

Abstract
Metastatic pancreatic adenocarcinoma remains one of the deadliest cancer diagnoses with 5-year survival rates as low as 3%. For decades, gemcitabine remained the mainstay of systemic therapy before the approvals of FOLFIRINOX and gemcitabine with nab-paclitaxel. Despite these advances in the early 2010s, almost all patients progress on systemic chemotherapy and significant effort is needed to identify novel therapeutic targets. A promising array of approaches is currently under investigation, enabled by deeper understanding of the immune system within the tumor microenvironment (TME) and of the key vulnerabilities in pathways essential for tumor survival. In this review, we will explore the different approaches to boost tumor immunity and to target tumor metabolic pathways that are currently under clinical investigation for systemic treatment, and highlight the promising therapeutic areas that may give rise to the next generation of therapies for pancreatic cancer.
Keywords: Pancreatic cancer, systemic therapy, immunotherapy, tumor microenvironment, metabolic reprogramming
A historical perspective to first-line systemic treatment
Approximately 50,000 people in the United States carry a diagnosis of pancreatic cancer and it accounts for 8% of all cancer deaths (ACS Cancer Facts and Figures, 2019) (1). The vast majority of diagnoses are exocrine tumors, namely pancreatic ductal adenocarcinomas (PDAC). Metastatic PDAC remains one of the deadliest diagnoses, with 5-year overall survival (OS) rates of 3% (SEER database). Chemotherapy has been the backbone of systemic therapy for metastatic PDAC for several decades, with the current standard of care treatments usually involving doublet (gemcitabine/nab-paclitaxel) or triplet (FOLFIRINOX) regimens.
Gemcitabine was first established as a systemic therapy option in a head-to-head trial with 5-FU in patients with advanced symptomatic pancreatic adenocarcinoma, notably with 72% and 76% of patients having Stage IV disease, respectively (2). Patients in the gemcitabine group had an advantageous OS of 5.65 months (vs. 4.41 months for 5-FU, P=0.0025) and progression-free survival (PFS) of 2.33 months (vs. 0.93 months for 5-FU, P=0.0002). On the basis of this, gemcitabine became the first-line option for advanced disease. Several trials thereafter using gemcitabine combinations with other agents did not show additional benefit compared to gemcitabine alone (3).
In 2011, almost 15 years after this initial study, the phase III ACCORD 11 trial established FOLFIRNOX (5-fluorouracil or 5-FU, leucovorin, irinotecan and oxaliplatin) as the new standard of care for advanced PDAC (4). The basis of this cocktail of treatments was early clinical evidence that irinotecan (5,6) and oxaliplatin combined with 5-FU (7) all had some benefit in PDAC, with synergism in preclinical studies (8–12) and little overlapping toxicities among the drugs. FOLFIRINOX was compared head-to-head to gemcitabine and found to be superior with a median OS of 11.1 months (vs. 6.8 months for gemcitabine, P<0.001) and median PFS of 6.4 months (vs. 3.3 months for gemcitabine, P<0.001) (4). Importantly, the objective response rate (ORR) with FOLFIRINOX was 31.6% (vs. 9.4% for gemcitabine, P<0.001), however, it was associated with significantly worse grade 3 or 4 toxicities (46% vs. 21% for gemcitabine), most notably diarrhea (13% vs. 2%), thrombocytopenia (9.1% vs. 3.6%), sensory neuropathy (9% vs. 0%) and febrile neutropenia (5.4% vs. 1.2%) (4).
The combination of gemcitabine with nab-paclitaxel was established as another first-line option for advanced PDAC in the IMPACT study published two years later, in 2013 (13). This combination was compared head-to-head with gemcitabine alone, and demonstrated an improved median OS of 8.5 months (vs. 6.7 months for gemcitabine, P<0.001) and median PFS of 5.5 months (vs. 3.7 months for gemcitabine, P<0.001) (13). The ORR for the combination was 23% (vs. 7% for gemcitabine, P<0.001), and notably, it was associated with a better toxicity profile than FOLFIRNOX, with 38% (vs. 27% for gemcitabine) of patients having grade 3 or 4 toxicities, most notably neuropathy (17% vs. 1% for gemcitabine), diarrhea (6% vs. 1%) and febrile neutropenia (3% vs. 1%) (13).
Despite these successes of the early 2010s, effective systemic options for advanced PDAC remain a major unmet need. The next frontier of systemic therapies is primed to go beyond more efficacious chemotherapies, with treatments to leverage our basic understanding of pancreatic tumors to alter tumor metabolism and the microenvironment (TME), including the immune system (Figure 1). The remainder of this review will focus on the next wave of treatment options for advanced PDAC.
Figure 1.
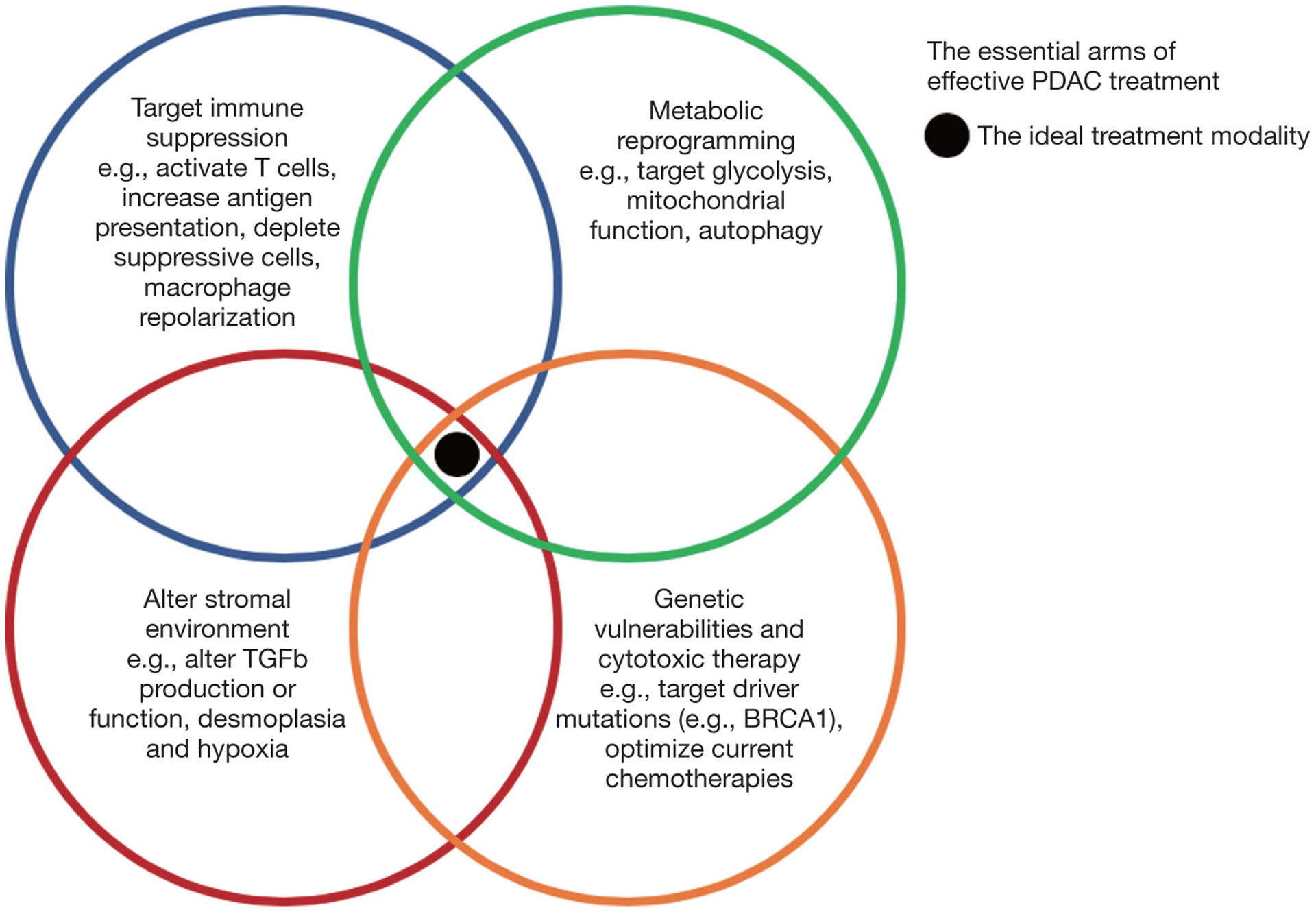
The essential arms of pancreatic cancer treatment. The ideal treatment modality or combination would target immune suppressive mechanisms, alter the stromal environment, target metabolic reprogramming by the tumor, and leverage genetic vulnerabilities in the tumor.
Chemotherapy regimens on the horizon
Current efforts to optimize chemotherapy regimens have included adding platinum-based chemotherapy to gemcitabine and nab-paclitaxel. Early phase Ib/II data of 25 patients has suggested that adding cisplatin to gemcitabine and nab-paclitaxel may be tolerable, with the most common grade 3 or 4 adverse events being thrombocytopenia (68%), anemia (32%), neutropenia (24%) and diarrhea (16%) (14,15). Promising efficacy was seen in this early trial with 8% of patients having a complete response, 62% a partial response, 17% stable disease and 12% progressive disease, with a median PFS of 10.1 months (14,15). A phase III study with this regimen is currently ongoing.
Other early studies are evaluating the role of liposomal irinotecan, 5-FU, leucovorin and oxaliplatin (NAPOX), an approved second-line treatment for metastatic PDAC after progression on gemcitabine (16), in the first-line setting for metastatic PDAC (17). Preliminary data from a phase I/II study suggested an ORR of 34% and disease control at 16 weeks achieved in 71.9% of patients. The data for median OS and PFS are yet to mature (17). Of note, grade 3 or higher treatment related toxicity was seen in 69.6% of patients, with the majority of those being neutropenia, and no complaints of grade 3 or higher fatigue or neuropathy were observed (17).
An evolving paradigm for maintenance and targeted therapy
The conventional approach to systemic treatment for metastatic PDAC is to continue treatment for as long as tolerable without progression, with little impetus to take breaks unless desired by the patient. More recently, this paradigm has been challenged in the PRODEGE35-PANOPTIMOX trial, a phase II study with the overall goal of reducing oxaliplatin exposure. Patients in this study were randomized to receive FOLFIRINOX until progression (arm A), FOLFIRINOX for 4 months followed by maintenance 5-FU until progression (arm B), or sequential treatment with gemcitabine and FOLFIRINOX every 2 months (arm C) (18). The 4-month ORR with continuous FOLFIRINOX (arm A) was 35% compared to 41% with maintenance 5-FU (arm B), with a median PFS of 6.3 vs. 5.7 months and median OS of 10.1 and 11.2 months, respectively (18). This therefore suggests that strategies for maintenance chemotherapy may be equivalent to continuous therapy in terms of overall survival, and warrant further investigation.
The clearest evidence for the role of maintenance therapy in metastatic PDAC was recently established in BRCA positive patients. As many as 4–7% of PDAC patients carry BRCA1 or BRCA2 mutations (19), which causes a deficiency in DNA double-strand break repair, and may confer increased sensitivity to chemotherapy (20–23). While there has yet to be comparative studies of platinum-based versus non-platinum-based first line chemotherapy options in advanced PDAC harboring BRCA mutations, the convention has been to favor FOLFIRINOX for these patients. This is supported by data from the phase III POLO trial, in which metastatic PDAC patients with BRCA mutations who had not progressed during at least 16 weeks of platinum based therapy were randomized to receive either olaparib, a PARP (polyadenosine diphosphate-ribose polymerase) inhibitor, or placebo (24). Among the patients who received olaparib, 22% remained progression free after 2 years (vs. 10% of patients who received placebo).
Cells that are more prone to DNA damage, such as those with BRCA mutations, are particularly sensitive to PARP inhibition, which leads to further double stranded DNA breaks and defective repair, and in turn to programmed tumor cell death (25). The POLO trial established olaparib as the first targeted therapy used in metastatic PDAC, and is now standard of care for maintenance in BRCA mutant PDAC patients who have not progressed on chemotherapy (24). Patients in the trial who received olaparib had an improved median PFS of 7.4 months (vs. 3.8 months for placebo, P=0.004), though the OS data has yet to mature (18.9 vs. 18.1 months at the time of analysis) (24).
A number of other genetic alterations have been identified in PDAC that may shed insight into targeted therapy approaches. The vast majority of PDAC patients harbor mutations in KRAS and TP53, and mutations in CDKN2A, SMAD4, ARID1A and TGFBR2 are also prevalent (26–28). Of note, however, as many as 44% of patients may harbor mutational signatures reflective of DNA damage repair (DDR) defects, with 20% of all patients harboring mutations in canonical DDR pathway genes including BRCA1, BRCA2, PALB2, ATM and CHEK2. These mutations might confer susceptibility to chemotherapy and be targetable by PARP inhibition (29). A small fraction of patients also harbor BRAF or other targetable mutations that potentiate MAPK signaling, and case reports have suggested vulnerability of these tumors to MEK inhibitors (29).
Immune modulating therapies
Checkpoint inhibitors
A hallmark of PDAC is its complex tumor microenvironment (TME) composed of cancer associated fibroblasts (CAFs), immune cells, endothelial cells, in addition to tumor cells (Figure 2) (30–33). In particular, CAFs, which comprise the bulk of most PDAC tumors (34,35), are important in the formation of a dense collagenous stroma that leads to a hypoxic microenvironment for tumor cells (36). Key mediators of CAF activation include sonic hedgehog (SHH), TGFb and cytokines including TNFa, IL1, IL6 and IL10, and once activated these cells play a critical role in producing a collagenous matrix (37–41). This matrix is composed of collagen, fibronectin and laminin along with non-collagenous proteins such hyaluronan (HA), among others, and is a key driver of the chemotherapeutic resistance of these tumors (42–45). Strategies targeting HA in particular have proven effective in pre-clinical models and will be discussed in further detail later in this review.
Figure 2.
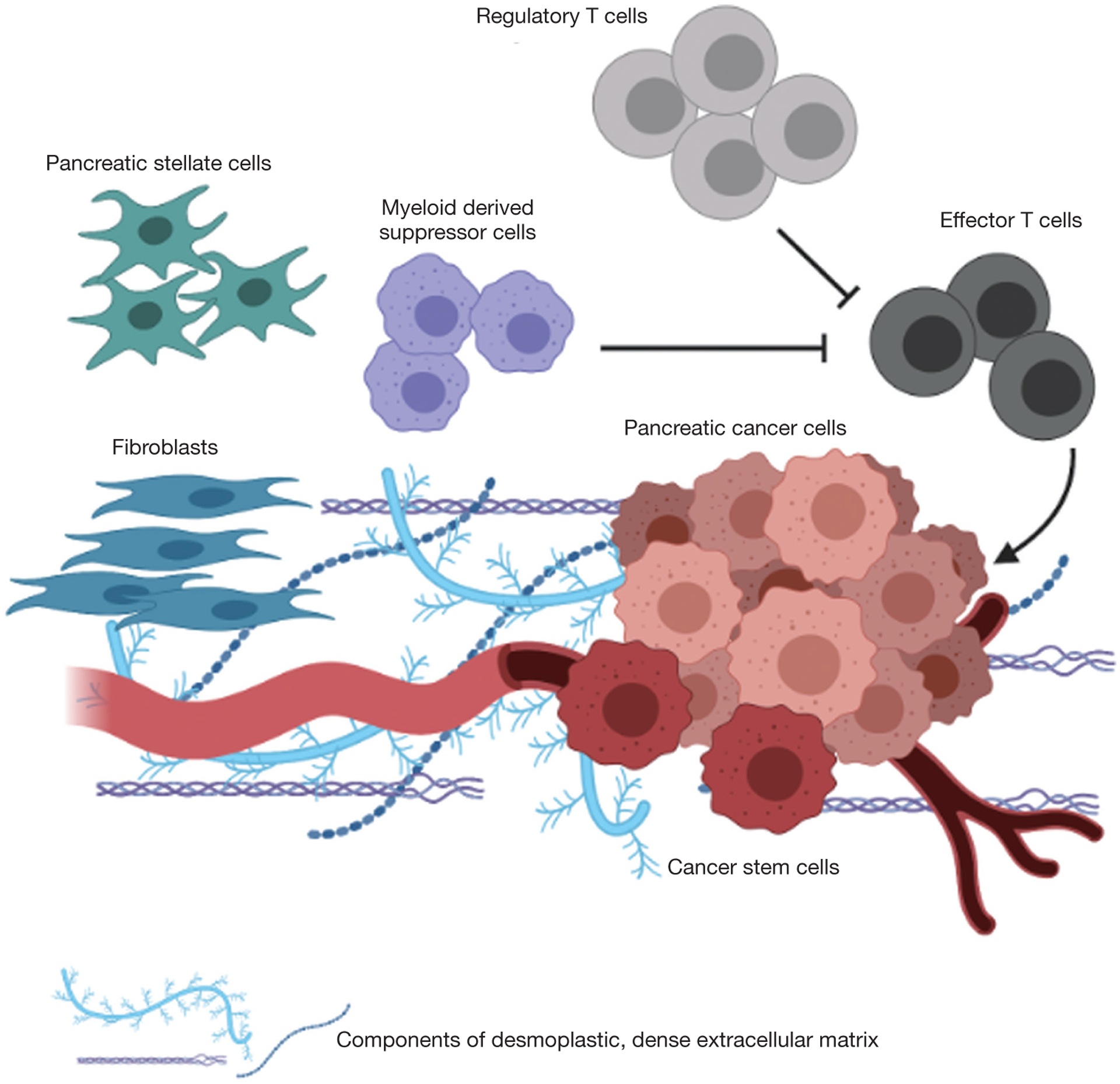
The pancreatic adenocarcinoma tumor microenvironment is composed of a dense, desmoplastic extracellular matrix that includes tumor cells, cancer associated fibroblasts, and immune cells, several of which have immunosuppressive functions including regulator T cells and myeloid derived suppressor cells.
CAFs also foster an immunosuppressive tumor environment (46,47) and promote metastasis (48,49) in part through their secretion of TGFb among other immune mediators (50,51). The majority of immune cells in the TME have immunosuppressive phenotypes, and include myeloid derived suppressor cells (MDSCs) and regulatory T cells (Tregs) (32,52). A role of GM-CSF secretion by tumor cells has also been implicated in promoting MDSC infiltration and antigen-specific T cell suppression (53). The phenotypic properties of these MDSCs are still being elucidated, and several studies have suggested they have a polymorphonuclear MDSC (PMN-MDSC) phenotype; that is, they are neutrophils that have migrated to the tumor with an immunosuppressive phenotype (54,55).
Programmed death ligand-1 (PD-L1), an inhibitory immune checkpoint, is expressed on myeloid cells within the TME and in tumor cells. Higher expression of PD-L1 in PDAC portends a poorer prognosis (56) and immune checkpoint blockade (ICB) has proven largely ineffective for metastatic PDAC, despite the successes in other tumor types (57,58). This is likely due in part to the suppressive immune environment of these tumors. The first trial using single-agent ICB was with ipilimumab (an anti-CTLA4 antibody) in a phase II study for locally advanced or metastatic PDAC (59). Among the 27 patients in the trial, there were no responders, though one patient was noted to have a delayed response after initial progression (59). Subsequent single-agent trials have followed suit, with only 2 out of 34 patients showing a partial response in a phase I trial of tremelimumab (anti-CTLA4 antibody) with gemcitabine (60), and none of the 14 patients showed a response in a phase I trial with BMS-936559 (anti-PD-L1 antibody) (61). A more recent effort seeks to evaluate the conventional chemotherapy regimen of gemcitabine and nab-paclitaxel with nivolumab (62).
To overcome immune exclusion of effector T cells in PDAC tumors, combination strategies of immune modulating agents may prove useful. Preclinical studies have suggested that CAFs are producers of CXCL12, and inhibition of CXCR4, the receptor of CXCL12, promotes effector T cell infiltration and synergy with ICB (63). This has been validated using human tissue using a live tumor slice culture system in which treatment consisting of CXCR4 and PD1 blockade leads to tumor cell killing by clonally expanded effector T cells (64). This combination strategy is now being tested in a phase II clinical trial with early reports suggesting a 34.5% disease control rate (65).
An exception to the rule of non-response to ICB in PDAC may be in the rare patients harboring tumors with mutations in mismatch repair (MMR) genes and high levels of microsatellite instability (MSI-H), in which pembrolizumab (an anti-PD1 antibody) has shown some efficacy in small trials of PDAC patients (66,67). In one report, among 8 MMR deficient PDAC patients, a 62% ORR and 75% disease control rate with pembrolizumab has been reported (66).
GVAX with ICB
The next wave of ICB approaches in PDAC supported by preclinical models has involved combination approaches with ICB to either boost the immune response of antigen presenting cells (APCs) or deplete suppressive cells within the TME (68). Several anti-PD1 antibodies have now been tested in clinical trials in combination with a regimen to optimize the microenvironment in PDAC, using cytoxan to deplete Tregs and GVAX to potentiate T cell activation (69). GVAX is either an autologous or allogeneic pancreatic cancer vaccine modified to express GM-CSF and irradiated to prevent further cell division. The motivation for using this is that GM-CSF, which is expressed by several immune cell types, is that it promotes influx of dendritic cells (DCs) and other APCs into the TME to prime T cell responses while also inducing production of granulocytes and monocytes by hematopoietic stem cells and polarizing macrophages in the TME to more inflammatory phenotypes (70–72). The best data to suggest the efficacy of GVAX in PDAC patients was in a phase Ib trial showing increased median OS of 5.7 months when given with ipilimumab compared to 3.6 months for ipilimumab alone (73).
Current ongoing efforts include combining GVAX and cytoxan with nivolumab and urelumab (an anti-CD137 antibody) (NCT02451982), with pembrolizumab and stereotactic body radiotherapy (SBRT) (NCT02648282), with ipilimumab, nivolumab and CRS-207 (a listeria-based vaccine) (NCT03006302) (74), and with pembrolizumab, CRS-207 and epacadostat (an IDO1 inhibitor) (NCT03006302). The complexity of these regimens, targeting multiple immune axes, represent the fundamental difficulty in activating anti-tumor immune responses in PDAC.
Targeting myeloid cells
Several strategies have also been employed to alter the phenotype of tumor myeloid cells or deplete suppressive myeloid cells. While an oversimplification, these cells are largely categorized as M1 (pro-inflammatory) macrophages and M2 (anti-inflammatory) macrophages, with some plasticity between these two extreme phenotypic states (75,76). Most macrophages in the tumor resemble an M2-like phenotype, which can support tumor progression and metastasis (77,78). A promising approach to altering polarization of M2-like cells to an M1 phenotype involves inhibition of CSF-1R, a myeloid growth factor receptor expressed on the surface of tumor-associated macrophages (TAMs) and MDSCs (76). Preclinical studies have demonstrated blockade of CSF-1R improves responses to ICB (68). Two current anti-CSF-1R antibodies are currently being tested in phase I clinical trials. The first, AMG820 has shown safety in patients with advanced solid tumors (79), and the second, IMC-CS4, is being tested with co-treatment of GVAX, cytoxan and pembrolizumab in borderline resectable PDAC (NCT03153410). A third approach, a small molecule inhibitor of CSF-1R, PLX3397, has also been evaluated in a phase I/II trial in combination with pembrolizumab for advanced solid tumors, including PDAC (80), and in a phase I study with durvalumab, however, further trials have not been initiated.
CCR2 and CXCR2, key myeloid chemokines, play important roles in recruitment of PMN-MDSCs the TME in PDAC, as evidenced, for example by the elevated expression of CXCR2 on these cells within the tumor (81,82). Preclinical studies have suggested that inhibition of the CCL2/CXCR2 axis can potentiate immune responses within pancreatic tumors (82,83). This strategy has been the basis for clinical testing of a small molecular CCR2 inhibitor, CCX872-B, in a phase Ib study in combination with FOLFIRINOX for non-metastatic PDAC, which showed an improved objective tumor response of 49% compared to FOLFIRINOX alone (0%), and a subsequent study in non-resectable and metastatic patients has demonstrated an OS of 29% at 18 months (84).
Anti-CD40 blockade
CD40 is a member of the TNF receptor family expressed on DCs, which when activated by CD40L (CD154), upregulates MHC class I, costimulatory molecules such as CD86, and other TNF superfamily members such as OX40, GITR and CD137. CD40-activated DCs are subsequently able to prime tumor-specific T cells independent of the STING or TLR pathways (85–90). CD40 agonist antibodies have thus been developed and have shown remarkable efficacy in preclinical PDAC models (91,92). A phase Ib study of newly diagnosed metastatic PDAC patients treated with gemcitabine, nab-paclitaxel and APX005M (a CD40 agonist antibody) with or without nivolumab has recently shown promising results (93). Of the 24 patients evaluable for dose-limiting toxicity in this study, 58% had a partial response and 33% had stable disease, though notably 54% of patients experienced an adverse event leading to discontinuation of treatment (93).
Radiation with ICB
Radiation therapy has the ability to potentiate tumor responses by activating the innate immune system. In particular, ionizing radiation causes the intracellular accumulation of reactive oxygen species (ROS) that then leads to DNA damage by the formation of toxic adducts, and single- and double-stranded DNA breaks (94). This DNA damage subsequently leads to the inhibition of the cell cycle in tumors, and triggers cell death pathways including apoptosis and autophagy (95). The DNA breaks from radiation are a potential vulnerability within tumors that may be leveraged with the treatment of PARP inhibitors, as described above for the treatment of tumors with mutations in DDR genes, and strategies to combine these two modalities are a direction of the future.
Importantly, cell death induced by radiation leads to the release of intracellular contents that activate innate immune pathways. Activation of the immune system within the TME increases DC infiltration and activation, including upregulation of MHC-I and antigen presentation, which is in part mediated by activation of the cGAS-STING pathway (96–100). This in turn leads to migration of DCs to tumor draining LNs to activate T cell responses (101). This innate immune activating properties of radiation therapy make it ripe for combination with ICB to bolster T cell immune responses.
The safety of combined radiotherapy and ICB has been established in several tumor types (102–106). Of note, evidence of abscopal effects have been prevalent in several case reports (106) and were noted to correlate with IFNγ-induced gene expression in a prospective phase I study performed in 73 patients with heavily pre-treated metastatic solid tumors who received extracranial SBRT followed by pembrolizumab (107). In this study, 26.9% of patients had a greater than 30% reduction of a non-irradiated lesion (107). In metastatic PDAC, the safety of radiation with ICB has been shown in a pilot study of durvalumab and tremelimumab given with SBRT, which showed this was tolerated well with no dose-limiting toxicities (108,109). The best evidence for efficacy of this combined modality approach in PDAC is from a phase II study of 25 metastatic patients who had progressed on one line of treatment that were treated with nivolumab and ipilimumab with radiation therapy (3 fractions of 8 Gy) given at cycle 2 (110). Among the 22 evaluable patients during an interim analysis, the ORR was 14% with a median PFS of 2.5 months (110).
Other approaches to microenvironment targeting
Pancreatic stellate cells, when activated, induce desmoplasia in PDAC, thus contributing to the dense, fibrous, hypovascular and consequently hypoxic microenvironment of these tumors (111,112). Together, this prevents adequate penetration of chemotherapy and contributes to an immunosuppressive environment and a failure of DCs to present antigens to tumor-specific T cells (113). A key driver of the desmoplastic reaction is the production of HA by fibroblasts (45,114). These findings have led to the development of PEGPH20, a PEGylated form of the recombinant human hyaluronidase PH20, which degrades HA in the stroma (115).
The combination of PEGPH20 with gemcitabine and nab-paclitaxel has been investigated in a phase II trial of patients with metastatic PDAC (116). In patients treated with this three-drug combination, the ORR was 40% compared to 33% for the gemcitabine and nab-paclitaxel arm of the study. However, HA levels were found to be predictive of response, and for patients with HA-high tumors (>50% of tumor surface), the ORR was 45% with the three drug combination compared to 31% with gemcitabine and nab-paclixel (116). In this latter subset, there was a trend towards improved median OS of 11.5 months compared to 8.5 months, respectively (116). This in turn has led to two phase III trials testing this combination on metastatic PDAC (NCT02175804). When PEGPH20 was combined with FOLFIRINOX, however, the combination was found to be detrimental and the study had to be stopped early due to toxicity in the investigational arm (117). A phase III trial will elucidate these conflicting results in the coming years (118). A phase II trial is also currently underway using combination treatment with PEGPH20 and pembrolizumab as second-line treatment for metastatic PDAC (NCT03634332).
Given the role of TGFb in promoting tumor growth, immunosuppression, and fibrosis of the TME, a natural therapeutic strategy may be inhibition of the TGFb signaling cascade (50,119). Thus far, attempts at systemic treatment with anti-TFGb agents have lead to concerning toxicities, however, recent therapies leveraging targeted delivery strategies provide some hope for the future. A phase I study of M7824, a bifunctional fusion protein composed of a monoclonal antibody against PD-L1 and a TGFb trap, in advanced solid tumors, including 5 PDAC patients, has shown promising safety (120). Among the PDAC patients treated, one showed a partial response and another had prolonged stable disease (120).
Angiotensin receptor blockade has been shown to decrease TGFb levels in several tumors. In particular, losartan has been shown to decrease stromal collagen and HA in desmoplastic tumor models by decreasing not just TGFb signaling, but also connective tissue growth factor, HA synthase 1 and 3, and endothelin-1 (121–123). This in turn mitigates the hypoxic TME, improving T cell infiltration and also the distribution and efficacy of therapies (123,124). In addition, a phase II study of neoadjuvant FOLIFIRINOX with losartan, which is known to decrease TGFb in the tumor (123,125), for locally advanced PDAC lead to an R0 resection rate of 61% (126), further supporting the notion that TGFb inhibition may provide therapeutic benefit. However, this combination has yet to be tested in the metastatic setting.
Altering metabolism and autophagy
As described above, the PDAC microenvironment is uniquely harsh and desmoplastic, with low vascularity creating hypoxic conditions and leading to nutrient deprivation (111,127–129). Survival in this environment requires cells to adapt their metabolic profile to survive, but opens the opportunity to exploit metabolic dependencies. KRAS mutations are found in almost all PDAC tumors, and lead to several metabolic derangements including increased glycolysis, which in turn leads to the accumulation of intermediates essential for rapid cell division such as metabolites for nucleotide synthesis (130–133).
Well-perfused, normal cells usually generate most of their energy, in the way of adenosine triphosphate (ATP), by oxidative phosphorylation in mitochondria, which involves the tricarboxylic acid cycle (TCA) and oxygen as the final acceptor of free radicals in the electron transport chain (128). Under oxygen-poor conditions, however, cells often resort to glycolysis, which in PDAC is further potentiated by KRAS mutations, in a process known as the Warburg effect, which is less efficient for ATP production (128). This leads to the accumulation of several metabolites that may further promote tumor growth and immunosuppression in the TME. For example, the accumulation of lactate lowers the pH in the TME and in turn impairs anti-tumor immune responses, enables an increased mutational burden and facilitates hydrolysis of extracellular proteins to promote metastasis (129).
A number of studies have suggested the increased dependence on glycolysis in PDAC, in part due to upregulation of glycolytic enzymes such as hexokinase 2 and lactate dehydrogenases (134–137), and the accumulation of glycolytic metabolites such as lactate (134,135). However, this only tells part of the story, and the balance of oxidative phosphorylation and glycolysis in PDAC is more nuanced. More recent studies have suggested that PDAC cells are especially dependent on oxidative phosphorylation even under low nutrient conditions and this may represent a unique vulnerability (138–141).
These basic findings have led to the exploration of therapeutic approaches to alter mitochondrial biology in PDAC. A meta-analysis of 11 studies, 2 of which were negative randomized-control trials, of PDAC patients on metformin have suggested that the addition of metformin may prolong OS, but only in localized and not metastatic disease (142–144). The results from a number of trials that are currently active or recently closed are pending, including combinations of metformin with gemcitabine and nab-paclitaxel (NCT02336087), with FOLIFIRNOX (NCT01666730), with rapamycin (NCT02048384) and with radiosurgery (NCT02153450).
A more promising recent approach has been the use of CPI-613, a lipoic acid analog that disrupts the activity of pyruvate dehydrogenase and alpha-ketoglutarate, two key mitochondrial enzymes (145,146). A phase I study of CPI-613 given with FOLFIRINOX in advanced PDAC has demonstrated a good safety profile with an ORR of 61% and complete response rate of 17% (147). A randomized phase III trial of CPI-613 with FOLFIRINOX compared to FOLFIRINOX is currently underway (NCT 03504423) (148) and phase I studies for the combination of CPI-613 with gemcitabine and nab-paclitaxel are also in process (NCT03435289) (149).
Another vulnerable pathway in PDAC is autophagy, which is a cytoplasmic cell recycling process that when inhibited disrupts oxidative phosphorylation and causes oxidative damage in these cells (150,151). Hydroxychloroquine (HCQ) is an inhibitor of autophagy that has been studied in a randomized phase II trial in combination with gemcitabine and nab-paclitaxel; however, it showed no benefit compared to gemcitabine and nab-paclitaxel alone in terms of 12-month OS and median PFS (152). More exploratory work on the vulnerabilities in the autophagy pathway in PDAC will need to be done before this could become a viable treatment strategy.
Targeting cancer stem cells
The WNT signaling pathway is critical in embryogenesis for morphogenesis and differentiation of many organs (153), and also plays a key role in oncogenesis in PDAC (154–156). However, unlike other cancers, where mutations in the WNT signaling pathway may be an initiating event, loss of function in PDAC is rare and not sufficient to drive development of the tumor (155). WNT signaling leads to proteasomal degradation of beta-catenin, causing cytoplasmic accumulation and nuclear localization (157). Preclinical studies have suggested that loss of beta-catenin can inhibit PDAC growth, thus creating the impetus to inhibit the WNT pathway in clinical studies (156). A phase Ib study of ipafricept (a WNT inhibitor) given with gemcitabine and nab-paclitaxel has demonstrated a tolerable safety profile, with PR in 34.6% of patients of patients, median PFS of 5.9 months and median OS of 9.7 months; however the program has been shut down by the sponsor (158).
Conclusions
Despite there being few FDA approved therapies that are effective in improving survival in PDAC patients, there are several lines of active investigation that may provide promising breakthroughs in the next decade. It is clear that the optimal approach to PDAC treatment will require a rationale combination of conventional chemotherapeutic approaches with multifaceted targeting of the vulnerabilities in the uniquely desmoplastic stroma, immunosuppressive TME, and metabolic physiology of PDAC tumors.
Acknowledgments
Dr. Hwang is supported by a Conquer Cancer Foundation/ASCO Young Investigator Award. Figures were created with BioRender.com.
Funding: None.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editor (Colin Weekes) for the focused issue “Systemic and Targeted Therapies for Pancreas Ductal Adenocarcinoma” published in Annals of Pancreatic Cancer. The article was sent for external peer review organized by the Guest Editor and the editorial office.
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/apc-2020-pda-05). The focused issue “Systemic and Targeted Therapies for Pancreas Ductal Adenocarcinoma” was commissioned by the editorial office without any funding or sponsorship. CW served as the unpaid Guest Editor of the focused issue. CW reports grants and personal fees from IPSEN, outside the submitted work. The other authors have no conflicts of interest to declare.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13. [DOI] [PubMed] [Google Scholar]
- 3.Di Marco M Metastatic pancreatic cancer: Is gemcitabine still the best standard treatment? (Review). Oncol Rep 2010;23:1183–92. [DOI] [PubMed] [Google Scholar]
- 4.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 2011;364:1817–25. [DOI] [PubMed] [Google Scholar]
- 5.Ueno H, Okusaka T, Funakoshi A, et al. A phase II study of weekly irinotecan as first-line therapy for patients with metastatic pancreatic cancer. Cancer Chemother Pharmacol 2007;59:447–54. [DOI] [PubMed] [Google Scholar]
- 6.Wagener DJT, Th Wagener DJ, Verdonk HER, et al. Phase II trial of CPT-11 in patients with advanced pancreatic cancer, an EORTC early clinical trials group study. Ann Oncol 1995;6:129–32. [DOI] [PubMed] [Google Scholar]
- 7.Ducreux M, Mitry E, Ould-Kaci M, et al. Randomized phase II study evaluating oxaliplatin alone, oxaliplatin combined with infusional 5-FU, and infusional 5-FU alone in advanced pancreatic carcinoma patients. Ann Oncol 2004;15:467–73. [DOI] [PubMed] [Google Scholar]
- 8.Azrak RG. Therapeutic Synergy Between Irinotecan and 5-Fluorouracil against Human Tumor Xenografts. Clin Cancer Res 2004;10:1121–9. [DOI] [PubMed] [Google Scholar]
- 9.Mans DRA, Grivicich I, Peters GJ, et al. Sequence-dependent growth inhibition and DNA damage formation by the irinotecan–5-fluorouracil combination in human colon carcinoma cell lines. Eur J Cancer 1999;35:1851–61. [DOI] [PubMed] [Google Scholar]
- 10.Mullany S, Svingen PA, Kaufmann SH, et al. Effect of adding the topoisomerase I poison 7-ethyl-10-hydroxycamptothecin (SN-38) to 5-fluorouracil and folinic acid in HCT-8 cells: elevated dTTP pools and enhanced cytotoxicity. Cancer Chemother Pharmacol 1998;42:391–9. [DOI] [PubMed] [Google Scholar]
- 11.Pavillard V, Formento P, Rostagno P, et al. Combination of irinotecan (CPT11) and 5-fluorouracil with an analysis of cellular determinants of drug activity. Biochemical Pharmacology 1998;56:1315–22. [DOI] [PubMed] [Google Scholar]
- 12.Zeghari-Squalli N, Raymond E, Cvitkovic E, et al. Cellular pharmacology of the combination of the DNA topoisomerase I inhibitor SN-38 and the diaminocyclohexane platinum derivative oxaliplatin. Clin Cancer Res 1999;5:1189–96. [PubMed] [Google Scholar]
- 13.Von Hoff DD, Ervin T, Arena FP, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 2013;369:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jameson GS, Borazanci EH, Babiker HM, et al. A phase Ib/II pilot trial with nab-paclitaxel plus gemcitabine plus cisplatin in patients (pts) with stage IV pancreatic cancer. J Clin Oncol 2017;35:341. [Google Scholar]
- 15.Jameson GS, Borazanci E, Babiker HM, et al. Response Rate Following Albumin-Bound Paclitaxel Plus Gemcitabine Plus Cisplatin Treatment Among Patients With Advanced Pancreatic Cancer. JAMA Oncol 2019;6:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang-Gillam A, Li CP, Bodoky G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet 2016;387:545–57. [DOI] [PubMed] [Google Scholar]
- 17.Wainberg Z, Boland P, Lieu C, et al. SO-005A phase 1/2, open-label, dose-expansion study of liposomal irinotecan (nal-IRI) plus 5-fluorouracil/leucovorin (5-FU/LV) and oxaliplatin (OX) in patients with previously untreated metastatic pancreatic cancer. Ann Oncol 2019. doi: 10.1093/annonc/mdz157.004. [DOI] [Google Scholar]
- 18.Dahan L, Phelip JM, Le Malicot K, et al. FOLFIRINOX until progression, FOLFIRINOX with maintenance treatment, or sequential treatment with gemcitabine and FOLFIRI.3 for first-line treatment of metastatic pancreatic cancer: A randomized phase II trial (PRODIGE 35-PANOPTIMOX). J Clin Oncol 2018;36:4000. [Google Scholar]
- 19.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fogelman D, Sugar EA, Oliver G, et al. Family history as a marker of platinum sensitivity in pancreatic adenocarcinoma. Cancer Chemother Pharmacol 2015;76:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011;16:1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo G, Lu Y, Jin K, et al. Pancreatic cancer: BRCA mutation and personalized treatment. Expert Rev Anticancer Ther 2015;15:1223–31. [DOI] [PubMed] [Google Scholar]
- 24.Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med 2019;381:317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connor MJ. Targeting the DNA Damage Response in Cancer. Molecular Cell 2015;60:547–60. [DOI] [PubMed] [Google Scholar]
- 26.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 27.Notta F, Chan-Seng-Yue M, Lemire M, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016;538:378–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu, Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguirre AJ, Nowak JA, Camarda ND, et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov 2018;8:1096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amedei A, Niccolai E, Prisco D. Pancreatic cancer: Role of the immune system in cancer progression and vaccine-based immunotherapy. Hum Vaccin Immunother 2014;10:3354–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apte MV, Wilson JS, Lugea A, et al. A Starring Role for Stellate Cells in the Pancreatic Cancer Microenvironment. Gastroenterology 2013;144:1210–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res 2007;67:9518–27. [DOI] [PubMed] [Google Scholar]
- 33.Erkan M, Hausmann S, Michalski CW, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol 2012;9:454–67. [DOI] [PubMed] [Google Scholar]
- 34.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009;324:1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 36.Go VLW. Tumors of the Pancreas (Atlas of Tumor Pathology Series 4). Pancreas 2007;35:388. [Google Scholar]
- 37.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008;455:406–10. [DOI] [PubMed] [Google Scholar]
- 38.Bailey JM, Swanson BJ, Hamada T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res 2008;14:5995–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Apte MV, Haber PS, Darby SJ, et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut 1999;44:534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erkan M, Michalski CW, Rieder S, et al. The Activated Stroma Index Is a Novel and Independent Prognostic Marker in Pancreatic Ductal Adenocarcinoma. Clin Gastroenterol Hepatol 2008;6:1155–61. [DOI] [PubMed] [Google Scholar]
- 41.Bachem MG, Schünemann M, Ramadani M, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005;128:907–21. [DOI] [PubMed] [Google Scholar]
- 42.Imamura T, Iguchi H, Manabe T, et al. Quantitative Analysis of Collagen and Collagen Subtypes I, III, and V in Human Pancreatic Cancer, Tumor-Associated Chronic Pancreatitis, and Alcoholic Chronic Pancreatitis. Pancreas 1995;11:357–64. [DOI] [PubMed] [Google Scholar]
- 43.Mollenhauer J, Roether I, Kern HF. Distribution of extracellular matrix proteins in pancreatic ductal adenocarcinoma and its influence on tumor cell proliferation in vitro. Pancreas 1987;2:14–24. [DOI] [PubMed] [Google Scholar]
- 44.Kohi S, Sato N, Cheng XB, et al. A novel epigenetic mechanism regulating hyaluronan production in pancreatic cancer cells. Clin Exp Metastasis 2016;33:225–30. [DOI] [PubMed] [Google Scholar]
- 45.Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013;62:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waghray M, Yalamanchili M, Dziubinski M, et al. GM-CSF Mediates Mesenchymal-Epithelial Cross-talk in Pancreatic Cancer. Cancer Discov 2016;6:886–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ene–Obong A, Clear AJ, Watt J, et al. Activated Pancreatic Stellate Cells Sequester CD8 T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013;145:1121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Z, Vonlaufen A, Phillips PA, et al. Role of Pancreatic Stellate Cells in Pancreatic Cancer Metastasis. Am J Pathol 2010;177:2585–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ligorio M, Sil S, Malagon-Lopez J, et al. Stromal Microenvironment Shapes the Intratumoral Architecture of Pancreatic Cancer. Cell 2019;178:160–75.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Principe DR, DeCant B, Mascarinas E, et al. TGF Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res 2016;76:2525–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004;432:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chellappa S, Hugenschmidt H, Hagness M, et al. Regulatory T cells that co-express RORγt and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. Oncoimmunology 2015;5:e1102828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012;21:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J, Byrne KT, Yan F, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49:178–93.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel S, Fu S, Mastio J, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol 2018;19:1236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamaki S, Yanagimoto H, Tsuta K, et al. PD-L1 expression in pancreatic ductal adenocarcinoma is a poor prognostic factor in patients with high CD8 tumor-infiltrating lymphocytes: highly sensitive detection using phosphor-integrated dot staining. Int J Clin Oncol 2017;22:726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med 2015;373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N Engl J Med 2013;369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Royal RE, Levy C, Turner K, et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J Immunother 2010;33:828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aglietta M, Barone C, Sawyer MB, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol 2014;25:1750–5. [DOI] [PubMed] [Google Scholar]
- 61.Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Firdaus I, Waterhouse DM, Gutierrez M, et al. nab-paclitaxel (nab-P) nivolumab (Nivo) ± gemcitabine (Gem) in patients (pts) with advanced pancreatic cancer (PC). J Clin Oncol 2016;34:TPS475. [Google Scholar]
- 63.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110:20212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seo YD, Jiang X, Sullivan KM, et al. Mobilization of CD8 T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin Cancer Res 2019;25:3934–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hidalgo M, Epelbaum R, Wolpin BM, et al. 1133PDA phase IIa trial to assess the safety and efficacy of BL-8040 and pembrolizumab in patients with metastatic pancreatic adenocarcinoma (PDAC). Ann Oncol 2018. doi: 10.1093/annonc/mdy288.006. [DOI] [Google Scholar]
- 66.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Soares KC, Rucki AA, Wu AA, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother 2015;38:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang AY, Golumbek P, Ahmadzadeh M, et al. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science 1994;264:961–5. [DOI] [PubMed] [Google Scholar]
- 71.Thomas AM, Santarsiero LM, Lutz ER, et al. Mesothelinspecific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med 2004;200:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Overmeire E, Stijlemans B, Heymann F, et al. M-CSF and GM-CSF Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the Tumor Microenvironment. Cancer Res 2016;76:35–42. [DOI] [PubMed] [Google Scholar]
- 73.Le DT, Lutz E, Uram JN, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother 2013;36:382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le DT, Crocenzi TS, Urum JN, et al. Randomized Phase II study of the safety, efficacy and immune response of GVAX pancreas (with cyclophosphamide) and CRS-207 with or without nivolumab in patients with previously treated metastatic pancreatic adenocarcinoma (STELLAR). J ImmunoTherapy Cancer 2015. doi: 10.1186/2051-1426-3-s2-p155. [DOI] [Google Scholar]
- 75.Jinushi M, Chiba S, Yoshiyama H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A 2011;108:12425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cui R, Yue W, Lattime EC, et al. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 2016;7:50735–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu H, Hang JJ, Han T, et al. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol 2016;37:8657–64. [DOI] [PubMed] [Google Scholar]
- 78.Meng F, Li C, Li W, et al. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life 2014;66:835–46. [DOI] [PubMed] [Google Scholar]
- 79.Papadopoulos KP, Gluck L, Martin LP, et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res 2017;23:5703–10. [DOI] [PubMed] [Google Scholar]
- 80.Wainberg ZA, Eisenberg PD, Sachdev JC, et al. Phase 1/2a study of double immune suppression blockade by combining a CSF1R inhibitor (pexidartinib/PLX3397) with an anti PD-1 antibody (pembrolizumab) to treat advanced melanoma and other solid tumors. J Clin Oncol 2016;34:TPS465. [Google Scholar]
- 81.Steele CW, Karim SA, Leach JDG, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016;29:832–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 2013;73:1128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanford DE, Belt BA, Panni RZ, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 2013;19:3404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Linehan D, Noel MS, Hezel AF, et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J Clin Oncol 2018;36:92. [Google Scholar]
- 85.Bennett SR, Carbone FR, Karamalis F, et al. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998;393:478–80. [DOI] [PubMed] [Google Scholar]
- 86.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4 T-helper and a T-killer cell. Nature 1998;393:474–8. [DOI] [PubMed] [Google Scholar]
- 87.Schoenberger SP, Toes RE, van der Voort EI, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998;393:480–3. [DOI] [PubMed] [Google Scholar]
- 88.French RR, Chan HT, Tutt AL, et al. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med 1999;5:548–53. [DOI] [PubMed] [Google Scholar]
- 89.Diehl L, den Boer AT, Schoenberger SP, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med 1999;5:774–9. [DOI] [PubMed] [Google Scholar]
- 90.Ma HS, Poudel B, Torres ER, et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol Res 2019;7:428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331:1612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res 2015;3:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.O’Hara MH, O’Reilly EM, Rosemarie M, et al. Abstract CT004: A Phase Ib study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine (Gem) and nab-paclitaxel (NP) with or without nivolumab (Nivo) in untreated metastatic ductal pancreatic adenocarcinoma (PDAC) patients. Clinical Trials 2019. doi: 10.1158/1538-7445.sabcs18-ct004. [DOI] [Google Scholar]
- 94.Ostheimer C, Evers C, Palm F, et al. Mortality after radiotherapy or surgery in the treatment of early stage non-small-cell lung cancer: a population-based study on recent developments. J Cancer Res Clin Oncol 2019;145:2813–22. [DOI] [PubMed] [Google Scholar]
- 95.Rouschop KMA, Ramaekers CHMA, Schaaf MBE, et al. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother Oncol 2009;92:411–6. [DOI] [PubMed] [Google Scholar]
- 96.Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 2019;20:657–74. [DOI] [PubMed] [Google Scholar]
- 97.Vanpouille-Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol 2015;33:445–74. [DOI] [PubMed] [Google Scholar]
- 99.Twyman-Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gameiro SR, Jammeh ML, Wattenberg MM, et al. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014;5:403–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodríguez-Ruiz ME, Vanpouille-Box C, Melero I, et al. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol 2018;39:644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barker CA, Postow MA, Kronenberg SA, et al. Concurrent Radiation Therapy (RT), Ipilimumab (Ipi) and/or Nivolumab (Nivo) on a Phase 1 Clinical Trial. Inter J Radiat Oncol Biol Physics 2015;93:S210–1. [Google Scholar]
- 103.Antonia SJ. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. Reply. N Engl J Med 2019;380:990. [DOI] [PubMed] [Google Scholar]
- 104.Levy A, Massard C, Soria JC, et al. Concurrent irradiation with the anti-programmed cell death ligand-1 immune checkpoint blocker durvalumab: Single centre subset analysis from a phase 1/2 trial. Eur J Cancer 2016;68:156–62. [DOI] [PubMed] [Google Scholar]
- 105.Liniker E, Menzies AM, Kong BY, et al. Activity and safety of radiotherapy with anti-PD-1 drug therapy in patients with metastatic melanoma. Oncoimmunology 2016;5:e1214788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hwang WL, Pike LRG, Royce TJ, et al. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat Rev Clin Oncol 2018;15:477–94. [DOI] [PubMed] [Google Scholar]
- 107.Luke JJ, Lemons JM, Karrison TG, et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients With Advanced Solid Tumors. J Clin Oncol 2018;36:1611–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gong J, Le TQ, Massarelli E, et al. Radiation therapy and PD-1/PD-L1 blockade: the clinical development of an evolving anticancer combination. J Immunother Cancer 2018;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duffy AG, Makarova-Rusher OV, Kleiner DE, et al. A pilot study of immune checkpoint inhibition in combination with radiation therapy in patients with metastatic pancreatic cancer. J Clin Oncol 2017;35:e15786. [Google Scholar]
- 110.Parikh A, Wo JYL, Ryan DP, et al. A phase II study of ipilimumab and nivolumab with radiation in metastatic pancreatic adenocarcinoma. J Clin Oncol 2019;37:391. [Google Scholar]
- 111.Whatcott CJ, Diep CH, Jiang P, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res 2015;21:3561–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Miyamoto H, Murakami T, Tsuchida K, et al. Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004;28:38–44. [DOI] [PubMed] [Google Scholar]
- 113.Lee CT, Mace T, Repasky EA. Hypoxia-driven immunosuppression: a new reason to use thermal therapy in the treatment of cancer? Int J Hyperthermia 2010;26:232–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kultti A, Zhao C, Singha NC, et al. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. Biomed Res Int 2014;2014:817613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hingorani SR, Zheng L, Bullock AJ, et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J Clin Oncol 2018;36:359–66. [DOI] [PubMed] [Google Scholar]
- 117.Ramanathan RK, McDonough S, Philip PA, et al. A phase IB/II randomized study of mFOLFIRINOX (mFFOX) pegylated recombinant human hyaluronidase (PEGPH20) versus mFFOX alone in patients with good performance status metastatic pancreatic adenocarcinoma (mPC): SWOG S1313 (NCT #01959139). J Clin Oncol 2018;36:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Doherty GJ, Tempero M, Corrie PG. HALO-109–301: a Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol 2018;14:13–22. [DOI] [PubMed] [Google Scholar]
- 119.Principe DR, Park A, Dorman MJ, et al. TGFβ Blockade Augments PD-1 Inhibition to Promote T-Cell–Mediated Regression of Pancreatic Cancer. Mol Cancer Ther 2019;18:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Strauss J, Heery CR, Schlom J, et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin Cancer Res 2018;24:1287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chauhan VP, Martin JD, Liu H, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun 2013;4:2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Diop-Frimpong B, Chauhan VP, Krane S, et al. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci 2011;108:2909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med 2017;9:eaan5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Incio J, Liu H, Suboj P, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov 2016;6:852–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Arnold SA, Rivera LB, Carbon JG, et al. Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFβ activation. PLoS One 2012;7:e31384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Murphy JE, Wo JY, Ryan DP, et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol 2019;5:1020–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Feig C, Gopinathan A, Neesse A, et al. The Pancreas Cancer Microenvironment. Clin Cancer Res 2012;18:4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vander Heiden MG, Vander Heiden MG, Cantley LC, et al. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009;324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Payen VL, Porporato PE, Baselet B, et al. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci 2016;73:1333–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Science Advances 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Waters AM, Der CJ. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med 2018;8:a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Santana-Codina N, Roeth AA, Zhang Y, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 2018;9:4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guillaumond F, Leca J, Olivares O, et al. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 2013;110:3919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mikuriya K, Kuramitsu Y, Ryozawa S, et al. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int J Oncol 2007;30:849–55. [PubMed] [Google Scholar]
- 136.Shi M, Cui J, Du J, et al. A novel KLF4/LDHA signaling pathway regulates aerobic glycolysis in and progression of pancreatic cancer. Clin Cancer Res 2014;20:4370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013;497:633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim Biophys Acta 2011;1807:552–61. [DOI] [PubMed] [Google Scholar]
- 139.Zheng J Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett 2012;4:1151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rossignol R, Gilkerson R, Aggeler R, et al. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res 2004;64:985–93. [DOI] [PubMed] [Google Scholar]
- 141.Birsoy K, Possemato R, Lorbeer FK, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014;508:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li X, Li T, Liu Z, et al. The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis. Sci Rep 2017;7:5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kordes S, Pollak MN, Zwinderman AH, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol 2015;16:839–47. [DOI] [PubMed] [Google Scholar]
- 144.Reni M, Dugnani E, Cereda S, et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin Cancer Res 2016;22:1076–85. [DOI] [PubMed] [Google Scholar]
- 145.Lycan TW, Pardee TS, Petty WJ, et al. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS One 2016;11:e0164244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Egawa Y, Saigo C, Kito Y, et al. Therapeutic potential of CPI-613 for targeting tumorous mitochondrial energy metabolism and inhibiting autophagy in clear cell sarcoma. PLoS One 2018;13:e0198940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alistar A, Morris BB, Desnoyer R, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol 2017;18:770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Philip A, Philip PA, Buyse ME, et al. Avenger 500, a phase III open-label randomized trial of the combination of CPI-613 with modified FOLFIRINOX (mFFX) versus FOLFIRINOX (FFX) in patients with metastatic adenocarcinoma of the pancreas. J Clin Oncol 2019;37:TPS479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Alistar AT, Morris B, Harrison L, et al. GA CPI 613: A single arm, open-label phase I study of CPI-613 in combination with gemcitabine and nab-paclitaxel for patients with locally advanced or metastatic pancreatic cancer. J Clin Oncol 2019;37:TPS459. [Google Scholar]
- 150.Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011;25:717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Guo JY, Chen HY, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011;25:460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karasic TB, O’Hara MH, Loaiza-Bonilla A, et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol 2019;5:993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Clevers H Wnt/β-Catenin Signaling in Development and Disease. Cell 2006;127:469–80. [DOI] [PubMed] [Google Scholar]
- 154.Zhang Y, Morris JP 4th, Yan W, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res 2013;73:4909–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morris JP 4th, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 2010;10:683–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pasca di Magliano M, Biankin AV, Heiser PW et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS One 2007;2:e1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takebe N, Harris PJ, Warren RQ, et al. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 2011;8:97–106. [DOI] [PubMed] [Google Scholar]
- 158.Dotan E, Cardin DB, Lenz HJ, et al. Phase Ib study of WNT inhibitor ipafricept (IPA) with nab-paclitaxel (Nab-P) and gemcitabine (G) in patients (pts) with previously untreated stage IV pancreatic cancer (mPC). J Clin Oncol 2019;37:369. [DOI] [PMC free article] [PubMed] [Google Scholar]