

Abstract
The microtubule-associated protein tau aggregates into distinct neurofibrillary tangles in brains afflicted with multiple neurodegenerative diseases such as Alzheimer’s disease and corticobasal degeneration (CBD). The mechanism of tau misfolding and aggregation is poorly understood. Determining the structure, dynamics, and water accessibility of tau filaments may give insight into the pathway of tau misfolding. Here, we investigate the hydration and dynamics of the β-sheet core of heparin-fibrillized 0N4R tau using solid-state NMR spectroscopy. This β-sheet core consists of the second and third microtubule-binding repeats, R2 and R3, which form a hairpin. Water-edited 2D 13C-13C and 15N-13C correlation spectra indicate that most residues in R2 and R3 domains have low water accessibility, indicating that this hairpin is surrounded by other proteinaceous segments. However, a small number of residues, especially S285 and S316, are well hydrated compared to other Ser and Thr residues, suggesting that there is a small water channel in the middle of the hairpin. To probe whether water accessibility correlates with protein dynamics, we measured the backbone N-H dipolar couplings of the β-sheet core. Interestingly, residues in the fourth microtubule-binding repeat, R4, show rigid-limit N-H dipolar couplings, even though this domain exhibits weaker intensities in the 2D 15N-13C correlation spectra. These results suggest that the R4 domain participates in cross-β hydrogen bonding in some of the subunits but exhibits dynamic disorder in other subunits. Taken together, these hydration and dynamics data indicate that the R2–R3 hairpin of 0N4R tau is shielded from water by other proteinaceous segments on the exterior, but contains a small water pore in the interior. This structural topology has various similarities with the CBD tau fibril structure, but also shows specific differences. The disorder of the R4 domain and the presence of a small water channel in the heparin-fibrillized 4R tau have implications for the structure of tau fibrils in diseased brains.
Graphical Abstract
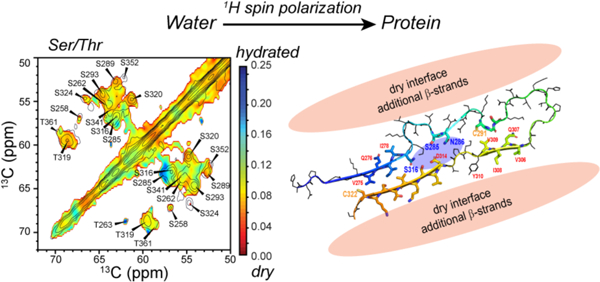
Introduction
Tau is an intrinsically disordered microtubule-associated protein that is abundant in the neurons of the human central nervous system. Due to the presence or absence of acidic inserts in the N-terminal portion of the protein (0N, 1N, or 2N tau) and the presence of three or four microtubule-binding repeats (3R or 4R), there are six tau isoforms in adult human brains.1 In a number of neurodegenerative diseases, tau aggregates into cross-β fibrils that appear as intraneuronal tangles in patient brains. Each disease’s fibrils are characterized by distinct tau isoform distributions. For example, Alzheimer’s disease (AD) and chronic traumatic encephalopathy (CTE) involve aggregates of both 3R and 4R tau; Pick’s disease has 3R tau aggregates; whereas corticobasal degeneration (CBD) involves 4R tau aggregates.2 There is significant evidence that tau propagates its pathological aggregated form in a prion-like fashion, by recruiting monomeric soluble tau into the same misfolded conformation.3–5 Cryo-electron microscopy (cryoEM) structures of tau fibrils obtained from patient brains6–10 have so far shown that each tauopathy has a unique β-sheet structure for the fibril core. This structure is conserved between patients with the same disease, whereas different tauopathies exhibit different fibril structures. These observations imply that each disease has a distinct pathway of tau misfolding. However, whether these conformational strains result from the tau isoform distributions in brains or from environmental factors is unknown.
Interaction of tau with water is one of the environmental factors that may play a significant role in shaping the misfolding of tau from its functional, dynamic and monomeric state to a partially ordered oligomeric state, and finally to a well-ordered β-sheet state. The hydration water of the soluble monomer must undergo pronounced reorganization in order to allow the protein to assemble into compact cross-β fibrils that contain dry interior regions packed with hydrophobic sidechains.11,12 The water reorganizational pathway cannot be unique, since the protein is able to adopt multiple morphologically and conformationally distinct fibrils. Moreover, two of the brain-derived tau fibrils contain unknown cofactors,8,9 whose interactions with the protein and with solvent molecules may have affected the fibril structure. Characterizing water interactions with tau is thus valuable for understanding the mechanism of tau misfolding as well as the structural polymorphism of tau filaments.
Because of the role of water in shaping protein misfolding, and because sufficiently large water pockets are potential sites for small molecule binding to detect and inhibit amyloid fibrils, water interactions with amyloid proteins are increasingly studied. Magnetic resonance spectroscopy is well suited for investigating amyloid hydration. Water interactions with the C-terminal half of tau (residues 255–441) in the initial stages of fibril assembly have been studied using Overhauser dynamic nuclear polarization (DNP) NMR.11 The data showed that the translational diffusivity of water that hydrates R3 residues slows significantly in the first three hours after the addition of heparin, when oligomer and spherical assemblies form. Water cavities in the mature fibrils of a transthyretin peptide,13 Aβ40,14,15 and FUS16 have been studied using solid-state NMR and cryoEM. The hydration water of Aβ40 fibrils has been found by relaxation NMR to be heterogeneously dynamic.15 Finally, distinct water exposures of surface residues versus interior steric-zipper residues have been reported for glucagon fibrils and a zinc-bound amyloid fibril.17,18 These water accessibility data provided important restraints for the three-dimensional structures of these amyloid peptides.
We recently conducted the first solid-state NMR study of a full-length tau fibril, 0N4R tau (UniProtKB P10636–6), assembled using heparin.19 13C and 15N chemical shifts measured from 2D and 3D correlation NMR spectra indicated a monomorphic rigid β-sheet core that spans the second and third microtubule-binding repeats. The rest of the protein shows a pronounced dynamic gradient: the R1 and R4 segments are semi-rigid and possess β-sheet character; the proline-rich domains are anisotropically mobile; while the N- and C-termini, containing about 150 residues, are nearly isotropically mobile. Based on long-range distance restraints, we proposed a structural model for the β-sheet core of 0N4R tau. This core is folded into a hairpin, with the R2 hexapeptide motif packed against residues around C322 in the R3 domain, while residues around C291 in the middle of the R2 is packed against the R3 hexapeptide motif. Because of the sparseness of the long-range distance restraints, this structural model does not include sidechains or the R1 and R4 domains. Determining the residue-specific water accessibility of the protein may provide information about the locations of the R1 and R4 domains relative to the R2–R3 hairpin. It may also shed light on the possible existence of internal water cavities in the R2–R3 hairpin. Since well hydrated residues usually occur on protein surfaces, which have fewer structural constraints, the dynamics of the microtubule-binding repeats, especially the elusive R1 and R4 domains, will also be important to characterize further.
Here we present a study of the site-specific hydration of heparin-fibrillized 0N4R tau and the dynamics of the four microtubule-binding repeats. We employ water 1H polarization transfer solid-state NMR experiments, which have been widely used to study the structural topology of membrane proteins20–24 and crystalline proteins.25,26 Preliminary water-edited 2D 13C-13C correlation spectra have been reported before.19 However they were measured under conditions where the signals of the rigid β-sheet residues were not well resolved from the signals of the dynamically disordered residues. We now employ more stringent dynamic filters to resolve the water accessibility of β-sheet residues. We find that most of the R2–R3 residues are well protected from water, except for a small number of residues, including S285 and S316. These data strongly suggest that the R2–R3 hairpin is surrounded by other proteinaceous segments on the exterior and contains a small water pore in the interior. We investigated the backbone mobility of the repeat domains by measuring 2D 15N-13Cα resolved N-H dipolar couplings. The data indicate that R4 residues have rigid-limit N-H dipolar couplings, even though their N-Cα correlation peaks are weakened by motion. This result suggests that R4 participates in cross-β hydrogen bonding in some of the subunits while being dynamically disordered in other subunits. These results shed new light on how the structure and dynamics of in vitro 4R tau fibrils compare with brain-derived 4R tau.
Materials and Methods
Expression, purification and fibrilization of 13C, 15N-labeled 0N4R tau
Recombinant 0N4R tau was expressed and purified as described previously.19 Briefly, the gene encoding 0N4R tau was cloned into a pET-28a vector and used to transform Rosetta (DE3) competent E. coli cells (Novagen). 1 L of LB was inoculated with an overnight starter culture, grown at 37°C until OD600 reached 0.8–0.9, then spun down at 3,000 g for 5 minutes. The cell pellet was resuspended in 1 L M9 minimal media containing 1.0 g 15NH4Cl, 3.0 g 13C-glucose, and 100 mg/L unlabeled glycine, glutamic acid and lysine. The cells were allowed to grow for 40 min before induction with 0.5 mM IPTG for 3 h.
After expression, cells were pelleted and resuspended in 50 mL lysis buffer containing 5 mM DTT, 1 mM EGTA, and cOmplete protease inhibitor tablets (Roche), then lysed using an M-110EH microfluidizer. The lysate was boiled for 20 min before being centrifuged at 127,000 g. The 0N4R tau-containing supernatant was first purified using a cation exchange column (SP Sepharose Fast Flow resin, GE Healthcare). It was then further purified on a reverse-phase HPLC using a semi-preparative column (250 mm length, 10 mm I.D., C4, 5 μm particle size, 300 Å pore size, Higgins analytical) and an acetonitrile/water gradient from 5% to 50% in 45 min. The elution containing 0N4R tau was pooled and lyophilized to yield the protein in powder form.
The protein powder (0.4 mg/mL) was dissolved in 1x PBS (pH 7.4) together with DTT (1 mM) and heparin (0.125 mg/mL, Santa Cruz Biotech, sc203075, 8,000–25,000 Da) in 1.5 mL Eppendorf tubes. The solution was shaken at 37°C and 1400 rpm for 3 days. The resulting fibril suspension was spun at 100,000 g and 4°C for 30 min. The supernatant was removed, and the pellets were pooled and washed twice with DI water to reduce the salt concentration. The solution was partially lyophilized to give a concentrated gel with ~4-fold excess water (by mass). About 17 mg fibrils and ~19 mg water were centrifuged into a thin-wall Revolution NMR pencil rotor with 3.2 mm diameter. The final free salt concentration was estimated to be 5 mM.
Solid-State NMR experiments
All solid-state NMR spectra were measured on a Bruker Avance 800 MHz (18.8 T) spectrometer using a BlackFox 3.2 mm HCN magic-angle-spinning (MAS) probe. The probe includes a crossed-coil dual-resonator low-E stator27 with high radiofrequency (rf) field homogeneity. The latter gives 3-fold higher sensitivity for 15N-13C cross polarization (CP) experiments compared to Bruker 3.2 mm Efree probes. 13C chemical shifts were referenced externally to the adamantane CH2 chemical shift at 38.48 ppm on the tetramethylsilane scale. 15N chemical shifts were referenced to the 15N peak of N-acetylvaline at 122.00 ppm on the liquid ammonia scale. Typical rf field strengths were 54–83 kHz for 1H, 16–50 kHz for 13C, and 25–36 kHz for 15N. Reported temperatures were direct readings from the probe thermocouple. The actual sample temperatures were 16–36 K higher in most experiments, based on the water 1H chemical shift of the sample.28, 29 The water 1H chemical shift of a hydrated DSS-containing POPC membrane sample was used as a secondary external standard to calibrate both the 1H chemical shift and the sample temperature.
Water-edited 2D 13C-13C correlation experiments were conducted under 10.5 kHz MAS at a set temperature of 260 K, with an estimated sample temperature of 290 K. For the equilibrium 2D spectrum, 70 μs 1H-13C CP and 23 ms Combined -Driven (CORD)30 spin diffusion were used. This CP contact time is much shorter than the previous value of 750 μs , and was instrumental for suppressing the random coil signals that overlap with the β-sheet signals in the spectra. The maximum t1 evolution time was 7.45 ms and the t2 acquisition time was 15.36 ms. The t1 increment was chosen to give a spectral width of 200 ppm for the indirect dimension. 1H decoupling during t1 and t2 periods was implemented using the two-pulse phase-modulated (TPPM)31 scheme at an rf field strength of 72 kHz. For each t1 slice, 64 scans were averaged.
To transfer the water 1H polarization to the protein, we applied a selective Hahn echo sequence that includes a hard 90° excitation pulse, a 950 μs Gaussian 180° pulse centered at 5.1 ppm, and a pair of 95 μs delays (Fig. S1a). This scheme differs from a 1H T2 filter with a hard 180° pulse,15, 17, 32 or a water-selective Gaussian 90° excitation pulse followed by a hard-180° T2 filter.33 We chose this scheme to maximally suppress the 1H signals of the many mobile residues in tau. The selected water 1H polarization was stored along the z-axis for magnetization transfer to the protein via chemical exchange and spin diffusion, and was then transferred to 13C for detection. The maximum 13C t1 evolution time was 3.73 ms. We measured water-edited 2D CC spectra with mixing times of 2 ms, 4 ms, 16 ms, and 36 ms. The number of scans per t1 increment was 880, 480, 288, and 160, respectively. Water-transfer buildup curves for selected Ser/Thr Cα and Cβ were least-square fit to a single exponential 1-exp(−t/τ), and presented in Fig. S2b.
We also measured a water-edited 2D 15N-13Cα correlation spectrum using a 2 ms 1H mixing time under 10.5 kHz MAS at a set temperature of 260 K. The same water selection and mixing module was inserted before the 1H-15N CP step (Fig. S1b). The equilibrium spectrum was measured without this module. Both spectra were measured using 1 ms 1H-15N CP with 70–100% amplitude on the 15N channel and 4 ms SPECIFICCP for 15N-13C polarization transfer.34, 35 1H TPPM and CW decoupling with an rf field strength of 83 kHz were used during the t1 and SPECIFICCP period, respectively. The maximum t1 evolution time was 4.8 ms while the t2 acquisition time was 15.4 ms. The number of scans per t1 slice was 160 for the equilibrium spectrum and 1536 were for the 2 ms water-edited spectrum.
We conducted a 13C and 15N dipolar-dephased MELODI-HETCOR-CORD 3D 1H-13C-13C correlation experiment (Fig. S1c) to measure the 1H chemical shifts of Ser and Thr hydroxyl groups.21 The experiment was carried out under 7 kHz MAS at a set temperature of 255 K, with an estimated sample temperature of 288 K. 13C-1H and 15N-1H dipolar dephasing was implemented using a 180° recoupling pulse on the 13C and 15N channels in the middle of each rotor period. The recoupling time was two rotor periods, during which an FSLG pulse train36 was used to decouple the 1H-1H dipolar interaction.21 The 1H transverse rf field strength for FSLG was 84 kHz, which was chosen to synchronize with the rotation period after accounting for a 7 μs 1H 180° pulse at the end of the first rotor period. This MELODI period was followed by 1H t1 evolution, during which the FSLG transverse field strength was set to 68.5 kHz. The t1 period was rotor-synchronized and measured to 50 rotor periods, corresponding to an FSLG-scaled effective maximum t1 of 4.1 ms. Subsequently, the 1H magnetization was transferred to the protein using 1 ms mixing. A 30 μs 1H-13C CP and a 2D 13C-13C correlation module with 23 ms CORD mixing were then used to detect and resolve the signals. The 13C t2 and t3 periods were 3.4 ms and 12.2 ms, respectively.
The 2D 15N-13Cα resolved 15N-1H dipolar-shift (DIPSHIFT) experiment was conducted under 7 kHz MAS at a set temperature of 302 K. At this high set temperature, the deviation from true sample temperature is small. We estimated the sample temperature to be 305 K based on the water 1H chemical shift. After 1 ms 1H-15N CP, the 15N magnetization was dephased by 1H-15N dipolar coupling during a 71 × 2 μs doubled-DIPSHIFT period (Fig. S1d).37 This dephasing was defined by an 15N 180° pulse in the middle of the rotor period. A control experiment without N–H dipolar dephasing was conducted by placing the 15N 180° pulse at the beginning of the rotor period (τ = 0.5 μs). The dephased 15N magnetization evolved during t1 up to 8.6 ms, then transferred to 13C by 4-ms SPECIFICCP for detection. The t2 acquisition time was 15.4 ms. The number of scans per t1 increment was 416 for the dephased experiment and 336 for the control experiment. 83-kHz TPPM and CW 1H decoupling were applied during the t1 evolution and SPECIFICCP, respectively. 1H homonuclear decoupling during DIPSHIFT was conducted using the FSLG sequence with a transverse rf field strength of 83 kHz.
Analysis of hydration maps and site-resolved N-H dipolar couplings
We analyzed the site-specific water accessibility of the protein in two ways. First, we calculated and plotted 2D hydration maps (Fig. 1, 2) using Python scripts that interface with NMRglue38 and matplotlib packages. The peak heights of the water-edited 2D CC and 2D NC spectra (S) were divided by the equilibrium intensities (S0) in a pointwise manner. The latter were taken to be the CP-based 2D spectra measured without water selection. Based on 1D 13C spectra, the water transferred magnetization is equilibrated after 100 ms, but the overall intensity is 1.61 times lower than the CP spectrum. Thus, we multiplied the S/S0 ratios by 1.61. The same scaling factor was also applied to the 2D NC spectral analysis. Peak picking in the 2D CC and NC spectra was based on chemical shift assignments from previous 3D NCC correlation spectra.19 Intensities in the S0 spectra that are below the root-mean-square (RMS) noise level were raised to one noise level, whereas intensities in the S spectra that are below 2–3 noise level were set to zero. This procedure reduces the number of abnormal contours in the heatmap.
Figure 1.
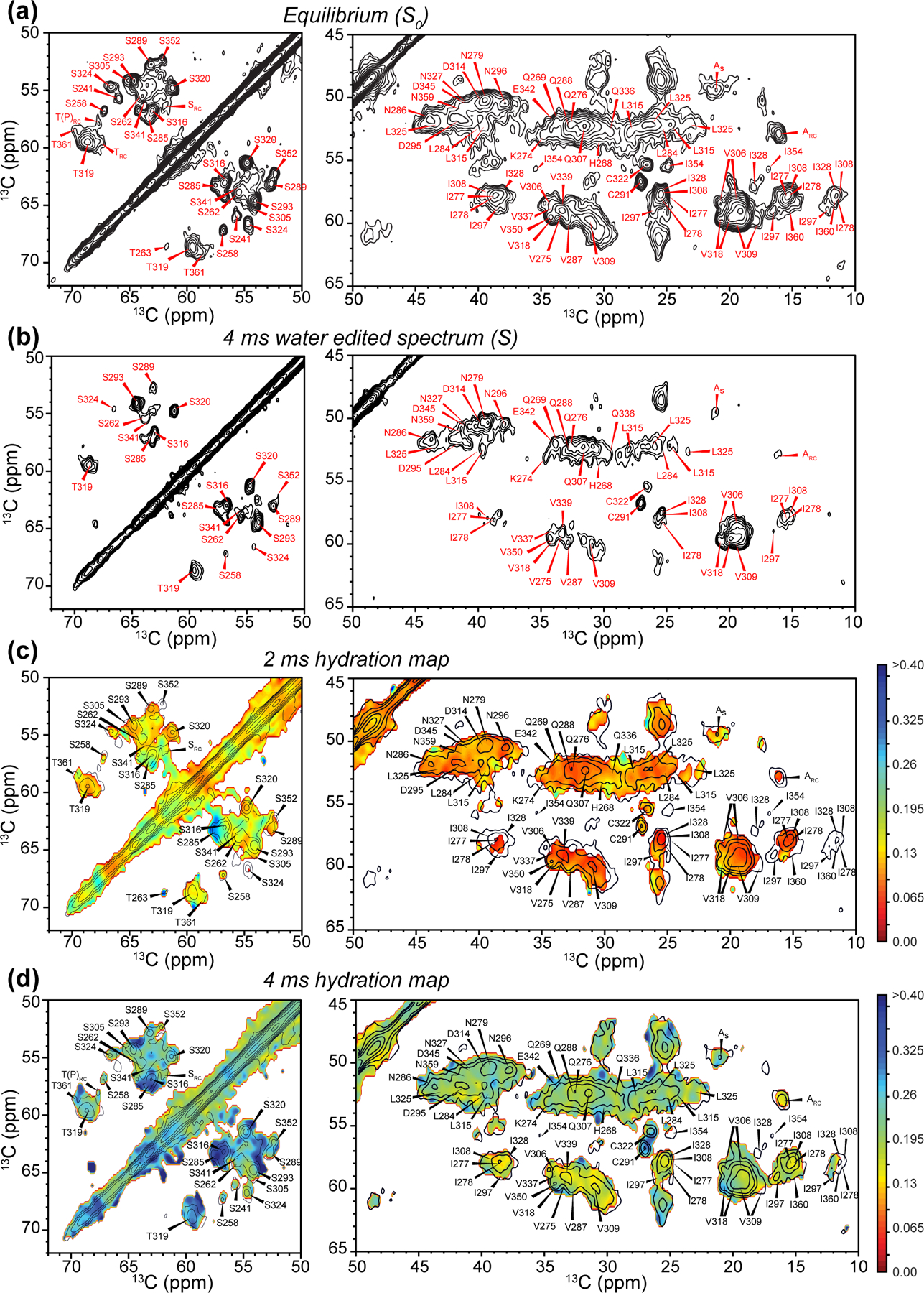
Water-edited 2D 13C-13C correlation spectra for investigating site-specific hydration. (a) Equilibrium 2D spectrum, measured with CP without water selection. The Ser/Thr region and the rest of the Cα-CX aliphatic region are shown. Assignments are based on previous 3D NCC correlation spectra.19 (b) 4 ms water-edited 2D spectrum. (c) Hydration map (pointwise S/S0 values) for the 2 ms water-edited spectrum, overlaid with the equilibrium spectrum, which is plotted with half the number of contour levels as in (a) for clarity. (d) Hydration map for the 4 ms water-edited spectrum. The same color scale as (c) is used to show the increased water polarization transfer compared to 2 ms mixing.
Figure 2.
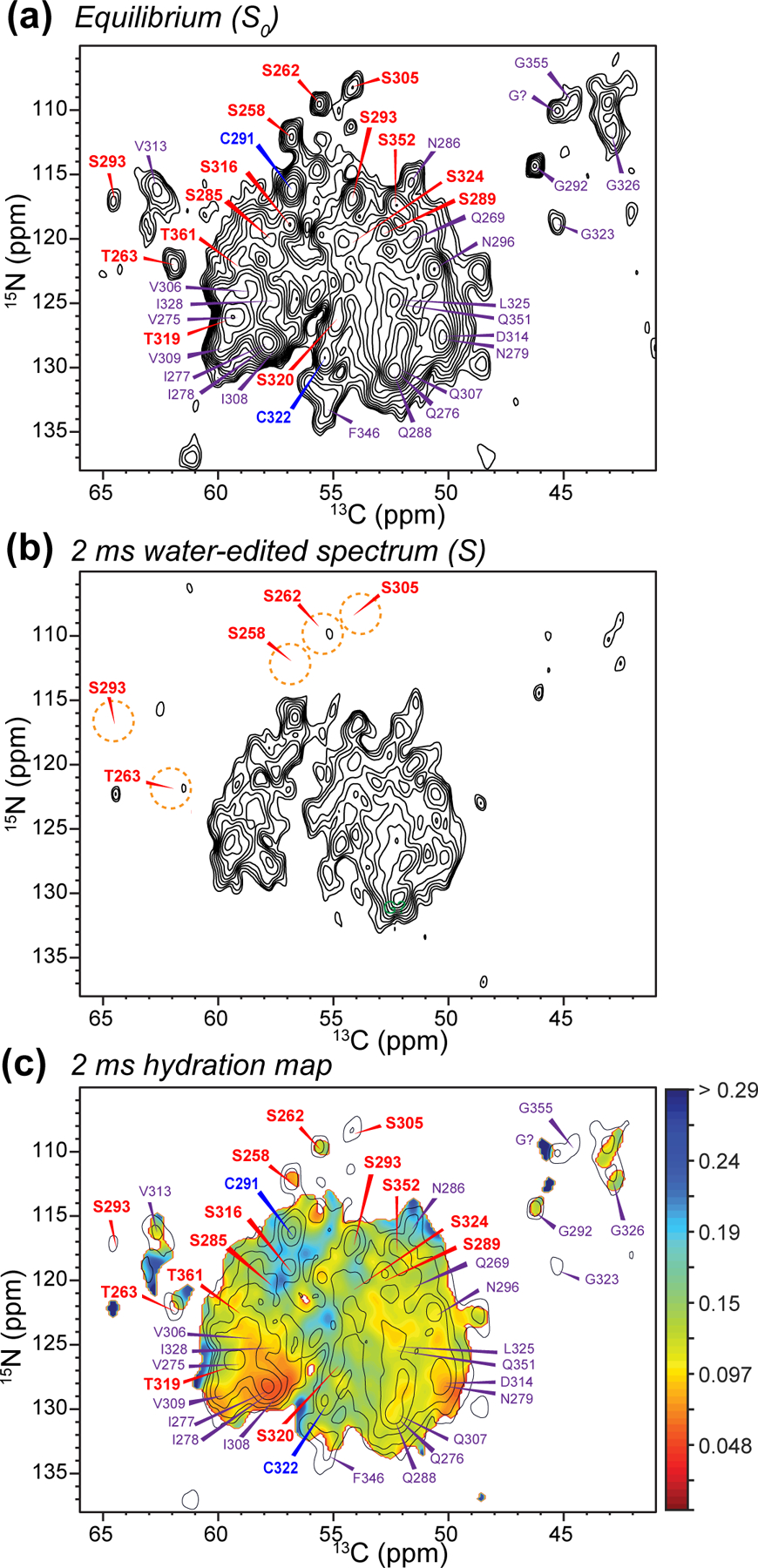
Water-edited 2D 15N-13C correlation spectra. (a) Equilibrium 2D spectrum, with assignments obtained from previous 3D NCC correlation spectra (Fig. S3). Ser, Cys, and other selected residues are assigned in red, blue, and purple, respectively. (b) 2 ms water-edited spectrum. (c) Hydration map (pointwise S/S0 values) for the 2 ms data, overlaid with the equilibrium spectrum, which is plotted with only half the number of contour levels as in (a) for clarity.
While the 2D hydration maps give a visual representation of the varying water accessibilities of the resolved peaks in the spectra, a comparison of hydration of residues along the protein sequence is better shown as S/S0 bar diagrams (Fig. 3). For the water-edited 2D CC spectra, we combined the intensity ratios of the 2 ms and 4 ms spectra to give higher signal-to-noise ratios. The 4 ms data was scaled to 51% to match the intensity of the 2 ms data before adding, so that the weighted average values represent the hydration at 2 ms mixing. This 2D CC data was then compared with the S/S0 values from the 2 ms water-edited 2D NC spectra.
Figure 3.
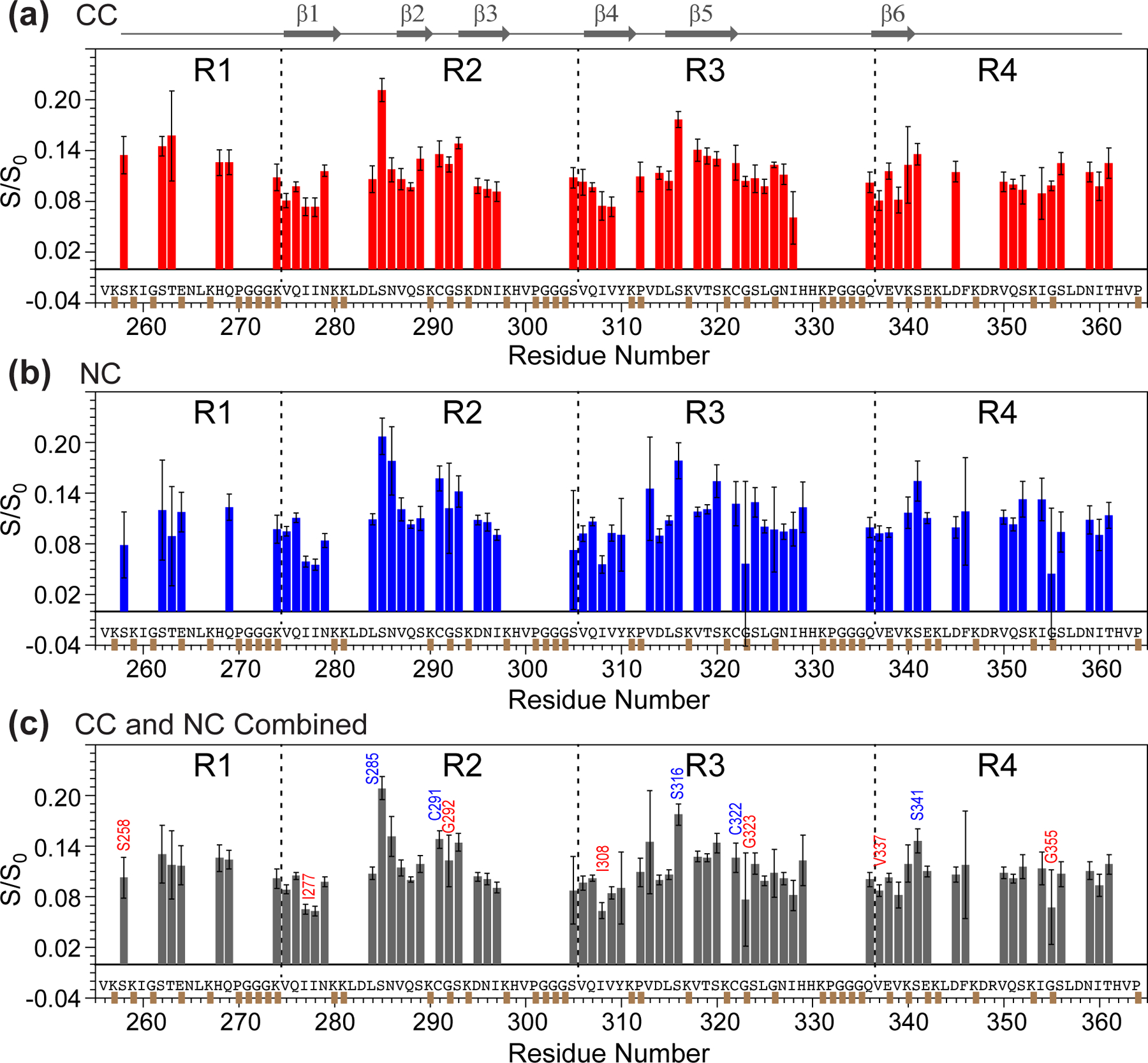
Residue-specific water accessibilities from 2D 13C-13C and 15N-13C correlation spectra. (a) Water accessibility obtained from the 2 ms and 4 ms 2D CC data. The Cα-Cβ and Cβ-Cα intensity values are averaged. The 4 ms S/S0 values were scaled to match the 2 ms S/S0 value. Thus, the bars reflect effective hydration at 2 ms. (b) Water accessibility obtained from the 2 ms N-Cα correlation spectra. (c) Combined water accessibility values at 2 ms, obtained from a weighted average of the 2D CC and NC data. Representative residues with high hydration are indicated in blue, whereas residues with low hydration are indicated in red.
For the 2D CC analysis, the S/S0 ratios of Ser and Thr residues were averaged between the Cα-Cβ and Cβ-Cα cross peaks on the two sides of the spectral diagonal. Error bars in the S/S0 represent the one standard error propagated from the signal-to-noise values of 1D cross-sections of the 2D spectra. We also calculated a whole-residue hydration value (Fig. 3c) by adding the S/S0 values of the 2D CC and NC data using a weighting factor of 43% and 57%, which minimize the random uncertainty.
The N-H dipolar order parameters (SNH) were obtained by comparing the experimentally measured intensity ratios between the half-rotor-period dephased (S) spectrum and the control spectrum (S0) with the calculated S/S0 values in simulated N-H DIPSHIFT curves.
GROMACS simulations of tau fibril hydration
We further investigated the water accessibility of 4R tau fibrils using molecular dynamics simulations in GROMACS. The five lowest-energy solid-state NMR structural models of 0N4R tau fibrils were simulated. These structural models vary due to the small number of long-range distance restraints known so far.19 For comparison, we also simulated water permeation into the CBD type-I tau filaments using the cryoEM structure.9 For each model, we created a 15-monomer fibril by duplicating and translating 3–5 monomer structures along the fibril axis to match the cross-β non-crystallographic symmetry. The 15 monomers were placed into periodic boundary water cubes of ~130 Å per side containing 100 mM NaCl using the CHARMM-GUI “Glycan Modeler” package. This package was also used to generate the GROMACS scripts.39–41 The structures were equilibrated, then molecular dynamics were simulated up to 100 ns using NMRbox.42 Snapshots of the simulated dynamics were extracted, and only the central monomer of the 15-monomer fibril and its nearby solvent molecules are shown.
Results
Residue-specific water accessibility of 0N4R tau from 2D NMR spectra.
To investigate the water accessibility of the rigid β-sheet core,19 we first measured water-edited 2D 13C-13C correlation spectra. Compared to previous spectra, which were measured using 750 μs 1H-13C CP,19 we now use a 70 μs CP contact time to selectively detect the most rigid β-sheet residues while suppressing the random coil signals (Fig. 1a). As a result, the 2D CC spectra are significantly simplified, allowing better resolution of residues such as S241, S289, S324 and S352. The application of a water-selective 1H T2 echo followed by a short 1H mixing period significantly attenuated the spectral intensities (Fig. 1b). At 4 ms mixing, strong signals such as C291 and C322 Cα-Cβ peaks still remain, but many signals of hydrophobic residues are suppressed. In comparison, the Ser and Thr region of the 2D spectrum shows higher intensities, even though many of these residues reside in the β-sheet core.19
The preferential retention of Ser and Thr intensities in these water-edited spectra indicates that proton exchange between water and sidechain OH facilitates 1H polarization transfer at the sample temperature of ~290 K of these experiments. The intrinsic proton exchange rate of Ser and Thr hydroxyl in water have been measured to be 700–900 s−1 at pH 7.0 at 36°C.43 This rate decreases with temperature, to ~100 s−1 at 20°C and ~10 s−1 at 4°C for Thr OH. For residues in the interior of folded proteins, hydrogen bonding and limited water exposure further slow chemical exchange, and cause structurally dependent variations in the exchange rates.16,25,26,44 Therefore, at our sample temperature of 290 K, the proton exchange rates of Ser and Thr OH groups are at most 100–200 s-1. For the short 1H mixing time of 2–4 ms, this chemical exchange favors the detection of Ser and Thr signals, but only modestly. At the same time, 1H spin diffusion is rapid in the rigid β-sheet segments of the protonated tau, which should reduce the difference between exchangeable and non-exchangeable residues.45–48 Indeed, NMR detection of chemical exchange often involves protein deuteration, to distinguish solvent-exposed and solvent-protected residues, or hydrogen-bonded and non-bonded sidechains.16,25,26,44 Taken together, these considerations suggest that in these protonated tau fibrils, the different water-edited spectral intensities between Ser/Thr residues and hydrophobic residues have a moderate contribution from chemical exchange, while the main difference should result from the water accessibility. Nevertheless, to ensure accurate interpretation, below we compare the hydration of Ser and Thr residues separately from the hydration of non-exchangeable hydrophobic residues.
To better visualize the relative water exposures of different residues and the time course of water polarization transfer, we divided the intensity of the water-edited 2D spectrum by the equilibrium intensity, and showed the result as hydration maps for the 2 ms and 4 ms 2D spectra in Fig. 1c,d. As expected, all residues exhibit higher water polarization transfer at the longer mixing time. Within each mixing time, the Ser and Thr residues display higher hydration than the other residues. Comparing among the Ser and Thr residues, we found three types of hydration behavior. First, S285 and S316 exhibit the highest water accessibility in both the 2 ms and 4 ms spectra, even though these two residues lie in the rigid R2 and R3 core. Second, the nearby S293 and S320 show lower water-transferred intensities compared to S285 and S316, but the intensities increase significantly from 2 ms to 4 ms, indicating intermediate hydration. Third, residues such as S289 and S324 are poorly hydrated in both the 2 ms and 4 ms spectra. Taken together, these data indicate that the water accessibility of the β-sheet core is not uniform, and small water cavities may be present within the β-sheet core.
Additional information about sidechain hydration can be obtained by comparing the Ser and Thr Cβ-Cα and Cα-Cβ cross peak intensities. As water 1H polarization transfer precedes the first 13C evolution period in the 2D experiment (Fig. S1a), a 13C-13C cross peak specifically indicates the water accessibility of the first carbon. Hence asymmetries in cross-peak intensities reveal whether the sidechain or backbone carbons are more water-exposed: if a sidechain extends towards water, then the Cβ-Cα cross peak should be more intense than the Cα-Cβ cross peak. Fig. S2 compares the Ser/Thr Cβ-Cα and Cα-Cβ cross peak buildup curves from 2 ms to 100 ms mixing. Residues such as S285, S316, and T319 exhibit higher Cβ-Cα intensities than Cα-Cβ intensities, consistent with their high water-transferred intensities at short mixing times. Thus, these residues are well hydrated and point their sidechains towards water. In comparison, residues such as S258 and S305 display higher Cα-Cβ cross peaks than Cβ-Cα cross peaks, suggesting that their sidechains point to a dry interior.
To compare the water exposure of all residues without the potential difference caused by exchangeable protons, and to specifically probe backbone HN hydration, we conducted the water-edited 2D N-Cα correlation experiment. Fig. 2 compares the equilibrium and 2 ms water-edited N-Cα spectra, where the assignment is based on the previous 3D spectra (Fig. S3). Similar to the 2D CC data, we observed non-uniform water polarization transfer. Specifically, S258, S262, T263, S293, and S305 exhibit lower water accessibility than other Ser and Thr residues. In comparison, residues such as C291, C322 and N286 have high intensities, indicating high water accessibility.
Taken together, these water-edited 2D CC and NC spectra indicate that S285 and S316 are the best hydrated residues in the β-sheet core, whereas other Ser, Thr and Cys residues are moderately or poorly hydrated. To compare the residue-specific water exposure quantitatively, we compared the water-transferred intensities (S/S0) from the 2D CC and NC spectra (Fig. 3). The S/S0 values range from 0.05 to 0.22. A whole-residue hydration value, reflecting both the sidechain and backbone, is obtained by a weighted average of the S/S0 values of the 2D CC and NC spectra (Fig. 3c). Several trends can be seen in this whole-residue hydration diagram. The R2 and R3 hexapeptide motifs, which span V275-K280 and V306-K311, exhibit the lowest S/S0 values, consistent with these segments being engaged in tight steric zippers. Second, the R1 and R4 domains do not have very different water accessibilities from R2 and R3 domains. This suggests that R1 and R4 are similarly surrounded by proteinaceous segments instead of being exposed to water. When the residue-specific water accessibilities are color-coded onto the 0N4R tau schematic and structural model (Fig. 4a, b), we find that the two most hydrated residues, S285 and S316, face each other across a central pocket that is lined by residues that were previously found to be disordered.19
Figure 4.
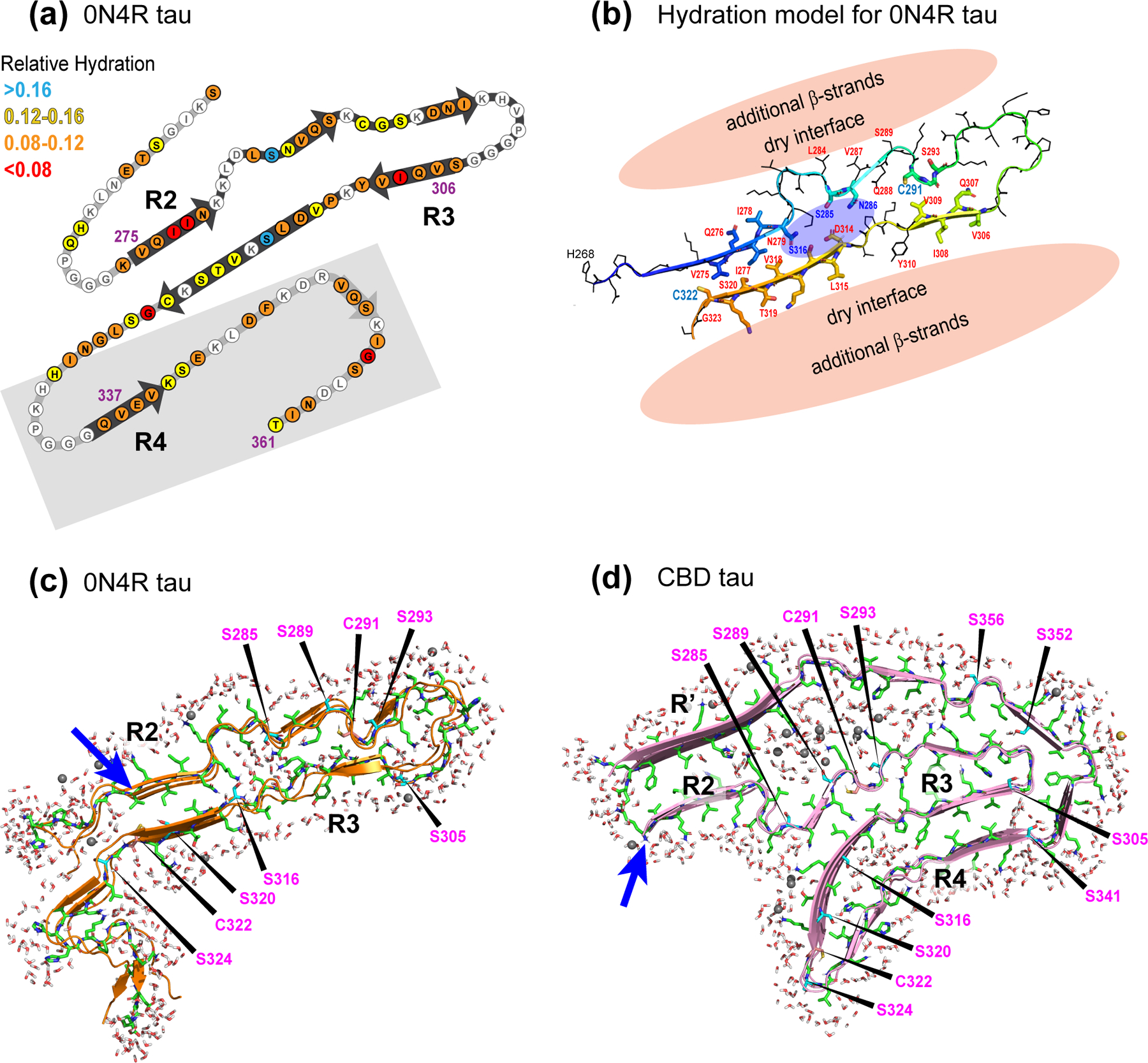
Measured and simulated hydration of 4R tau fibrils. (a) Schematic of the solid-state NMR structural model of heparin-fibrillized 0N4R tau, where the measured whole-residue water-accessibility values are indicated in color. Grey shaded area indicates segments whose 3D fold is not known. (b) Structural model (#1) of the β-sheet core of heparin-fibrillized 0N4R tau and the proposed hydration. A water pocket flanked by S285 and S316 is proposed, and the exterior of the R2 and R3 domains is surrounded by proteinaceous density and is thus dry. (c) Simulated water permeation into 0N4R tau fibrils after 100 ns. Structural model #1 was used for the simulation. (d) Simulated water permeation into CBD tau fibrils after 50 ns. Blue arrows indicate the beginning of the R2 domain in the two structural models, and key Ser and Cys residues are indicated. In both simulations, the water molecules on the external surfaces of the β-sheet core should not be interpreted as real, because no information about the structure of the disordered protein segments is known for either tau proteins. Only the internal water density should be interpreted.
We also investigated whether there are Ser and Thr residues that do not exchange with water, by implementing a 3D 1H-13C-13C MELODI-HETCOR experiment,21 This experiment selectively suppresses the magnetization of protons that are directly bound to rigid 13C or 15N by dipolar dephasing, then encodes the 1H chemical shifts of the mobile water, hydroxyl, and methyl groups (Fig. S1c). This 1H magnetization is allowed to diffuse to neighboring protons during a 1 ms mixing period before being transferred to 13C for detection. The 2D CC plane at the water 1H chemical shift of 5.1 ppm has a similar pattern to the 2D water-edited spectra (Fig. S4), confirming that the source of the 1H magnetization is indeed water. In 2D planes away from the water and methyl 1H chemical shifts, we did not detect strong Ser and Thr cross peaks. We attribute this absence to a combination of limited sensitivity and rapid 1H spin diffusion in the protonated protein, which equilibrates the magnetization of most protons. It is noteworthy that hydroxyl groups with non-water 1H chemical shifts were recently reported in the FUS fibrils,16 which have a similarly polar amino acid composition as tau, and which contains similarly large disordered domains. There, perdeuterated FUS that was back-exchanged with OH and NH was used to detect the presence of water-protected sidechain OH moieties.
Molecular dynamics simulation of tau hydration
To gain further insight into residue-specific hydration of the tau fibrils, we conducted molecular dynamics simulations of water permeation into the β-sheet core of both heparin-fibrillized 0N4R tau19 and CBD 4R tau.9 We recently proposed five structural models for in vitro 0N4R tau.19 The coexistence of several models is due to the small number of experimentally measured long-range distance contacts. We simulated water permeation using all five models, with model 1 (Fig. 4c) having the lowest energy. As these simulations only included the structurally constrained microtubule-binding repeats, we only focus on the number and locations of water molecules within the fibril core. Fig. 4c, d and Fig. S5 show snapshots of the various hydrated tau structures. In most in vitro structural models, a small number of water molecules is seen to permeate the interior of the R2–R3 hairpin. Model 2 and model 4 exhibited higher water densities in the hairpin, but the structures were unstable beyond 2 ns: cross-β hydrogen bonds fell apart along large stretches of the 15 monomer filaments during the simulations. In comparison, the CBD tau structure9 remained stable during the simulations, and a large number of water molecules permeated into the core. Interestingly, high water densities are found near residues K290, K294 and K370, the position that is occupied by a cofactor in the cryoEM structure. This cofactor was not included in the simulation. Water also infiltrated several pockets around residues S316 and S341.
N-H dipolar couplings suggest that the semi-rigid R4 domain has cross-β character
We recently investigated the backbone and sidechain dynamics of 0N4R tau using 2D-resolved C-H dipolar couplings. An independent probe of backbone mobility is the N-H dipolar coupling, which can be resolved in 2D N-Cα correlation spectra. A fully immobilized N-H group should exhibit strong dipolar dephasing that corresponds to an order parameter SNH of 1, whereas motions faster than tens of microseconds should partially average the dipolar couplings and cause less dipolar dephasing. We measured two 15N-13Cα correlation spectra with 0 and 71 μs DIPSHIFT dephasing, the latter corresponding to half a rotor period at the MAS rate of 7 kHz. The calculated N-H DIPSHIFT curves at this MAS rate indicate S/S0 values of 0.40–0.95 at half a rotor period for SNH values of 1.0–0.2 (Fig. 5a). The measured dipolar-dephased 2D 15N-13Cα spectrum shows clearly weaker intensities than the control spectrum (S0) (Fig. 5b, c), indicating that most residues in the β-sheet core are immobilized. However, a number of residues, including S305, V313 and H329, show higher intensities in the dephased spectrum that correspond to SNH values of 0.5–0.7 (Fig. 5c, d). Thus, these residues have a more dynamic backbone. Residue S305, located between the 301PGGG304 segment and the hexapeptide motif 306VQIVY310, manifests the highest residual intensity, indicating significant dynamic disorder.
Figure 5.
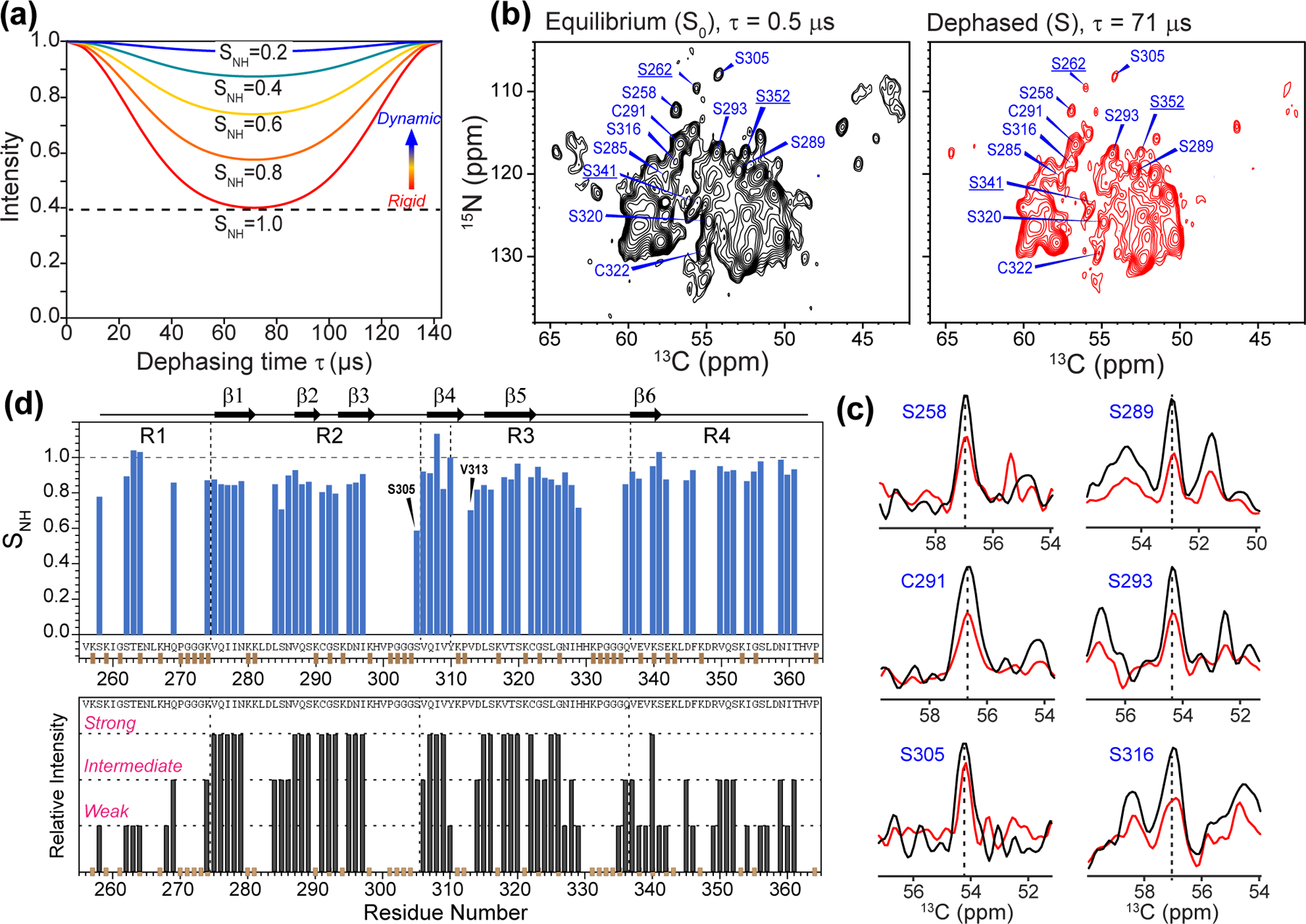
Mobility of 0N4R tau from site-resolved N-H dipolar couplings. (a) Simulated N-H doubled DIPSHIFT curves under 7 kHz MAS for a range of order parameters. The rigid-limit dipolar coupling is 12.1 kHz after accounting for FSLG scaling of 0.577 and doubling due to doubled DIPSHIFT. (b) 2D 15N-13C correlation spectra without N-H dipolar dephasing and with half of a rotor period of dipolar dephasing. The spectra were measured under 7 kHz MAS. For clarity, only Ser and Cys residues are assigned. Underlined residues are from the R1 and R4 domains that show significant dipolar dephasing, indicating that they are rigid. (c) Selected 1D cross sections from the control (black) and dipolar-dephased (red) spectrum. More mobile residues such as S305 show less dipolar dephasing. (d) N-H dipolar order parameters obtained from the DIPSHIFT spectra. Interestingly, R4 residues show large SNH values, indicating that the CP-detected R4 residues are well ordered. For comparison, the previously reported site-resolved peak intensities from 3D NCC spectra are shown below.19 Many R1 and R4 residues that show weak intensities in the 3D correlation spectra display high N-H order parameters.
Interestingly, the assigned β-sheet R4 residues also display large N-H order parameters (Fig. 5d), implying that the R4 domain is similarly involved in the hydrogen-bonded cross-β fibrils. This result is unexpected, because R4 residues previously showed weaker intensity in 3D NCACX and NCOCX spectra compared to R2 and R3 residues, and also showed lower intensities at high temperature than R2 and R3 residues19. These observations led to the previous conclusion that the R4 domain is semi-rigid. However, the current N-H dipolar couplings were measured after the 1H-15N CP and before the 15N-13C CP (Fig. S1d), both of which selectively detect rigid residues. Therefore, the high N-H order parameters observed here are not necessarily contradictory to the previous data, but indicate that a fraction of proteins contain well-ordered and rigid R4 residues while the rest contain a dynamic R4 domain.
Discussion
The interior and exterior of the fibril core from water accessibility data
These hydration and dynamics data provide new insights into the structure and disorder of heparin-fibrillized 0N4R tau fibrils. Our data indicate that the exterior of all four microtubule-binding repeats is surrounded by extensive proteinaceous densities, while the interior of the R2–R3 hairpin contains a small water channel. This conclusion results from three lines of evidence. First, the R2 and R3 hexapeptide motifs, 275VQIINK280 and 306VQIVYK311, form tight steric zippers based on long-range contacts and strong β-sheet signals.19 Thus, these sidechains must zigzag and point to opposite sides of the backbone. If these segments were exposed to solvent on one side and engaged in the steric zipper on the other side, then half of the sidechains should be hydrated while the other half should be dry. Such an alternating water-accessibility pattern has been observed in glucagon fibrils17 and a de novo designed fibril.18 Instead, the water-edited 2D spectra show that both hexapeptide motifs are uniformly poorly hydrated (Fig. 3), indicating that the residues that are not involved in the steric zipper face other protein chains rather than water. Second, V306 and V309 in the R3 hexapeptide motif exhibit two resolved Cγ chemical shifts. The two methyl groups in Val residues normally undergo rapid rotameric jumps as long as they are not sterically hindered.49 This would give rise to a single averaged Cγ chemical shift. V306 and V309 are part of a rigid cross-β strand, thus their sidechains lie on opposite sides of the backbone. Yet both residues display two distinct Cγ chemical shifts: 19.4 and 20.6 ppm for V306 and 19.7 and 17.7 ppm for V309.19 Hence V306 and V309 must each pack against other sidechains, which also explain their low water accessibilities (Fig. 3c). Third, the R1 and R4 domains, which are less rigid than R2 and R3 based on their weaker N-Cα spectral intensities,19 do not show higher water accessibility. This suggests that these two repeat domains are surrounded by other protein residues.
If the exterior of the four repeat domains is surrounded by other proteinaceous density rather than by water, then the presence of several well hydrated residues, especially S285 and S316, implies the presence of a small water pore inside the R2–R3 hairpin. This water pocket is local, because the water-edited S285 signal is significantly higher than the water-edited signal of the neighboring S289 (Fig. 3c). A localized and “rugged” hydration landscape for tau is overall consistent with emerging data about the hydration of other amyloid fibrils. In the three-fold Aβ40 fibril, Met35 but not the neighboring hydrophobic residues point toward a central water pore.15 The FUS fibril, which has a similarly polar amino acid sequence as tau, also lacks large water channels.50 Moreover, a number of Ser and Thr residues in FUS form water-excluded sidechain hydrogen bonds, as evidenced by the fact that their OH chemical shifts are well resolved from the bulk water 1H chemical shift. The fibrils formed by a transthyretin peptide (residues 105–115) contain a large number of hydration water.13 These water molecules lie in an elongated cavity that separates multiple protofilaments, whereas each protofilament contains dry steric zippers. In the glucagon fibril, a pair of antiparallel β-strands along the fibril axis alternately point the even and odd-numbered residues toward water.17 This unusual symmetry may be caused by the polar and diverse amino acid sequence of the peptide, which does not give a strong preference for half of the residues to reside in a dry steric zipper. These structural data suggest that the presence or absence of water pockets in cross-β amyloid fibrils, as well as the size of these water pockets, may depend strongly on whether hydrophobic or polar interactions provide the dominant stabilization energy for the β-sheet structure, and on how protofilaments assemble together. We speculate that large water channels may form more readily in hydrophobic amyloid fibrils whereas small water pockets may be more associated with polar amyloid proteins.
The overall dryness of the 0N4R tau fibril core has implications for therapeutic interventions. It suggests that small-molecule drugs and imaging agents may not be able to intercalate into the R2–R3 hairpin. Instead, the semi-rigid R1 and R4 domains, or regions outside the four repeat domains, may be better target sites for drug binding.
The hydrophilicity of protein sidechains may also play a significant role in the folding pathway of an amyloid protein. The formation of small oligomers51 as nucleation sites for fibrils depends on the free energies of the monomer/water system relative to the oligomer/water mixture. Changes of water dynamics during fibril formation have been investigated for tau using neutron scattering, MD simulations,52 Overhauser DNP and EPR.11 Quasi-elastic neutron scattering data showed that tau fibrils have more dynamic hydration water compared to tau monomers. This increased water dynamics was suggested to result from the polar fuzzy coat of the protein, which may form a two-layered polyelectrolyte brush with a geometry that facilitates water diffusion.52 When water molecules associated with the R3 domain are probed using nitroxide spin labeled residues and the Overhauser DNP effect, Han and coworkers found that the R3-associated water slowed down its translational diffusion in the first three hours of fibril assembly, whereas the dynamics of water associated with the C-terminus of the protein is largely unaffected.11 These results suggest that water interaction with amyloid proteins is sensitive to the conformation and hydrophilicity of the protein residues. Systematic investigations of the water accessibility and water dynamics of amyloid proteins in their monomeric, oligomeric, and fibrillar states should provide a more comprehensive understanding of the folding process and structural polymorphism of these proteins.
Our MD simulations (Fig. 4) show that the number of water molecules inside the R2–R3 hairpin is relatively small. This result is broadly consistent with the experimentally measured low water accessibilities. Since the 0N4R tau structural model is still low-resolution, these water simulations should be taken qualitatively, to be refined by future experimental data. Nevertheless, in membrane-embedded ion channels such as the influenza M2 protein, a small pore diameter of ~8 Å can already accommodate multiple water molecules, as seen in high-resolution crystal structures.53,54 Relaxation-filtered solid-state NMR experiments similar to the ones conducted here show that water 1H polarization can be transferred to various M2 sidechains.20,32,33,55 Therefore, the number of water molecules in protein interior does not need to be large for water to acquire the dynamics that is necessary to survive the T2 relaxation filter and to transfer polarization to protein residues on the millisecond timescale.
Similarities and differences between heparin-fibrilized 0N4R tau and CBD tau
Our data indicate that the structure of heparin-fibrillized 0N4R tau has a remarkably high degree of similarity with the CBD tau structure, although the two fibril structures are distinct (Fig. 4d).9 First, both heparin-fibrillized 0N4R tau and CBD tau exhibit a hairpin between the R2 and R3 domains, with the turn spanning the 301PGGG304 segment. In CBD tau, C291 in the 291CGS293 motif forms a steric zipper with V313 after the end of the R3 hexapeptide, 306VQIVYK311. In heparin-fibrillized 0N4R tau, C291 stacks against V309 (Fig. S6). Thus, there is a four-residue shift in sidechain packing. Second, the CBD tau structure exhibits a significant kink between the R2 hexapeptide motif and S285, which is also observed in heparin-fibrillized 0N4R tau. Third, in CBD tau, the R3 domain continues onward to form a second hairpin with the R4 domain, such that the half of the R3 residues that do not contact the R2 sidechains form steric zippers with R4 (Fig. S6). In heparin-fibrillized 0N4R tau, the R4 position is not yet known, but the current hydration data indicate that the R3 exterior is protected from water, and the R4 segment is rigid and participates in hydrogen bonding in some of the subunits. The most likely explanation of these observations is that that R3 and R4 are packed together, likely through sidechain steric zippers, in agreement with the CBD tau fold. Likewise, the enclosure of the R2 segment by the R’ domain in the CBD tau structure is consistent with the current observation that R2 in heparin-fibrillized tau does not have a solvent-exposed exterior.
An apparent difference between the heparin-fibrillized 0N4R tau and CBD tau is that the former does not include R4 and R’ domains in the rigid β-sheet core,19 while the latter’s β-sheet core spans R2-R4 and the first ten residues of R’.9 However, in the CBD tau structure, the R4 and R’ domains have lower resolution (4.5 Å) than the R2–R3 domain (2.5–3.0 Å). This disorder is in excellent agreement with the NMR-detected dynamic disorder of the R4 domain.19 Helical reconstruction of CBD tau structure used ~33% of all particle images from six samples, with some samples giving only 10% high-quality particle images. The small fraction of particles used for image analysis indicates the presence of static disorder in the full population of CBD tau samples. For comparison, the NMR data reflect the structure and dynamics of the full ensemble of protein molecules. Therefore, the NMR data of heparin-fibrillized 0N4R tau and the cryoEM data of brain-derived CBD tau are consistent in showing that the R4 domain is less ordered than the R2–R3 domains.
In principle, two types of disorder could exist for the R4 domain. Either all protein molecules exhibit the same dynamic disorder in R4, or some protein molecules contain rigid and well-ordered R4 while other molecules have a dynamic R4 domain. The 2D 15N-13C resolved N-H dipolar couplings (Fig. 5) indicate that the second scenario is true: R4 residues that are sufficiently rigid to contribute to the 2D 15N-13C spectra have rigid-limit N-H dipolar coupling. Thus, some protein subunits have a well ordered R4 domain that interacts with R3 through steric zippers, whereas other subunits have a dynamically disordered R4 domain that does not interact strongly with R3, especially at high temperature. Those subunits that contain a well ordered R4 manifest their signals in the NCA and NCO spectra, and their rigid-limit N-H dipolar couplings suggest that these R4 segments participate in cross-β hydrogen bonding along the fibril axis. In comparison, those subunits that have a dynamic R4 domain do not contribute significant intensities to the NCA and NCO spectra. Thus their N-H order parameters are not measured in the experiment.
While the global three-dimensional fold of in vitro and in vivo 4R tau is similar, our water accessibility data indicate some specific differences in the sidechain structures. For example, in 0N4R tau, S285 is well hydrated while in CBD tau, the S285 sidechain is protected from water by the neighboring L282 and V287. In 0N4R tau, S289 is poorly hydrated, while in CBD tau, S289 points directly to the cofactor pocket, which is most likely well hydrated.
In conclusion, the present hydration and dynamics data indicate that the exterior of the R2–R3 β-sheet hairpin of heparin-fibrillized 0N4R tau is protected from water by other protein segments, while the interior of the R2–R3 hairpin contains a small water pore. The R4 domain most likely interacts with the R3 sidechains, as in CBD tau. The R4 domain participates in cross-β hydrogen bonding in some of the subunits while showing dynamic disorder in other subunits. Within the R2–R3 β-sheet core, the sidechain stacking of in vitro 0N4R tau has site-specific differences from the sidechain stacking of CBD tau. These water accessibility and dynamics data thus give insights into the three-dimensional fold of both the ordered and disordered regions of the microtubule-binding repeats in 4R tau fibrils. The small size of the water pore in the R2–R3 hairpin and the poor hydration of most of the rigid core suggests that small-molecule drugs should preferably target regions outside these two repeats.
Supplementary Material
Acknowledgement
This work is supported by NIH grant AG059661 to M.H. The authors are grateful to Dr. Haifan Wu for purification and fibrilization of 0N4R tau. The authors thank Sjors Scheres for sharing the cryoEM coordinates of the CBD tau fibrils. The GROMACS simulations used the NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by NIH grant P41GM111135 (NIGMS).
Footnotes
Accession code
0N4R Tau, UniProtKB P10636–6
Supporting Information Available
Additional water-edited spectra, water accessibility analysis, NMR pulse sequence diagrams, MD simulations, and schematic models of 0N4R tau and CBD tau, are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- [1].Wang Y, and Mandelkow E (2016) Tau in physiology and pathology, Nat. Rev. Neurosci 17, 5–21. [DOI] [PubMed] [Google Scholar]
- [2].Goedert M, Eisenberg DS, and Crowther RA (2017) Propagation of Tau Aggregates and Neurodegeneration, Annu. Rev. Neurosci 40, 189–210. [DOI] [PubMed] [Google Scholar]
- [3].Morozova OA, March ZM, Robinson AS, and Colby DW (2013) Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein, Biochemistry 52, 6960–6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, Jackson S, O’Neill MJ, Isaacs AM, Hutton ML, Szekeres PG, Goedert M, and Bose S (2015) Conformation determines the seeding potencies of native and recombinant Tau aggregates, J. Biol. Chem 290, 1049–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Goedert M, Masuda-Suzukake M, and Falcon B (2017) Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration, Brain 140, 266–278. [DOI] [PubMed] [Google Scholar]
- [6].Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease, Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, and Goedert M (2018) Structures of filaments from Pick’s disease reveal a novel tau protein fold, Nature 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M, and Scheres SHW (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules, Nature Epub ahead of print [DOI] [PMC free article] [PubMed]
- [9].Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T, Murayama S, Ghetti B, Hasegawa M, Goedert M, and Scheres SHW (2020) Novel tau filament fold in corticobasal degeneration, a four-repeat tauopathy, Nature 580, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN, Seyfried NT, Petrucelli L, and Fitzpatrick AWP (2020) Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains, Cell 180, 633–644.e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pavlova A, Cheng CY, Kinnebrew M, Lew J, Dahlquist FW, and Han S (2016) Protein structural and surface water rearrangement constitute major events in the earliest aggregation stages of tau, Proc. Natl. Acad. Sci. U. S. A 113, E127–E136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Daidone I, Ulmschneider MB, Di Nola A, Amadei A, and Smith JC (2007) Dehydration-driven solvent exposure of hydrophobic surfaces as a driving force in peptide folding, Proc. Natl. Acad. Sci. U. S. A 104, 15230–15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fitzpatrick AW, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Muller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TP, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, and Dobson CM (2013) Atomic structure and hierarchical assembly of a cross-b amyloid fibril, Proc. Natl. Acad. Sci. USA 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, and Tycko R (2013) Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue, Cell 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang T, Jo H, DeGrado WF, and Hong M (2017) Water Distribution, Dynamics, and Interactions with Alzheimer’s β-Amyloid Fibrils Investigated by Solid-State NMR, J. Am. Chem. Soc 139, 6242–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Murray DT, and Tycko R (2020) Side Chain Hydrogen-Bonding Interactions within Amyloid-like Fibrils Formed by the Low-Complexity Domain of FUS: Evidence from Solid State Nuclear Magnetic Resonance Spectroscopy, Biochemistry [DOI] [PMC free article] [PubMed]
- [17].Gelenter MD, Smith KJ, Liao S-Y, Mandala VS, Dregni AJ, Lamm MS, Tian Y, Xu W, Pochan DJ, Tucker TJ, Su Y, and Hong M (2019) The peptide hormone glucagon forms amyloid fibrils with two coexisting β-strand conformations, Nat. Struc. Mol. Biol 26, 592–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee M, Wang T, Makhlynets OV, Wu Y, Polizzi NF, Wu H, Gosavi PM, Stöhr J, Korendovych IV, DeGrado WF, and Hong M (2017) Zinc-binding structure of a catalytic amyloid from solid-state NMR, Proc. Natl. Acad. Sci. U. S. A 114, 6191–6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dregni AJ, Mandala VS, Wu H, Elkins MR, Wang HK, Hung I, DeGrado WF, and Hong M (2019) In vitro 0N4R tau fibrils contain a monomorphic β-sheet core enclosed by dynamically heterogeneous fuzzy coat segments, Proc. Natl. Acad. Sci. U.S.A 116, 16357–16366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Luo W, and Hong M (2010) Conformational changes of an ion channel membrane protein detected through water-protein interactions using solid-state NMR spectroscopy, J. Am. Chem. Soc 132, 2378–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li S, Su Y, Luo W, and Hong M (2010) Water-protein Interactions of an Arginine-Rich Membrane Peptide in Lipid Bilayers Investigated by Solid-State Nuclear Magnetic Resonance Spectroscopy, J. Phys. Chem. B 114, 4063–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ader C, Schneider R, Seidel K, Etzkorn M, Becker S, and Baldus M (2009) Structural rearrangements of membrane proteins probed by water-edited solid-state NMR spectroscopy, J. Am. Chem. Soc 131, 170–176. [DOI] [PubMed] [Google Scholar]
- [23].Chen Y, Zhang Z, Tang X, Li J, Glaubitz C, and Yang J (2014) Conformation and Topology of Diacylglycerol Kinase in E.coli Membranes Revealed by Solid-state NMR Spectroscopy, Angew. Chem. Int. Edit 53, 5624–5628. [DOI] [PubMed] [Google Scholar]
- [24].Wang S, Shi L, Kawamura I, Brown, Leonid S, and Ladizhansky V (2011) Site-Specific Solid-State NMR Detection of Hydrogen-Deuterium Exchange Reveals Conformational Changes in a 7-Helical Transmembrane Protein, Biophys. J 101, L23–L25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Böckmann A, Juy M, Bettler E, Emsley L, Galinier A, Penin F, and Lesage A (2005) Water-protein hydrogen exchange in the micro-crystalline protein crh as observed by solid state NMR spectroscopy, J. Biomol. NMR 32, 195–207. [DOI] [PubMed] [Google Scholar]
- [26].Agarwal V, Linser R, Fink U, Faelber K, and Reif B (2010) Identification of hydroxyl protons, determination of their exchange dynamics, and characterization of hydrogen bonding in a microcrystallin protein, J. Am. Chem. Soc, 132, 3187–3195. [DOI] [PubMed] [Google Scholar]
- [27].McNeill SA, Gor’kov PL, Shetty K, Brey WW, and Long JR (2009) A low-E magic angle spinning probe for biological solid state NMR at 750 MHz, J. Magn. Reson 197, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Böckmann A, Gardiennet C, Verel R, Hunkeler A, Loquet A, Pintacuda G, Emsley L, Meier BH, and Lesage A (2009) Characterization of different water pools in solid-state NMR protein samples, J. Biomol. NMR 45, 319–327. [DOI] [PubMed] [Google Scholar]
- [29].Cavanagh J, Fairbrother WJ III, A. G. P., and Skelton NJ (1996) Protein NMR spectroscopy: principles and practice, Academic Press, San Diego. [Google Scholar]
- [30].Hou GJ, Yan S, Trebosc J, Amoureux JP, and Polenova T (2013) Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids, J. Magn. Reson 232, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bennett AE, Rienstra CM, Auger M, Lakshmi KV, and Griffin RG (1995) Heteronuclear decoupling in rotating solids, J. Chem. Phys 103, 6951–6958. [Google Scholar]
- [32].Williams JK, and Hong M (2014) Probing membrane protein structure using water polarization transfer solid-state NMR, J. Magn. Reson 247, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mandala VS, Gelenter MD, and Hong M (2018) Transport-Relevant Protein Conformational Dynamics and Water Dynamics on Multiple Time Scales in an Archetypal Proton Channel: Insights from Solid-State NMR, J. Am. Chem. Soc 140, 1514–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baldus M, Petkova AT, Herzfeld J, and Griffin RG (1998) Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems, Mol. Phys 95, 1197–1207. [Google Scholar]
- [35].Hung I, and Gan ZH (2015) Spin-locking and cross-polarization under magic-angle spinning of uniformly labeled solids, J. Magn. Reson 256, 23–29. [DOI] [PubMed] [Google Scholar]
- [36].Levitt MH, Kolbert AC, Bielecki A, and Ruben DJ (1993) High-Resolution H-1-Nmr in Solids with Frequency-Switched Multiple-Pulse Sequences, Solid State Nuc. Magn. Reson 2, 151–163. [DOI] [PubMed] [Google Scholar]
- [37].Hong M, Gross JD, Rienstra CM, Griffin RG, Kumashiro KK, and Schmidt-Rohr K (1997) Coupling Amplification in 2D MAS NMR and Its Application to Torsion Angle Determination in Peptides, J. Magn. Reson 129, 85–92. [DOI] [PubMed] [Google Scholar]
- [38].Helmus JJ, and Jaroniec CP (2013) Nmrglue: an open source Python package for the analysis of multidimensional NMR data, J. Biomol. NMR 55, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jo SW, Cheng X, Islam SM, Huang L, Rui H, Zhu A, Lee HS, Qi YF, Han W, Vanommeslaeghe K, MacKerell AD, Roux B, and Im W (2014) CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues, Biomolecular Modelling and Simulations 96, 235–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Park SJ, Lee J, Patel DS, Ma HJ, Lee HS, Jo S, and Im W (2017) Glycan Reader is improved to recognize most sugar types and chemical modifications in the Protein Data Bank, Bioinformatics 33, 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, and Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maciejewski MW, Schuyler AD, Gryk MR, Moraru II, Romero PR, Ulrich EL, Eghbalnia HR, Livny M, Delaglio F, and Hoch JC (2017) NMRbox: A Resource for Biomolecular NMR Computation, Biophys. J 112, 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liepinsh E, and Otting G (1996) Proton exchange rates from amino acid side chains--implications for image contrast, Magn. Reson. Med 35, 30–42. [DOI] [PubMed] [Google Scholar]
- [44].Takeda M, Jee J, Ono AM, Terauchi T, and Kainosho M (2011) Hydrogen exchange study on the hydroxyl groups of serine and threonine residues in proteins and structure refinement using NOE restraints with polar side-chain groups, J. Am. Chem. Soc 133, 17420–17427. [DOI] [PubMed] [Google Scholar]
- [45].Schmidt-Rohr K, and Spiess HW (1994) Multidimensional Solid-State NMR and Polymers, 1st ed., Academic Press, San Diego. [Google Scholar]
- [46].Huster D, Yao XL, and Hong M (2002) Membrane Protein Topology Probed by 1H Spin Diffusion from Lipids Using Solid-State NMR Spectroscopy, J. Am. Chem. Soc 124, 874–883. [DOI] [PubMed] [Google Scholar]
- [47].Kumashiro KK, Schmidt-Rohr K, Murphy OJ, Ouellette KL, Cramer WA, and Thompson LK (1998) A novel tool for probing membrane protein structure: solid-state NMR with proton spin diffusion and X-nucleus detection, J. Am. Chem. Soc 120, 5043–5051. [Google Scholar]
- [48].Hong M, and Schmidt-Rohr K (2013) Magic-angle-spinning NMR techniques for measuring long-range distances in biological macromolecules, Acc. Chem. Res 46, 2154–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hong M, Mishanina TV, and Cady SD (2009) Accurate measurement of methyl 13C chemical shifts by solid-state NMR for the determination of protein sidechain conformation: the influenza M2 transmembrane peptide as an example, J. Am. Chem. Soc, 131, 7806–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Murray DT, Kato M, Lin Y, Thurber KR, Hung I, McKnight SL, and Tycko R (2017) Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains, Cell 171, 615–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Barnes CA, Robertson AJ, Louis JM, Anfinrud P, and Bax A (2019) Observation of β-Amyloid Peptide Oligomerization by Pressure-Jump NMR Spectroscopy, J. Am. Chem. Soc 141, 13762–13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fichou Y, Schiro G, Gallat FX, Laguri C, Moulin M, Combete J, Zamponi M, Hartlein M, Picart C, Mossou E, Lortat-Jacob H, Colletier JP, Tobias DJ, and Weik M (2015) Hydration water mobility is enhanced around tau amyloid fibers, Proc. Natl. Acad. Sci. U. S. A 112, 6365–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Acharya A, Carnevale V, Fiorin G, Levine BG, Polishchuk A, Balannick V, Samish I, Lamb RA, Pinto LH, DeGrado WF, and Klein ML (2010) Structural mechanism of proton transport through the influenza A M2 protein, Proc. Natl. Acad. Sci. U. S. A 107, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Thomaston JL, Alfonso-Prieto M, Woldeyes RA, Fraser JS, Klein ML, Fiorin G, and DeGrado WF (2015) High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction, Proc. Natl. Acad. Sci. U. S. A 112, 14260–14265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hong M, Fritzsching KJ, and Williams JK (2012) Hydrogen-bonding partner of the proton-conducting histidine in the influenza M2 proton channel revealed from 1H chemical shifts, J. Am. Chem. Soc 134, 14753–14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.