

Abstract
Abdominal aortic aneurysm (AAA), commonly occurring in the aged population, is a degenerative disease that dilate and weaken infrarenal aorta due to progressive degeneration of aortic wall integrity. Vinpocetine, a derivative of alkaloid vincamine, has long been used for cerebrovascular disorders and cognitive impairment in the aged population. Recent studies have indicated that vinpocetine antagonizes occlusive vascular disorders such as intimal hyperplasia and atherosclerosis. However, its role in vascular degenerative disease AAA remains unexplored. Herein, we determined the effect of vinpocetine on the formation of AAA as well as the intervention of pre-existing moderate AAA. AAA was induced by periaortic elastase application in C57BL/6J mice. Systemic vinpocetine treatment was applied daily via intraperitoneal injection. We showed that vinpocetine pre-treatment remarkably attenuated aneurysmal dilation assessed by diameter and volume. More importantly, vinpocetine also significantly suppressed the progression of pre-existing moderate AAA in a post-intervention model. Vinpocetine improved multiple cellular and molecular changes associated with AAA, such as elastin degradation, media smooth muscle cell depletion, collagen fibers remodeling and macrophage infiltration in aneurysmal tissues. Vinpocetine potently suppressed TNF-α-induced NF-κB activation and proinflammatory mediator expression in primary cultured macrophages in vitro, as well as in the aorta wall in vivo, suggesting vinpocetine conferred anti-AAA effect at least partially via the inhibition of inflammation. Taken together, our findings reveal a novel role of vinpocetine in AAA formation, development, and progression. Given the excellent safety profile of vinpocetine, this study suggests vinpocetine may be a novel therapeutic agent for AAA prevention and treatment.
Keywords: abdominal aortic aneurysm, vinpocetine, elastase
INTRODUCTION
An abdominal aortic aneurysm (AAA) is permanent weakening and dilatation of the infrarenal abdominal aorta that most commonly occurs in men >65 years of age.1 AAA is defined by exceeding the normal aorta diameter by 50%, or >3 cm.2 It is a multifactorial disease with risk factors including smoking, male sex, advanced age and genetics.3 If left untreated, the aortic wall continues to weaken and dilate. Eventually, the infrarenal aorta becomes unbale to withstand the blood pressure, resulting in AAA rupture and abdominal hemorrhage. Until now, no proven pharmaceutical treatment is available to AAA, which drives an unmet need to develop drug treatments that limit or prevent the progression of aneurysm.1 AAA is a degenerative disease. The pathogenesis of AAAs is complex, attributing to progressive compromised changes in the integrity of the aortic wall. In the healthy aorta wall, elastic fibers deposited in between smooth muscle cells (SMCs) in the media of aortic wall are responsible for the viscoelastic property of aorta. Whereas in AAA, the aortic wall is characterized by extracellular matrix degradation, media degeneration and inflammatory cells infiltration.3, 4 On histological level, proteolytic fragmentation of the media elastin, destruction of matrix collagen fibers, loss of media SMC, extensive transmural infiltration by macrophages and lymphocytes are hallmark features in AAA.3–5 The exact order of these pathological events and their underlying mechanisms yet largely remain to be identified.
Vinpocetine is a synthetic derivative from vincamine, an alkaloid extracted from the periwinkle plant, Vinca minor.6, 7 Since the discovery in 1978, vinpocetine has been widely used in many Asian and European countries for treatment of cognitive impairment in aged population.8 Vinpocetine-containing memory enhancers, such as Ginkgo Biloba with Vinpocetine (brand trunature), Vinpocetine-Triple Strength (brand Swanson) are also currently used as dietary supplements in the United States. So far, no significant side effects, toxicity, or contraindications at therapeutic dose regimen of vinpocetine has been reported.6 Vinpocetine appears to have multiple molecular targets. Vinpocetine has been first found as an inhibitor for Ca2+/calmodulin-activated cyclic nucleotide phosphodiesterase 1 (PDE1).6 Vinpocetine has been also found to block voltage dependent Na+ channels.9 Recently, vinpocetine has been shown to be an IkB kinase (IKK) inhibitor.10 In response to external inflammatory stimuli, activated IKK complex cause degradation of IκBα, leading to liberation and nuclear translocation of NF-κB which activates transcription of inflammatory molecules.11 Vinpocetine has been found to suppress NF-κB-dependent inflammation in a variety of cell types and multiple inflammatory animal models.10, 12–16 We and others have previously explored the roles of vinpocetine in a number of occlusive vascular diseases. For example, vinpocetine attenuated mouse carotid artery intima hyperplasia induced by blood-flow cessation.17 Vinpocetine also suppressed high-fat diet induced atherosclerosis in ApoE deficient mice.18, 19 However, the role of vinpocetine in degenerative vascular disease such as AAA remains unknown. Given the facts that AAA is an aging related vascular disease and vinpocetine is currently used in the aged population, it is of great interest to understand the effects of vinpocetine in the AAA development. In the present study, we examine the effect of vinpocetine in prevention as well as intervention of AAA development and vascular pathologies, using the experimental mouse model induced by periaortic elastase in combination with 3-aminopropionitrile fumarate salt (BAPN) treatment to block elastin/collagen formation.
METHODS
Reagents
The source and catalog number for all reagents used in this study are listed in Online Table 1.
Mouse model of AAA
All wild type C57BL/6 mice were bred and housed in animal facility of our institute under a 12:12 hour light-dark cycle. Standard chow and water ad libitum were available before experiment. During experiment, 13-week-old wild type C57BL/6 male mice were treated with 0.2% BAPN (w/v) in drinking water. BAPN treatment started from 2 days before surgery until the end of study.20 For surgery, mice were anesthetized with inhaled isoflurane. Buprenorphine was used for analgesia. Periaortic application of porcine pancreas elastase were blinded performed as previously described with slight modifications.20 Briefly, the connective tissue surrounding abdominal aorta was cleaned off from approximately 2 mm below the left renal artery to the bifurcation. A 6mm × 9mm piece of whatman paper were placed on the exposed aortic adventitia for 10 minutes and 40ul of elastase (7.6 mg protein/mL, 4 units/mg protein) were applied directly to the whatman paper. After that, the exposed area was washed gently with saline twice and the abdomen was closed routinely in layers. For sham groups, heat deactivated elastase (100°C for 30 minutes), instead of active elastase, was applied topically to the aorta. The rest of the surgical procedure was identical to the active elastase groups. Two studies were investigated about the effect of vinpocetine treatment on AAA formation and progression. (1) To study effect of vinpocetine on AAA development, mice were randomly separated into four groups: saline/sham (n=6), vinpocetine/sham (n=6), saline/elastase (n=10), vinpocetine/elastase (n=7). Mice were intraperitoneally injected with 5mg/kg vinpocetine or saline daily, as described previously, starting from 2 days before surgery until the end of study.10, 17, 18 (2) To examine the effect of vinpocetine on the progression and expansion of pre-existing AAAs, abdominal aorta size were assessed by ultrasound imaging before surgery as baseline and on the 6th day following surgery of periaortic application of elastase. These mice were then randomly separated into two groups: saline/elastase (n=9), vinpocetine/elastase (n=9). Mice were treated with vinpocetine or saline daily starting from the 6th day post-surgery till harvest. In both studies all mice were euthanized at 28 days post-surgery. We chose male mice because male gender is a major risk factor for AAA. AAA prevalence in males is 4 times greater than females in human patients.21 In rodent experimental models, such as Angiotensin II-induced AAAs in apolipoprotein E −/− mice or elastase perfusion-induced AAA, females are also protected from AAA formation.21 All animals were used in accordance with the guidelines of the National Institutes of Health and American Heart Association for the care and use of laboratory animals. The procedures were performed in accordance with experimental protocols that were approved by the University Committee on Animal Resources at the University of Rochester.
Ultrasound imaging
A Vevo 2100 ultrasound imaging platform (FUJIFILM VisualSonics) was utilized to assess aneurysm progression in periaortic elastase induced AAA model. Mice were anesthetized with isoflurane, via nosecone, placed on a 37°C warming pad, in a supine position, and feet restrained. Body temperature, respiration rate and ECG were monitored. Heart rate was kept at a consistent range (500–550 beats per minute). Hair was removed from the abdomen and Aquasonic 100 ultrasound transmission gel was placed on abdomen to increase probe contact. The probe was applied on long axis to locate the aorta. Pulse wave (PW mode) doppler was used to confirm aortic flow, and M mode images were taken at the widest part of the aneurysm. Data for diastole internal diameter were shown for individual animals.
Morphometric analysis of maximal aortic width and aneurysmal area
At the end of the experiments, mice were anesthetized via intraperitoneal injection of ketamine (100 mg/kg), midazolam (5mg/kg) and heparin (1600 units/kg). Mice were euthanized via cervical dislocation when lost toe pinch response. Aortas were perfused with saline and fixed with 10% phosphate-buffered formalin (NBF) for 2 minutes. Whole aorta was dissected from the surrounding connective tissue and fixed with 10% NBF for 24 hours at 4°C. Subsequently aortas were further cleaned with a dissecting microscope. Pictures were taken with a digital camera with a ruler set aside. The adventitial circumferences at the maximal expanded portion of the aneurysm were quantified as the maximal abdominal aortic diameter.22 The maximum diameter of the abdominal aorta was analyzed using Image J software after adjusting the scale according to the ruler in aorta pictures. At least 3 measurements of the maximal expanded portion of the infrarenal aorta for each mouse were averaged before calculating the mean of each experimental group. The total aneurysmal area was defined as the outline of intrarenal aorta under renal artery and up to iliac artery as described previously.23 Aortas were subsequently embedded in paraffin. Aortic cross sections (5 um each) were collected serially from proximal to distal abdominal aorta in levels.
Van Gieson elastin staining
Paraffin sections were stained for elastic fibers using van Gieson Elastic Stain kit according to the manufacturer’s instructions. The section from the largest diameter of AAA for each animal was selected for staining. Briefly, deparaffinized and hydrated sections were stained in elastic stain solution for 30 minutes and decolorize in differentiating solution. Following that sections were rinsed in sodium thiosulfate solution shortly and stained in Van Gieson stain solution for 15 minutes. Sections were viewed with a BX51 upright microscope (Olympus, Japan) using Olympus CellSens Standard acquisition software. The surface area occupied by elastic fibers staining was quantified using image J as described previously.24 Eight visual fields (magnification 200) evenly distributed at media of every lesion section were included to quantify the amount of elastin staining. All images were set to the same hue, saturation and brightness and measured for positive staining area. The elastin content is expressed as a percentage of elastic fiber area in media area.
Sirius red/Fast green collagen staining
Paraffin sections were stained for collagen fibers using Sirius red and Fast green as described previously.25 The section from the largest diameter of AAA for each animal was selected for staining. Briefly, sections were pre-incubated with 0.04% Fast green and stained with 0.1% Fast green and 0.04% Sirius red for 30 minutes. Collagen exhibited red fibers, while the non-collagen cellular proteins were green. Sections were viewed with a BX51 microscope.
Alcian Blue staining
Paraffin sections were stained for extracellular proteoglycans using Alcian Blue staining as described previously.26 Sections were stained in Alcian Blue solution for 30 minutes and counterstained with nuclear fast red. Eight visual fields (magnification 200) evenly distributed at media of every lesion section were included to quantify the amount of positive staining (area of staining per media area). All images were quantified with image J software by setting to the same hue, saturation and brightness. Three sections located at 300um intervals from aneurysmal center segment (segment of largest diameter) were analyzed for each animal. The Alcian Blue positive staining area per media area were standardized to the averaged amount of staining in saline/sham control group. Sections were viewed with a BX51 microscope.
Immunohistochemistry staining
Aortic cross sections were deparaffinized, followed by treatment with citrate buffer for antigen retrieval and 3%H2O2. The sections were blocked with Dako serum-free blocking solution and incubated with α-SMA (1:500), F4/80 (1:200), p65 (1:300) primary antibody diluted in Dako antibody diluent overnight at 4°C. Subsequently, the sections were incubated with biotinylated secondary IgG antibodies for 1 hour. Avidin-biotinylated enzyme complex and a diaminobenzidine substrate chromogen system were used for detection. Matched IgG was used in place of the primary antibody as a negative control. Sections were viewed with a BX51 upright microscope. Slides were viewed with EVOS® FL Auto microscope (Life technologies, AMAFD1000). All immunohistochemistry staining were analyzed with NIH image J software. The F4/80 and p65 staining is expressed as a percentage of the positive staining area over total section area. All images of each staining were set to the same hue, saturation and brightness and measured for positive staining area. The α-SMA staining was analyzed by calculating the integration optical density value of positive staining in media. Media region was drawn with the freehand selection tool around the entire aorta. Three sections located at 300um intervals from aneurysmal center segment (segment of largest diameter) were analyzed for each animal. The positive staining in each experimental group were standardized to the averaged amount of staining in saline/sham control group.
Immunofluorescence staining
Abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples were double stained of p65 and Mac2 immunofluorescence. Sections were deparaffinized, followed by heat treatment with citrate buffer for antigen retrieval. Following that, sections were permeabilized by 0.3% Triton X-100/PBS for 10 minutes. Nonspecific binding sites were blocked with Dako serum-free blocking solution at room temperature for 1 hour, and incubated with anti-p65 primary antibody (1:100) and anti-Mac2 primary antibody (1:200) overnight at 4°C. Subsequently, the sections were incubated with Alexa Fluror-594 conjugated anti-rabbit (for p65) or Alexa Fluror-488 conjugated anti-rat (for Mac2) secondary antibody for 1 hour at room temperature. Sections were subsequently incubated with DAPI (4′,6-diamidino-2-phenylindole) for nuclei staining and mounted with ProLong™ Gold Antifade Mountant. Slides were then viewed with a confocal microscope (Olympus FV1000-IX81) using 60X objective. Matched IgG was used in place of the primary antibody as a negative control. Colocalization analysis were performed in Fiji software using Colocalization.java plugin27 with same threshold set for both red and green channels, and colocalized points are shown in white. Two points are considered as colocalized if their respective intensities are strictly higher than the threshold of their channels, and if their ratio of intensity is strictly higher than the ratio setting value.
Isolation of primary mouse resident peritoneal macrophages
Resident peritoneal macrophages were isolated by PBS lavage and purified by adhesion as described previously.28, 29 Briefly, C57BL/6 mice were sacrificed by CO2 asphyxiation. 5ml ice-cold sterile PBS with 1mM EDTA was injected into the peritoneal cavity. Mouse peritoneum was gently massaged to dislodge any attached cells into the PBS solution. The PBS containing resident peritoneal cells were slowly withdrawn by a syringe. Cell suspension was stored on ice until seeding to plastic culture plates or glass coverslip in RPMI1640 medium supplemented with 10% fetal bovine serum (vol/vol) and 0.5mg/ml penicillin-streptomycin. After 2 hours incubation in standard condition (5% CO2 in air in a humidified environment at 37°C), plates were washed three times with PBS to remove nonadherent cells. More than 90% of the adherent monolayer should be macrophages. After overnight culture, macrophages were used for experiments. Macrophages were starved in serum free RPMI1640 medium for 4 hours. Cells were subsequently pretreated with 30uM vinpocetine for 60min before treatment with or without murine TNF-α (10ng/ml) for 30 minutes for p65 immunostaining or 6 hours for Real-time PCR in the continued presence or absence of vinpocetine (30uM). When performing vinpocetine treatment, vinpocetine was added to culture medium in test tube and mixed thoroughly before plating.
Immunocytochemistry staining
Cells were fixed in 4% paraformaldehyde for 10 minutes, followed by permeabilization in PBS/0.1% (v/v) triton-100 for 10min. Cells were subsequently blocked with Dako serum-free blocking solution for 60min and incubated with p65 primary antibody (1:700) diluted in Dako antibody diluent overnight at 4°C. Cells were then incubated in Alexa Fluor Plus 594 secondary antibody for 1 hour at room temperature. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Staining was visualized using an Olympus IX-81 microscope with 60X objective with FV10-ASW 4.2 Viewer software. p65 nuclear translocation was evaluated using NIH ImageJ software. Quantification of p65 subcellular distribution was expressed as the percentage of p65 integrated intensity in nucleus over total cell.30 At least 200 cells were quantified for each experimental group from 3 separate experiments.
Real-time PCR
RNA was extracted from macrophages lysate or infrarenal aorta lysate using RNeasy kit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized with iScriptDNA synthesis kit (Bio-Rad). qPCR amplification was performed by using IQ SYBR Green Supermix (Bio-Rad) according to the manufacturer’s instructions. Each reaction was performed in duplicate. The result was expressed as the ratio of target gene versus GAPDH. Primers for qPCR were as follows: mTNF-α: 5′-TCT TCT CAT TCC TGC TTG TGG-3′(Forward) 5′-GGT CTG GGC CAT AGA ACT GA-3′(Reverse); mIL-1β: 5′-GAG TGT GGA TCC CAA GCA AT-3′(Forward) 5′-ACG GAT TCC ATG GTG AAG TC-3′(Reverse); mGAPDH: 5′-AGG TCG GTG TGA ACG GAT TTG-3′(Forward) 5′-TGT AGA CCA TGT AGT TGA GGT CA-3′(Forward).
LDH cytotoxicity assay
Primary mouse resident peritoneal macrophages were isolated to determine optimum cell number to use for lactate dehydrogenase (LDH) cytotoxicity assay (CyQUANT LDH Cytotoxicity Assay kit, Invitrogen) according to manufacturer’s instructions. Cells were seeded in a serial dilution in two sets of triplicate wells in a 96-well tissue culture plate. Triplicate wells of medium-only were included for background measurement. After wash and overnight culture, the number of macrophages were counted. One set of 0–10,000 cells series was used to determine the Spontaneous LDH Release. Another set 0–10,000 cells series was used to determine the Maximum LDH release. LDH activity were measured according to manufacturer’s instructions. Briefly, 10μL of sterile water was added to each well of the Spontaneous LDH Release dilution series, while 10μL of 10X Lysis Buffer was added to the Maximum LDH Release dilution series to lyse cells. Plate was incubated in an incubator at 37°C for 45 minutes. 50 μL of each sample medium was then transferred to a 96-well plate in triplicate wells. 50μL of Reaction Mixture was then added to each sample well, and plate was incubated at room temperature for 30 minutes. 50μL of Stop Solution was added to each well to stop reaction. Absorbance were then measured at 490 nm and background was subtracted from each absorbance value. The Maximum LDH Release absorbance and Spontaneous LDH Release absorbance were plotted versus macrophage cell number, with the greatest difference between the Spontaneous and Maximum determining the optimal cell number to use in the following experiments. 10,000 macrophages in triplicate wells in a 96-well tissue culture plate were used for experimental groups (Vehicle control, Vinpocetine, TNFα, TNFα and Vinpocetine), Spontaneous LDH Activity group and Maximum LDH Activity group. Medium only were included. Experiment were performed using same experiment condition as in Fig. 6 C and D. Briefly, after overnight culture, cells of experimental groups were starved in serum free RPMI1640 medium for 4 hours. After that, cells were pretreated with 30uM vinpocetine for 60min before treatment with or without murine TNF-α (10ng/ml) for 6 hours in the continued presence or absence of vinpocetine (30uM). Cells of Spontaneous LDH Activity and Maximum LDH Activity were starved at the same time without any treatment. After that, LDH activity were measured. Background was subtracted from absorbance of experimental groups, Spontaneous LDH Activity group and Maximum LDH Activity group. % Cytotoxicity of experimental groups were calculated by using the following formula:
Data are presented as % cell viability by subtracting % cytotoxicity from 100.
Figure 6.
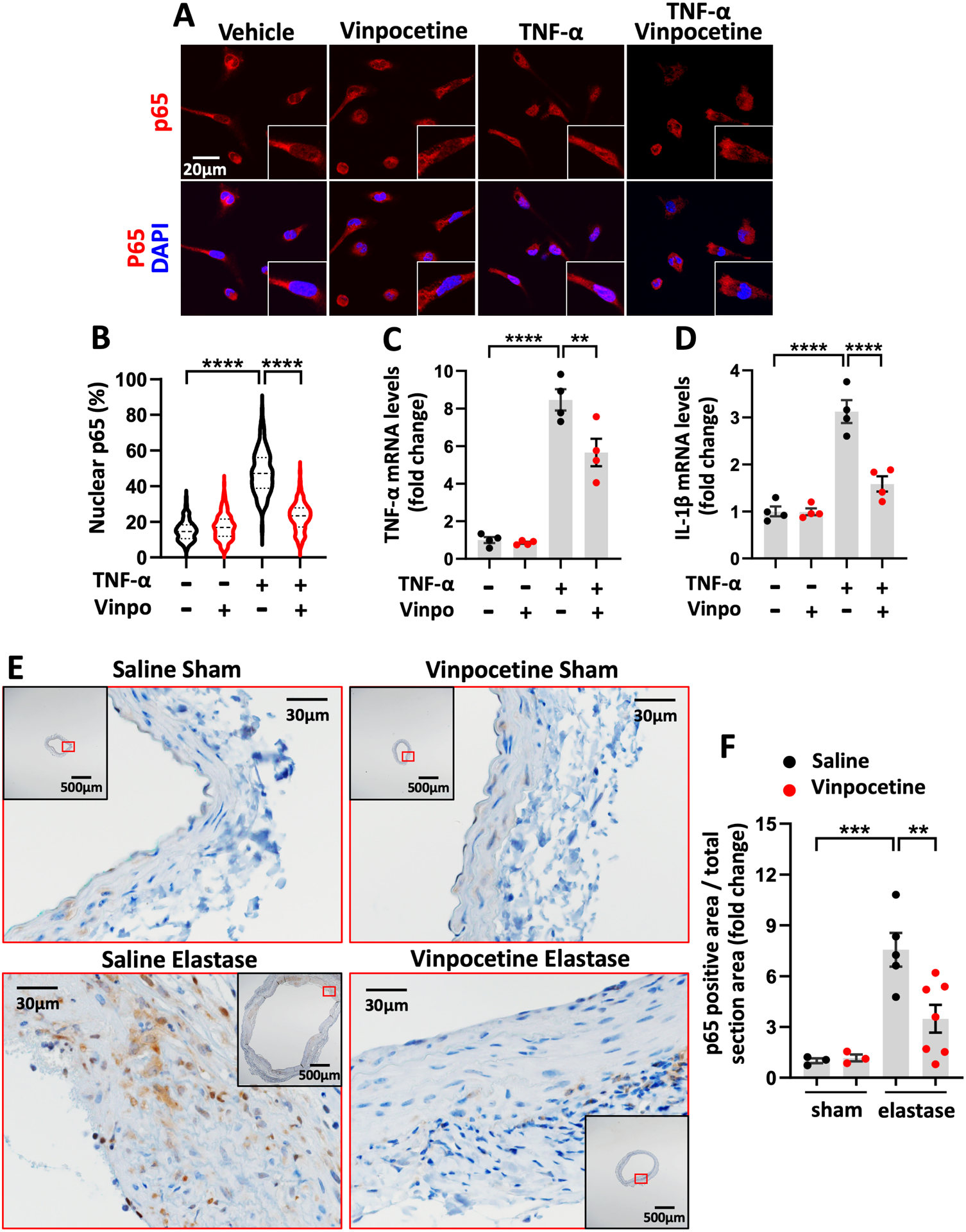
Vinpocetine suppressed NF-κB mediated inflammation in primary macrophages and in AAA. A through D, Mouse resident peritoneal macrophages were starved for 4 hours. Cells were subsequently pretreated with 30uM vinpocetine for 60min before treatment with or without murine TNF-α (10ng/ml) for 30 minutes (A and B) or 6 hours (C and D) in the continued presence or absence of vinpocetine. A, p65 immunocytochemistry staining in macrophages. B, Quantification of p65 subcellular distribution, expressed as the percentage of p65 integrated intensity in nucleus over total cell. At least 200 cells were quantified for each experimental group from 3 separate experiments. C through D, mRNA levels of TNF- α, IL-1β in macrophages by real-time PCR. n=4 separate experiments. E, Immunostaining of p65 on abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples. The inset is the 4x image of the whole section. Image outlined red is the magnification of the area highlighted in red box in inset. F, Percentage of p65 positive staining area in total section area, normalized to the averaged amount of staining in saline/sham control group. Three sections located at 300um intervals from aneurysmal center segment (segment of largest diameter) were analyzed for each animal. Each dot represents one animal. Saline/sham (n=3), vinpocetine/sham (n=3), saline/elastase (n=5), vinpocetine/elastase (n=7). Statistics were performed with parametric Welch ANOVA with Games-Howell post-hoc test (B), parametric one-way ANOVA with Holm-Sidak’s post-hoc test (C, D, F). Data are mean ± SEM. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Statistical analysis
Assumptions of normality and equal variance were tested using R (version 3.6.2, https://www.R-project.org/).31 Shapiro-Wilk test were used to test for normal distribution. Brown-Forsythe test was used to test for equality of variances. Detailed statistical methods were listed in Online Table 2. All tests were two-sided, with a significance level for two-sided tests set at 5%. Statistical analyses and plotting was conducted using GraphPad Prism 8 software. All data are presented as mean ± SEM.
RESULTS
Vinpocetine Pretreatment Reduced AAA Dilatation
To study effects of vinpocetine on AAA development, vinpocetine or saline were provided to C57BL/6 mice starting from 2 days before surgery until the end of study as shown in Fig. 1A. Mice were treated with 5mg/kg vinpocetine daily via intraperitoneal injection (i.p.) as described previously.10, 17, 18 Meanwhile, mice were provided with low dose of BAPN in drinking water (0.2%) daily.20 BAPN is an irreversible inhibitor of lysyl oxidase (LOX) which is critical in maintaining homeostasis of the elastic lamina.32 For the surgery, mice were subjected to periaortic elastase treatment or sham operation in the infrarenal part of the abdominal aorta. Mice were randomly separated into four groups: saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase. Mouse aortas were dissected on the 28th days post-surgery for macroscopic examination. Aneurysm formation was defined as the increase in the external width of the infrarenal aorta by 50% or greater compared to that in the sham group. Compared to saline/sham controls (0.77 ± 0.02mm), elastase induced remarkable dilatation in maximal aortic width (3.50 ± 0.25mm), which was around 355% of increase (Fig. 1B–D). Vinpocetine attenuated AAA dilation (2.49 ± 0.24mm), which had 28.85% of decrease in aortic width compared to the saline AAA group (Fig. 1B–D). Consistent with aortic width data, the AAA lesion area induced by elastase was also significantly reduced by vinpocetine (Fig. 1E). The images of AAA from all animals are shown in Fig. S1.
Figure 1.
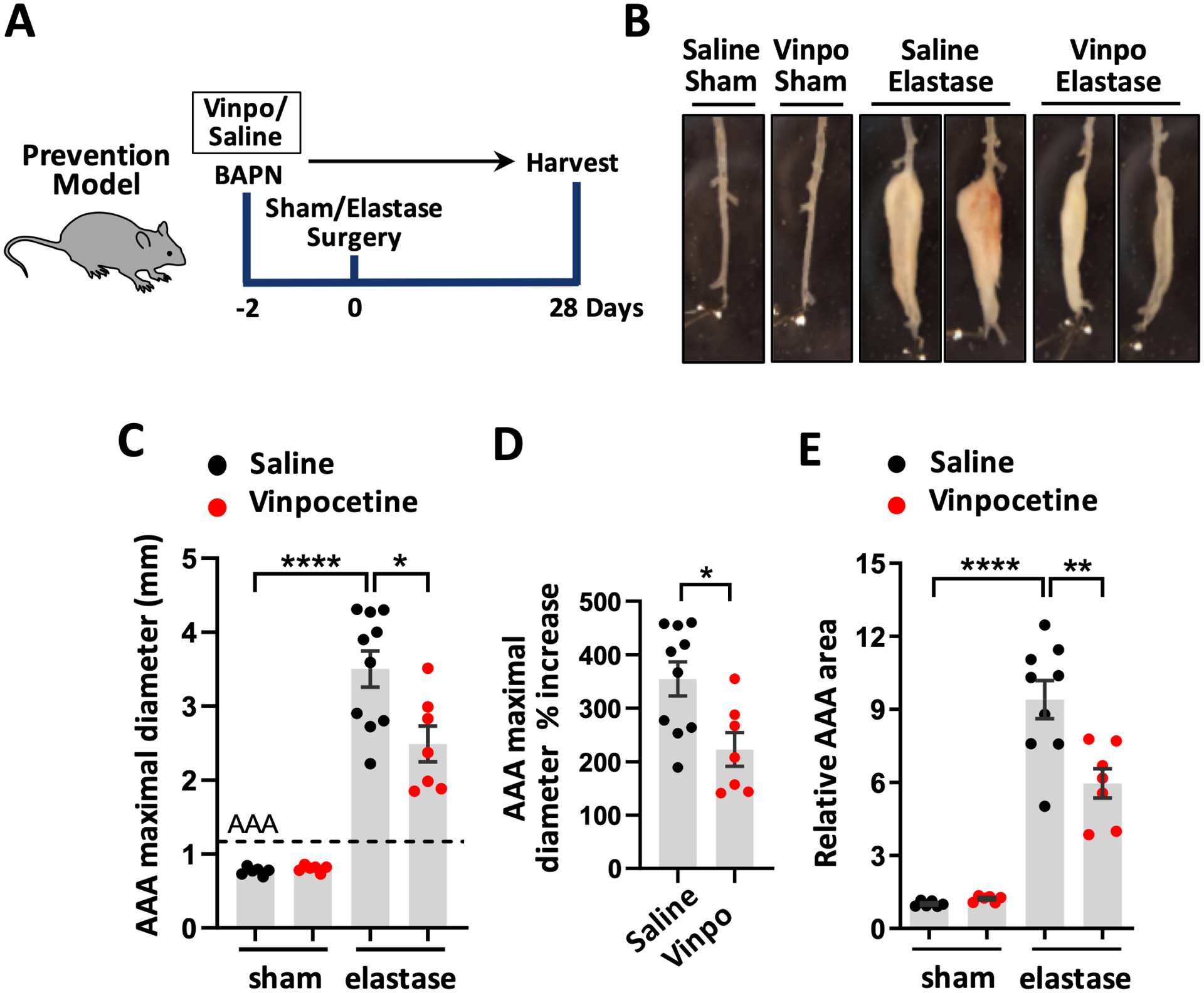
Vinpocetine attenuated AAA Dilatation. A, Experiment design to test the effect of vinpocetine pre-treatment in AAA formation. Vinpocetine (5mg/kg, ip.) or saline were provided to mice starting from 2 days before surgery. Mice were randomly separated into four groups: saline/sham (n=6), vinpocetine/sham (n=6), saline/elastase (n=10), vinpocetine/elastase (n=7). AAAs were harvested on the 28th day post-surgery. B, Representative infrarenal aortas showing the maximal aortic width. C through E, quantification of the maximal aortic width (C), percentage increase of maximal AAA width in saline/elastase group and vinpocetine/elastase group, relative to the averaged aortic width in saline/sham control (D), total aneurysmal area (E). Each dot represents one animal. Vinpo indicates vinpocetine. Dash line indicates 50% increase from the averaged aortic width of saline/sham mice, which defines AAA. Statistics were performed with parametric Welch ANOVA with Dunnett’s T3 post-hoc test (C, E), parametric unpaired Student’s t-test (D). Data are mean ± SEM. *P < 0.05, **P < 0.01, and ****P < 0.0001.
Post-Intervention Treatment of Vinpocetine Limited AAA Expansion
Next, we examined the effect of vinpocetine on the progression and expansion of pre-existing AAAs. As described in Fig. 2A, BAPN drinking water were provided to mice starting from 2 days before surgery until the end of study. Abdominal aorta size were assessed by ultrasound imaging before surgery as baseline. Mice were subjected to surgery with active elastase application. On the 6th day following surgery, compared to the baseline, the diastole internal diameter of aorta by ultrasound imaging exceeded a 50% dilation of normal abdominal aorta diameter, indicating AAA occurrence (Fig. 2B and 2C). These mice were then randomly grouped and treated with vinpocetine or saline daily starting from the 6th day post-surgery till harvest on the 28th days post-surgery. Macroscopic assessment of maximal aortic width and AAA area showed that vinpocetine significantly attenuated AAA progression, though to a lesser extent compared to the prevention model (Fig. 2D–F). The difference in maximal aortic diameter between prevention study (Fig. 1C) and post-intervention study (Fig. 2E) was due to use of two different lots of elastase enzyme. Since there is natural difference in elastase efficacy between different lots, usage of a single lot of elastase within each individual study (prevention or post-intervention) was ensured. All microscopic images of AAA samples are shown in Fig. S2.
Figure 2.
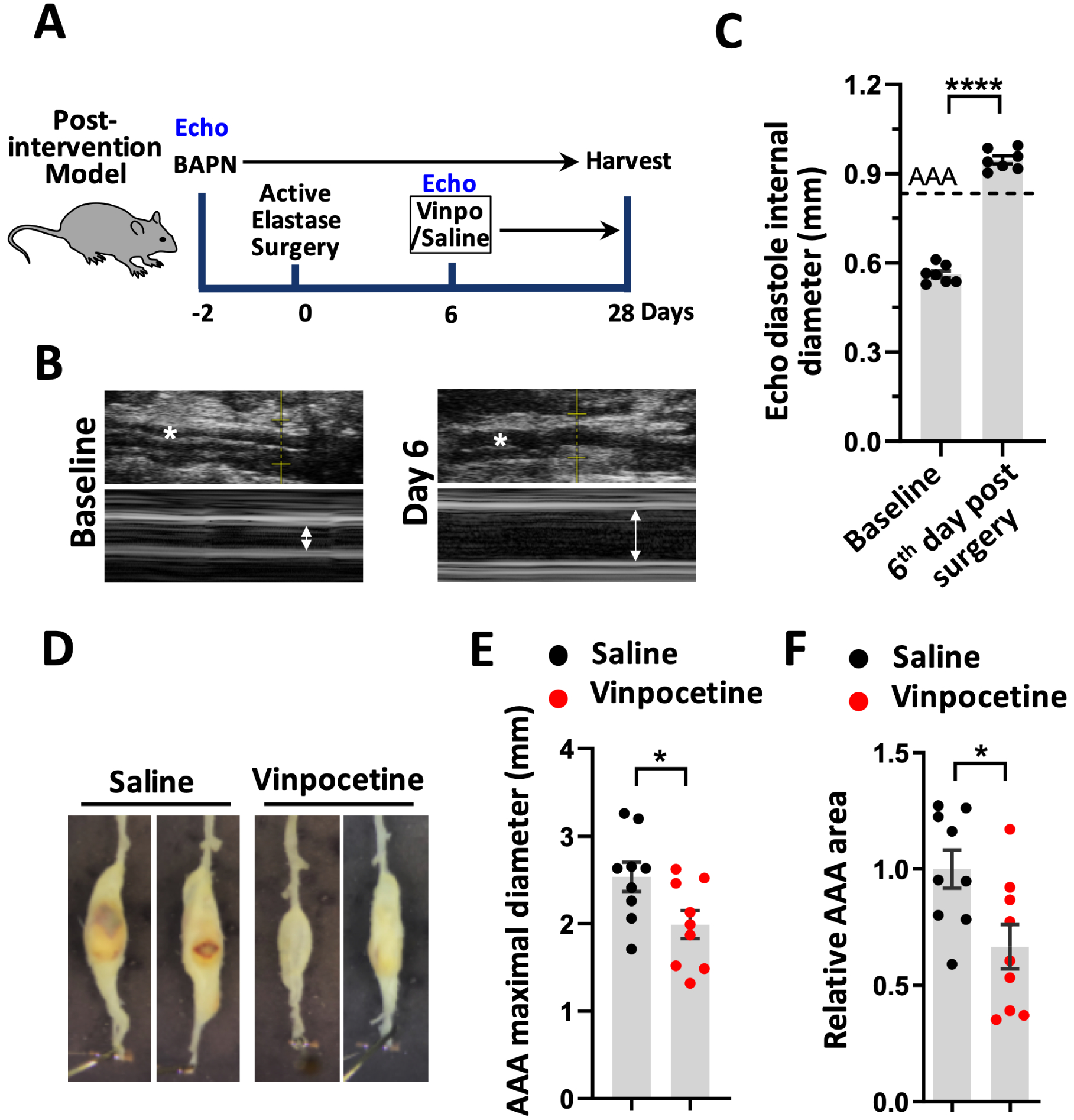
Post-intervention treatment of vinpocetine reduced AAA progression. A, Experiment design of a post-intervention model to test the effect of vinpocetine treatment on the progression and expansion of pre-existing AAA. Ultrasound imaging (echo) were performed to assess abdominal aorta size before surgery as baseline as well as on the 6th day following surgery. These mice were then randomly grouped into two groups: saline/elastase, vinpocetine/elastase. Treatment with vinpocetine (5mg/kg, ip.) or saline daily started from the 6th day post-surgery until harvest. Vinpo indicates vinpocetine. B and C, M-mode (B) and diastole internal diameter (C) of aorta at baseline and on the 6th day following surgery assessed by Echo. * indicates abdominal aorta. Dash line indicates 50% dilation compared to normal abdominal aortic diameter at baseline. n=7 mice. D, Representative infrarenal aortas. E and F, Quantification of the maximal aortic width (E) and total aneurysmal area (F). Each dot represents one animal. Saline/elastase (n=9), vinpocetine/elastase (n=9). Statistics were performed with parametric paired Student’s t-test (C), parametric unpaired Student’s t-test (E, F). Data are expressed as mean ± SEM. *P < 0.05, and ****P < 0.0001.
Vinpocetine Attenuated Elastin Degradation and Media Smooth Muscle Cell Depletion
The extracellular matrix proteins, such as elastic and fibrillar collagen fibers, support the hemodynamic load of the aorta. Progressive degradation of the matrix by proteolysis is considered as the most influential mechanism for AAA expansion. This results in gradual imbalance between the synthesis and degradation of elastin and collagen, leading to permanent diffusive expansion of matrix structure in aneurysmal wall.3 The degradation of elastin in media of aneurysmal wall is thought to be an early step in AAA formation. Dramatic degradation of elastic fibers was observed in elastase induced AAA, while sham groups had no appreciable elastin degradation (Fig. 3A). Much less elastin fragmentation was observed in the vinpocetine/elastase group compared to the saline/elastase group (Fig. 3A). Quantification of elastic fibers content revealed an 81.85% loss of elastin in elastase group, and vinpocetine treated group displayed a lower (54.35%) loss of elastin (Fig. 3B). All AAA samples with staining for elastin that are subjected for quantification are shown in Fig. S3. Hematoxylin and eosin staining of these samples are shown in Fig. S4.
Figure 3.
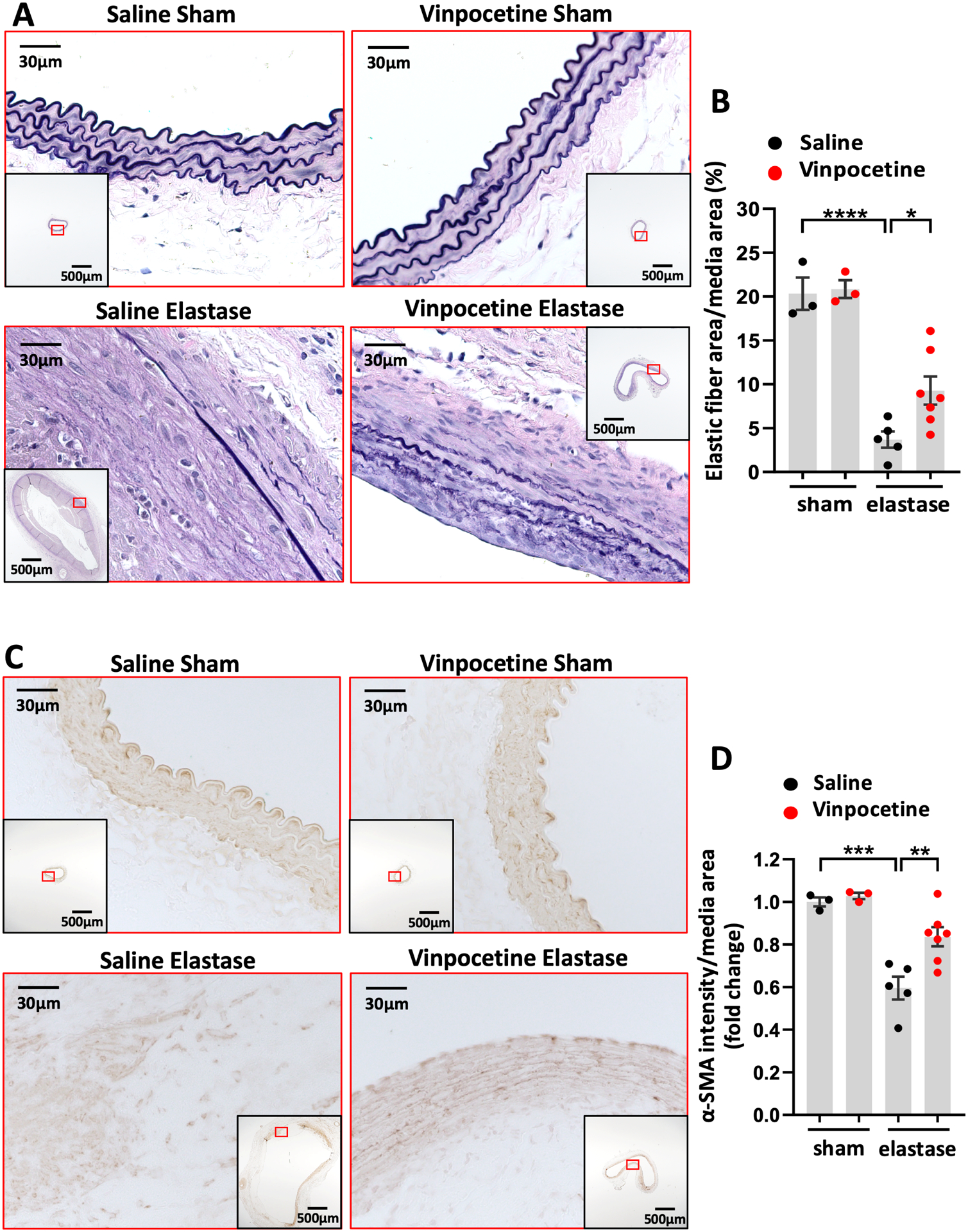
Vinpocetine attenuated elastic fiber degradation and media smooth muscle cell depletion in AAA. A, Van Gieson staining of elastin on abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples. The inset is the 4x image of the whole section. Image outlined red is the magnification of the area highlighted in red box in inset. B, Elastic fiber content in media area. Eight visual fields (magnification 200) of each section were included to quantify the amount of elastin staining in media. C, Immunostaining of α-SMA on abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples. The inset is the 4x image of the whole section. Image outlined red is the magnification of the area highlighted in red box in inset. D, Staining intensity of α-SMA in media, normalized to the averaged amount of staining in saline/sham control group. Three sections located at 300um intervals from aneurysmal center segment (segment of largest diameter) were analyzed for each animal. B and D, Each dot represents one animal. Saline/sham (n=3), vinpocetine/sham (n=3), saline/elastase (n=5), vinpocetine/elastase (n=7). Statistics were performed with parametric one-way ANOVA with Holm-Sidak’s post-hoc test. Data are mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001, and ****P < 0.0001.
SMC are predominant cells constituting the media layer of normal aortic wall. They are important for aorta to respond to the dynamic pressure from the pulsatile blood flow as well as in maintaining the homeostasis of extracellular matrix synthesis and degradation.33 Loss of SMCs in the medial layer is regarded as a key event in the loss of structural integrity in the aneurysmal wall.3, 5 We performed immunostaining of α-smooth muscle actin (α-SMA), a SMC marker, in AAA samples. In elastase induced AAA, SMCs in the media lost their circular and longitudinal layer arrangement. In some regions along the aneurysmal wall, there were complete loss of SMC depicted by media breaks (Fig. 3C). In contrast, the vinpocetine treated group manifested less destruction in the medial SMC layer (Fig. 3C). Quantification of the α-SMA staining intensity in media revealed that vinpocetine protected AAA from a severe medial SMC depletion (Fig. 3D). Negative control of α-SMA immunostaining is shown in Fig. S5. All AAA samples with α-SMA immunostaining included in quantification are shown in Fig. S6.
Vinpocetine Alleviated Collagen Fiber Remodeling and Proteoglycan Accumulation in AAA
Degradation of medial elastin transfers the tensile stress to collagen fibers, therefore they are important for the resistance of aorta in the absence of medial elastin in late stage.33–35 In abdominal aorta of control group, collagen fibers were present in media aligning elastic lamina as well as in adventitia. Elastase induced collagen remodeling in AAA was depicted by breaks of collagen in media and disarranged collagen structure in adventitia (Fig. 4A). Vinpocetine administration relatively preserved collagen structure which resembled the controls (Fig. 4A). All AAA samples subjected to Sirius red/Fast green collagen staining are shown in Fig. S7.
Figure 4.
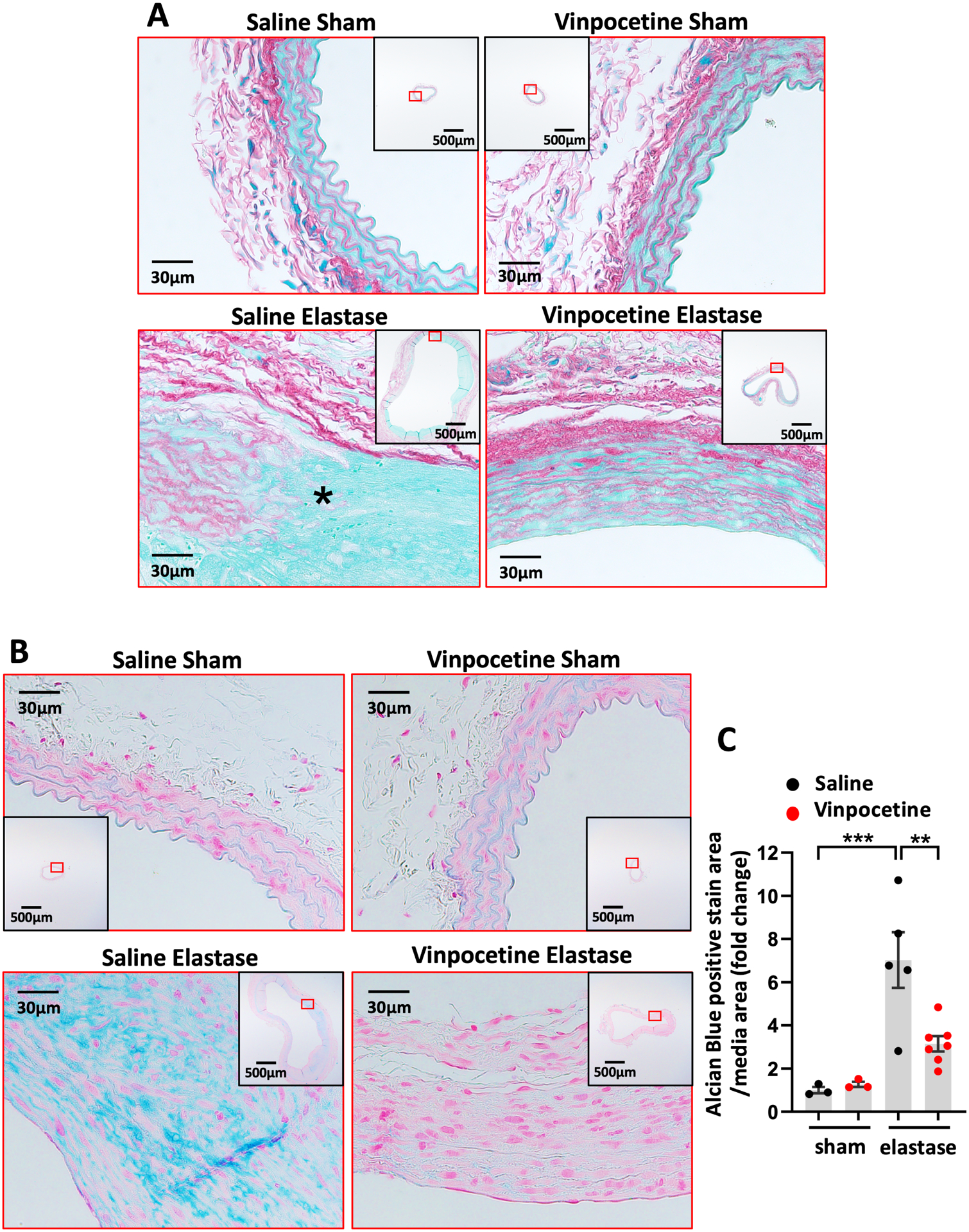
Vinpocetine alleviated collagen fiber remodeling and proteoglycan accumulation in AAA. A, Sirius red/Fast green staining of abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples. * indicates breakage of media collagen fibers. The inset is the 4x image of the whole section. Image outlined red is the magnification of the area highlighted in red box in inset. B, Alcian blue staining of abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples. The inset is the 4x image of the whole section. Image outlined red is the magnification of the area highlighted in red box in inset. C, Relative amount of proteoglycan in media. Eight visual fields (magnification 200) of each section were included to quantify the amount of proteoglycan positive staining (blue). Three sections located at 300um intervals from aneurysmal center segment (segment of largest diameter) were analyzed for each animal. The Alcian Blue positive staining area per media area were normalized to the averaged amount of staining in saline/sham control group. Saline/sham (n=3), vinpocetine/sham (n=3), saline/elastase (n=5), vinpocetine/elastase (n=7). Each dot represents one animal. Statistics were performed with parametric one-way ANOVA with Holm-Sidak’s post-hoc test. Data are mean ± SEM. **P < 0.01, and ***P < 0.001.
Beside elastic and collagen fibers, proteoglycans are also implicated in the organization of aortic wall.34 Proteoglycan accumulation is a hallmark of medial degeneration and has been observed in abdominal and thoracic aneurysms.36–38 Pathological accumulation of proteoglycans in aortic wall may generate an interstitial swelling pressure that deleteriously impact extracellular matrix hemostasis and mechanosensing of SMC.39, 40 We therefore performed Alcian blue staining to assess proteoglycan accumulation in AAA tissues. In contrast to modest intralamellar staining in control tissues, we observed an increased level of proteoglycans in aneurysmal media with degenerative lesions as well as some distorted proteoglycan configuration (Fig. 4B). Vinpocetine treatment ameliorated the accumulation of proteoglycans in media and preserved its structure (Fig. 4B and 4C). All AAA samples with Alcian blue staining included in quantification are shown in Fig. S8.
Vinpocetine Inhibited Macrophage Infiltration in AAA
Numerous studies in animals and humans indicate that inflammation is an important hallmark in AAA development.1 Macrophages are the major cells mediate transmural inflammation in damaged AAA wall.41 Vinpocetine has been shown to be an potent anti-inflammatory agent due to its ability to inhibit NF-κB signaling10, therefore we have focused on studying the effect of vinpocetine on macrophage inflammation in AAA in this study. We examined macrophage infiltration in AAA by staining F4/80, a specific macrophage marker.42 In elastase induced AAA, macrophage infiltration was drastically observed in aortic wall, which were significantly decreased by vinpocetine treatment (Fig. 5A and 5B). Negative control of F4/80 immunostaining is shown in Fig. S9. All AAA samples with F4/80 immunostaining included in quantification are shown in Fig. S10.
Figure 5.
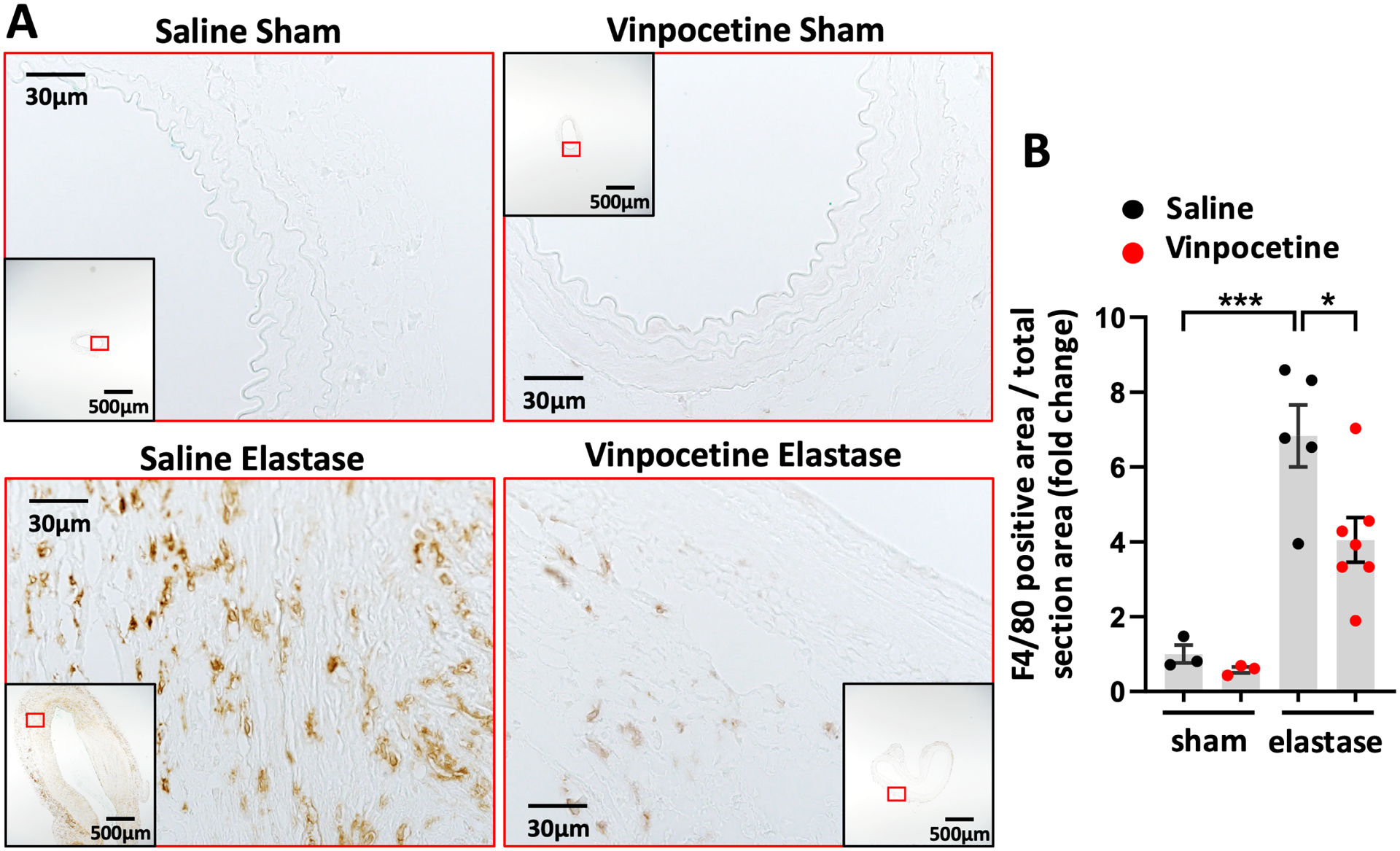
Vinpocetine inhibited macrophage infiltration in AAA. A, Immunostaining of F4/80 on abdominal aortic cross sections from saline/sham, vinpocetine/sham, saline/elastase, vinpocetine/elastase samples. The inset is the 4x image of the whole section. Image outlined red is the magnification of the area highlighted in red box in inset. B, Percentage of F4/80 positive staining area in total section area, normalized to the averaged amount of staining in saline/sham control group. Three sections located at 300um intervals from aneurysmal center segment (segment of largest diameter) were analyzed for each animal. Each dot represents one animal. Saline/sham (n=3), vinpocetine/sham (n=3), saline/elastase (n=5), vinpocetine/elastase (n=7). Statistics were performed with parametric one-way ANOVA with Holm-Sidak’s post-hoc test. Data are mean ± SEM. *P < 0.05, ***P < 0.001.
Vinpocetine Suppressed NF-κB Mediated Inflammation in Macrophages and in AAA
Vinpocetine has been previously found to inhibit NF-κB dependent transcription activity of proinflammatory mediators in rat aortic smooth muscle cells, mouse macrophage cell line and human monocyte lymphoma cell line.10, 19 In this study, we used primary mouse resident peritoneal macrophages and treated cells with TNF-α as one representative inflammatory stimulus. TNF-α can be synthesized by activated macrophages and play autocrine and paracrine effects. TNF-α cytokine level has been reported to be increased after surgery in perivascular elastase induced AAA in mice.20 In our hand, TNF-α mRNA level was also increased in elastase-induced AAA (Fig. S11). We found that vinpocetine treatment of 30 minutes suppressed TNF-α induced nuclear translocation of p65 in macrophages (Fig. 6A and 6B), a critical step in the activation of NF-kB- dependent transcription. Negative control of p65 immunocytochemistry is shown in Fig. S12. In addition, Vinpocetine treatment for 6 hours reduced the proinflammatory molecule expression, such as TNF-α and IL-1β at the mRNA level (Fig. 6C and 6D). Cell viability analysis showed that vinpocetine did not have a significant effect on cell viability (Fig. S13). Consistently, immunostaining of p65 in elastase induced AAA showed evident nuclear localization and increased protein expression, which were ameliorated by vinpocetine treatment (Fig. 6E and 6F). Negative control of p65 immunohistochemistry is shown in Fig. S14. Using Mac2 as a macrophage marker, double immunofluorescence staining of p65 and Mac2 were performed in AAA samples. Fig. S15A shows cross sections from the same level used for p65 immunohistochemistry (Fig. 6E) at the same area. There are fair amounts of p65 positive macrophages in elastase induced AAA, whereas vinpocetine treatment decreased the co-staining of p65 and Mac2, suggesting that vinpocetine at least partially attenuated NF-kB activation in macrophages of aneurysmal tissues. Here we chose to stain macrophages with Mac2 antibody instead of F4/80 was because the host species of p65 primary antibody is the same as F4/80 (rabbit), whereas different from Mac2 (rat). Negative control of p65 and Mac2 immunostaining is shown in Fig. S15B.
DISCUSSION
In the present study, we demonstrated that vinpocetine, when applied systematically, reduced periaortic elastase induced AAA dilatation in mice in vivo. Notably, vinpocetine was also able to attenuate the progression of existing moderate AAA. This protective effect was associated with alleviated degradation and destruction of matrix fibers and less medial SMC depletion. Consistent with the anti-inflammatory effects of vinpocetine in previously reported models10, 12–16, 19, we also found that vinpocetine suppresses inflammatory responses in the aneurysmal tissues, including reduced macrophage contents and nuclear NF-kB levels. In primary cultured peritoneal macrophages, vinpocetine similarly reduced NF-kB nuclear translocation and inflammatory molecule expression. Thus, this anti-inflammatory mechanism of action at least partially contributes to the anti-AAA effect in vivo. Given the excellent safety profile of vinpocetine for long-term use, our findings suggest that vinpocetine may represent as a novel and attractive therapeutic agent for AAA.
There are several different mouse models that have been widely used to study AAA pathogenesis, including subcutaneous Angiotensin II infusion via the osmotic mini-pump, intraluminal elastase perfusion in abdominal aorta, and periaortic treatment with elastase or calcium chloride.1, 43, 44 The AAA induced by the periaortic elastase in combination with BAPN treatment is a reliable animal model that provides a stable, advanced-stage infrarenal AAA.20 It undergoes progressive expansion of the infrarenal aorta for up to 14 weeks after initial induction.20 In particular, the reproducibility and consistency of this model is advantageous for studying drug efficacy. This model has been reported to resemble many features of human AAA, including long term progressive expansion of aneurysm, elastin fragmentation and degradation, loss of media SMC, elevation of MMPs proteolytic capacity, influx of a range of inflammatory cells to the aorta, intraluminal thrombus formation and aortic rupture.20, 45, 46 We indeed observed remarkable breaks and degradation of elastic fibers in media (Fig. 3A), depletion of medial SMCs (Fig. 3C), collagen structure remodeling (Fig. 4A) and macrophage infiltration (Fig. 5). Despite the technical advantage and similarity of this model to human AAA, validation of the effect of vinpocetine in other AAA animal models will be also of great interest.
Of the known mechanisms, the anti-inflammatory function of vinpocetine may largely contribute to its inhibitory role in AAA growth. Human and animal AAA studies have both demonstrated that chronic inflammation is a key factor in AAA pathology.41 Initial arterial injury may produce elastin breakdown products, that recruit macrophages to the injury site.47–50 Recruited macrophages get activated and produce more inflammatory mediators, such as TNF-α, IL-1β, which if left uncontrolled, turning on an autoregulatory loop to enhance ongoing local inflammation.41, 51 Chronic inflammation in AAA represents an unresolved inflammation over time. Vinpocetine has been shown to attenuate NF-κB–dependent inflammatory response using macrophage cell lines in vitro as well as in various inflammatory animal models in vivo.10, 13, 19, 52 The present study also revealed similar mechanism of action of vinpocetine in primary macrophages as well as observed less macrophage infiltration and NF-κB activation in AAA tissues. In this regard, vinpocetine may combat AAA by limiting initial inflammatory insult and benefit chronic inflammation resolution. In addition to macrophages, vinpocetine has been previously reported to inhibit NF-κB mediated inflammation in vascular smooth muscle cells and endothelial cells, therefore it is possible that vinpocetine might also act to against inflammation of other vascular cell types in AAAs.
CONCLUSIONS
This study demonstrates that vinpocetine pretreatment or post-intervention reduced AAA diameter and volume in an experimental mouse model induced by periaortic elastase application. Vinpocetine improved multiple cellular and molecular changes associated with AAA, such as elastin degradation, media smooth muscle cell depletion, collagen fibers remodeling and macrophage infiltration in aneurysmal tissues. Mechanistically, vinpocetine potently suppressed NF-κB-dependent inflammation in primary macrophage and diminished inflammatory response in the aorta wall in vivo, suggesting vinpocetine protects against AAA via anti-inflammatory effect.
Supplementary Material
Clinical Perspectives.
A current challenge is the lack of effective pharmacological therapeutics to prevent abdominal aortic aneurysm (AAA) progression. AAA is a degenerative vascular disease that is more frequently associated with aged male population. Vinpocetine has long been used to treat cognitive impairment in aged population. Therefore, it is of great interest and significance to demonstrate the effect of vinpocetine on AAA development and intervention.
Vinpocetine pretreatment or post-intervention reduced AAA diameter and volume in an experimental mouse model induced by periaortic elastase application. This occurred in association with improved cellular and molecular changes, such as elastin degradation, media smooth muscle cell depletion and macrophage infiltration in aneurysmal tissues. Its mechanism was related to inhibition of NF-κB-dependent inflammation in primary macrophage in vitro and diminished inflammatory response in the aorta wall in vivo.
The findings suggest that vinpocetine may be a novel therapeutic agent for AAA prevention and treatment.
Funding
This work was funded by National Institute of Health (NIH) HL134910 and HL088400 (to C. Y.). and American Heart Association 20PRE35210148 (to C.Z.).
Abbreviations
- AAA
abdominal aortic aneurysm
- SMCs
smooth muscle cells
- PDE1C
phosphodiesterase 1
- IKK
IkB kinase
- BAPN
3-aminopropionitrile fumarate salt
- LOX
lysyl oxidase
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- p65
transcription factor p65
- TNF-α
tumor necrosis factor alpha
- IL-1β
interleukin 1 beta
Footnotes
Data Availability Statement
Upon manuscript acceptance, the data generated during the current study will be deposited to Figshare repository.
REFERENCE
- 1.Golledge J Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2019;16(4):225–42. [DOI] [PubMed] [Google Scholar]
- 2.Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8(2):92–102. [DOI] [PubMed] [Google Scholar]
- 3.Sakalihasan N, Michel JB, Katsargyris A, Kuivaniemi H, Defraigne JO, Nchimi A, et al. Abdominal aortic aneurysms. Nat Rev Dis Primers. 2018;4(1):34. [DOI] [PubMed] [Google Scholar]
- 4.Davis FM, Daugherty A, Lu HS. Updates of Recent Aortic Aneurysm Research. Arterioscler Thromb Vasc Biol. 2019;39(3):e83–e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuivaniemi H, Ryer EJ, Elmore JR, Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13(9):975–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang YS, Li JD, Yan C. An update on vinpocetine: New discoveries and clinical implications. Eur J Pharmacol. 2018;819:30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang C, Yan C. Updates of Recent Vinpocetine Research in Treating Cardiovascular Diseases. J Cell Immunol. 2020;2(5):211–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Bagoly E, Feher G, Szapary L. [The role of vinpocetine in the treatment of cerebrovascular diseases based in human studies]. Orv Hetil. 2007;148(29):1353–8. [DOI] [PubMed] [Google Scholar]
- 9.Bonoczk P, Gulyas B, Adam-Vizi V, Nemes A, Karpati E, Kiss B, et al. Role of sodium channel inhibition in neuroprotection: effect of vinpocetine. Brain Res Bull. 2000;53(3):245–54. [DOI] [PubMed] [Google Scholar]
- 10.Jeon KI, Xu X, Aizawa T, Lim JH, Jono H, Kwon DS, et al. Vinpocetine inhibits NF- B-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proceedings of the National Academy of Sciences. 2010;107(21):9795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;1999(5):RE1. [DOI] [PubMed] [Google Scholar]
- 12.Liu RT, Wang A, To E, Gao J, Cao S, Cui JZ, et al. Vinpocetine inhibits amyloid-beta induced activation of NF-kappaB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp Eye Res. 2014;127:49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz-Miyazawa KW, Pinho-Ribeiro FA, Zarpelon AC, Staurengo-Ferrari L, Silva RL, Alves-Filho JC, et al. Vinpocetine reduces lipopolysaccharide-induced inflammatory pain and neutrophil recruitment in mice by targeting oxidative stress, cytokines and NF-kappaB. Chem Biol Interact. 2015;237:9–17. [DOI] [PubMed] [Google Scholar]
- 14.Lourenco-Gonzalez Y, Fattori V, Domiciano TP, Rossaneis AC, Borghi SM, Zaninelli TH, et al. Repurposing of the Nootropic Drug Vinpocetine as an Analgesic and Anti-Inflammatory Agent: Evidence in a Mouse Model of Superoxide Anion-Triggered Inflammation. Mediators Inflamm. 2019;2019:6481812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komatsu K, Nam DH, Lee JY, Yoneda G, Yan C, Li JD. Vinpocetine Suppresses Streptococcus pneumoniae-Induced Inflammation via Inhibition of ERK1 by CYLD. J Immunol. 2020;204(4):933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fattori V, Borghi SM, Guazelli CFS, Giroldo AC, Crespigio J, Bussmann AJC, et al. Vinpocetine reduces diclofenac-induced acute kidney injury through inhibition of oxidative stress, apoptosis, cytokine production, and NF-kappaB activation in mice. Pharmacol Res. 2017;120:10–22. [DOI] [PubMed] [Google Scholar]
- 17.Cai Y, Knight WE, Guo S, Li JD, Knight PA, Yan C. Vinpocetine suppresses pathological vascular remodeling by inhibiting vascular smooth muscle cell proliferation and migration. J Pharmacol Exp Ther. 2012;343(2):479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai Y, Li JD, Yan C. Vinpocetine attenuates lipid accumulation and atherosclerosis formation. Biochem Biophys Res Commun. 2013;434(3):439–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuang J, Peng W, Li H, Lu Y, Wang K, Fan F, et al. Inhibitory effects of vinpocetine on the progression of atherosclerosis are mediated by Akt/NF-kappaB dependent mechanisms in apoE−/− mice. PLoS One. 2013;8(12):e82509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu G, Su G, Davis JP, Schaheen B, Downs E, Roy RJ, et al. A novel chronic advanced stage abdominal aortic aneurysm murine model. J Vasc Surg. 2017;66(1):232–42 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hannawa KK, Eliason JL, Upchurch GR Jr. Gender differences in abdominal aortic aneurysms. Vascular. 2009;17 Suppl 1:S30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ Res. 2016;119(10):1076–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng J, Koenig SN, Kuivaniemi HS, Garg V, Hans CP. Pharmacological inhibitor of notch signaling stabilizes the progression of small abdominal aortic aneurysm in a mouse model. J Am Heart Assoc. 2014;3(6):e001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiraya S, Miyake T, Aoki M, Yoshikazu F, Ohgi S, Nishimura M, et al. Inhibition of development of experimental aortic abdominal aneurysm in rat model by atorvastatin through inhibition of macrophage migration. Atherosclerosis. 2009;202(1):34–40. [DOI] [PubMed] [Google Scholar]
- 25.Segnani C, Ippolito C, Antonioli L, Pellegrini C, Blandizzi C, Dolfi A, et al. Histochemical Detection of Collagen Fibers by Sirius Red/Fast Green Is More Sensitive than van Gieson or Sirius Red Alone in Normal and Inflamed Rat Colon. PLoS One. 2015;10(12):e0144630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osada H, Kyogoku M, Matsuo T, Kanemitsu N. Histopathological evaluation of aortic dissection: a comparison of congenital versus acquired aortic wall weakness. Interact Cardiovasc Thorac Surg. 2018;27(2):277–83. [DOI] [PubMed] [Google Scholar]
- 27.ImageJ. Plugins 2020. [cited 2020]; Available from: https://imagej.nih.gov/ij/plugins/index.html.
- 28.Mukhopadhyay S, Pluddemann A, Hoe JC, Williams KJ, Varin A, Makepeace K, et al. Immune inhibitory ligand CD200 induction by TLRs and NLRs limits macrophage activation to protect the host from meningococcal septicemia. Cell Host Microbe. 2010;8(3):236–47. [DOI] [PubMed] [Google Scholar]
- 29.Davies JQ, Gordon S. Isolation and culture of murine macrophages. Methods Mol Biol. 2005;290:91–103. [DOI] [PubMed] [Google Scholar]
- 30.Ingles-Esteve J, Morales M, Dalmases A, Garcia-Carbonell R, Jene-Sanz A, Lopez-Bigas N, et al. Inhibition of specific NF-kappaB activity contributes to the tumor suppressor function of 14-3-3sigma in breast cancer. PLoS One. 2012;7(5):e38347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2019. [Google Scholar]
- 32.Siegel RC, Martin GR. Collagen cross-linking. Enzymatic synthesis of lysine-derived aldehydes and the production of cross-linked components. J Biol Chem. 1970;245(7):1653–8. [PubMed] [Google Scholar]
- 33.Thompson RW. Reflections on the pathogenesis of abdominal aortic aneurysms. Cardiovasc Surg. 2002;10(4):389–94. [DOI] [PubMed] [Google Scholar]
- 34.Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. The Lancet. 2005;365(9470):1577–89. [DOI] [PubMed] [Google Scholar]
- 35.Dobrin PB, Mrkvicka R. Failure of elastin or collagen as possible critical connective tissue alterations underlying aneurysmal dilatation. Cardiovasc Surg. 1994;2(4):484–8. [PubMed] [Google Scholar]
- 36.Cikach FS, Koch CD, Mead TJ, Galatioto J, Willard BB, Emerton KB, et al. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight. 2018;3(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schriefl AJ, Collins MJ, Pierce DM, Holzapfel GA, Niklason LE, Humphrey JD. Remodeling of intramural thrombus and collagen in an Ang-II infusion ApoE−/− model of dissecting aortic aneurysms. Thromb Res. 2012;130(3):e139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melrose J, Whitelock J, Xu Q, Ghosh P. Pathogenesis of abdominal aortic aneurysms: possible role of differential production of proteoglycans by smooth muscle cells. J Vasc Surg. 1998;28(4):676–86. [DOI] [PubMed] [Google Scholar]
- 39.Roccabianca S, Ateshian GA, Humphrey JD. Biomechanical roles of medial pooling of glycosaminoglycans in thoracic aortic dissection. Biomech Model Mechanobiol. 2014;13(1):13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roccabianca S, Bellini C, Humphrey JD. Computational modelling suggests good, bad and ugly roles of glycosaminoglycans in arterial wall mechanics and mechanobiology. J R Soc Interface. 2014;11(97):20140397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol. 2015;35(8):1746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue T, Plieth D, Venkov CD, Xu C, Neilson EG. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int. 2005;67(6):2488–93. [DOI] [PubMed] [Google Scholar]
- 43.Senemaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB, Coscas R. Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2017;37(3):401–10. [DOI] [PubMed] [Google Scholar]
- 44.Lysgaard Poulsen J, Stubbe J, Lindholt JS. Animal Models Used to Explore Abdominal Aortic Aneurysms: A Systematic Review. Eur J Vasc Endovasc Surg. 2016;52(4):487–99. [DOI] [PubMed] [Google Scholar]
- 45.Romary DJ, Berman AG, Goergen CJ. High-frequency murine ultrasound provides enhanced metrics of BAPN-induced AAA growth. Am J Physiol Heart Circ Physiol. 2019;317(5):H981–H90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peshkova IO, Aghayev T, Fatkhullina AR, Makhov P, Titerina EK, Eguchi S, et al. IL-27 receptor-regulated stress myelopoiesis drives abdominal aortic aneurysm development. Nat Commun. 2019;10(1):5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hance KA, Tataria M, Ziporin SJ, Lee JK, Thompson RW. Monocyte chemotactic activity in human abdominal aortic aneurysms: role of elastin degradation peptides and the 67-kD cell surface elastin receptor. J Vasc Surg. 2002;35(2):254–61. [DOI] [PubMed] [Google Scholar]
- 48.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, et al. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116(3):753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo G, Booms P, Halushka M, Dietz HC, Ney A, Stricker S, et al. Induction of macrophage chemotaxis by aortic extracts of the mgR Marfan mouse model and a GxxPG-containing fibrillin-1 fragment. Circulation. 2006;114(17):1855–62. [DOI] [PubMed] [Google Scholar]
- 50.Guo G, Munoz-Garcia B, Ott CE, Grunhagen J, Mousa SA, Pletschacher A, et al. Antagonism of GxxPG fragments ameliorates manifestations of aortic disease in Marfan syndrome mice. Hum Mol Genet. 2013;22(3):433–43. [DOI] [PubMed] [Google Scholar]
- 51.Mechanisms Nathan C. and modulation of macrophage activation. Behring Inst Mitt. 1991(88):200–7. [PubMed] [Google Scholar]
- 52.Wang H, Zhang K, Zhao L, Tang J, Gao L, Wei Z. Anti-inflammatory effects of vinpocetine on the functional expression of nuclear factor-kappa B and tumor necrosis factor-alpha in a rat model of cerebral ischemia-reperfusion injury. Neurosci Lett. 2014;566:247–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.