

Summary
N6-methyladenosine (m6A) is the most abundant mRNA modification, which is installed by the METTL3-METTL14-WTAP methyltransferase complex. Although the importance of m6A methylation in mRNA dynamics has been well-documented recently, the regulation of m6A machinery remains obscure. Through a genome-wide CRISPR screen, we identify the ERK pathway and USP5 as positive regulators of the m6A pathway. We find that ERK phosphorylates METTL3 at S43/S50/S525 and WTAP at S306/S341, followed by deubiquitination by USP5, resulting in stabilization of the m6A methyltransferase complex. Lack of METTL3/WTAP phosphorylation reduces decay of m6A-labeled pluripotent factors and traps mouse embryonic stem cells in the pluripotent state. The same phosphorylation can also be found in ERK-activated human cancer cells and contribute to tumorigenesis. Overall, our study reveals an unrecognized function of ERK in regulating m6A methylation.
Keywords: ERK, METTL3, WTAP, m6A, USP5
Graphical Abstract
eTOC Blurb
Sun et al. demonstrate that activation of the ERK pathway promotes m6A methylation by phosphorylation of METTL3 and WTAP. Phosphorylation of METTL3 increases interaction with USP5, thereby decreasing ubiquitination to stabilize the m6A methyltransferase complex. Activation of the ERK-METTL3/WTAP signaling axis promotes stem cell differentiation and tumorigenesis.
Introduction
Recent studies have shown that messenger RNA (mRNA) modifications play a critical role in regulating biological and pathological processes (Frye et al., 2018). Among over 150 known RNA modifications, N6-methyladenosine (m6A) is an evolutionarily conserved and the most abundant internal mRNA modification in eukaryotic mRNA. m6A is reversibly, site-selectively installed on mRNA transcripts by “writers,” with a portion that can be removed by “erasers”. The m6A methylation is mediated by a core complex of three components: METTL3, METL14, and WTAP (Liu et al., 2014). The crystal structure of the METTL3 and METTL14 complex suggests that METTL3 is the catalytic component while METTL14 contributes to substrate RNA binding. WTAP, on the other hand, recruits METTL3 and METTL14 to nuclear speckles (Ping et al., 2014; Wang et al., 2014b). Meanwhile, “eraser” proteins FTO and ALKBH5 remove m6A modification (Jia et al., 2011; Zheng et al., 2013).
Increasing evidence suggests that the m6A modification is involved in the regulation of RNA splicing, localization, stability, and translation (Shi et al., 2017; Wang et al., 2014a; Wang et al., 2015; Xiao et al., 2016; Zhou et al., 2019). It has been found that m6A affects numerous physiological and pathological processes. For example, loss of METTL3 in mouse embryonic cells (mESCs) depletes m6A and increases stability of certain transcripts such as Nanog (Batista et al., 2014; Wang et al., 2014b). This impedes decay of pluripotency factors, thereby delaying proper lineage priming and fate transition, leading to early embryo lethality (Geula et al., 2015). Furthermore, METTL3 knockdown is known to induce apoptosis (Dominissini et al., 2012) and METTL3 overexpression can promote tumorigenesis in multiple cancer types (Barbieri et al., 2017; Cai et al., 2018; Chen et al., 2018; Cheng et al., 2019; Dahal et al., 2019; Hua et al., 2018; Li et al., 2019; Lin et al., 2016; Liu et al., 2019; Miao et al., 2019; Taketo et al., 2018; Visvanathan et al., 2018; Vu et al., 2017).
The importance of m6A methylation has been well described, yet gaps in our understanding of how this process is regulated remain. We therefore used a genome-wide CRISPR screen to identify regulators of m6A methylation. The biological importance of these regulations was further studied in mESCs and relevant cancer cells.
Results:
ERK Activation Promotes mRNA m6A Methylation
To identify regulators of m6A RNA methylation, we employed a circular RNA GFP reporter containing a consensus GGACU motif in HeLa cells. The GFP pre-mRNA transcript is assembled by back-splicing to generate a circular RNA that joins two exon fragments of GFP, as depicted in Figure 1A. m6A methylation of the GGACU motifs on the circular RNA can drive translation initiation of the GFP transcript, producing GFP fluorescence signal. Consequently, the GFP signal from this circular RNA reporter can be used as a readout of m6A methylation. Indeed, consistent with a previous report (Yang et al., 2017), circRNA containing GGACT was translated into GFP, whereas mutation to GGTCT reduced GFP levels. Co-expression of METTL3 increased GFP expression from the GGACT reporter but not the GGTCT control (Figure S1A). Furthermore, signal from the GGACT reporter was decreased by catalytic mutant D395A METTL3, as well as siMETTL3, siMETTL14, and siWTAP, but was increased by siFTO and siALKBH5 (Figure S1A).
Figure 1. ERK Activation Promotes mRNA m6A Methylation.

(A) Schematic diagram of a circular RNA (circRNA) translation reporter consisting of a single exon and two introns with complementary sequences. The exon containing GGACU can be back-spliced to generate circRNAs that drive GFP translation.
(B) Overview of the CRISPR screen. Cas9 KO libraries are packaged into lentivirus and then transduced into HeLa cells contain circRNA GFP reporters. Cells with the top and bottom 5% GFP expression were collected by flow cytometry. The sgRNA were amplified from genomic DNA and then sequenced, followed by statistical analyses to identify candidate genes.
(C) Positive regulators for the m6A pathway identified in the screen using circular GGACU-GFP reporters.
(D) Pathway analysis of sgRNA enriched in the bottom 5% GFP cells with circRNA GFP reporters.
Next, we performed a CRISPR knockout-based genomic screen targeting 19,050 genes and 1,864 miRNA (Joung et al., 2017). Combining a CRISPR knockout library with a circular RNA m6A-GFP reporter allowed us to screen for possible regulators of m6A methylation (Figure 1B). Knockout of genes that promote or suppress m6A methylation would decrease or increase translation of the GFP transcript, respectively. Cells with the top and bottom 5% of GFP expression were therefore collected, followed by high-throughput sequencing in order to identify negative and positive regulators of m6A methylation, respectively. We compared the genes that were enriched in the low-GFP-expressing and the high-GFP-expression populations (Table S1). As expected, knockout of METTL3 led to low GFP signal in the screen (Figure 1C). Interestingly, pathway enrichment analysis of the gRNAs in low-GFP-expressing cells identified genes involved in the RAS and MAPK signaling pathways (Figure 1C–D and Table S2). Several hits which are known to activate ERK, including SHC1, CDC42, MAP3K1, PTPN11, and GRB2, decreased GFP signal from the GGACT reporter significantly more than the control GGTCT reporter (Figure S1B).
To determine how the RAS/MAPK pathway affects m6A methylation, we investigated the status of the m6A methyltransferase complex during MAPK pathway activation. A phos-tag gel (Kinoshita et al., 2006) revealed that constitutively active MEK S218D/S222D, BRAF V600E, or HER2 V659E, increased the phosphorylation-dependent mobility shift of METTL3 and WTAP, but not METTL14 (Figure S1C). We further co-transfected a panel of 13 oncogenic kinases, including ATM, ATR, IKK-α, IKK-β, IKK-ε, AKT, GSK-3β, mTOR, MEK, CDC2, FAK, EGFR, and HER2 with METTL3 in 293T cells. As shown in Figure S1D, MEK and HER2, which activate ERK, induced the most significant phosphorylation-dependent mobility shift of METTL3. We also employed NANOG 3’ UTR, which contains three m6A consensus RRACT motif sites that mediate the methylation-dependent decay of NANOG (Zhang et al., 2016), as a readout of the cellular m6A methylation activity. Mutation of the adenosine residue (AAACT to AATCT, and GGACT to GGTCT) resulted in increased luciferase activity, suggesting that the mutation prevented methylation and thereby increased the stability of the luciferase-NANOG 3’-UTR fusion mRNA. We noticed that overexpression of the m6A methyltransferase complex (METTL3-METTL14-WTAP) decreased wild type RRACT but not mutant RRTCT reporter expression. Overexpression of the ERK activators alone decreased WT NANOG 3’UTR reporter expression, and the effect was enhanced with the m6A methyltransferase complex (Figure S1E). Together, our results show that the activation of MAPK pathway promotes mRNA m6A methylation.
ERK Phosphorylates METTL3 and WTAP
To determine how ERK activates m6A methylation, we first tested whether ERK interacts with the mRNA m6A methyltransferase complex. Co-immunoprecipitation showed that METTL3 associates with ERK1 and ERK2 upon BRAF transfection (Figure S2A). Considering that ERK1 and ERK2 are highly similar and possess identical substrate specificity in vitro, we focused on ERK2 hereafter because ERK2 expression exceeds ERK1 in most cells. The interaction between ERK2 and WTAP was also observed after RAF activation (Figure S2B). The interaction of endogenous ERK2 with METTL3 and WTAP was observed in A375 cells, a human melanoma cell line with constitutively active ERK due to a BRAF V600E mutation (Fig 2A). After MEK stimulation, activated ERK translocates into the cell nucleus to activate nuclear substrates, or forms a dimer to activate cytoplasmic substrates (Casar et al., 2009). As shown in Figure S2C, ERK activated by BRAF co-localizes with METTL3 and WTAP in the nucleus, suggesting that METTL3 complex could be a nuclear substrate of ERK.
Figure 2. ERK Interacts and Phosphorylates METTL3 and WTAP.

(A) A375 cells were treated with/without 0.1 μM trametinib for 1 hr. Lysates were analyzed by IP and IB as indicated.
(B) Sequence alignment of the conserved D-domain on METTL3 and WTAP predicted by the ELM website. The D domain possesses a consensus binding sequence of (Lys/Arg)0–2-(X)1–6-Φ-X-Φ; where Φ is a hydrophobic residue such as Leu, Ile, Val, Phe, and X is any amino acid.
(C) Interaction between wild-type (WT) or mutant METTL3, WTAP, and ERK2 in lysates from BRAF-expressing 293T cells transfected as indicated was examined by co-IP. EE, R415E/R416E METTL3 or R71E/R72E WTAP.
(D) Sequence alignment of the conserved serine residues on METTL3 that are phosphorylated by ERK.
(E) Mass spectrometry detected S43, S50 and S525 phosphorylation in METTL3 in 293T cells co-transfected with BRAF V600E.
(F) Phos-tag SDS-PAGE showing the phosphorylation status of WT or non-phosphorylatable alanine mutants of METTL3 in 293T cells co-transfected with BRAF. 2A, T43A/S50A; 3A, S43A/S50A/S525A.
(G) Sequence alignment of the serine/threonine-proline (S/T-P) motif on WTAP.
(H) Phos-tag SDS-PAGE showing the phosphorylation status of WT or non-phosphorylatable alanine mutants of human WTAP in 293T cells co-transfected with BRAF. 2A, S306A/S341A.
See also Figure S2.
ERK displays a specificity for phosphorylation at the serine/threonine-proline (S/T-P) motif. Since the S/T-P motif is found in many proteins, ERK either uses a common docking domain (Matsuoka et al.) to bind to a D domain (K/R0–2-X1–6-φ-X-φ) or uses the F-site recruitment site (FRS) to bind to the F-site (FX-F/Y-P) (Roskoski, 2012). Analysis using the Eukaryotic Linear Motif database (http://elm.eu.org) revealed residues 415–421 in METTL3 and residues 71–77 in WTAP as potentially conserved D domains (Figure 2B). We found that a CD mutant (321N) form of ERK2, but not an FRS mutant (263A) form, abolished its interaction with METTL3 and WTAP (Figure 2C). Mutational analysis of the putative D domain residues of METTL3 and WTAP abolished the interaction. Co-IP showed that ERK2 only binds to the myc-tagged domain where the D-domain is located (Figure S2D). These results support the interaction of ERK with METTL3 and WTAP.
Given the physical interaction between ERK and METTL3, and that calf intestinal alkaline phosphatase can eliminate the mobility shift induced by ERK (Figure S2E), we examined whether METTL3 is a physiological substrate of ERK. In vitro kinase assay suggests that ERK directly phosphorylates METTL3 (Figure S2F). We noticed that p38 and JNK, but not ERK5, phosphorylate METTL3, although not as strongly as ERK2. Mass spectrometry analysis showed that ERK phosphorylates METTL3 at three highly conserved residues S43, S50, and S525 (Figure 2D–E). Mutational analysis further confirmed these three sites as main ERK phosphorylation sites (Figure 2F). There is no available structure for WTAP, and published studies of the METTL3 structure focus on the SAM-binding and methyltransferase domains, which cannot provide information for S43 and S50. However, we noticed that S525 lies on the same face and close to the D Domain of METTL3 (Fig S2H). This suggests that the phosphorylation occurs near the ERK binding region.
To investigate METTL3 phosphorylation by ERK inside cells, we raised a polyclonal antibody that targets S43-phosphorylated METTL3. This antibody recognizes S43-phosphorylated METTL3 but not a mutant form of METTL3, 3A METTL3, in which all three phosphorylation serine sites are replaced with alanine (Figure S2F). This P-S43 antibody was then used as a tool to monitor METTL3 phosphorylation. The endogenous METTL3 phosphorylation can be detected in A375 (BRAF-V600E mutant) and HCT116 (K-Ras-G12D mutant) and abrogated by MEK inhibitor treatment for 1 hour (PD0325901 or trametinib) (Fig S2I–J). Although this antibody is not suitable for immunoprecipitation (data not shown), phos-tag gels suggested about 80% of endogenous METTL3 was phosphorylated in A375 and HCT116 and decreased to 40% upon addition of MEK inhibitors.
To determine the phosphorylation sites of WTAP, we examined whether mutations of the S/T-P motif affect the ERK-induced phosphorylation. Among the three S/T-P motifs in human WTAP (Figure 2G), we found that S306 and S341 are the main ERK phosphorylation sites of human WTAP (Figure 2H). We noticed that S306 is not conserved in mouse and rat WTAP orthologs; however, there is a unique S/T-P motif at T298 in mouse and rat WTAP, which can also be phosphorylated by ERK (Figure S2K). In conclusion, we show that ERK interacts with and phosphorylates METTL3 and WTAP.
USP5 is Required for ERK-Mediated METTL3 Stabilization
Next, we investigated how ERK-induced phosphorylation increases RNA m6A methyltransferase complex activity. We noticed that ERK activation increased wild-type (WT) but not 3A METTL3 expression (Figure 2F), and that WT METTL3 stable transfectants maintained consistently higher expression levels than those of 3A METTL3 in both mouse ESCs (mESCs) and human A375 cells. (Figure 3A). This observation suggested a model in which METTL3 phosphorylation by ERK stabilizes the protein, which could explain the higher METTL3 protein level and elevated m6A methylation activity observed with ERK activation. We then investigated whether ERK activation could affect METTL3 stability and found that inhibition of ERK by PD0325901 increased its ubiquitination at 8-hour treatment (Figure S3A). Furthermore, the degradation of METTL3 induced by inhibition of ERK activity was restored by addition of a proteasome inhibitor MG132 (Figure S3B). The ubiquitination level of 3A METTL3 was also higher than that of WT METTL3 (Figure 3B). To assess more directly the effects of ERK on METTL3 stability, cycloheximide was used to suppress protein synthesis and the degradation of METTL3 protein was monitored. As shown in Figure 3C, ERK activation increased the stability of WT but not non-phosphorylatable 3A METTL3; meanwhile, phospho-mimetic 3E METTL3 showed an increase in stability compared to WT METTL3.
Figure 3. USP5 is Required for ERK-Mediated METTL3 Stabilization.

(A) Comparison of METTL3 and WTAP protein levels in mESCs and A375 stable transfectants by immunoblotting (IB).
(B) 293T cells transfected as indicated were treated with MG-132 (10 μM, 8hr) followed by IP/IB.
(C) 293T cells transfected as indicated for 48 hr, followed by cycloheximide (CHX) 10 μg/ml for 0–12 h. Lysates were used for IB to measure the protein levels of METTL3. Density of METTL3 expression was quantified by ImageJ and the relative fold compared to the untreated WT was indicated and plotted at the right panel.
(D) BRAF expression promotes METTL3-USP5 interaction. Lysates of 293T cells transfected as indicated for 48 hr were subjected to IP with anti-myc antibody followed by IB.
(E) USP5 decreases ubiquitination of METTL3. Lysates of 293T cells transfected as indicated for 48 hr were subjected to IP with anti-myc antibody followed by IB.
(F) Knockdown of SPOP, TRIM28, and ANAPC1 attenuated USP5 inhibition-induced degradation of METTL3. A375 cells were transfected with siRNA for 72 hr or treated with 10 μM EOAI3402143 (EOAI) for 8 hr before IB analysis.
Since METTL14 is known to stabilize METTL3 (Wang et al., 2016), we investigated whether phosphorylation of METTL3 by ERK affects METTL3-METTL14 complex formation. The interaction between METTL3 and METTL14 was not obviously affected by ERK inhibition treatment for 1 hour (Figure S3C). Moreover, 3A METTL3 also interacts with METTL14 normally (Figure S3D). Interestingly, we noticed that ERK activation increased the interaction between METTL3 and WTAP, which became weaker with 3A METTL3 and was further attenuated with non-phosphorylatable WTAP S306A/S341A (2A) (Figure S3D). It has been shown that WTAP depletion does not affect METTL3 complex stability, but rather reduces nuclear localization of METTL3 (Kobayashi et al., 2018; Ping et al., 2014). Consistent with previous reports, knockdown of WTAP decreased nuclear METTL3 (Figure S3E). Considering the interaction between METTL3 and WTAP was attenuated by non-phosphorylatable mutant forms, cellular fractionation and immunostaining was used to examine METTL3. As shown in Figure S3F–G, nuclear METTL3 was markedly reduced in cells expressing 3A METTL3 and 2A WTAP.
To gain further insight into how ERK phosphorylation decreases METTL3 ubiquitination, we examined whether any ubiquitin ligases or deubiquitinases were identified in our CRISPR-based genomic screen. Since the top hits identify regulators of GGACT reporter m6A methylation rather than just METTL3, not all hits—namely, USP43, USP15, or USP7 (Table S1, top 1000 sgRNAs)—affected METTL3 expression level (Figure S4A). However, we found knockdown of USP5 and USP1 (Rank 2845 and 2339) decreased METTL3 in A375 (Figure S4B). Commercially available USP1 inhibitor SJB3–019A and USP5 inhibitor EOAI3402143 (EOAI) also decreased METTL3 (Figure S4C). Considering USP5 is implicated in a wide range of pathological processes and had the most pronounced effect on METTL3, we chose to further investigate it.
We first investigated whether phosphorylation of METTL3 in the presence of activated BRAF affects the METTL3-USP5 interaction. We noticed that ERK activation increased the interaction between METTL3 and USP5, which became weaker with phospho-defective mutant S43A, S50A, S525A, and fully attenuated when all three sites were mutated (Figure 3D). We also noticed that 3E METTL3 binds to USP5 more strongly. Interestingly, BRAF expression further promoted 3E METTL3-USP5 interaction and increased its expression. This suggests that BRAF may also affect USP5 activity. It has been demonstrated that USP5 activity was increased in cells expressing BRAF V600E (Potu et al., 2014). We further find that ERK activation by BRAF promoted USP5 translocation into the nucleus to colocalize with METTL3 (Figure S4D). USP5 is a large protein composed of five specific domains, including the cryptic ZnF domain, ZnF domain, C-box domain, UBA1/UBA2 domain, and H-box domain. To define which domain was critical for the action of USP5 on METTL3, constructs of these domains were co-transfected with METTL3. The IP assay suggested that METTL3 binds more strongly to the cryptic ZnF domain compared to the C-box domain and H-box domain (Figure S4E). On the other hand, USP5 binds to the methyltransferase domain of METTL3 (Figure S4F). Reciprocal IP confirmed the endogenous METTL3-USP5 interaction, which was inhibited by MEK inhibitor (Figure S4G). Lastly, ERK-phosphorylated or phosphomimetic 3E METTL3 displayed greater stability upon USP5 inhibitor EOAI treatment (Figure S4H). Taken together, these results suggest that ERK activation translocates USP5 to the cell nucleus, which interacts with phosphorylated METTL3 to promote its stability.
Because USP5 is an enzyme that could prevent protein ubiquitination, we further examined whether USP5 stabilizes METTL3 through deubiquitination (Figure 3E). Overexpression of wild-type but not catalytically dead C335A USP5 decreased ubiquitination and stabilized METTL3. Furthermore, in vitro addition of USP5 reduced EOAI-induced METTL3 ubiquitination (Figure S4I), suggesting that METTL3 is a direct substrate of USP5. To identify ubiquitin ligases that contribute to USP5 inhibition-induced degradation of METTL3, we searched consensus motif and physical association databases, as well as our CRISPR-based genomic screen. METTL3 contains an SPOP-binding consensus motif and COP1-binding destruction motif (http://elm.eu.org). It also interacts with TRIM28, HUWE1, and UBR5 (https://thebiogrid.org), and may be ubiquitinated by SMURF1 (http://ubibrowser.ncpsb.org). Finally, we identified FBXW8, FBXW12, SPOPL, TRIM2, and ANAPC1 as negative regulators of m6A methylation from our CRISPR screen. To find ubiquitin ligases involved in METTL3 degradation, A375 cells with USP5 knockdown were further transfected with siRNA targeting ubiquitin ligases. Knockdown of SPOP, TRIM28, or ANAPC1 partially abolished USP5 inhibition-mediated METTL3 degradation (Figure 3F). Because SPOP is a well-known tumor suppressor and localizes to nuclear speckles (where the m6A methyltransferase localizes), we tested an SPOP inhibitor, SPOP-IN-6b, which was able to partially reverse EOAI-induced ubiquitination and degradation of METTL3 (Figure S4J–K). Lastly, we were interested in what type of ubiquitin linkages that the USP5 inhibition induced. USP5 has been shown to cleave multiple types of polyubiquitin linkage, including K6, K11, K29, K48 and K63 (Avvakumov et al., 2012; Raasi et al., 2005). We observed K11, K48 and K63-linked polyubiquitination on METTL3 after USP5 knockdown. Furthermore, knockdown of ANAPC1 and SPOP decreased K11 and K48 ubiquitination, respectively (Figure S4L). In summary, these results suggest that USP5 stabilizes METTL3 through deubiquitination.
Phosphorylation of METTL3/WTAP by ERK Facilitates Resolution of Pluripotency
Autocrine FGF4 is reported to be the major stimulus for ERK signaling in mESCs. Interference with FGFR or ERK activity impeded the ability of mESCs to undergo differentiation and retain expression of pluripotency factors including Nanog. We observed that p-43 METTL3 phosphorylation was enhanced by FGF4 and reduced by MEK inhibitor PD0325901 or FGFR1 inhibitor PD173074 (Figure S5A). Because both ERK activation and METTL3 expression have been reported to be required for mESCs to exit the pluripotent state upon differentiation, we further investigated whether phosphorylation of METTL3/WTAP affects mESCs fate. We introduced TetOn-shWTAP into METTL3-KO mESCs to tune endogenous WTAP expression. Cells were then transduced with WT or a phospho-inactive mutant of CuO-METTL3-T2A-WTAP constructs. With 50 μg/ml cumate, the expression level of exogenous R-WT METTL3 is comparable to that of endogenous METTL3 and we noticed that 3A METTL3 expression is consistently lower than that of WT (Figure S5B). Quantification of m6A by LC-MS/MS showed a synergistic reduction of 3A METTL3 and 2A WTAP (Figure 4A). We then examined whether pluripotency of mESCs expressing R-3A2A was affected. R-3A2A mESCs exhibited higher stage specific embryonic antigen 1 (SSEA-1) expression (Figure S5C) and increased proliferation (Figure 4B). These observations support the notion that loss of METTL3 and WTAP phosphorylation trap mESCs in the pluripotent state.
Figure 4. Phosphorylation of METTL3/WTAP by ERK Facilitates Resolution of Pluripotency.

(A) LC-MS/MS quantification of the m6A/A ratio in mRNA of mESCs stable transfectants. *p < 0.05, **p < 0.01, ***p < 0.001.
(B) Cell growth of R-WT and R-3A2A mESCs were measured by sulforhodamine B dye (SRB assay). Data are presented as relative to Day 1. ***p < 0.001.
(C) MeRIP-qPCR of pluripotency transcripts in mESCs stable transfectants. *p < 0.05, **p < 0.01.
(D) qPCR analysis of pluripotency genes in mESCs stable transfectants. *p < 0.05, **p < 0.01. (E) qPCR analysis for pluripotency and differentiation markers expression after 8 days of embryonic body induction. *p < 0.05, **p < 0.01, ***p < 0.001.
Unless otherwise indicated, all data in this figure contain n = 3 replicates per group, and represent mean ± SEM. All p-values were also calculated by Student’s t test.
See also Figure S5.
Mettl3-deficient mESCs fail to exit pluripotency despite differentiation cues, likely because loss of m6A impedes the degradation of pluripotency-promoting transcripts. We subsequently examined reported m6A-methylated pluripotency factor transcripts, including Nanog, Zfp42, Klf2, Sox2, and Lefty1 (Batista et al., 2014; Bertero et al., 2018; Geula et al., 2015). Pou5f1, which does not harbor any m6A modification, was also used as a negative control. m6A-RIP-qPCR confirmed decreased m6A (Figure 4C) and RT-qPCR indicated upregulation (Figure 4D) of these m6A-labeled pluripotency transcripts in R-3A2A mESCs. Furthermore, after transcription arrest by actinomycin D treatment, these transcripts showed delayed turnover in R-3A2A (Figure S5D). These findings suggest that METTL3 phosphorylation controls the level of critical pluripotency regulators. Considering ERK activation is the primary stimulus for mESCs to exit self-renewal and acquire competence of differentiation (Kunath et al., 2007), we then compared the capacity for differentiation by transferring mESCs to differentiation media for embryoid bodies (EBs). R-3A2A mESCs generated smaller EB spheres (Figure S5E), failed to repress pluripotent genes, and adequately up-regulated developmental markers (Figure 4E). Lastly, because ERK activation controls the transition from the primitive ectoderm-like cell state to a neural progenitor cell state, we tested whether METTL3 phosphorylation could affect differentiation towards a neural lineage. As shown in Figure S5F, even upon induction of differentiation, R-3A2A mESCs continued to express Nanog and failed to upregulate primary neural markers Sox1 and Nestin. These results support the notion that ERK-dependent phosphorylation of METTL3 and WTAP promotes mESC differentiation.
Transcripts Affected by Phosphorylation of Methyltransferase Complex in mESCs
To gain further insight into how the phosphorylation of the m6A methyltransferase complex affects the m6A-modified transcripts, we mapped the m6A methylome in mESCs. Comparison of the R-WT with R-3A2A mESCs revealed a global loss of methylation sites (Figure 5A). Consistent with previous m6A-seq results (Dominissini et al., 2012; Meyer et al., 2012), the m6A peaks identified are enriched near the start and stop codons and were characterized by the canonical GGACU motifs (Figure S6A–B). Using our in-house R-package “MeRIPtools,” which tests for m6A-IP enrichment using a binomial-distribution-based model, we found 7,591 m6A peaks that exhibited a significant decrease in the R-3A2A cells compared to the R-WT cells (Figure 5B, Table S3), such as modification sites in Nanog, Lefty1, and Zfp219 (Figure 5C). The genes showing decreased m6A methylation significantly overlap with those in functional gene sets important for pluripotency, including targets of NANOG and MYC (Figure S6C). The transcripts exhibiting differential methylation were consistent between replicates (Figure S6D). Furthermore, the transcripts showing decreased m6A methylation (Figure 5D) and differential expression (Figure 5E) were enriched for gene ontology (GO) terms related to pluripotency and mRNA processing. Importantly, many of the genes involved in pluripotency showed reduced m6A methylation in R-3A2A when compared with R-WT mESCs (Figure 5F).
Figure 5. Transcripts Affected by Phosphorylation of Methyltransferase Complex in mESCs.

(A) Cumulative distribution function of log2 peak intensity of m6A-modified sites in R-WT and R-3A2A mESCs.
(B) Volcano plot for peaks with differential m6A intensity between R-WT and R-3A2A mESCs. Fold change (FC) is the ratio of IP over Input for R-WT and R-3A2A.
(C) Coverage plots of the m6A peaks of Nanog, Lefty1, and Zfp219 comparing R-WT and R-3A2A mESCs. Plotted coverages are the medians of three replicates.
(D) Gene enrichment analysis with WikiPathway terms of differentially m6A methylated peaks in R-WT and R-3A2A mESCs for molecular functions.
(E) Gene enrichment analysis with WikiPathway terms of differentially expressed genes (p < 0.05).
(F) A histogram showing relative m6A peak enrichment of R-3A2A compared to R-WT mESCs, indicating higher m6A methylation in pluripotency genes (PluriNetwork) for R-WT mESCs.
To expand our observation of pathways that are enriched when comparing R-WT and R-3A2A mESCs, we performed a functional class scoring approach (gene-set enrichment analysis, GSEA) in addition to GO analysis. GSEA showed enrichment of histone binding proteins (Figure S6E, Table S4). Considering that it has been reported that m6A regulates histone modifications in part by destabilizing mRNA of histone-modifying enzymes (Wang et al., 2018), we used an ELISA kit to compare 21 different Histone H3 modifications. H3K27me3 showed the most dramatic changes (Figure S6F) and we detected reduced m6A peaks in several components of the PRC2 complex mRNA (Figure S6G). These results suggest phosphorylation of the m6A methyltransferase complex decreases H3K27me3 partially by regulating the PRC2 complex.
Phosphorylation of the m6A Methyltransferase Complex May Affect Tumorigenesis
As one of the most frequently mutated signaling pathways in cancer, the RAS/RAF/MEK/ERK signaling cascade has long been viewed as a promising target for cancer therapy (Liu et al., 2018a). Given that phosphorylation of the m6A methyltransferase complex by ERK facilitates resolution of pluripotency in mESCs, we further investigated whether the m6A methyltransferase complex can be similarly regulated in certain cancer cells. Using CancerMine (Lever et al., 2019), a literature-mined resource, we summarized that METTL3 could behave as an oncogene in many cancer types (Figure S7A).
We first examined melanoma due to the high prevalence of constitutively active BRAF V600E mutation (50–60%) and clinical success with BRAF and MEK inhibitors (Schadendorf et al., 2018). The m6A levels on mRNA are higher in MEL-624 and A375 cells, which harbor a BRAF V600E mutation (Fig S7B). As expected, the stability of the m6A methyltransferase complex was reduced in the R-3A2A A375 cells (Figure 6A), which contributed to the overall lower m6A level on mRNA (Figure 6B). MEK inhibitors PD0325901 and trametinib were found to reduce the protein levels of the m6A methyltransferase complex at 8 hours (Figure 6C) and the overall mRNA m6A levels at 48 hours (Figure 6D) in A375 melanoma cells. In addition, these two MEK inhibitors also decreased m6A methyltransferase complex level in HCT-116 cells, which is a colon cancer line that possesses the most common KRAS mutation (G12D) (Figure S7C).
Figure 6. Phosphorylation of the m6A Methyltransferase Complex May Affect Tumorigenesis.

(A) Lysates of A375 stable transfectants harvested at different time points after treatment with cycloheximide (CHX) 10 μg/ml were analyzed by IB.
(B) LC-MS/MS quantification of the m6A/A ratio in mRNA of A375 stable transfectants. **p < 0.01.
(C) After 8 hr treatment with 10 μM PD0325901 or 0.1 μM trametinib, lysates from A375 cells were analyzed by IB.
(D) LC-MS/MS quantification of the m6A/A ratio in mRNA of A375 cells treated with 10 μM PD0325901 or 0.1 μM trametinib for 48 hr. **p < 0.01, ***p < 0.001.
(E) After 8 hr treatment with 10 μM EOAI3402143 (EOAI) or 30 μM vialinin A, cell lysates from A375 cells were analyzed by immunoblot.
(F) A375 stable transfectants as indicated were treated with 3 μM EOAI3402143 (EOAI) or 10 μM vialinin A before measuring cell viability by SRB assay. Data are presented as relative to the R-WT cells without drug treatment. *p < 0.05, **p < 0.01, ***p < 0.001.
(G) Immunofluorescence analysis of METTL3 (green) in SKBR3 cells treated with 1 μM tucatinib and 1 μM lapatinib for 8 hr. DAPI (blue) was used to mark the nucleus. Scale bars, 10 μm.
(H) LC-MS/MS quantification of the m6A/A ratio in mRNA of SKBR3 and BT474 cells treated with 1 μM tucatinib and 1 μM lapatinib for 48 hr. *p < 0.05, **p < 0.01, ***p < 0.001.
Unless otherwise indicated, all data with error bars in this figure represent n = 3 replicates per group, and represent mean ± SEM. All p-values were also calculated by Student’s t test.
See also Figure S7.
Because knockdown of USP5 increases METTL3 in A375 melanoma cells (Figure S5B), we assessed the potential clinical relevance of USP5. Interestingly, melanoma patients with high USP5 had shorter overall survival (Figure S7D). Two structurally unrelated USP5 inhibitors, EOAI3402143 and vialinin, were employed to evaluate the effect of USP5 on the METTL3 level in melanoma cells. We found that these two USP5 inhibitors increased ubiquitination of METTL3, resulting in decreased METTL3 protein level (Figure 6E and S7E–F). Furthermore, MEK inhibition, R-3A2A, or METTL3-WTAP knockdown can sensitize melanoma and colon cancer cells to USP5 inhibition (Figure 6F and S7G), supporting the connection between USP5 and METTL3.
Lastly, considering that HER2 expression phosphorylates METTL3 and WTAP (Figure S1C and S7H) and m6A levels are higher in the HER2-overexpressed SKBR3 and BT474 cells (Figure S7I), we investigated whether inhibition of HER2 could affect m6A methylation. Two HER2 inhibitors, tucatinib and lapatinib, could reduce METTL3 protein level at 8 hours and cellular mRNA m6A methylation at 48 hours in HER2-positive breast cancer (Figure 6G and 6H). Overall, our data support that ERK-dependent METTL3 stabilization affects cellular mRNA m6A methylation which could contribute to tumorigenesis. More focused cancer studies are required to assess the effects and scope of such a regulatory mechanism in the future.
Discussion
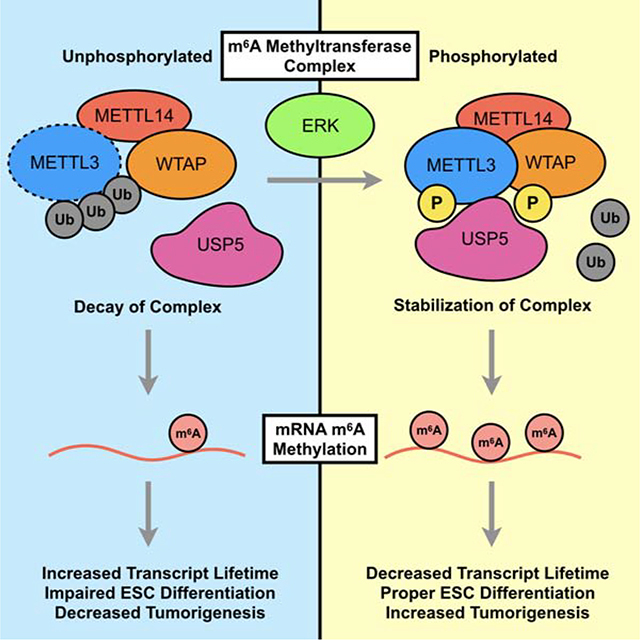
m6A RNA methylation plays crucial roles in regulating RNA metabolism. While many studies have shown the importance of METTL3 (Batista et al., 2014; Choe et al., 2018; Geula et al., 2015; Lin et al., 2016; Liu et al., 2018b; Wang et al., 2014b), few have shown how the methyltransferase complex itself is regulated. Here, we identify an ERK-METTL3/WTAP signaling axis that regulates mESCs differentiation and potentially affects tumorigenesis. Initially, we deployed a genome-wide CRISPR screen using an m6A methylation-dependent GFP reporter. RAS and MAPK pathways were identified as the top pathways in the positive regulation of m6A methylation. Biochemical studies showed that ERK proteins could phosphorylate METTL3 on S43/S50/S525 and WTAP at S306/S341. We also found that phosphorylation of METTL3 decreases METTL3 ubiquitination through interaction with USP5. These findings explain elevated m6A levels on mRNA upon ERK activation. This pathway underlines a previously unrecognized effect of ERK activation through RNA methylation during differentiation of pluripotent mESCs (Figure 7).
Figure 7.

A Schematic Model of the Role of the m6A Methyltransferase Phosphorylation by ERK.
Phosphorylation of METTL3 and Potential Effects
Our studies reveal METTL3 phosphorylation by ATM and ATR (Figure S1D). Phosphoproteomic studies of ATM/ATR substrates have previously uncovered that, in response to DNA damage, METTL3 was phosphorylated at residues S350 and T356 in an ATM-dependent manner (Matsuoka et al., 2007). Furthermore, phosphorylation of METTL3 was found to be an early responder to DNA damage (Bennetzen et al., 2010). Considering that m6A rapidly accumulates at UV-irradiated sites (Xiang et al., 2017) and lack of METTL3 catalytic activity delays DNA repair and Pol κ-mediated response to UV DNA damage, it would be interesting to explore the role of ATM/ATR-induced METTL3 phosphorylation in the future. In addition, in vitro kinase assay showed that certain stress-activated MAP kinase, p38 and JNK, can also phosphorylate METTL3. Interestingly, METTL3 has previously been linked to MAPK signaling. In dendritic cells, dental pulp stem cells, or intestinal epithelia, depletion of METTL3 leads to reduced phosphorylation of p38, ERK, and JNK (Feng et al., 2018; Wang et al., 2019; Zong et al., 2019); whereas phosphorylation is increased in osteoblasts and colorectal cancer cells (Deng et al., 2019; Zhang et al., 2019a). Although effects of METTL3 on MAPKs may vary based on cell type, taken together, these data suggest potential feedback loop mechanisms between METTL3 and MAPK pathways.
Our phosphorylation studies estimated the relative levels of phosphorylated and non-phosphorylated METTL3. We noticed that trametinib was more effective than PD0325901 in decreasing the mobility shift of METTL3 in A375 cells, although these two MEK inhibitors inhibited ERK activation and METTL3 S43 phosphorylation equally well (Figure S3H). Trametinib is known to inhibit the proliferation of BRAF V600E cells with lower IC50 than PD0325901, even though they had similar potency against MEK1 in vitro. Part of the reason is that trametinib inhibits the CRAF-driven signaling more effectively than PD0325901 (Lito et al., 2014). We observed that CRAF phosphorylates METTL3 and leads to an additional band in the 3A METTL3 mutant, when comparing to MEK and BRAF expression (data not shown). Therefore, we propose that the more potent effect of trametinib could be partially due to CRAF inhibition. Although the RAF family of enzymes are canonically depicted as activators of MEK, they carry out additional functions as well. BRAF is the most potent MEK activator in vivo, whereas ARAF and CRAF interact with several other proteins in a MEK-independent manner (Desideri et al., 2015; Leicht et al., 2007). Interestingly, ARAF is also detected in our CRISPR screen (Figure 1C). How ARAF and CRAF affect METTL3 through MEK-independent signaling pathway still needs future investigation.
Ubiquitin Ligases and Ubiquitin Linkages of METTL3
To systemically study the regulation of METTL3 ubiquitination, we plan to construct a reporter encoding both DsRed and EGFP-METTL3, which allows us to use EGFP:DsRED ratio as an indicator of the stability of METTL3 in the future (Yen et al., 2008). CRISPR activation (CRISPRa) and CRISPR inhibition (CRISPRi) screens, which focus on proteostasis, could be used to identify ubiquitin ligases and deubiquitinases for METTL3. In the current study, we found that SPOP, ANAPC1, and TRIM28 promote degradation of METTL3, while USP5 reverses this process. A previous study showed that TRIM28 immunoprecipitated with METTL3, METTL14, and WTAP (Yue et al., 2018), which raised the possibility that TRIM28 could be a component of the m6A writer complex. SPOP is known to mediate K48-linked ubiquitination and proteasome degradation of various substrates, such as estrogen receptors, PTEN, BRD4, MyD88, and PD-L1 (Wei et al., 2018). Interestingly, when SPOP oligomerizes with increased cellular level, it localizes to nuclear speckles (Marzahn et al., 2016), where METTL3-WTAP is located.
We observed K11, K48 and K63-linked polyubiquitination on METTL3 after USP5 knockdown. We also found that K11 ubiquitination of METTL3 can be elevated by USP5 inhibition and attenuated by ANAPC1 (subunit of anaphase-promoting complex, APC) knockdown. APC is the major E3 ligase that assembles K11-linked ubiquitin chain to drive proteasomal degradation and mitotic exit (Wickliffe et al., 2011). The abundance of K11 linkage strongly increases when APC is active during mitosis. In accordance with our observation, recently it was found that METTL3 is significantly downregulated during M phase (Fei et al., 2020). Elucidation of the post-translational and proteolysis degradation of METTL3 during cell cycle may shed further insights on how METTL3 contributes to normal cell differentiation and tumorigenesis.
METTL3/WTAP Phosphorylation by ERK is Important for mESCs Differentiation
Consistent with previous observations (Batista et al., 2014; Geula et al., 2015), our m6A-seq data revealed extensive mRNA m6A methylation in mESCs. Differentially methylated transcripts upon loss of METTL3/WTAP phosphorylation are enriched for genes involved in pluripotency and RNA processing, like those found in METTL3 KO studies. Our report suggests that ERK activation increases m6A methylation on key pluripotent transcripts, thus contributing to their decay. Tuning the phosphorylation state of METTL3 could be an effective post-translational way to adjust global mRNA m6A methylation.
While we found that phosphorylation of METTL3 affects its interaction with WTAP and USP5, interaction with other binding partners could also be affected. For instance, SMAD2/3 interacts with the METTL3-METT14-WTAP complex to promote m6A binding to particular transcripts in mESCs (Bertero et al., 2018). ZFP217 has also been found to sequester METTL3, thereby restricting m6A methylation of certain transcripts in ESCs (Aguilo et al., 2015). It would be interesting to determine whether METTL3 phosphorylation affects interaction with SMAD2/3 or ZFP217 in mESCs, which could further explain how phosphorylation of m6A writer proteins can affect methylation of core pluripotency factor transcripts.
Other Pathways that May Regulate RNA m6A Methylation
While our study sheds light on ERK phosphorylation of METTL3, other questions remain. First, what are other pathways that could regulate m6A methylation? Pathways identified from Gene Ontology enrichment analysis for genes that promote m6A include transcriptional regulation by TP53 and HIF-1 signaling. The same analysis for genes that apparently suppress m6A include sphingosine-1-phosphate receptor signaling and protein targeting to mitochondria (data not shown). Modulation of activity for these pathways may reveal more regulators of m6A methylation.
Second, how does the interplay between different post-translational modifications of METTL3 affect its activity? METTL3 is not only phosphorylated but also SUMOylated (Du et al., 2018). SUMOylation of METTL3 reduces its methyltransferase activity, thereby lowering m6A abundance. Intriguingly, MAPK activation can modulate SUMOylation; specifically, ERK activation de-SUMOylates the Elk-1 transcription factor (Yang et al., 2003; Yang and Sharrocks, 2010). It would be interesting to further explore whether METTL3 phosphorylation affects SUMOylation level.
Overall, our study sheds light on a signaling relationship between ERK pathway and m6A methylation. Several challenges remain in understanding how cells regulate m6A methylation spatiotemporally, as well as which transcripts are methylated. The post-translational regulation of m6A writers, readers, and erasers may provide insight for these questions and could be an important factor in affecting the m6A epitranscriptome.
Limitations
While our results support a model in which ERK phosphorylates m6A methyltransferase components METTL3 and WTAP, leading to stabilization by USP5, our study is not without potential caveats. For instance, although we were able to identify ERK as an m6A regulator, we noticed that not all known m6A regulatory proteins were among the top-ranked gene in our genome-wide CRISPR screen. Part of the reason might be due to the use of a circular RNA reporter in our screen. How m6A is installed on circRNA and how it affects translation could be different from mRNA. For example, YTHDF1 is not involved in the translation of circRNA (Di Timoteo et al., 2020). However, our targeted knockdown of METTL3, METTL14, WTAP, ALKBH5, and FTO did show expected reporter responses (Figure S1A).
We also showed that METTL3 can bind to USP5, and that this binding is promoted by ERK-mediated phosphorylation. This may be an example of how phosphorylation sites act as switches to regulate protein-protein interactions (Jin and Pawson, 2012). Based on expression of different USP5 protein domains, METTL3 appears to bind most strongly to the cryptic ZnF domain, which possesses multiple positively charged residues on the domain surface. Although there is no structure for WTAP and currently available structural data of METTL3 do not contain S43 and S50, prediction software (Sanchez-Garcia et al., 2019) suggests that METTL3 R523 is involved in the interaction with USP5. Its proximity to S525 likely suggests that S525 phosphorylation may increase binding affinity to a positively charged region of USP5. These findings would be strengthened with structural studies showing the binding site for USP5 on METTL3, as well as the phospho-sites of the METTL3-METTL14-WTAP structure.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Chuan He (chuanhe@uchicago.edu).
Materials Availability
Antibodies targeting the Ser43 phosphorylation of METTL3 were generated in collaboration with Lifetein LLC. There are restrictions to the availability of the anti-Ser43 phosphorylated METTL3 antibody due to the lack of an external centralized repository for its distribution and our need to maintain the stock. We are glad to share the antibody with reasonable compensation by requestor for its processing and shipping. All other unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The CRISPR screening and m6A-seq data generated during this study are available at GSE138776. The human data for the skin cutaneous melanoma (SKCM) was derived from the The Cancer Genome Atlas (TCGA).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
mESC culture and differentiation
mESCs were generated, maintained, and differentiated essentially as previously described (Geula et al., 2015). METTL3 knockout mESCs were kindly provided by Dr. Howard Y. Chang (Stanford University) and regularly tested negatively for mycoplasma contamination. Established ESC clones were genotyped by PCR and validated as METTL3-deficient by qPCR and Western blot. mESCs were cultured on mitomycin C-treated mouse embryonic fibroblasts in ES medium containing DMEM supplemented with 15% FBS, 1 mM L-glutamine, 0.1 mM mercaptoethanol, 1% non-essential amino acid, 1% penicillin/streptomycin, nucleosides 1,000 U /ml leukemia inhibitory factor, 3 μM CHIR99021 and 1 μM PD0325901. To observe the phenotype of METTL3-KO derived stable transfectants, 2μg/ml doxycycline were added to knock down WTAP through TetOn-shWTAP, 50 μg/ml cumate were added to induce CuO-METTL3-T2A-WTAP expression, and PD0325901 was removed from the medium to be able to compare the difference between WT and 3A2A. For embryoid body (EB) differentiation, 5×106 ESC were disaggregated with trypsin and transferred to non-adherent suspension culture dishes and cultured in MEF medium (DMEM supplemented with 1% L-Glutamine, 1% Non-essential amino acids, 1% penicillin/streptomycin and 15% FBS) for 8 days. The serum-free neural induction protocol was applied as described (Ying et al., 2003). ES cells were plated in 6-well plates at a density of 1.5X105 cells/well in N2B27 medium with LIF (100 units/ml). The next day (day 0), the medium was changed to N2B27 without LIF. Medium was renewed daily thereafter.
Cell Culture
HeLa, 293T, 293TN, A375, CHL-1, MEL-624 cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. MCF-7, T47D, SKBR3, and HCT-116 cells were maintained in RPMI supplemented with 10% FBS and 1% penicillin/streptomycin. BT474 cells were maintained in RPMI supplemented with 20% FBS and 1% penicillin/streptomycin. To observe the phenotype of stable transfectants, 2 μg/ml doxycycline was added to knock down WTAP and METTL3, while 50 μg/ml cumate was added to induce CuO-METTL3-T2A-WTAP expression.
METHOD DETAILS
Plasmids
The circRNA reporters containing split GFP with a m6A motif were kindly provided by Z. Wang (Chinese Academy of Science, Shanghai, China) and subcloned into pCDH-CMV-MCS-EF1α-RFP (System Biosciences, CD512B-1). The CRISPR knockout pooled library (#1000000048), METTL3 (#53739), METTL14 (#53740), WTAP (#53741), pKMyc (#19400), Flag-ATM (#31985), ATR (#31611), Flag-IKKε (#26201), HA-GSK-3β (#14754), ERK1 (#23509), ERK2 (#23498), B-Raf V600E (#17544), HA-Ubiquitin (#17608), Ubiquitin-KO (#17603), Ubiquitin-K6 (#22900), Ubiquitin-K11 (#22901), Ubiquitin-K27 (#22902), Ubiquitin-K29 (#22903), Ubiquitin-K33 (#17603), Ubiquitin-48 (#17605), Ubiquitin-K63 (#17606), pMD2.G (#12259) and psPAX2 (#12260) were ordered from Addgene. Flag-IKKα, Flag-IKKβ, HA-AKT, Flag-mTOR, HA-MEKDD, HA-CDC2, FAK, EGFR, HER2 V659E, pCMV5-HA, and pCMV5-Flag were kindly provided by M.C. Hung (China Medical University, Taichung, Taiwan). pLightSwitch R01_3’UTR and NANOG 3’UTR were ordered from Switchgear Genomics. Mouse METTL3 (MR209093), mouse WTAP (MR216877), and human USP5 (RC224191) were purchased from OriGene. METTL3 (human and mice), METTL14, and WTAP were subcloned into pKMyc, METTL14 was subcloned into pCMV5-HA, and WTAP (human and mice), ERK1, ERK2, and USP5 were cloned into pCMV5-Flag. All mutants were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The annealed shMETTL3 (TRCN0000289742), shWTAP (TRCN0000231424-human, TRCN0000124351-mice) specific targeted sequence was inserted into Tet-pLKO-puro (Addgene, #21915). Myc-METTL3-T2A-Flag-WTAP was cloned into pCDH-CuO-MCS-EF1α-RFP (System Biosciences, QM512B-1). pCDG-EF1α-CymR-T2A-Neo (QM400PA-2) for the cumate suppressor was ordered from System Biosciences. Human METTL3 was subcloned into the pLenti-DsRed_IRES_SNCA:EGFP lentiviral reporter (Addgene, #92195), after removing the SNCA gene from the plasmid in order to form a pLenti-DsRed-IRES-METTL3-EGFP reporter.
Transfection and Virus Production
For transient transfection, cells were transfected by Lipofectamine 2000 as previously described (Lee et al., 2007). Briefly, cells were seeded 24 hours before transfection, and the plasmid(s) and Lipofectamine reagent were combined in Opti-MEM media. Media was then replaced with regular media without antibiotics 6 hours later. For lentivirus production, a lentiviral construct (pCDH-CMV-MCS-EF1α-RFP plasmids for overexpressing circRNA-GFP, Tet-pLKO-puro for inducible knockdown of METTL3 or WTAP, pCDG-EF1α-CymR-T2A-Neo for cumate repressor, pCDH-CuO-MCS-EF1α-RFP for inducible overexpression of METTL3-T2A-WTAP, or pLenti-DsRed-IRES-METTL3-EGFP as METTL3 protein stability reporter) together with pMD2.G and psPAX2 were co-transfected into 293TN cells (System Biosciences). Viruses were concentrated by the PEG-it Virus Precipitation Solution and used for infecting cells in the presence of TransDux (System Biosciences). Pools of stable transfectants were selected by antibiotics or sorted by flow cytometry. Doxycycline (0.5 μg/mL) was used to induce shRNA while cumate (50 μg/mL) was used to induce shRNA-resistant cDNA expression.
Luciferase Reporter Assay
The luciferase plasmid LightSwitch 3’UTR Reporter, containing the NANOG 3’UTR or random negative control R01_3’UTR (Switchgear Genomics) was co-transfected with the m6A writer complex and ERK-activated kinase into HeLa cells for two days. NANOG wild type RRACT 3’UTR reporter was mutated at 46A, 397A, and 743A (44AAACT48, 395GGACT399, 741AAACT745) to form a mutant RRTCT 3’UTR reporter. Luciferase expression was measured using the Luciferase Assay System according to the commercial protocol (Promega). NANOG 3’UTR luciferase activity was normalized to cells transfected with R01_3’UTR.
Flow Cytometry
Flow cytometry analysis was conducted on BD LSR Fortessa, and cell sorting was conducted on BD FACSAria Fusion. For SSEA-1 expression, cells were disaggregated with trypsin, blocked with TruStain FcX (BioLegend) then incubated with anti-SSEA-1 (BioLegend) in cell staining buffer (BioLegend).
CRISPR Screen
The genome-wide CRISPR-Cas9 gene knockdown screen was accomplished using GeCKOv2 gene knockout library following a published protocol (Joung et al., 2017). Briefly, the GeCKOV2 library was amplified in Endura electrocompetent cells (Lucigen) then co-transfected with pMD2.G and psPAX2 into 293TN cells to produce a lentiviral library. HeLa-circGFP cells were infected at 0.3 MOI for 3 days, then selected with 2 μg/ml puromycin for 1 week before flow cytometry sorting. Genomes of harvested cells were extracted by Quick-gDNA MidiPrep (Zymo). sgRNA after PCR amplification was sent to the University of Chicago Genomics Facility to be sequenced on Illumina HiSeq 4000 in single-end read mode. RIGER was used to analyze the sequencing results. To obtain the ranked difference plot, sgRNAs were ranked according to the difference between number of reads in low and high GFP populations. The top 1% of the sgRNAs that ranked with the greatest difference were selected for gene ontology enrichment analysis.
Immunoprecipitation, immunoblotting, in vitro kinase and deubiquitination assay
Immunoprecipitation (Jin et al.) and immunoblotting (IB) were performed as previously described (Sun et al., 2016). In brief, protein samples were isolated from respective cells by lysis in RIPA buffer (1% Triton X-100, 150 mM NaCl, 20 mM Na2PO4, pH 7.4) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Subsequently, a BCA assay (Thermo Scientific) was used to determine the protein concentrations. For IP, indicated antibody and protein A/G magnetic beads (Thermo Scientific) were incubated with lysate at 4°C overnight, followed by washing and elution with sample buffer (Bio-Rad). Equal amounts of protein were separated by SDS-PAGE followed by wet transfer to PVDF membranes. Blots were blocked with 5% non-fat milk or BSA and incubated with respective primary antibody at 4°C overnight. Primary antibodies were detected by HRP-linked secondary antibodies (Cell Signaling) together with SuperSignal West Pico Plus chemiluminescent substrate (Thermo Scientific) and imaged in a FluorChem R system (ProteinSimple). Densitometry calculations are calculated using ImageJ. To measure protein stability, cells were treated with 50 μg/mL cycloheximide (CHX) and harvested at 0, 6, and 12 hr.
Phosphate-affinity gel electrophoresis was performed in gels containing 60 μM MnCl2, and 30 μM acrylamide-pendant Phos-tag ligand (AAL-107, Wako Chemicals). For in vitro kinase assays, recombinant full-length kinase expressed in E. coli cells with an N-terminal GST tag, and N-terminal GST-tagged human METTL3/METTL14 complex expressed in Sf9 insect cells were purchased from SignalChem. Active kinase was diluted in Kinase Dilution Buffer III (SignalChem) and incubated with METTL3/METTL14 at 30°C for 15 min. The reaction was stopped by the addition of the sample buffer then analyzed by IB. For in vitro deubiquitination, 293T cells transfected with HA-ubiquitin were treated with 10 μM MG-132 and 10 μM EOAI3402143 for 8 hr. IP-purified METTL3 were incubated without or with purified USP5 (SignalChem) in deubiquitination buffer (SignalChem) following commercial protocol.
Confocal Microscopy
For confocal microscopy, cells after treatments were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, blocked with 5% bovine serum albumin, incubated with primary antibodies overnight at 4°C followed by incubation with the appropriate secondary antibody tagged with Alexa 488 or Alexa 568 (Molecular Probes). Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI) before mounting. Confocal fluorescence images were captured using Olympus FV1000 confocal spectral microscope.
Mass Spectrometry
To identify phosphorylation sites of METTL3, METTL3 precipitated from 293T cells co-transfected with myc-METTL3 and B-Raf V600E was analyzed by Coomassie Blue staining. The protein band corresponding to METTL3 was excised and subjected to in-gel digestion with tryspin and chymotrypsin. Samples were analyzed by Ultimate Capillary LC system (Dionex) directly coupled to LTQ Orbitrap Mass Analyzer (Thermo Scientific) using the TopTenTM method. The data were searched on MASCOT (MassMatrix) against the human Swiss-Prot database. All the identified phospho-peptides were further confirmed by manually checking the results.
RNA Extraction and Real-Time qPCR
Total RNA was isolated using TRIzol (Invitrogen), and 200 ng of RNA was reversed transcribed into cDNA using PrimeScript RT Reagent Kit (Takara). Real-time qPCR was performed using the FastStart Essential DNA Green Master (Roche). HPRT1 was used as an internal control for normalization. Primers used in this study are listed in Table S5. For measuring RNA stability, cells were treated with 5 μg/ml actinomycin D and harvested at 0, 6, and 12 hr to determine the half-life of target mRNAs.
LC-MS/MS m6A quantification of poly(A) RNA.
mRNA was extracted from the total RNA using 2 rounds of the Dynabeads mRNA Purification Kit (Thermo Scientific). 100 ng of mRNA was digested by nuclease P1 (1U) in 20 μl of buffer containing 20 mM NH4OAc (pH = 5.3) at 42°C for 2 hr, followed by dephosphorylation with the addition of FastAP Thermosensitive Alkaline Phosphatase (1U) and FastAP buffer at 37°C for 2 hr. The sample was then diluted to 50 μl, and filtered (0.22 μm pore size, 4 mm diameter, Millipore). 5 μl of the solution was separated by reverse phase ultra-performance liquid chromatography on a C18 column, followed by online mass spectrometry detection using an Agilent 6410 QQQ triple-quadrupole LC mass spectrometer in positive electrospray ionization mode. The nucleosides were quantified by using the nucleoside-to-base ion mass transitions of 282 to 150 (m6A) and 268 to 136 (A). Quantification was carried out by comparison with a standard curve obtained from pure nucleoside. The ratio of m6A to A was calculated based on the calibrated concentrations (Liu et al., 2018b).
m6A-IP and m6A-seq
m6A-IP was performed using the EpiMark N6-Methyladenosine enrichment kit (NEB). Full length purified mRNA was used in m6A-IP-qPCR. For m6A-seq, mRNA was adjusted to 15 ng/μl in 100 μl and fragmented using a BioRuptor Ultrasonicator (Diagenode) with 30 s on/off for 30 cycles. Input and RNA eluted from m6A-IP were used to prepare libraries with TruSeq Stranded mRNA Library Prep Kit (Illumina). Sequencing was carried out at the University of Chicago Genomics Facility on Illumina HiSeq 4000 in single-end read mode with 50 bp reads per read. Reads were aligned to the mycoplasma genome to assess contamination, followed by alignment to mouse genome version 10 (mm10) with HISAT2 v2.1.0 (Kim et al., 2015) with parameter –k 1.
The input library of m6A sequencing was used for comparing gene expression levels. DESeq2 (Love et al., 2014) was applied for differential expression between R-WT and R-3A2A mESCs with FDR < 0.05 cutoff. m6A-seq data were analyzed as described before (De Jesus et al., 2019). m6A peak calling was performed using exomePeak R/Bioconductor package v 3.7 (Meng et al., 2013). Significant peaks with false discovery rate less than 0.05 were annotated to the RefSeq database (mm10). Homer v4.9.1 (Heinz et al., 2010) was used to search for the enriched motif in the m6A peak region where random peaks of 200 bp were used as background sequences for motif discovery. m6A peak distribution on the metagene was plotted by the R package Guitar (Cui et al., 2016).
Differential analysis of m6A methylation was performed using the R package RADAR and MeRIPtools (Zhang et al., 2019b). To summarize and visualize the m6A methylome data, principal component analysis (PCA) was performed using singular value decomposition approach implemented in R function (prcomp) on the log-transformed m6A-IP data. Pathway and gene ontology enrichment analysis were performed using WebGestalt (Liao et al., 2019) with default settings. Pathway enrichment terms were determined using WikiPathway and KEGG terms.
Cell Proliferation Assay
Cells were seeded in 96-well plates. The cell proliferation was assessed by SRB assay (Vichai and Kirtikara, 2006) at various time points. Briefly, cells after treatments were fixed with 10% TCA then stained with 0.05% SRB. After washing 3–4 minutes with water, bound SRB was solubilized with 10 mM Trizma base and measured at 515nm.
Quantification of Histone Modifications
Histones were prepared from fresh cell pellets using Total Histone Extraction Kit (Epigentek). The efficiency of histone extraction was controlled by Coomassie Blue staining and IB with anti-H3 antibody. Histone posttranslational modifications were quantified using the Histone H3 Modification Multiplex Assay Kit (Epigentek) following commercial protocol. Each histogram corresponds to the mean of 2 independent experiments and each measure was obtained using a pool of 100 ng of total histones from 2 independent extractions.
Structural Analysis of Protein
The atomic coordinates and structure factors for the reported crystal structures were obtained from the Protein Data Bank (PDB) with the accession code 5IL0 for the ligand-free form of the METTL3-METTL14 complex and 3IHP for the covalent Ubiquitin-USP5 complex. All figures representing structures were prepared with PyMOL. The prediction of partner-specific protein interfaces was performed using the xgBoost based Interface Prediction of Specific Partner Interactions (BIPSPI) web server (Sanchez-Garcia et al., 2019). Predictions were performed using structural data using PDB atomic coordinates as described in the structural analysis of proteins.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each experiment was performed at least three times, and representative data are shown. Data in the bar graphs are given as the mean ± SEM. Means were checked for statistical difference using Student’s t test, and p-values < 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001). For survival analysis, Kaplan-Meier analysis and a log rank test were applied.
Supplementary Material
Table S1. List of top 1000 sgRNA Identified in the Low and High GFP populations, related to Figure 1
Table S3. List of the Genes with Significant m6A Peak Changed Comparing R-WT and R-3A2A mESCs. Coordinates of the Peak, in Mouse Genome mm10, the Beta Regression Coefficient, and the p-value are Displayed, related to Figure 5.
Table S4. Summary of the Genes Identified by GSEA Related to Histone Binding Proteins, related to Figure 5.
Table S2. List of sgRNA Identified Related to Ras and MAPK Pathways, related to Figure 1
Table S5. Primers for RT-qPCR of mESCs, related to Figure 4.
Table S6. Information for siRNA, related to Figures 2 and 3.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Myc-tag (9B11) Mouse mAb | Cell Signaling | 2276 |
| Flag (M2) Mouse mAb | Sigma-Aldrich | F1804 |
| HA-tag (C29F4) Rabbit mAb | Cell Signaling | 3724 |
| B-Raf (D9T6S) Rabbit mAb | Cell Signaling | 14814 |
| HER2/ErbB2 (D8F12) XP Rabbit mAb | Cell Signaling | 4290 |
| p-ERK1/2 (Thr202/Tyr204) (D13.14.4E) XP Rabbit mAb | Cell Signaling | 4370 |
| GAPDH (D16H11) XP Rabbit mAb | Cell Signaling | 5174 |
| β-Actin (8H10D10) Mouse mAb | Cell Signaling | 3700 |
| METTL3 (EPR18810) Rabbit mAb | Abcam | ab195352 |
| p-METTL3 (S43) | Lifetein | customized |
| METTL14 (D8K8W) Rabbit mAb | Cell Signaling | 51104 |
| WTAP (4A10G9) Mouse mAb | Proteintech | 60188 |
| USP5 (EPR10454) Rabbit mAb | Abcam | Ab154170 |
| IgG Isotype control (DA1E) Rabbit mAb | Cell signaling | 3900 |
| Necleolin (D4C7O) Rabbit mAb | Cell signaling | 14574 |
| ERK2 Rabbit Ab | Cell signaling | 9108 |
| ERK5 (D315V) Rabbit mAb | Cell signaling | 12950 |
| p38 (D13E1) Rabbit mAb | Cell signaling | 8690 |
| JNK2 (56G8) Rabbit mAb | Cell signaling | 9258 |
| Alexa 488- SSEA-1 (MC480) Mouse mAb | BioLegend | 125609 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phos-tag Acrylamide | Wako | AAL-107 |
| cycloheximide | Sigma-Aldrich | 01810 |
| Actinomycin D | Sigma-Aldrich | A1410 |
| MG-132 | Sigma-Aldrich | 474787 |
| dabrafenib | Selleck Chemicals | S2807 |
| PLX-4720 | Selleck Chemicals | S1152 |
| PD0325901 | Selleck Chemicals | S1036 |
| Trametinib | Selleck Chemicals | S2673 |
| EOAI3402143 | MedchemExpress | HY-111408 |
| Vialinin A | Cayman Chemical | 10010519 |
| tucatinib | Selleck Chemicals | S8362 |
| Lapatinib | Selleck Chemicals | S1028 |
| SJB3–019A | MedchemExpress | HY-80012 |
| SPOP-IN-6b | MedchemExpress | HY-122615 |
| PD173074 | Selleck Chemicals | S1264 |
| DAPI | Sigma-Aldrich | D9542 |
| Active ERK2 | SignalChem | M28–10G |
| Active p38a | SignalChem | M39–10BG |
| Active ERK5 | SignalChem | M32–10G |
| Active JNK2 | SignalChem | M34–10BG |
| Active USP5 | SignalChem | U505–380H |
| METTL3/METTL14 | SignalChem | M323–380G |
| Nuclease P1 from Penicillium citrinum | Sigma-Aldrich | N8630 |
| Calf Intestinal Alkaline Phosphatase | New England Biolabs | M0290 |
| Mice FGF-4 | R&D systems | 5846-F4 |
| Cumate | Sigma-Aldrich | 268402 |
| FastAP Thermosensitive Alkaline Phosphatase | Thermo Scientific | EF0654 |
| Critical Commercial Assays | ||
| Dynabeads mRNA DIRECT Kit | ThermoFisher | 61006 |
| EpiMark N6-Methyladenosine Enrichment Kit | New England Biolabs | E1610S |
| TruSeq Stranded mRNA Library Prep | Illumina | 20020594 |
| Luciferase Assay System | ProMega | E1500 |
| PrimeScript RT Reagent Kit | Takara | RR037B |
| QuikChange Site-Directed Mutagenesis Kit | Strategene | 200521 |
| EpiQuik Histone H3 Modification Multiplex Assay Kit | Epigentek | P-3100–96 |
| Deposited Data | ||
| Mendeley dataset | This study | 10.17632/nxgryg682r.1 |
| CRISPR Screening, m6A-MeRIP-seq, and RNA-seq | This study | GSE138776 |
| Experimental Models: Cell Lines | ||
| HeLa | American Type Culture Collection ATCC (ATCC) |
CCL-2 |
| 293T | ATCC | CRL-3216 |
| 293TN | System Biosciences | LV900A-1 |
| Mouse ESC METTL3KO | Provided by Howard Y. Chang | N/A |
| A375 | Provided by Dr. Yu-Ying He | CRL-1619 |
| CHL-1 | ATCC | CRL-9446 |
| MEL-624 | Provided by Dr. Yu-Ying He | N/A |
| HCT116 | ATCC | CCL-247 |
| MCF7 | ATCC | HTB-22 |
| T47D | ATCC | HTB-133 |
| SKBR3 | ATCC | HTB-30 |
| BT474 | ATCC | HTB-20 |
| Oligonucleotides | ||
| For siRNAs, please see supplemental table S6 | Qiagen | N/A |
| Recombinant DNA | ||
| Circular RNA-GFP reporter | Provided by Z. Wang Laboratory | N/A |
| pCDH-CMV-MCS-EF1α-RFP | System Biosciences | CD512B-1 |
| pCDH-CMV-circRNA-GGACT-EF1α-RFP | Generated in this study | N/A |
| pCDH-CMV- circRNA-GGTCT -EF1α-RFP | Generated in this study | N/A |
| CRISPR KO pooled library | Addgene | 1000000048 |
| Human METTL3 | Addgene | 53739 |
| Human METTL14 | Addgene | 53740 |
| Human WTAP | Addgene | 53741 |
| pKMyc | Addgene | 19400 |
| Human Flag-ATM | Addgene | 31985 |
| Human ATR | Addgene | 31611 |
| Human Flag-IKKε | Addgene | 26201 |
| Human HA-GSK-3β | Addgene | 14754 |
| Human ERK1 | Addgene | 23509 |
| Human ERK2 | Addgene | 23498 |
| Human B-Raf V600E | Addgene | 17544 |
| Human HA-Ubiquitin | Addgene | 17608 |
| Human Ubiquitin-KO | Addgene | 17603 |
| Human Ubiquitin-K6 | Addgene | 22900 |
| Human Ubiquitin-K11 | Addgene | 22901 |
| Human Ubiquitin-K27 | Addgene | 22902 |
| Human Ubiquitin-K29 | Addgene | 22903 |
| Human Ubiquitin-K33 | Addgene | 17603 |
| Human Ubiquitin-K48 | Addgene | 16705 |
| Human Ubiquitin-K63 | Addgene | 17606 |
| pMD2.G | Addgene | 12259 |
| psPAX2 | Addgene | 12260 |
| Human Flag-IKKα | Provided by M.C. Hung Laboratory | N/A |
| Human Flag-IKKβ | Provided by M.C. Hung Laboratory | N/A |
| Human HA-AKT | Provided by M.C. Hung Laboratory | N/A |
| Human Flag-mTOR | Provided by M.C. Hung Laboratory | N/A |
| Human HA-MEKDD | Provided by M.C. Hung Laboratory | N/A |
| Human HA-CDC2 | Provided by M.C. Hung Laboratory | N/A |
| Human FAK | Provided by M.C. Hung Laboratory | N/A |
| Human EGFR | Provided by M.C. Hung Laboratory | N/A |
| Human HER2 V659E | Provided by M.C. Hung Laboratory | N/A |
| Human pCMV5-HA | Provided by M.C. Hung Laboratory | N/A |
| Human pCMV5-Flag | Provided by M.C. Hung Laboratory | N/A |
| pLightSwitch R01_3’UTR | Switchgear Genomics | S890001 |
| pLightSwitch NANOG_3’UTR | Switchgear Genomics | S807466 |
| Mouse METTL3 | OriGene | MR209093 |
| Mouse WTAP | OriGene | MR216877 |
| Human USP5 | OriGene | RC224191 |
| Human pKMyc-METTL3 | Generated in this study | N/A |
| Mouse pKMyc-METTL3 | Generated in this study | N/A |
| Human pCMV5-HA-METTL14 | Generated in this study | N/A |
| Human pCMV5-Flag-WTAP | Generated in this study | N/A |
| Mouse pCMV5-Flag-WTAP | Generated in this study | N/A |
| Human pCMV5-Flag-ERK1 | Generated in this study | N/A |
| Human pCMV5-Flag-ERK2 | Generated in this study | N/A |
| Human pCMV5-Flag-USP5 | Generated in this study | N/A |
| Tet-pLKO-puro | Addgene | 21915 |
| Human Tet-On-shMETTL3 | Generated in this study | N/A |
| Human Tet-On-shWTAP | Generated in this study | N/A |
| pCDH-CuO-MCS-EF1α-RFP | System Biosciences | QM512B-1 |
| pCDG-EF1α-CymR-T2A-Neo | System Biosciences | QM400PA-2 |
| pLenti-DsRed_IRES_SNCA:EGFP | Addgene | 92195 |
| Human pLenti-DsRed-IRES-METTL3-EGFP reporter | Generated in this study | N/A |
| Software and Algorithms | ||
| FlowJo | Treestar | https://www.flowjo.com |
| RIGER | Broad Institute | https://software.broadinstitute.org/GENE-E/download.html |
| HISAT2 | CCB JHU | https://ccb.jhu.edu/software/hisat2/index.shtml |
| DESeq2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| exomePeak R | Bioconductor | https://bioconductor.org/packages/release/bioc/html/exomePeak.html |
| Guitar | Bioconductor | https://bioconductor.org/packages/release/bioc/html/Guitar.html |
| RADAR | GitHub | https://scottzijiezhang.github.io/RADARmanual/Mannual.htm |
| MeRIPtools | GitHub | https://scottzijiezhang.github.io/MeRIPtoolsManual/index.html |
| Homer | UCSD | http://homer.ucsd.edu/homer/motif/ |
| Gene set enrichment analysis (GSEA) | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| GenePattern | N/A | https://cloud.genepattern.org/gp/pages/index.jsf |
Highlights.
Activation of the ERK pathway phosphorylates METTL3 and WTAP.
METTL3 phosphorylation increases USP5-mediated deubiquitination to stabilize METTL3.
Phosphorylation of METTL3 promotes mESC differentiation.
METTL3 phosphorylation in ERK-activated tumor cells contributes to tumorigenesis.
Acknowledgements
We thank the Genomics Facility, Integrated Light Microscopy Core Facility, and Flow Cytometry Core Facility at the University of Chicago for technical support. We also thank the Mass Spectrometry Facility at Ohio State University. This work was supported by NIH grants F32 CA221007 (B.T.H.), CA247175 (A.C.Z.), and HG008935 (C.H.). C.H. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
C.H. is a scientific founder and a member of the scientific advisory board of Accent Therapeutics, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, Lee DF, Chen CH, Rengasamy M, Andino B, et al. (2015). Coordination of m(6)A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell 17, 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov GV, Walker JR, Xue S, Allali-Hassani A, Asinas A, Nair UB, Fang X, Zuo X, Wang YX, Wilkinson KD, et al. (2012). Two ZnF-UBP domains in isopeptidase T (USP5). Biochemistry 51, 1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, and Andersen JS (2010). Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics 9, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de Los Mozos IR, Sadee C, et al. (2018). The SMAD2/3 interactome reveals that TGFbeta controls m(6)A mRNA methylation in pluripotency. Nature 555, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casar B, Pinto A, and Crespo P (2009). ERK dimers and scaffold proteins: unexpected partners for a forgotten (cytoplasmic) task. Cell Cycle 8, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al. (2018). mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X., Wei Z., Zhang L., Liu H., Sun L., Zhang SW., Huang Y., and Meng J. (2016). Guitar: An R/Bioconductor Package for Gene Annotation Guided Transcriptomic Analysis of RNA-Related Genomic Features. Biomed Res Int 2016, 8367534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jesus DF, Zhang Z, Kahraman S, Brown NK, Chen M, Hu J, Gupta MK, He C, and Kulkarni RN (2019). m6A mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nature Metabolism 1, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, Liu H, Deng Q, Wu X, Lan P, et al. (2019). m(6)A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Onco Targets Ther 12, 4391–4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desideri E, Cavallo AL, and Baccarini M (2015). Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 161, 967–970. [DOI] [PubMed] [Google Scholar]
- Di Timoteo G, Dattilo D, Centron-Broco A, Colantoni A, Guarnacci M, Rossi F, Incarnato D, Oliviero S, Fatica A, Morlando M, et al. (2020). Modulation of circRNA Metabolism by m(6)A Modification. Cell Rep 31, 107641. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, Li L, Chen R, Wang Y, Deng R, et al. (2018). SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res 46, 5195–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Q, Zou Z, Roundtree IA, Sun HL, and He C (2020). YTHDF2 promotes mitotic entry and is regulated by cell cycle mediators. PLoS Biol 18, e3000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Li Q, Meng R, Yi B, and Xu Q (2018). METTL3 regulates alternative splicing of MyD88 upon the lipopolysaccharide-induced inflammatory response in human dental pulp cells. J Cell Mol Med 22, 2558–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Harada BT, Behm M, and He C (2018). RNA modifications modulate gene expression during development. Science 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Guillamot M, Ouazia D, Dolgalev I, Yeung ST, Kourtis N, Dai Y, Corrigan K, Zea-Redondo L, Saraf A, Florens L, et al. (2019). The E3 ubiquitin ligase SPOP controls resolution of systemic inflammation by triggering MYD88 degradation. Nat Immunol 20, 1196–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, and Pawson T (2012). Modular evolution of phosphorylation-based signalling systems. Philos Trans R Soc Lond B Biol Sci 367, 2540–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Williamson A, Banerjee S, Philipp I, and Rape M (2008). Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, and Zhang F (2017). Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12, 828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, and Koike T (2006). Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5, 749–757. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Ohsugi M, Sasako T, Awazawa M, Umehara T, Iwane A, Kobayashi N, Okazaki Y, Kubota N, Suzuki R, et al. (2018). The RNA Methyltransferase Complex of WTAP, METTL3, and METTL14 Regulates Mitotic Clonal Expansion in Adipogenesis. Mol Cell Biol 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath T, Saba-El-Leil MK, Almousailleakh M, Wray J, Meloche S, and Smith A (2007). FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 134, 2895–2902. [DOI] [PubMed] [Google Scholar]
- Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, et al. (2007). IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455. [DOI] [PubMed] [Google Scholar]
- Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, and Tzivion G (2007). Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta 1773, 1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever J, Zhao EY, Grewal J, Jones MR, and Jones SJM (2019). CancerMine: a literature-mined resource for drivers, oncogenes and tumor suppressors in cancer. Nat Methods 16, 505–507. [DOI] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Choe J, Du P, Triboulet R, and Gregory RI (2016). The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell 62, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, Saborowski M, Kastenhuber E, Fellmann C, Ohara K, et al. (2014). Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell 25, 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Yang X, Geng M, and Huang M (2018a). Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B 8, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al. (2018b). m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol 20, 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzahn MR, Marada S, Lee J, Nourse A, Kenrick S, Zhao H, Ben-Nissan G, Kolaitis RM, Peters JL, Pounds S, et al. (2016). Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J 35, 1254–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- Meng J, Cui X, Rao MK, Chen Y, and Huang Y (2013). Exome-based analysis for RNA epigenome sequencing data. Bioinformatics 29, 1565–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potu H, Peterson LF, Pal A, Verhaegen M, Cao J, Talpaz M, and Donato NJ (2014). Usp5 links suppression of p53 and FAS levels in melanoma to the BRAF pathway. Oncotarget 5, 5559–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raasi S, Varadan R, Fushman D, and Pickart CM (2005). Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol 12, 708–714. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2012). ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res 66, 105–143. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia R, Sorzano COS, Carazo JM, and Segura J (2019). BIPSPI: a method for the prediction of partner-specific protein-protein interfaces. Bioinformatics 35, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, van Akkooi ACJ, Berking C, Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A, and Ugurel S (2018). Melanoma. Lancet 392, 971–984. [DOI] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, and He C (2017). YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 27, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HL, Cui R, Zhou J, Teng KY, Hsiao YH, Nakanishi K, Fassan M, Luo Z, Shi G, Tili E, et al. (2016). ERK Activation Globally Downregulates miRNAs through Phosphorylating Exportin-5. Cancer Cell 30, 723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V, and Kirtikara K (2006). Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wang H., Hu X., Huang M., Liu J., Gu Y., Ma L., Zhou Q., and Cao X. (2019). Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun 10, 1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, and Nam Y (2016). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell 63, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014a). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014b). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, et al. (2018). N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat Neurosci 21, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Fried J, Li Y, Hu L, Gao M, Zhang S, and Xu B (2018). Functional roles of Speckle-Type Poz (SPOP) Protein in Genomic stability. J Cancer 9, 3257–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Williamson A, Meyer HJ, Kelly A, and Rape M (2011). K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol 21, 656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. (2017). RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell 61, 507–519. [DOI] [PubMed] [Google Scholar]
- Yang SH, Jaffray E, Hay RT, and Sharrocks AD (2003). Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol Cell 12, 63–74. [DOI] [PubMed] [Google Scholar]
- Yang SH, and Sharrocks AD (2010). MAP Kinase: SUMO pathway interactions. Methods Mol Biol 661, 343–367. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y, et al. (2017). Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res 27, 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, and Elledge SJ (2008). Global protein stability profiling in mammalian cells. Science 322, 918–923. [DOI] [PubMed] [Google Scholar]
- Ying QL, Stavridis M, Griffiths D, Li M, and Smith A (2003). Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat Biotechnol 21, 183–186. [DOI] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, and Semenza GL (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A 113, E2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gu X, Li D, Cai L, and Xu Q (2019a). METTL3 Regulates Osteoblast Differentiation and Inflammatory Response via Smad Signaling and MAPK Signaling. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhan Q, Eckert M, Zhu A, Chryplewicz A, De Jesus DF, Ren D, Kulkarni RN, Lengyel E, He C, et al. (2019b). RADAR: differential analysis of MeRIP-seq data with a random effect model. Genome Biol 20, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou KI., Shi H., Lyu R., Wylder AC., Matuszek Z., Pan JN., He C., Parisien M., and Pan T. (2019). Regulation of Co-transcriptional Pre-mRNA Splicing by m(6)A through the Low-Complexity Protein hnRNPG. Mol Cell 76, 70–81 e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong X, Zhao J, Wang H, Lu Z, Wang F, Du H, and Wang Y (2019). Mettl3 Deficiency Sustains Long-Chain Fatty Acid Absorption through Suppressing Traf6-Dependent Inflammation Response. J Immunol 202, 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of top 1000 sgRNA Identified in the Low and High GFP populations, related to Figure 1
Table S3. List of the Genes with Significant m6A Peak Changed Comparing R-WT and R-3A2A mESCs. Coordinates of the Peak, in Mouse Genome mm10, the Beta Regression Coefficient, and the p-value are Displayed, related to Figure 5.
Table S4. Summary of the Genes Identified by GSEA Related to Histone Binding Proteins, related to Figure 5.
Table S2. List of sgRNA Identified Related to Ras and MAPK Pathways, related to Figure 1
Table S5. Primers for RT-qPCR of mESCs, related to Figure 4.
Table S6. Information for siRNA, related to Figures 2 and 3.
Data Availability Statement
The CRISPR screening and m6A-seq data generated during this study are available at GSE138776. The human data for the skin cutaneous melanoma (SKCM) was derived from the The Cancer Genome Atlas (TCGA).