

Abstract
The development of organ fibrosis has garnered rising attention as multiple diseases of increasing and/or high prevalence appear to progress to the chronic stage. Such is the case for heart, kidney, liver, and lung where diseases such as diabetes, idiopathic/autoimmune disorders, and nonalcoholic liver disease appear to notably drive the development of fibrosis. Noteworthy is that the severity of these pathologies is characteristically compounded by aging. For these reasons, research groups and drug companies have identified fibrosis as a therapeutic target for which currently, there are essentially no effective options. Although a limited body of published studies are available, most literature indicates that in multiple organs, premenopausal women are protected from developing severe forms of fibrosis suggesting an important role for sex hormones in mitigating this process. Investigators have implemented relevant animal models of organ disease linked to fibrosis supporting in general, these observations. In vitro studies and transgenic animals models have also been used in an attempt to understand the role that sex hormones and related receptors play in the development of fibrosis. However, in the setting of chronic disease in some organs such as the heart older (postmenopausal) women within a few years can quickly approach men in disease severity and develop significant degrees of fibrosis. This review summarizes the current body of relevant literature and highlights the imperative need for a major focus to be placed on understanding the manner in which sex and the presence or absence of related hormones modulates cell phenotypes so as to allow for fibrosis to develop.
INTRODUCTION
Organ fibrosis commonly represents a late stage of disease development that substantially contributes to decrease/loss of function.1 As such, the development of organ fibrosis has garnered rising attention as multiple diseases of increasing and/or high prevalence appear to prominently progress to the chronic stage. Such is the case for heart, kidney, liver, and lung where diseases such as diabetes, idiopathic/autoimmune disorders, and nonalcoholic liver disease appear to notably drive the development of fibrosis. As many of these diseases can evolve over a period of decades, aging can further compound the severity of fibrosis.2 For example, untreated or poorly controlled hypertension in a middle age subject, can promote the hypertrophic remodeling of the myocardium that is accompanied by excessive collagen deposition, which over time (decades) can lead to severe tissue scarring and eventually, heart failure (HF).3 For these reasons, multiple research groups and drug companies have identified fibrosis as a therapeutic target for which currently, there are very few effective options. However, as implied above, the targeting of fibrosis is inherently challenging as an effective compound would likely need to reverse a process that evolved over the long-term.
Sex based differences in chronic disease development and treatment has emerged as an important factor to consider given the general aging profile of the population. For many chronic diseases, a large body of epidemiological data indicates clear sex-based differences in disease presentation and evolution.4 In the case of cardiometabolic diseases, premenopausal women are typically protected from developing severe forms of the pathologies and as such, fibrosis.5 However, upon menopause, many women within a few years, quickly approach men in disease severity and in some cases supersede them.5 As an example, a disease where older women comprise ~two-thirds of the patient population, is HF with preserved ejection fraction (HFpEF) where fibrosis is a suspect mechanism for disease development.6 Sex-based differences in disease prevalence strongly suggest an important physiological role for estrogens or other ovarian hormones in protection (during the reproductive phase) and evolution in the postmenopausal phase.7 These facts imply that to truly understand sex-based differences in the pathophysiology of a given disease, preclinical studies would need to factor sex and age as variables in their design. Historically, this has not been the case as the great majority of studies use young male animals. To address this shortcoming, the NIH implemented a requirement to use both sexes in the experimental design of funded research or properly justify the exclusion of one sex. Thus, there are decades of work that would need to be performed in properly designed and controlled research using female cells and/or animals to even up with that of males.
As fibrosis represents a prominent pathological component of many chronic diseases a recognition of sex-based differences (Fig 1) is warranted. This review aims to summarize the current state of knowledge for sex-based differences in several of the most common organs where fibrosis can prominently factor in disease severity and uses the heart as an example to provide a greater in depth-perspective on challenges that will need to be addressed by those attempting to understand the fundamentals of fibrosis and develop effective therapies.
Fig. 1.
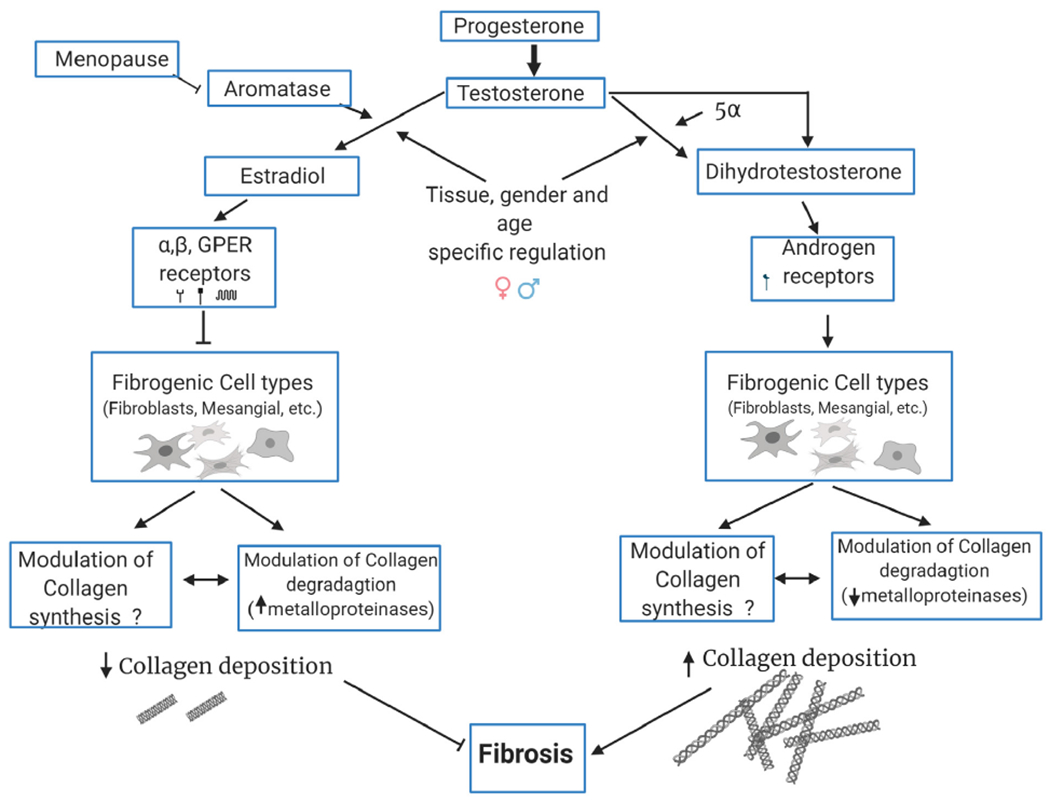
Cell biological mechanisms responsible for sex-specific effects in the pathogenesis of organ fibrosis.
FIBROSIS
Fibrosis is the excessive accumulation of extracellular matrix (ECM) that often occurs as a wound healing response to repeated or chronic tissue injury and can lead to the disruption of organ architecture and loss of function.8 Fibrosis occurs when the synthesis of new collagen by ECM producing cells exceeds the rate at which it is degraded, such that the total amount of collagen increases over time, leaving nonfunctional scar tissue.1 Fibrosis can occur as a spontaneous process but is most commonly, the end result of chronic inflammatory reactions induced by a variety of stimuli such as trauma, ischemia, oxidative stress, autoimmune reactions among others.1 Most chronic fibrotic disorders have in common a persistent irritant/insult that sustains the production of growth factors, proteolytic enzymes, angiogenic factors and fibrogenic cytokines, which stimulate the excess deposition of connective tissue elements that progressively remodels and/or replaces normal tissue architecture which has been irreversibly damaged.1
Normal collagen density (ie, area fraction) in organs such as skeletal muscle and myocardium of small and large young mammals including humans is ~2%–3%. With aging it doubles to ~4%–6%.9 It is not readily apparent that in normal, young or older mammals and in humans, tissue/organ collagen density varies in any significant manner as per sex however, relevant literature is scant. As mentioned above, in context of disease development and long-term progression, sex-based differences in many chronic diseases are well recognized. In general terms, estrogens are considered to confer protection to premenopausal women in, for example, the development of ischemic heart disease.10 However, such protection is greatly diminished after menopause.10 Thus, the comparison for the propensity/presence of fibrosis in humans has to be discussed in the context of the analysis of data as per the hormonal status. Although the principal function of sex steroid action is to regulate reproductive functions, studies in diverse fields have unequivocally established that sex steroids also act on multiple nonreproductive tissues include immune, central nervous, cardiovascular, and skeletal systems, as well as in cells from liver, skin, and kidneys.11 For example, immune cell function is modulated by sex hormones at many levels. Immune cells express receptors for sex steroids including estrogen, progesterone, and testosterone.12 While the exact molecular mechanisms of how sex hormones regulate the immune system are yet to be completely understood, studies show that they control development, homeostasis, gene expression, and signaling processes of T and B lymphocytes.13 In general, adult females mount stronger innate and adaptive immune responses than males.14 Whereas estrogens in general are considered to be immune-stimulatory, androgens are considered immunosuppressive.14 This results in faster clearance of pathogens and greater vaccine efficacy in females than in males but also contributes to their increased susceptibility to inflammatory and autoimmune diseases and associated fibrosis. Autoimmune diseases impact ~8% of the population, but 78% of those affected are women.15 Women are 3 times more likely than men to develop these types of diseases.15
Although sex hormones are likely to exert either a direct or indirect action over fibrogenesis, their role has been poorly explored. As discussed in the sections below, estradiol appears to yield protective effects, whereas androgens are “more permissive” of fibrosis progression.16 This paradigm fits with the approach taken by modern medicine, which has a sex perspective and accept that they may have different experiences and manifestations of the same disease. However, currently clinical and basic research falls dramatically short in their attempt to address more accurately this sex dichotomy as in including more women in clinical trials or develop animal and cellular models using both sexes where age is also factored in the experimental design. An attempt to address this issue comes from the recent NIH directive to require the use of both sexes in funded research and properly justify the focus of the study to only one. Ultimately, the understanding of the mechanisms by which the presence or absence of hormones impact fibrosis-driven organ failure in humans will allow for the development of treatment paradigms that may better target a specific patient population. Below, we review evidence for differences in organ fibrosis as per sex at the epidemiological/clinical, organ and cell levels. Where clinical evidence regarding sex-based differences in organ fibrosis is either weak or absent, we rely on secondary “functional” indicators that most likely reflect on this pathology.
RENAL FIBROSIS
Epidemiological studies.
Chronic kidney disease (CKD) has reached epidemic proportions worldwide with 2+ million patients affected by end-stage renal disease (ESRD).17,18 The progression to ESRD is most commonly associated with the development of glomerulosclerosis (fibrosis) secondary to diabetes.19 There is a limited body of clinical studies where renal fibrosis is rigorously documented using biopsies with no studies available where comparisons are made by sex. However, there is a well-recognized preponderance of CKD prevalence in male over female patients.20 Men progress to ESRD faster than premenopausal women in pathologies such as autoimmune glomerulonephritis, hypertensive glomerulosclerosis, and polycystic kidney disease.16 According to the 2015 United States Renal Data System, 62% of CKD patients that reached ESRD were men, whereas 38% were women.20 Silbiger and Neugarten reported that the ratio between men and women that reaches ESRD due to hypertensive nephropathy or glomerulonephritis is 1.6 men per affected woman.21 The possible protective role of estrogens on the progression of renal disease is further evidenced by the lower prevalence of CKD in premenopausal women compared with age-matched men. Interestingly, this renoprotection observed in females disappears after the beginning of menopause22,23 suggesting that estrogens may play a critical protective role.16 However, it is still not clear whether the lower prevalence of CKD in females is due to a hormonal renoprotection promoted by estrogens or due to the absence of the profibrotic effects caused by testosterone24,25 or of its more active metabolite dihydrotestosterone.
A recent study reported on sex-based risk assessment for kidney disease. Lifetime risk is the cumulative risk of experiencing an outcome between a disease-free index age and death. Turin estimated the lifetime risk of ESRD in a cohort of ~3 million adults without ESRD from 1997 to 2008. The study estimated the sex- and index age-specific lifetime risk of incident ESRD and accounted for the competing risk of death. Among those individuals without ESRD at age 40 years, the lifetime risk of ESRD was 2.66% for men and 1.76% for women. The risk was higher in persons with reduced kidney function – for eGFR = 44–59 mL/min, the lifetime risk of ESRD was 7.51% for men and 3.21% for women, whereas men and women with relatively preserved kidney function (eGFR = 60–89 mL/min) had lifetime risks of ESRD of 1.01% and 0.63%, respectively. The lifetime risk of ESRD was consistently higher for men at all ages and eGFR strata vs women. The study concluded that ~1 in 40 men and 1 in 60 women of middle age will develop ESRD during their lifetimes.26
Animal models.
Sex differences in renal disease progression has been examined in several models of renal failure in rodents. Lombet demonstrated that male rats submitted to a 5/6 renal ablation injury exhibited a reduction of the progressive course of the disease after castration.27 In agreement with these findings, Tomiyoshi showed that castration attenuated proteinuria and glomerulosclerosis (electron microscopy/histology) and ameliorated glucose tolerance in spontaneously hyperglycemic Otsuka Long-Evans Tokushima fatty male rats.28 More recently, Verzola reported that testosterone stimulates tubular epithelial cell apoptosis, leading to renal damage.29
The impact of sex on the development of experimental CKD induced by chronic nitric oxide (NO) inhibition through L-NAME was recently studied in rats.20 Long-term L-NAME administration leads to vasoconstriction, resulting in severe hypertension, albuminuria, renal ischemia, glomerulosclerosis (periodic acid-Schiff staining), interstitial expansion, and macrophage infiltration. After 30 days male L-NAME rats exhibited albuminuria, augmented cortical histological damage, interstitial inflammation and fibrosis (Immunohistochemistry/ Immunofluorescence). In contrast, age-matched female NAME rats showed significantly lower albuminuria, glomerular ischemia, glomerulosclerosis (periodic acid-Schiff staining), expression of α–smooth muscle actin (SMA) in kidney interstitial cells. The investigators proposed that female renoprotection could be provided by both the estrogen anti-inflammatory activity and/or by the relative lack of testosterone or DTH.20
In a study using a renal wrap model where the right kidney is removed while the contralateral kidney is made ischemic despite similar levels of mean arterial pressure and renal function, male rats had a greater degree of renal damage 9 weeks after injury vs females.23 Apparently, gonadal steroids could explain these sex differences as orchidectomy was renal protective while ovariectomy exacerbated the degree of glomerular and tubular damage. Furthermore, estradiol treatment of ovariectomized renal wrap females prevented the aggravating effects of ovariectomy suggesting that the hormone is protective. Also, female rats produce lower amounts of reactive oxygen species in the kidney in response to injury and appears to be associated with their ability to maintain greater endothelial nitric oxide synthase (eNOS) activity vs males.23 Indeed, Reckelhoff demonstrated that females have greater expression of kidney eNOS vs males. Renal levels of eNOS mRNA and protein were 80% higher in kidneys from females vs males.30
Podocyte damage and apoptosis are thought to be important in the development of glomerulosclerosis. Female estrogen receptor knockout mice develop glomerulosclerosis (immunohistochemistry desmin/nephrin)at 9 months of age because of their elevated blood testosterone levels that are 8 times higher than those of their female littermates.31 Using this model, Doublier examined the pathogenesis of glomerulosclerosis to determine whether testosterone and/or 17β-estradiol impact the function and survival of podocytes.31 Glomerulosclerosis was associated with the expression of desmin and the loss of nephrin, markers of podocyte damage and apoptosis. Ovariectomy preserved the function and survival of podocytes by eliminating the source of endogenous testosterone production. In contrast, testosterone supplementation induced podocyte apoptosis in ovariectomized wild-type mice. Importantly, podocytes express functional androgen and estrogen receptors, which, upon stimulation by their respective ligands, have opposing effects and may explain the observations noted above.31 Dixon demonstrated that through the modulation of ECM via TGF-β and downstream regulatory proteins in female rats, estradiolcan attenuate diabetic renal disease progression once developed.32
It has also been postulated that an interaction between estrogen and the renin-angiotensin system may confer renoprotection. Brosnihan demonstrated that estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of the system.33 Studies developed by Komukai also indicate that female sex hormones decreased renin levels, angiotensin-converting enzyme activity, angiotensin II receptor-1 expression, and aldosterone production, thus contributing to renoprotection against glomerular damage and inflammation.34 Conversely, Katz and Roper demonstrated that testosterone can stimulate the activity of the system.35
In vitro models.
As noted above, estradiol appears as a key player in sex related differences noted in renal injury. Published studies have reported on the potential renoprotective effects of estrogen in the kidney by decreasing mesangial cell proliferation as well as decreasing the synthesis and accumulation of mesangial ECM, thus reducing the development of glomerulosclerosis.31,36–39 In cultured male mouse mesangial cells, estradiol also inhibits apoptosis and TGF-β expression and activity,40 a result which was also replicated by Silbiger.24
Mesangial cells derived from adult male and female Wistar rat kidneys appear to exhibit sex-specific profibrotic and proinflammatory phenotypes.41 Specifically, male mesangial cells express higher baseline fibronectin and proinflammatory cytokine levels (TNFα and IL-1β) vs female cells. Treatment with estradiol down-regulates baseline fibronectin levels in female but has no effect on male cells. In female cells, estradiol decreases TNFα levels and increases IL-1β levels, while testosterone increases them. These data would suggest that male mesangial cells inherently exhibit greater profibrotic and proinflammatory characteristics vs female cells. Thus, sexual dimorphism in mesangial cells may play a contributory role in the faster rate of progression to ESRD vs males.41 Kwan also studied the effects of estrogens and testosterone on male mesangial cell proliferation and collagen synthesis. Estradiol at 10 and 100 nM had a modest proliferative effect on cultured mesangial cells and the effect was reversed by tamoxifen (1 μM). At higher doses (1 μM and 10 μM) estradiol markedly suppressed total collagen synthesis (types I and IV) while testosterone had no effect. The effects of estradiol on mesangial cell collagen generation may help explain the slower development of glomerulosclerosis in women. In the presence of estradiol (0.01 – 100 nM) there was a progressive increase in mRNA levels of ERα and ERβ. ERα protein levels increased approximately 2.5-fold after 24 hours and up to approximately 5.4-fold after 72 hours while ERβ protein levels increased approximately 2.1-fold after 24 hours. Thus, estradiol positively regulated the expression of the ER subtypes thus, maintaining cell responsiveness to estrogens.42
The effects of estrogens on matrix metalloproteinases (MMP) were also studied. Estradiol increased MMP-9 mRNA and activity. Increased MMP activity may also be an important mechanism by which estrogens influence ECM turnover and protect against glomerulosclerosis.43 As noted above, podocyte damage may be crucial to the development of glomerulosclerosis. Doublier reported that testosterone can induce mesangial podocyte apoptosis in vitro by androgen receptor activation, but independent of the TGF-β1 signaling pathway. Pretreatment with estradiol prevented testosterone-induced podocyte apoptosis, an effect mediated by ERK and protected podocytes from TGF-β1 or TNFα-induced apoptosis.31
LIVER FIBROSIS
Epidemiological studies.
In a review by Guy and Peters, it was reported that women more commonly present with acute liver failure, autoimmune hepatitis, benign liver lesions, primary biliary cirrhosis, and toxin-mediated hepatotoxicity. Women less commonly have malignant liver tumors, primary sclerosing cholangitis, and viral hepatitis and there is a decreased rate of decompensated cirrhosis with hepatitis C virus infection. However, men are 2-fold more likely to die from chronic liver disease and cirrhosis vs women. Also, liver transplants occur less commonly in women vs men.44 The natural history of liver disease in women can vary according to etiology. Although women have slower progression of fibrosis and decreased incidence of cirrhosis pretransplantation, after liver transplantation, women have a higher risk of advanced fibrosis and graft loss in hepatitis C related disease. Women have a 31% increased risk of advanced recurrent disease vs men. In patients transplanted for hepatitis related disease, women have a 14% increased risk of death at 5 years vs men.44 According to a 2005 analysis by the National Center for Health Statistics, men are 2-fold more likely to die from chronic liver disease and cirrhosis vs women. Women represent ~30% of liver transplant recipients. However, women appear more likely than men to die on the waiting list in the Model for End-Stage Liver Disease era vs the pre–Model for End-Stage Liver Disease era and the reasons for these differences remain unknown. Once transplanted, women have better long-term survival after liver transplant vs men.44
Animal models.
In a study by Xu, liver fibrosis was induced in male, female, and ovariectomized rats bycarbon tetrachloride (CCL4) administration. All groups were treated with estradiol. Tamoxifen was given to male animals. Estradiol treatment reduced collagen synthesis and content, decreased the areas of hepatic stellate cells positive for αSMA in both sexes including ovariectomized rats whereas, tamoxifen had the opposite effect. Altogether, the fibrotic response of the female liver to CCL4 was significantly weaker vs males.45 In intact and ovariectomized female hepatofibrotic rats, estradiol treatment reduced aspartate aminotransferase, alanine aminotransferase, hyaluronic acid, and type IV collagen in sera while suppressing hepatic collagen content and stellate cell SMA levels.46 There was also a negative correlation between the percentage of fibrotic area of liver tissue and serum estradiol levels.46 The effects of estradiol on hepatic fibrosis was examined by Yasuda in male and female rats by the administration of a single dose of dimethylnitrosamine.47 The fibrotic response of the male liver after treatment was significantly stronger vs females. In males, estradiol reduced hepatic mRNA for type I and III procollagens and the tissue inhibitor of metalloproteinase-1 (TIMP-1), as well as deposition of type I and III collagen protein total hepatic collagen and malondialdehyde, a product of lipid peroxidation. Concomitant administration of a neutralizing antibody against estradiol enhanced fibrogenesis, as judged by the same parameters. Ovariectomy in the female model had a fibrogenic effect, inducing the hepatic expression of both types of procollagen and TIMP-1. In addition, the number of SMA positive cells increased. Estradiol replacement was fibrosuppressive in ovariectomized rats. These findings suggest that estradiol suppressed the induction of hepatic fibrosis, and may in part, underlie the more rapid progression in males of hepatic fibrosis.47
In vitro models.
Yasuda also studied rat hepatic stellate cells in culture (sex not specified). Estradiol treatment reduced cell number, type I collagen production and SMA expression.48 Liu examined normal female rat hepatic stellate cell phenotype in response to estrogen treatments. 17 β-estradiol, 2-hydroxyestradiol or 2-methoxyestradiol were separately added to cells. 17 β-estradiol and its metabolites concentration-dependently inhibited cell proliferation and collagen synthesis while at 100 nm, they inhibited SMA expression.46 Fibrosis was induced in rats by administration of CCL4, and activation was monitored as the level of collagen I mRNA or SMA.49 Both male and female rats were studied. Stellate cell activation, rather than collagen synthesis, proved to be the target of both HOE 077 and Safironil (antifibrotic compounds which were designed as competitive inhibitors of collagen protein synthesis) in the intact liver. In culture, the drugs not only prevented the activation of stellate cells but also accelerated their deactivation. Interestingly, the response of cells from females was greater vs male cells, leading to the conclusion that stellate activation is sexually dimorphic. This finding may be relevant to the observation that fibrosis in chronic viral hepatitis progresses less rapidly and that hepatocellular carcinoma is less frequent in females than in males.49
PULMONARY FIBROSIS
Epidemiological studies.
A considerable body of epidemiologic data indicates that the incidence and pathogenesis of a variety of lung diseases are influenced by sex.50 While genetic and environmental factors clearly promote chronic obstructive pulmonary disease, asthma, lung fibrosis, lung cancer, and other respiratory ailments have been reported to be influenced in some manner by sex.50 Fibrotic interstitial pneumonias are more prevalent in males vs women of advancing age, although little is known about the underlying mechanisms.51
In idiopathic pulmonary fibrosis (IPF), incidence is higher in males however, females have better survival.50,52 Kalafatis et al., examined sex differences at presentation of patients included in the Swedish IPF registry over a 3-year period from its 2014 launch. Analysis was performed for data concerning demographics, lung function, and quality of life.53 Three hundred forty-eight patients (250 [72%] males, 98 [28%] females, median age 72 years in both genders) were included. Baseline lung function (Forced vital capacity, 68.9% ± 14.4 vs 73.0% ± 17.7, P < 0.05; Total lung capacity, 62.2% ± 11.8 vs 68.6% ± 11.3%, P < 0.001) were lower at presentation in male vs females. Results reported poor quality of life, but no difference was found between genders in this and other measured parameters. Thus, this study demonstrates that registry female IPF patients have greater preserved lung function than males at inclusion.53
Animal models.
A common means to experimentally induce pulmonary fibrosis is based on the use of bleomycin. Studies report that female rats have higher mortality rates and more severe fibrosis vs male, as indicated by higher levels of lung collagen deposition and fibrogenic cytokine expression.54 In ovariectomized female rats treated with either estradiol or vehicle, results showed diminished fibrosis in the ovariectomized rats without estradiol. Estradiol replacement restored the fibrotic response.54 In contrast, using a similar model of bleomycin-induce fibrosis, Redente evaluated the contributions of age and sex to the development of pulmonary fibrosis. Aged (52–54 week) male mice developed more severe lung disease, indicated by increased mortality, increased collagen deposition, and neutrophilic alveolitis vs female counterparts and young mice (8–12 week) independent of sex. Young male mice developed more fibrotic disease vs females regardless of age. There was no difference in fibrosis between young and aged female mice.51 This adverse effect on lung function was found to be due to male sex hormones, as castrated males exhibited a female-like response to bleomycin whereas the opposite happens when giving testosterone to females.55 Male mice had significantly higher basal static lung compliance than females and a more pronounced decline in static compliance after bleomycin administration.56 No differences between sex appear in immune cell infiltration into the lung or in total lung collagen content. Furthermore, castrated male mice exhibited a female-like response to bleomycin while female mice given exogenous androgen exhibited a male-like response. These data indicate that androgens play an exacerbating role in decreased lung function after bleomycin administration.56 Altogether, results using the bleomycin model appear unclear as to the role played by sex and related sex hormones.
In vitro models.
A very limited amount of work comparing sex differences, or the effects of sex-related hormones has been reported using lung fibroblasts in culture. In the study by Gharaee-Kermani lung fibroblasts isolated from bleomycin-treated female rats exhibited increased responsiveness to estradiol treatment, causing dose-dependent increases in procollagen-1 and TGF-β1 mRNA expression levels. These findings suggest that the effect of estradiol is selective and female rats may develop an exaggerated response to lung injury relative to male rats.54 Elliot, reported that mRNA levels of ERα were selectively upregulated in lung tissue and cultured myofibroblasts from male IPF patients as well as in male mice treated with bleomycin. IPF myofibroblasts exhibited increased responsiveness to estradiol compared with controls (non-IPF fibroblasts) and ERα receptor antagonists diminished this effect. The in vivo inhibition of ERα attenuated bleomycin-induced lung fibrosis in mice. Further experiments demonstrated that ERα antagonism exerted its antifibrotic properties through negative regulation of profibrotic kinase-controlled signal transduction pathways, including Smad2 and AKT (protein kinase B).57
CARDIAC FIBROSIS
Epidemiological studies.
It is well-established that premenopausal women have a decreased incidence of cardiovascular disease (CVD) vs age-matched males as they are largely protected from developing coronary artery disease (CAD), myocardial infarction (MI), hypertension, and pathological cardiac remodeling.58 However, upon menopause, CVD becomes more prevalent in women. The CONFIRM long-term registry study showed that while CAD and MI were more prevalent in men (43% men vs 27% women), women tended to acquire heart disease ~10 years later. On average, women who develop CAD were older (62 vs 59 years in men), experienced more hypertension and concentric left ventricular (LV) hypertrophy. Death due to CAD shows a consistent 2.5–4.5 male to female ratio. However, differences vanish once women reach menopause such that CVD and overall mortality become independent of sex. It is therefore proposed that in premenopausal women, estrogen and associated genetic and epigenetic modifications exert protective effects on the cardiovascular system.59
Initial reports pertaining to sex-based differences in cardiac structure and function in patients appeared in the 1980’s. However, it was not until the 1990s that studies emerged that intended to systematically compare sex-based differences. Villari reported-on differences in LV structure/function in 56 aortic stenosis patients denoting in men greater LV chamber enlargement, endocardial fibrosis (with abnormal collagen architecture) and stiffness while accompanied by lower EF. Petrov compared 92 aortic stenosis patients indicating that women demonstrated more LV hypertrophy than men (86% vs 56%) with less fibrosis.60 However, following valve replacement surgery this process reversed more frequently in women.61 In 2 2014 follow-up aortic stenosis studies by the same group, they also reported that women with maladaptive LV hypertrophy (which was less common than men) had worse survival vs men. Men also developed greater levels of fibrosis that could also be documented at the level of transcriptome analysis.62 In the setting of atrial fibrillation, aggravation of fibrosis in women (ages 55–60) in comparison to men counterparts, may be causal for the low success rate of atrial fibrillation catheter ablation.63 In support of the estrogen hypothesis, studies indicate that normalized heart mass differences (which on average a higher in women vs men) become greater after menopause.64,65 The overall impact of cardiac hypertrophy on morbidity and mortality is higher in women.66 Several studies have reported that women receiving hormone replacement therapy (HRT) have a lower LV mass vs woman without HRT.67,68 HRT has also been shown to attenuate the development of LV hypertrophy in hypertensive postmenopausal women.69 However, no direct evidence is available on the effects of HRT on cardiac fibrosis.
The CONFIRM registry also indicated that in patients with CAD, 37% of women develop HFpEF vs 23% of men. Multiple epidemiological reports indicate that HFpEF is more predominant in postmenopausal female patients by ~2:1 vs men.6,70 The causes for this unequal distribution remain unclear. However, HFpEF in women is closely associated with the development of hypertension and concentric LV hypertrophy.6,70 The pathophysiology of HFpEF is likely complex and involves various elements of chamber remodeling such as concentric hypertrophy, microvascular dysfunction, altered ventricular-vascular coupling, and calcium cycling.71 However, increased myocardial stiffness is recognized as a key factor.72 Passive myocardial stiffness is regulated both within myocytes (by cytoskeletal elements such as titin) and by components of the ECM, in particular, fibrillar collagens type I and III.72 Normal collagen content in healthy young myocardium ranges from 2% to 3%. As a function of normal aging, collagen content increases, and this process is known to be aggravated by hypertension.9 In failing hearts, collagens can comprise up to 30% of the total myocardial mass dramatically increasing tissue stiffness.73 The “Cardiovascular Health Study” quantified associations between serum markers of fibrosis and HF in the elderly. Interestingly, in HFpEF patients, women had significantly higher plasma levels of the propeptide for type I collagen (PIP) vs men, suggesting greater levels of production of this critical form of fibrillar collagen.74 As part of the Multi-Ethnic Study of Atherosclerosis (MESA), an evaluation of age-related myocardial fibrosis was performed in 1231 participants (51% women, age 54–93 years) using magnetic resonance imaging.75 In women, lower LV chamber volume over a 10-year period was strongly related to diffuse interstitial fibrosis. Increased diffuse myocardial fibrosis with fewer myocytes (related to aging and remodeling) may explain the enhanced prevalence of diastolic dysfunction and HFpEF seen in older women. These data align with reports from a cross-sectional sample of Olmsted County, Minnesota, residents (≥45 years, n = 2042), where clinical data, echocardiography, and blood pressure measurements were obtained. Advancing age and female gender were associated with increases in vascular and ventricular systolic and diastolic stiffness even in the absence of cardiovascular disease.76
Cardiac fibrosis is widely recognized as one of the hallmarks of pathological chamber remodeling as seen with hypertrophy, and can promote disease progression.77–81 The development of diffuse fibrosis adversely impacts the material properties and thus, myocardial diastolic function. In many cases, diastolic function fails as the filling and relaxation dynamics become dysfunctional.82 In HFpEF patients the LV cannot fully relax, chamber filling is impaired and cardiac output is reduced even though EF is normal. Altogether, evidence provided by the abovementioned studies suggest a role for excess fibrosis in the development of HFpEF, which is more common in postmenopausal women.72
Animal models.
Very few animal studies have focused on sex-based differences in cardiac fibrosis in response to CVD. However, as noted above, cardiac fibrosis is closely linked to chamber remodeling and a limited number of studies have examined sex-based differences and/or the roles played by ovarian hormones/estrogen in remodeling. As implied in the section above, for animal models to best recapitulate cardiac diseases in humans, aging would need to be incorporated and only few studies include it in their design.
In spontaneously hypertensive rats (SHR), female had better indices of systolic function and smaller LV dimensions vs male SHR, which also developed LV dysfunction and HF after 1 year.83 In the aortic banding-induced pressure overload model male but not female rats showed LV chamber dilation, loss of concentric remodeling, fibrosis and elevated wall stress 20 weeks after surgery.84 In line with this study, Weinberg reported that, in comparison with males, females developed higher LV pressures in the isolated heart 6 weeks after banding, despite a similar degree of LV hypertrophy and systolic wall stress.85 Cardiac models of pressure overload hypertrophy report differences between sexes. In general, females develop concentric hypertrophy vs males, who develop eccentric hypertrophy.58,86 Gene expression profiling also revealed that male hearts had a stronger induction of ECM-related genes and a stronger repression of mitochondrial genes vs female hearts.86 In a rat model of volume overload induced by aortocaval fistula, female but not male rats showed minimal mortality and no significant LV dilatation after fistulation.87 In a mouse model of hypertrophic cardiomyopathy associated with the mutation R403Q of α-myosin heavy chain, male mice developed progressive LV dilatation and reduced systolic function. In contrast, female mice with a similar degree of cardiac hypertrophy showed preserved function without chamber dilation.88 Sex also impact cardiac remodeling after MI. Mortality and LV rupture during the first week after MI were higher in males vs females.89 Three months after MI, males showed worse LV function with more cardiac chamber dilation and hypertrophy. Females had 3 times lower mortality despite similar infarct size and showed a better functional outcome.89 Wu found that female mice with MI underwent less LV remodeling vs males, with better preserved systolic function.90 Administration of estrogens in ovariectomized rats reduces the development of aortic banding-induced cardiac hypertrophy by 30%.91 Furthermore, administration of an ERβ agonist in ovariectomized SHR rats lowered blood pressure and prevented hypertrophy.92 Estrogen treatment promotes myocyte survival in infarcted mice.93 Together, these results imply that estrogens exert cardioprotective effects.
Aging models have used various rat strains (mostly male) to characterize changes in cardiac structure/function.94–97 When using ~2 year old female Fischer 344 rats, aging results in LV hypertrophy and diastolic dysfunction, while males from the same strain/age develop eccentric remodeling, mitral regurgitation, interstitial fibrosis, and impaired systolic function.98 Hybrid Brown Norway/Fischer 344 rats (F344BN) have also been used to study the effects of aging. Female rats of up to 30 months of age show development of LV hypertrophy, dilatation, and diastolic dysfunction,99 while decreases in collagen area fraction have been reported.100 Bustamante recently reported on the effects of aging in female rats of 18–21 months of age. While echocardiography did not detect major changes in LV structure/function direct catheter-based hemodynamics documented the prolongation of relaxation, a modest but significant reduction in CO while maintaining EF in the setting of hypertension. Histology demonstrated an approximate doubling of collagen area fraction in the absence of chamber stiffening as revealed by ex vivo LV passive pressure-volume curves and epicardial strains.101
The high prevalence of HFpEF in older women suggests a strong link between low estrogen levels and the disease.102 As discussed above, published studies suggest the development of greater levels of myocardial fibrosis in postmenopausal women vs men with normal aging or with HF.74,75 In rats, unlike menopausal women, estradiol levels during aging can be near their younger counterpart even past 20 months of age.99 Thus, ovariectomy is commonly used to examine the role that low estrogen levels play in altering cardiac structure/function.103 Unfortunately, only an extremely limited number of studies have used this approach. Using young male and female rats undergoing aortic banding, a lesser degree of LV chamber remodeling, loss of function and fibrosis was noted in female animals.84 However, the protective role of estrogens in rodent models of pressure overload was greatly diminished with ovariectomy.104 Stice reported that ovariectomy in 20-month-old Norway Brown rats decreases LV fractional shortening from 50% (observed in control 4-month-old animals) to 40% and was restored by estradiol supplementation. These changes were associated with the activation of regulators of inflammation in isolated myocytes, which were also suppressed by estradiol.105 In a recent study regarding the influence of aging and estrogen depletion on LV structure/function a hybrid female F344BN rats was implemented.106 A subgroup of animals at 18 months of age underwent an ovariectomy for 2 months and relevant endpoints examined. In their study, ovariectomy did not alter blood pressure, systolic function or lead to the development of hypertrophy or additional interstitial fibrosis beyond that noted for aging. Significant alterations were only noted in LV relaxation and increases in filling pressure.106 In the study by Bustamante, a large (~30%) significant loss in stroke volume index, cardiac output index, and increased arterial elastance in the presence of preserved EF was reported in ovariectomized rats. While ovariectomy did not lead to further increases in collagen area fraction as per aging, it did appear to uniquely influence the distribution of myocardial fibrosis toward the endocardium (vs epicardium). Interestingly, papillary fibrosis was notably increased with ovariectomy vs aging and may have important implications for disease development in the presence of mitral valve dysfunction.101
Several studies have explored the role played by selective estrogen receptors. In the setting of transverse aortic constriction in male and female ERβ knockout (ERβKO) mice, the role of estrogen/ERβ on myocardial hypertrophy becomes more evident.86 ER deletion augments the transverse aortic constriction -induced increase in cardiomyocyte diameter in both sexes.86 Moreover ERβKO females develop an increased degree of hypertrophy compared with WT females, whereas αERKO mice respond in a manner identical to WT females.58 Also, hearts of bilateral ovariectomized hypertensive female mice, showed worse patterns of LV remodeling, diastolic dysfunction, inflammation, and fibrosis than their sham counterparts.107,108 In a study performed in 1-year old female rats, ovariectomy triggered an increase in collagen type I/III ratio and decreased MMP-2 activity. Interestingly, in this model, estrogens prevented the effects of ovariectomy on heart remodeling and fibrosis.109
The G protein-coupled estrogen receptor (GPER) also known as G protein-coupled receptor 30 (GPR30) also mediates the effects of estrogen in multiple organs including the heart. In an ischemia-reperfusion model treatment with G1 a GPER agonist, decreased infarct size of male treated animals.110 In a female rat model of aging with ovariectomy, GPER activation by G1 was able to mitigate adverse LV remodeling, myocardial relaxation, and interstitial fibrosis, therefore delaying the development of diastolic dysfunction.106,111 Sex-differences have also been identified in the setting of a GPER knockout (KO) mice model, where male mice but not females demonstrate impaired LV function.112 Using cardiomyocyte specific GPR30 KO mice, KO’s males show greater adverse alterations in cardiac structure and impaired systolic and diastolic function vs KO’s females.113
In vitro models.
An extremely limited number of in vitro studies have examined for phenotypic differences in cardiac fibroblasts isolated from male or female animal or human donors and/or the effects of estrogens. Lee reported on the enhanced proliferation of cardiac fibroblast isolated from female rats in response to 10–20 nM estradiol stimulation. Zhou reported that neonatal rat cardiac fibroblasts stimulated with 100 nM angiotensin II increased cell proliferation, collagen types I and III synthesis and production of fibronectin and vimentin. Angiotensin II up-regulated AT1 receptor mRNA and down-regulated AT2 receptor levels. Estradiol at 100 nM prevented increases in proliferation and attenuated collagen synthesis in response to angiotensin II.114,115 The increased AT1 receptor mRNA levels and decreased AT2 receptor mRNA levels were also partially reversed. A similar study was performed by Pedram in neonatal cells where 10 nM estradiol also blocked 100 nM angiotensin II induced fibronectin, vimentin via effects on cAMP signaling.107 In female rat cardiac fibroblasts, treatment with 10 nM estradiol led to a significant down-regulation of basal collagen I and III expression, whereas both collagens were up-regulated in male cells. These effects were replicated in human cardiac fibroblasts.116 In a study by Griffin, whereas female rat cardiac fibroblasts were resistant to hypoxia-induced inhibition in DNA synthesis while male cells were susceptible. In female cells, the inclusion of estrogen led to DNA synthesis inhibition whereas in male cells it partially reversed the effects of hypoxia.117
CONCLUSIONS
The presence of sex-based differences in organ fibrosis, strongly suggest an important role for sex hormones in modulating disease progression (see Table I), in particular over the long-term (ie, in the setting of chronic diseases such as diabetes and hypertension). Apparently, estrogens confer a highly effective degree of protection toward the development of tissue fibrosis. Unfortunately, women have been rather underrepresented in clinical trials particularity, during their premenopausal stage, mainly, because in women’s physiology there are so many variations during their lifetime driven by hormonal cycles that complicate the establishment of reliable models. Aging and menopause also need to be considered as determinant factors in disease progression. Furthermore, preclinical studies have predominantly been performed using male animals and cells derived from young male donors. In consequence, obtaining an in-depth understanding of the roles that sex hormones such as estrogen and testosterone play in developing fibrosis has been hampered by these limitations. The testing of promising novel antifibrotic therapies will need to take into account these disease modifiers. Looking into the future studies will need to be designed incorporating male and female subjects and/or patients (even of different age brackets) such that a broader appreciation of disease progression and more specific effects of therapy can be evaluated.
Table I.
Sex related differences in pathogenesis of organ fibrosis
| Adult female | Adult female with reduced estrogen levels | Adult male | |
|---|---|---|---|
| Kidney | H: Slower development of glomerulosclerosis38 A: Female rats produce lower amounts ROS in response to injury24 C: Competitive inhibitors of collagen protein synthesis on mesangial cells exhibited a greater response on females vs. male cells49 |
H: Become more susceptible to renal diseases after menopause17 A: E2 treatment of ovariectomized female rats prevented the aggravating renal damage24 |
H: Men progress to end stage renal disease (ESRD) faster than premenopausal women17 H: Had a greater degree of glomerular and tubular damage24 C: Mesangial cells inherently exhibit greater profibrotic and proinflammatory characteristics 42 A: Castration attenuated glomerulosclerosis in fatty male rats 28 |
| Liver | H: Have slower progression of fibrosis and decreased incidence of cirrhosis pretransplantation44 A: Fibrotic response of the female liver to CCL4 was significantly weaker vs males46 |
A: Ovariectomy in the female model had a fibrogenic effect, inducing the hepatic expression of both types of procollagen and TIMP-148 | H: Men are 2-fold more likely to die from chronic liver disease and cirrhosis vs women44 H: The fibrotic response of the male liver is significantly stronger vs females44 |
| Lungs | A: Female rats may have an exaggerated response to lung injury relative to male rat54 | A: Female rats have higher mortality rates and more severe fibrosis vs male54 | A: Idiopathic pulmonary fibrosis, incidence is higher in males50,116 H, A: mRNA levels of ERα) were selectively upregulated in lung tissue and cultured myofibroblasts from male IPF patients as well as in male mice treated with bleomycin57 |
| Heart | H: Women demonstrated more LV hypertrophy than men with less fibrosis61 C: Female rat cardiac fibroblasts, treatment with estradiol (10–8M) led to a significant downregulation of basal collagen I and III expression114 |
H: Women show greater aggravation of fibrosis in atrial fibrillation (ages 55–60)63 A: Advancing age and female gender were associated with increases in vascular and ventricular systolic and diastolic stiffness75 A: In a rat model of aging with ovariectomy, GPER activation was able to mitigate adverse LV remodeling and interstitial fibrosis104,109 |
H: Men greater endocardial fibrosis and stiffness61 A: Gene expression profiling also revealed that male hearts had a stronger induction of ECM-related genes and a stronger repression of mitochondrial genes85 C: Using cardiomyocyte specific GPER KO mice, males show greater adverse alterations in cardiac structure and impaired systolic and diastolic function vs. KO’s females111 |
Abbreviations: A, Animal, C, Cells; H, Human.
ACKNOWLEDGMENTS
All authors read the journal’s authorship agreement and the manuscript was reviewed and approved by all authors. Drs. Villarreal is a cofounder and stockholder (Dr Ceballos) of Cardero Therapeutics, Inc. All authors read the journal’s policy on disclosure of potential conflicts of interest. This work was supported by the Department of Defense DoD PR150090, National Institute of Health, NIH DK98717, AG47326, and VA I01BX3230 to Dr. Villarreal.
Abbreviation:
- α-SMA
Alpha Smooth Muscle Actin
- AT
Angiotensin Receptor
- CAD
Coronary Artery Disease
- cAMP
Cyclic Adenosine Triphosphate
- CCL4
Carbon Tetrachloride
- ADL
Activity of Daily Living
- CONFIRM Registry
Coronary CT Angiography Evaluation for Clinical Outcomes: AnInternational Multicenter
- CVD
Cardiovascular Disease
- E2
Estradiol = 17β estradiolECM: Extracellular Matrix
- EF
Ejection Fraction
- ENOS
Endothelial Nitric Oxide Synthase
- ER
Estrogen Receptor
- ERK
Extracellular Signal-Regulated Kinase
- ERKO
ER Knockout
- ESRD
End Stage Renal Disease
- F344BN
Hybrid Brown Norway/Fischer 344 Rats
- GPER
G Protein-Coupled Estrogen Receptor
- HF
Heart Failure
- HFpEF
Heart Failure with Preserved Ejection Fraction
- HRT
Hormone Replacement Therapy
- IL-1β
Interleukin 1 Beta
- L-NAME
Nitro-L-Arginine Methyl Ester
- LV
Left Ventricle
- MELD
Model for End-Stage Liver Disease
- MESA
Multi-Ethnic Study of Atherosclerosis
- MI
Myocardial Infarction
- MMP
Matrix Metalloproteinases
- MRI
Magnetic Resonance Imaging
- NIH
National Health Institute
- PINP
Pro-Peptide for Type I Collagen
- SHR
Spontaneously Hypertensive Rats
- TAC
Transverse Aortic Constriction
- TGF-β
Transforming Growth Factor Beta
- TIMP-1
Tissue Inhibitor of Metalloproteinase-1
- TNF
Tumor Necrosis Factor Alpha
Footnotes
Conflicts of Interest: All authors have read the journal’s policy on disclosure of potential conflicts of interest and have none to declare.
REFERENCES
- 1.Wynn TA Cellular and molecular mechanisms of fibrosis. J Pathol 2008;46:26–32. 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hecker L, Logsdon NJ, Kurundkar D, et al. Reversal of persistent fibrosis in aging by targeting nox4-Nrf2 redox imbalance. 2015;6. doi: 10.1126/scitranslmed.3008182. Reversal [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomek J, Bub G. Hypertension-induced remodelling: on the interactions of cardiac risk factors. J Physiol 2017;595:4027–36. 10.1113/JP273043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regitz-Zagrosek V Sex and gender differences in health. Science & society series on sex and science. EMBO Rep 2012;13:596–603. 10.1038/embor.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulman-Marcus J, Hartaigh Bó, Gransar H, et al. Sex-specific associations between coronary artery plaque extent and risk of major adverse cardiovascular events: from the CONFIRM long-term registry. Physiol Behav 2017;176:139–48. 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borlaug BA. Sex, load, and relaxation: are women more susceptible to load-dependent diastolic dysfunction. J Am Coll Cardiol 2011;57:1234–6. 10.1016/j.jacc.2010.10.033. [DOI] [PubMed] [Google Scholar]
- 7.Iorga A, Cunningham CM, Moazeni S, Ruffenach G, Umar S, Eghbali M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol Sex Differ 2017;8:33 10.1186/s13293-017-0152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Il Jun J, LF Lau. Resolution of organ fibrosis. J Clin Invest 2018;128:97–107. 10.1172/JCI93563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis 2011;2:158–73. [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia M, Mulvagh SL, Merz CNB, Buring JE, Manson JAE. Cardiovascular disease in women: clinical perspectives. Circ Res 2016;118:1273–93. 10.1161/CIRCRESAHA.116.307547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan D, Ansar Ahmed S. The immune system is a natural target for estrogen action: opposing effects of estrogen in two prototypical autoimmune diseases. Front Immunol 2016;6 (JAN):1–8. 10.3389/fimmu.2015.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furman D Sexual dimorphism in immunity: improving our understanding of vaccine immune responses in men. Expert Rev Vaccines 2015;14:461–71. 10.1586/14760584.2015.966694. [DOI] [PubMed] [Google Scholar]
- 13.Moulton VR. Sex hormones in acquired immunity and autoimmune disease. Front Immunol 2018;9(OCT):1–21. 10.3389/fimmu.2018.02279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol 2016;16:626–38. 10.1038/nri.2016.90. [DOI] [PubMed] [Google Scholar]
- 15.Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol 2008;173:600–9. 10.2353/ajpath.2008.071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yanes LL, Sartori-Valinotti JC, Reckelhoff JF. Sex steroids and renal disease. Hypertension 2008;51:976–81. 10.1161/hypertensionaha.107.105767. [DOI] [PubMed] [Google Scholar]
- 17.El Nahas M, Barsoum R, Eknoyan G, et al. The global challenge of chronic kidney disease. Kidney Int 2005;68:2918–29. 10.1111/j.1523-1755.2005.00774.x. [DOI] [PubMed] [Google Scholar]
- 18.Couser WG, Remuzzi G, Mendis S, Tonelli M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int 2011;80:1258–70. 10.1038/ki.2011.368. [DOI] [PubMed] [Google Scholar]
- 19.Cobo G, Hecking M, Port FK, et al. Sex and gender differences in chronic kidney disease: progression to end-stage renal disease and haemodialysis. Clin Sci 2016;130:1147–63. 10.1042/CS20160047. [DOI] [PubMed] [Google Scholar]
- 20.Fanelli C, Dellê H, Cavaglieri RC, Dominguez WV, Noronha IL. Gender differences in the progression of experimental chronic kidney disease induced by chronic nitric oxide inhibition. Biomed Res Int 2017. 10.1155/2017/2159739 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silbiger SR, Neugarten J. The impact of gender on the progression of chronic renal disease. Am J Kidney Dis 1995;25:515–33. 10.1016/0272-6386(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 22.Hecking M, Bieber BA, Ethier J, et al. Sex-specific differences in hemodialysis prevalence and practices and the male-to-female mortality rate: the Dialysis Outcomes and Practice Patterns Study (DOPPS). PLoS Med 2014;11 10.1371/journal.pmed.1001750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandberg K Mechanisms underlying sex differences in progressive renal disease. Gend Med 2008;5:10–23. 10.1016/S1550-8579(08)80004-6. [DOI] [PubMed] [Google Scholar]
- 24.Silbiger S, Lei J, Neugarten J. Estradiol suppresses type I collagen synthesis in mesangial cells via activation of activator protein-1. Kidney Int 1999;55:1268–76. 10.1046/j.1523-1755.1999.00376.x. [DOI] [PubMed] [Google Scholar]
- 25.Neugarten J, Acharya A, Silbiger SR. Effect of gender on the progression of nondiabetic renal disease: a meta-analysis. J Am Soc Nephrol 2000;11:319–29. [DOI] [PubMed] [Google Scholar]
- 26.Turin TC, Tonelli M, Manns BJ, et al. Lifetime Risk of ESRD. J Am Soc Nephrol 2012;23:1569–78. 10.1681/asn.2012020164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lombet JR, Adler SG, Anderson PS, Nast CC, Olsen DR, Glassock RJ. Sex vulnerability in the subtotal nephrectomy model of glomerulosclerosis in the rat. J Lab Clin Med 1989;114:66–74. 10.5555/uri pii:0022214389901601. [DOI] [PubMed] [Google Scholar]
- 28.Tomiyoshi Y, Sakemi T, Aoki S, Miyazono M. Different effects of castration and estrogen administration on glomerular injury in spontaneously hyperglycemic Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Nephron 2002;92:860–7. 10.1159/000065442. [DOI] [PubMed] [Google Scholar]
- 29.Verzola D, Gandolfo MT, Salvatore F, et al. Testosterone promotes apoptotic damage in human renal tubular cells. 65; 2004. doi: 10.1111/j.1523-1755.2004.00497.x [DOI] [PubMed] [Google Scholar]
- 30.Reckelhoff JF, Hennington BS, Glover Moore A, Blanchard EJ, Cameron J. Gender differences in the renal nitric oxide (NO) system. Am J Hypertens 1998;11:97–104. [DOI] [PubMed] [Google Scholar]
- 31.Doublier S, Lupia E, Catanuto P, et al. Testosterone and 17p-estradiol have opposite effects on podocyte apoptosis that precedes glomerulosclerosis in female estrogen receptor knockout mice. 2016;25:289–313. doi: 10.1007/s11065-015-9294-9.Functional [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixon A, Maric C. 17β-Estradiol attenuates diabetic kidney disease by regulating extracellular matrix and transforming growth factor-β protein expression and signaling. Am J Physiol Physiol 2007;293:F1678–90. 10.1152/ajprenal.00079.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brosnihan B, LiP, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS.; 1997. http://ajpregu.physiology.org/. Accessed January 3, 2020. [DOI] [PubMed]
- 34.Komukai K, Mochizuki S, Yoshimura M. Gender and the renin-angiotensin-aldosterone system. Fundam Clin Pharmacol 2010;24:687–98. 10.1111/j.1472-8206.2010.00854.x. [DOI] [PubMed] [Google Scholar]
- 35.Katz FH, Roper EF. Testosterone effect on renin system in rats. Proc Soc Exp Biol Med 1977;155:330–3. 10.3181/00379727-155-39800. [DOI] [PubMed] [Google Scholar]
- 36.Gross ML, Adamczak M, Rabe T, et al. Beneficial effects of estrogens on indices of renal damage in uninephrectomized SHRsp rats. J Am Soc Nephrol 2004;15:348–58. 10.1097/01.ASN.0000105993.63023.D8. [DOI] [PubMed] [Google Scholar]
- 37.Mankhey RW, Bhatti F, Maric C. 17β-Estradiol replacement improves renal function and pathology associated with diabetic nephropathy. Am J Physiol Ren Physiol 2005;288 10.1152/ajprenal.00195.2004. [DOI] [PubMed] [Google Scholar]
- 38.Kwan G, Neugarten J, Sherman M, et al. Effects of sex hormones on mesangial cell proliferation and collagen synthesis. Kidney Int 1996;50:1173–9. 10.1038/ki.1996.425. [DOI] [PubMed] [Google Scholar]
- 39.Guccione M, Silbiger S, Lei J, Neugarten J. Estradiol upregulates mesangial cell MMP-2 activity via the transcription factor AP-2. Am J Physiol Physiol 2002;282:F164–9. 10.1152/ajprenal.0318.2000. [DOI] [PubMed] [Google Scholar]
- 40.Negulescu O, Bognar I, Lei J, Devarajan P, Silbiger S, Neugarten J. Estradiol reverses TGF-β induced mesangial cell apoptosis by a casein kinase 2-dependent mechanism. Kidney Int 2002;62:1989–98. 10.1046/j.1523-1755.2002.00679.x. [DOI] [PubMed] [Google Scholar]
- 41.Pawluczyk IZA, Tan EKC, Harris KPG. Rat mesangial cells exhibit sex-specific profibrotic and proinflammatory phenotypes. Nephrol Dial Transplant 2009;24:1753–8. 10.1093/ndt/gfn714. [DOI] [PubMed] [Google Scholar]
- 42.Kwan G, Neugarten J, Sherman M, et al. Effects of sex hormones on mesangial cell proliferation and collagen synthesis. Kidney Int 1996;50:1173–9. 10.1038/ki.1996.425. [DOI] [PubMed] [Google Scholar]
- 43.Potier M, Elliot SJ, Tack I, et al. Expression and regulation of estrogen receptors in mesangial cells: influence on matrix metalloproteinase-9. J Am Soc Nephrol 2001;12:241–51 http://www.ncbi.nlm.nih.gov/pubmed/11158214. [DOI] [PubMed] [Google Scholar]
- 44.Guy J, Peters MG. Liver disease in women: the influence of gender on epidemiology, natural history, and patient outcomes. Gastroenterol Hepatol 2013;9:633–9. [PMC free article] [PubMed] [Google Scholar]
- 45.Xu JW, Gong J, Chang XM, et al. Estrogen reduces CCL4-induced liver fibrosis in rats. World J Gastroenterol 2002;8 (5):883–7. 10.3748/wjgs.v8.i5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu QH, Li DG, Huang X, Zong CH, Xu QF, Lu HM. Suppressive effects of 17β-estradiol on hepatic fibrosis in CCI4-induced rat model. World J Gastroenterol 2004;10:1315–20. 10.3748/wjg.v10.i9.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yasuda M, Shimizu I, Shiba M, Ito S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999;29:719–27. 10.1002/hep.510290307. [DOI] [PubMed] [Google Scholar]
- 48.Yasuda M, Shimizu I, Shiba M, Ito S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999;29:719–27. 10.1002/hep.510290307. [DOI] [PubMed] [Google Scholar]
- 49.Wang YJ, Wang SS, Bickel M, Guenzler V, Gerl M, Bissell DM. Two novel antifibrotics, HOE 077 and Safironil, modulate stellate cell activation in rat liver injury: differential effects in males and females. Am J Pathol 1998;152:279–87. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9422545. [PMC free article] [PubMed] [Google Scholar]
- 50.Card JW, Zeldin DC. Hormonal influences on lung function and response to environmental agents: lessons from animal models of respiratory disease. Proc Am Thorac Soc 2009;6:588–95. 10.1513/pats.200904-020rm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Redente EF, Jacobsen KM, Solomon JJ, et al. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Cell Mol Physiol 2011;301:L510–8. 10.1152/ajplung.00122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han MK, Murray S, Fell CD, et al. Sex differences in physiological progression of idiopathic pulmonary fibrosis. Eur Respir J 2008;31:1183–8. 10.1183/09031936.00165207. [DOI] [PubMed] [Google Scholar]
- 53.Kalafatis D, Gao J, Pesonen I, Carlson L, Sköld CM, Ferrara G. Gender differences at presentation of idiopathic pulmonary fibrosis in Sweden. BMC Pulm Med 2019;19:222 10.1186/s12890-019-0994-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gharaee-Kermani M, Hatano K, Nozaki Y, Phan SH. Gender-based differences in bleomycin-induced pulmonary fibrosis. Am J Pathol 2005;166:1593–606. 10.1016/S0002-9440(10)62470-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Redente EF, Jacobsen KM, Solomon JJ, et al. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 2011;301:510–8. 10.1152/ajplung.00122.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Voltz JW, Card JW, Carey MA, et al. Male sex hormones exacerbate lung function impairment after bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2008;39:45–52. 10.1165/rcmb.2007-0340OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elliot S, Periera-Simon S, Xia X, et al. MicroRNA let-7 down-regulates ligand-independent estrogen receptor–mediated male-predominant pulmonary fibrosis. Am J Respir Crit Care Med 2019;200:1246–57. 10.1164/rccm.201903-0508OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Skavdahl M, Steenbergen C, Clark J, et al. Estrogen receptor-mediates male-female differences in the development of pressure overload hypertrophy. 2019;27709:469–476. doi: 10.1152/ajpheart.00723.2004. [DOI] [PubMed] [Google Scholar]
- 59.Kessler EL, Rivaud MR, Vos MA, Van Veen TAB. Sex-specific influence on cardiac structural remodeling and therapy in cardiovascular disease. Biol Sex Differ 2019;10:1–11. 10.1186/s13293-019-0223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villari B, Campbell SE, Schneider J, Vassalli G, Chiariello M, Hess OM. Sex-dependent differences in left ventricular function and structure in chronic pressure overload. Eur Heart J 1995;16:1410–9. 10.1093/oxfordjournals.eurheartj.a060749. [DOI] [PubMed] [Google Scholar]
- 61.Petrov G, Regitz-Zagrosek V, Lehmkuhl E, et al. Regression of myocardial hypertrophy after aortic valve replacement: faster in women. Circulation 2010;122(11 SUPPL. 1):23–8. 10.1161/CIRCULATIONAHA.109.927764. [DOI] [PubMed] [Google Scholar]
- 62.Petrov G, Dworatzek E, Schulze TM, et al. Maladaptive remodeling is associated with impaired survival in women but not in men after aortic valve replacement. JACC Cardiovasc Imaging 2014;7:1073–80. 10.1016/j.jcmg.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 63.Li Z, Wang Z, Yin Z, et al. Gender differences in fibrosis remodeling in patients with long-standing persistent atrial fibrillation. Oncotarget 2017;8:53714–29. 10.18632/oncotarget.16342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LEVY D, ANDERSON KM, SAVAGE DD, KANNEL WB, CHRISTIANSEN JC, CASTELLI WP. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors in Thai elderly men and women. J Med Assoc Thail 1988;83:1082–94. [Google Scholar]
- 65.Dannenberg AL, Levy D, Garrison RJ. Impact of age on echocardiographic left ventricular mass in a healthy population (the Framingham study). Am J Cardiol 1989;64:1066–8. 10.1016/0002-9149(89)90816-3. [DOI] [PubMed] [Google Scholar]
- 66.Liao Y, Cooper RS, Mensah GA, McGee DL. Left ventricular hypertrophy has a greater impact on survival in women than in men. Circulation 1995;92:805–10. 10.1161/01.CIR.92.4.805. [DOI] [PubMed] [Google Scholar]
- 67.Lim WK, Wren B, Jepson N, Roy S, Caplan G. Effect of hormone replacement therapy on left ventricular hypertrophy. Am J Cardiol 1999;83:1132–4. 10.1016/S0002-9149(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 68.Pines A, Fisman EZ, Levo Y, et al. Menopause-induced changes in left ventrictilar wall thickness complete membranous obstruction of the inferior vena cava. 1993;72:240–241. [DOI] [PubMed] [Google Scholar]
- 69.Modena MG, Muia N, Aveta P, Molinari R, Rossi R. Effects of transdermal 17β-estradiol on left ventricular anatomy and performance in hypertensive women. Hypertension 1999;34:1041–6. 10.1161/01.HYP.34.5.1041. [DOI] [PubMed] [Google Scholar]
- 70.Scantlebury DC, Borlaug BA. Why are women more likely than men to develop heart failure with preserved ejection fraction. Curr Opin Cardiol 2011;26:562–8. 10.1097/HCO.0b013e32834b7faf. [DOI] [PubMed] [Google Scholar]
- 71.Bhuiyan T, Maurer MS. HFpEF diagnosis. 2011;5:440–449. doi: 10.1007/s12170-011-0184-2.Heart [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J 2011;32:670–9. 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weber KT. Extracellular matrix remodeling in heart failure. Circulation 1997;96:4065–82. 10.1161/01.CIR.96.11.4065. [DOI] [PubMed] [Google Scholar]
- 74.Barasch E, Gottdiener JS, Aurigemma G, et al. Association between elevated fibrosis markers and heart failure in the elderly the cardiovascular health study. Circ Hear Fail 2009;2:303–10. 10.1161/CIRCHEARTFAILURE.108.828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu CY, Liu YC, Wu C, et al. Evaluation of age-related interstitial myocardial fibrosis with cardiac magnetic resonance contrast-enhanced T1 mapping: MESA (Multi-Ethnic Study of Atherosclerosis). J Am Coll Cardiol 2014;62:1280–7. 10.1016/j.jacc.2013.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Redfield MM, Jacobsen SJ, Borlaug BA, Rodeheffer RJ, Kass DA. Age- and gender-related ventricular-vascular stiffening: a community-based study. Circulation 2005;112:2254–62. 10.1161/CIRCULATIONAHA.105.541078. [DOI] [PubMed] [Google Scholar]
- 77.Mann DL, Barger PM, Burkhoff D. myocardial recovery and the failing heart. J Am Coll Cardiol 2012;60:2465–72. 10.1016/j.jacc.2012.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mewton N, Liu CY, Croisille P, Bluemke D, Lima JAC. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol 2011;57:891–903. 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991;83:1849–65. [DOI] [PubMed] [Google Scholar]
- 80.Weber KT, Brilla CG, Janicki JS, Reddy HK, Campbell SE. Myocardial fibrosis: role of ventricular systolic pressure, arterial hypertension, and circulating hormones. Basic Res Cardiol 1991;3(86 Suppl):25–31. 10.1007/978-3-662-30769-4_3. [DOI] [PubMed] [Google Scholar]
- 81.Wong TC, Piehler KM, Kang IA, et al. Myocardial extracellular volume fraction quantified by cardiovascular magnetic resonance is increased in diabetes and associated with mortality and incident heart failure admission. Eur Heart J 2014;35:657–64. 10.1093/eurheartj/eht193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Part II. Causal mechanisms and treatment. Circulation 2002;105:1503–8. 10.1161/hc1202.105290. [DOI] [PubMed] [Google Scholar]
- 83.Pfeffer JM, Pfeffer MA, Fletcher P. Favorable effects of therapy on cardiac performance in spontaneously hypertensive rats. Am J Physiol Hear Circ Physiol 1982;11 10.1152/ajpheart.1982.242.5.h776. [DOI] [PubMed] [Google Scholar]
- 84.Douglas PS, Katz SE, Weinberg EO, Chen MH, Bishop SP, Lorell BH. Hypertrophic remodeling: gender differences in the early response to left ventricular pressure overload. J Am Coll Cardiol 1998;32:1118–25. 10.1016/S0735-1097(98)00347-7. [DOI] [PubMed] [Google Scholar]
- 85.Weinberg EO, Thienelt CD, Katz SE, et al. Gender differences in molecular remodeling in pressure overload hypertrophy. J Am Coll Cardiol 1999;34:264–73. 10.1016/S0735-1097(99)00165-5. [DOI] [PubMed] [Google Scholar]
- 86.Fliegner D, Schubert C, Penkalla A, et al. Female sex and estrogen receptor- attenuate cardiac remodeling and apoptosis in pressure overload. 2019;1597–1606. doi: 10.1152/ajpregu.00825.2009. [DOI] [PubMed] [Google Scholar]
- 87.Gardner JD, Brower GL, Janicki JS. Gender differences in cardiac remodeling secondary to chronic volume overload. J Card Fail 2002;8:101–7. 10.1054/jcaf.2002.32195. [DOI] [PubMed] [Google Scholar]
- 88.Berul CI, Christe ME, Aronovitz MJ, Seidman CE, Seidman JG, Mendelsohn ME. Electrophysiological abnormalities and arrhythmias in alpha MHC mutant familial hypertrophic cardiomyopathy mice. 1997;99:570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cavasin MA, Tao Z, Menon S, Yang XP. Gender differences in cardiac function during early remodeling after acute myocardial infarction in mice. Life Sci 2004;75:2181–92. 10.1016/j.lfs.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 90.Wu JC, Nasseri BA, Bloch KD, Picard MH, Scherrer-Crosbie M. Influence of Sex on Ventricular Remodeling after Myocardial Infarction in Mice. J Am Soc Echocardiogr 2003;16:1158–62. 10.1067/S0894-7317(03)00648-5. [DOI] [PubMed] [Google Scholar]
- 91.Van Eickels M, Grohé C, Cleutjens JPM, Janssen BJ, Wellens HJJ, Doevendans PA. 17B-Estradiol Attenuates the Development of Pressure-Overload Hypertrophy. Circulation 2001;104:1419–23. 10.1161/hc3601.095577. [DOI] [PubMed] [Google Scholar]
- 92.Jazbutyte V, Arias-Loza PA, Hu K, et al. Ligand-dependent activation of ERβ lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovasc Res 2008;77:774–81. 10.1093/cvr/cvm081. [DOI] [PubMed] [Google Scholar]
- 93.Patten RD, Pourati I, Aronovitz MJ, et al. 17 Beta-estradiol differentially affects left ventricular and cardiomyocyte hypertrophy following myocardial infarction and pressure overload. J Card Fail 2008;14:245–53. 10.1016/j.cardfail.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Valero-Muñoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a “fishing expedition. JACC Basic to Transl Sci 2017;2:770–89. 10.1016/j.jacbts.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pacher P, Mabley JG, Liaudet L, et al. Left ventricular pressure-volume relationship in a rat model of advanced aging-associated heart failure. Am J Physiol Heart Circ Physiol 2004;287:H2132–7. 10.1152/ajpheart.00405.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pacher P, Nagayama T, Mukhopadhyay P, Bátkai S, Kass DA. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat Protoc 2008;3:1422–34. 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boluyt MO. Echocardiographic assessment of age-associated changes in systolic and diastolic function of the female F344 rat heart. J Appl Physiol 2004;96:822–8. 10.1152/japplphysiol.01026.2003. [DOI] [PubMed] [Google Scholar]
- 98.Forman DE, Cittadini A, Azhar G, Douglas PS, Wei JY. Cardiac morphology and function in senescent rats: gender-related differences. J Am Coll Cardiol 1997;30:1872–7. 10.1016/S0735-1097(97)00411-7. [DOI] [PubMed] [Google Scholar]
- 99.Fannin J, Rice KM, Thulluri S, et al. Age-associated alterations of cardiac structure and function in the female F344xBN rat heart. Age (Omaha) 2014;36 10.1007/s11357-014-9684-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hacker TA. Age-related changes in cardiac structure and function in Fischer 344 x Brown Norway hybrid rats. AJP Hear Circ Physiol 2005;290:H304–11. 10.1152/ajpheart.00290.2005. [DOI] [PubMed] [Google Scholar]
- 101.Bustamante M, Garate-Carrillo A, Ito B R, et al. Unmasking of oestrogen-dependent changes in left ventricular structure and function in aged female rats: a potential model for pre-heart failure with preserved ejection fraction. J Physiol 2019;597:1805–17. 10.1113/JP277479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao Z, Wang H, Jessup JA, Lindsey SH, Chappell MC, Groban L. Role of estrogen in diastolic dysfunction. AJP Hear Circ Physiol 2014;306:H628–40. 10.1152/ajpheart.00859.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Knowlton AA, Lee AR. Estrogen and the cardiovascular system. Pharmacol Ther 2012;135:54–70. 10.1016/j.pharmthera.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bhuiyan MS, Shioda N, Fukunaga K. Ovariectomy augments pressure overload-induced hypertrophy associated with changes in Akt and nitric oxide synthase signaling pathways in female rats. Am J Physiol Endocrinol Metab 2007;293:E1606–14. 10.1152/ajpendo.00246.2007. [DOI] [PubMed] [Google Scholar]
- 105.Stice JP, Chen L, Kim SC, et al. 17B-Estradiol, aging, inflammation, and the stress response in the female heart. Endocrinology 2011;152:1589–98. 10.1210/en.2010-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alencar AK, da Silva JS, Lin M, et al. Effect of age, estrogen status, and late-life GPER activation on cardiac structure and function in the Fischer344×Brown Norway female rat. J Gerontol A Biol Sci Med Sci 2017;72:152–62. 10.1093/gerona/glw045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pedram A, Razandi M, O’Mahony F, Lubahn D, Levin ER. Estrogen receptor-β prevents cardiac fibrosis. Mol Endocrinol 2010;24:2152–65. 10.1210/me.2010-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mori T, Kai H, Kajimoto H, et al. Enhanced cardiac inflammation and fibrosis in ovariectomized hypertensive rats: a possible mechanism of diastolic dysfunction in postmenopausal women. Hypertens Res 2011;34:496–502. 10.1038/hr.2010.261. [DOI] [PubMed] [Google Scholar]
- 109.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 2001;280:53–60. 10.1152/ajpcell.2001.280.Lc53. [DOI] [PubMed] [Google Scholar]
- 110.Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol - Hear Circ Physiol 2010;298:16–23. 10.1152/ajpheart.00588.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang H, Jessup JA, Lin MS, Chagas C, Lindsey SH, Groban L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc Res 2012;94:96–104. 10.1093/cvr/cvs090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Delbeck M, Golz S, Vonk R, et al. Impaired left-ventricular cardiac function in male GPR30-deficient mice. Mol Med Rep 2010;4:37–40. 10.3892/mmr.2010.402. [DOI] [PubMed] [Google Scholar]
- 113.Wang H, Sun X, Chou J, et al. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: a sex-specific gene profiling analysis. Biochim Biophys Acta Mol Basis Dis 2017;1863:1870–82. 10.1016/j.bbadis.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee H, Eghbali-webb M.Estrogen enhances proliferative capacity of Cardiac fibroblasts by estrogen receptor- and mitogen-activated protein kinase-dependent pathways. 1998;1368:1359–1368. [DOI] [PubMed] [Google Scholar]
- 115.Zhou L, Shao Y, Huang Y, Yao T, Lu LM. 17β-Estradiol inhibits angiotensin II-induced collagen synthesis of cultured rat cardiac fibroblasts via modulating angiotensin II receptors. Eur J Pharmacol 2007;567:186–92. 10.1016/j.ejphar.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 116.Dworatzek E, Mahmoodzadeh S, Schriever C, et al. Sex-specific regulation of collagen i and III expression by 17β-Estradiol in cardiac fibroblasts: Role of estrogen receptors. Cardiovasc Res 2019;115:315–27. 10.1093/cvr/cvy185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Griffin M, Lee HW, Zhao L, Eghblai-Webb M. Gender-related differences in proliferative response of cardiac fibroblasts to hypoxia: effects of estrogen. Mol Cell Biochem 2000;215:21–30. 10.1023/A:1026585420021. [DOI] [PubMed] [Google Scholar]