

Abstract
BACKGROUND:
Idiopathic pulmonary fibrosis (IPF) is a common subtype of interstitial lung disease (ILD). Information about the associated comorbidities and predictors of survival among Saudi patients with IPF is limited.
AIMS:
The aim of the study was to determine the clinical characteristics, associated comorbidities, and prognostic factors that impact IPF survival.
METHODS:
Consecutive IPF patients diagnosed in our ILD center were included. The information analyzed included demographics, physiological parameters, and associated comorbidities, among others. Cox regression models were used to identify independent predictors of survival.
RESULTS:
The data of 212 patients with IPF were available for the analysis. The mean age was 66.4 years, and 70.8% were male. The mean time between the onset of symptoms and diagnosis was 11.6 months (range: 1–48 months). Common comorbid conditions noted in the IPF cohort included pulmonary hypertension (49.6%), diabetes mellitus (43.2%), hypertension (42.2%), osteoporosis (40.4%), and gastroesophageal reflux disease (32.1%). Acute exacerbation (AE) was noted in 21.2% of the IPF patients. AE, final saturation <85%, walking distance <300 m, and antifibrotic therapy were independent predictors of survival.
CONCLUSIONS:
In our IPF cohort, we found that there was a significant delay between the onset of symptoms and diagnosis. Moreover, we identified multiple comorbidities associated with IPF, which increases the burden on both IPF patients and clinicians. Importantly, AE and the use of antifibrotic therapy were independent predictors of survival. It is of paramount importance for clinicians to diagnose IPF at an early stage, refer patients to experienced centers, recognize comorbidities, and initiate antifibrotic therapy regardless of the underlying disease severity.
Keywords: Acute exacerbation, comorbidities, idiopathic pulmonary fibrosis, survival
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive fibrosing lung disease of unknown etiology that primarily occurs in older adults and is associated with a poor prognosis.[1] Although IPF is rare, population studies on interstitial lung disease (ILD) show that IPF is one of the most common forms of fibrotic lung disease.[2,3,4]
In IPF, challenging situations arise for patients and clinicians. First, dyspnea and cough are commonly reported symptoms among IPF patients; thus, misdiagnosis with more common conditions that have similar symptoms, such as asthma, chronic obstructive lung disease, heart failure, and others, is quite common, resulting in a delay in referral to specialized centers, a delay in diagnosis, and ultimately, a delay in initiating the appropriate treatment.[5,6] Second, the natural history of IPF is highly variable, and it is difficult to predict the clinical course in each patient, which makes it more challenging for clinicians to provide proper care than for specialized centers dedicated to IPF management. Third, because IPF occurs in the elderly, there are comorbidities that need to be recognized and aggressively managed to improve quality of life and survival. Finally, after the approval of antifibrotic therapies, namely nintedanib and pirfenidone, as treatments for IPF, several studies have confirmed that both the medications result in sustained reductions in the decline in lung function, thus reducing the rate of IPF progression and stabilizing the disease.[7,8,9] Unfortunately, many hospitals across the Kingdom of Saudi Arabia did not include antifibrotic therapies as a formulary medication or they have applied restrictions to the number of patients who can receive antifibrotic therapy, thus creating another challenge for both physicians and IPF patients.
In this context, the aim of the present study was to determine the clinical characteristics and the prevalence of comorbidities associated with IPF. In addition, we sought to determine prognostic factors associated with an increased risk of mortality.
Methods
The present study is a retrospective review of the ongoing prospective database collected at the ILD center at King Saud University Medical City. Consecutive patients diagnosed with IPF between March 2013 and December 1, 2019, were included in the study. This study was approved by the Institutional Research Board at the College of Medicine, King Saud University, Riyadh, Saudi Arabia (approval number E-20-4608). The need to obtain written informed consent was waived because of the retrospective nature of the current study. All other types of ILD were excluded.
A standard form was used to collect clinical information from the first ILD clinic visit, including general symptoms, pulmonary symptoms, smoking history, medication use, reported medical history, environmental history, occupational history, family history, and physical findings. In addition, associated comorbidities present at the time of initial evaluation or developed during the follow-up period, including acute exacerbation (AE) as previously described,[10] were collected.
Pulmonary function tests) (MGC Diagnostics Corporation, St. Paul, Minnesota, USA) were performed using standard methodologies, including spirometry, plethysmography, and measurement of the diffusion capacity of the lung for carbon monoxide (DLco).[11,12,13] The 6-min walk test (6MWT) was conducted in accordance with the American Thoracic Society guidelines.[14] High-resolution computed tomography (HRCT) scans were evaluated by a chest radiologist experienced in the interpretation of diffuse lung disease. Computed tomography (CT) findings are described using the nomenclature recommendations of the Fleischner Society.[15] The radiological pattern of usual interstitial pneumonia (UIP) was categorized as previously described.[16,17] When available, surgical lung biopsy (n = 14) was reviewed by an experienced pathologist. Histopathological evidence of the UIP pattern was determined as previously described.[1,18] IPF was diagnosed according to the established guidelines.[1,18] When pulmonary hypertension (PH) was suspected, right-heart catheterization (RHC) was performed. PH was defined as a mean pulmonary artery pressure (mPAP) 21–24 mmHg with pulmonary vascular resistance ≥3 WU or mPAP ≥25 mmHg, as previously described.[19] A multidisciplinary approach involving various specialties, including pulmonology, rheumatology, radiology and pathology, was implemented for all ILD patients, and a management plan was adopted after the multidisciplinary consensus recommendation.
Statistical analysis
Descriptive statistics are presented as the mean ± standard deviation or number (percentage) to describe the quantitative and categorical study variables. Group comparisons were assessed by Student's t-test or the Mann–Whitney U-test, as appropriate. To study survival in the IPF cohort, Kaplan–Meier survival curves and the log-rank test were used to investigate the time from the first diagnosis to death, transplant, loss to follow-up, or end of the study period (i.e., disease duration). Survival time was censored on December 1, 2019, or at the time a patient underwent lung transplant or was lost to follow-up. Unadjusted hazard ratios (HRs) were obtained for all study parameters using a Cox proportional hazard regression analysis. Univariate parameters with P < 0.05 were considered for inclusion in multivariate models to identify the independent predictors of mortality in IPF patients. Two-sided P < 0.05 and 95% confidence intervals (CIs) are used to report the statistical significance and precision of our results, respectively. All of the analyses were performed using the Statistical Package for the Social Sciences software (SPSS, version 18.0; SPSS Inc., Chicago, IL, USA).
Results
Two hundred and twelve consecutive IPF patients were identified. The mean age was 66.4 years, and 70.8% were male. A history of tobacco use was noted in 42.4% of the cohort. The mean time between the onset of symptoms and diagnosis was 11.6 months (range: 1–48 months). During the follow-up period, AE was noted in 21.2% of the IPF patients [Table 1]. Common comorbid conditions noted in the IPF cohort were PH (49.6%), diabetes mellitus (43.2%), hypertension (42.2%), osteoporosis (40.4%), and gastroesophageal reflux disease (32.1%). Thirty-five (16.5%) patients had no associated comorbidities, 54 (25.2%) patients had one, 52 (24.5%) had two, 47 (22.2%) had three, and 24 (11.3%) had four or more comorbidities.
Table 1.
Demographic characteristics of the study cohort
| Variable | IPF (n=212) |
|---|---|
| Age | 66.4±11.7 |
| Female sex | 62 (29.2) |
| Ever smoker | 89 (42.4) |
| Disease duration, months | 32.0±24.9 |
| BMI (kg/m2) | 27.5±5.6 |
| Surgical lung biopsy | 14 (6.6) |
| Acute exacerbations | 45 (21.2) |
| Comorbidities | |
| Pulmonary hypertension† | 65 (49.6) |
| Diabetes mellitus | 89 (43.2) |
| Hypertension | 87 (42.2) |
| Osteoporosis‡ | 59 (40.4) |
| GERD* | 68 (32.1) |
| Ischemic heart disease | 38 (18.4) |
| Emphysema | 25 (11.8) |
| Hiatus hernia | 14 (6.6) |
| Pneumothorax | 11 (5.2) |
| Pulmonary embolism | 6 (2.8) |
| Sleep apnea | 5 (2.4) |
| Lung cancer | 4 (1.8) |
Data are presented as the means±SD or n (%). †Defined by right-heart catheterization (n=131), ‡Measured by bone mineral density (n=146), *Based on barium swallow test. BMI=Body mass index, GERD=Gastroesophageal reflux disease, IPF=Idiopathic pulmonary fibrosis, SD=Standard deviation
Baseline PFT and 6MWT results are shown in Table 2. Sixty-five (30.6%) patients received antifibrotic therapy. The reported adverse events due to antifibrotic therapies are shown in Table 3. The most common adverse events associated with nintedanib or pirfenidone therapy are gastrointestinal symptoms and fatigue [Table 3]. Among patients receiving nintedanib therapy, diarrhea was managed by dose reduction to 100 mg twice daily in five patients or concomitant therapy with loperamide in eight patients. Furthermore, an additional 21 (9.9%) patients were not able to tolerate either nintedanib or pirfenidone therapy (mostly due to gastrointestinal adverse events) and refused to continue using the medication despite temporary discontinuation or dose reduction.
Table 2.
Clinical characteristics of the study cohort
| Variable | IPF (n=212) |
|---|---|
| Pulmonary function test | |
| FVC, % predicted | 57.3±20.0 |
| FEV1, % predicted | 66.9±21.9 |
| FEV1/FVC, ratio | 88.6±9.5 |
| TLC, % predicted | 58.6±16.4 |
| DLCO, % predicted† | 43.0±20.5 |
| 6-minute walk test‡ | |
| Initial BORG score | 0.9±1.1 |
| Final Borg score | 3.2±2.2 |
| Initial SpO2, % | 94.4±7.3 |
| Final SpO2, % | 85.3±7.4 |
| Distance, meters | 296.9±122.7 |
| Treatment | |
| Antifibrotic | 64 (30.2) |
| Oxygen supplementation | 114 (53.8) |
†n=172; ‡n=194. Data are presented as the means±SD or n (%). FVC=Forced vital capacity, FEV1=Forced expiratory volume in one second, TLC=Total lung capacity, DLco=Diffusion capacity of the lung for carbon monoxide, SpO2=Oxygen saturation by pulse oximetry, IPF=Idiopathic pulmonary fibrosis, SD=Standard deviation
Table 3.
Reported adverse events among patients using antifibrotic therapy
| Event | Pirfenidone (n=40)* | Nintedanib (n=24)* |
|---|---|---|
| Anorexia | 21 (52.5) | 4 (16.7) |
| Fatigue | 14 (35) | 2 (8.3) |
| Weight loss | 10 (25) | - |
| Nausea | 9 (22.5) | 2 (8.3) |
| Liver enzyme elevation | 1 (2.5) | 1 (4.2) |
| Diarrhea | - | 5 (20.8) |
| Photosensitivity | - | - |
| Skin reaction | 1 (2.5) | - |
Data are presented as the n (%). *Patients could have multiple positive adverse events
Survival analysis of the idiopathic pulmonary fibrosis cohort
There were 68 patients (32%) who died during the follow-up period and 3 (1.4%) who underwent lung transplantation. After stratifying the IPF cohort based on the development of AE, patients without exacerbation had significantly better survival than those with exacerbation (P = 002) [Figure 1]. Moreover, severe oxygen desaturation at the end of the 6MWT (final saturation <85%) and walking distance <300 m were strong predictors of mortality (P ≤ 0.0001 and P = 0.002, respectively) [Figures 2 and 3, respectively]. Furthermore, the use of antifibrotic therapy was associated with significantly improved survival (P = 0.001) [Figure 4].
Figure 1.
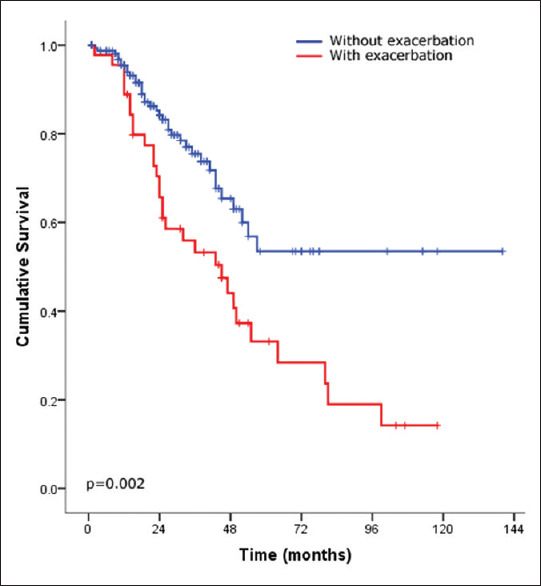
Kaplan–Meier survival curves of idiopathic pulmonary fibrosis after stratifying patients by the presence of exacerbation (presence, red line and absence, and blue line)
Figure 2.
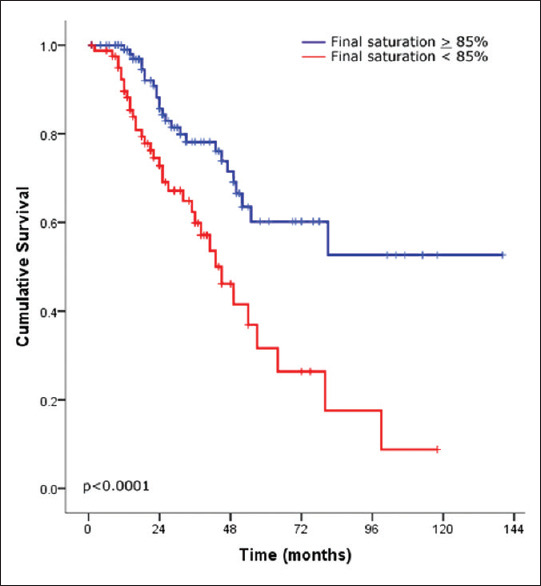
Kaplan–Meier survival curves of idiopathic pulmonary fibrosis after stratifying patients according to the 6-min walk test final oxygen saturation by pulse oximetry (<85%, red line and ≥85%, blue line)
Figure 3.
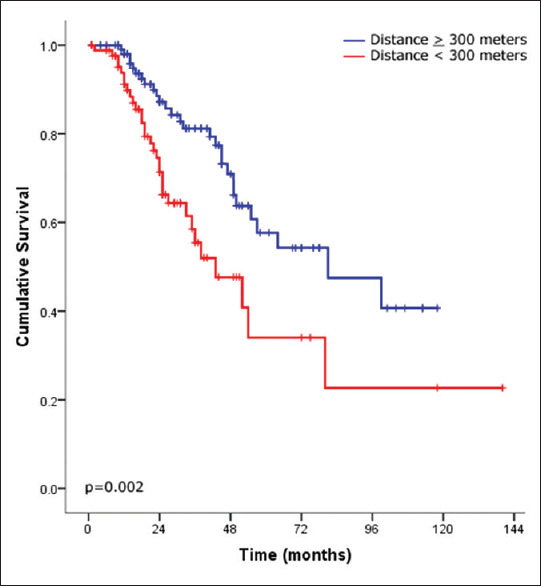
Kaplan–Meier survival curves of idiopathic pulmonary fibrosis after stratifying patients according to the 6-min walk test distance (<300 M, red line and ≥300 M, blue line)
Figure 4.
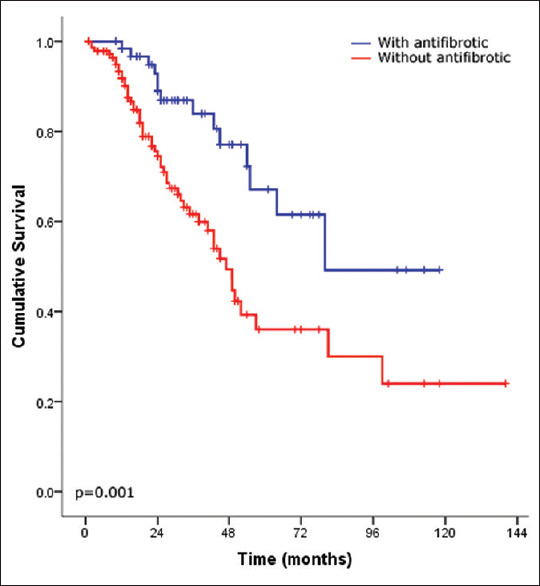
Kaplan–Meier survival curves of idiopathic pulmonary fibrosis after stratifying patients according to the use of antifibrotic therapy (use, blue line and no use, red line)
In univariate cox regression [Table 4], multiple variables were associated with an increased risk of mortality in the IPF cohort, including AE (HR, 2.227; 95% CI, 1.371–3.618; P = 0.001), final saturation <85% (HR, 2.640; 95% CI, 1.577–4.418; P < 0.0001), and walking distance <300 m (HR, 2.229; 95% CI, 1.331–3.731; P = 0.002). In contrast, variables associated with improved survival were the use of antifibrotic therapy (HR, 0.420; 95% CI, 0.236–0.749; P = 0.003), a higher body mass index (for every one unit increase, HR, 0.936; 95% CI, 0.893–0.980; P = 0.005), a higher total lung capacity (for each 1% increase from the mean, HR, 0.963; 95% CI, 0.946–0.980; P ≤ 0.0001), a higher forced vital capacity (for each 1% increase from the mean, HR, 0.972; 95% CI, 0.959–0.985; P ≤ 0.0001), and a higher DLco (for each 1% increase from the mean, HR, 0.986; 95% CI, 0.973–1.00; P = 0.047).
Table 4.
Variables predicting survival in patients with idiopathic pulmonary fibrosis
| Variable | Unadjusted |
Adjusted |
||
|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | |
| Acute exacerbation | 2.227 (1.371-3.618) | 0.001 | 4.134 (2.194-7.787) | <0.0001 |
| 6MWT final SpO2 <85% | 2.640 (1.577-4.418) | <0.0001 | 2.805 (1.572-5.005) | <0.0001 |
| Antifibrotic therapy | 0.420 (0.236-0.749) | 0.003 | 0.227 (0.106-0.484) | <0.0001 |
| 6MWTD <300 m | 2.229 (1.331-3.731) | 0.002 | 2.647 (1.417-4.946) | 0.002 |
| BMI (kg/m2) | 0.936 (0.893-0.980) | 0.005 | 0.948 (0.896-1.003) | 0.064 |
| TLC, % predicted | 0.963 (0.946-0.980) | <0.0001 | 0.990 (0.963-1.018) | 0.496 |
| FVC, % predicted | 0.972 (0.959-0.985) | <0.0001 | 0.995 (0.975-1.016) | 0.644 |
| DLCO, % predicted | 0.986 (0.973-1.000) | 0.047 | ||
| Age | 1.008 (0.986-1.029) | 0.487 | ||
| Female sex | 0.663 (0.373-1.176) | 0.160 | ||
| Ever smoker | 1.275 (0.782-2.078) | 0.330 | ||
| Pulmonary hypertension | 1.073 (0.611-1.885) | 0.806 | ||
HR=Hazard ratio, and 95% CI=95% confidence interval, 6MWT=6-min walk test, SpO2=Oxygen saturation by pulse oximetry, 6MWTD=6-min walk test distance, BMI=Body mass index, TLC=Total lung capacity, FVC=Forced vital capacity, DLco=Diffusion capacity of the lung for carbon monoxide
A multivariate analysis using a Cox regression model [Table 4] revealed that AE, final saturation <85%, and walking distance <300 m were independent predictors of decreased survival. However, the use of antifibrotic therapy was the only variable independently associated with improved survival.
Discussion
Here, we describe the clinical characteristics of consecutive IPF patients diagnosed in our ILD center. We show that it primarily occurs in the elderly population and predominantly in males; IPF is associated with multiple comorbidities. Importantly, the development of AE and the use of antifibrotic therapy were independently associated with survival.
When IPF is suspected, promptly referring patients to specialized centers experienced in the diagnosis and management of IPF is of paramount importance. Many types of ILD significantly overlap with IPF and resources, including serological tests, HRCT, and other tests needed to establish the diagnosis of IPF, are not readily available in every hospital. In addition, identifying and managing comorbidities and discussing every ILD case in a multidisciplinary meeting that involves experts in various specialties can be very challenging for clinicians not experienced in managing ILD cases. In our center, all tests needed to establish the diagnosis are performed within 5 days of the initial evaluation if ILD is suspected. Moreover, a multidisciplinary approach involving various specialties, including pulmonology, rheumatology, radiology, and pathology, was implemented for all ILD patients before a final diagnosis was rendered. In our study, we found that the mean delay between the onset of symptoms and diagnosis was nearly 1 year, and in some patients, it was as long as 4 years. Collard et al.[5] noted that 55% of IPF patients reported a delay of more than 1 year between the onset of symptoms and diagnosis. Lamas et al.[20] noted that the median delay between the onset of dyspnea and referral to a tertiary care center was 2.2 years. In another study, they noted that the time from onset of symptoms until the IPF patient received the correct diagnosis ranged between 4 and 5 years.[6] Such a delayed diagnosis will result in an increased risk of mortality among IPF patients independent of disease severity.[20] Collectively, the findings in our study and the cited studies imply that it is very important to make a correct diagnosis of IPF as early as possible, which can be achieved by increasing awareness among clinicians and expediting referrals to specialized ILD centers.
A significant variation between studies[21,22] was noted with regard to comorbidities in IPF patients. Different methodologies, sample sizes, lifestyles, smoking habits, races, and other factors may explain the observed variation in comorbidities among the IPF populations studied. In the present study, we identified many comorbidities associated with IPF. Importantly, 33% of our IPF cohort had three or more associated comorbidities, which increased the burden on patients, clinicians, and the health-care system. PH was the most prevalent (49.6%) comorbidity in our IPF cohort, which reflects the aggressive screening program for PH in our center and the easy accessibility to perform RHC when needed. Although there are currently no approved therapies for the treatment of PH in IPF patients, identifying PH in a specialized center ensures that the patient is followed up regularly, disease progression is monitored more closely, and patients participate in clinical trials and are considered for lung transplantation.
When sudden worsening of respiratory symptoms arises in IPF patients in the absence of identifiable causes, the diagnosis of AE should be promptly considered. The exact cause of exacerbation is unknown; however, possible precipitating triggers, including infection, aspiration, surgery, drugs, and others, have been implicated.[10] Since exacerbation is associated with significant morbidity and mortality,[23,24] it creates significant challenges for patients, families, and clinicians. Moreover, although exacerbation is commonly noted in patients with advanced-stage lung fibrosis, it can occur even in patients with well-preserved lung function parameters.[10,25] Thus, the natural course of IPF is highly variable and unpredictable. In the present study, the prevalence of AE was 21.2%. Importantly, the development of AE in our cohort was the most powerful predictor of mortality, highlighting the urgent need to understand the etiology of IPF exacerbation and the best management strategy that can be offered, which would ultimately have a positive impact on IPF patient survival.
In 2015, the international treatment guidelines conditionally recommended the use of nintedanib and pirfenidone as treatments for IPF.[26] Subsequently, many studies have provided further evidence that both the drugs are effective at reducing disease progression and are associated with disease stabilization.[7,8,9] Moreover, the effects of nintedanib and pirfenidone have consistently been observed across ages, sexes, races, and importantly, a wide spectrum of disease severities, including mild, moderate, and severe lung fibrosis.[8,25,27,28] Our data provide further evidence that the use of antifibrotic therapy is independently associated with improved survival. In addition, in agreement with the results from clinical trials, gastrointestinal adverse events were the most common adverse event noted in patients taking nintedanib and pirfenidone; however, they were mild to moderate in severity and did not lead to the discontinuation of therapy in the majority of the patients.
Previous studies have identified physiological parameters derived from the PFT and 6MWT as markers of mortality in IPF patients.[29,30,31] In the present study, multivariate analysis with Cox proportional hazards regression analysis showed that a 6MWT final saturation <85% and walking distance <300 m were associated with a nearly threefold increased risk of mortality. This highlights the importance of 6MWT parameters as surrogate markers of disease severity among Saudi patients with IPF.
The present study had several limitations. The retrospective review of a database from a single center may have introduced selection bias. However, our data were collected prospectively for each patient at the time of initial evaluation and subsequently during the follow-up. Moreover, the present IPF cohorts are derived from highly specialized centers dedicated to the diagnosis and management of ILD, thus introducing institutional bias.
Conclusions
We describe a large cohort of consecutive IPF patients diagnosed in one center. Importantly, we show that multiple comorbidities occur in IPF patients. Furthermore, we show that AE was associated with a fourfold increased risk of mortality. Moreover, the use of antifibrotic therapy in our cohort was independently associated with improved survival. It is of paramount importance for clinicians to diagnose IPF at an early stage, refer patients to experienced centers when possible, recognize significant comorbidities, and initiate antifibrotic therapy regardless of the underlying disease severity. We hope that the results of our study will be used to develop institutional and professional guidelines to help change the current practice with regard to managing and treating IPF patients.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alhamad EH. Interstitial lung diseases in Saudi Arabia: A single-center study. Ann Thorac Med. 2013;8:33–7. doi: 10.4103/1817-1737.105717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, Sanyal S, Brillet PY, Brauner M, et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J. 2017;50:1–12. doi: 10.1183/13993003.02419-2016. [DOI] [PubMed] [Google Scholar]
- 4.Dhooria S, Agarwal R, Sehgal IS, Prasad KT, Garg M, Bal A, et al. Spectrum of interstitial lung diseases at a tertiary center in a developing country: A study of 803 subjects. PLoS One. 2018;13:e0191938. doi: 10.1371/journal.pone.0191938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collard HR, Tino G, Noble PW, Shreve MA, Michaels M, Carlson B, et al. Patient experiences with pulmonary fibrosis. Respir Med. 2007;101:1350–4. doi: 10.1016/j.rmed.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Hewson T, McKeever TM, Gibson JE, Navaratnam V, Hubbard RB, Hutchinson JP. Timing of onset of symptoms in people with idiopathic pulmonary fibrosis. Thorax. 2017:683–5. doi: 10.1136/thoraxjnl-2017-210177. [DOI] [PubMed] [Google Scholar]
- 7.Brunnemer E, Wälscher J, Tenenbaum S, Hausmanns J, Schulze K, Seiter M, et al. Real-world experience with nintedanib in patients with idiopathic pulmonary fibrosis. Respiration. 2018;95:301–9. doi: 10.1159/000485933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crestani B, Huggins JT, Kaye M, Costabel U, Glaspole I, Ogura T, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir Med. 2019;7:60–8. doi: 10.1016/S2213-2600(18)30339-4. [DOI] [PubMed] [Google Scholar]
- 9.Zurkova M, Kriegova E, Kolek V, Lostakova V, Sterclova M, Bartos V, et al. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir Res. 2019;20:16. doi: 10.1186/s12931-019-0977-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working Group Report. Am J Respir Crit Care Med. 2016;194:265–75. doi: 10.1164/rccm.201604-0801CI. [DOI] [PubMed] [Google Scholar]
- 11.Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 12.Wanger J, Clausen JL, Coates A, Pedersen OF, Brusasco V, Burgos F, et al. Standardisation of the measurement of lung volumes. Eur Respir J. 2005;26:511–22. doi: 10.1183/09031936.05.00035005. [DOI] [PubMed] [Google Scholar]
- 13.Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26:720–35. doi: 10.1183/09031936.05.00034905. [DOI] [PubMed] [Google Scholar]
- 14.ATS statement: Guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166:111–7. doi: 10.1164/ajrccm.166.1.at1102. [DOI] [PubMed] [Google Scholar]
- 15.Hansell DM, Bankier AA, MacMahon H, McLoud TC, Müller NL, Remy J. Fleischner Society: Glossary of terms for thoracic imaging. Radiology. 2008;246:697–722. doi: 10.1148/radiol.2462070712. [DOI] [PubMed] [Google Scholar]
- 16.Travis WD, Costabel U, Hansell DM, King TE, Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–48. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sverzellati N, Lynch DA, Hansell DM, Johkoh T, King TE, Jr, Travis WD. American Thoracic Society-European Respiratory Society Classification of the Idiopathic Interstitial Pneumonias: Advances in knowledge since 2002. Radiographics. 2015;35:1849–71. doi: 10.1148/rg.2015140334. [DOI] [PubMed] [Google Scholar]
- 18.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 19.Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53:1–15. doi: 10.1183/13993003.01914-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: A cohort study. Am J Respir Crit Care Med. 2011;184:842–7. doi: 10.1164/rccm.201104-0668OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oldham JM, Collard HR. Comorbid conditions in idiopathic pulmonary fibrosis: Recognition and management. Front Med (Lausanne) 2017;4:123. doi: 10.3389/fmed.2017.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caminati A, Lonati C, Cassandro R, Elia D, Pelosi G, Torre O, et al. Comorbidities in idiopathic pulmonary fibrosis: An underestimated issue. Eur Respir Rev. 2019;28:1–11. doi: 10.1183/16000617.0044-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur Respir J. 2011;37:356–63. doi: 10.1183/09031936.00159709. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki A, Kondoh Y, Brown KK, Johkoh T, Kataoka K, Fukuoka J, et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology. 2020;25:525–34. doi: 10.1111/resp.13682. [DOI] [PubMed] [Google Scholar]
- 25.Kolb M, Richeldi L, Behr J, Maher TM, Tang W, Stowasser S, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72:340–6. doi: 10.1136/thoraxjnl-2016-208710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015;192:e3–19. doi: 10.1164/rccm.201506-1063ST. [DOI] [PubMed] [Google Scholar]
- 27.Albera C, Costabel U, Fagan EA, Glassberg MK, Gorina E, Lancaster L, et al. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur Respir J. 2016;48:843–51. doi: 10.1183/13993003.01966-2015. [DOI] [PubMed] [Google Scholar]
- 28.Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, Stowasser S, et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med. 2016;193:178–85. doi: 10.1164/rccm.201503-0562OC. [DOI] [PubMed] [Google Scholar]
- 29.Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, Battle E, et al. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest. 2011;140:221–9. doi: 10.1378/chest.10-2572. [DOI] [PubMed] [Google Scholar]
- 30.Mura M, Porretta MA, Bargagli E, Sergiacomi G, Zompatori M, Sverzellati N, et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: A 3-year prospective study. Eur Respir J. 2012;40:101–9. doi: 10.1183/09031936.00106011. [DOI] [PubMed] [Google Scholar]
- 31.du Bois RM, Albera C, Bradford WZ, Costabel U, Leff JA, Noble PW, et al. 6-Minute walk distance is an independent predictor of mortality in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2014;43:1421–9. doi: 10.1183/09031936.00131813. [DOI] [PubMed] [Google Scholar]