

Abstract
Sepsis remains a major public health problem with no major therapeutic advances over the last several decades. The clinical and biological heterogeneity of sepsis have limited success of potential new therapies. Accordingly, there is considerable interest in developing a precision medicine approach to inform more rational development, testing, and targeting of new therapies. We previously developed the Pediatric Sepsis Biomarker Risk Model (PERSEVERE) to estimate mortality risk and proposed its use as a prognostic enrichment tool in sepsis clinical trials; prognostic enrichment selects patients based on mortality risk independent of treatment. Here, we show that PERSEVERE has excellent performance in a diverse cohort of children with septic shock with potential for use as a predictive enrichment strategy; predictive enrichment selects patients based on likely response to treatment. We demonstrate that the PERSEVERE biomarkers are reliably associated with mortality in mice challenged with experimental sepsis, thus providing an opportunity to test precision medicine strategies in the preclinical setting. Using this model, we tested two clinically feasible therapeutic strategies, guided by the PERSEVERE-based enrichment, and found that mice identified as high risk for mortality had a greater bacterial burden and could be rescued by higher doses of antibiotics. The association between higher pathogen burden and higher mortality risk was corroborated among critically ill children with septic shock. This bedside to bench to bedside approach provides proof of principle for PERSEVERE-guided application of precision medicine in sepsis.
INTRODUCTION
Precision medicine aims to align therapeutic interventions with underlying biology (1). Septic shock is a major cause of morbidity and mortality worldwide, including in the pediatric population (2, 3). Despite years of research and development, there have been no major therapeutic breakthroughs for septic shock and there is no precision medicine approach. Treatment for septic shock has remained essentially unchanged for decades, relying primarily on care bundles, protocols, antibiotics, and intensive care unit–based organ support. An important barrier for developing new therapies for septic shock is lack of a unifying, targetable biological mechanism applicable to all patients with septic shock (2). Given the extensive biological heterogeneity observed in septic shock, it is perhaps expected that countless therapies have failed at the phase 3 clinical trial stage despite seemingly robust preclinical data. Consequently, there is considerable interest in developing precision medicine approaches, and there have been calls for prognostic and predictive enrichment strategies for more rational development, testing, and targeting of new therapies (1, 2, 4, 5).
Prognostic enrichment refers to the selection of patients for study based on their expected disease-related event rate, such as mortality, whereas predictive enrichment refers to the selection of patients more likely to respond to a therapeutic intervention based on underlying biology (4). Predictive enrichment requires a firm understanding of the underlying biological basis of disease, which is comparatively absent in septic shock.
We previously developed a prognostic enrichment tool for pediatric septic shock, the Pediatric Sepsis Biomarker Risk Model (PERSEVERE), using a multistep approach (6). The initial step involved agnostic, discovery-oriented transcriptomic studies, coupled with machine learning, to identify gene expression patterns associated with mortality from pediatric septic shock. From about 80 candidate genes identified, we subsequently reduced the list to 12 genes based on two a priori criteria. The first criterion was biological plausibility linking the gene to sepsis biology on the basis of existing knowledge. The second criterion was the ability to measure the protein product of the gene in the serum compartment. These 12 initial PERSEVERE biomarkers were subsequently reduced to 5 biomarkers using classification and regression tree (CART) methodology to develop a model estimating the risk of 28-day mortality among children with septic shock. The five PERSEVERE biomarkers, each of which reflects sepsis biology, include C-C chemokine ligand 3 (CCL3), interleukin 8 (IL8), heat shock protein 70 kDa 1B (HSPA1B), granzyme B (GZMB), and matrix metallopeptidase 8 (MMP8). PERSEVERE-based risk assignment occurs within the first 24 hours of a septic shock diagnosis, which is a relevant time frame for a clinically useful prognostic enrichment strategy in acute illness (6–8).
We subsequently developed PERSEVERE II, which represents a calibration of PERSEVERE and the addition of admission platelet count as a predictor variable (9). Because PERSEVERE and PERSEVERE II include objective biomarker data, we posited that this information might reflect septic shock biology and hence inform prediction, beyond just being prognostic for mortality risk.
Here, we took a bedside to bench to bedside translational research approach to further develop the clinical utility of PERSEVERE. We confirmed the performance of PERSEVERE and PERSEVERE II in a prospectively enrolled, heterogeneous cohort of children with septic shock and subsequently tested the ability of the PERSEVERE biomarkers to estimate baseline risk of mortality in a murine model of sepsis. We showed that the association between the PERSEVERE biomarkers and outcome from sepsis is conserved in mice and designed an experimental model of the biological links between clinically relevant biomarkers and the pathways to poor outcome from sepsis. Last, we assessed the utility of this model for testing experimental therapeutic interventions and demonstrated that PERSEVERE can function not only as a prognostic enrichment tool but also as a predictive enrichment tool with delineated underlying biology amenable to intervention.
RESULTS
Prospective testing of PERSEVERE and PERSEVERE II
The cohort for comparing PERSEVERE and PERSEVERE II consisted of 461 patients with a 28-day mortality rate of 12.6%. Table S1 describes the cohort, which is characterized by substantial heterogeneity with respect to age, comorbidity burden, and causative pathogens. We used PERSEVERE to assign each study subject a baseline mortality probability (fig. S1); prognostic performance for 28-day mortality was modest, with an area under the receiver operating curve (AUROC) of 0.67 [95% confidence interval (CI), 0.58 to 0.75]. Figure 1 shows the classification of the test cohort according to PERSEVERE II. In contrast to PERSEVERE, PERSEVERE II had excellent performance with an AUROC of 0.83 (95% CI, 0.77 to 0.88) for discriminating between survivors and nonsurvivors at 28 days. The AUROC of PERSEVERE II was greater than that of the Pediatric Risk of Mortality (PRISM) III score (0.74; 95% CI, 0.66 to 0.81; P = 0.03). Test characteristics of PERSEVERE and PERSEVERE II are shown in Table 1, and Fig. 2 shows 28-day survival curves for high-, intermediate-, and low-risk groups defined according to PERSEVERE II. These further demonstrate the utility of PERSEVERE II for prognosis.
Fig. 1. Classification of the test cohort patients according to PERSEVERE II.
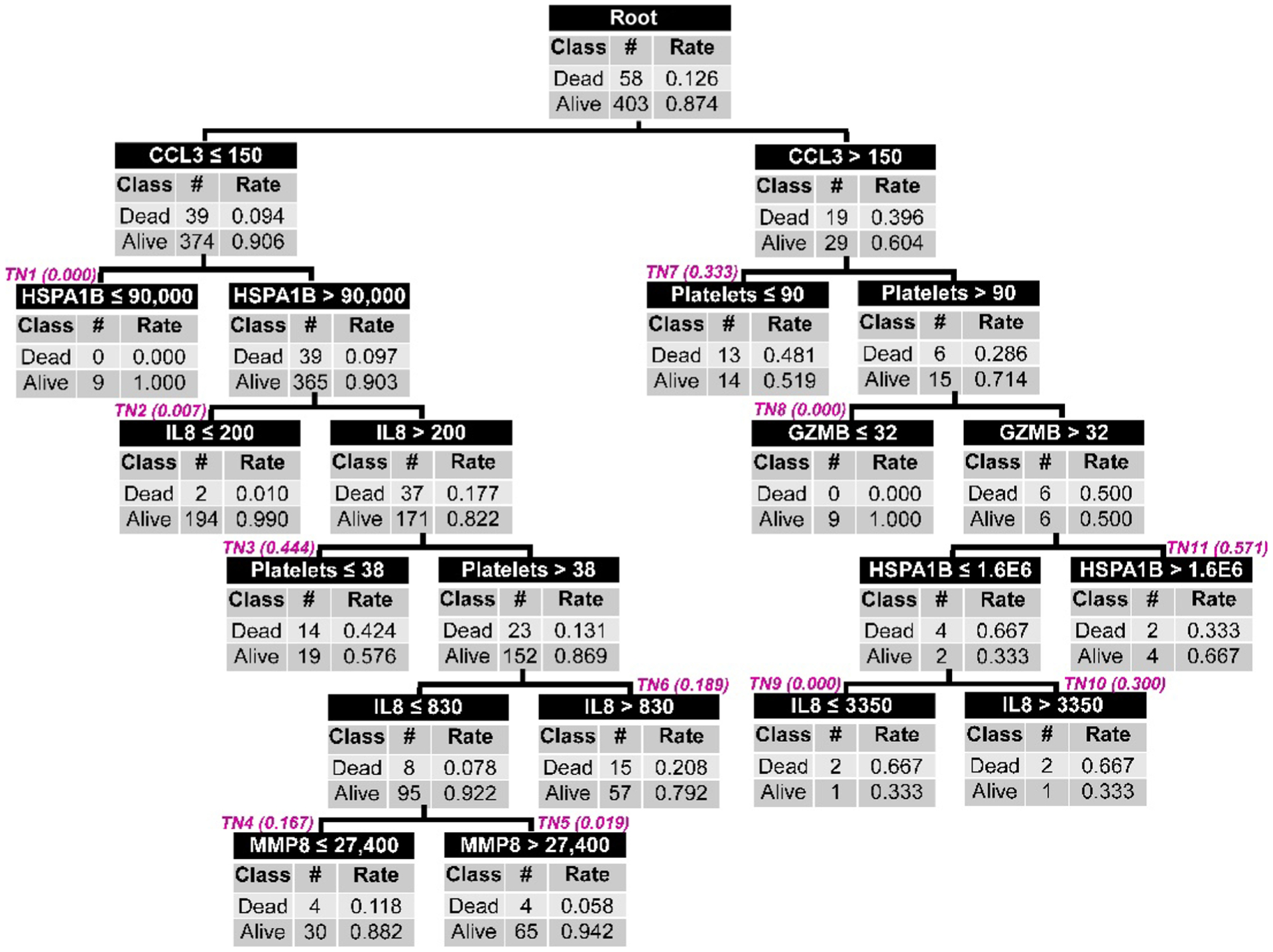
All patients (n = 461) are included in the root node at the top of the figure, with the corresponding number of 28-day survivors and nonsurvivors and the respective rates. Patients are subsequently allocated to daughter nodes using a biomarker-based criterion as indicated in the top row of each node. All biomarker data are shown as picograms per milliliter, and platelet data are shown as the number of platelets per microliter. Each daughter node provides the number of survivors and nonsurvivors allocated to that node and the respective rates. Subsequent daughter nodes are generated, ending in terminal nodes (TNs) indicated by magenta (italic font). The terminal nodes are used to assign a baseline mortality risk to a patient classified to a given terminal node. The baseline mortality risk corresponding to each terminal node is indicated in parentheses next to the TN and is derived from the published PERSEVERE II model (9). These baseline mortality risks are used for construction of the AUROC. For calculation of the diagnostic test characteristics, the mortality probability is dichotomized into those who are predicted to survive and those who are predicted to not survive by 28 days. Patients allocated to TN1, TN2, TN5, TN8, and TN9 (mortality risk, 0.000 to 0.019) are classified as predicted survivors. Patients allocated to TN3, TN4, TN6, TN7, TN10, and TN11 are classified as predicted nonsurvivors (mortality risk, 0.167 to 0.571).
Table 1. Test characteristics of PERSEVERE and PERSEVERE II.
AUC, area under the curve.
| Variable | PERSEVERE | PERSEVERE II | ||
|---|---|---|---|---|
| Value | 95% CI | Value | 95% CI | |
| AUC | 0.67 | 0.58–0.75 | 0.83 | 0.77–0.88 |
| True positives (n) | 35 | — | 50 | — |
| True negatives (n) | 303 | — | 278 | — |
| False positives (n) | 100 | — | 125 | — |
| False negatives (n) | 23 | — | 8 | — |
| Sensitivity | 60% | 47–73 | 86% | 74–93 |
| Specificity | 75% | 71–79 | 69% | 64–73 |
| Positive predictive value | 26% | 19–34 | 29% | 22–36 |
| Negative predictive value | 93% | 89–95 | 97% | 94–99 |
| (+) Likelihood ratio | 2.4 | 1.9–3.2 | 2.8 | 2.3–3.3 |
| (−) Likelihood ratio | 0.5 | 0.4–0.7 | 0.2 | 0.1–0.4 |
Fig. 2. The 28-day survival curves for patients stratified into three PERSEVERE II–based mortality risk strata.
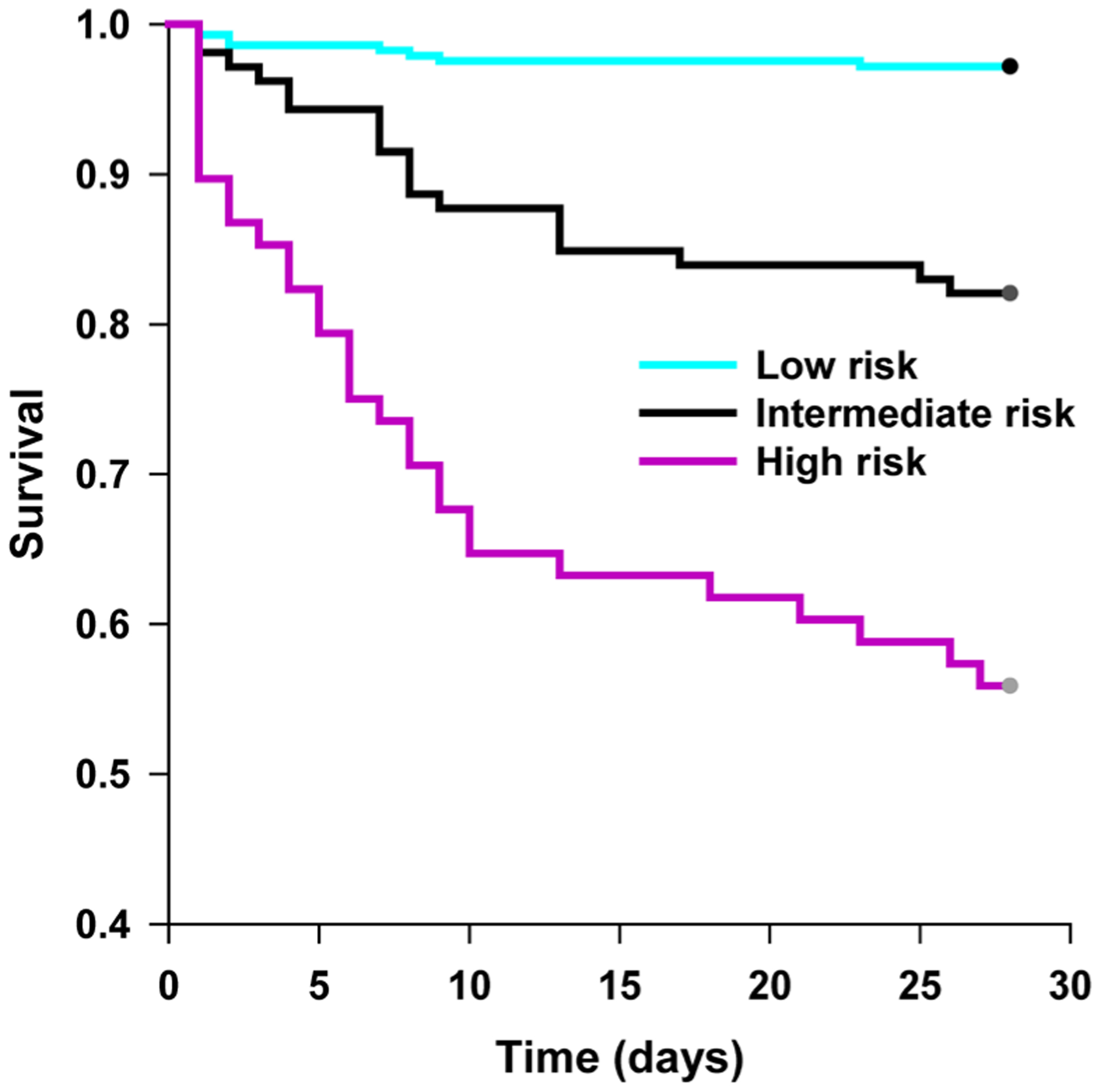
Patients were grouped into one of three PERSEVERE II–based mortality risk strata: low risk, reflecting patients classified to TN1, TN2, TN5, TN8, or TN9 (mortality risk, 0.000 to 0.019); intermediate risk, reflecting patients classified to TN4 or TN6 (mortality risk, 0.167 to 0.189); and high risk, reflecting patients allocated to TN3, TN7, TN10, or TN11 (mortality risk, 0.300 to 0.571). We then generated 28-day survival curves for patients within each stratum. P < 0.001 for all pairwise comparisons, and log rank test with Holm-Sidak method for multiple comparisons.
As a test of biological plausibility and internal validity, we compared the true-negative patients (those correctly predicted to survive) and false-positive patients (those incorrectly predicted to die by 28 days), as classified by PERSEVERE II. We reasoned that if the false-positive patients were indeed at higher baseline mortality risk but that the risk was mitigated by clinical interventions, then the false-positive patients should have a greater degree of sepsis-related illness severity when compared to the true-negative patients. Compared to the true-negative patients, the false-positive patients had higher median PRISM III scores and greater organ failure burden (table S2). False-positive patients did not have a significantly different length of stay in the pediatric intensive care unit (PICU) or number of PICU-free days. Collectively, these data support the concept that PERSEVERE II reliably identifies greater illness severity among children with septic shock.
We note that ≤1% of patients were classified to terminal node 9 (three patients), 10 (three patients), or 11 (six patients) using PERSEVERE II (Fig. 1), raising the possibility of an over fit model. Figure 3 shows a pruned model in which these patients are now reclassified to a new terminal node 9, informed by a GZMB cut-point previously associated with a mortality risk of 0.297 (9). This pruned version of PERSEVERE II increases the AUROC to 0.86 (95% CI, 0.81 to 0.90), reflecting an increased sensitivity for mortality of 90% (95% CI, 78 to 96) without a change in specificity.
Fig. 3. The PERSEVERE II decision tree after pruning.
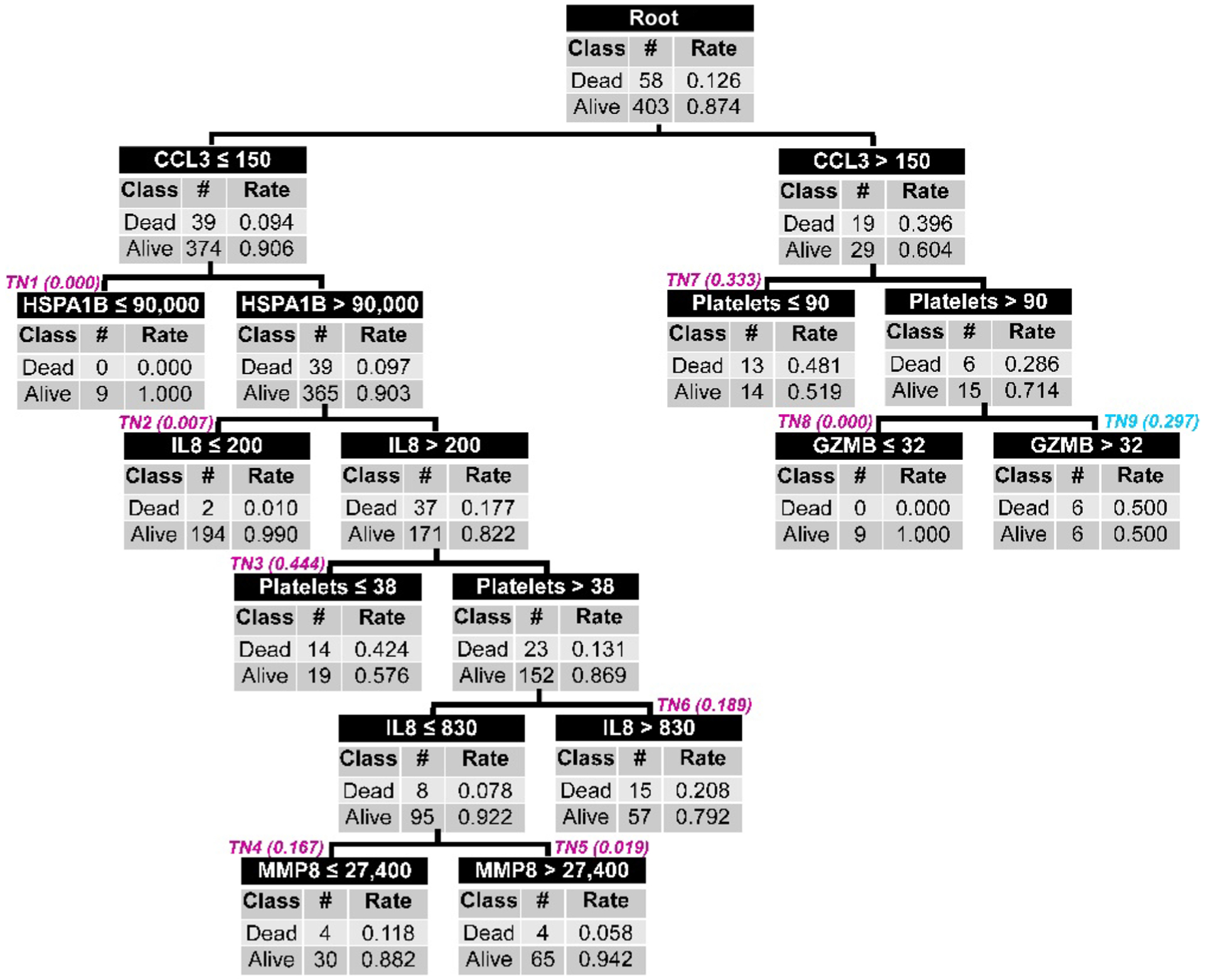
TN9, TN10, and TN11 from the PERSEVERE II decision tree (see Fig. 1) were pruned and replaced by new TN9 informed by a GZMB decision rule and highlighted by cyan italics. See the main text for the diagnostic test characteristics of the pruned PERSEVERE II decision tree.
Derivation and testing of a murine PERSEVERE model
We next determined whether the association between the PERSEVERE biomarkers and poor outcome from sepsis is conserved in experimental mice challenged with sepsis, because this would provide a model to begin testing precision medicine strategies for sepsis. In 94 mice, we measured the murine homologs of the PERSEVERE biomarkers 8 hours after experimental sepsis induced by a cecal ligation and perforation (CLP) procedure. Analogous to the approach used for PERSEVERE, we used CART analysis to derive a murine version of PERSEVERE (mPERSEVERE). Figure 4 shows the derived mPERSEVERE decision tree. The top-level decision node is informed by CCL3, with subsequent decision nodes informed by keratinocyte chemoattractant (KC) and MMP8. The AUROC of the derived decision tree for discriminating surviving mice from dead mice at 10 days after CLP was 0.86 (95% CI, 0.78 to 0.94), with a sensitivity for mortality of 100% (95% CI, 91 to 100) and a specificity of 59% (95% CI, 43 to 73). In a separate test cohort of mice challenged with CLP (n = 18), mPERSEVERE had an AUROC of 0.83 (95% CI, 0.63 to 1.00) for discriminating between surviving and dead mice at 10 days after CLP (fig. S2). Table 2 shows the other diagnostic test characteristics for mPERSEVERE in the derivation and test cohorts.
Fig. 4. The derived mPERSEVERE decision tree.
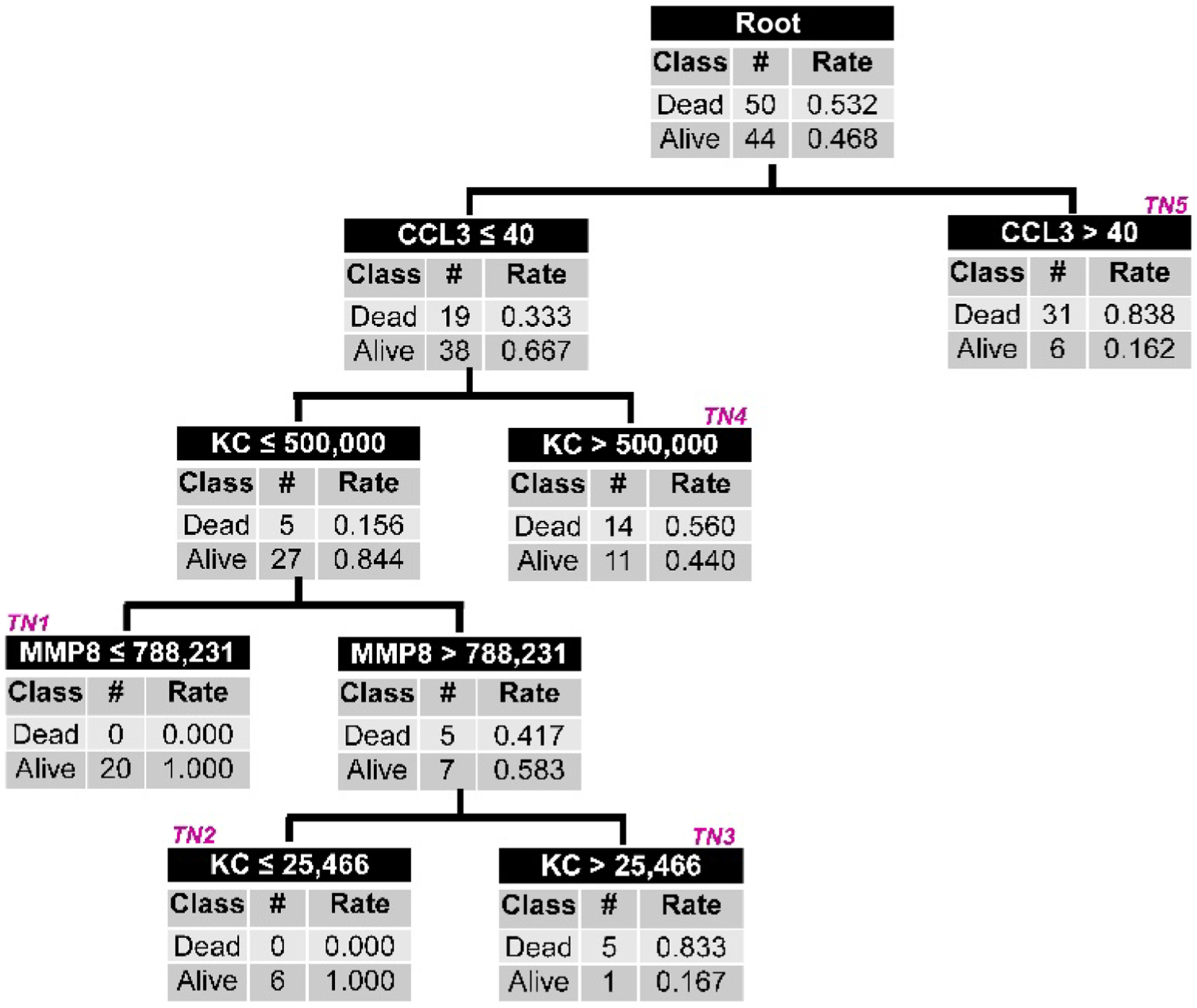
All mice subjected to CLP (n = 94) are included in the root node at the top of the figure, with the corresponding number of 10-day survivors and nonsurvivors and the respective rates. Mice are subsequently allocated to daughter nodes using a biomarker-based criterion as indicated in the top row of each node. All biomarker data are shown as picograms per milliliter. Each daughter node provides the number of survivors and nonsurvivors allocated to that node and the respective rates. Subsequent daughter nodes are generated, ending in terminal nodes (TNs) indicated by magenta (italic font). The terminal nodes estimate the mortality probability for a mouse classified to a given terminal node, and these values are used to calculate the AUROC. For calculation of diagnostic test characteristics, the mortality probability is dichotomized into mice that are predicted to survive and those that are predicted to not survive by 10 days. Mice allocated to TN1 and TN2 (mortality risk, 0.000) are classified as predicted survivors. Mice allocated to TN3, TN4, and TN5 are classified as predicted nonsurvivors (mortality risk, 0.560 to 0.838).
Table 2.
Test characteristics of the mPERSEVERE model in the derivation and test cohorts.
| Variable | Derivation cohort | Test cohort | ||
|---|---|---|---|---|
| Value | 95% CI | Value | 95% CI | |
| N | 94 | — | 18 | — |
| AUC | 0.86 | 0.78–0.94 | 0.83 | 0.63–1.00 |
| True positives (n) | 50 | — | 8 | — |
| True negatives (n) | 26 | — | 5 | — |
| False positives (n) | 18 | — | 3 | — |
| False negatives (n) | 0 | — | 2 | — |
| Sensitivity | 100% | 91–100 | 80% | 44–96 |
| Specificity | 59% | 43–73 | 63% | 26–90 |
| Positive predictive value | 74% | 61–83 | 73% | 39–93 |
| Negative predictive value | 100% | 84–100 | 71% | 30–95 |
| (+) Likelihood ratio | 2.4 | 1.7–3.5 | 2.1 | 0.8–5.5 |
| (−) Likelihood ratio | — | — | 0.3 | 0.1–1.3 |
Initial characterization of high- and low-risk mice
We measured inflammation and bacterial burden and compared these between mice classified as low risk and those characterized as high risk using mPERSEVERE. Because the lung is a key target organ for inflammation and injury during sepsis, we measured lung myeloperoxidase (MPO) activity 24 hours after CLP as a measure of tissue inflammation. Lung MPO activity was greater in the high-risk mice than in the low-risk mice (Fig. 5A). We measured serum IL6 concentrations as an indicator of systemic inflammation; IL6 was increased 24 hours after CLP in high-risk mice when compared to low-risk mice (Fig. 5B). Bacterial burden, measured as colony counts in the peritoneal compartment 24 hours after CLP, was also greater in the high-risk mice than in the low-risk mice (Fig. 5C). We note that in these experiments, we were unable to determine whether MPO, IL6, or bacterial burden was associated with predictive accuracy of mPERSEVERE because the methods required sacrifice of the mice at 24 hours. In combination, these data indicate that high-risk mice are characterized by more inflammation and greater bacterial burden when compared to the low-risk mice.
Fig. 5. Characterization of experimental mice subjected to CLP and stratified into low and high risk of mortality according to mPERSEVERE.
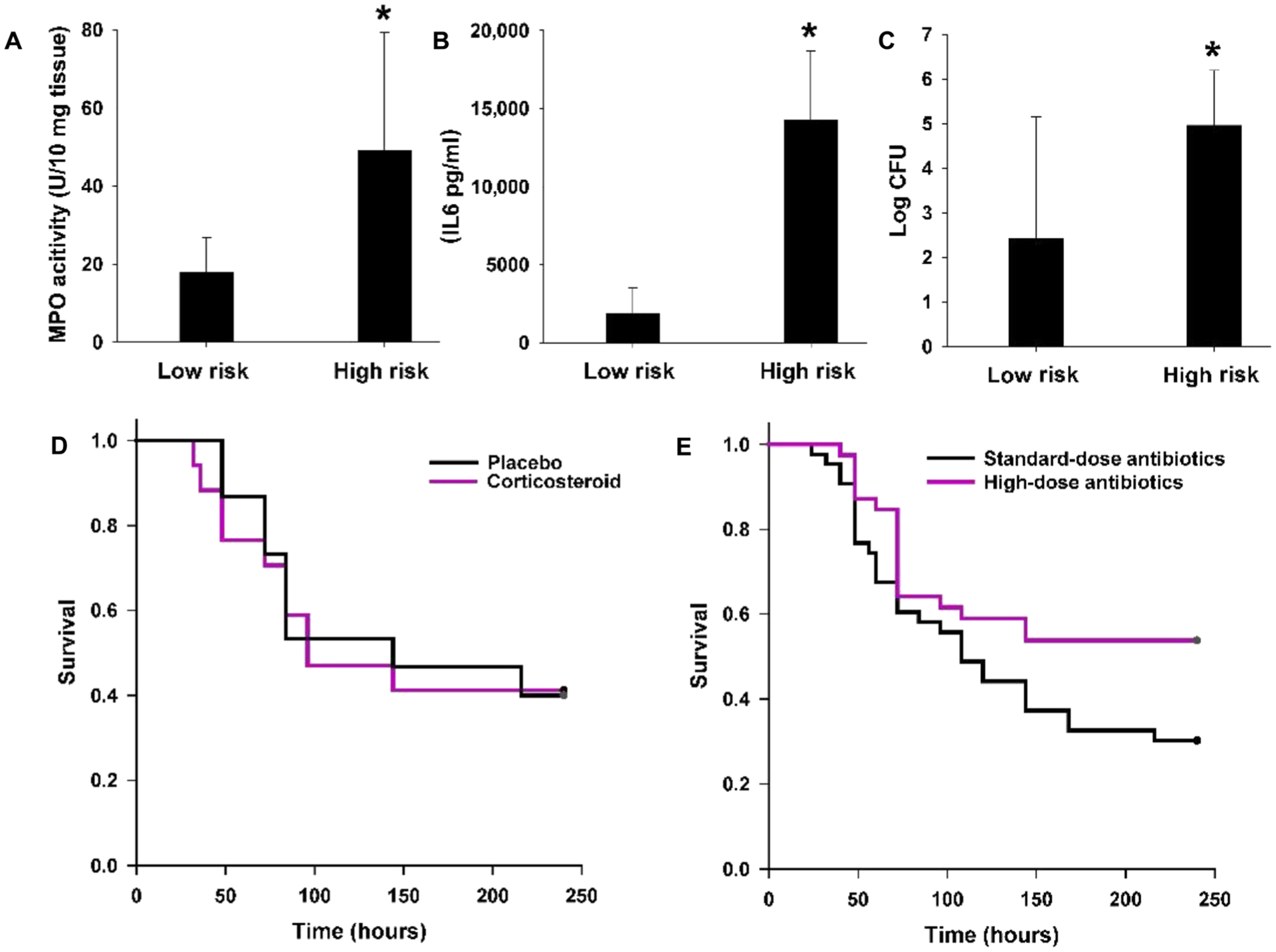
(A) Lung MPO activity in low-risk (n = 20) versus high-risk (n = 20) mice 24 hours after CLP (*P < 0.05 versus low-risk mice). (B) Serum IL6 concentrations in low-risk (n = 20) versus high-risk mice (n = 20) 24 hours after CLP (*P < 0.05 versus low-risk mice). (C) Log-transformed bacterial colony-forming units (CFU) from the peritoneal cavity of low-risk (n = 20) versus high-risk mice (n = 20) 24 hours after CLP (*P < 0.05 versus low-risk mice). (D) Ten-day survival curves of high-risk mice randomized to placebo (n = 15) or dexamethasone (n = 17) in a blinded manner (P = 0.910, log rank survival). (E) Ten-day survival curves of high-risk mice randomized to standard-dose antibiotics (n = 43) or high-dose antibiotics (n = 39) in a blinded manner (P = 0.040, log rank survival).
Experimental therapeutic interventions guided by mPERSEVERE-based enrichment
The finding that high-risk mice have a greater degree of inflammation and a greater bacterial burden suggests two broad possibilities consistent with current clinical paradigms of sepsis pathophysiology (2). One possibility is that the high-risk mice primarily have excessive sepsis-induced inflammation that is damaging to host tissues but is otherwise relatively ineffective at clearing the bacterial burden in the peritoneal compartment. An alternative possibility is that high-risk mice have a greater bacterial burden in the peritoneal compartment despite antibiotic treatment, resulting in excessive inflammation as a secondary biological response to a high bacterial burden.
We tested the hypothesis that excessive inflammation is the primary cause of poor outcome in the high-risk mice by treating them with a broad-acting anti-inflammatory drug, dexamethasone (10). In these experiments, mice were randomly assigned to receive placebo or dexamethasone in a blinded manner. Figure 5D shows that dexamethasone did not improve survival in high-risk mice compared to placebo, suggesting that excessive inflammation is not the primary pathological mechanism in the high-risk mice.
We then tested the hypothesis that a greater bacterial burden is a primary cause of poor outcome in the high-risk mice by doubling the dose of antibiotics administered after CLP. In these experiments, mice were randomly assigned to receive standard-dose antibiotics or high-dose antibiotics in a blinded manner. The antibiotic dosing regimen was repeated 24 hours later, again in a blinded manner. Figure 5E shows that in the high-risk mice, a higher dose of antibiotics improved survival when compared to the standard dose of antibiotics.
These experiments in mice are consistent with the utility of mPERSEVERE-based enrichment in identifying animals with higher risk of mortality that can be partially mitigated with higher doses of antibiotics. Higher doses of antibiotics did not completely rescue the high-risk mice, reflecting either bacterial resistance despite the high doses of antibiotics or that a greater bacterial burden alone does not fully account for the higher mortality rate in this group.
To begin testing whether the PERSEVERE biomarkers are directly involved in the mechanism of poor outcome, mice were randomly assigned to receive either an anti-CCL3 antibody or an isotype control antibody in a blinded manner. The rationale for these studies is that CCL3 informs the top-level decision node of mPERSEVERE, and higher concentrations of CCL3 are associated with increased risk of mortality after CLP in mice. As shown in fig. S3A, the anti-CCL3 antibody did not modify survival in the entire cohort of un-stratified mice. Among high-risk mice, the 10-day survival rates for those treated with the anti-CCL3 antibody or the isotype control antibody were 58 and 35%, respectively, but the survival curves were not significantly different (P = 0.149, log rank survival; fig. S3B). Among the low-risk mice, the 10-day survival rates for those treated with the anti-CCL3 antibody or the isotype control antibody were 33 and 63%, respectively, but the survival curves were not significantly different (P = 0.287, log rank test; fig. S3C). These data provide a rationale to further pursue an anti-CCL3 strategy in experimental sepsis, guided by mPERSEVERE-based predictive enrichment.
Evidence of greater bacterial burden among high-risk patients
Given the finding of higher pathogen burden in the high-risk mice, we revisited our clinical data for corroborating evidence. The patient cohort was divided into the three PERSEVERE II risk strata, as described above, and we determined the proportion of patients in each stratum with a positive culture, as determined by the clinical laboratory. Figure 6A shows that the proportion of patients with any pathogen isolated from any normally sterile site increased with increasing PERSEVERE II–based risk. Figure 6B shows that among patients with positive cultures, the proportion of patients with positive blood cultures also increased with increasing PERSEVERE II–based risk. Among the eight patients who were classified by PERSEVERE II as low risk but did not survive, only two had positive cultures. Both of these positive cultures were from the lung compartment rather than the blood compartment. This indicates a potential misdiagnosis of sepsis and not misclassification as being at high risk of increased pathogen burden by PERSEVERE II. These data are consistent with our findings in mice, demonstrated that increased pathogen burden is associated with increased PERSEVERE II–based mortality risk among children with septic shock, and support the potential utility of our experimental model for testing precision medicine strategies in sepsis.
Fig. 6. Estimation of pathogen burden among patients stratified into three PERSEVERE II–based mortality risk strata.
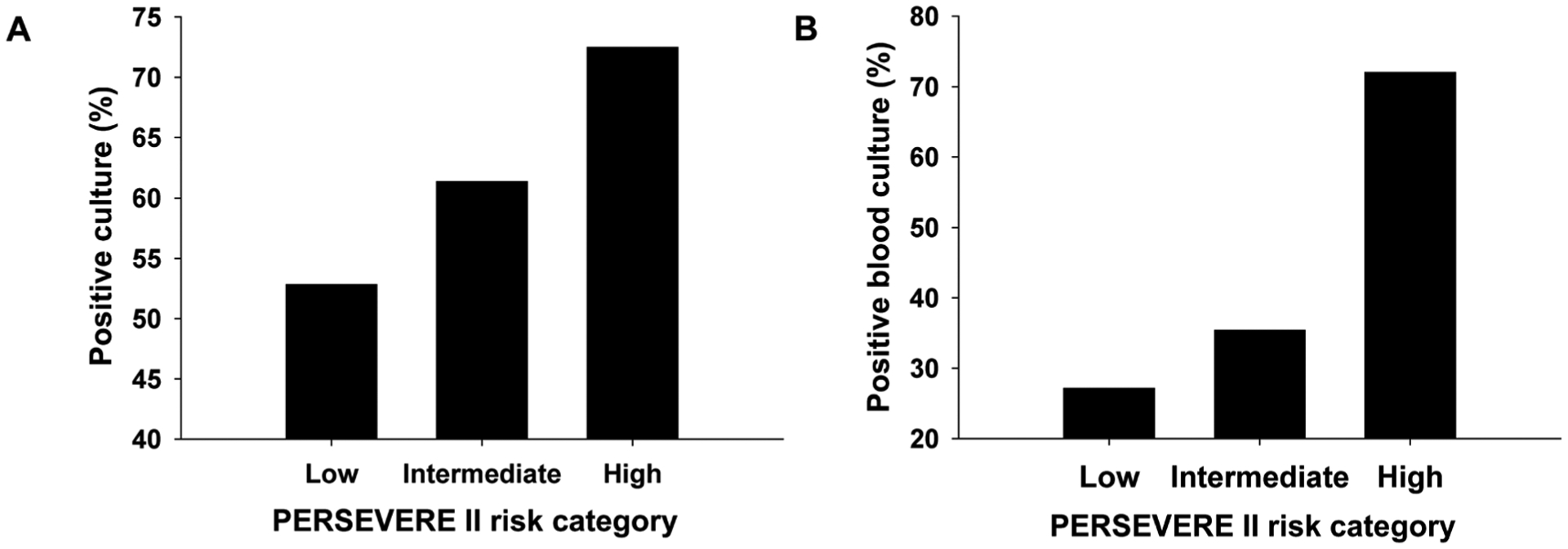
Patients were grouped into one of three PERSEVERE II–based mortality risk strata: low risk (n = 286; mortality risk, 0.000 to 0.019), intermediate risk (n = 106; mortality risk, 0.167 to 0.189), and high risk (n = 69; mortality risk, 0.300 to 0.571). (A) Proportion of patients in each risk stratum having any positive pathogen culture from any normally sterile site (P < 0.05, χ2 test, two degrees of freedom). (B) Among patients with positive cultures, proportion of patients in each risk stratum having a positive blood culture (P < 0.05, χ2 test, two degrees of freedom).
Assuming that a high bacterial burden is indeed a pathological feature of PERSEVERE II–defined high-risk patients and that it can be targeted with higher dose antibiotics, we explored the potential clinical benefit of treating high-risk patients with high-dose antibiotics by calculating the number needed to treat (NNT) to rescue one high-risk patient from 28-day mortality. The mortality rate for the 69 high-risk patients was 45%. Assuming an absolute risk reduction ranging from 45% (complete rescue) to 5% (10% relative risk reduction), table S3 shows that the NNT ranges from 2 to 22. In this way, the potential clinical benefit of different treatment effect sizes can be weighed against the risks of high-dose antibiotics.
DISCUSSION
Prediction models such as PERSEVERE are often tested using holdout samples or other cross-validation approaches. A substantially more rigorous approach to assess model validity is prospective testing in an independent cohort, wherein the model rules are applied a priori without any modifications, as was done in the current study. This approach is essential for assessing generalizability and reliability and is much more rigorous than cross-validation approaches. Whereas PERSEVERE had just modest performance, PERSEVERE II had excellent performance. This is consistent with our previous observation that PERSEVERE II appears to have greater generalizability across a broad range of children with septic shock (9). In particular, the negative likelihood ratio for mortality generated by PERSEVERE II has clinical utility for identifying those who are at the lowest risk for mortality (11, 12).
The overall accuracy of PERSEVERE II is consistent with an upper limit for the ability to predict 28-day outcome from clinical septic shock, as recently suggested by Sweeney et al. (13). This upper limit reflects the challenges inherent with predicting 28-day outcomes from baseline PERSEVERE biomarkers, given the multiple clinical interventions that can occur during the intervening period and that can modify risk. In addition, the upper limit reflects that 100% accuracy in predicting outcome is not likely to be achieved because this would imply that death from septic shock is largely predetermined and not modifiable by clinical interventions. These assertions are supported by our comparisons of the true-negative and false-positive patients identified by PERSEVERE II. Compared to the true-negative patients, the false-positive patients had greater illness severity, as measured by PRISM III, and greater organ failure burden, suggesting that PERSEVERE II was reliable in identifying patients with a greater degree of sepsis-related illness severity and a corresponding higher risk of mortality, but perhaps, clinical interventions mitigated the risk.
We further tested the biological relevance of the PERSEVERE biomarkers by measuring them in a murine model of sepsis, at a time point early in the course of sepsis, analogous to the biomarker measurements for PERSEVERE II. Recognizing the limitations inherent to animal models of sepsis, the murine homologs of the PERSEVERE biomarkers reliably estimated mortality risk in mice challenged with CLP. We noted that CCL3 informs the first-level decision rule for mPERSEVERE. This same biomarker informs the first-level decision rule for both PERSEVERE and PERSEVERE II (6, 9). In addition, KC, the murine homolog of IL8 in humans, appears to have predictive capacity for mPERSEVERE, as does IL8, a principal chemokine for recruitment and activation of neutrophils, for PERSEVERE II (9, 14). Although it is likely that other biomarkers can estimate the risk of mortality in experimental mice challenged with sepsis, these studies indicate that the links between the PERSEVERE biomarkers and the biological pathways to poor outcome from sepsis are conserved between mice and humans, providing an experimental model that is underpinned with clinically relevant biological pathways.
mPERSEVERE provides an opportunity to begin exploring the underlying biology reflected by the PERSEVERE biomarkers and to begin testing mPERSEVERE-guided treatment strategies in mice. This will allow accelerated evaluation of preclinical approaches that are currently impeded by the challenges of conducting interventional studies involving critically ill children with septic shock.
Our initial studies suggested that high-risk mice are characterized by a greater degree of inflammation and a higher bacterial burden. To address the possibility of a primary hyperinflammatory state, we administered dexamethasone using a dosing regimen previously reported to decrease mortality in mice with presumed excessive sepsis-induced inflammation (10). Dexamethasone did not improve survival in mPERSEVERE-defined high-risk mice, which is consistent with our previous observational data demonstrating that PERSEVERE by itself does not identify those children with septic shock who are more likely to benefit from corticosteroids (15, 16). The association between PERSEVERE biomarkers and the treatment effect of adjunctive corticosteroids is being explored in a randomized clinical trial among children with septic shock (clinicaltrials.gov: NCT03401398).
In contrast to dexamethasone, a higher dose of antibiotics did improve survival in high-risk mice. This demonstrates with experimental data that a higher bacterial burden in mice is a primary driver of poor outcome, consistent with our observational clinical data.
Admittedly, other pathological mechanisms are likely to be operative in high-risk mice, including dysregulated inflammation not mitigated by the dexamethasone regimen used in our experiments, because a higher dose of antibiotics did not completely rescue them. Our preliminary studies suggest that targeting CCL3 might provide an opportunity to mitigate sepsis-related dysregulated inflammation. Such studies need to be guided by mPERSEVERE-based stratification because a previous study demonstrated that the complete absence of CCL3, via genetic ablation, increases mortality among mice challenged with CLP (17). Additional research will be needed to further dissect the direct role of the PERSEVERE biomarkers in sepsis biology.
We acknowledge the inherent limitations in comparing experimental microbiological data from mice and clinical microbiological data from our pediatric cohort. Although more than 70% of the PERSEVERE II–defined highest-risk patients were culture positive, some were not. This may reflect that there are likely multiple confounders that we did not measure, that there are additional potential mechanisms reflected in the PERSEVERE biomarkers, such as CCL3, or that sepsis may not be correctly diagnosed as previously discussed. It is also possible this reflects that we relied on clinical laboratory data that do not directly quantify bacterial burden; it is generally accepted that clinical tests for pathogen detection require a sufficient pathogen burden to be positive, particularly in the blood compartment. Because we show that the proportion of patients with positive pathogen cultures increases with PERSEVERE II mortality risk, we posit that this corroborates, at least in part, our findings in experimental mice.
Although typical antibiotic dosing regimens for children are based on weight, dosing of antibiotics for clinical sepsis, in both children and adults, is based on population data rather than genuine individualized dosing. Existing dosing regimens reflect a balance between what is thought to be appropriate for pathogen eradication and the potential toxicity associated with higher doses of antibiotics. It is becoming increasingly apparent, however, that antibiotic pharmacokinetics are often substantially altered in critically ill patients with sepsis (18), so standard dosing regimens based on population data, even if weight based, are likely insufficient to adequately treat all patients with sepsis, particularly those with a higher pathogen burden (18–21). Formal therapeutic drug monitoring programs for patients with septic shock and other forms of critical illness (21, 22) are being implemented to address this challenge, but the resources required for such programs are considerable. Furthermore, the concept of treating with higher doses of antibiotics needs to be balanced against the increased risk of antibiotic-associated toxicity. On the basis of our findings, we suggest that PERSEVERE II might identify a subgroup of children with septic shock who will benefit most from targeted therapeutic drug monitoring to ensure optimal dosing of antibiotics. In addition, the estimated NNT may be clinically justifiable depending on the effect size of high-dose antibiotics. This is one example of how PERSEVERE II is moving beyond prognosis to predictive enrichment, becoming a tool for precision medicine in septic shock.
Previous phase 3 trials in sepsis, the vast majority of which have failed, have not applied any effective prognostic or predictive enrichment strategies. The enrollment procedures for these previous trials have been informed by broad clinical criteria. That approach is flawed because it generates highly heterogeneous cohorts, with different baseline mortality risk and different underlying biology. With this degree of heterogeneity in both baseline risk and underlying biology, any potential benefit of an experimental intervention is diluted by subgroups of patients who derive no benefit from the intervention or who are harmed by the intervention. The approach highlighted by our current study is fundamentally different because it applies enrichment strategies to demonstrate that an experimental intervention is beneficial only in subgroups defined by the enrichment strategies, albeit in experimental mice. We provide proof of principle supporting the concept that interventional trials in sepsis can and must be conducted in a more rational and efficient manner through the use of enrichment strategies.
In summary, PERSEVERE II demonstrated excellent discriminative ability in a prospectively enrolled and heterogeneous cohort of children with septic shock, confirming its reliability and clinical utility as a prognostic enrichment tool. Experimental studies in a murine model of sepsis suggest that a high pathogen burden is a primary driver of mortality, which can be partially mitigated by higher doses of antibiotics. Consistent with this, we show that children with septic shock and a high PERSEVERE II mortality risk have a high pathogen burden. PERSEVERE II has the potential to extend beyond prognosis, becoming a predictive enrichment tool, for example, to identify patients with sepsis who most warrant therapeutic drug monitoring to optimize antibiotic dosing. Other potential interventions such as pharmacologic modulation of CCL3 and KC, guided by mPERSEVERE-based enrichment, also require investigation.
MATERIALS AND METHODS
Study design
Human studies involved critically ill children with septic shock. The study was an observational cohort with prespecified outcomes of interest and the procurement of biological specimens. Other than blood draws, there were no study-related interventions, and all clinical care was at the discretion of the clinical teams caring for the study subjects. Protocol details are provided below.
Murine studies involved an established model of sepsis induced by CLP. All studies involved C57BL/6 male mice, 8 to 10 weeks of age. All mice received antibiotics and fluid resuscitation after CLP. For interventional studies, mice were randomized and treated with the experimental intervention in a blinded manner. Protocol details are provided below.
Study subjects and data collection
Study subjects were enrolled from January 2015 to December 2018 in an observational study ongoing at multiple PICUs across the United States. The study protocol was approved by the local Institutional Review Board of each participating institution and was previously described in detail (6, 23). Briefly, children between the ages of 1 week and 18 years of age admitted to the PICU and meeting pediatric-specific consensus criteria for septic shock (24) were enrolled after obtaining informed consent from parents or legal guardians. Blood samples were obtained within 24 hours of a septic shock diagnosis for isolation of serum. Clinical and laboratory data were collected daily while in the PICU. Mortality was tracked for 28 days after enrollment. Organ failure was tracked over the first 7 days after enrollment and defined using pediatric specific consensus criteria (24). PICU-free days were calculated by subtracting the number of days in the PICU from a theoretical maximum of 28 days. Patients who remained in the PICU longer than 28 days or died by 28 days were assigned a value of zero PICU-free days.
PERSEVERE biomarkers and baseline mortality risk assignment
Serum concentrations of the PERSEVERE biomarkers were measured using a multiplex magnetic bead platform (MILLIPLEX MAP) designed for this project by the EMD Millipore Corporation and a Luminex 100/200 System (Luminex Corporation), according to the manufacturers’ specifications. Assay performance data were previously published (6).
Biomarker concentrations were used to assign each patient a baseline mortality risk using the predefined criteria of the published PERSEVERE (6) and PERSEVERE II (9) decision trees. In addition to the PERSEVERE biomarkers, PERSEVERE uses age, and PERSEVERE II uses admission platelet count as predictor variables.
Murine model of sepsis
Studies involving mice complied with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (eighth edition, 2011) and met approval of the Institutional Animal Care and Use Committee of the Cincinnati Children’s Research Foundation. Wild-type C57BL/6 male mice, 8 to 10 weeks of age, were obtained from Charles Rivers Laboratories. All mice were fed standard rodent chow and maintained on 12-hour light/ 12-hour dark cycle.
Sepsis was induced using the CLP model, as previously described (25). Briefly, mice were anesthetized with isoflurane inhalation, a midline laparotomy was performed, and the cecum was identified. A 3–0 silk suture was used to ligate the cecum about 5 mm distal to the ileocecal valve, and two punctures were made through-and-through using a 21-gauge needle. A small amount of fecal content was expressed from each puncture site, and the cecum was returned to the abdominal cavity, which was closed in an interrupted fashion. The skin was closed using GLUture Topical Adhesive (Abbott Laboratories), and mice received a 1-ml subcutaneous injection of normal saline to compensate for fluid loss during the procedure. Mice were subsequently placed on a heating pad and allowed to recover from anesthesia. For all experiments, except the high-dose antibiotic experiments described below, mice received standard-dose antibiotics 8 hours after CLP: metronidazole (12.5 mg/kg) and ceftriaxone (25 mg/kg). Antibiotics were intraperitoneally redosed 24 hours later. For survival experiments, mice were followed up for 10 days.
PERSEVERE biomarkers in experimental mice
The murine homologs of the PERSEVERE biomarkers were measured from blood samples obtained 8 hours after CLP. Blood samples (100 μl) were obtained via a submandibular puncture and bleed, as previously described (26). Concentrations of the PERSEVERE biomarkers were obtained using a custom magnetic bead multiplex assay (R&D Systems) and a Luminex 100/200 System (Luminex Corporation), according to the manufacturers’ specifications. The murine version of the PERSEVERE biomarker panel consisted of CCL3, MMP8, GZMB, and KC (a murine homolog of IL8). We were unable to include the murine homolog of HSPA1B in the multiplex assay because of the lack of a suitable anti-HSPA1B antibody for multiplexing.
Assays of inflammation and bacterial burden
Twenty-four hours after CLP, a group of mice was euthanized for tissue and blood procurement to conduct the following assays. MPO activity was measured from lung tissue samples using a colorimetric activity assay (Sigma-Aldrich) and reported as MPO activity units/10 mg of lung tissue. Serum IL6 concentrations were measured using antibody-coated magnetic beads corresponding to murine IL6 (R&D Systems) and a Luminex instrument. To quantify bacterial burden in the peritoneal cavity, peritoneal lavage was performed by injecting 500 μl of sterile phosphate-buffered saline (PBS) into the peritoneal cavity using a 27-gauge needle, and aspirating back 150 μl after the animal was manipulated to ensure uniform distribution of the injected fluid within the peritoneal cavity. Serial dilutions of the recovered peritoneal fluid were plated onto sheep blood agar plates and incubated at 37°C for 48 hours. Plated dilutions with 30 to 300 colonies were used to quantify bacterial burden. Colony counts were conducted blinded to mortality risk and were corrected by the respective dilution factor and log transformed for analyses.
mPERSEVERE-guided therapeutic interventions in experimental mice
We trialed two clinically feasible therapeutic interventions in mice subjected to CLP and analyzed their effects after stratification using mPERSEVERE. As a general anti-inflammatory strategy, one group of mice intraperitoneally received dexamethasone (2.5 mg/kg; Sigma-Aldrich) 8 hours after CLP and a second dose 24 hours later, as previously reported (10). Placebo-treated mice received an equal volume of PBS at both time points. The second strategy involved a doubling of the antibiotic dose [high-dose antibiotics, metronidazole (25 mg/kg) and ceftriaxone (50 mg/kg)] intraperitoneally administered 8 hours after CLP and a second dose 24 hours later. Comparator mice received standard-dose antibiotics [metronidazole (12.5 mg/kg) and ceftriaxone (25 mg/kg)] at both time points.
We also trialed an experimental therapeutic intervention seeking to neutralize one of the PERSEVERE biomarkers, CCL3 (27). One group of mice intraperitoneally received 10 μg per mouse of a goat polyclonal anti-CCL3 antibody (AB-450-NA, R&D Systems) 8 hours after CLP. Placebo-treated mice received a normal goat immunoglobulin G isotype control antibody (AB-108-C, R&D Systems) using the same dose and schedule as the anti-CCL3 antibody group.
These therapeutic interventions in experimental mice were randomized and blinded. To achieve randomization and blinding, a research assistant prepared individual syringes of the therapeutic interventions and labeled them using a predefined code corresponding to the interventions. A second research assistant, blinded to the code, injected the coded solutions and monitored the mice over the next 10 days for survival.
Modeling and statistical analyses
Consistent with our previous approach to deriving PERSEVERE and PERSEVERE II, we used CART analysis to derive mPERSEVERE (Salford Predictive Modeler v.8.0, Salford Systems) (6, 8, 9, 28). The primary outcome variable for the modeling procedures was 10-day mortality. Predictor variables included the four murine homologs of the PERSEVERE biomarkers, described above. We pruned terminal nodes having <5% of the mice in the root node and terminal nodes that did not improve classification based on the class probability method. Weighting of cases and costs for misclassification was not used.
All other statistical analyses used SigmaStat Software (Systat Software Inc.). Descriptive data are reported using medians, inter-quartile ranges, frequencies, and percentages. Comparisons between groups used the Mann-Whitney U test, t test, or χ2 test, as appropriate. Performance of PERSEVERE and PERSEVERE II was evaluated by calculating the respective AUROC with 95% CIs, as well as diagnostic test characteristics with 95% CIs. AUROC curves were compared using the method of Hanley and McNeil for paired samples (28).
Supplementary Material
Acknowledgments:
We thank K. Harmon and P. Lahni for technical assistance in the conduct of these studies.
Funding: This work was supported by the NIH R35GM126943 (to H.R.W.) and Research Innovation in Support of Excellence Award from Cincinnati Children’s Hospital Medicine (J.T.C.).
Footnotes
Competing interests: The Cincinnati Children’s Research Foundation and H.R.W. hold U.S. patents for the PERSEVERE biomarkers (PCT/US2013/025223, Multi-Biomarker-Based Outcome Risk Stratification Model for Pediatric Septic Shock; PCT/US13/25221, Multi-Biomarker-Based Outcome Risk Stratification Model for Adult Septic Shock; PCT/US14/67438, Temporal Pediatric Sepsis Biomarker Risk Model; and PCT/US08/06172, Biomarkers for Septic Shock Patients). C.J.L. is named as a co-inventor on the patents. H.R.W. also serves on the scientific advisory boards for Inflammatix, Endpoint Health, and Eccrine Systems Inc. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/11/518/eaax9000/DC1
Fig. S1. Classification of the test cohort patients according to PERSEVERE.
Fig. S2. Classification of the test cohort mice according to the derived mPERSEVERE.
Fig. S3. Ten-day survival curves of mice challenged with CLP and then randomized to treatment with an anti-CCL3 antibody or an isotype control antibody.
Table S1. Demographic and clinical characteristics of the study cohort according to 28-day survival.
Table S2. Comparison of illness severity measures among true-negative and false-positive subjects.
Table S3. Calculation of the NNT.
REFERENCES AND NOTES
- 1.Atreya MR, Wong HR, Precision medicine in pediatric sepsis. Curr. Opin. Pediatr 31, 322–327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen J, Vincent J-L, Adhikari NKJ, Machado FR, Angus DC, Calandra T, Jaton K, Giulieri S, Delaloye J, Opal S, Tracey K, van der Poll T, Peflrene E, Sepsis: A roadmap for future research. Lancet Infect. Dis 15, 581–614 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Weiss SL, Fitzgerald JC, Pappachan J, Wheeler D, Jaramillo-Bustamante JC, Salloo A, Singhi SC, Erickson S, Roy JA, Bush JL, Nadkarni VM, Thomas NJ, Global epidemiology of pediatric severe sepsis: The sepsis prevalence, outcomes, and therapies study. Am. J. Respir. Crit. Care Med 191, 1147–1157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prescott HC, Calfee CS, Thompson BT, Angus DC, Liu VX, Toward smarter lumping and smarter splitting: Rethinking strategies for sepsis and acute respiratory distress syndrome clinical trial design. Am. J. Respir. Crit. Care Med 194, 147–155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stanski NL, Wong HR, Prognostic and predictive enrichment in sepsis. Nat. Rev. Nephrol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong HR, Salisbury S, Xiao Q, Cvijanovich NZ, Hall M, Allen GL, Thomas NJ, Freishtat RJ, Anas N, Meyer K, Checchia PA, Lin R, Shanley TP, Bigham MT, Sen A, Nowak J, Quasney M, Henricksen JW, Chopra A, Banschbach S, Beckman E, Harmon K, Lahni P, Lindsell CJ, The pediatric sepsis biomarker risk model. Crit. Care 16, R174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaplan JM, Wong HR, Biomarker discovery and development in pediatric critical care medicine. Pediatr. Crit. Care Med 12, 165–173 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong HR, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, Bigham MT, Weiss SL, Fitzgerald JC, Checchia PA, Meyer K, Quasney M, Hall M, Gedeit R, Freishtat RJ, Nowak J, Raj SS, Gertz S, Grunwell JR, Lindsell CJ, Improved risk stratification in pediatric septic shock using both protein and mRNA biomarkers. PERSEVERE-XP. Am. J. Respir. Crit. Care Med 196, 494–501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong HR, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, Bigham MT, Weiss SL, Fitzgerald JC, Checchia PA, Meyer K, Quasney M, Hall M, Gedeit R, Freishtat RJ, Nowak J, Raj SS, Gertz S, Howard K, Harmon K, Lahni P, Frank E, Hart KW, Nguyen TC, Lindsell CJ, Pediatric sepsis biomarker risk model-II: Redefining the pediatric sepsis biomarker risk model with septic shock phenotype. Crit. Care Med 44, 2010–2017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osuchowski MF, Connett J, Welch K, Granger J, Remick DG, Stratification is the key: Inflammatory biomarkers accurately direct immunomodulatory therapy in experimental sepsis. Crit. Care Med 37, 1567–1573 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gallagher EJ, Clinical utility of likelihood ratios. Ann. Emerg. Med 31, 391–397 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Grimes DA, Schulz KF, Refining clinical diagnosis with likelihood ratios. Lancet 365, 1500–1505 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Sweeney TE, Perumal TM, Henao R, Nichols M, Howrylak JA, Choi AM, Bermejo-Martin JF, Almansa R, Tamayo E, Davenport EE, Burnham KL, Hinds CJ, Knight JC, Woods CW, Kingsmore SF, Ginsburg GS, Wong HR, Parnell GP, Tang B, Moldawer LL, Moore FE, Omberg L, Khatri P, Tsalik EL, Mangravite LM, Langley RJ, A community approach to mortality prediction in sepsis via gene expression analysis. Nat. Commun 9, 694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffith JW, Sokol CL, Luster AD, Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol 32, 659–702 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Atkinson SJ, Cvijanovich NZ, Thomas NJ, Allen GL, Anas N, Bigham MT, Hall M, Freishtat RJ, Sen A, Meyer K, Checchia PA, Shanley TP, Nowak J, Quasney M, Weiss SL, Banschbach S, Beckman E, Howard K, Frank E, Harmon K, Lahni P, Lindsell CJ, Wong HR, Corticosteroids and pediatric septic shock outcomes: A risk stratified analysis. PLOS ONE 9, e112702 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong HR, Atkinson SJ, Cvijanovich NZ, Anas N, Allen GL, Thomas NJ, Bigham MT, Weiss SL, Fitzgerald JC, Checchia PA, Meyer K, Quasney M, Hall M, Gedeit R, Freishtat RJ, Nowak J, Raj SS, Gertz S, Lindsell CJ, Combining Prognostic and Predictive Enrichment Strategies to Identify Children With Septic Shock Responsive to Corticosteroids. Crit. Care Med 44, e1000–e1003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi H, Tashiro T, Miyazaki M, Kobayashi M, Pollard RB, Suzuki F, An essential role of macrophage inflammatory protein 1α/CCL3 on the expression of host’s innate immunities against infectious complications. J. Leukoc. Biol 72, 1190–1197 (2002). [PubMed] [Google Scholar]
- 18.Richter DC, Heininger A, Brenner T, Hochreiter M, Bernhard M, Briegel J, Dubler S, Grabein B, Hecker A, Kruger WA, Mayer K, Pletz MW, Storzinger D, Pinder N, Hoppe-Tichy T, Wieterer S, Zimmermann S, Brinkman A, Weigand MA, Lichtenstern C, Bacterial sepsis: Diagnostics and calculated antibiotic therapy. Anaesthesist 68, 40–62 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Abdul-Aziz MH, Sulaiman H, Mat-Nor M-B, Rai V, Wong KK, Hasan MS, Abd Rahman AN, Jamal JA, Wallis SC, Lipman J, Staatz CE, Roberts JA, Beta-Lactam Infusion in Severe Sepsis (BLISS): A prospective, two-centre, open-labelled randomised controlled trial of continuous versus intermittent beta-lactam infusion in critically ill patients with severe sepsis. Intensive Care Med. 42, 1535–1545 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Udy AA, Lipman J, Jarrett P, Klein K, Wallis SC, Patel K, Kirkpatrick CMJ, Kruger PS, Paterson DL, Roberts MS, Roberts JA, Are standard doses of piperacillin sufficient for critically ill patients with augmented creatinine clearance? Crit. Care 19, 28 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veiga RP, Paiva J-A, Pharmacokinetics–pharmacodynamics issues relevant for the clinical use of beta-lactam antibiotics in critically ill patients. Crit. Care 22, 233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jager NGL, van Hest RM, Lipman J, Taccone FS, Roberts JA, Therapeutic drug monitoring of anti-infective agents in critically ill patients. Expert Rev. Clin. Pharmacol 9, 961–979 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Wong HR, Shanley TP, Sakthivel B, Cvijanovich N, Lin R, Allen GL, Thomas NJ, Doctor A, Kalyanaraman M, Tofil NM, Penfil S, Monaco M, Tagavilla MA, Odoms K, Dunsmore K, Barnes M, Aronow BJ; Genomics of Pediatric SIRS/Septic Shock Investigators, Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol. Genomics 30, 146–155 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein B, Giroir B, Randolph A; International Consensus Conference on Pediatric Sepsis, International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics. Pediatr. Crit. Care Med 6, 2–8 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Atkinson SJ, Nolan M, Klingbell L, Harmon K, Lahni P, Zingarelli B, Wong HR, Intestine-derived matrix metalloproteinase-8 is a critical mediator of polymicrobial peritonitis. Crit. Care Med 44, e200–e206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golde WT, Gollobin P, Rodriguez LL, A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim. 34, 39–43 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Di Liberto D, Locati M, Caccamo N, Vecchi A, Meraviglia S, Salerno A, Nebuloni N, Caceres N, Cardona P-J, Dieli F, Mantovani A, Role of the chemokine decoy receptor D6 in balancing inflammation, immune activation, and antimicrobial resistance in Mycobacterium tuberculosis infection. J. Exp. Med 205, 2075–2084 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanley JA, McNeil BJ, A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 148, 839–843 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.