

Abstract
Understanding genetic and epigenetic changes that underlie abnormal proliferation of hematopoietic stem and progenitor cells is critical for development of new approaches to monitor and treat leukemia. The unfolded protein response (UPR) is a conserved adaptive signaling pathway that governs protein folding, secretion and energy production and serves to maintain protein homeostasis in various cellular compartments. Deregulated UPR signaling, which often occurs in hematopoietic stem cells and leukemias, defines the degree of cellular toxicity and perturbs protein homeostasis, at the same time offering a novel therapeutic target. Here we review current knowledge related to altered UPR signaling in leukemias and highlight possible strategies for exploiting the UPR as treatment for this disease.
Keywords: Myeloid leukemia, lymphoid leukemia, hematological malignancy, unfolded protein response (UPR), hematopoietic stem cell (HSC), ER stress
The UPR in Hematopoietic Stem Cell Biology
Hematopoietic stem cells (HSCs) (see Glossary) give rise to numerous progeny over their life span and maintain hematopoiesis under normal or stress conditions [1, 2]. Under steady-state conditions, HSCs remain dormant and rely in part on low protein synthesis rates to sustain self-renewal capacity and protect HSC integrity [3, 4]. However, proliferating HSCs show increased protein synthesis to allow expansion of the stem cell pool [5]. Due to poor protein folding capacity, HSCs tend to accumulate more unfolded/misfolded proteins, which in turn promote endoplasmic reticulum (ER) stress. Re-establishment of ER homeostasis in response to this stress requires activation of the unfolded protein response (UPR). UPR components sense protein misfolding in the ER and initiate a cellular response that both transcriptionally and non-transcriptionally aims to alleviate cellular stress, or triggers apoptosis if stress cannot be resolved (Figure 1) [6, 7]. The UPR governs cellular protein homeostasis (proteostasis) by modulating protein translation, degradation, and transcriptionally regulates genes functioning in protein folding, protein quality control, and ER associated degradation (ERAD) programs [8], which collectively regulate cellular responses to ER stress. Crosstalk among these pathways, including mitogen-activated protein kinases (MAPK) signaling and redox signaling, determines cell’s ability to cope with consequences of impaired ER function, resulting in autophagy, cell death or survival programs [9] (Figure 1). Therefore, the UPR is proposed to sustain a healthy homeostatic balance, in part through clearing individual cells after ER stress.
Figure 1: Molecular Mechanisms of the UPR.
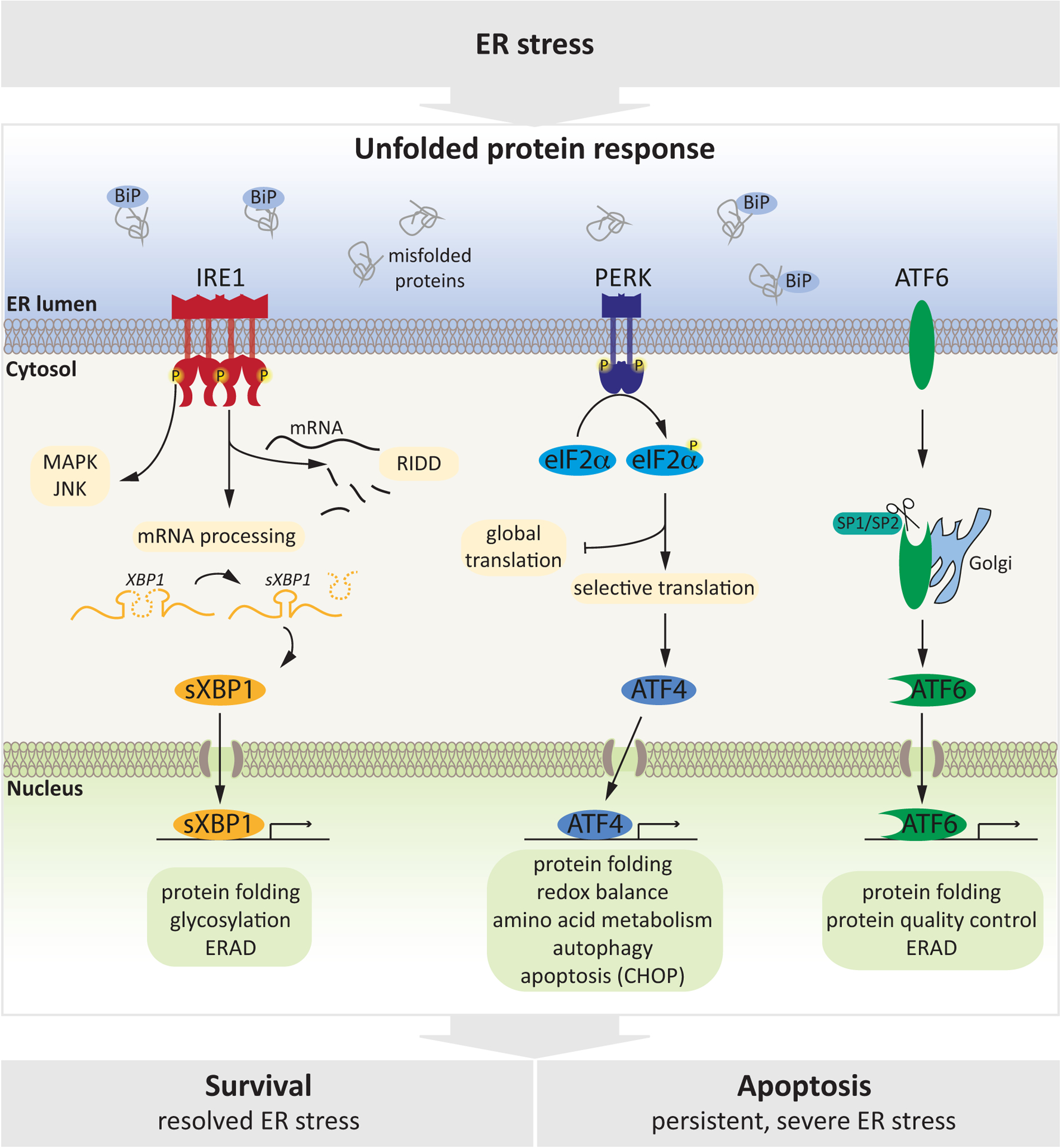
ER stress, triggered by accumulation of un- or mis-folded proteins in the ER lumen is sensed by three transmembrane proteins (IRE1, PERK, ATF6) that serve as ER stress sensors and cooperatively coordinate the UPR. IRE1α and PERK are type I ER transmembrane proteins and are activated by oligo/dimerization and autophosphorylation. IRE1α has a cytoplasmic kinase and endoribonuclease domain and once activated, it catalyzes excision of a 26-nucleotide intron from the mRNA encoding X-box binding protein-1 (XBP-1) which is translated to form the transcription factor spliced XBP1 (sXBP1), and also degrades a subset of ER-associated RNAs through a process called IRE1 dependent decay (RIDD). Through its kinase domain, IRE1 activates further stress-related pathways to modulate response to stress. PERK is composed of an ER luminal stress sensor and a cytosolic protein kinase domain. Activated PERK attenuates global protein synthesis by phosphorylating serine 51 of the initiation factor eukaryotic translation initiator factor 2α (eIF2α), globally decreasing initiation of mRNA translation while enabling selective translation of stress-related mRNAs including activating transcription factor 4 (ATF4). Finally, ATF6 is a transmembrane protein whose cytosolic domain has transcriptional activity. Following ER stress, ATF6 is transported to the Golgi apparatus where it is processed by site 1 protease (S1P) and S2P, releasing a transcriptionally active ATF6 protein which then enters the nucleus. All three arms of the UPR mediate a transcriptional response (by activating sXBP1, ATF4 or ATF6) to increase ER capacity to resolve stress, by upregulating target genes implicated in protein folding, quality control and ERAD. Upon severe or prolonged stress, the UPR orchestrates a pro-apoptotic response that eliminates damaged cells.
The interplay between pro-survival and pro-apoptotic signaling of the UPR (Box 1) is crucial for maintaining organismal homeostasis, under both steady state and stress conditions, especially in HSCs to prevent propagation of damaged HSCs and, ultimately, leukemogenesis [10]. Indeed, under steady-state and mild ER stress conditions (such as hypoxia and increased metabolic demand), the inositol-requiring enzyme 1 alpha (IRE1α) and the activating transcription factor 6 (ATF6) arms of the UPR reportedly promote HSC survival and stemness potential [11–13]. For example, Estradiol (E2), the most active type of estrogen, specifically upregulates the IRE1α-sXBP1 axis in HSCs by directly activating IRE1α transcription. That activity increases HSC capacity to reconstitute the hematopoietic system upon transplantation in lethally irradiated mice and accelerates hematopoietic regeneration [11]. Consistently, IRE1α pathway activation has been shown to promote HSC survival and stemness potential under ER stress conditions (such as accumulation of misfolded protein and oxidative stress), and HSCs with high IRE1α activity exhibit increased reconstitution potential in vivo compared to HSCs with low IRE1α activity [13, 14]. In addition, activating transcription factor 4 (ATF4)-mediated responses in HSCs subjected to low stress conditions, such as amino acid deprivation, exhibit cyto-protection and facilitate HSC persistence [15]. However, under severe ER stress conditions, prolonged activation of the protein kinase R-like endoplasmic reticulum kinase (PERK)-ATF4 axis induces expression of pro-apoptotic factors such as C/EBP homologous protein (CHOP), a transcription factor that controls genes mediating apoptosis [8, 9, 16–22]. To this end, gene expression analysis has revealed enrichment of UPR components in a mixed population of HSC and progenitor cells (HSPCs) as compared to downstream progenitors, including components of the PERK-ATF4-CHOP axis [7]. Relative to effects seen in progenitor populations, elevated PERK signaling in HSPCs confers greater sensitivity to ER stress-induced apoptosis, such as following treatment with the ER-inducing drug tunicamycin (Tm) [7]. Consequently, overexpression of the ER co-chaperone ERDJ4 [7], or treatment with chemical chaperones [23], protects HSPCs from proteotoxic stress and increases their reconstitution capacity in xenograft assays by blocking upregulation of stress-related genes, such as CHOP. These results suggest that the PERK pathway plays an important role in maintaining HSC integrity, by eliminating damaged stem cells.
Box 1: The UPR in Cell Survival and Death Programs.
The mammalian UPR has evolved into a dynamic and flexible network of signaling events that responds to various inputs over a wide range of basal metabolic states. During ER stress conditions, activation of the UPR reduces unfolded protein load through several pro-survival mechanisms. However, when ER stress is not mitigated and homeostasis is not restored, the UPR triggers apoptosis [9]. Two distinct UPR-related responses, the adaptive response and/or apoptotic response, are observed over time in cells undergoing ER stress. In the adaptive phase, PERK inhibits general protein translation through eIF2α phosphorylation [111], and IRE1α activation leads to selective degradation of mRNAs encoding for certain ER-located proteins through regulated IRE1-dependent decay (RIDD) [20]. Autophagy is also activated by ER stress to eliminate accumulation of protein aggregates and possibly damaged ER (a process termed ER-phagy) through the lysosomal pathway [112]. Together, these mechanisms reduce the influx of proteins into the ER to allow adaptive and repair mechanisms that re-establish ER homeostasis. Simultaneously, a massive gene-expression response is initiated through at least three distinct UPR transcription factors: ATF4, sXBP1 and ATF6. These transcription factors promote adaptive responses that aim to restore ER function and maintain cell survival including amino acid transport and synthesis, redox signaling, protein folding and ERAD [113]. Unresolved ER stress results in apoptosis which depends on the core mitochondrial apoptosis pathway regulated by the B cell lymphoma 2 (BCL-2) protein family [114]. When activated at the transcriptional or post-translational level, BCL-2 homology 3 (BH3)-only proteins regulate the activation of BAX and/or BH antagonist or killer (BAK) to trigger apoptosis. Sustained PERK/ATF4 activation induces expression of pro-apoptotic factors such as CHOP, which downregulates the anti-apoptotic protein BCL-2 and induces the expression of some BH3-only genes mediating apoptosis [18, 21]. Under certain conditions, IRE1α can also induce cell death by activating the pro-apoptotic IRE1-TRAF2-JNK pathway, with subsequent downstream engagement of the BCL-2 family members, concomitant with the activation of RIDD, resulting in the degradation of mRNAs encoding key mediators of protein folding [72, 115]. Additional complementary mechanisms are proposed to induce cell death under chronic ER stress, including activation of the BH3-only protein BH3-interacting domain death agonist (BID) by caspase 2, as well as ER calcium release, which may sensitize mitochondria to activate apoptosis [116]. Therefore, it is essential to understand how UPR sensors shift their signaling output to determine divergent cell fate decisions.
Current analysis suggests that distinct arms of the UPR serve dedicated functions in HSCs: sustained PERK activation is associated with programmed cell death, and the IRE1α-sXBP1 arm is mainly linked to survival and regeneration. Additional studies are needed to define how both arms interact to sustain HSC proliferation and self-renewal status. In this review, we summarize how this conserved response to stress is hijacked by leukemic cells to enable their survival during leukemogenesis and leukemia progression as well as to promote resistance to therapy. We summarize current evidence for elevated UPR signaling in chronic and acute leukemias, providing a rationale for targeting the UPR in a heterogeneous group of these malignancies.
UPR Function in Leukemogenesis
Leukemias emerge from accumulation of genetic alterations in HSCs or lineage-restricted progeny [24, 25]. Leukemic cells often hijack stem cell programs to fuel leukemogenesis and progression, and myeloid leukemias are, like normal hematopoiesis, maintained by leukemic stem cells (LSCs) [2, 25] (Box 2). Given the importance of the UPR in regulating normal HSC survival and self-renewal, activation of these processes in LSCs is expected to confer a clonal advantage enabling LSCs to survive the increased metabolic demands associated with increased proliferation (Figure 2). Indeed, the UPR, and in particular the IRE1α-XBP1 axis have been demonstrated to promote survival of HSCs during early transformation steps in experimental systems [13]. In conditional knock-in mice bearing the NRASG12D-mutated allele in HSCs, constitutive NRAS signaling transforms HSCs into pre-leukemic stem cells (pre-LSCs), leading to a broad spectrum of myeloid neoplasms. In this model, IRE1α-sXBP1 signaling is activated, protecting pre-LSCs from ER-stress-induced apoptosis and enhancing both their ability to compete for nutrients with other cells and their self-renewal capacity [13]. Thus, like normal HSCs, pre-LSCs utilize the UPR to adapt to harsh conditions of the bone marrow niche that trigger ER stress and to cope with increased metabolic demands that underlie oncogene activation and proliferation [13]. Interestingly, expression of other driver mutations, such as the internal tandem duplication in FMS-like tyrosine kinase 3 (FLT3-ITD), one of the most frequent mutations in acute myeloid leukemia (AML) and usually associated with poor outcome [26], does not confer a survival advantage following treatment with ER stress inducers [13], suggesting that different driver mutations utilize distinct mechanisms to protect pre-LSCs from stress. One example for alternate stress adaption is the decrease in protein synthesis rates by downregulating ribosome biogenesis observed in RUNX1-mutated pre-LSCs [27]. RUNX1 is a transcription factor mutated in de novo and therapy-related leukemias [28]. Low biosynthetic activity in RUNX1-mutant pre-LSCs is accompanied by reduced UPR and p53 signaling and consequently resistance to genotoxic and ER stress. This stress resistance provides a selective advantage to pre-LSC and allows them to expand in bone marrow and outcompete normal HSPCs [27]. These studies highlight the importance of maintaining ER homeostasis during HSC transformation to leukemia.
Box 2: Classification of Leukemias.
Leukemia is a blood cancer caused by the rapid production of abnormal white blood cells and is the most common cancer in children and adolescents. Clinically and pathologically, leukemia is subdivided into four major subtypes: ALL (acute lymphoblastic leukemia), CLL (chronic lymphoblastic leukemia), AML (acute myeloid leukemia), and CML (chronic myeloid leukemia). [117]. Classification is based in part on the type of blood cell affected and includes both lymphoblastic and myeloid leukemias. In the former, malignant changes occur in the type of marrow cell that becomes lymphocytes, which function in the immune system. With myeloid leukemia, malignancy occurs in cells that usually mature into red blood cells, other types of white cells, and platelets [118, 119]. Classification is also based on whether leukemias take an acute or chronic form based on rate of progression. In acute leukemias, abnormal blood cells are blasts and usually remain poorly differentiated and largely non-functional, leading eventually to defective hematopoiesis. Acute leukemias often rapidly progress and require immediate treatment. In chronic leukemias, some blast cells may remain and may mature and function normally [118, 119]. Thus, progression of chronic leukemias is usually slower and may not require treatment as aggressive as that used against acute leukemias. Overall, etiology, clinical features, treatment, and survival of leukemia cells differ significantly, requiring type-specific analysis.
Figure 2: UPR Signaling in Leukemia Initiation, Progression and Therapy Response.
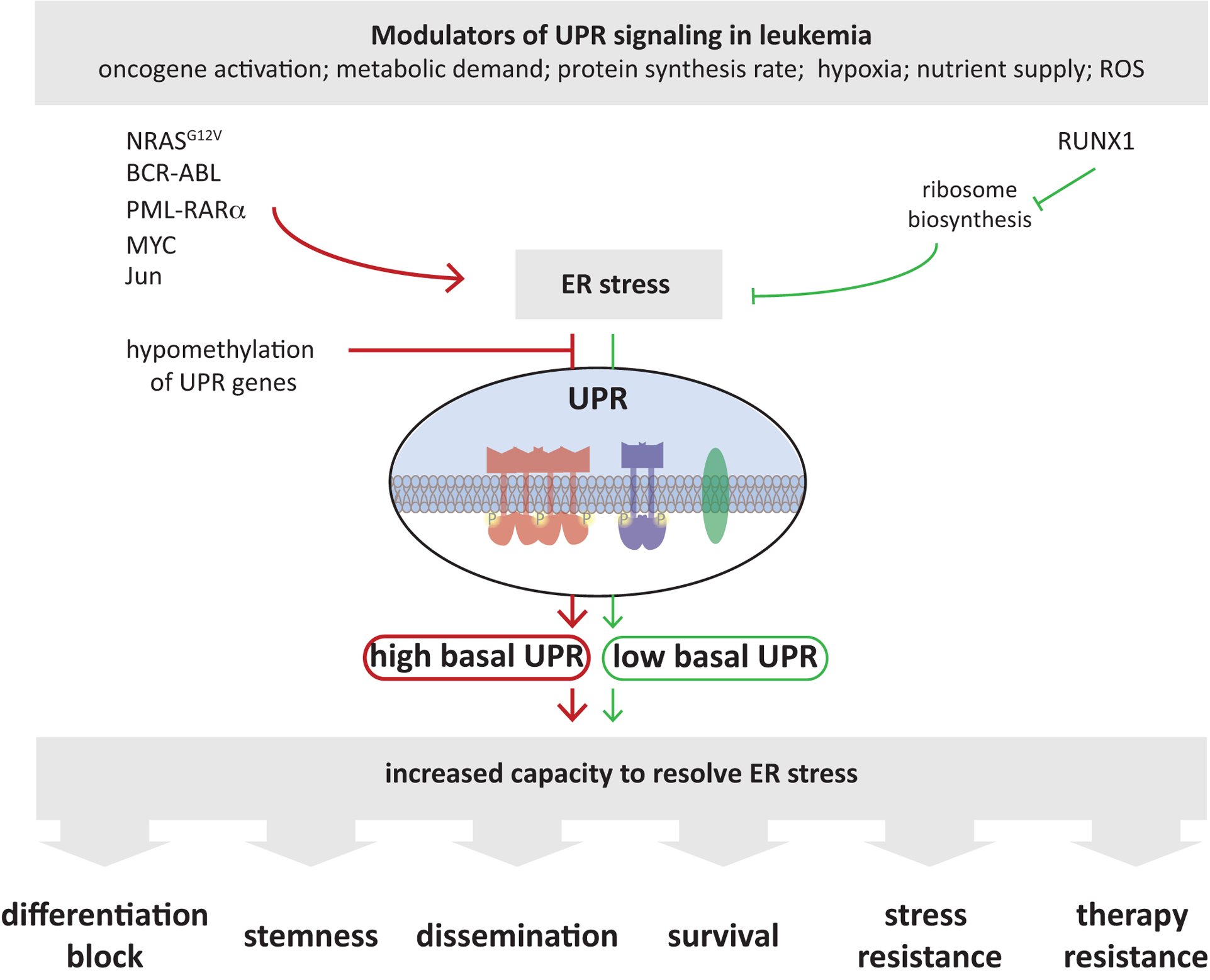
Leukemic and pre-leukemic cells are exposed to harsh environmental conditions (hypoxia, ROS) that increase protein misfolding and trigger ER stress. Genetic alterations commonly observed in leukemia, such as BCR-ABL, PML-RARα or MYC mutations as well as other oncogenic signaling events, such as increased c-Jun levels, contribute to elevated UPR signaling in leukemias. UPR components can also be upregulated, putatively independent of ER stress in leukemia, e.g. by hypomethylation of UPR genes. All these events (red arrows) culminate to promote UPR signaling and leukemia cells are often characterized by high basal UPR signaling to increases the capacity of cells to cope with stress and restore ER homeostasis. The adaptive signaling events orchestrated by the UPR promotes leukemogenesis and progression by altering diverse cellular processes, from affecting cellular differentiation, as occurs in AML, to increased cell survival and therapy resistance in multiple disease types.
Exemplary alternate routes to maintain ER homeostasis are displayed (green arrows), where RUNX1 mutations decrease ribosome biogenesis, resulting in reduced ER protein load, thereby increasing the capacity of the ER to cope with stressful insults, which also endows RUNX1-mutated cells with increased survival and therapy resistance. Targeting the UPR thus could prevent leukemia progression and overcome resistance to standard therapeutic regimens.
The first study addressing UPR function in leukemogenesis was performed in the leukemic cell line NB4, which harbors the t(15;17) (q24.1;q21.2) translocation, a cytogenetic hallmark of acute promyelocytic leukemia (APL). This translocation enables expression of the oncogenic fusion protein promyelocytic leukemia-retinoic acid receptor alpha (PML-RARα). In normal cells, wild-type RARα forms heterodimers with a member of the soluble nuclear receptor co-repressor 1 (N-CoR) family of transcriptional co-repressors [29]. Oncogenic PML-RARα binds to N-CoR protein in the cytosol immediately after translation inducing an abnormal N-CoR conformation that triggers UPR-mediated clearance of abnormal N-CoR by the ERAD system [30]. This leads to decrease levels of soluble N-CoR protein in the nucleus and may underlie uncontrolled proliferation seen in APL. UPR cues may thus impact N-CoR localization and expression levels and define the extent of transcriptional repression on specific promoters affecting myeloid differentiation.
Taken together, aberrant expression of diverse oncogenic drivers in leukemia results in the activation of UPR components, highlighting the importance of UPR signaling in the etiology of this disease.
The Role of the UPR in Leukemia Cell Survival
Normal and leukemic hematopoietic cells are exposed to harsh environmental conditions in the bone marrow such as hypoxia, elevated reactive oxygen species (ROS) levels, and nutrient deprivation, which results in increased cellular metabolism and protein synthesis, culminating in ER stress. Leukemic cells accommodate ER stress by activating the UPR, allowing them to escape cell death and continue proliferation. While somatic mutations in UPR components were not identified so far in leukemia patients, activated UPR signaling clearly contributes to leukemia progression and survival, as discussed below. Correspondingly, activation of the IRE1α-XBP1 branch of the UPR, reflected in increased sXBP1, BiP and calreticulin expression, was found in ~18% of AML patients [31, 32].
The transcription factor c-Jun was recently found to be overexpressed in multiple AML subtypes and required for AML cell survival and progression in vivo [33]. Upon ER stress, c-Jun induces transcription of UPR target genes (such as XBP1 and ATF4) by direct binding to their promotors, allowing leukemic cells to resolve ER stress through a cytoprotective UPR. Correspondingly, c-Jun inhibition blocks UPR activation and the ability of AML cells to cope with ER stress. Of note, in different AML subtypes [inv(16), t(8;21) and 11q23] there is a positive correlation between c-Jun expression and that of numerous UPR genes, including ATF3, ATF4, CHOP, HSPA5 and PPP1R15B, confirming c-Jun’s importance in the UPR and the etiology of this cancer type. c-Jun is also implicated in chronic myeloid leukemia (CML) stem cell survival [34] and has been shown to be a downstream component of signaling by the heparin-binding growth factor, pleiotrophin (PTN), which is necessary for CML pathogenesis and initiation. PTN promotes CML stem cell growth, survival, and resistance to tyrosine kinase inhibitors via induction of c-Jun and the UPR [34] (Figure 2).
Notably, the ER is the cells major calcium store and calcium is a key player in regulating early and late stages cell death [35, 36]. Calcium release from the ER leads to rapid uptake and accumulation in mitochondria, promoting the mitochondrial apoptotic pathway, characterized by mitochondrial swelling, perturbation of the outer membrane, and release of apoptosis-inducing factors into the cytosol [35, 37]. This calcium-dependent ER-initiated apoptotic pathway is inactivated in BCR-ABL-expressing cells, representing a pro-survival mechanism and potential opportunity for therapeutic targeting [38]. BCR-ABL overexpression in immortalized murine 32D cells reportedly decreases calcium release from ER stores and correspondingly reduces capacitative calcium entry (CCE) [38]. Conversely, treatment with imatinib, which blocks BCR-ABL tyrosine kinase activity, reverses CCE and restores calcium release from ER stores. Accordingly, cells expressing high BCR-ABL levels are resistant to classical inhibitors of the mitochondrial apoptotic pathway [38].
Adaptive UPR signaling and tight control of secretory mechanisms is of major importance in B cells, which secrete immunoglobulins. Indeed, promoters of genes encoding several UPR components, including ERN-1 (IRE1), HSPA5 (BiP), PRDM1 (BLIMP-1) and XBP-1, are hypomethylated in pre-B acute lymphoblastic leukemia (ALL) cells relative to either normal pre-B cells or mature B-cell lymphoma [39]. This epigenetic state results in increased expression of these genes in the pre-B ALL, compared with mature B cells or non-Hodgkin lymphomas. Among these genes, Hspa5 is required for survival and malignant transformation of pre-B cells. Hspa5 deletion induces cell cycle arrest in pre-B cells in vitro and decreases leukemia burden in vivo, resulting in prolonged survival of ALL transplant recipient mice [39]. The importance of elevated UPR activity to ALL etiology is also reflected in analysis of pre-B-ALL, in which elevated XBP1 expression at diagnosis coincides with poor patient outcome. Indeed, Xbp1 deletion in a mouse model of BCR-ABL-transformed pre-B ALL increased expression of the UPR components Hsp90b1, Del3 and Atf6 and reduced expression of genes functioning in the secretory pathway and in the Golgi apparatus (such as Sirpa and B3gnt5). Xbp1-deficient mice also show downregulation of immunoglobulin (Ig) light chain assembly genes as well as genes functioning in B cell antigen expression [39]. Similar to its role in B-ALL, XBP1 has also an important function in chronic lymphoblastic leukemia (CLL) etiology. XBP1 deletion in CLL cells (in the Eμ-TCL1 transgenic mouse model) is sufficient to attenuate leukemia development and prolong animal survival [40]. This phenotype is consistent with observed decreases in phosphorylation of Syk and Btk, key B-cell receptor (BCR) signaling components required for CLL cell survival. The authors of this study also noted marked decreases in synthesis of secretory μ heavy chains and decreased IgM secretion in Xbp1-deleted CLL model mice [40]. Notably, stimulation of CLL cells with sIgM in vitro using anti IgM antibodies increased expression of UPR components, an effect blocked by BTK inhibitors. Lastly, immunohistochemistry and gene expression analysis has demonstrated relatively high levels of UPR components in lymph nodes of CLL patients [41]. Overall, these studies suggest that IRE1α inhibitors may offer a novel therapeutic modality for ALL and CLL.
Hyperactivation of the Myc oncogene promotes robust protein synthesis leading to ER stress induction and UPR activation [42, 43] (Figure 2). Recent findings confirm that Myc regulates UPR activation in T-ALL cells via increasing transcription of the ubiquitin fusion degradation 1 (UFD1) gene. UFD1 is a component of the ERAD complex and facilitates ubiquitin-dependent degradation of misfolded/unfolded proteins. UFD1 inhibition in human T-ALL cells exacerbates ER stress, decreases cell growth, and induces apoptosis, in part by activating PERK-mediated proapoptotic signaling [44]. These findings point to Myc/UFD1 signaling as a key driver of the ER stress response in T-ALL and suggest that this pathway could be exploited therapeutically [44]. Accordingly, PERK activity was recently shown to promote dissemination of leukemic cells into peripheral blood and lymph nodes and thus may play an important role in progression of Myc-driven leukemias [45].
Interactions between leukemic cells and the BM microenvironment mediated by extracellular vesicles (EVs) promote leukemia cell survival [46]. Such transmission of UPR and ER stress factors by leukemic cells via EVs has been implicated in remodeling of the bone marrow niche in AML, which alters composition and function of the BM microenvironment [47]. The ability of EVs to transmit ER stress in vivo from AML xenografts to bone marrow stroma upregulates expression of core UPR components and promotes subsequent differentiation of mesenchymal stem cells, a process requiring cell-cell transfer of Bone Morphogenic Protein 2 (BMP2) by AML-EVs [47]. Notably, transmissible ER stress was previeously indentified as a source of chemoresistance in solid tumors [48].
Taken together, these studies confirm the importance of UPR activation for leukemic cell survival and highlight possible therapeutic strategies for exploiting the UPR as a novel target to ameliorate leukemia patient outcomes.
The Role of the UPR in Resistance to Therapy
Chemotherapy and targeted therapies are critical modes of cancer treatment. However, their efficacy is compromised by tumor cells’ ability to develop intrinsic and acquired resistance [49]. Among resistance mechanisms are blocking drug uptake, alteration of the drug target, induction of drug-detoxification mechanisms, repair of drug-induced damage, and activation of anti-apoptotic and pro-survival pathways [49, 50]. LSCs play important roles in leukemia relapse and drug resistance, hampering complete cure of the disease [51]. Clinical evidence and in vitro studies have linked UPR activation to drug resistance in several cancer types [52, 53]. For example, CML treatment improved significantly following development of ABL tyrosine kinase inhibitors (TKIs), such as imatinib, dasatinib, nilotinib, bosutinib, and ponatinib. However, resistance to TKI or cancer recurrence after TKI discontinuation remains an obstacle to cure. Interestingly, PERK-eIF2α phosphorylation positively correlates with resistance to these inhibitors in CML [54]. Ectopic expression of BCR-ABL in 32D myeloid cells or its expression in CML lines (such as K562 and BV173) coincides with elevated levels of ER stress and concomitant activation of the PERK-eIF2α pathway [54]. Along these lines, increased PERK and eIF2α protein expression and phosphorylation is seen in CD34+ cells from peripheral blood of CML patients resistant to imatinib or other TKIs. Correspondingly, expression of dominant negative forms of either PERK or eIF2α sensitizes CML cells to imatinib-mediated cell death. Chemoresistance can also be attributed in part to autophagy [55], a protective mechanism essential for CML cell survival, leukemogenesis and imatinib resistance [55–57]. Indeed, therapy combining imatinib with the autophagy inhibitor hydroxychloroquine decreases CML resistance to imatinib [58].
The importance of UPR signaling to CML therapy-resistance is also supported by reports on ATF6 pathway overactivation in CML cells, which has been attributed to high expression of protein disulfide isomerase A5 (PDIA5) [12]. Those authors showed that PDIA5 was necessary for ATF6 activation upon ER stress by catalyzing rearrangement of ATF6 disulfide bonds under stress conditions in a manner promoting ATF6 export from the ER and activation of its target genes. Genetic and pharmacological inhibition of the PDIA5/ATF6 axis restored imatinib sensitivity in imatinib-resistant K562 leukemia cells [12]. These results are in line with independent studies showing the important role of eIF2α phosphorylation and ATF6 activity in protecting cancer cells from chemotherapeutic drugs [59–61].
A recent study also highlights PERK/NRF2 and autophagy pathways as functioning in resistance to histone methyltransferase G9a inhibition in AML LSCs [62]. G9a regulates transcription of multiple genes primarily by catalyzing dimethylation of histone H3 lysine 9 (H3K9me2) [63]. These activities consequently induce changes in cellular redox homeostasis, decreasing ROS production [64, 65]. Pharmacological and genetic targeting of G9a inhibits AML cell proliferation and reduces LSC frequency in a mouse model and in human AML cell lines [66, 67]. G9a inhibition by either BIX-01294 or siRNA activates the pro-survival PERK/NRF2 pathway and suppresses ROS generation. Inhibition of PERK/NRF2 or autophagy increases ROS generation and enhances G9a-induced apoptosis of LSCs [62]. This data suggests that treatment with inhibitors of either PERK/NRF2 or autophagy may overcome resistance to G9a inhibition and eliminate LSCs and may serve as treatment for AML.
Overall, leukemic cells hijack UPR signaling as a mean to promote drug resistance. Therapeutic approaches that target UPR sensors and their downstream effectors may be useful to overcome drug resistance phenotypes.
Targeting the UPR in Leukemias
Targeting the UPR and ER stress is a promising approach in novel anti-cancer therapies (reviewed in [68]). Nonetheless, as noted above, a plethora of cellular functions are controlled by these signaling cascades and could be targeted in various ways. Below is a summary of interventions or drugs either in development or proposed to target UPR-associated signaling pathways relevant to leukemia (Table 1).
Table 1:
List of compounds used to exploit the UPR for leukemia therapy
| Therapeutic drug | Classification/Mechanism | Disease | Development stage | Reference |
|---|---|---|---|---|
| MKC-3946 | IRE1α inhibitor | AML | Preclinical studies | 26934650 |
| STF-083010 | IRE1α inhibitor | ALL, CLL | Preclinical studies | 24821775, 22692508 |
| B-I09 | IRE1α inhibitor | CLL | Preclinical studies | 24812669 |
| A106 | IRE1α inhibitor | ALL, CLL | Preclinical studies | 24821775, 22692508 |
| GSK2606414, GSK2656157 | PERK inhibitors | APL | Preclinical studies | 28776567 |
| Eeyarestatin I (EerI) | ERAD inhibitor | ALL, CLL | Preclinical studies | 19164757 |
| Epigallocatechin gallate (EGCG) | BiP inhibitor | ALL | Preclinical studies | 21517817 |
| Pep42 | BiP inhibitor | ALL | Preclinical studies | 21517817 |
| BMTP-78 | BiP inhibitor | AML | Preclinical studies | 29205207 |
| Bortezomib | Proteasome inhibitor | AML | Phase I-III clinical trials | NCT01861314, NCT04173585, NCT01371981 |
| ALL | Phase III clinical trials | NCT02112916 |
Targeting UPR Sensors (IRE1α and PERK)
To date, drug development targeting the UPR as leukemia treatment has mainly focused on inhibiting pro-survival signaling mediated by IRE1α-XBP1 or PERK-ATF4 signals (Figure 3). Two catalytic domains within IRE1α, the RNase domain and the ATP binding site of the kinase domain, have been exploited in developing inhibitors, and pioneering studies in multiple myeloma (MM) proved their anti-cancer activity [69, 70]. These strategies have also led to development of several compounds, such as STF-083010, MKC-3646, B-I09, and hydroxyl-aryl-aldehydes (HAA), that inhibit the catalytic core of the IRE1α RNase domain [69, 70], blocking its ability to cleave XBP1 mRNA and consequently reducing levels of sXBP1. MKC-3646 in fact has a potent anti-proliferative effect against MM [70] and cytotoxic activity against AML cell lines and primary cells derived from AML patients [71]. Pharmacological inhibition of IRE1α using STF-083010 also promotes apoptosis of primary B-ALL cells and prolongs survival of B-ALL bearing mice in vivo [39]. Notably, inhibition of the IRE1α RNase domain predominantly blocks XBP1 splicing, without affecting RIDD, suggesting RNase and RIDD domains constitute distinct targets [72]. Pharmacological inhibition of IRE1 using B-I09 also has anti-tumor effects in CLL cells and in tumor-bearing Eμ-TCL1 mice [40]. Inhibition of the IRE-1/XBP-1 pathway decreases phosphorylation of BTK, and synergizes with ibrutinib in suppressing human CLL cell growth in vitro [40], providing a rationale to assess this combination against chronic B cell leukemia. More recently, IRE1 kinase inhibitors have been developed and evaluated in mouse models of MM [73, 74]. This class of inhibitors stabilize IRE1 in the inactive state and prevent activation of the RNase domain [74]. However, the efficacy and specificity of these inhibitors in leukemia require further studies.
Figure 3: Therapeutic Targeting of the UPR in Leukemia.
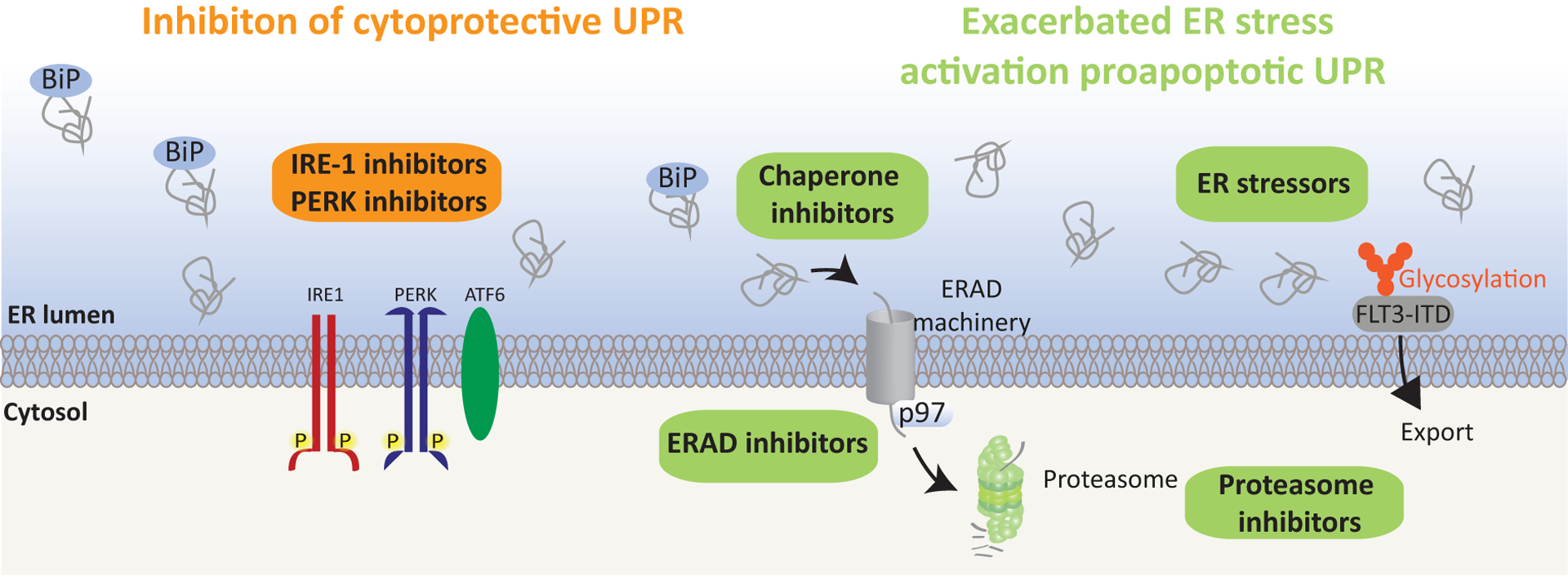
Given the dual ability of the UPR to either promote cell survival or induce death, two differing strategies could be used to target the ER stress response in leukemia. The first (left) includes inhibiting the UPR cytoprotective arms associated with the IRE1/XBP1or PERK axes. The goal of the second approach (right) would be to exacerbate ER stress to promote pro-apoptotic signaling. The rationale is that cancer cells display higher basal ER stress than do normal cells and will thus display increased susceptibility to ER stress-inducing drugs. Among these types of drugs tested preclinically against leukemia are chaperone inhibitors, as indicated, which prevent sequestration of un-/mis-folded proteins in the ER, thereby increasing ER stress. Moreover, ERAD inhibitors, which prevent retro-translocation of un-/misfolded proteins from the ER to the cytosol for proteasomal degradation, may interfere with the ER’s ability to resolve stress. Finally, treatment with drugs which prevents protein glycosylation and thus protein export from the ER (right), promotes protein retention in the ER, thereby inducing stress. FLT3-ITD positive AML may be especially vulnerable to such treatment, as in addition to inducing ER stress, these drugs prevent FLT3-ITD glycosylation and export from the ER, thereby decreasing FLT3-ITD oncogenic signaling [81]. Candidate UPR inhibitors are listed in Table 1.
GSK2606414 and GSK2656157, originally designed as selective ATP-competitive inhibitors, are among the most widely studied PERK inhibitors and have high potency and pharmacokinetic properties suitable for use in vivo [75, 76]. Both inhibitors have anti-proliferative activity in multiple cancer models in vivo including MM [77]. Moreover, combining PERK inhibition with retinoic acid (RA) or arsenic trioxide (ATO) treatment is reportedly effective against APL [78]. Correspondingly, induction of differentiation (by RA) of APL cell lines or primary human APL cells sensitizes them to ER stress. Indeed, combining ATO with Tm has a synergistic cytotoxic effect in both RA-sensitive and RA-resistant APL cell lines. Such sensitivity can be attributed to the PERK pathway, as inhibition of PERK by GSK2606414 enhances toxicity of Tm and ATO [78].
Exaggerating ER Stress to Trigger UPR-mediated Apoptosis
Therapeutic induction of ER stress emerges as a promising treatment for FLT3-ITD positive leukemia. FLT3-ITD is particularly interesting in the context of the UPR as it is a misfolded protein mostly retained in the ER due to impaired glycosylation [79]. Therefore, drugs that impact FLT3-ITD glycosylation reportedly have anti-proliferative effects on FLT3-ITD-positive AML cells (Figure 3) [80]. Among them, Tm inhibits FLT3-ITD glycosylation resulting in ER-stress-induced apoptosis and synergizes with FLT3-ITD kinase inhibitors [81]. Fluvastatin, a clinically approved inhibitor of mevalonate synthesis, also inhibits FLT3 signaling by inhibiting FLT3-ITD glycosylation and thus prolongs survival of mice with FLT3-ITD leukemia [82]. Lastly, low levels of drugs that generate either ER (Tm) or oxidative (arsenic trioxide) stress combined with retinoic acid was reported to kill AML cells characterized by MLL fusion proteins or the FLT3-ITD mutation [83]. Similar findings have been reported in T-ALL and B-ALL settings, as treatment of ALL cells with 2-deoxy-D-glucose (2-DG), Tm, or metformin leads to UPR induction and ER stress/UPR-mediated apoptosis [84, 85]. Noteworthy, the effects of Tm on a large plethora of cellular components, resulting in global cellular changes, should be considered when evaluating this drug for therapy.
Targeting the ERAD machinery to disrupt proteostasis is another approach to exploit ER stress for therapeutic purposes. The concept is mainly studied in secretory cells such as plasma cells, which -as antibody-producing cells- are strongly dependent on a well-developed secretory system and are pruned to potential protein overload. Here, proteasomal degradation represents the main pathway for ERAD. This led to development of the proteasome inhibitor bortezomib which blocks the 20S proteasome with great clinical success in MM and mantle cell lymphoma [86–89]. Bortezomib is now being studied in combination with other standard chemotherapy drugs for the treatment of relapsed or refractory ALL, newly diagnosed pediatric patients with T-cell ALL and AML [90–92]. Another approach is targeting the AAA-ATPase p97, a key ERAD component [93] (Figure 3). p97 (also known as Valosin-containing protein) is an abundant and conserved ATPase that functions in diverse activities, including protein quality control, chromatin remodeling, autophagy, and DNA repair [93, 94]. Recent efforts to develop small molecule p97 inhibitors have identified several ATP-competitive and allosteric inhibitors, including Eeyarestatin I (EerI) [95–98]. p97 inhibition induces ER stress and cell death in solid tumors [98] and in MM [29], with promising activity and tolerability in in vivo models. Eer1 treatment of hematological cancer cells elicits an integrated stress response program in the ER to activate the UPR transcription factors ATF3 and ATF4, which together activate expression of the BH3-only protein NOXA and induce cell death [99]. Thus, ERAD inhibitors represent a novel class of anticancer drugs targeting the UPR. However, p97 inhibitors should be distinguished from the general proteasome inhibitors as many of the observed effects of p97 inhibition differ from those triggered by proteasome inhibition. This could be explained by the fact that p97 inhibitors activity may not be limited to the ER, as p97 is implicated in control of misfolded proteins at other subcellular locations, including polysomes or nuclear transcriptional complexes, and may also inhibit co-translational protein transport across the ER membrane by inactivating the Sec61complex [93, 100–102].
The neural precursor cell expressed, developmentally down-regulated (NEDD8) conjugation pathway, implicated in the regulation of cullin-RING ligases (CRLs) dependent protein ubiquitination, represents another regulatory component which impacts the UPR with concomitant effects on cell death programs in ALL [103]. CRLs are an integral part of the multi-protein E3 ubiquitin ligase complex SCF. Selective inhibition of NEDD8 with the small molecule Pevonedistat attenuates CRL activity and induces cell death in AML, CLL, and ALL. How CRLs control UPR signaling remains unclear, however, this could be explained by accumulation of proteins which were not efficiently ubiquitinated or degraded due to reduced CRL activity. Since the SCF complex is also implicated in the regulation of ATF4 ubiquitination and degradation [104], limiting SCF activity via NEDD8 inhibitors may limit leukemia progression. Along these lines, NEDD8 inhibitors sensitize ALL and CML cells to ABL kinase inhibitors [105].
Another proposed leukemia treatment involves targeting chaperones that function in maintenance of ER homeostasis, among them, BiP. The peptidomimetic drug 78 (BMTP-78) targets BiP and induces apoptosis of several leukemia and lymphoma lines, as well as primary AML cells derived from patients (Figure 3). However, BMTP-78 has exhibited unacceptable toxicity in vivo, potentially attributable to effects on the tumor microenvironment [106]. Epigallocatechin gallate (EGCG), a natural compound from green tea [107], also targets the BiP ATP-binding domain, promoting a conformational change that inactivates the protein and attenuates its anti-apoptotic function. Interestingly, EGCG treatment reportedly induces apoptosis of different B-lineage ALL lines and sensitizes B-ALL cells to eradication by vincristine, a chemotherapy drug commonly used against B-ALL [108]. Although the anti-tumor potential of EGCG has been extensively studied [109, 110], its use as a natural compound component of combination therapies remains untested.
Concluding Remarks
Studies summarized here reflect emerging appreciation for the importance of the UPR in leukemia. Indeed, in response to extrinsic and intrinsic cues, activation of the UPR network represents an important step in oncogenic transformation and influences several activities relevant to leukemic cells including development, progression, and chemoresistance. Different UPR arms serve distinct purposes: the PERK pathway may function in autophagy and apoptosis, while the ATF6 and IRE1 arms likely govern cell survival. A balance between UPR arms requires further studies to define changes occurring during transformation, which may reveal mechanisms targetable at early stages of leukemia development (see Outstanding Questions). Moreover, genetic models enabling analysis of UPR components in hematopoietic malignancies, while available, have not been used to assess the impact of targeting either specific UPR arms or their combination. Although targeting ER stress/UPR signaling has great potential as an intervention in leukemias, further studies are warranted to answer unresolved issues. Therefore, further mapping of regulatory components that fine tune UPR sensors should enable one to achieve and exploit a more refined assessment of select UPR signaling in each of the different leukemias.
Outstanding Questions.
What signals activate the UPR in different leukemia subtypes?
How does the UPR impact tumor metabolism and interaction with the tumor microenvironment?
Can modulators of downstream UPR components reverse the course of leukemia development ?
Would fine-tuning UPR signaling redirect the course of leukemia development or counter treatment resistance?
Can we develop genetic models to study the role of UPR signaling in leukemias?
Would precision medicine approaches using advanced computer modeling and analysis tools allow us to define UPR-related treatment modalities? (we did not speak about it in the review at all – should mention?)
Could transcriptomic and proteomic data from leukemia patients allow us to map new paths underlying UPR dysregulation, in order to monitor and treat the disease?
Highlights.
Activated UPR contributes to leukemia development and progression
Intrinsic and extrinsic cues govern/modulate activity of UPR components in leukemia
Specific UPR components may serve as markers of leukemia development and therapy response
Targeting the UPR may overcome leukemia resistance to therapy
Acknowledgments
We thank Ani Deshpande and Daniela Senft for their valuable comments. Support by NCI grants R35 CA197465 and P01 CA128814 (ZR) is gratefully acknowledged.
Glossary
- Autophagy
Is an evolutionary conserved homeostatic process that involves degradation and recycling of cytosolic components by the lysosomes
- Chaperones
Is a ubiquitous family of proteins that functions in protein folding and proteostasis by contributing to the correct folding of polypeptides or their assembly into oligomeric structures
- ER-associated degradation (ERAD)
Constitutes a cellular pathway which targets misfolded proteins of the ER for ubiquitination and subsequent degradation by the ubiquitin proteasome system
- Fusion-protein
Is a protein product of two or more genes that originally coded for separate proteins and were fused by chromosomal rearrangements, resulting in protein product for both genes (or part of them)
- Hematopoietic stem cells (HSCs)
Are cells characterized by their unique ability to self-renew and replenish all blood cell types in the body
- Hypoxia
Is a state or a condition marked by low oxygen levels (<3%)
- Leukemic stem cells (LSCs)
Are a subpopulation of leukemia cells that possess stem cell properties distinct from the bulk leukemia cells, including self-renewal and drug resistance
- Proteostasis (Protein homeostasis)
Reflects complex pathways that controls the biogenesis, folding, trafficking and degradation of proteins in cells
- Progenitors
Are descendants of stem cells, which are more constrained in their differentiation potential and capacity for self-renewal
- Reactive oxygen species (ROS)
Are unstable and highly reactive chemicals containing oxygen which can cause cellular damage and stress
- Self-renewal
Presents a process by which stem cells divide to make more stem cells, perpetuating the stem cell pool throughout life, while maintaining their undifferentiated state
- Mitochondrial apoptotic pathway
Is regulated by pro-and anti-apoptotic members of the Bcl-2 protein family and characterized by mitochondrial membrane permeabilization and subsequent release of cytochrome c into the cytoplasm to activate caspases
References
- 1.Seita J and Weissman IL (2010) Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2 (6), 640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laurenti E and Göttgens B (2018) From haematopoietic stem cells to complex differentiation landscapes. Nature 553 (7689), 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Signer RA et al. (2014) Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509 (7498), 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hidalgo San Jose L et al. (2020) Modest Declines in Proteome Quality Impair Hematopoietic Stem Cell Self-Renewal. Cell Rep 30 (1), 69–80.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigurdsson V et al. (2016) Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 18 (4), 522–32. [DOI] [PubMed] [Google Scholar]
- 6.Walter P and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334 (6059), 1081–6. [DOI] [PubMed] [Google Scholar]
- 7.van Galen P et al. (2014) The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510 (7504), 268–72. [DOI] [PubMed] [Google Scholar]
- 8.Hwang J and Qi L (2018) Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci 43 (8), 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senft D and Ronai ZA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40 (3), 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigurdsson V and Miharada K (2018) Regulation of unfolded protein response in hematopoietic stem cells. International Journal of Hematology 107 (6), 627–633. [DOI] [PubMed] [Google Scholar]
- 11.Chapple RH et al. (2018) ERalpha promotes murine hematopoietic regeneration through the Ire1alpha-mediated unfolded protein response. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higa A et al. (2014) Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol Cell Biol 34 (10), 1839–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu L et al. (2019) Adaptive endoplasmic reticulum stress signalling via IRE1alpha-XBP1 preserves self-renewal of haematopoietic and pre-leukaemic stem cells. Nat Cell Biol 21 (3), 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheller-Wendorff M and Muller-Tidow C (2019) IRE1alpha maintains HSC stemness under ER-stress. Nat Cell Biol 21 (3), 297–298. [DOI] [PubMed] [Google Scholar]
- 15.van Galen P et al. (2018) Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Cell Reports 25 (5), 1109–1117.e5. [DOI] [PubMed] [Google Scholar]
- 16.Chang TK et al. (2018) Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol Cell 71 (4), 629–636 e5. [DOI] [PubMed] [Google Scholar]
- 17.Han J et al. (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15 (5), 481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hetz C et al. (2015) Proteostasis control by the unfolded protein response. Nat Cell Biol 17 (7), 829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollien J et al. (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186 (3), 323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollien J and Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313 (5783), 104–7. [DOI] [PubMed] [Google Scholar]
- 21.Marciniak SJ et al. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18 (24), 3066–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maurel M et al. (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci 39 (5), 245–54. [DOI] [PubMed] [Google Scholar]
- 23.Miharada K et al. (2014) Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep 7 (5), 1381–1392. [DOI] [PubMed] [Google Scholar]
- 24.Holyoake TL and Vetrie D (2017) The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood 129 (12), 1595–1606. [DOI] [PubMed] [Google Scholar]
- 25.Vetrie D et al. (2020) The leukaemia stem cell: similarities, differences and clinical prospects in CML and AML. Nature Reviews Cancer. [DOI] [PubMed] [Google Scholar]
- 26.Kindler T et al. (2010) FLT3 as a therapeutic target in AML: still challenging after all these years. Blood 116 (24), 5089–102. [DOI] [PubMed] [Google Scholar]
- 27.Cai X et al. (2015) Runx1 Deficiency Decreases Ribosome Biogenesis and Confers Stress Resistance to Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 17 (2), 165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mangan JK and Speck NA (2011) RUNX1 mutations in clonal myeloid disorders: from conventional cytogenetics to next generation sequencing, a story 40 years in the making. Crit Rev Oncog 16 (1–2), 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonard DM et al. (2007) Nuclear receptor coregulators and human disease. Endocr Rev 28 (5), 575–87. [DOI] [PubMed] [Google Scholar]
- 30.Khan MM et al. (2004) The fusion oncoprotein PML-RARalpha induces endoplasmic reticulum (ER)-associated degradation of N-CoR and ER stress. J Biol Chem 279 (12), 11814–24. [DOI] [PubMed] [Google Scholar]
- 31.Schardt JA et al. (2009) Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin Cancer Res 15 (11), 3834–41. [DOI] [PubMed] [Google Scholar]
- 32.Schardt JA et al. (2011) Activation of the unfolded protein response in human acute myeloid leukemia. Methods Enzymol 489, 227–43. [DOI] [PubMed] [Google Scholar]
- 33.Zhou C et al. (2017) JUN is a key transcriptional regulator of the unfolded protein response in acute myeloid leukemia. Leukemia 31 (5), 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Himburg HA et al. (2020) Chronic myeloid leukemia stem cells require cell-autonomous pleiotrophin signaling. J Clin Invest 130 (1), 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monteith GR et al. (2017) The calcium-cancer signalling nexus. Nat Rev Cancer 17 (6), 367–380. [DOI] [PubMed] [Google Scholar]
- 36.Giorgi C et al. (2018) Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol 28 (4), 258–273. [DOI] [PubMed] [Google Scholar]
- 37.Giorgi C et al. (2008) Ca2+ signaling, mitochondria and cell death. Curr Mol Med 8 (2), 119–30. [DOI] [PubMed] [Google Scholar]
- 38.Piwocka K et al. (2006) Bcr-Abl reduces endoplasmic reticulum releasable calcium levels by a Bcl-2-independent mechanism and inhibits calcium-dependent apoptotic signaling. Blood 107 (10), 4003–10. [DOI] [PubMed] [Google Scholar]
- 39.Kharabi Masouleh B et al. (2014) Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 111 (21), E2219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang CH et al. (2014) Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest 124 (6), 2585–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krysov S et al. (2014) Stimulation of surface IgM of chronic lymphocytic leukemia cells induces an unfolded protein response dependent on BTK and SYK. Blood 124 (20), 3101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagy P et al. (2013) Myc-driven overgrowth requires unfolded protein response-mediated induction of autophagy and antioxidant responses in Drosophila melanogaster. PLoS Genet 9 (8), e1003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hart LS et al. (2012) ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest 122 (12), 4621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huiting LN et al. (2018) UFD1 contributes to MYC-mediated leukemia aggressiveness through suppression of the proapoptotic unfolded protein response. Leukemia 32 (11), 2339–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gui J et al. (2019) The PKR-Like Endoplasmic Reticulum Kinase Promotes the Dissemination of Myc-Induced Leukemic Cells. Mol Cancer Res 17 (7), 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butler JT and Kurre P (2019) Transmissible ER stress shapes the leukemic microenvironment. Oncotarget 10 (41), 4080–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doron B et al. (2019) Transmissible ER stress reconfigures the AML bone marrow compartment. Leukemia 33 (4), 918–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rodvold JJ et al. (2017) Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci Signal 10 (482). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holohan C et al. (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13 (10), 714–26. [DOI] [PubMed] [Google Scholar]
- 50.Vasan N et al. (2019) A view on drug resistance in cancer. Nature 575 (7782), 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ravandi F and Estrov Z (2006) Eradication of leukemia stem cells as a new goal of therapy in leukemia. Clin Cancer Res 12 (2), 340–4. [DOI] [PubMed] [Google Scholar]
- 52.Bahar E et al. (2019) Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling. Cancers (Basel) 11 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avril T et al. (2017) Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 6 (8), e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kusio-Kobialka M et al. (2012) The PERK-eIF2alpha phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 11 (21), 4069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bellodi C et al. (2009) Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 119 (5), 1109–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altman BJ et al. (2011) Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 30 (16), 1855–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ianniciello A et al. (2017) Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget 8 (57), 96984–96992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horne GA et al. (2020) A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mounir Z et al. (2011) Akt determines cell fate through inhibition of the PERK-eIF2alpha phosphorylation pathway. Sci Signal 4 (192), ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peidis P et al. (2010) HDAC pharmacological inhibition promotes cell death through the eIF2alpha kinases PKR and GCN2. Aging (Albany NY) 2 (10), 669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peidis P et al. (2011) Doxorubicin bypasses the cytoprotective effects of eIF2alpha phosphorylation and promotes PKR-mediated cell death. Cell Death Differ 18 (1), 145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jang JE et al. (2020) PERK/NRF2 and autophagy form a resistance mechanism against G9a inhibition in leukemia stem cells. J Exp Clin Cancer Res 39 (1), 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casciello F et al. (2015) Functional Role of G9a Histone Methyltransferase in Cancer. Front Immunol 6, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riahi H et al. (2019) The histone methyltransferase G9a regulates tolerance to oxidative stress-induced energy consumption. PLoS Biol 17 (3), e2006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park SE et al. (2016) Inhibition of EHMT2/G9a epigenetically increases the transcription of Beclin-1 via an increase in ROS and activation of NF-kappaB. Oncotarget 7 (26), 39796–39808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kondengaden SM et al. (2016) Discovery of novel small molecule inhibitors of lysine methyltransferase G9a and their mechanism in leukemia cell lines. Eur J Med Chem 122, 382–393. [DOI] [PubMed] [Google Scholar]
- 67.Lehnertz B et al. (2014) The methyltransferase G9a regulates HoxA9-dependent transcription in AML. Genes Dev 28 (4), 317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hetz C et al. (2019) Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol 15 (8), 764–775. [DOI] [PubMed] [Google Scholar]
- 69.Papandreou I et al. (2011) Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117 (4), 1311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mimura N et al. (2012) Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood 119 (24), 5772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun H et al. (2016) Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 7 (14), 18736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tam AB et al. (2014) Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep 9 (3), 850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harnoss JM et al. (2019) Disruption of IRE1alpha through its kinase domain attenuates multiple myeloma. Proc Natl Acad Sci U S A 116 (33), 16420–16429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L et al. (2012) Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat Chem Biol 8 (12), 982–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Axten JM et al. (2013) Discovery of GSK2656157: An Optimized PERK Inhibitor Selected for Preclinical Development. ACS Med Chem Lett 4 (10), 964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Axten JM et al. (2012) Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}−2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55 (16), 7193–207. [DOI] [PubMed] [Google Scholar]
- 77.Atkins C et al. (2013) Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73 (6), 1993–2002. [DOI] [PubMed] [Google Scholar]
- 78.Masciarelli S et al. (2018) Retinoic acid and arsenic trioxide sensitize acute promyelocytic leukemia cells to ER stress. Leukemia 32 (2), 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmidt-Arras DE et al. (2005) Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol 25 (9), 3690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu X and Chen F (2019) Targeting on glycosylation of mutant FLT3 in acute myeloid leukemia. Hematology 24 (1), 651–660. [DOI] [PubMed] [Google Scholar]
- 81.Tsitsipatis D et al. (2017) Synergistic killing of FLT3ITD-positive AML cells by combined inhibition of tyrosine-kinase activity and N-glycosylation. Oncotarget 8 (16), 26613–26624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Williams AB et al. (2012) Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood 120 (15), 3069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masciarelli S et al. (2019) Retinoic acid synergizes with the unfolded protein response and oxidative stress to induce cell death in FLT3-ITD+ AML. Blood Adv 3 (24), 4155–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DeSalvo J et al. (2012) Inhibition of Akt potentiates 2-DG-induced apoptosis via downregulation of UPR in acute lymphoblastic leukemia. Mol Cancer Res 10 (7), 969–78. [DOI] [PubMed] [Google Scholar]
- 85.Leclerc GM et al. (2013) Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS One 8 (8), e74420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kane RC et al. (2006) United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res 12 (10), 2955–60. [DOI] [PubMed] [Google Scholar]
- 87.Kane RC et al. (2007) Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res 13 (18 Pt 1), 5291–4. [DOI] [PubMed] [Google Scholar]
- 88.Moreau P et al. (2015) Frontline therapy of multiple myeloma. Blood 125 (20), 3076–84. [DOI] [PubMed] [Google Scholar]
- 89.Raedler L (2015) Velcade (Bortezomib) Receives 2 New FDA Indications: For Retreatment of Patients with Multiple Myeloma and for First-Line Treatment of Patients with Mantle-Cell Lymphoma. Am Health Drug Benefits 8 (Spec Feature), 135–40. [PMC free article] [PubMed] [Google Scholar]
- 90.Messinger YH et al. (2012) Bortezomib with chemotherapy is highly active in advanced B-precursor acute lymphoblastic leukemia: Therapeutic Advances in Childhood Leukemia & Lymphoma (TACL) Study. Blood 120 (2), 285–90. [DOI] [PubMed] [Google Scholar]
- 91.Ishitsuka K et al. (2015) A phase II study of bortezomib in patients with relapsed or refractory aggressive adult T-cell leukemia/lymphoma. Cancer Sci 106 (9), 1219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bertaina A et al. (2017) The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br J Haematol 176 (4), 629–636. [DOI] [PubMed] [Google Scholar]
- 93.Meyer H et al. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol 14 (2), 117–23. [DOI] [PubMed] [Google Scholar]
- 94.Meyer H and Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127 (Pt 18), 3877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang Q et al. (2008) Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem 283 (12), 7445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chou TF et al. (2011) Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 108 (12), 4834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chou TF et al. (2013) Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem 8 (2), 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Magnaghi P et al. (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9 (9), 548–56. [DOI] [PubMed] [Google Scholar]
- 99.Wang Q et al. (2009) ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A 106 (7), 2200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Braunstein I et al. (2015) Proteasomal degradation of preemptive quality control (pQC) substrates is mediated by an AIRAPL-p97 complex. Mol Biol Cell 26 (21), 3719–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Franz A et al. (2016) Ring of Change: CDC48/p97 Drives Protein Dynamics at Chromatin. Front Genet 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cross BC et al. (2009) Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J Cell Sci 122 (Pt 23), 4393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leclerc GM et al. (2016) The NEDD8-activating enzyme inhibitor pevonedistat activates the eIF2alpha and mTOR pathways inducing UPR-mediated cell death in acute lymphoblastic leukemia. Leuk Res 50, 1–10. [DOI] [PubMed] [Google Scholar]
- 104.Lassot I et al. (2001) ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol Cell Biol 21 (6), 2192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bahjat M et al. (2019) The NEDD8-activating enzyme inhibitor MLN4924 induces DNA damage in Ph+ leukemia and sensitizes for ABL kinase inhibitors. Cell Cycle 18 (18), 2307–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Staquicini DI et al. (2018) Therapeutic targeting of membrane-associated GRP78 in leukemia and lymphoma: preclinical efficacy in vitro and formal toxicity study of BMTP-78 in rodents and primates. Pharmacogenomics J 18 (3), 436–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ermakova SP et al. (2006) (−)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res 66 (18), 9260–9. [DOI] [PubMed] [Google Scholar]
- 108.Uckun FM et al. (2011) Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br J Haematol 153 (6), 741–52. [DOI] [PubMed] [Google Scholar]
- 109.Shanafelt TD et al. (2009) Phase I trial of daily oral Polyphenon E in patients with asymptomatic Rai stage 0 to II chronic lymphocytic leukemia. J Clin Oncol 27 (23), 3808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shanafelt TD et al. (2013) Phase 2 trial of daily, oral Polyphenon E in patients with asymptomatic, Rai stage 0 to II chronic lymphocytic leukemia. Cancer 119 (2), 363–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Harding HP et al. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6 (5), 1099–108. [DOI] [PubMed] [Google Scholar]
- 112.Kroemer G et al. (2010) Autophagy and the integrated stress response. Mol Cell 40 (2), 280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13 (2), 89–102. [DOI] [PubMed] [Google Scholar]
- 114.Tait SW and Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11 (9), 621–32. [DOI] [PubMed] [Google Scholar]
- 115.Han D et al. (2009) IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138 (3), 562–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tabas I and Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13 (3), 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vardiman JW et al. (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114 (5), 937–51. [DOI] [PubMed] [Google Scholar]
- 118.Cancer I.A.f.R.o. (2014) World Cancer Report (International Agency for Research on Cancer), 2014 edn., World Health Organization; 2014 ed. edition (April 1, 2014). [Google Scholar]
- 119.Board W.C.o.T.E. et al. (2017) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Medicine).