation to the regulation of rRNA expression and the H19 Factor5 had pathways enriched in cell cycle regulation and response to cellular stress.
We then assessed the association between birth weight or PI, as the surrogate for fetal growth, and each factor that showed significant association with PM2.5 exposure (Table S8, Additional file 1). In girls a SD increase in IGF2 Factor5 is associated with 0.05 g/cm3 decrease in PI (p-value = 0.026). Mediation analysis was performed assuming the causality in the relationship among PM2.5 exposure, IGF2 Factor5 and PI in girls, despite that PM2.5 exposure and PI was not significantly associated, which is tolerable in mediation analysis [49]. We took the average value of the PM2.5 concentrations from the last three gestational weeks as the exposure variable since we have found significant association between PM2.5 and IGF2 Factor5 during the last 3 weeks in girls. However, we did not notice the factor scores of IGF2 Factor5 to be mediating the relationship between fetal growth and air pollution (Table S9, Additional file 1).
Discussion
Both IGF2 and H19 are imprinted genes integrally important in the transfer of nutrients from mother to offspring and involved in fetal and postnatal growth. To assess the importance of the epigenetic regulation of this gene cluster and the influence by in utero particulate air pollution exposure, we tested the association between DNA methylation of the IGF2/H19 cluster in cord blood and prenatal air pollution and identified vulnerable exposure windows during gestation. The DNA methylation levels in the genes were integrated into common factors and the relationship between the factor scores and exposure was estimated through the DLNM cross-basis. The associations were found to be sex-specific. We also have addressed the functionality and biological significance of the two genes by assessing the association between fetal growth and the methylation levels, and mapping to functional pathways via DNA transcriptome.
Early-life health status have been shown to be associated with methylation and expression of imprinted genes, which are susceptible to environmental changes during fetal growth. Thus, identifying environmental determinants for early-life imprinted gene methylation profile is of importance. A schematic illustration of IGF2 and H19 activation is shown in Figure S3, Additional file 1. The two genes share one enhancer downstream of H19. DMRs in the promoters of the two genes interact with the enhancer, determining which allele of which gene segment to be transcribed. Studies have shown that methylation on loci in the DMR is important for activating or blocking the transcription [51–54]. No study yet has shown whether the changes in DNA methylation level of the enhancer regions has an influence on molecular pathways or health status in human, although it has been reported that deletion of the enhancer reduced 20% of the birth size in mice [55]. The present study found an association between prenatal PM2.5 exposure and methylation of H19 Factor2 and Factor5, which are factors highly correlated to loci close to the enhancer. In addition, the heatmap in Fig. 1a suggests that H19 gene body was generally hypermethylated across samples, while the region neighboring the TSS, which included H19 Factor 2 and Factor5 CpGs, showed much more variation. In the meanwhile, the most variable region in IGF2 included a fraction of gene body (IGF2 Factor 5) and a fraction of TSS regions (both IGF2 factors). Our findings might suggest unrevealed mechanisms underlying the transcription regulation mechanism of IGF2/H19.
The CpG loci were found to be highly correlated, calling for multivariate techniques to model the correlation structure. By applying factor analysis to the CpG loci we reduced the dimension of the methylation data and removed CpG loci with low correlations. The extracted factors were independent to each other, allowing the control of FWER. In addition, the varimax rotation of factor axes brought an easier interpretation of the latent factors since each CpG had a high loading on one factor and near zero on other factors, which made the factors to carry non-overlapping information. Factor analysis has been applied to omics data in previous studies [56–58], but the use in an epidemiological study on methylation data is rather new. Most of the CpG loci which mapped to one factor located within each other’s close neighborhood and all factors distributed across the whole length of the genes, justifying our choice of the factors to represent all the CpG loci mapping to the genes.
The direction of the association between PM2.5 and H19 Factor5 was positive while H19 Factor2, IGF2 Factor1 and Factor5 were inversely associated with the gestational PM2.5 exposure. Since almost all the loadings of the most relevant CpGs on factors were positive, the behavior of factors is consistent in direction with the behavior of the majority of the selected CpG loci, and a positive/negative association of a factor to PM2.5 in general could be translated into a local hypermethylation/hypomethylation related to the exposure. This is the situation for IGF2 Factor5, H19 Factor 2 and Factor5. An exception was IGF2 Factor1, which included one CpG (cg26913576) inside gene body with negative loading on this factor. This indicated an inverse correlation between this individual CpG and the other CpGs that were relevant to this same factor, which is also observable in the mean beta-value at cg26913576 (hypermethylated) and the other CpGs (hypomethylated). Therefore, the inverse association between boys’ IGF2 Factor1 and prenatal PM2.5 exposure in weeks 38–40 indicated an inverse association in the majority of CpGs of this factor and simultaneously a positive association at cg26913576. These findings based on factors are only partly comparable to epigenome-wide association studies (EWAS), because although changes in methylation at single CpGs are reflected through the loadings, it is not necessary that individual CpGs are significantly associated with the exposure and the correlation between CpGs is stressed which is not available in EWAS.
Previous studies have shown that placental expression level of IGF2/H19 was inversely associated with maternal PM2.5 exposure during pregnancy [22] and that PM2.5 exposure was in association with smaller birth size [14, 40]. It has also been reported that hypermethylation in the DMR of H19 was associated with lower weight-for-length [59] and higher transcription level of H19 was linked to larger length-of-gestational-age [60]. Our results of positive correlation between PM2.5 exposure and H19 Factor5, as well as the inverse association between IGF2 Factor5 and girls’ PI might add to these. On the other hand, the negative association between PM2.5 exposure and H19 Factor2, IGF2 Factor1 and Factor5 methylation is consistent with a study on smoking showing lower H19 methylation in cord blood was associated with maternal smoking during pregnancy and partially mediated newborns’ being small for gestational age [61]. In the mediation analysis, we did not identify IGF2 Factor5 as a mediator significantly in the association between PM2.5 exposure and fetal growth in girls. Indeed, the mediation effect should be found in multiple factors since both PM2.5 exposure and fetal growth are associated with multi-omic alterations, and IGF2 Factor5 might take up only a small fraction. However, the regulation pathway of IGF2/H19 is not completely understood yet. Complexity also comes from that the enhancer is shared by the two genes that are activated in different alleles, but the methylation array provided no clue to distinguish between the methylation on paternal and maternal alleles. Functional interpretation of the factors might find its reflection in the results of pathway analysis. The pathway analysis was performed indirectly via correlating to transcripts in the same cord blood samples, and was expected to suggest potential pathways involved in the epigenetic alteration associated with prenatal PM2.5 exposure, which might include trans regulation of IGF2/H19 imprinting, or multi-omic alterations occurred in parallel to DNA methylation. Methylation in the two genes reflected different cellular functions: IGF2 Factor1 was related to hemoglobin metabolism while H19 factors were mostly related to cell cycle and response to cellular stress. Through studying the factors of the IGF2/H19 gene cluster, we might find indication in the influence on metabolism and even cellular ageing [62] by prenatal exposure to fine particulate matters.
The use of DLNM models allowed us to investigate more detailed gestational exposure windows with higher temporal resolution (weeks) while previous studies on exposures during pregnancy mostly focused on trimester averages or on whole pregnancy averages. DLNM has been typically used to investigate the triggering effect of exposures on health outcomes such as mortality, taking delayed effects into account up to, for instance, 1 month after the exposure. Only recently, has the DLNM approach been introduced to prenatal and perinatal epidemiology [41, 63]. The application of robust linear mixed-effects model is also relatively new, through which we reduced the influence of outliers on different levels and improved the detection of the association.
Despite the novelties and strengths of the present study, restrictions existed in the study design since our results are based on pollutant concentration at the maternal residence, and potential misclassification may be present because we could not account for other exposure sources that contribute to personal exposure, such as exposure during commuting or at work. However, our high-resolution model of residential exposure has recently been shown to be associated with internal exposure to nanosized particles black carbon in urine [10] as well as in placental tissue [11]. Moreover, we do not know the persistence of the current findings as our methylation were only studied at one time point (birth). The sample size of this study was relatively small which might have not provided sufficient statistical power. This is especially clear when performing sex-specific analysis. As shown in Fig. 2 for H19 Factor 5, the associations in all observations, girls and boys were in the same shape. However, the significant association was only detected in all observations but not in either subset.
Conclusion
Our study showed that alterations in methylation of two imprinted genes known to be important in fetal growth were associated with in utero PM2.5 exposure. Our findings added to the growing body of evidence that prenatal exposure to fine particulate matters impacts newborns on molecular level. The identification of vulnerable exposure windows and the specific relevant CpG loci or genetic regions might contribute to further investigation on the mechanism underlying the effects of particulate matters on fetal growth, as well as the unravelling of the molecular pathway of IGF2/H19 imprinting.
Supplementary Information
Additional file 1. Supplementary Methods S1-S4. Supplementary Tables S3-S9. Supplementary Figures S1-S3.
Additional file 2. Supplementary Tables S1-S2.
Disclaimer
Where authors are identified as personnel of the International Agency for Research on Cancer / World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer / World Health Organization.
Abbreviations
- BMI
Body mass index
- DF
Degree of freedom
- DLM
Distributed lag model
- DLNM
Distributed lag non-linear model
- DMR
Differentially methylated region
- DOHaD
Developmental Origins of Health and Diseases
- ENVIRONAGE
Environmental Influence on Ageing in Early Life birth cohort
- EWAS
Epigenome-wide association study
- FWER
Family-wise error rate
- IGF2
Insulin-like growth factor 2
- LRT
Likelihood ratio test
- ML
Maximum-likelihood
- ORA
Overrepresentation analysis
- PI
Ponderal index
- PM
Particulate matter
- REML
Restricted maximum likelihood
- SD
Standard deviation
- TSS
Transcription starting site
Authors’ contributions
TSN coordinates the ENVIRONAGE birth cohort. TSN, BC and CW designed the research hypotheses. AG and ZH performed the methylation array. CW performed the statistical analyses. RA provided guidance to perform overrepresentation analysis. CW, BC and TSN prepared the first draft of the manuscript. CW, BC, TSN, RA and MP were involved in data interpretation. All authors contributed in critical revision of the manuscript. The author(s) read and approved the final manuscript.
Funding
The ENVIRONAGE birth cohort is supported by the EU program Ideas (ERC-2012-StG 310898) and the Flemish Scientific Fund (FWO, G073315N) and ‘Kom op Tegen Kanker’. B.C. is a postdoctoral fellow of the FWO (12Q0517N). The methylation assay was supported by the FP7 project EXPOSOMICS (308610).
Availability of data and materials
Data available on request.
Ethics approval and consent to participate
The study protocol was approved by the Ethical Committee of Hasselt University and East-Limburg Hospital in Genk (Belgium) and has been carried out according to the declaration of Helsinki. Written informed consent was obtained from all participating mothers.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Congrong Wang, Email: congrong.wang@uhasselt.be.
Michelle Plusquin, Email: michelle.plusquin@uhasselt.be.
Akram Ghantous, Email: GhantousA@iarc.fr.
Zdenko Herceg, Email: HercegZ@iarc.fr.
Rossella Alfano, Email: rossella.alfano@uhasselt.be.
Bianca Cox, Email: bianca.cox@uhasselt.be.
Tim S. Nawrot, Email: tim.nawrot@uhasselt.be
References
- 1.Lambertini L, et al. Imprinted gene expression in fetal growth and development. Placenta. 2012;33(6):480–6. doi: 10.1016/j.placenta.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Constancia M, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417(6892):945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- 3.Lau MM, et al. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994;8(24):2953–2963. doi: 10.1101/gad.8.24.2953. [DOI] [PubMed] [Google Scholar]
- 4.Gabory A, et al. The H19 gene: regulation and function of a non-coding RNA. Cytogenet Genome Res. 2006;113(1–4):188–193. doi: 10.1159/000090831. [DOI] [PubMed] [Google Scholar]
- 5.Adkins RM, et al. Association of birth weight with polymorphisms in the IGF2, H19, and IGF2R genes. Pediatr Res. 2010;68(5):429–434. doi: 10.1203/PDR.0b013e3181f1ca99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.St-Pierre J, et al. IGF2 DNA methylation is a modulator of newborn's fetal growth and development. Epigenetics. 2012;7(10):1125–1132. doi: 10.4161/epi.21855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics. 2012;7(7):735–746. doi: 10.4161/epi.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoyo C, et al. Association of cord blood methylation fractions at imprinted insulin-like growth factor 2 (IGF2), plasma IGF2, and birth weight. Cancer Causes Control. 2012;23(4):635–645. doi: 10.1007/s10552-012-9932-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng S, et al. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol Environ Saf. 2016;128:67–74. doi: 10.1016/j.ecoenv.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 10.Saenen ND, et al. Children’s Urinary Environmental Carbon Load. A Novel Marker Reflecting Residential Ambient Air Pollution Exposure? Am J Respir Crit Care Med. 2017;196(7):873–881. doi: 10.1164/rccm.201704-0797OC. [DOI] [PubMed] [Google Scholar]
- 11.Bové H, et al. Ambient black carbon particles reach the fetal side of human placenta. Nat Commun. 2019;10(1):3866. doi: 10.1038/s41467-019-11654-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wick P, et al. Barrier capacity of human placenta for nanosized materials. Environ Health Perspect. 2010;118(3):432–436. doi: 10.1289/ehp.0901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah PS, Balkhair T. Air pollution and birth outcomes: a systematic review. Environ Int. 2011;37(2):498–516. doi: 10.1016/j.envint.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen M, et al. Ambient air pollution and low birthweight: a European cohort study (ESCAPE) Lancet Respir Med. 2013;1(9):695–704. doi: 10.1016/S2213-2600(13)70192-9. [DOI] [PubMed] [Google Scholar]
- 15.Zhang M, et al. Maternal exposure to ambient particulate matter </=2.5 microm during pregnancy and the risk for high blood pressure in childhood. Hypertension. 2018;72(1):194–201. doi: 10.1161/HYPERTENSIONAHA.117.10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madhloum N, et al. Neonatal blood pressure in association with prenatal air pollution exposure, traffic, and land use indicators: an ENVIRONAGE birth cohort study. Environ Int. 2019;130:104853. doi: 10.1016/j.envint.2019.05.047. [DOI] [PubMed] [Google Scholar]
- 17.Saenen ND, et al. Child's buccal cell mitochondrial DNA content modifies the association between heart rate variability and recent air pollution exposure at school. Environ Int. 2019;123:39–49. doi: 10.1016/j.envint.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee A, et al. Prenatal fine particulate exposure and early childhood asthma: effect of maternal stress and fetal sex. J Allergy Clin Immunol. 2018;141(5):1880–1886. doi: 10.1016/j.jaci.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genc S, et al. The adverse effects of air pollution on the nervous system. J Toxicol. 2012;2012:782462. doi: 10.1155/2012/782462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sunyer J, et al. Association between traffic-related air pollution in schools and cognitive development in primary school children: a prospective cohort study. PLoS Med. 2015;12(3):e1001792. doi: 10.1371/journal.pmed.1001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barker DJ. The fetal and infant origins of adult disease. Bmj. 1990;301(6761):1111. doi: 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kingsley SL, et al. Maternal residential air pollution and placental imprinted gene expression. Environ Int. 2017;108:204–211. doi: 10.1016/j.envint.2017.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen BG, et al. Cohort Profile: The ENVIRonmental influence ON early AGEing (ENVIRONAGE): a birth cohort study. Int J Epidemiol. 2017;46(5):1386–1387. doi: 10.1093/ije/dyx033. [DOI] [PubMed] [Google Scholar]
- 24.Association, W.M World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 25.Landmann E, et al. Ponderal index for discrimination between symmetric and asymmetric growth restriction: percentiles for neonates from 30 weeks to 43 weeks of gestation. J Matern Fetal Neonatal Med. 2006;19(3):157–160. doi: 10.1080/14767050600624786. [DOI] [PubMed] [Google Scholar]
- 26.Lefebvre W, et al. Presentation and evaluation of an integrated model chain to respond to traffic- and health-related policy questions. Environ Model Softw. 2013;40:160–170. doi: 10.1016/j.envsoft.2012.09.003. [DOI] [Google Scholar]
- 27.Maiheu B., et al.,Bepaling van de best beschikbare grootschalige concentratiekaarten luchtkwaliteit voor België (Identifying the best available large-scale concentration maps for air quality in Belgium).2012.
- 28.Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Aryee MJ, et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinhold L, et al. A statistical model for the analysis of beta values in DNA methylation studies. BMC Bioinformatics. 2016;17(1):480. doi: 10.1186/s12859-016-1347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fortin JP, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15(12):503. doi: 10.1186/s13059-014-0503-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price EM, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6(1):4. doi: 10.1186/1756-8935-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaunt TR, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016;17(1):61. doi: 10.1186/s13059-016-0926-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor SL, et al. Effects of imputation on correlation: implications for analysis of mass spectrometry data from multiple biological matrices. Brief Bioinform. 2017;18(2):312–320. doi: 10.1093/bib/bbw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 36.Zhang W, et al. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 2015;16(1):14. doi: 10.1186/s13059-015-0581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franklin SB, et al. Parallel analysis: a method for determining significant principal components. J Veg Sci. 1995;6(1):99–106. doi: 10.2307/3236261. [DOI] [Google Scholar]
- 38.O’connor BPJBRM. Instruments, and computers, SPSS and SAS programs for determining the number of components using parallel analysis and Velicer’s MAP test. Behav Res Methods Instrum Comput. 2000;32(3):396–402. doi: 10.3758/BF03200807. [DOI] [PubMed] [Google Scholar]
- 39.Gu Z, et al. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19):2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 40.Winckelmans E, et al. Fetal growth and maternal exposure to particulate air pollution--more marked effects at lower exposure and modification by gestational duration. Environ Res. 2015;140:611–618. doi: 10.1016/j.envres.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Martens DS, et al. Prenatal air pollution and Newborns' predisposition to accelerated biological aging. JAMA Pediatr. 2017;171(12):1160–1167. doi: 10.1001/jamapediatrics.2017.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hochstenbach K, et al. Global gene expression analysis in cord blood reveals gender-specific differences in response to carcinogenic exposure in utero. Cancer Epidemiol Biomark Prev. 2012;21(10):1756–1767. doi: 10.1158/1055-9965.EPI-12-0304. [DOI] [PubMed] [Google Scholar]
- 43.Winckelmans E, et al. Newborn sex-specific transcriptome signatures and gestational exposure to fine particles: findings from the ENVIRONAGE birth cohort. Environ Health. 2017;16(1):52. doi: 10.1186/s12940-017-0264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gasparrini A, Armstrong B, Kenward MG. Distributed lag non-linear models. Stat Med. 2010;29(21):2224–2234. doi: 10.1002/sim.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson A, et al. Bayesian distributed lag interaction models to identify perinatal windows of vulnerability in children's health. Biostatistics. 2017;18(3):537–552. doi: 10.1093/biostatistics/kxx002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koller M. robustlmm: An R Package for Robust Estimation of Linear Mixed-Effects Models. J Stat Softw. 2016;75(6):24. doi: 10.18637/jss.v075.i06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu G, He Q-Y. ReactomePA: an R/bioconductor package for reactome pathway analysis and visualization. Mol BioSyst. 2016;12(2):477–479. doi: 10.1039/C5MB00663E. [DOI] [PubMed] [Google Scholar]
- 48.Vrijens K, et al. Placental hypoxia-regulating network in relation to birth weight and ponderal index: the ENVIRONAGE birth cohort study. J Transl Med. 2018;16(1):2. doi: 10.1186/s12967-017-1375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valeri L, Vanderweele TJ. Mediation analysis allowing for exposure-mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol Methods. 2013;18(2):137–150. doi: 10.1037/a0031034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabachnick B, Fidell LS. Using Multivarite Statistics. 2007. p. 980. [Google Scholar]
- 51.Thorvaldsen JL, et al. Developmental profile of H19 differentially methylated domain (DMD) deletion alleles reveals multiple roles of the DMD in regulating allelic expression and DNA methylation at the imprinted H19/Igf2 locus. Mol Cell Biol. 2006;26(4):1245–1258. doi: 10.1128/MCB.26.4.1245-1258.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hark AT, et al. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405(6785):486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 53.Engel N, et al. Antagonism between DNA hypermethylation and enhancer-blocking activity at the H19 DMD is uncovered by CpG mutations. Nat Genet. 2004;36(8):883–8. doi: 10.1038/ng1399. [DOI] [PubMed] [Google Scholar]
- 54.Sasaki H, Ishihara K, Kato R. Mechanisms of Igf2/H19 imprinting: DNA methylation, chromatin and long-distance gene regulation. J Biochem. 2000;127(5):711–5. doi: 10.1093/oxfordjournals.jbchem.a022661. [DOI] [PubMed] [Google Scholar]
- 55.Leighton PA, et al. An enhancer deletion affects both H19 and Igf2 expression. Genes Dev. 1995;9(17):2079–2089. doi: 10.1101/gad.9.17.2079. [DOI] [PubMed] [Google Scholar]
- 56.Argelaguet R, et al. Multi-Omics Factor Analysis—a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. 2018;14(6):e8124. doi: 10.15252/msb.20178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu L, et al. Association between brain gene expression, DNA methylation, and alteration of ex vivo magnetic resonance imaging transverse relaxation in late-life cognitive decline. JAMA Neurology. 2017;74(12):1473–1480. doi: 10.1001/jamaneurol.2017.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rijlaarsdam J, et al. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: a model approach for replication. Epigenetics. 2016;11(2):140–149. doi: 10.1080/15592294.2016.1145329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalez-Nahm S, et al. DNA methylation of imprinted genes at birth is associated with child weight status at birth, 1 year, and 3 years. Clin Epigenetics. 2018;10:90. doi: 10.1186/s13148-018-0521-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kappil MA, et al. Placental expression profile of imprinted genes impacts birth weight. Epigenetics. 2015;10(9):842–9. doi: 10.1080/15592294.2015.1073881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bouwland-Both MI, et al. Prenatal parental tobacco smoking, gene specific DNA methylation, and newborns size: the generation R study. Clin Epigenetics. 2015;7:83. doi: 10.1186/s13148-015-0115-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catic A. Cellular metabolism and aging. Prog Mol Biol Transl Sci. 2018;155:85–107. doi: 10.1016/bs.pmbts.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu H, et al. Associations between maternal weekly air pollutant exposures and low birth weight: a distributed lag non-linear model. Environ Res Lett. 2018;13(2):024023. doi: 10.1088/1748-9326/aaa346. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary Methods S1-S4. Supplementary Tables S3-S9. Supplementary Figures S1-S3.
Additional file 2. Supplementary Tables S1-S2.
Data Availability Statement
Data available on request.