

Abstract
Ca2+ is an essential signal for pancreatic β-cell function. Ca2+ plays critical roles in numerous β-cell pathways such as insulin secretion, transcription, metabolism, endoplasmic reticulum function and the stress response. Therefore, β-cell Ca2+ handling is tightly controlled. At the plasma membrane Ca2+ entry primarily occurs through voltage-dependent Ca2+ channels (VDCCs). VDCC activity is dependent on orchestrated fluctuations in the plasma membrane potential or voltage, which are mediated via the activity of many ion channels. During the pathogenesis of type-2 diabetes (T2D) the β-cell is exposed to stressful conditions, which result in alterations of Ca2+ handling. Some of the changes in β-cell Ca2+ handling that occur under stress result from perturbations in ion channel activity, expression or localization. Defective Ca2+ signaling in the diabetic β-cell alters function, limits insulin secretion and exacerbates hyperglycemia. In this review, we focus on the β-cell ion channels that control Ca2+ handling and how they impact β-cell dysfunction in T2D.
Keywords: calcium, insulin, glucagon, glucose, β-cells, secretion, metabolism
Graphical Abstract
Introduction
The islets of Langerhans are micro organs embedded in the pancreas and contain endocrine cells which secrete hormones to regulate blood glucose. In rodents the most abundant cell type in the islet is the β-cell which secretes insulin to lower blood glucose and is located in the core of the islet. This is followed by the α-cell which is found in the mantle of the islet and secretes glucagon to elevate blood glucose levels. Other minor cell types, including δ-cells, ε-cells, and PP cells secrete somatostatin, ghrelin, and pancreatic polypeptide respectively. These hormones are released into the circulation to coordinate glucose storage and utilization in target tissues but also act in an autocrine or paracrine manner within the islets for feedback regulation to achieve glucose homeostasis in changing metabolic environment. Here, it is worth noting that compared to rodent islets, human islets have lower β- to α-cell ratios and β-cells form clusters that are interspersed with non-β cells rather than residing in the core; these distinct architectures likely impact signaling between cell types in the different species.
The secretory activity of each endocrine cell type is controlled mostly by ion channels. These ion channels control K+, Na+, Cl−, and Ca2+ fluxes to generate electrical and Ca2+ signals needed for secretion. The ionic mechanisms underlying secretion regulation is best understood in β-cells, as they are the sole source of insulin and indispensable for glucose regulation. β-cells are equipped with a large repertoire of channels that can be regulated at the level of transcription, translation, post-translational modifications and intracellular trafficking to respond to extracellular and intracellular signals and fine tune their secretory activities. Over sixty different ion channels are known to be expressed. Their roles in insulin secretion in physiological and pathological conditions are of primary interest in the islet biology field.
Type 2 diabetes mellitus is a complex chronic disease often associated with obesity. Initiation and progression of the disease likely involve many genetic and environmental factors. It is clear that both insulin resistance and failure of β-cells to secrete sufficient amount of insulin to curtail hyperglycemia are the two main drivers of T2D. In T2D, plasma glucose and insulin secretion become decoupled. Studies of extreme phenotypes such as monogenic forms of diabetes, genome-wide association studies, and profiling gene expression changes have implicated many ion channels and their regulatory proteins in the cause or acceleration of the disease. Understanding the ionic mechanisms that underlie this pathology is critically important for identifying therapeutic targets for the prevention and treatment of T2D.
This review will discuss ion channels that have been shown or proposed to contribute to β-cell dysfunction and T2D based on genetic and functional evidence from animal and human studies. In particular, we highlight ion channels that control Ca2+ entry necessary for secretion and how metabolic changes associated with insulin resistance alter ion channel regulation and Ca2+ handling to cause defective secretion. The primary focus will be on β-cells but other islet cell types and their involvement in T2D resulting from ion channel defects are also considered.
Ion channels in secretagogue-stimulated electrical excitability
β-cell plasma membrane electrical excitability is required for physiological glucose-stimulated Ca2+ entry and insulin secretion from β-cells. Electrical excitability results from changes in β-cell membrane potential (Vm) that is tightly controlled by the orchestrated opening and closing of many ion channels during glucose-stimulation (Figure 1). At rest the β-cell membrane sits near - 70 mV due to the activities of KATP channels and a slight sodium leak through other small conductance channels. During glucose-induced increases in the ATP/ADP ratio KATP channel closure leads to depolarization due to cessation of K+ efflux but continued Na+ leak[1]. Glucose metabolism also results in β-cell swelling that activates the volume regulatory Cl− current through SWELL1, leading to Cl− efflux and further Vm depolarization[2]. In human β-cells depolarization activates voltage-dependent Ca2+ channels (VDCCs) and voltage-gated Na+ channels (VGSCs) resulting in the upstroke of the action potential (AP); the AP is the primary electrical signal of the β-cell. The AP upstroke depolarizes the Vm and allows even more Ca2+ entry via VDCC activation activating big conductance K+ (BK) channels, which repolarizes the AP. During glucose-stimulation the Vm sits at a depolarized plateau potential where APs can rapidly fire leading to sustained Ca2+ entry and insulin secretion. A drop in blood glucose levels results in a reduction in the β-cell ATP/ADP ratio leading to activation of KATP channels, which causes Vm hyperpolarization and terminates electrical excitability.
Figure 1:

Ion channels that contribute to the human β-cell action potential. Glucose is taken up and metabolized in β-cells resulting in both an increase in the ATP/ADP ratio as well as cell swelling. The increase in the ATP/ADP ratio leads to KATP closure whereas swelling activates SWELL-1 Cl- efflux. Reduced KATP activity together with the depolarizing influences of SWELL1 mediated Cl- efflux and Na+ influx through Na+ leak channels results in Vm depolarization. Vm depolarization activates voltage-dependent Ca2+ channels (VDCCs) and voltage-gated Na+ channels (VGSCs) that are responsible for the upstroke of the action potential (AP); a representative human β-cell AP is shown on the right side of the β-cell. The rapid Vm depolarization as well as Ca2+ entry that occurs during the AP upstroke activates K+ channels including BK, KV, SK and IK. BK channels are voltage and calcium activated, which provide the largest conductance and fastest activating K+ current that is responsible for the initiation of AP repolarization. KV channels are activated by Vm depolarization and also contribute to AP repolarization. Finally, the Ca2+ activated K+ channels including SK and IK are smaller conductance K+ currents that stay active for longer following AP repolarization leading to afterhyperpolarization and a reduction in AP frequency.
β-cell excitotoxicity
Insulin resistance results in increased β-cell electrical excitability and insulin secretion during the pathogenesis of diabetes. Too much electrical excitability and excessive Ca2+ entry causes disruptions in β-cell Ca2+ handling resulting in a stressed condition which is referred to here as β-cell excitotoxicity. During β-cell excitotoxicity the resulting changes in Ca2+ homeostasis leads to changes in ion channel activity as well as significant changes in many signaling cascades (e.g. mitochondrial dysfunction). These stressful conditions result in elevated cytokine secretion (such as from immune cells) that further impact β-cell ion channel activity and Ca2+ handling, which negatively impacts insulin secretion. Moreover, the elevations in cytoplasmic Ca2+ during excitotoxicity result in transcriptional changes that also impact ion channel activity and thus Ca2+ handling. Single cell transcriptome analysis has found that the transcription of mouse β-cell ion channels changes dramatically in response to short term insulin resistance associated with a high fat diet [3]. For example, VDCCs such as Cacna1d that encodes Cav1.3 are significantly reduced [3]. Whereas K+ channel transcripts such as KCNK16 and Kcnb1 that encode TALK-1 and Kv2.1 respectively are increased[3]. β-cell transcriptome studies have also been performed on humans with or without T2D and ion channels transcript abundance are also different between these groups[4, 5]. As the human transcriptomes are from patients with long standing diabetes, the changes in β-cell ion channel transcript abundance are different from short term insulin resistance changes in rodents. For example the Transient Receptor Potential Cation Channel Subfamily M Member 3 (TRPM3) is significantly downregulated in human β-cells from T2D patients[4]. The stressful conditions of elevated β-cell electrical excitability in T2D lead to many changes that eventually impact the failure of β-cells to meet the increased demand for insulin[6]. Importantly, β-cell excitotoxicity is not only due to changes in ion channel function but also due to changes in β-cell nutrient utilization and ER stress that occur during the pathogenesis of T2D.
β-cell ion channels that control and disrupt calcium entry in T2D
KATP Channels
KATP channels are weakly inwardly rectifying K+ channels present in the β-cell plasma membrane. Activities of KATP channels are controlled by intracellular ATP and ADP, the ratio of which fluctuates according to blood glucose levels. This enables the channel to serve as a metabolic sensor and couples serum glucose to insulin secretion. Channel response to changing ATP/ADP ratios is coordinated by the two constituent protein subunits: an inward rectifier Kir6.2 and an ABC transporter protein sulfonylurea receptor 1 (SUR1). Recent high resolution cryo-EM structures show that the channel is a hetero-octamer with four Kir6.2 subunits in the center forming the K+ conducting pore and four SUR1 subunits in the periphery[7–9]. Each SUR1 protein interacts with a Kir6.2 via its N-terminal transmembrane domain (TMD0) which is connected to SUR1’s ABC transporter core structure consisting of two transmembrane domains and two nucleotide binding domains by a long intracellular loop (L0) (Figure 2).
Figure 2:
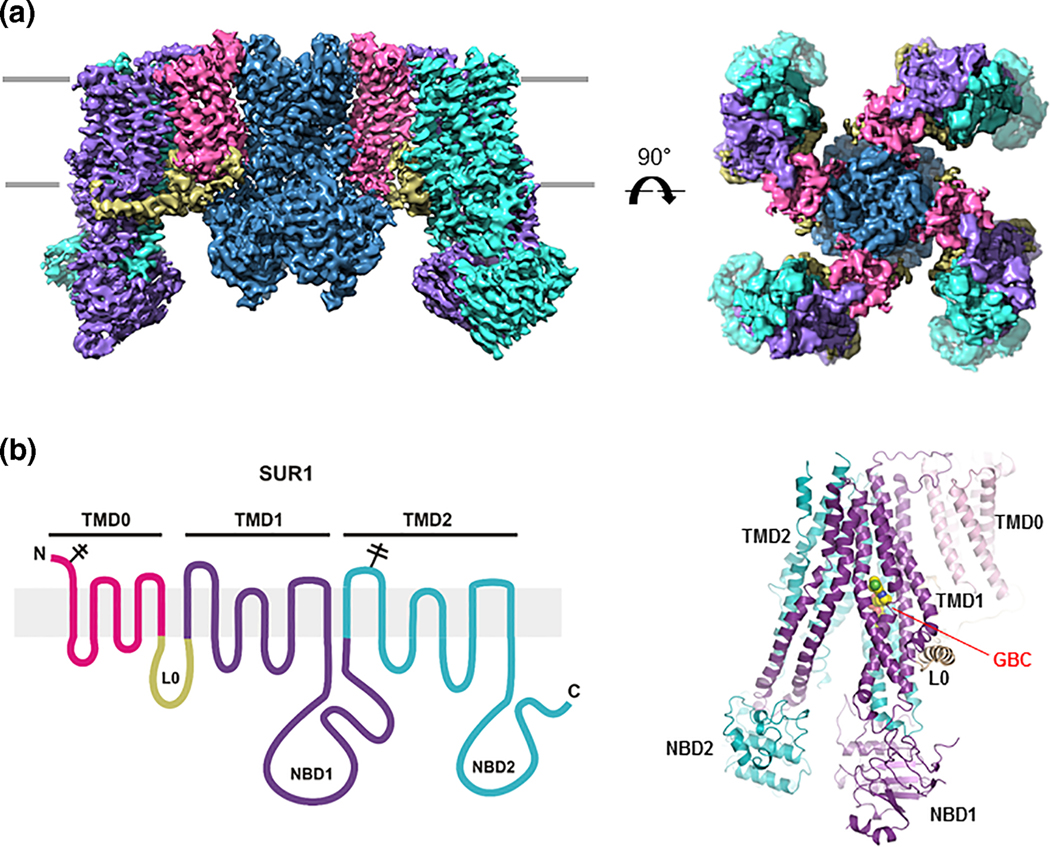
(A) CryoEM structure of the KATP channel viewed from the side (left) and the top (right). (B) Left: SUR1 topology showing the different domains. Right: Structure of SUR1 built from cryoEM density map showing the glibenclamide (GBC) binding site above NBD1 in the transmembrane bundle formed by transmembrane helices from both TMD1 and TMD2.
Both Kir6.2 and SUR1 interact with intracellular adenine nucleotides to regulate channel activity[10, 11]. ATP, and to a much lesser extent ADP, binds to a non-canonical nucleotide binding pocket formed by the N- and C-terminal cytoplasmic domains of two adjacent Kir6.2 subunits and closes the channel [12, 13]. The inhibitory effect of ATP occurs in isolated membrane patches even in the absence of Mg2+ thus does not involve ATP hydrolysis[10]. ATP and ADP also interact with the two nucleotide binding domains (NBDs) of SUR1 to stimulate channel activity in an Mg2+-dependent manner[14]. In the intracellular milieu where most ATP and ADP are in complex with Mg2+, binding of MgATP at NBD1 and MgADP at NBD2 causes dimerization of the two NBDs[9]. This conformational change is transduced to the Kir6.2 pore and antagonizes the ATP inhibition at Kir6.2, resulting in a net gain of channel activity. Thus, channel activities resulting from the tug-of-war between ATP inhibition at Kir6.2 and MgADP stimulation at SUR1 reflect the changes in ATP/ADP ratios in β-cells as the blood glucose concentration swings. It is worth noting that although NBD2 is capable of binding and hydrolyzing MgATP[15, 16], there is evidence that MgATP hydrolysis to MgADP at this site is not required for NBD dimerization and channel stimulation[17, 18]. Thus the mere increased occupancy of MgADP at NBD2 when MgADP concentrations increase following a fall of blood glucose likely underlies the MgADP stimulation effect.
The importance of KATP channels in insulin secretion and glucose regulation is evident from genetic variations in the channel genes (KCNJ11 for Kir6.2 and ABCC8 for SUR1) associated with a spectrum of insulin secretion disorders[19]. Loss-of-function KATP mutations were linked to congenital hyperinsulinism soon after the channel was cloned in 1995[20]. Although it was predicted that gain-of-function mutations would cause the opposite clinical phenotype, i.e. diabetes, direct demonstration did not come until almost ten years later[21]. A pioneer study showing that overexpressing an ATP-insensitive variant of Kir6.2 in mice resulted in a neonatal diabetes phenotype[22] paved the way for subsequent studies that identified many mutations in the Kir6.2 and SUR1 genes in human neonatal diabetes patients. Neonatal diabetes associated mutations in Kir6.2 typically reduce channel inhibition by ATP whereas those in SUR1 tend to increase channel stimulation by MgADP but some have also been shown to reduce ATP inhibition by allosteric mechanisms[23, 24]. In addition to neonatal diabetes, some mutations can also cause neurological defects including developmental delay and epilepsy, referred to as DEND syndrome[23]. This is most likely due to overactivity of these mutant channels in neurons[25] known to express KATP channels rather than secondary effect of hyperglycemia although detailed mechanisms are still not well understood. Aside from causing congenital diabetes, genetic variants in KATP channels have also been associated with MODY (maturity onset diabetes of the young) and one polymorphism in KCNJ11 (rs5219; resulting in E23K; Table 1) is associated with increased risk of T2D in multiple GWAS studies[26, 27]. The effects of the K allele on channel activity and insulin secretion assessed by glucose tolerance tests have been investigated in a number of studies[26, 28–30]. Although some reported a small effect, others failed to detect significant difference compared to the E allele. There is now increased appreciation that tiny effect sizes of many common risk alleles likely contribute to complex diseases such as T2D[31]. Despite strong genetic evidence, proof of genotype-phenotype association may be challenging.
Table 1.
Genetic Variations in Ion Channels Implicated in Diabetes 1
| Gene Name | Common Name | Proposed function in β-cells | Polymorphisms or mutations | Functional effects on the channel | Impact on blood glucose | References |
|---|---|---|---|---|---|---|
| KCNJ11 | Kir6.2 (KATP pore subunit) | setting resting Vm | p.E23K | Gain of function | Increased risk of T2D | [26, 27] |
| ABCC8 | SUR1 (KATP regulatory subunit) | setting resting Vm | p.R370S | Loss of function | Associated with hyperinsulinemia early in life but glucose intolerance or diabetes in adult carriers | [34] |
| ABCC8 | SUR1 (KATP regulatory subunit) | setting resting Vm | p.E1507K | Loss of function | Associated with hyperinsulinemia early in life but glucose intolerance or diabetes in adult carriers | [32, 33] |
| KCNK16 | TALK-1 | Vm; ER K+ flux | p.A277E | Gain of function | Increased risk of T2D | [58, 59, 70] |
| KCNH6 | hERG2 (Kv11.2) | AP repolarization? | P235L | Loss of function | Hyperinsulinemia hypoglycemia early in life and hyperinsulinemia and diabetes in adults | [103] |
| KCNJ9 | Kir3.3 or GIRK3 | GPCR activation induced Vm hyperpolarization | p.V366A | Not determined | Increased risk of T2D in PIMA Indians | [124] |
| SCN9A | Nav1.7 | AP | p.I739V | Gain of function | Two thirds of these patients developed diabetes | [152] |
Note numerous gain of function mutations causing neonatal diabetes have been identified in KCNJ11 and ABCC8; they are not listed in this table.
Worth noting, two dominant inactivating ABCC8 mutations, E1507K and R370S, have been associated with hyperinsulinism early in life but glucose intolerance or diabetes in adult carriers[32–34]. The glucose intolerance may be attributed to glucose-blindness (i.e. impaired insulin secretion responses to acute or ramp glucose stimulations), a phenotype well documented in mice lacking KATP channels[35, 36]. The impaired insulin secretion response to glucose may be a result of chronic elevations of cytosolic Ca2+. Curiously, later large scale studies of dominant congenital hyperinsulinsm-causing mutations did not find significantly increased risk for diabetes later in life[37]. An interesting possibility is that the different findings are due to differences in how the patients were treated for their hyperinsulinemia and hypoglycemia, for example with diazoxide (a KATP channel opener) versus glucose feeding, which would affect Ca2+ levels, hence β-cell function.
KATP channels have been a major therapeutic target for T2D for more than half a century[38]. Sulfonylureas such as glibenclamide (glyburide) and glinides such as repaglinide (prandin) inhibit KATP channel activity to stimulate insulin secretion[39] and remain a first-line T2D treatment, especially in low-income settings[40]. Although long-term use of sulfonylureas frequently results in accelerated β-cell failure likely caused by chronic hyperexcitability and ER stress[41–43], studies in mice suggest this failure is readily reversible without permanent damage to β-cells[44]. Whether periodic stops in sulfonylurea therapy can prolong drug effect in human patients remains to be seen[45]. Since the discovery that gain-of-function KATP mutations underlie neonatal diabetes, glibenclamide has also become a mainstay for treating KATP-neonatal diabetes and DEND syndrome[46]. In addition to sulfonylureas and glinides, carbamazepine, an anticonvulsant best known for its effect to block Nav channels[47], was recently shown to also inhibit KATP channel activity[48]. Carbamazepine competes for glibenclamide binding to KATP channels with an estimated Kd of ~25 nM[49] and abolishes channel response to MgADP[50]. Structural studies using cryoEM have revealed that glibenclamide and carbamazepine as well as repaglinide share a common binding pocket in the SUR1 protein, consistent with these drugs acting via a similar mechanism to inhibit KATP channels[13, 51–53]. Given the similarity between carbamazepine and sulfonylureas in their effects on KATP channels, it would be predicted that carbamazepine would also stimulate insulin secretion. However, since carbamazepine also inhibits Nav channels and may have other actions such as maintaining functional β-cell mass[54], its effect on insulin secretion will require further investigation.
Two-Pore-Domain Potassium (K2P) Channels
K2P channels are small conductance K+ channels that are active at all membrane potentials and thus termed “leak” K+ channels. Human β-cells show high expression of many K2P channels including: TASK-1, TALK-1, TALK-2, and TWIK-1. The K2P channels expressed in islets show outward rectification with small K+ currents at hyperpolarized potentials that increase during depolarization. During glucose-induced inhibition of KATP channels most K2P channels stabilize the plateau potential from where APs fire[35, 55, 56]. Therefore, K2P channels help to regulate the Vm and Ca2+ entry during GSIS (Figure 3)[57]. Although the roles of K2P channels in islet biology are still being elucidated, K2P channel polymorphisms and mutations have been identified that negatively impact β-cell function. For example, a GOF polymorphism in KCNK16 that encodes TALK-1(rs1535500 resulting in A277E) is associated with an increased risk of developing T2D[58, 59]. This is likely due to enhanced β-cell Vm polarization, which is predicted to reduce β-cell Ca2+ entry and insulin secretion. Furthermore, rs1535500 also results in increased expression of the downstream gene KCNK17 in human islets, which encodes TALK-2[60]. This suggests that higher levels of TALK-2 together with greater function of TALK-1 lead to a hyperpolarizing impact on β-cell Vm, which would limit VDCC activity and reduce insulin secretion. Interestingly a gain-of-function mutation in TALK-2 that results in severe cardiac conduction disorder does not cause overt diabetes[61]; however, patients with this mutation would be predicted to show decreased GSIS. Because KCNK16 (that encoded TALK-1) is the most abundant and most β-cell restricted K+ channel transcript, mutations that cause extensive TALK-1 channel gain-of-function would be predicted to cause neonatal diabetes or maturity onset diabetes of the young (MODY).
Figure 3:

Ion channels that control where the β-cell Vm sits. At rest the activity of KATP channels holds the β-cell Vm below the activation threshold for VDCCs. When blood glucose concentrations are elevated closure of KATP leads to Vm depolarization. During glucose-induced KATP closure the constitutive activity of small conductance K2P potassium channels such as TALK-1 and TASK-1 balance the depolarizing conductance from SWELL-1 and Na+ leak channels at a depolarized plateau potential from where VDCCs and VGSCs can efficiently activate, allowing AP firing. Ca2+ entry which occurs during VDCC activation leads to Ca2+ release from the endoplasmic reticulum (ER), which results in activation of Ca2+ activated K+ currents (Kslow) that hyperpolarize the Vm leading to VDCC closure. Kslow currents eventually inactivate in response to the resulting low Ca2+ levels caused by hyperpolarization-induced closure of VDCCs, which results in another wave of depolarization and Ca2+ influx. These β-cell oscillations in Vm and Ca2+ are responsible for pulsatile insulin secretion. K2P channels such as TALK-1 also form functional complexes on the ER membrane. ER TALK-1 channels provide a countercurrent for Ca2+ ions leaving the ER and potentiate ER Ca2+ release. This is important for increasing Kslow activation and therefore loss of TALK-1 channels impairs Ca2+ ER release limiting the Kslow current and increasing Ca2+ oscillation frequency. Other K2P channels expressed in the human β-cell (e.g. TWIK-1 and TALK-2) also presumably play roles in regulating Vm or ER Ca2+ handling.
Interestingly, TALK-1 and TASK-1 channels also form function complexes on the endoplasmic reticulum (ER) membrane (Figure 3). Maintenance of ER Ca2+ (Ca2+ER) homeostasis requires that Ca2+ movement across the ER membrane is balanced with a simultaneous K+ flux in the opposite direction [62–64]. Similar to other ER K+ channels such as the trimeric intracellular cation channels, TRIC-A or TRIC-B [65, 66], TALK-1 and TASK-1 control Ca2+ movement across the ER membrane by balancing Ca2+ER release with K+ flux in the opposite direction, [62–64]. Without this K+ countercurrent, Ca2+ release from the ER rapidly generates a negative charge on the inside of the ER membrane, inhibiting further Ca2+ER release [67–69]. TALK-1 control of Ca2+ER release also impacts the β-cell Vm response to glucose[70]. Glucose-induced Ca2+ entry results in Ca2+ER release leading to activation of Ca2+-activated K+ (KCa2+) channels that repolarize the Vm during the silent phases between oscillations of membrane depolarization (these KCa2+ currents are collectively termed Kslow)[70]. Therefore, TALK-1 channel control of Ca2+ER release slows β-cell Ca2+ oscillation frequency and thus is predicted to tune pulsatile insulin secretion. This suggests that polymorphisms or mutations that impact TALK-1 function would be predicted to impact Ca2+ER handling as well as Vm. Indeed the T2D associated GOF polymorphism (rs1535500, TALK-1-A277E) in KCNK16 results in less Ca2+ER[70]. TALK-1-A277E-induced reduction in Ca2+ER storage also causes an increased ER stress response. Importantly, Ca2+ER homeostasis becomes perturbed during the pathogenesis of T2D as well as T1D, which leads to β-cell dysfunction[71–74]. Furthermore, certain mutations cause monogenic neonatal diabetes by increasing ER stress (e.g. INS-1, and WFS-1)[75, 76]. Therefore, enhanced TALK channel activity at the ER membrane with rs1535500 would be predicted to contribute to ER stress during the pathogenesis of T2D. Other K2P channels may also help control the ER stress response by tuning β-cell Ca2+ER handling.
Although K2P channels are K+ selective ion channels, one of the β-cell K2P channels, TWIK-1, is not only permeable to K+ but undergoes pore collapse under acidic conditions becoming permeable also to Na+ [1]. In β-cells, TWIK-1 channels rapidly cycle between endosomes and the plasma membrane. When TWIK-1 channels reach the plasma membrane from acidic endosomes they are still in a conformation permeable to Na+. Interestingly, because conformational recovery from the pore collapse upon subsequent exposure to physiological extracellular pH (>7) as the channels arrive the plasma membrane is a slow process[1], the TWIK-1 channels are recycled back to the endosomes before they ever lose Na+ permeability and become permeable exclusively to K+ at the plasma membrane [1]. Under low glucose during conditions when KATP channels are active the TWIK-1 Na+ permeability does not significantly influence the β-cell Vm. However, when KATP is inhibited during high glucose conditions the TWIK-1 Na+ permeability helps to depolarize the β-cell Vm. Thus, mouse β-cells deficient for TWIK-1 sit at a more hyperpolarized Vm under stimulatory glucose conditions and are predicted to have less VDCC activity and reduced insulin secretion[1]. This suggests that TWIK-1 could also provide an important Na+ leak current in human β-cells.
Calcium-activated K+ channels (KCa2+)
Calcium-activated K+ (KCa2+) channels play an important role in the repolarizing phase of the β-cell action potential. The KCa2+ channel with the highest expression in human β-cells is the BK channel, which is activated by both depolarization as well as Ca2+ binding. The BK channels are the largest and most rapidly activating K+ current that initiates during the AP upstroke and therefore these channels play an important role repolarizing the β-cell AP. Inhibition of BK channels results in increased human β-cell AP amplitude as well as increased depolarization-induced insulin secretion (during KATP inhibition)[77]. BK channels are also expressed in neurons and LOF mutations in these channels cause epilepsy with cerebellar atrophy due to increased neuronal electrical excitability[78], which may also be the case in β-cells of patients with BK channel LOF; this would be predicted to improve GSIS. Interestingly, GOF mutations in BK also cause epilepsy due to faster AP firing frequency[79], which has been suggested to be due to faster AP repolarization and increased ability to fire subsequent APs[79]. While the role of BK channel GOF mutations on glucose tolerance has not been determined, if BK GOF mutations cause greater AP firing frequency in β-cells as they do in neurons, then this would be predicted to lead to an improvement of glucose tolerance due to greater electrical excitability and Ca2+ entry following glucose stimulation.
Other KCa2+ channels are also expressed in human β-cells including the small-conductance Ca2+-activated potassium channel, SK3, and the intermediate conductance Ca2+-activated K+ channel, IK[80, 81]. These channels limit the frequency of β-cell APs similar to their action in neurons[82–85]. SK3 and IK activation occurs as a consequence of Ca2+ influx during the AP and remains open after termination of the AP leading to after-hyperpolarization (AHP)[86, 87]. AHP limits the firing of another AP and thus reduced AP firing frequency[87]. KCNN3 and KCNN4 the genes that code for SK3 and IK1 are expressed at fairly low levels in human β-cells[80]. Thus, human islet insulin secretion is only modestly increased by IK inhibition and does not change with SK3 inhibition[88]. Therefore, these channels may not contribute substantially to dysfunction of Ca2+ handling in T2D. Gain-of-function mutations in IK channels cause hereditary xerocytosis (HX)[89]. Although certain types of HX are associated with diabetes and overactivity of IK would be predicted to limit insulin secretion[88], HX-associated diabetes is primarily due to pancreatic hemochromatosis and iron deposition[90]. It has also been found that islet SK3 expression is increased in rats following intrauterine growth restriction; a condition which increases the risk of T2D development in adulthood[91]. Therefore, conditions that upregulate β-cell SK3 levels could potentially negatively impact β-cell function by reducing β-cell electrical excitability.
Kv channels
Many voltage-gated K+ (Kv) channels are expressed in human islets[4, 80]. Although Kv currents can readily be recorded from human β-cells, their role in regulating electrical excitability and insulin secretion is controversial[77, 92]. Kv2.1 and Kv2.2 currents are responsible for 65–80% of human β-cell Kv activity and can be inhibited with toxins[77, 92]. Interestingly, stromatoxin inhibition of both Kv2.2 and Kv2.1 has been shown to have little impact on human β-cell electrical activity and GSIS55. However, another Kv2.1 and Kv2.2 toxin inhibitor (guangxitoxin-1E) causes substantial increases of human islet GSIS[92]. Furthermore, a small molecule (RY796) inhibitor of both Kv2.1 and Kv2.2 also enhances human islet GSIS[92]. While the exact roles of Kv2.1 and Kv2.2 channel activity on human β-cell GSIS remain to be determined, the KCNB2 gene that encodes Kv2.2 shows much higher expression (nearly 10 fold greater) levels in β-cells when compared to KCNB1 that encodes Kv2.1[4, 80]. This suggests that Kv2.2 channels may be more important in controlling human β-cell electrical activity; selective Kv2.2 knockdown in human β-cells will help uncover how this channel impacts electrical activity. Interestingly, the expression of human islet Kv2.1 is reduced in T2D[93]. However, as the KCNB1 is expressed at a low level compared to other K+ channel transcripts (e.g. encoding BK, Kir6.2 or TALK-1)[4, 80], the K+ conductance of Kv2.1 would be expected to only modestly impact human β-cell electrical activity[77]. Mutations in KCNB1 have been identified, which result in infantile epilepsy[94–97]. These mutations cause dominant negative inhibition of Kv2.1 or loss of K+ selectivity of the Kv2.1 channel complex[94–97]; both of which result in increased neuronal excitability that leads to generalized or focal seizures[94–97]. Importantly, a mutation in Kv2.1 that results in a constitutively activated[95], nonselective cation channel that is not voltage activated would be predicted to result in a large enhancement of insulin through increased Ca2+ influx. While this has yet to be assessed in serum from these patients, the outstanding resource for patients with KCNB1 mutations (KCNB1.org) could allow a clear assessment of the function or lack of function of Kv2.1 currents during human insulin secretion.
Other Kv channels are also expressed in the human β-cell, which presumably contribute to AP repolarization and could impact β-cell function in T2D patients. For example, the gene that encodes the Kv1.7 channel is in a region on chromosome 19 (19q13.3) that has SNPs linked to an increased susceptibility for developing T2D[98]. Furthermore, blocking Kv1.7 in rodent islets increases AP firing frequency and insulin secretion[99]. Other Kv channels that are expressed in and regulate human β-cell function are the human ether-á-go-go-(HERG) and related gene encoding Kv11.1 and Kv11.2 respectively[4, 80], which are also important cardiac or neuron repolarizing K+ channels[100, 101]. Importantly, loss-of-function Kv11.1 mutations that cause long-QT syndrome result in increased insulin secretion and hypoglycemia[102]. This suggests that Kv11.1 inhibits GSIS presumably through polarizing influences on the human β-cell Vm[102]. Interestingly, however, loss-of-function mutations in KCNH6 the gene encoding Kv11.2 causes maturity onset diabetes of the young[103]. The diabetic phenotype occurs in adults and individuals with these mutations also show hyperinsulinemia as children[103, 104]. Similarly, mice with a Kv11.2 LOF mutation show hyperinsulinemia after birth followed by hypoinsulinemia as adults[103]. Thus, Kv11.2 LOF induces enhanced Ca2+ entry into β-cells that results in increased insulin secretion as well as ER stress over time leading to β-cell failure and diabetes[103]. As Kv11.x GOF mutations would be predicted to limit VDCC activity and reduce insulin secretion, these mutations would be predicted to also result in a diabetic phenotype.
Kv proteins have also been shown to have roles independent of K+ flux. For example, Kv2.1 and Kv2.2 channels interact with vesicle-associated membrane protein-associated proteins isoforms A and B (VAPA and VAPB) to enable close association of the ER and plasma membrane[105, 106], which presumably could control Ca2+ handling and/or influence actin dynamics of the human β-cell. Furthermore, Kv2.1 has also been shown to interact with SNARE machinery (e.g. Syntaxin-1A and SNAP-25)[107, 108], recruit insulin granules to the plasma membrane, and enhance GSIS[109]. SNARE machinery such as Syntaxin-1A has also been shown to bind many other ion channels such as VDCCs and the KATP channel complex to modulate their function or surface expression, which may also impact insulin secretion[110, 111]. Although KCNB1 the gene encoding Kv2.1 is expressed at much lower levels than the other β-cell ion channels that bind syntaxin-1A, reduced T2D islet KCNB1 has been suggested to negatively impact human β-cell insulin secretion through reduced Kv2.1 syntaxin-1A interactions[93]. This suggests patients with a truncation of the C-terminus of Kv2.1 (such as chr20:47991009delG,p.S363fs*13; which does not contain a sytaxin-1A binding site) may show significantly impaired GSIS and a predisposition for developing diabetes[112](KCNB1.org).
G protein-gated inwardly rectifying K+ (GIRK) channels
GIRK channels are activated by Gi/o-coupled receptors when they release Gβγ[113, 114]. Two primary types of Gi-coupled receptors found in human β-cells are the somatostatin receptors (SSTRs) and the α2 adrenoreceptors (α2-ADRs)[115–118]. Therefore, somatostatin and epinephrine activate β-cell GIRK channels[119], which cause β-cell Vm hyperpolarization and reduced VDCC activity[113, 120]. The importance of GIRK control of β-cell Ca2+ handling is exemplified by the increase in Ca2+ entry and insulin secretion that occurs during Gi receptor signaling inhibition with pertussis toxin (PTX; also termed islet-activating protein)[121, 122]. Although GIRK has not been directly linked to T2D, a polymorphism in the α2A-ADR encoding gene ADRA2A (rs553668), is associated with an increased risk of developing T2D[123]. Human islets from carriers of the rs553668 polymorphism exhibit reduced insulin secretion and greater levels of α2A-ADR[123], which causes enhanced GIRK activation and thus hyperpolarizes the β-cell reducing VDCC activity. Mutations and polymorphisms in the genes that encode GIRK channels have also been identified, which may impact glucose homeostasis. For example, single nucleotide polymorphisms in KCNJ9 the gene encoding GIRK3 show significant correlation with increased risk of developing diabetes in a PIMA Indian population[124]. Furthermore, a gain-of-function mutation in KCNJ5 that encodes the GIRK4 channel causes Familial Sinus Node Disease[125] but has not yet been shown to disrupt glucose homeostasis in these patients. As GIRK4 is expressed in human β-cells[80, 120], over activity of this channel would be predicted to inhibit insulin secretion.
GIRK channels are also regulated by signals other than Gβγ such as phosphatidylinositides, cholesterol and intracellular Na+ [126–128]. Thus their activity changes under conditions associated with T2D may also lead to perturbations in β-cell function. For example phosphatidylinositol 4,5-bisphosphate (PIP2) is required for GIRK channel opening[129–131]. As insulin resistance that occurs during the pathogenesis of T2D leads to inhibition of insulin-induced activation of the PI3K/AKT pathway[132], this may reduce GIRK activity. Furthermore certain GIRK channels activity is enhanced during conditions of high intracellular Na+ [126, 133, 134]. Because intracellular Na+ concentration increases during excessive electrical excitation that occurs during the excitotoxic conditions associated with T2D, Na+-mediated activation of GIRK may eventually lead to β-cell polarization and limit VDCC activity[6]. On the other hand, GIRK activation under excitotoxic circumstances may also help protect β-cells from apoptosis by reducing the excessive Ca2+ entry[135]. Finally, cholesterol activates GIRK conductance and thus changes in cholesterol typically observed in patients with T2D could result in overactive β-cell GIRK, which would also reduce VDCC activity and insulin secretion[136].
Ca2+ channels
Voltage-dependent Ca2+ channels are the primary entry point for β-cell Ca2+ during GSIS. As Ca2+ entry is essential for β-cell GSIS, blocking VDCCs impairs insulin secretion[137]. The VDCCs that have been implicated in GSIS include: the L-type channels CaV1.2 and CaV1.3; the P/Q-type channel CaV2.1; and the T-type channel CaV3.2[138]. Indeed, gain-of-function mutations in VDCCs have been shown to result in hyperinsulinemia and hypoglycemia. For example a GOF mutation in the CaV1.2 channel associated with Timothy syndrome results in intermittent hypoglycemia, which may be due to enhanced insulin secretion[139]. Furthermore, a gain-of-function mutation in the CaV1.3 channel also results in hyperinsulinemia and hypoglycemia[140]. As enhanced VDCC activation causes increased insulin secretion, reduced VDCC activity would be predicted to reduce insulin secretion and result in diabetic phenotypes. Indeed, VDCC transcript abundance has been shown to be reduced in T2D. Expression of the CACNA1D gene that encodes CaV1.3 is reduced in T2D islets, which correlates with reduced insulin secretion[141]. This is due to polymorphisms in the promoter of CACNA1D that result in reduced expression of the CACNA1D gene[141]. The CACNA1D gene shows open chromatin structure in human islets and changes in transcription factor binding to this promotor are proposed to reduce CaV1.3 levels[141, 142]. As reduced CaV1.3 levels are associated with T2D, it would be predicted that a loss-of-function mutation in CaV1.3 would also disrupt insulin secretion and result in a diabetic phenotype[143]. Although CaV1.3 LOF mutations cause deafness, how CaV1.3 LOF mutations impact glucose homeostasis has not been assessed[143].
Aside from genetic contributions, during the pathogenesis of diabetes signals that regulate VDCC activity change and likely impact β-cell Ca2+ influx and insulin secretion. For example, PIP2 β-cell increases VDCC activity[144]. As detailed above, PIP2 signaling becomes disrupted under the insulin resistant conditions of T2D due to reduced insulin-induced activation of the β-cell PI3K/AKT pathway[132]. Therefore, reductions in β-cell PIP2 levels would be predicted to impair VDCC activity in T2D. Furthermore, inflammatory cytokines are known to increase in T2D and cytokines impact VDCC activity. Short term exposure of β-cells to cytokines has been shown to increase low-voltage activated VDCC currents[145], which would be expected to actually enhance insulin secretion. However, elevated basal β-cell Ca2+ levels on a background of insulin hypersecretion may also have negative impacts such as enhancing β-cell ER-stress and mitochondrial dysfunction causing deterioration of β-cell health[146]. Indeed, VDCC inhibition with verapamil has been found to protect β-cells from the stressful conditions of T2D. While this is due in-part to verapamil-mediated repression of thioredoxin-interacting protein expression that results in reduced oxidative stress[135], other agents such as diazoxide that also limit VDCC activity by hyperpolarizing the β-cell Vm protect β-cells from failure under T2D conditions[147, 148]. It is known that during the early stage of T2D development, β-cells compensate for insulin resistance by secreting more insulin through elevated total Ca2+ influx. Thus, the relationship between VDCC activity and β-cell function in T2D may depend on the stage of the disease[6].
While changes in VDCC activity can result in T2D [6, 141], there are also changes in location of VDCCs to the insulin granule that are negatively impacted during the pathogenesis of T2D[149]. During insulin secretion VDCCs cluster near granule release sites[149]. β-cell L-type VDCC clusters of 15 to 20 channel complexes form near insulin granules during secretion[149]. VDCC clustering allows for greater depolarization-induced Ca2+ entry near docked insulin granules, which helps to promote insulin granule fusion with the plasma membrane[149]. In T2D β-cells Ca2+ influx near granules is disrupted and total depolarization-induced Ca2+ entry is reduced[149]. Ca2+ entry at the plasma membrane micro domains near insulin granules in response to membrane depolarization is also significantly reduced in T2D β-cells[149]. Furthermore, insulin granule release does not preferentially occur near Ca2+ entry sites in T2D[149]. Therefore, both Ca2+ handling and granule location to hot spots for Ca2+ entry following secretagogue stimulation are perturbed in T2D β-cells.
Na+ Channels
Voltage-gated Na+ channels play an important role in controlling action potential firing in human β-cells. Thus, inhibition of VGSCs results in reduced human β-cell AP amplitude, diminished Ca2+ entry, and reduced insulin secretion [77]. β-cell ablation of rodent VGSC encoding α-subunits (e.g. Scn3a) or β-subunits (e.g. Scn1b) also leads to reduced GSIS[150, 151]. While mutations or polymorphisms in VGSC encoding genes have not been shown to increase diabetes risk, a gain-of-function mutation in Nav1.7 (I739V) has been identified that causes painful neuropathy[152]. Two thirds of the patients identified with this GOF Nav1.7 (I739V) mutation also developed diabetes[152], which suggests that increased VGSC may result in a diabetic phenotype but further studies are required to support this. As blocking VGSCs causes reduces insulin secretion[77], it seems counterintuitive that more activity of VGSCs could cause β-cell dysfunction. However, blockade of VGSCs with carbamazepine has been shown to protect β-cells from inflammation associated β-cell destruction[153]. VGSC inhibition may reduce β-cell excitotoxicity in response to the increased electrical excitability of β-cells required to meet increasing insulin demand in T2D; indeed this has been shown in T2D after reducing β-cell electrical excitability with the KATP activator diazoxide[147]. However, there are many off target influences of carbamazepine such as VDCCs[154], which may also play a role in the β-cell protective influences of carbamazepine. Interestingly, there is heterogeneous expression of Scn3a in human islet β-cells, which potentially implicates that the different amounts of β-cell VGSCs could provide variability in β-cell electrical excitability and GSIS[155]. Heterogeneity of human β-cell Scn3a expression may also impact sensitivity to excitotoxicity that occurs during the pathogenesis of T2D.
Swell-Activated Cl− Channels (SWELL)
During glucose stimulation β-cells swell leading to activation of volume-regulated anion current (VRAC), which is also termed the swell-activated Cl− current (ICl-,SWELL)[156–158]. VRAC/SWELL activation allows for the regulatory volume decrease (RVD) that occurs following exposure of β-cells to hypoosmotic conditions[156]. Although VRAC/SWELL would be predicted to produce RVD following glucose-induced swelling, β-cells maintain an increased volume during the duration of glucose stimulation[156]. Thus, VRAC/SWELL currents activated during glucose-induced swelling remain active until euglycemia has been reached[156]. The leucine-rich repeat–containing (LRRC8) proteins form the channels termed SWELL1, which are responsible for the VRAC/SWELL currents in β-cells[159]. When SWELL1 is activated during glucose-induced β-cell swelling Cl− efflux through these channels enhance depolarization leading to greater Ca2+ influx through VDCCs[156–158]. Although SWELL1 serves as a critical ion channel that controls glucose-stimulated β-cell depolarization, this channel complex has also been shown to transport neurotransmitters such as glutamate and γ-aminobutyric acid (GABA)[160, 161]. Because ionotropic glutamate and GABA receptors play an important role in regulating β-cell electrical excitability (detailed below), SWELL1 may contribute to controlling the activities of these receptors by influencing ligand release from β-cells. SWELL1 activity may change under diabetic conditions; for example, the GLUT1, GLUT-2 and glucokinase are reduced in T2D islets[162] and the resulting reduction in glucose transport and utilization in T2D β-cells could lead to reduced swelling as well as activation of SWELL1. Therefore, reduced SWELL1 in T2D could reduce glucose-stimulated depolarization, VDCC activation and insulin secretion.
Ionotropic Receptors
Neuronal innervation as well as paracrine mediated neurotransmitter release modulates islet cell function in part through ionotropic receptors, which becomes disrupted during the pathogenesis of diabetes. The main ionotropic receptors expressed in the islet are: GABA receptors, glycine receptors, purinergic receptors and glutamate receptors(Figure 4) [163–167], as we discuss below.
Figure 4:
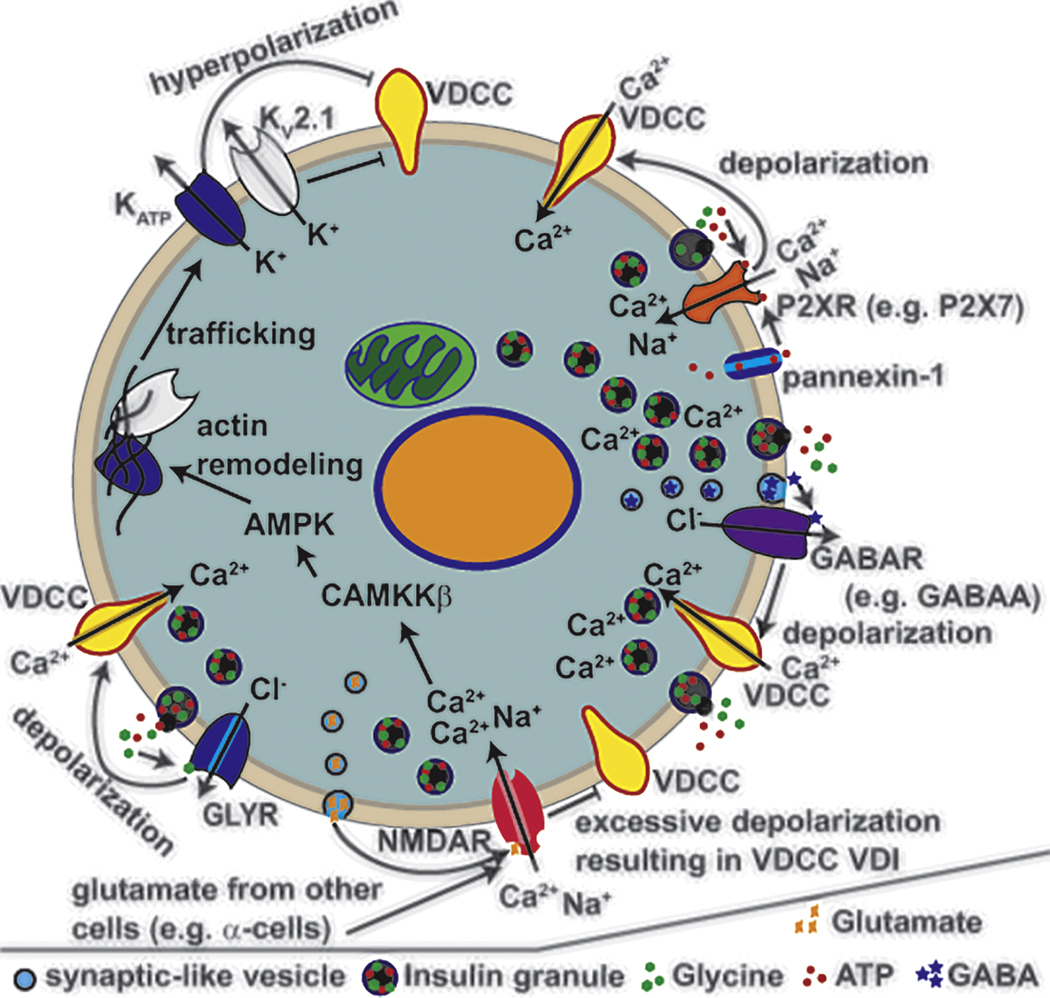
Ionotropic receptors that control β-cell insulin secretion. Ionotropic receptors expressed in human β-cells are activated by ligands and selective to specific ions. Upon activation the ionotropic receptors modulate VDCC activity by controlling the Vm. Ionotropic receptors that are permeable to Cl− such as the glycine receptors (GLYRs) and GABA receptors (GABAARs) lead to Vm depolarization upon activation due to Cl− efflux, which results in enhanced VDCC activation and insulin secretion. Ionotropic receptors that are permeable to Ca2+ and Na+ such as the NMDARs and P2XRs lead to both Ca2+ entry and Vm depolarization upon activation. While P2XRs enhance Ca2+ entry and GSIS, NMDARs cause the opposite. Two possible mechanisms for NMDAR inhibition of GSIS are through excessive depolarization causing voltage-dependent inactivation of VDCCs or through enhanced trafficking of K+ channels (including KATP and KV2.1) to the plasma membrane leading to Vm hyperpolarization. Ionotropic receptor ligands are released from the β-cell (autocrine) or are released from other nearby cells (paracrine) to control β-cell function.
GABA receptors
GABA receptors (GABARs) are also expressed on human β-cells. GABARs are chloride channels that when activated depolarize the β-cell and have been shown to stimulate VDCC activity leading to insulin secretion[163]. Depolarization by GABARs is due to Cl− efflux from the β-cell (Figure 4); a high intracellular concentration of Cl− in β-cells allows for Cl− efflux at the Vm from where electrical excitability occurs[168]. However, there is also evidence that activation of GABARs can inhibit insulin secretion; this results either due to voltage-dependent inactivation of VGSCs and VDCCs or potentially via Cl− influx through GABARs instead of efflux[169]. β-cells that are depolarized above the reversal potential of Cl− (~−40mV) would be expected to show Cl− influx through GABARs and this could be the case under conditions of excessive depolarization, which may occur under certain conditions such as T2D patients treated with sulfonylureas. Islets from T2D donors also show reduced expression of ionotropic GABA(A)Rs, which would be predicted to reduce GABA signaling[170]. GABA is produced and secreted from β-cells. GABA levels are reduced in T2D islets[169]. The enzyme GAD65 that produces GABA from glutamate shows reduced activity under the stressful conditions associated with T2D, which can be due to changes in transcription of GAD65 as well as perturbations in palmitoylation that change the intracellular localization of GAD65[171, 172]. Reduced GAD65 activity leads to reduced β-cell GABA in human islets from patients with T2D[169]. To compensate for the reduced GABA levels the islet GABARs show increased open probability and current in T2D β-cells when compared to nondiabetic (ND) β-cells[169]. This is not due to expression differences in the genes that encode GABARs as these receptors show equivalent expression in non-diabetic and T2D islets[169].
GABA has also been reported to have protective impacts on β-cell function, stimulate β-cell replication, as well as promoting α-cell neogenesis and conversion into β-cells[173–177]. GABA stimulated β-cell depolarization causes Ca2+ entry, which can activate PI3K and Akt signaling pathways that promote growth and survival[178]. While this may be one mechanism that promotes β-cell replication in response to GABA, it has also been shown that GABA promotes α-cell neogenesis and transdifferentiation to increase β-cell mass[174]. GABA induced β-cell formation through α-cells would be predicted to incorporate GABA-induced changes in Vm potentially through hyperpolarization induced inhibition of glucagon secretion, which could lead to α-cell hyperplasia that occurs during conditions of reduced liver glucagon signaling[179]. The positive impacts of GABA on β-cell number and function indicate that increasing GABA or GABA activity in islets during the pathogenesis of T2D would limit β-cell dysfunction and maintain β-cell mass[173–176]. For example, positive allosteric modulators that enhance GABA activity on GABARs result in enhanced human β-cell replication and GABAR activators (e.g. muscimol) stimulate human β-cell calcium entry and insulin secretion[180]. However, it is worth keeping in mind that recent studies call into question whether activation of GABARs enhance β-cell mass or function[181, 182]. Thus, more studies are needed to fully elucidate the functional role of GABAR in β-cells.
Glycine receptors
Human β-cells express ionotropic glycine receptors (GlyRs) including GlyRα1 and GlyRα3[164]. Glycine is also found in insulin secretory granules and can be released during GSIS[183]. GlyR activation by glycine results in β-cell Cl- efflux leading to Vm depolarization and increased electrical excitability (Figure 4)[164]. Glycine-induced elevations of human β-cell Ca2+ enhance insulin-secretion and thus glycine provides autocrine feedback to enable greater GSIS. Furthermore, supplementation with glycine improves human glucose tolerance[184–188]. Patients with T2D show decreased expression of the GlyRα1 transcript as well as protein abundance[5, 164]. Reduced GlyRα1 in T2D β-cells would be predicted to reduce glycine-induced depolarization and the resulting amplification of insulin secretion. Indeed glycine induced Cl- currents are reduced by ~50% in β-cells from T2D donors when compared to nondiabetic β-cells[164]. Moreover, T2D patients treated with conditions that increase plasma glycine levels show improved glucose handling and a delay in diabetes onset155; 156; 157; 158; 159. Interestingly, insulin also stimulates GlyR activity[164, 189] and therefore the insulin resistance associated with T2D is predicted to reduce GlyR activation during glucose-stimulation. Taken together, these data suggest that glycine control of β-cell Ca2+ entry through activation of GlyRs is perturbed in T2D and contributes to reductions in β-cell Ca2+ handling and insulin secretion.
Purinergic receptors
The P2X purinoceptors (P2XRs) are activated by purinergic ligands such as ATP and are primarily permeable to Ca2+ (Figure 4). There are many P2X receptors expressed in human β-cells including P2X2 P2X3 P2X4 P2X6 and P2X7[165]. All P2X receptors would contribute to ATP-induced Ca2+ entry and enhancement of insulin secretion (Figure 4)[166]. As ATP is released from insulin granules during GSIS[190], P2XR activation augments glucose-stimulated Ca2+ entry and thus GSIS[166]. While the exact role for each P2X receptor of the human β-cell has yet to be conclusively established, changes in P2X receptor protein levels have been observed in T2D islets[165]. Specifically, P2X7 levels are reduced in β-cells of patients with T2D[165]. Interestingly, P2XR7 expression and P2X7 protein levels are also controlled by glucose and saturated fatty acid levels[165]. When glucose and palmitic acid are elevated human islets show increased P2X7 levels in the short term[165]. This suggests that increased P2X7 levels can help β-cells adapt to the toxic conditions that eventually cause T2D. As P2X7 levels eventually drop in T2D, reduced P2X7 during prolonged stress may lead to reductions in GSIS due to reduced Ca2+ entry.
Polymorphisms near or in the P2XR7 gene are also associated with changes in glucose homeostasis[191]. A GWAS study in a Chinese population identified many polymorphisms near P2XR7 that is associated with an increased risk of developing T2D[192]. Certain polymorphisms in or near P2XR7 were also found associated with changes in insulin sensitivity and variations in glucose homeostasis[191]. Moreover, a missense polymorphism (rs1718119) that results in A348T within P2RX7 was identified that is associated with increased insulin sensitivity and greater insulin secretion. Importantly, the P2RX7 A348T results in a gain-of-function that increases channel activity in response to ATP and is thus predicted to enhance β-cell Ca2+ entry and insulin secretion[191, 193]. On the other hand, rodents with a P2RX7 variant (P451L) that results in a loss-of channel function show impaired glucose tolerance and insulin resistance compared to controls (with a proline at position 451), which suggests a potential impairment in islet function[191]. Therefore, P2RX7 plays an important role in enhancing β-cell insulin secretion and reduced P2RX7 function leads to islet dysfunction and insulin resistance, which increases the risk of T2D.
Glutamate receptors
Many ionotropic glutamate receptors are expressed in the islet (reviewed in[194]). They are known to be responsible for controlling α-cell Ca2+ handling and glucagon secretion as well as δ-cell somatostatin secretion[195, 196]. In the β-cell the primary ionotropic glutamate receptor that controls membrane potential are N-methyl-d-aspartate receptors (NMDAR), which are composed of GluN subunits including the GluN1 obligatory subunit. Interestingly, NMDAR activation limits glucose-stimulated Ca2+ influx in human and mouse β-cells, which has been proposed to be due to activation of K+ conductance that leads to inhibition of VDCC activity[167]. Recently, NMDARs have been reported to regulate the trafficking of KATP channels and Kv2.1 channels in INS-1 and dispersed human β-cells[197]. Stimulation of NMDARs either directly by agonists or by leptin acting on β-cell leptin receptors increases the abundance of KATP and Kv2.1 channels at the cell surface and results in membrane hyperpolarization, which would explain how activation of NMDARs inhibits GSIS.
Patients with T2D show elevations in glutamate levels[198, 199]. This change has to do with differences in consumption but also changes in liver production of glutamate169; 170. Thus, elevations in glutamate in obese individuals during the pathogenesis of T2D may lead to over activation of NMDARs. Moreover, hyperglycemic conditions have been shown to lead to increased β-cell glutamate release, which could also contribute to impaired β-cell function in T2D. Importantly, inhibition of NMDARs with dextromethorphan leads to both enhanced insulin secretion as well as protecting β-cells from failure in response to the stressful conditions associated with T2D[167]. Interestingly the enhanced insulin secretion effect by NMDAR inhibitors is dependent on the presence of KATP channels as mouse islets from Kir6.2 knockout animals failed to show such response, suggesting NMDAR inhibition stimulates insulin secretion by blocking NMDAR-dependent KATP channel trafficking to the β-cell membrane. Although NMDAR inhibition allows greater glucose-stimulated Ca2+ entry into β-cells stimulating GSIS, elevated NMDAR activation also promotes Ca2+ permeability through these ionotropic receptors. Thus, continual NMDAR Ca2+ permeability in T2D may perturb Ca2+ handling, which is predicted to reduce both insulin secretion as well as negatively impact β-cell health. This is supported by experiments on rodent β-cells that show reduced cell viability and decreased GSIS with chronic NMDAR activation. Treating diabetic rodents with an NMDAR antagonist (memantine) improved β-cell function and glucose homeostasis[198]. Together, these studies show that human β-cell NMDAR activity plays an important role in Ca2+ handling and suggest NMDARs as a therapeutic target for T2D.
Conclusions and Future Perspectives
Recent advances in human genetics, transcriptome and proteome analyses of normal and T2D islet cells reaching single cell resolution, and mechanistic analysis of ion channel regulations in cell lines, isolated islets and animal models have markedly improved our understanding of the role of ion channels in islet biology and how ion channel dysregulation contributes to dysfunction of the islets, in particular the ability of β-cells to secrete sufficient insulin in T2D. Despite the progress, many questions remain and translating the knowledge gained into clinical practice for the prevention and treatment T2D is still challenging.
From the genetic study perspective, while GWAS data offer potential T2D risk candidates they often need to be followed up by careful mechanistic studies to demonstrate causality. In this regard, studying human extreme phenotypes such as monogenic diabetes including neonatal diabetes and maturity onset diabetes of the young (MODY) have proven fruitful in identifying ion channels that are critical for insulin secretion. However, even when there is evidence for causality, genetic variations of a specific ion channel gene alone are unlikely to serve as a strong predictor of disease propensity since genetic background, epigenetic and environmental factors could impact how genetic variations manifest. As an example, genetic association studies in different ethnic populations often uncover different genetic risk factors. Most genetic studies thus far have focused on linkage with increased risk of T2D. It may be informative to also analyze genetic traits pertaining to ion channels of the islets that are protective against T2D. This approach has been successfully employed to identify genetic variants of proteins important for cardiovascular function, specifically in controlling cholesterol levels.
Mechanistic studies probing the functional role of ion channels have often been conducted in cell lines and transgenic mouse models. Results from these studies may or may not translate to humans as there is clear evidence that β-cell ion channel compositions are not identical in rodents and humans. Further, findings from mouse models can be clouded by the genetic background and strategies of genetic manipulations used. There is also the added caution of metabolic differences between rodents and humans. Efforts to study human islets are also met with potential variations from donors of different gender, age, ethnicity, and in the case of T2D, stage of the disease and other metabolic indicators such as obesity.
A central theme highlighted in this review is the importance of Ca2+ homeostasis in β-cell function and health. Intracellular Ca2+ is handled by not only ion channels present in the plasma membrane but also intracellular membranes including the ER, the mitochondria, and lysosomes. The coordinated actions of ion channels involved in Ca2+ handling and their regulation by signaling molecules afford β-cells the ability to adapt to increased insulin demands such as during pregnancy or mild insulin resistance. However, while enhanced Ca2+ signals are expected to augment insulin secretion and improve glucose tolerance, prolonged and unchecked Ca2+ signals can harm β-cells, resulting in β-cell excitotoxicity that reduces insulin secretion and causes β-cell demise. Therefore, strategies which enhance insulin secretion without causing permanent cell damage are needed. Better understanding of Ca2+ handling with increased temporal and spatial resolutions under physiological and pathological conditions will be important for such efforts.
Finally, pharmacological agents targeting β-cell ion channels are an essential part of the therapeutic development efforts for T2D. Currently available drugs or toxins have largely been discovered by serendipity or screening compound libraries. Rational drug design based on ion channel mechanisms has been limited by the difficulty in obtaining high resolution structures of these membrane proteins. Recent advances in single-particle cryo-electron microscopy techniques have made it possible to obtain high resolution structures of many ion channels at an astonishing pace. This presents an exciting opportunity for designing new and improved drugs that target β-cell ion channels for the treatment of T2D and other insulin secretion diseases.
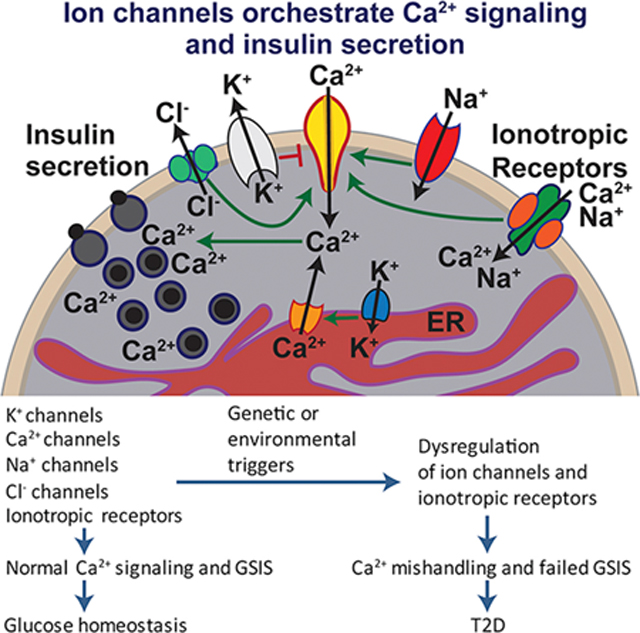
Ca2+ is an essential signal for pancreatic β-cell function including insulin secretion, transcription, metabolism, and the stress response.
β-cell Ca2+ signals are governed by orchestrated activities of ion channels.
In type 2 diabetes, disruption of ion channel function and regulation leads to β-cell excitotoxicity and Ca2+ mishandling to impair glucose-induced insulin secretion response.
Understanding the ionic mechanisms that regulate insulin secretion will help identify therapeutic modalities for diabetes that target the excitation-secretion pathway.
Acknowledgements
This work was supported by grants from the National Institute of Health (DK097392 and DK115620 for DAJ, and DK057699 and DK066485 to SLS).
Footnotes
Conflict of Interest Statement: The authors have no competing financial interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Chatelain FC, Bichet D, Douguet D, Feliciangeli S, Bendahhou S, Reichold M, et al. TWIK1, a unique background channel with variable ion selectivity. Proc Natl Acad Sci U S A. 2012;109:5499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhang Y, Xie L, Gunasekar SK, Tong D, Mishra A, Gibson WJ, et al. SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol. 2017;19:504–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dusaulcy R, Handgraaf S, Visentin F, Howald C, Dermitzakis ET, Philippe J, et al. High-fat diet impacts more changes in beta-cell compared to alpha-cell transcriptome. PLoS One. 2019;14:e0213299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Segerstolpe A, Palasantza A, Eliasson P, Andersson EM, Andreasson AC, Sun X, et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016;24:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, et al. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016;24:608–15. [DOI] [PubMed] [Google Scholar]
- [6].Chen C, Chmelova H, Cohrs CM, Chouinard JA, Jahn SR, Stertmann J, et al. Alterations in beta-Cell Calcium Dynamics and Efficacy Outweigh Islet Mass Adaptation in Compensation of Insulin Resistance and Prediabetes Onset. Diabetes. 2016;65:2676–85. [DOI] [PubMed] [Google Scholar]
- [7].Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, et al. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li N, Wu JX, Ding D, Cheng J, Gao N, Chen L. Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell. 2017;168:101–10 e10. [DOI] [PubMed] [Google Scholar]
- [9].Lee KPK, Chen J, MacKinnon R. Molecular structure of human KATP in complex with ATP and ADP. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–6. [DOI] [PubMed] [Google Scholar]
- [11].Puljung MC. Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. The Journal of general physiology. 2018;150:653–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. The EMBO journal. 2005;24:229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng SL. Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].de Wet H, Mikhailov MV, Fotinou C, Dreger M, Craig TJ, Venien-Bryan C, et al. Studies of the ATPase activity of the ABC protein SUR1. The FEBS journal. 2007;274:3532–44. [DOI] [PubMed] [Google Scholar]
- [16].Masia R, Enkvetchakul D, Nichols CG. Differential nucleotide regulation of KATP channels by SUR1 and SUR2A. J Mol Cell Cardiol. 2005;39:491–501. [DOI] [PubMed] [Google Scholar]
- [17].Ortiz D, Voyvodic P, Gossack L, Quast U, Bryan J. Two neonatal diabetes mutations on transmembrane helix 15 of SUR1 increase affinity for ATP and ADP at nucleotide binding domain 2. The Journal of biological chemistry. 2012;287:17985–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ortiz D, Gossack L, Quast U, Bryan J. Reinterpreting the action of ATP analogs on K(ATP) channels. The Journal of biological chemistry. 2013;288:18894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. The Journal of clinical investigation. 2005;115:2047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268:426–9. [DOI] [PubMed] [Google Scholar]
- [21].Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. The New England journal of medicine. 2004;350:1838–49. [DOI] [PubMed] [Google Scholar]
- [22].Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell. 2000;100:645–54. [DOI] [PubMed] [Google Scholar]
- [23].Ashcroft FM, Puljung MC, Vedovato N. Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends in endocrinology and metabolism: TEM. 2017;28:377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Balamurugan K, Kavitha B, Yang Z, Mohan V, Radha V, Shyng SL. Functional characterization of activating mutations in the sulfonylurea receptor 1 (ABCC8) causing neonatal diabetes mellitus in Asian Indian children. Pediatric diabetes. 2019;20:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Clark RH, McTaggart JS, Webster R, Mannikko R, Iberl M, Sim XL, et al. Muscle dysfunction caused by a KATP channel mutation in neonatal diabetes is neuronal in origin. Science. 2010;329:458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nielsen EM, Hansen L, Carstensen B, Echwald SM, Drivsholm T, Glumer C, et al. The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes. 2003;52:573–7. [DOI] [PubMed] [Google Scholar]
- [27].Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–72. [DOI] [PubMed] [Google Scholar]
- [28].Schwanstecher C, Meyer U, Schwanstecher M. K(IR)6.2 polymorphism predisposes to type 2 diabetes by inducing overactivity of pancreatic beta-cell ATP-sensitive K(+) channels. Diabetes. 2002;51:875–9. [DOI] [PubMed] [Google Scholar]
- [29].Sakura H, Wat N, Horton V, Millns H, Turner RC, Ashcroft FM. Sequence variations in the human Kir6.2 gene, a subunit of the beta-cell ATP-sensitive K-channel: no association with NIDDM in while Caucasian subjects or evidence of abnormal function when expressed in vitro. Diabetologia. 1996;39:1233–6. [DOI] [PubMed] [Google Scholar]
- [30].Florez JC, Burtt N, de Bakker PI, Almgren P, Tuomi T, Holmkvist J, et al. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004;53:1360–8. [DOI] [PubMed] [Google Scholar]
- [31].Ingelsson E, McCarthy MI. Human Genetics of Obesity and Type 2 Diabetes Mellitus: Past, Present, and Future. Circulation Genomic and precision medicine. 2018;11:e002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, et al. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. The Journal of clinical investigation. 2000;106:897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M. A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet. 2003;361:301–7. [DOI] [PubMed] [Google Scholar]
- [34].Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S, Perry A, Abu-Libdeh A, Glaser B, et al. Novel de novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes. 2008;57:1935–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem. 2000;275:9270–7. [DOI] [PubMed] [Google Scholar]
- [36].Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, et al. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. The Journal of biological chemistry. 2002;277:37176–83. [DOI] [PubMed] [Google Scholar]
- [37].Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, et al. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. The Journal of clinical investigation. 2008;118:2877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Henquin JC. Misunderstandings and controversies about the insulin-secreting properties of antidiabetic sulfonylureas. Biochimie. 2017;143:3–9. [DOI] [PubMed] [Google Scholar]
- [39].Inagaki N, Gonoi T, Clement JPt, Namba N, Inazawa J, Gonzalez G, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–70. [DOI] [PubMed] [Google Scholar]
- [40].Mohan V, Cooper ME, Matthews DR, Khunti K. The Standard of Care in Type 2 Diabetes: Re-evaluating the Treatment Paradigm. Diabetes therapy : research, treatment and education of diabetes and related disorders. 2019;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].van Raalte DH, Verchere CB. Improving glycaemic control in type 2 diabetes: Stimulate insulin secretion or provide beta-cell rest? Diabetes, obesity & metabolism. 2017;19:1205–13. [DOI] [PubMed] [Google Scholar]
- [42].Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced beta-cell apoptosis in cultured human islets. The Journal of clinical endocrinology and metabolism. 2005;90:501–6. [DOI] [PubMed] [Google Scholar]
- [43].Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. The New England journal of medicine. 2006;355:2427–43. [DOI] [PubMed] [Google Scholar]
- [44].Remedi MS, Nichols CG. Chronic antidiabetic sulfonylureas in vivo: reversible effects on mouse pancreatic beta-cells. PLoS medicine. 2008;5:e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Erion K, Corkey BE. beta-Cell Failure or beta-Cell Abuse? Frontiers in endocrinology. 2018;9:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ashcroft FM. New uses for old drugs: neonatal diabetes and sulphonylureas. Cell metabolism. 2010;11:179–81. [DOI] [PubMed] [Google Scholar]
- [47].Willow M, Catterall WA. Inhibition of binding of [3H]batrachotoxinin A 20-alpha-benzoate to sodium channels by the anticonvulsant drugs diphenylhydantoin and carbamazepine. Molecular pharmacology. 1982;22:627–35. [PubMed] [Google Scholar]
- [48].Chen PC, Olson EM, Zhou Q, Kryukova Y, Sampson HM, Thomas DY, et al. Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. The Journal of biological chemistry. 2013;288:20942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Devaraneni PK, Martin GM, Olson EM, Zhou Q, Shyng SL. Structurally distinct ligands rescue biogenesis defects of the KATP channel complex via a converging mechanism. The Journal of biological chemistry. 2015;290:7980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhou Q, Chen PC, Devaraneni PK, Martin GM, Olson EM, Shyng SL. Carbamazepine inhibits ATP-sensitive potassium channel activity by disrupting channel response to MgADP. Channels (Austin). 2014;8:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wu JX, Ding D, Wang M, Kang Y, Zeng X, Chen L. Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell. 2018;9:553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ding D, Wang M, Wu JX, Kang Y, Chen L. The Structural Basis for the Binding of Repaglinide to the Pancreatic KATP Channel. Cell reports. 2019;27:1848–57 e4. [DOI] [PubMed] [Google Scholar]
- [53].Martin GM, Sung MW, Yang Z, Innes LM, Kandasamy B, David LL, et al. Mechanism of pharmacochaperoning in a mammalian KATP channel revealed by cryo-EM. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lee JTC, Shanina I, Chu YN, Horwitz MS, Johnson JD. Carbamazepine, a beta-cell protecting drug, reduces type 1 diabetes incidence in NOD mice. Scientific reports. 2018;8:4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dufer M, Haspel D, Krippeit-Drews P, Aguilar-Bryan L, Bryan J, Drews G. Oscillations of membrane potential and cytosolic Ca(2+) concentration in SUR1(−/−) beta cells. Diabetologia. 2004;47:488–98. [DOI] [PubMed] [Google Scholar]
- [56].Gopel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol. 1999;521 Pt 3:717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279:F793–801. [DOI] [PubMed] [Google Scholar]
- [58].Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nature genetics. 2011;44:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Vierra NC, Dadi PK, Jeong I, Dickerson M, Powell DR, Jacobson DA. Type 2 Diabetes-Associated K+ Channel TALK-1 Modulates beta-Cell Electrical Excitability, Second-Phase Insulin Secretion, and Glucose Homeostasis. Diabetes. 2015;64:3818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, et al. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:2301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Friedrich C, Rinne S, Zumhagen S, Kiper AK, Silbernagel N, Netter MF, et al. Gain-of-function mutation in TASK-4 channels and severe cardiac conduction disorder. EMBO molecular medicine. 2014;6:937–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].McKinley D, Meissner G. Evidence for a K+, Na+ permeable channel in sarcoplasmic reticulum. J Membr Biol. 1978;44:159–86. [DOI] [PubMed] [Google Scholar]
- [63].Kuum M, Veksler V, Kaasik A. Potassium fluxes across the endoplasmic reticulum and their role in endoplasmic reticulum calcium homeostasis. Cell Calcium. 2015;58:79–85. [DOI] [PubMed] [Google Scholar]
- [64].Guo T, Nani A, Shonts S, Perryman M, Chen H, Shannon T, et al. Sarcoplasmic reticulum K(+) (TRIC) channel does not carry essential countercurrent during Ca(2+) release. Biophys J. 2013;105:1151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M, et al. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature. 2007;448:78–82. [DOI] [PubMed] [Google Scholar]
- [66].Yamazaki D, Tabara Y, Kita S, Hanada H, Komazaki S, Naitou D, et al. TRIC-A channels in vascular smooth muscle contribute to blood pressure maintenance. Cell Metab. 2011;14:231–41. [DOI] [PubMed] [Google Scholar]
- [67].Dickerson MT, Vierra NC, Milian SC, Dadi PK, Jacobson DA. Osteopontin activates the diabetes-associated potassium channel TALK-1 in pancreatic beta-cells. PLoS One. 2017;12:e0175069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vierra NC, Dadi PK, Milian SC, Dickerson MT, Jordan KL, Gilon P, et al. TALK-1 channels control beta cell endoplasmic reticulum Ca2+ homeostasis. Science signaling. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vierra NC, Dadi PK, Milian SC, Dickerson MT, Jordan KL, Gilon P, et al. TALK-1 channels control beta cell endoplasmic reticulum Ca(2+) homeostasis. Sci Signal. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kono T, Ahn G, Moss DR, Gann L, Zarain-Herzberg A, Nishiki Y, et al. PPAR-gamma activation restores pancreatic islet SERCA2 levels and prevents beta-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol Endocrinol. 2012;26:257–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol Metab. 2011;22:266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Meneur C, Bernal-Mizrachi E. Natural history of beta-cell adaptation and failure in type 2 diabetes. Mol Aspects Med. 2015;42:19–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wang W, Liu C, Jimenez-Gonzalez M, Song WJ, Hussain MA. The undoing and redoing of the diabetic beta-cell. J Diabetes Complications. 2017;31:912–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A. 2007;104:15040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].De Franco E, Flanagan SE, Yagi T, Abreu D, Mahadevan J, Johnson MB, et al. Dominant ER Stress-Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes. 2017;66:2044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, et al. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes. 2008;57:1618–28. [DOI] [PubMed] [Google Scholar]
- [78].Tabarki B, AlMajhad N, AlHashem A, Shaheen R, Alkuraya FS. Homozygous KCNMA1 mutation as a cause of cerebellar atrophy, developmental delay and seizures. Hum Genet. 2016;135:1295–8. [DOI] [PubMed] [Google Scholar]
- [79].Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–8. [DOI] [PubMed] [Google Scholar]
- [80].Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, et al. Novel Observations From Next-Generation RNA Sequencing of Highly Purified Human Adult and Fetal Islet Cell Subsets. Diabetes. 2015;64:3172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Molecular metabolism. 2016;5:449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Shepard PD, Bunney BS. Repetitive firing properties of putative dopamine-containing neurons in vitro: regulation by an apamin-sensitive Ca(2+)-activated K+ conductance. Experimental brain research. 1991;86:141–50. [DOI] [PubMed] [Google Scholar]
- [83].Deignan J, Lujan R, Bond C, Riegel A, Watanabe M, Williams JT, et al. SK2 and SK3 expression differentially affect firing frequency and precision in dopamine neurons. Neuroscience. 2012;217:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Dufer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic beta-cells. Diabetes. 2009;58:1835–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. The Journal of physiology. 2010;588:3525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, et al. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–14. [DOI] [PubMed] [Google Scholar]
- [87].Pennefather P, Lancaster B, Adams PR, Nicoll RA. Two distinct Ca-dependent K currents in bullfrog sympathetic ganglion cells. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3040–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Henquin JC, Dufrane D, Gmyr V, Kerr-Conte J, Nenquin M. Pharmacological approach to understanding the control of insulin secretion in human islets. Diabetes, obesity & metabolism. 2017;19:1061–70. [DOI] [PubMed] [Google Scholar]
- [89].Rapetti-Mauss R, Lacoste C, Picard V, Guitton C, Lombard E, Loosveld M, et al. A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood. 2015;126:1273–80. [DOI] [PubMed] [Google Scholar]
- [90].Imashuku S, Muramatsu H, Sugihara T, Okuno Y, Wang X, Yoshida K, et al. PIEZO1 gene mutation in a Japanese family with hereditary high phosphatidylcholine hemolytic anemia and hemochromatosis-induced diabetes mellitus. International journal of hematology. 2016;104:125–9. [DOI] [PubMed] [Google Scholar]
- [91].Rashid CS, Lien YC, Bansal A, Jaeckle-Santos LJ, Li C, Won KJ, et al. Transcriptomic Analysis Reveals Novel Mechanisms Mediating Islet Dysfunction in the Intrauterine Growth-Restricted Rat. Endocrinology. 2018;159:1035–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Li XN, Herrington J, Petrov A, Ge L, Eiermann G, Xiong Y, et al. The role of voltage-gated potassium channels Kv2.1 and Kv2.2 in the regulation of insulin and somatostatin release from pancreatic islets. The Journal of pharmacology and experimental therapeutics. 2013;344:407–16. [DOI] [PubMed] [Google Scholar]
- [93].Fu J, Dai X, Plummer G, Suzuki K, Bautista A, Githaka JM, et al. Kv2.1 Clustering Contributes to Insulin Exocytosis and Rescues Human beta-Cell Dysfunction. Diabetes. 2017;66:1890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Saitsu H, Akita T, Tohyama J, Goldberg-Stern H, Kobayashi Y, Cohen R, et al. De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Scientific reports. 2015;5:15199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Thiffault I, Speca DJ, Austin DC, Cobb MM, Eum KS, Safina NP, et al. A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. The Journal of general physiology. 2015;146:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Latypova X, Matsumoto N, Vinceslas-Muller C, Bezieau S, Isidor B, Miyake N. Novel KCNB1 mutation associated with non-syndromic intellectual disability. Journal of human genetics. 2017;62:569–73. [DOI] [PubMed] [Google Scholar]
- [97].Marini C, Romoli M, Parrini E, Costa C, Mei D, Mari F, et al. Clinical features and outcome of 6 new patients carrying de novo KCNB1 gene mutations. Neurology Genetics. 2017;3:e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kalman K, Nguyen A, Tseng-Crank J, Dukes ID, Chandy G, Hustad CM, et al. Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. The Journal of biological chemistry. 1998;273:5851–7. [DOI] [PubMed] [Google Scholar]
- [99].Finol-Urdaneta RK, Remedi MS, Raasch W, Becker S, Clark RB, Struver N, et al. Block of Kv1.7 potassium currents increases glucose-stimulated insulin secretion. EMBO molecular medicine. 2012;4:424–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Cheng YJ, Yao H, Ji CC, Chen XM, Fan J, Liu LJ, et al. A Heterozygous Missense hERG Mutation Associated with Early Repolarization Syndrome. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2018;51:1301–12. [DOI] [PubMed] [Google Scholar]
- [101].Shi W, Wymore RS, Wang HS, Pan Z, Cohen IS, McKinnon D, et al. Identification of two nervous system-specific members of the erg potassium channel gene family. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:9423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hylten-Cavallius L, Iepsen EW, Wewer Albrechtsen NJ, Svendstrup M, Lubberding AF, Hartmann B, et al. Patients With Long-QT Syndrome Caused by Impaired hERG-Encoded Kv11.1 Potassium Channel Have Exaggerated Endocrine Pancreatic and Incretin Function Associated With Reactive Hypoglycemia. Circulation. 2017;135:1705–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Yang JK, Lu J, Yuan SS, Asan, Cao X, Qiu HY, et al. From Hyper- to Hypoinsulinemia and Diabetes: Effect of KCNH6 on Insulin Secretion. Cell Rep. 2018;25:3800–10 e6. [DOI] [PubMed] [Google Scholar]
- [104].Proverbio MC, Mangano E, Gessi A, Bordoni R, Spinelli R, Asselta R, et al. Whole genome SNP genotyping and exome sequencing reveal novel genetic variants and putative causative genes in congenital hyperinsulinism. PloS one. 2013;8:e68740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Johnson B, Leek AN, Sole L, Maverick EE, Levine TP, Tamkun MM. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:E7331–E40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kirmiz M, Vierra NC, Palacio S, Trimmer JS. Identification of VAPA and VAPB as Kv2 Channel-Interacting Proteins Defining Endoplasmic Reticulum-Plasma Membrane Junctions in Mammalian Brain Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2018;38:7562–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].MacDonald PE, Wang G, Tsuk S, Dodo C, Kang Y, Tang L, et al. Synaptosome-associated protein of 25 kilodaltons modulates Kv2.1 voltage-dependent K(+) channels in neuroendocrine islet beta-cells through an interaction with the channel N terminus. Mol Endocrinol. 2002;16:2452–61. [DOI] [PubMed] [Google Scholar]
- [108].Michaelevski I, Chikvashvili D, Tsuk S, Singer-Lahat D, Kang Y, Linial M, et al. Direct interaction of target SNAREs with the Kv2.1 channel. Modal regulation of channel activation and inactivation gating. The Journal of biological chemistry. 2003;278:34320–30. [DOI] [PubMed] [Google Scholar]
- [109].Dai XQ, Manning Fox JE, Chikvashvili D, Casimir M, Plummer G, Hajmrle C, et al. The voltage-dependent potassium channel subunit Kv2.1 regulates insulin secretion from rodent and human islets independently of its electrical function. Diabetologia. 2012;55:1709–20. [DOI] [PubMed] [Google Scholar]
- [110].Leung YM, Kwan EP, Ng B, Kang Y, Gaisano HY. SNAREing voltage-gated K+ and ATP-sensitive K+ channels: tuning beta-cell excitability with syntaxin-1A and other exocytotic proteins. Endocrine reviews. 2007;28:653–63. [DOI] [PubMed] [Google Scholar]
- [111].Chen PC, Bruederle CE, Gaisano HY, Shyng SL. Syntaxin 1A regulates surface expression of beta-cell ATP-sensitive potassium channels. American journal of physiology Cell physiology. 2011;300:C506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].de Kovel CGF, Syrbe S, Brilstra EH, Verbeek N, Kerr B, Dubbs H, et al. Neurodevelopmental Disorders Caused by De Novo Variants in KCNB1 Genotypes and Phenotypes. JAMA Neurol. 2017;74:1228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kailey B, van de Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic beta- and alpha-cells. Am J Physiol Endocrinol Metab. 2012;303:E1107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Dascal N, Kahanovitch U. The Roles of Gbetagamma and Galpha in Gating and Regulation of GIRK Channels. International review of neurobiology. 2015;123:27–85. [DOI] [PubMed] [Google Scholar]
- [115].Lacey RJ, Chan SL, Cable HC, James RF, Perrett CW, Scarpello JH, et al. Expression of alpha 2- and beta-adrenoceptor subtypes in human islets of Langerhans. The Journal of endocrinology. 1996;148:531–43. [DOI] [PubMed] [Google Scholar]
- [116].Robertson RP, Porte D Jr. Adrenergic modulation of basal insulin secretion in man. Diabetes. 1973;22:1–8. [DOI] [PubMed] [Google Scholar]
- [117].Dollinger HC, Raptis S. Inhibition of exocrine and endocrine pancreatic function by somatostatin in man. Zeitschrift fur Gastroenterologie Verhandlungsband. 1976:56–9. [PubMed] [Google Scholar]
- [118].Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An atlas and functional analysis of G-protein coupled receptors in human islets of Langerhans. Pharmacology & therapeutics. 2013;139:359–91. [DOI] [PubMed] [Google Scholar]
- [119].Ferrer J, Nichols CG, Makhina EN, Salkoff L, Bernstein J, Gerhard D, et al. Pancreatic islet cells express a family of inwardly rectifying K+ channel subunits which interact to form G-protein-activated channels. The Journal of biological chemistry. 1995;270:26086–91. [DOI] [PubMed] [Google Scholar]
- [120].Iwanir S, Reuveny E. Adrenaline-induced hyperpolarization of mouse pancreatic islet cells is mediated by G protein-gated inwardly rectifying potassium (GIRK) channels. Pflugers Archiv : European journal of physiology. 2008;456:1097–108. [DOI] [PubMed] [Google Scholar]
- [121].Araki Y, Yoshida T, Nakamura N, Koyama K, Nakamura Y, Nakano K, et al. Effect of islet-activating protein (IAP) upon insulin secretion from human pancreatic islets. Endocrinologia japonica. 1981;28:139–43. [DOI] [PubMed] [Google Scholar]
- [122].Katada T, Ui M. Perfusion of the pancreas isolated from pertussis-sensitized rats: potentiation of insulin secretory responses due to beta-adrenergic stimulation. Endocrinology. 1977;101:1247–55. [DOI] [PubMed] [Google Scholar]
- [123].Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ, et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science. 2010;327:217–20. [DOI] [PubMed] [Google Scholar]
- [124].Wolford JK, Hanson RL, Kobes S, Bogardus C, Prochazka M. Analysis of linkage disequilibrium between polymorphisms in the KCNJ9 gene with type 2 diabetes mellitus in Pima Indians. Mol Genet Metab. 2001;73:97–103. [DOI] [PubMed] [Google Scholar]
- [125].Kuss J, Stallmeyer B, Goldstein M, Rinne S, Pees C, Zumhagen S, et al. Familial Sinus Node Disease Caused by a Gain of GIRK (G-Protein Activated Inwardly Rectifying K(+) Channel) Channel Function. Circ Genom Precis Med. 2019;12:e002238. [DOI] [PubMed] [Google Scholar]
- [126].Petit-Jacques J, Sui JL, Logothetis DE. Synergistic activation of G protein-gated inwardly rectifying potassium channels by the betagamma subunits of G proteins and Na(+) and Mg(2+) ions. The Journal of general physiology. 1999;114:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Bukiya AN, Durdagi S, Noskov S, Rosenhouse-Dantsker A. Cholesterol up-regulates neuronal G protein-gated inwardly rectifying potassium (GIRK) channel activity in the hippocampus. The Journal of biological chemistry. 2017;292:6135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kim D, Bang H. Modulation of rat atrial G protein-coupled K+ channel function by phospholipids. The Journal of physiology. 1999;517 ( Pt 1):59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 1998;391:803–6. [DOI] [PubMed] [Google Scholar]
- [130].Sui JL, Petit-Jacques J, Logothetis DE. Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zhang H, He C, Yan X, Mirshahi T, Logothetis DE. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nature cell biology. 1999;1:183–8. [DOI] [PubMed] [Google Scholar]
- [132].Kolic J, Manning Fox JE, Chepurny OG, Spigelman AF, Ferdaoussi M, Schwede F, et al. PI3 kinases p110alpha and PI3K-C2beta negatively regulate cAMP via PDE3/8 to control insulin secretion in mouse and human islets. Molecular metabolism. 2016;5:459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Ho IH, Murrell-Lagnado RD. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. The Journal of biological chemistry. 1999;274:8639–48. [DOI] [PubMed] [Google Scholar]
- [134].Ho IH, Murrell-Lagnado RD. Molecular mechanism for sodium-dependent activation of G protein-gated K+ channels. The Journal of physiology. 1999;520 Pt 3:645–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Xu G, Chen J, Jing G, Shalev A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes. 2012;61:848–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bukiya AN, Blank PS, Rosenhouse-Dantsker A. Cholesterol intake and statin use regulate neuronal G protein-gated inwardly rectifying potassium channels. Journal of lipid research. 2019;60:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiological reviews. 1981;61:914–73. [DOI] [PubMed] [Google Scholar]
- [138].Yang SN, Berggren PO. The role of voltage-gated calcium channels in pancreatic beta-cell physiology and pathophysiology. Endocrine reviews. 2006;27:621–76. [DOI] [PubMed] [Google Scholar]
- [139].Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. [DOI] [PubMed] [Google Scholar]
- [140].Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H, et al. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes. 2017;18:320–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Reinbothe TM, Alkayyali S, Ahlqvist E, Tuomi T, Isomaa B, Lyssenko V, et al. The human L-type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia. 2013;56:340–9. [DOI] [PubMed] [Google Scholar]
- [142].Bysani M, Agren R, Davegardh C, Volkov P, Ronn T, Unneberg P, et al. ATAC-seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes. Sci Rep. 2019;9:7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. [DOI] [PubMed] [Google Scholar]
- [144].Xie B, Nguyen PM, Gucek A, Thonig A, Barg S, Idevall-Hagren O. Plasma Membrane Phosphatidylinositol 4,5-Bisphosphate Regulates Ca(2+)-Influx and Insulin Secretion from Pancreatic beta Cells. Cell chemical biology. 2016;23:816–26. [DOI] [PubMed] [Google Scholar]
- [145].Wang L, Bhattacharjee A, Zuo Z, Hu F, Honkanen RE, Berggren PO, et al. A low voltage-activated Ca2+ current mediates cytokine-induced pancreatic beta-cell death. Endocrinology. 1999;140:1200–4. [DOI] [PubMed] [Google Scholar]
- [146].Ramadan JW, Steiner SR, O’Neill CM, Nunemaker CS. The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes. Cell Calcium. 2011;50:481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Greenwood RH, Mahler RF, Hales CN. Improvement in insulin secretion in diabetes after diazoxide. Lancet. 1976;1:444–7. [DOI] [PubMed] [Google Scholar]
- [148].Bjorklund A, Lansner A, Grill VE. Glucose-induced [Ca2+]i abnormalities in human pancreatic islets: important role of overstimulation. Diabetes. 2000;49:1840–8. [DOI] [PubMed] [Google Scholar]
- [149].Gandasi NR, Yin P, Riz M, Chibalina MV, Cortese G, Lund PE, et al. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. The Journal of clinical investigation. 2017;127:2353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ernst SJ, Aguilar-Bryan L, Noebels JL. Sodium channel beta1 regulatory subunit deficiency reduces pancreatic islet glucose-stimulated insulin and glucagon secretion. Endocrinology. 2009;150:1132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJ, et al. Na+ current properties in islet alpha- and beta-cells reflect cell-specific Scn3a and Scn9a expression. J Physiol. 2014;592:4677–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Hoeijmakers JG, Faber CG, Merkies IS, Waxman SG. Channelopathies, painful neuropathy, and diabetes: which way does the causal arrow point? Trends Mol Med. 2014;20:544–50. [DOI] [PubMed] [Google Scholar]
- [153].Yang YH, Vilin YY, Roberge M, Kurata HT, Johnson JD. Multiparameter screening reveals a role for Na+ channels in cytokine-induced beta-cell death. Mol Endocrinol. 2014;28:406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ambrosio AF, Silva AP, Malva JO, Soares-da-Silva P, Carvalho AP, Carvalho CM. Carbamazepine inhibits L-type Ca2+ channels in cultured rat hippocampal neurons stimulated with glutamate receptor agonists. Neuropharmacology. 1999;38:1349–59. [DOI] [PubMed] [Google Scholar]
- [155].Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human islets contain four distinct subtypes of beta cells. Nat Commun. 2016;7:11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Miley HE, Sheader EA, Brown PD, Best L. Glucose-induced swelling in rat pancreatic beta-cells. The Journal of physiology. 1997;504 ( Pt 1):191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Stuhlmann T, Planells-Cases R, Jentsch TJ. LRRC8/VRAC anion channels enhance beta-cell glucose sensing and insulin secretion. Nature communications. 2018;9:1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kang C, Xie L, Gunasekar SK, Mishra A, Zhang Y, Pai S, et al. SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nature communications. 2018;9:367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 2014;157:447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ. Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. Journal of cell science. 2017;130:1122–33. [DOI] [PubMed] [Google Scholar]
- [161].Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron. 2019;102:813–27 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–35. [DOI] [PubMed] [Google Scholar]
- [163].Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, et al. Gamma-aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes. 2010;59:1694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Yan-Do R, Duong E, Manning Fox JE, Dai X, Suzuki K, Khan S, et al. A Glycine-Insulin Autocrine Feedback Loop Enhances Insulin Secretion From Human beta-Cells and Is Impaired in Type 2 Diabetes. Diabetes. 2016;65:2311–21. [DOI] [PubMed] [Google Scholar]
- [165].Glas R, Sauter NS, Schulthess FT, Shu L, Oberholzer J, Maedler K. Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia. 2009;52:1579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Jacques-Silva MC, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, et al. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:6465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Marquard J, Otter S, Welters A, Stirban A, Fischer A, Eglinger J, et al. Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nature medicine. 2015;21:363–72. [DOI] [PubMed] [Google Scholar]
- [168].Eberhardson M, Patterson S, Grapengiesser E. Microfluorometric analysis of Cl-permeability and its relation to oscillatory Ca2+ signalling in glucose-stimulated pancreatic beta-cells. Cellular signalling. 2000;12:781–6. [DOI] [PubMed] [Google Scholar]
- [169].Korol SV, Jin Z, Jin Y, Bhandage AK, Tengholm A, Gandasi NR, et al. Functional Characterization of Native, High-Affinity GABAA Receptors in Human Pancreatic beta Cells. EBioMedicine. 2018;30:273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, et al. gamma-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia. 2012;55:1985–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kanaani J, Diacovo MJ, El-Husseini Ael D, Bredt DS, Baekkeskov S. Palmitoylation controls trafficking of GAD65 from Golgi membranes to axon-specific endosomes and a Rab5a-dependent pathway to presynaptic clusters. Journal of cell science. 2004;117:2001–13. [DOI] [PubMed] [Google Scholar]
- [172].Hou J, Li Z, Zhong W, Hao Q, Lei L, Wang L, et al. Temporal Transcriptomic and Proteomic Landscapes of Deteriorating Pancreatic Islets in Type 2 Diabetic Rats. Diabetes. 2017;66:2188–200. [DOI] [PubMed] [Google Scholar]
- [173].Li J, Casteels T, Frogne T, Ingvorsen C, Honore C, Courtney M, et al. Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell. 2017;168:86–100 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell. 2017;168:73–85 e11. [DOI] [PubMed] [Google Scholar]
- [175].Liu W, Son DO, Lau HK, Zhou Y, Prud’homme GJ, Jin T, et al. Combined Oral Administration of GABA and DPP-4 Inhibitor Prevents Beta Cell Damage and Promotes Beta Cell Regeneration in Mice. Frontiers in pharmacology. 2017;8:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Feng AL, Xiang YY, Gui L, Kaltsidis G, Feng Q, Lu WY. Paracrine GABA and insulin regulate pancreatic alpha cell proliferation in a mouse model of type 1 diabetes. Diabetologia. 2017;60:1033–42. [DOI] [PubMed] [Google Scholar]
- [177].Untereiner A, Abdo S, Bhattacharjee A, Gohil H, Pourasgari F, Ibeh N, et al. GABA promotes beta-cell proliferation, but does not overcome impaired glucose homeostasis associated with diet-induced obesity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2019;33:3968–84. [DOI] [PubMed] [Google Scholar]
- [178].Soltani N, Qiu H, Aleksic M, Glinka Y, Zhao F, Liu R, et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I, et al. Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes. 2013;62:1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Purwana I, Zheng J, Li X, Deurloo M, Son DO, Zhang Z, et al. GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes. 2014;63:4197–205. [DOI] [PubMed] [Google Scholar]
- [181].Ackermann AM, Moss NG, Kaestner KH. GABA and Artesunate Do Not Induce Pancreatic alpha-to-beta Cell Transdifferentiation In Vivo. Cell metabolism. 2018;28:787–92 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].van der Meulen T, Lee S, Noordeloos E, Donaldson CJ, Adams MW, Noguchi GM, et al. Artemether Does Not Turn alpha Cells into beta Cells. Cell metabolism. 2018;27:218–25 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Gammelsaeter R, Froyland M, Aragon C, Danbolt NC, Fortin D, Storm-Mathisen J, et al. Glycine, GABA and their transporters in pancreatic islets of Langerhans: evidence for a paracrine transmitter interplay. J Cell Sci. 2004;117:3749–58. [DOI] [PubMed] [Google Scholar]
- [184].Tulipani S, Griffin J, Palau-Rodriguez M, Mora-Cubillos X, Bernal-Lopez RM, Tinahones FJ, et al. Metabolomics-guided insights on bariatric surgery versus behavioral interventions for weight loss. Obesity (Silver Spring). 2016;24:2451–66. [DOI] [PubMed] [Google Scholar]
- [185].Gralka E, Luchinat C, Tenori L, Ernst B, Thurnheer M, Schultes B. Metabolomic fingerprint of severe obesity is dynamically affected by bariatric surgery in a procedure-dependent manner. Am J Clin Nutr. 2015;102:1313–22. [DOI] [PubMed] [Google Scholar]
- [186].Magkos F, Bradley D, Schweitzer GG, Finck BN, Eagon JC, Ilkayeva O, et al. Effect of Roux-en-Y gastric bypass and laparoscopic adjustable gastric banding on branched-chain amino acid metabolism. Diabetes. 2013;62:2757–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Laferrere B, Reilly D, Arias S, Swerdlow N, Gorroochurn P, Bawa B, et al. Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci Transl Med. 2011;3:80re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Glynn EL, Piner LW, Huffman KM, Slentz CA, Elliot-Penry L, AbouAssi H, et al. Impact of combined resistance and aerobic exercise training on branched-chain amino acid turnover, glycine metabolism and insulin sensitivity in overweight humans. Diabetologia. 2015;58:2324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Ster J, Colomer C, Monzo C, Duvoid-Guillou A, Moos F, Alonso G, et al. Insulin-like growth factor-1 inhibits adult supraoptic neurons via complementary modulation of mechanoreceptors and glycine receptors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:2267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Braun M, Wendt A, Karanauskaite J, Galvanovskis J, Clark A, MacDonald PE, et al. Corelease and differential exit via the fusion pore of GABA, serotonin, and ATP from LDCV in rat pancreatic beta cells. The Journal of general physiology. 2007;129:221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Todd JN, Poon W, Lyssenko V, Groop L, Nichols B, Wilmot M, et al. Variation in glucose homeostasis traits associated with P2RX7 polymorphisms in mice and humans. The Journal of clinical endocrinology and metabolism. 2015;100:E688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Ma RC, Hu C, Tam CH, Zhang R, Kwan P, Leung TF, et al. Genome-wide association study in a Chinese population identifies a susceptibility locus for type 2 diabetes at 7q32 near PAX4. Diabetologia. 2013;56:1291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Sun C, Chu J, Singh S, Salter RD. Identification and characterization of a novel variant of the human P2X(7) receptor resulting in gain of function. Purinergic signalling. 2010;6:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Otter S, Lammert E. Exciting Times for Pancreatic Islets: Glutamate Signaling in Endocrine Cells. Trends in endocrinology and metabolism: TEM. 2016;27:177–88. [DOI] [PubMed] [Google Scholar]
- [195].Cabrera O, Jacques-Silva MC, Speier S, Yang SN, Kohler M, Fachado A, et al. Glutamate is a positive autocrine signal for glucagon release. Cell metabolism. 2008;7:545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Muroyama A, Uehara S, Yatsushiro S, Echigo N, Morimoto R, Morita M, et al. A novel variant of ionotropic glutamate receptor regulates somatostatin secretion from delta-cells of islets of Langerhans. Diabetes. 2004;53:1743–53. [DOI] [PubMed] [Google Scholar]
- [197].Wu Y, Fortin DA, Cochrane VA, Chen PC, Shyng SL. NMDA receptors mediate leptin signaling and regulate potassium channel trafficking in pancreatic beta-cells. The Journal of biological chemistry. 2017;292:15512–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Huang XT, Li C, Peng XP, Guo J, Yue SJ, Liu W, et al. An excessive increase in glutamate contributes to glucose-toxicity in beta-cells via activation of pancreatic NMDA receptors in rodent diabetes. Sci Rep. 2017;7:44120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]