

Abstract
Chromatin in higher eukaryotic nuclei is extensively bound by various RNA species. We recently developed a method for in situ capture of global RNA interactions with DNA by deep sequencing (GRID-seq) of fixed permeabilized nuclei that allows identification of the entire repertoire of chromatin-associated RNAs in an unbiased manner. The experimental design of GRID-seq is related to those of two recently published strategies (MARGI (mapping RNA–genome interactions) and ChAR-seq (chromatin-associated RNA sequencing)), which also use a bivalent linker to ligate RNA and DNA in proximity. Importantly, however, GRID-seq also implements a combined experimental and computational approach to control nonspecific RNA–DNA interactions that are likely to occur during library construction, which is critical for accurate interpretation of detected RNA–DNA interactions. GRID-seq typically finds both coding and non-coding RNAs (ncRNAs) that interact with tissue-specific promoters and enhancers, especially super-enhancers, from which a global promoter–enhancer connectivity map can be deduced. Here, we provide a detailed protocol for GRID-seq that includes nuclei preparation, chromatin fragmentation, RNA and DNA in situ ligation with a bivalent linker, PCR amplification and high-throughput sequencing. To further enhance the utility of GRID-seq, we include a pipeline for data analysis, called GridTools, into which key steps such as background correction and inference of genomic element proximity are integrated. For researchers experienced in molecular biology with minimal bioinformatics skills, the protocol typically takes 4–5 d from cell fixation to library construction and 2–3 d for data processing.
Introduction
The recent decade of functional genomics research has revealed that eukaryotic genomes are more active in transcription than previously thought1. Besides protein-coding mRNAs, a large repertoire of ncRNAs is now known to be transcribed pervasively in the genome2,3. Increasing evidence suggests that RNAs function in different layers of gene expression regulation and, in many cases, through actively engaging with specific chromatin regions2,4. Several techniques, such as ChIRP5, CHART6 and RAP-DNA7, that focus on a single RNA species have been developed to profile RNA–chromatin interactions. Given the broad participation of regulatory RNAs in chromatin with different RNAs preferentially acting in cis or trans, it has become desirable to develop technologies for comprehensive definition of the global RNA–chromatin interaction landscape in eukaryotic genomes. Such technology would enable the identification of potential regulatory RNAs and assignment of their action sites in an unbiased manner.
We have recently developed a genome-wide approach for in situ GRID-seq, which provides a strategy for de novo identification of the full spectrum of chromatin-interacting RNAs8. This technology effectively detects the association of all RNA species of sufficient abundance with chromatin and assigns their specific chromatin contacts. Other groups have independently developed related technologies around the same period of time9,10. To construct a library for deep sequencing, these methods all use a ‘bivalent’ linker, which is designed to carry a bipartite sequence with a single-stranded RNA (ssRNA) or DNA (ssDNA) sequence on one side for ligation to RNA and a double-stranded DNA (dsDNA) on the other side for ligation to restriction-digested genomic DNA (gDNA). GRID-seq and the associated bioinformatics pipeline have additional specific built-in features, such as a background model to evaluate nonspecific interactions and a proximity model to infer the special relationships of distant regulatory DNA elements, such as enhancers, to specific gene promoters.
In this article, we systemically describe both experimental and bioinformatics protocols for GRID-seq in detail, and emphasize the great care required for evaluating data quality before deriving conclusions about functional connections or biological significance of specific RNA–DNA interactions. The GRID-seq method can be broadly applied to most nucleated cell types under diverse cellular environments or disease conditions, and the data will substantially increase our understanding of the immensely complex nature of gene regulation in the context of chromatin architecture.
Overview of the GRID-seq procedure
The experimental steps in GRID-seq library construction are illustrated in Fig. 1a. In essence, the technology uses a biotinylated bivalent linker (Fig. 1b) to ligate RNA and DNA in situ on formaldehyde and disuccinimidyl glutarate (DSG) double-fixed nuclei. The use of such a biotinylated linker enables affinity purification of ligated products for downstream steps. We first perform RNA ligation and then convert ligated RNA into cDNA, using one of the DNA strands in the linker as primer. We then ligate the linker to nearby gDNA ends that are digested by the frequent cutter AluI. After removing proteins, we purify linker-ligated products by biotin selection. To eliminate single-end-ligated products (i.e., linker–gDNA or linker–cDNA, see Fig. 2a), we digest the products with MmeI to generate paired-end tags and purify size-defined products for library construction. Next, Y-shaped adaptors are ligated to the paired-end tags for subsequent PCR amplification, generating the final DNA library for high-throughput sequencing (Fig. 2b,c). Typically, GRID-seq library construction takes 4–5 d, and we emphasize five quality-control checkpoints (QC1–5) during the process. QC1 and QC2 are optional and are described in Boxes 1 and 2, respectively. The expected results of the other three checkpoints are shown in Fig. 2a–c.
Fig. 1 |. Schematics of the GRID-seq experiment.
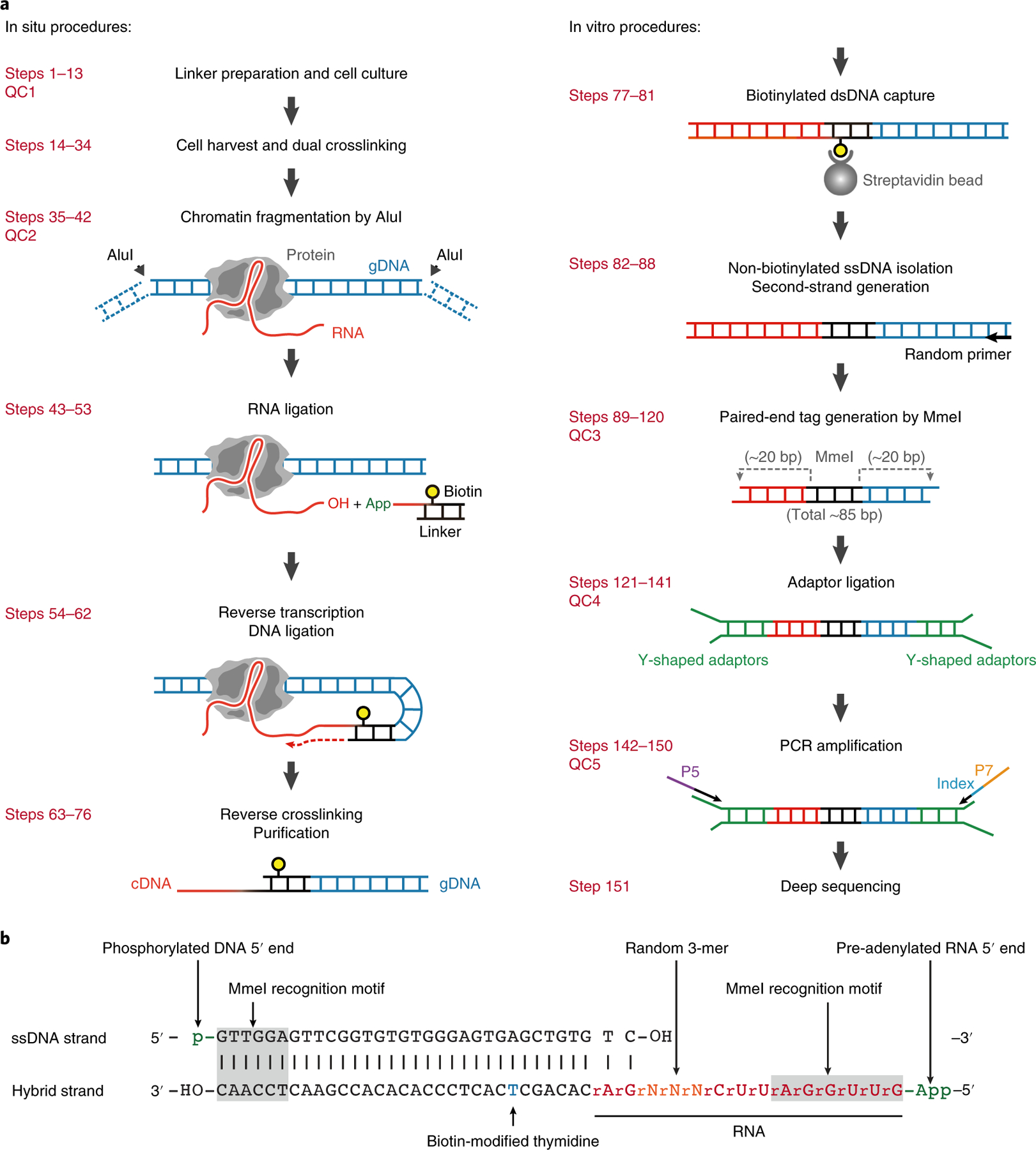
a, Left, GRID-seq experimental steps performed in situ on fixed nuclei. A bivalent linker is used to ligate chromatin-associated RNA (with the ssRNA stretch on the linker) and fragmented genomic DNA crosslinked in proximity. Right, steps performed in solution. b, The structure of the bivalent GRID-seq Linker. The top strand is a 5′-phosphorylated DNA sequence (black) and the bottom strand is a hybrid consisting of both DNA and RNA bases (red) with a biotin-modified thymidine (blue). Both ends of the linker carry an MmeI restriction site (gray-shaded). App, pre-adenylation.
Fig. 2 |. Exemplary gel images of library products.
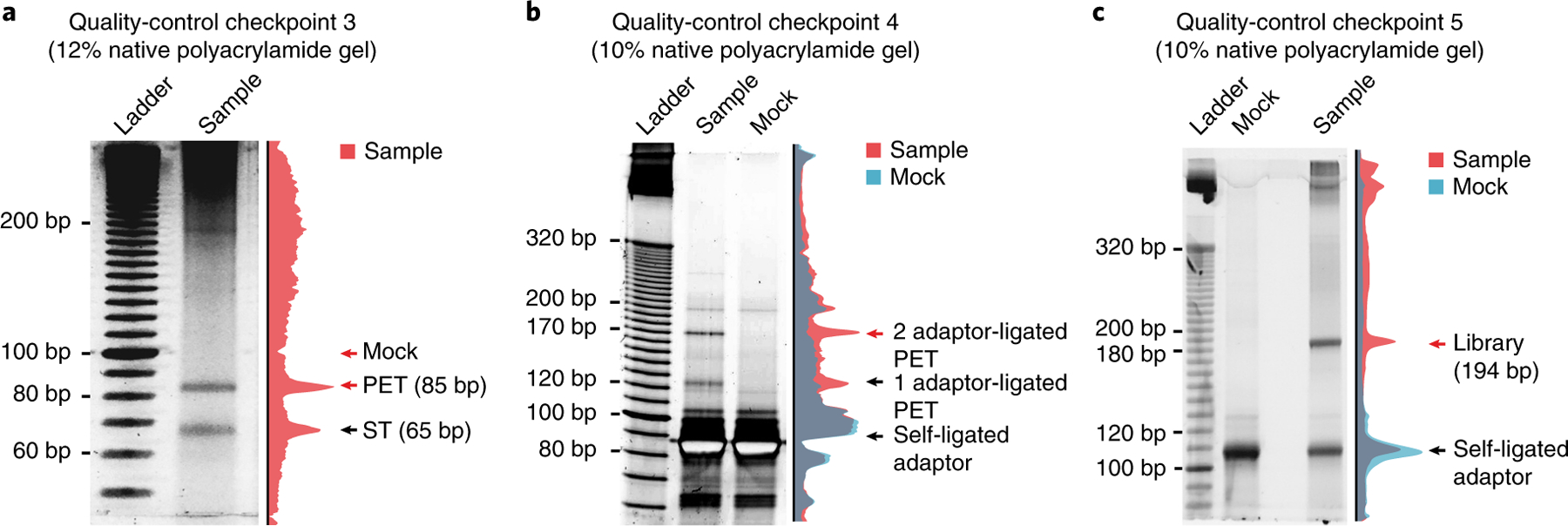
a, Polyacrylamide gel image of library products after paired-end tagging by MmeI. Red arrows indicate the locations where gel slices are cut and DNA is recovered. Although lacking discernable bands, the slice that was cut at ~100 bp for the mock sample (see Step 114 for details) contains adequate background DNA to serve as a control sample for downstream steps. Image adapted with permission from Li et al.8, Springer Nature. b, Polyacrylamide gel image of library products after adaptor ligation. The two distinct bands in the sample lane are adaptor-ligated products. The red arrow indicates the band containing the product ligated at both ends that needs to be recovered for downstream steps in library construction. c, Polyacrylamide gel image of library products after PCR amplification. The final library products, indicated by the red arrow, all have a precise size of 194 bp. The sample lanes of the gel images in a, b and c are quantified, and the normalized intensity is illustrated on the right side of each gel. PET, paired-end tag that denotes a linker simultaneously ligated with RNA and DNA on each side; ST, singleton tag that denotes one-side-ligated linker.
Box 1 |. Quality-control checkpoint 1 for linker integrity ● Timing 3 h.
Procedure
-
1
Take 4 pmol of the annealed linkers and dilute them with nuclease-free water to make a 20-µl solution. Split the sample into two parallel tubes.
-
2
Mix one linker sample with 1 µg of RNase A and incubate for 5 min at room temperature.
-
3
Add 2 µl each of the 6× loading buffer to the two samples and load them into an 18% (wt/vol) native polyacrylamide gel. Load 2 μl of 10-bp DNA ladder in parallel.
-
4
Perform gel electrophoresis in 1× TBE buffer at 100 V for 15 min and then at 120 V for ~1.5 h.
▲ CRITICAL STEP The 6× loading buffer contains purple dye and blue dye as size indicators. End the electrophoresis when the purple dye reaches the end of the gel.
-
5
Place the gel in 30 ml of 1× TBE buffer containing 1 μl of SYBR Gold, and stain for 10 min on a shaker in a dark room.
-
6
Visually check the gel with a Dark Reader transilluminator. A typical result is shown in the gel photo below.
▲ CRITICAL STEP The control sample containing the linker without pre-adenylation should be set up in a manner similar to that used for the pre-adenylated sample described in Steps 1–10, but omitting the addition of the Mth RNA ligase. We recommend including this control only when testing a newly purchased batch of linker.
▲ CRITICAL STEP If the untreated linker migrates to a position similar to that of the RNase-digested linker, it indicates that the RNA stretch of the linker may be degraded and thus the linker should not be used.
? TROUBLESHOOTING
Results
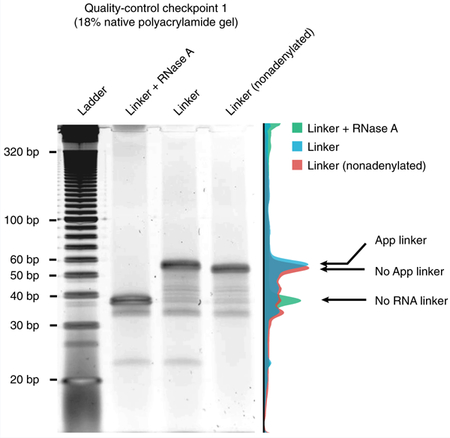
The figure shows the adenylated linker on a representative image of an 18% native polyacrylamide gel image. The first lane contains RNase A-digested linker; the second lane contains linker molecules that were successfully pre-adenylated (App linker); the third lane contains linker without pre-adenylation (No App linker). The three sample lanes of the gel image are quantified, and the normalized intensity is illustrated on the right side. Note the subtle size shift as a result of the adenylation.
Box 2 |. Quality-control checkpoint 2 for AluI digestion efficiency ● Timing 3 h.
Procedure
-
1
Take 20 µl of the AluI-digested nuclei suspension and dilute it with 120 µl of nuclease-free water.
-
2
Add 150 µl of 2× proteinase K buffer and 10 µl of proteinase K.
-
3
Incubate the reaction for 1 h at 65 °C in a thermal mixer C with intermittent mixing at 800 r.p.m.
-
4
Transfer all the solution to a MaXtract high-density tube and mix with 300 μl of phenol/chloroform/isoamyl alcohol by shaking.
! CAUTION Phenol/chloroform/isoamyl alcohol is highly toxic. Always handle it in a fume hood.
-
5
Centrifuge the tube for 5 min at 16,000g at 4 °C.
-
6
Transfer the supernatant to a new 1.5-ml tube. Add 2 μl of GlycoBlue and 30 μl of 3 M NaOAc, and mix well by pipetting.
-
7
Add 900 μl of 100% (vol/vol) ethanol and vortex thoroughly.
-
8
Allow the solution to precipitate for at least 30 min at −80 °C.
-
9
Centrifuge the solution for 30 min at 16,000g at 4 °C. Remove and discard the supernatant and air-dry the pellet for 3 min in a SpeedVac dryer.
-
10
Dissolve the pellet in 20 μl of nuclease-free distilled water.
-
11
Add 1 µg of RNase A and incubate for 5 min at room temperature.
-
12
Add 2 µl of the 6× loading buffer to the sample and load it on a 1.5% (wt/vol) agarose gel. Load 2 μl of 100-bp DNA ladder in parallel.
-
13
Perform gel electrophoresis in 1× TBE buffer at 100 V for 20 min.
▲ CRITICAL STEP The 6× loading buffer contains purple dye and blue dye as size indicators. End the electrophoresis when the purple dye reaches the end of the gel.
-
14
Stain the gel with SYBR Gold for 10 min on a shaker in a dark room.
-
15
Visually check the gel with a Dark Reader transilluminator. A typical result is shown in the gel photo below.
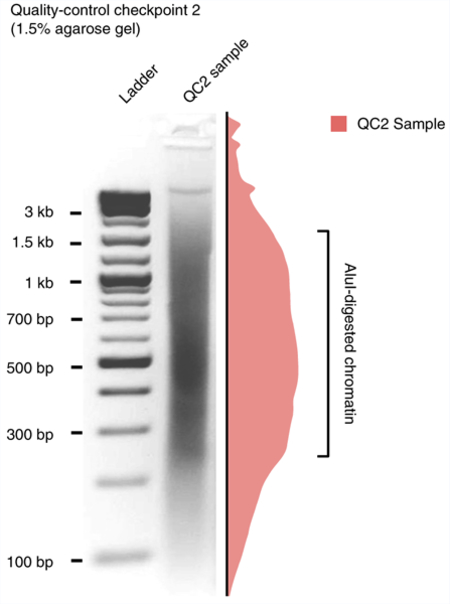
The figure shows an AluI-digested genome run on a 1.5% (wt/vol) agarose gel. The sample lane of the gel image is quantified, and the normalized intensity is illustrated on the right side. The highlighted portion of the smear that harbors the most abundant DNA fragments should ideally be within 200–1,500 bp.
? TROUBLESHOOTING
It generally takes 1 d to produce the sequencing reads from a GRID-seq library using the single-end 100-cycle kit on an Illumina HiSeq-2500 sequencer. Sequencing 100 bp across the linker (45 bp) ensures correct identification of RNA and DNA mate reads (~20 bp each) on the basis of the linker orientation. The GRID-seq reads are then processed using a customized pipeline diagramed in Fig. 3. First, raw reads are trimmed and scanned for the linker sequence. Reads are then split into RNA and DNA mate-pair reads, which are independently mapped to the reference genome. Only those with uniquely aligned loci are kept for downstream analysis in our current analysis. Next, chromatin-associated RNAs are identified and a collection of ‘trans-acting signals’ (defined by mRNAs that interact with all chromosomes except the chromosomes from which they are transcribed) is utilized to infer the nonspecific background. Linked DNA reads of each chromatin-associated RNA are then normalized, producing a background-corrected interactome matrix to be used for functional analysis to address specific biological questions.
Fig. 3 |. Flowchart of the GRID-seq data-processing pipeline.
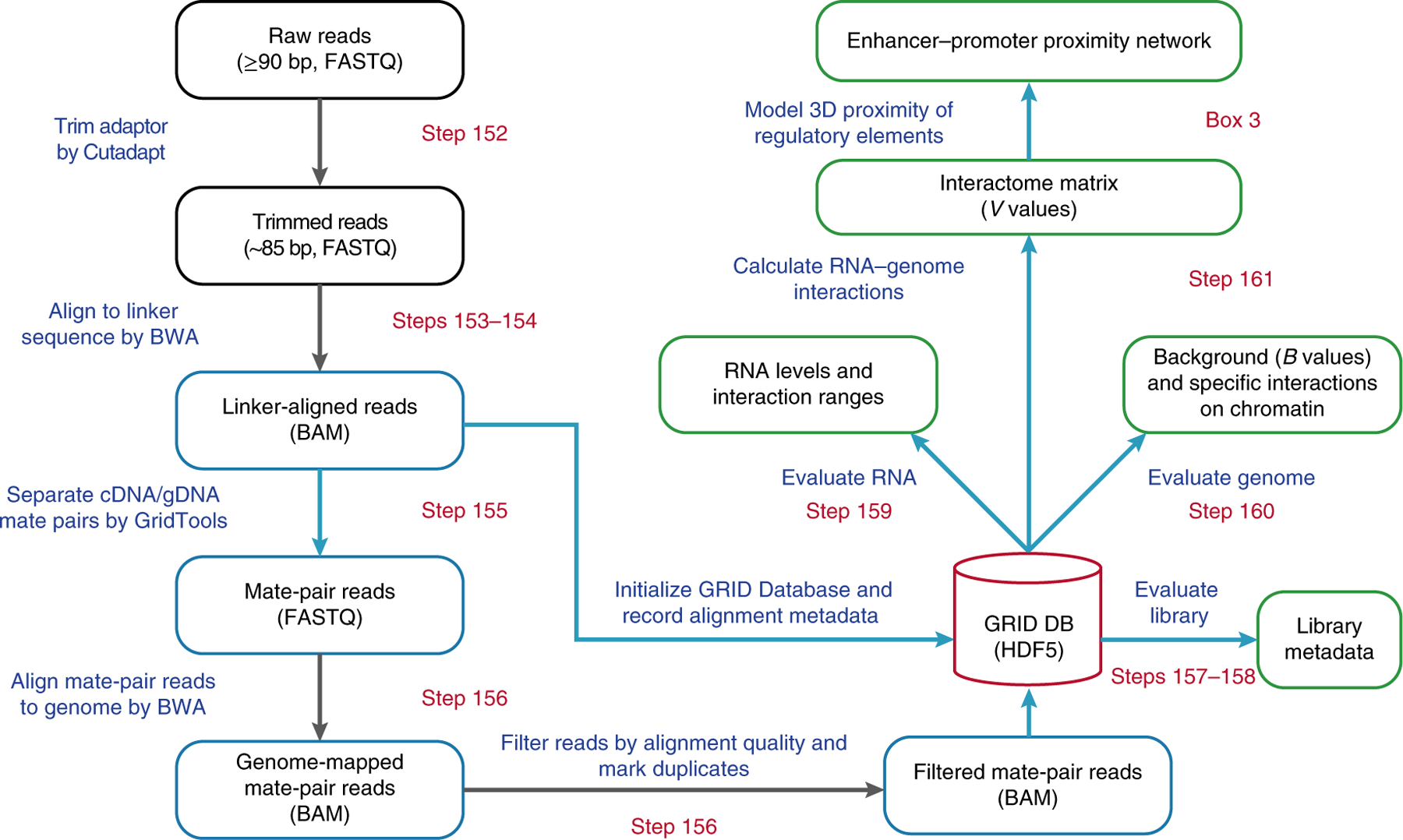
Stages of the pipeline are outlined, and key steps are shown in the boxes. Starting from the raw sequencing reads, the adaptor sequences are trimmed off. The resulting sequence length is denoted in the parentheses. The position of the linker is identified by BWA alignment, and the flanking sequences are split into mate pairs of RNA and DNA reads, based on the orientation of the linker. The mate-pair reads are next aligned to the reference genome. Only uniquely mapped read-pairs are stored in the GRID Database, which also stores various metadata generated during the pipeline as a hub for further analyses (green boxes). By evaluating the captured RNAs and genomic loci, a genome-wide interactome is calculated as a matrix, reporting specific interaction signals of all chromatin-associated RNAs on the binned genome. Steps processed by the in-house software, GridTools, are labeled with blue boxes and arrows. File formats are denoted in parentheses for certain steps.
Advantages of GRID-seq
A related technology, MARGI, has been designed to detect genome-wide RNA–chromatin interactomes9. MARGI first uses a bivalent linker to ligate RNA and DNA, and after reverse transcription to convert the RNA portion to cDNA, followed by biotin selection, released DNA is circularized for library construction. As products ligated to either DNA or RNA could also be circularized, a fraction of MARGI reads might not be genuine RNA–DNA mates. One strategy to identify and filter out such noise, as we have implemented in the GRID-seq data analysis pipeline, is to ensure that RNA reads can be mapped only to the transcribed strand in the genic regions while not requiring DNA reads to have such strand specificity.
ChAR-seq is based on the same design principle as GRID-seq, thus resulting in similar RNA–DNA contacts detected in Drosophila cells10. The most significant difference between GRID-seq and ChAR-seq lies in the strategy for defining ‘nonspecific’ interactions. Theoretically, once a transcribed RNA is released from its site of transcription, it may act like a ‘trans-acting’ RNA to target other specific chromatin regions or explore all accessible chromatin regions in a nonspecific manner. Technically, fragmented free RNAs and DNAs produced during experimental procedures can also come into contact with open chromatin regions nonspecifically. Collectively, signals from these nonspecific contacts can obscure the interpretation of biologically meaningful interactions. ChAR-seq addressed this issue by spiking a reference RNA into the fixed and permeabilized nuclei before RNA ligation to evaluate the false-positive rate of RNA–DNA contacts, which was estimated to be ~0.5%10. Although this approach provides a practical estimate of nonspecific ligation to fragmented RNAs during library construction, the information, however, has not yet been used to develop a statistical model for defining specific RNA–DNA interactions in a locus-specific manner across the genome. By contrast, GRID-seq has implemented a combined experimental and computational approach to define background RNA–DNA interactions8. Our experimental strategy is to construct a library on mixed cells from different species, such as mammalian and Drosophila cells, thus allowing the assessment of cross-species RNA–DNA interactions during library construction. This experimentally derived background is next compared with a computationally derived background based on collective DNA contacts by all expressed mRNAs released from their transcribing chromosomes. After normalizing to equal counts, we found that the experimental background matches the computationally derived background, indicating that such nonspecific contacts overwhelmed specific contacts by genuine trans-acting RNAs. Importantly, this observation enables the use of endogenous transcripts to develop a statistical model for inferring specific RNA–DNA interactions across the genome.
Limitations of GRID-seq
A key limitation of GRID-seq lies in its nature in detecting all-to-all RNA–DNA interactions, which requires rather deep sequencing to generate a robust contact map. For example, we typically generate ~150 million–200 million raw reads for each biological replicate GRID-seq library (Fig. 4a). Unlike DNA–DNA interactions detected by Hi-C (high-throughput chromatin conformation capture), the GRID-seq signals are in general proportional to RNA abundance in the cell. Thus, many RNAs of low abundance may escape detection. More than 50% of detected chromatin-associated RNA species are of 104 to 106 reads from a typical GRID-seq library. Such density is considerably sparser compared to those generated by single-capture methods such as ChIRP or CHART, which are normally in the range of 106–108 reads, as demonstrated earlier5,6. Thus, while GRID-seq offers a strategy for comprehensive analysis of RNA–DNA interactions, it is effective only for RNAs with sufficient expression levels. Technically, moderately increasing sequencing depth alone may not be sufficient to detect low-abundant RNAs because most of the increased sequencing depth would be largely consumed by these highly abundant RNAs according to our saturation analysis (Supplementary Fig. 1a, b). In addition, the resulting RNA–DNA contact map does not provide any information on the specific proteins involved. One further strategy to address such experimental needs is to couple GRID-seq (or related technologies) with a capture method for specific RNAs or proteins of interest, analogous to the recently developed HiChIP11 and PLAC-seq12.
Fig. 4 |. GRID-seq data quality and features.
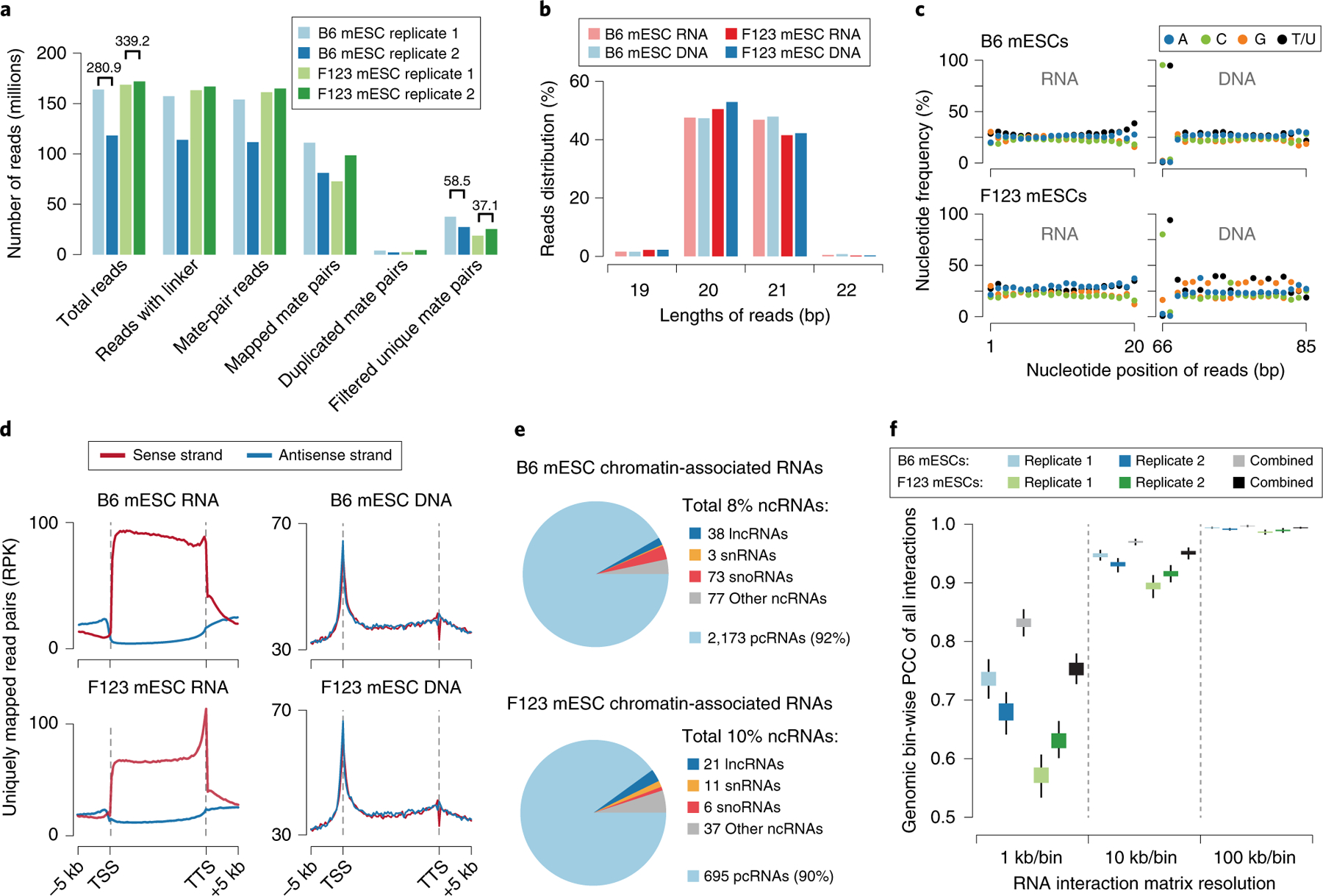
a, Summary of sequenced GRID-seq libraries constructed on two mouse embryonic stem cell (mESC) lines (B6 and F123). Shown are numbers of reads and read mate pairs remaining after individual data-processing steps. The filtered unique mate pairs are used for interaction density calculation. The numbers above the bars represent the replicate-combined number of reads. b, Summary of the RNA/DNA read length of each mESC library. c, Nucleotide frequency of RNA (left) and DNA (right) reads. Note the specific dinucleotide as part of the AluI recognition site at the end of DNA reads, but the lack of nucleotide bias in any position of RNA reads. d, Metagene profiles showing RNA or DNA read coverage on regions from 5 kb upstream of the transcription start site (TSS) to 5 kb downstream of the transcription termination site (TTS) of all annotated genes in the mouse genome. Strand-specific alignment of uniquely mapped RNA (left) and DNA (right) reads relative to gene annotation. Note that mapped RNA reads have the same strand specificity as their transcripts, but this is not the case for DNA. e, Types of chromatin-associated RNAs identified in the two libraries. The summarized percentages of all non-coding RNAs (ncRNAs) are shown in the legend. Among these, the numbers of identified species of long non-coding RNAs (lncRNAs), small nuclear RNAs (snRNAs), small nucleolus RNAs (snoRNAs), and other types of ncRNAs are shown to the side of their respective categories. The percentage and number of identified protein-coding RNAs (pcRNAs) are also shown at the bottom. f, Pearson’s correlation coefficient (PCC) of GRID-seq interaction density of each chromosome at decreasing resolution (increasing bin size) across the mouse genome in the two mESC libraries. Boxes range from the lower quartile to the upper quartile of the data, and the whiskers extend to a 1.5× interquartile range beyond the boxes. RPK, reads per kilobase.
The current version of GRID-seq relies on restriction digestion with a type IIS restriction enzyme (MmeI). The advantage of this approach is to ensure that the linker is ligated to both DNA and RNA, which yields products of a defined size (45-bp linker and 20–21-nt RNA and DNA at each end). Such relatively small sequence length, however, prevents efficient mapping of certain reads back to the reference genome, especially in repeat-rich regions. Typically, ~80% of RNA sequences could be uniquely mapped to the genome, whereas a slightly lower mapping rate of ~70% was observed with mated DNA sequences. By requiring that mated reads contain uniquely mapped sequences on both the RNA and DNA sides, we found that only 20% of raw reads meet such a requirement. In comparison, although such limitation is less severe with MARGI and ChAR-seq, as these technologies use randomly fragmented DNA for library construction, the trade-off is that a fraction of reads may contain only ligated DNA or RNA, and as a result, a similar percentage (~20%) of total ChAR-seq reads could be uniquely aligned to the reference Drosophila genome10. It is important to point out that many mated reads with multiple maps from either the RNA or DNA side may reflect biologically meaningful RNA–DNA interactions, which is a subject for future studies. In any case, future users of GRID-seq can adapt such random fragmentation strategy for library construction to increase the mapping power for mated RNA and DNA sequences. In adapting such a strategy, we suggest that it is still useful to perform MmeI digestion to evaluate the suitability of a library for costly deep sequencing because MmeI digestion produces a diagnostic 85-bp band if the linker is ligated to both RNA and DNA and a ~65-bp band if the linker is ligated to either RNA or DNA, which allows evaluation of a library that contains a significant fraction of desired products. If any of the steps in library construction fails, no band will be seen.
Applications of GRID-seq
We have demonstrated that GRID-seq is able to detect different classes of RNAs on chromatin8. Most RNAs interact with chromatin near their sites of transcription, but we also detect many trans-acting RNAs on remote chromosomal locations or on different chromosomes. Interestingly, our data from different cell lines exhibit considerable tissue-specificity in both identified RNA classes and their chromatin binding sites. We found that nascent RNAs frequently interact with specific promoters and enhancers, leading to an important concept of RNA ‘clouds’ on transcription hubs. We are able to infer global promoter–enhancer connectivity from interactions of nascent RNAs with promoters and enhancers. Strikingly, we found that RNA can link promoters to enhancers that are several mega-bases away in linear distance, and each transcription hub anchors on average four promoters, a single super-enhancer (a genomic region 10–40 kb in size that contains multiple active enhancers, as defined earlier13) and multiple typical enhancers. These findings showcase the power of the GRID-seq technology in studying functional genomic interactions in the 3D genome.
Before the development of GRID-seq and related technologies, Hi-C was a main approach for studying 3D genome organization. Although powerful in describing proximal chromatin structures, the majority of Hi-C-detected DNA–DNA contacts are invariant among different cell types14,15, suggesting its limitation in studying cell-type-specific gene regulation in the context of chromatin structures. By contrast, GRID-seq signals are mostly cell type specific because they are derived from expressed RNAs, which in most cases are not confined within topologically associated domains (TADs) defined by Hi-C8. Therefore, in addition to direct applications in identifying specific regulatory RNAs on chromatin to pursue their functions on an individual basis, GRID-seq provides a complementary approach to Hi-C in studying chromatin architecture tightly associated with specific regulated gene expression events.
Experimental design
Cell culture
GRID-seq experiments were performed in two strains of mouse embryonic stem (ES) cells that were cultured under different conditions, which are presented as examples in the current study to illustrate the experimental and bioinformatical procedures of GRID-seq. C57BL/6 mouse ES cells (B6 mESCs) were cultured in so-called ‘2i/LIF’ (two kinase inhibitor–leukemia inhibitory factor) medium and the F1 Mus musculus castaneus × S129/SvJae mouse ES cells (F123 mESCs) were grown in LIF medium without 2i (see ‘Reagents’ for media composition). F123 mESCs were routinely maintained on 0.1% gelatin-coated plates with mitomycin C–treated mouse embryonic fibroblasts. Cells were passaged twice on 0.1% gelatin-coated feeder-free plates before harvesting after trypsin treatment. Both cell lines were subsequently passaged upon reaching 80% confluency. The origins, authentication and mycoplasma-testing methods of the cell lines used in the current study are listed in the Life Sciences Reporting Summary. Biological replicates of independently cultured cells were set up for each cell line to ensure robustness and reproducibility in generating GRID-seq libraries.
Linker design
The structure of the linker is critical to achieving efficient in situ ligation and thus key to successful GRID-seq library construction. One strand of the linker is a 5′-phosphorylated DNA sequence, and the other strand is a hybrid consisting of both DNA and RNA bases with a biotin-modified thymidine in the middle. Both ends of the linker carry an MmeI restriction site for the restriction enzyme to cut ligated DNA fragments 19–23 bp distal to the linker, a procedure called paired-end tagging. The RNA end is phosphorylated by synthesis and pre-adenylated during the linker preparation step. The pre-adenylated base permits ligation in the absence of ATP, which prevents ligation among endogenous RNAs. A 3-mer randomized RNA base serves as a unique identifier for identifying PCR duplicates in the library (Fig. 1b, Table 1). We experimented with many different versions of linker design before deciding on the final linker configuration, which has a 12-nt RNA overhang and a 33-bp DNA backbone. Other linkers we tested include a linker with an 8-nt RNA overhang that showed inefficient RNA ligation, a linker with a shorter (18 bp) backbone that ligated efficiently but resulted low yield at the MmeI-digestion step, and a longer linker with a 41-bp backbone length that showed decreased in situ ligation. This gives the impression that linker length is critical to efficient ligation to RNA and DNA, which clearly indicates room for further improvement in linker design in future studies.
Table 1 |.
Required oligos in GRID-seq
| Oligo | Sequence (5′–3′) | Modification |
|---|---|---|
| L1 | /5Phos/rGrUrUrGrGrArUrUrCrNrNrNrGrACACAGC/iBiodT/CACTCCACACACCGAACTCCAAC | 5Phos: 5′ phosphorylation; rN: random ribonucleotide; iBiodT: biotin-modified thymidine |
| L2 | /5Phos/GTTGGAGTTCGGTGTGTGGGAGTGAGCTGTGTC | 5Phos: 5′ phosphorylation |
| A1 | /5Phos/AGATCGGAAGAGCACACGTCT | 5Phos: 5′ phosphorylation |
| A2 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNN | N: random nucleotide |
| Primer1 | AATGATACGGCGACCACCGAGATCTACAC/*****/ACACTCTTTCCCTACACGACGCTCTTCCGATCT | *: 5-nt index for multiplexing libraries |
| Primer2 | CAAGCAGAAGACGGCATACGAGACGTGTGCTCTTCCGATCT |
Crosslinking and nuclei isolation
We performed GRID-seq on a variety of mammalian and Drosophila cell lines. Because GRID-seq is performed on isolated nuclei, the technology can in principle be implemented in adherent cells, cells grown in suspension or fixed tissues. We tested multiple conditions for crosslinking cells. In Drosophila, stronger crosslinking (e.g., 2 mM DSG + 3% (wt/vol) formaldehyde) improves the identification of small RNAs, typically producing more signals for all chromatin-interacting RNAs, as compared with the data generated under weaker crosslinking conditions (e.g., 1% (wt/vol) formaldehyde only) (X.L., B.Z. and X.-D.F., data not shown). We speculate that strong crosslinking conditions may help stabilize long-range interactions. However, such strong crosslinking conditions also tend to make isolation of nuclei difficult and decrease their permeability to linker and enzyme, not allowing them to enter the matrix in subsequent steps. We also recommend conducting pilot experiments to test and optimize SDS concentration at the nuclei isolation step by checking nuclei integrity and the percentage of cytoplasmic membrane-free nuclei under a microscope before performing GRID-seq on a new cell type. Some cells, such as HepG2 and MCF7 cells, are difficult to lyse after crosslinking and thus require a higher SDS concentration (up to 0.5% (wt/vol)) to help with isolation of fixed nuclei. For certain cell types, however, a high SDS concentration may destroy the nuclei preparation.
Chromatin fragmentation and in situ ligation
The permeabilized nuclei are incubated with the AluI restriction enzyme to fragment gDNA in situ. T4 polynucleotide kinase is then used to phosphorylate the 5′ ends of both DNA and RNA, while dephosphorylating their 3′ ends. Next, the nuclei are incubated with the pre-adenylated bivalent linker and RNA ligase in an ATP-free reaction to ligate the 3′ end of the nuclear RNA to the ssRNA stretch of the linker. cDNA is subsequently generated by reverse transcriptase, extending the complementary strand of the linker into the ligated RNA. After washing the nuclei to remove excessive linker, the dsDNA end of the linker is ligated to nearby AluI-fragmented gDNA. At this point, the RNA–chromatin contacts in the nuclei are covalently joined by the linker.
Paired-end tag generation by MmeI
A biotinylated nucleotide is present in the DNA–RNA hybrid strand of the linker. Thus, the cDNA-containing strand can be eluted after biotin selection, followed by conversion to dsDNA by random priming using the Klenow fragment, which has primer extension and displacement capability but lacks the 3′ to 5′ exonuclease activity. Next, the resulting dsDNA is digested by MmeI to generate paired-end tags with fixed lengths (~85 bp) to eliminate single-end-ligated products, similar to LongSAGE16 and ChIA-PET17.
Adaptor ligation and amplification
Each resulting product after MmeI digestion contains a 2-base 3′ overhang. To preserve the overhanging bases for maximal genome alignment efficiency during data analysis, we ligate the products with a Y-shaped adaptor that has a 2-base 3′ overhang of random nucleotides (Table 1). To prevent self-ligation, alkaline phosphatase is used to dephosphorylate the DNA before adaptor ligation. DNA ligation, 5′-phosphorylation and nick-sealing are subsequently carried out by T4 DNA ligase. Ligated DNAs are size-selected for amplification by a low number of PCR cycles. Multiple samples can be individually barcoded at this point by using PCR primers with different index sequences for pooled sequencing.
Data processing
To better elucidate the procedure for evaluating the general quality of GRID-seq data, we herein use the previously published GRID-seq data on B6 mESCs and newly generated GRID-seq data on F123 mESCs for demonstration. Both libraries consist of two biological replicates and are sequenced at an average depth of 160 million reads per replicate. After genome alignment and filtering, uniquely mapped mate-pair reads are used to construct RNA–DNA interactomes. The in-house software—GridTools—generates metadata, providing statistics, including read counts, length distribution, nucleotide frequency and strand specificity, to initially evaluate the data quality. In the demonstration, the F123 mESC data originally contained more reads but resulted in fewer uniquely mapped reads for interactome calculation, as compared with the B6 mESC data (Fig. 4a). Read lengths of both datasets are as expected (Fig. 4b), but the nucleotide frequency appears slightly biased in the F123 mESC DNA reads (Fig. 4c). Because the cytosine frequency at the 66-bp position was expected to be ~100% due to AluI digestion, its deviation indicates that this bias in the newly generated data on F123 mESCs may be introduced by contaminated gDNA. In both datasets, the RNA reads show the same strand orientation as the original transcripts, whereas the DNA reads lack any strand specificity (Fig. 4d), validating the respective origins of the reads. An apparent 3′-end bias in the RNA reads from the F123 library might result from more exposed 3′ ends for ligation with the linker at their polyA sites in certain cell types.
Next, RNA density is evaluated to identify chromatin-associated RNAs. Specifically, we first ranked RNA levels by calculating RNA read-mate density normalized by gene length (reads per kilobase (RPK) value), similar to RNA-seq analysis. We set a cutoff close to the inflection point at RPK ≥100 to exclude genes that express low amounts of RNA captured on chromatin (Supplementary Fig. 1a,b). Then, we summarized the coverage of DNA reads linked with each RNA species on the 1-kb-binned genome and ranked RNA species by their maximal mate DNA density. We set another cutoff of maximal DNA density at RPK ≥10 to exclude the genes that express RNAs that do not aggregate into significant chromatin-interaction peaks, but rather spread out over the genome (Supplementary Fig. 1c,d). Finally, the RNA species that simultaneously met these two criteria were defined as chromatin-associated RNAs (Supplementary Fig. 1e,f and Fig. 4e). The collective DNA reads that are linked with chromatin-associated RNA in trans are used for background construction.
To further estimate the potential complexity of a GRID-seq library, random subsampling simulations can be performed to test whether saturation has been reached at the current sequencing depth. A demonstration in B6 mESC datasets showed that the 58.5 million useable read mates probably have not covered all chromatin-associated RNAs in the cell, as indicated by the saturation curve, where the number of identified chromatin-associated RNAs still increases linearly at 55 million reads depth (Supplementary Fig. 1g). On the other hand, the chromatin regions where these RNAs interact have been mostly covered at the 10-RPK level, as suggested by the plateauing DNA saturation curve (Supplementary Fig. 1h). Similar conclusions were also reached with the F123 mESC datasets. Thus, in these cases, a substantial increase in sequencing depth would be required to increase the detection power of those low-abundant chromatin-associated RNAs and their interaction sites across the genome. As mentioned earlier, many low-abundant RNAs may still escape detection because abundant RNAs will consume most sequence reads. Thus, future users of GRID-seq would have to consider the trade-off between the detection power for low-abundant RNAs and the associated cost.
As in analysis of Hi-C data, it is key to first define the resolution of a given GRID-seq dataset. In general, a higher resolution generates reproducible peaks that are more distinct but requires higher data density and more intensive computational power. GridTools makes a balanced call for an optimal resolution on a given dataset by intensively evaluating the bin-wise Pearson’s correlation coefficient (PCC) in each chromosome between pairs of the two halves of a randomly divided dataset under different bin sizes ranging from 1 kb to 1 Mb. The randomly halved GRID-seq datasets, simulating two independent repeats, were used to calculate the Pearson’s correlation in each chromosome, and the final PCC score was summarized from the average of resampling the datasets ten times. At a 1-kb/bin resolution, for example, B6 mESC data had a median PCC of ~0.7 for all chromosomes, whereas F123 mESC data scored lower at 0.6, indicating more robustness of RNA–chromatin interactions in the B6 mESC data. Thus, functional analysis of individual replicates of F123 mESC data could be reliably conducted only at a lower resolution. However, the combined datasets from replicates produced improved PCCs in almost all GRID-seq datasets (Fig. 4f). We thus recommend combining data to obtain interactome maps at the highest possible resolution. In general, a resolution with a PCC >0.8 is considered optimal. However, such a threshold is empirical, and users should always consider the trade-off between signal reliability and sensitivity when choosing a map resolution that is suitable for specific conclusions.
Using GridTools, background and foreground signals can be generated (Fig. 5a). Upon correction against the background, the identified RNA–DNA interaction signals can next be used to construct the interactome matrix, which serves as a resource for many types of downstream analyses, including global interactome profiling (Fig. 5b), differential interactions of individual RNAs under different biological contexts (Fig. 5c, note the relatively lower read density across the illustrated chromosome region with the F123 mESC dataset as compared with the B6 mESC dataset, indicating either a lower background with the F123 data or the presence of certain dominant signals in the F123 dataset that reduce most other signals after data normalization), and functional analysis of specific RNAs (Fig. 5d). The discrepancy between the interactomes from the two ES cell lines may be of both a technical and a biological nature. F123 mESCs are heterozygous (129/sv × Cast) compared to the reference genome, which hinders mapping of many reads to certain genome loci. This technical caveat may contribute to a generally reduced mapping rate in the F123 as compared with the B6 mESC samples. More importantly, considering that the B6 and F123 cell lines were cultured in medium with and without two small-molecule inhibitors (2i), respectively, the observed differences in the resultant RNA–DNA interactomes may reflect biological changes in genome organization, gene expression, and global interactome elicited by different culture conditions. Thus, these two mES cell lines are expected to display differences in RNA–chromatin interactomes. Finally, we developed z-score-based statistics to infer chromatin proximity8, which can be used to deduce global enhancer–promoter connectivity (Box 3). The main programs and parameters used throughout the analyses are listed in Table 2.
Fig. 5 |. Statistical models and GRID-seq derived RNA–chromatin interaction matrix.
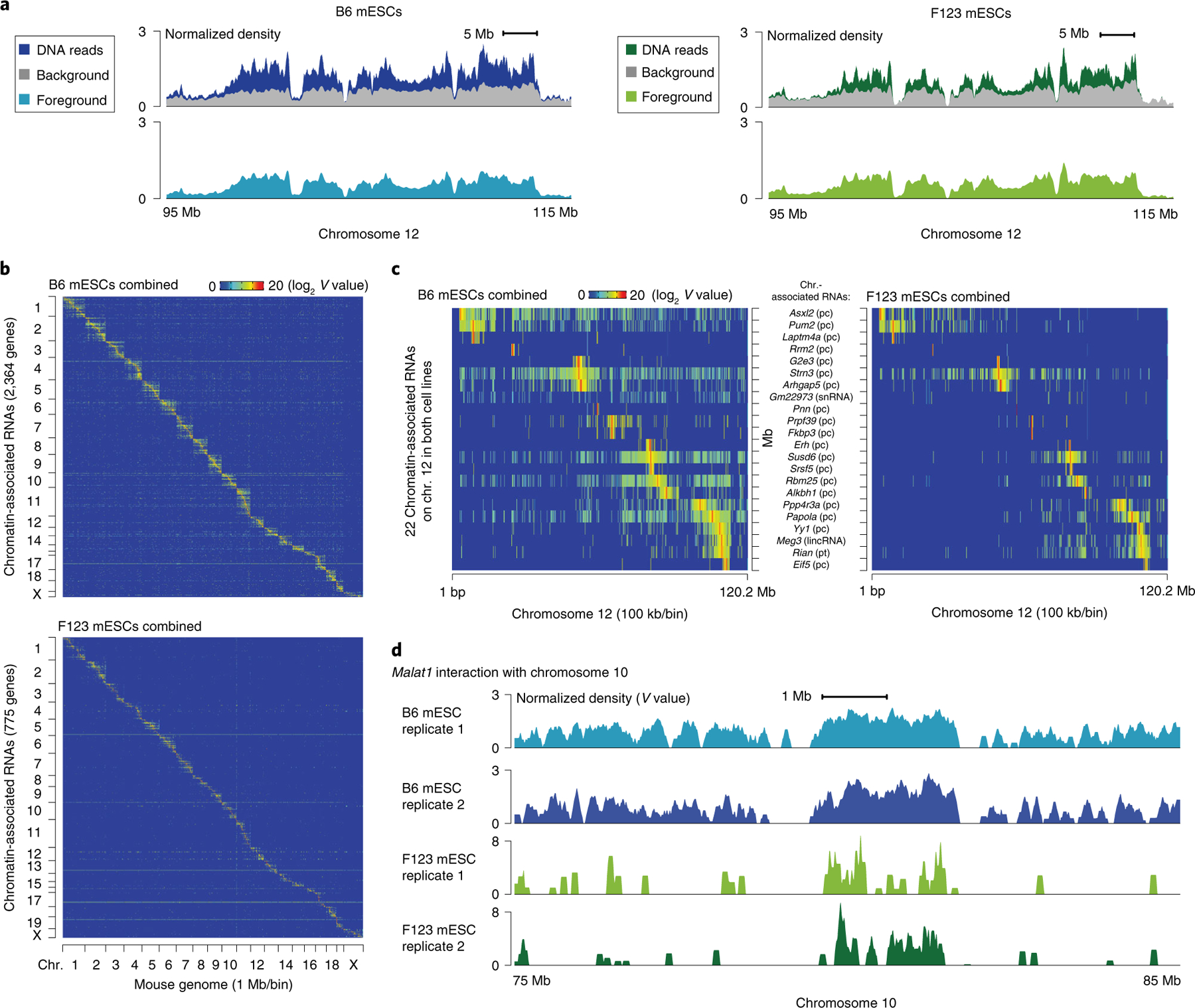
a, Foreground (light blue and light green) and background (gray) in a highlighted region of chromosome 12 relative to the GRID-seq raw signals (dark blue and dark green). The background is derived from the collection of trans-acting protein-coding mRNAs in the two mouse ES cell lines, B6 mESCs and F123 mESCs. Foreground and background are shown in normalized density for comparison. b, Heatmap showing chromatin-associated RNAs across the whole mouse genome in B6 (top) and F123 mESCs (bottom), obtained from combining two biological replicates. Row: chromatin-associated RNAs ordered by their gene locations. The total numbers of chromatin-associated RNA species are denoted in the parentheses in the y-axis label. Column: mouse genome in 1 Mb/bin resolution. c, Two enlarged representative regions on chromosome 12, showing only common chromatin-associated RNAs in both mESC lines that are transcribed from chromosome 12. Cell-specific RNA–chromatin interaction patterns in B6 (left) and F123 mESCs (right) are shown at 100 kb/bin resolution. RNA names are shown in the middle, followed by their respective types in parentheses. d, Detailed Malat1–chromatin interaction peaks in a representative region of chromosome 10 in replicates of the two mESC datasets. chr., chromosome; pc, protein-coding; pt, processed transcript; V, interaction density.
Box 3 |. Modeling the network of enhancer–promoter proximity ● Timing 3 h.
Procedure
-
1Once the RNA–chromatin contact matrix is generated, an enhancer–promoter proximity network can be modeled using GridTools.py model, if enhancer annotation is provided as a BED file.
GridTools.py model -k 100 -x 10 -z 2 -e mm10.enhancers.bed test.h5 | gzip -c > test.net.gz
▲ CRITICAL STEP The required BED file must be in a format with at least four columns. Although genomic intervals are not limited to active chromatin elements, the proximity-modeling parameters are optimized for enhancers and active promoters.
-
2
Because the collective trans signals from mRNAs are statistically unlikely to reflect proximal interactions, any cis signal that rejects the null distribution at a stringent significance level would most likely reflect chromatin proximity between the active genomic elements8. The generated network file consists of a column of z scores for all trans and cis interactions. The distribution of trans and cis z scores can be visualized as a density plot to estimate the appropriate z score cutoff (panel a of the figure).
▲ CRITICAL STEP z-score distributions vary considerably among samples. Thus, it is beneficial to evaluate the cutoff case by case. A less stringent z-score cutoff might introduce noise and decrease the specificity of inferred proximity, whereas a highly stringent one might leave too few signals to construct meaningful transcription hubs.
-
3
On the basis of the observed distribution, we choose z ≥ 3 as a threshold for the B6 mESC dataset. The filtered cis signals are subsequently imported into Cytoscape for visualization (panel b of the figure). The network can be rendered by self-organized layout algorithms such as Edge-Repulsive Spring-Electric Layout (built into Cytoscape v.3.0 or later) that simulate connections between a pair of nodes as a mechanic spring model in which both attractive and repulsive forces constrain the global layout to the optimal equilibrium.
-
4
Transcription hubs consisting enhancers, promoters and other elements can be isolated from the network for further functional study (panel c of the figure).
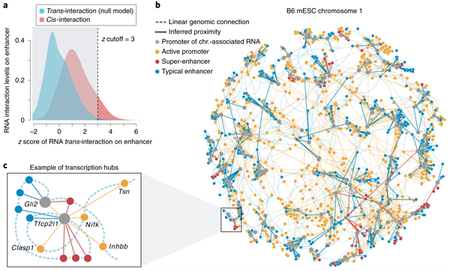
The figure depicts a z-score-based statistical model for RNA–chromatin interactions in spatial proximity. Panel a shows density distributions of cis- and trans-chromosomal RNA interaction levels on enhancers in B6 mESCs; x axis: z scores of all trans-chromosomal RNA interactions (blue). Z ≥ 3 was used to identify significant RNA interactions between individual promoter and enhancer elements. Signals below the cutoff (gray shaded) were excluded from the visualization. In panel b, a self-organized map for visualization of the chromosome 1 network in B6 mESCs is shown. Panel c represents an individual transcription hub on chromosome 1. Color labels are the same as in b, and gene names of the promoters are shown. Note that the linear genomic connections in b and c are not to scale.
Table 2 |.
Main programs and parameters used throughout the analyses
| Program | Function | Parameter and description | Steps |
|---|---|---|---|
| cutadapt | cutadapt trims and filters reads to expected length and quality | 152 | |
| -l trims the sequence to length (bp) | |||
| --max-n discards reads with more than n N bases | |||
| bwa | index | bwa index builds the index of reference sequences | 153, 156 |
| -p prefix is used for output files | |||
| mem | bwa mem maps reads to the indexed linker or reference genome | 154, 156 | |
| -p specifies the paired-end mode | |||
| -k specifies minimum seed length, matches shorter than specified will be missed | |||
| -w specifies band width; gaps larger than it will be omitted | |||
| -T specifies minimum score; alignment scored less than it will not be output | |||
| -L specifies clipping penalty; used for the score calculation | |||
| -B specifies mismatch penalty; used for the score calculation | |||
| -O specifies gap open penalty; used for the score calculation | |||
| samtools | view | samtools view reads, filters and transforms SAM/BAM files | 154, 156 |
| -u specifies output of uncompressed BAM files | |||
| -b specifies output in the BAM format | |||
| -f bits flag; alignments with all bits present will be output | |||
| sort | samtools sort sorts alignments by coordinates or read names | 154, 156 | |
| -n specifies sort by read name rather than by chromosomal coordinates | |||
| -m specifies maximum memory (in KB, MB or GB) per thread | |||
| fixmate | samtools fixmate fills in mate coordinates and related flags from a name-sorted | 156 | |
| alignment | |||
| -p disables FR (forward–reverse orientation) proper pair check | |||
| -m adds ms (mate score) tags | |||
| markdup | samtools markdup marks duplicate alignments from a coordinate sorted file | 156 | |
| GridTools.py | matefq | GridTools.py matefq parses the reads mapped to the GRID-seq linker in the BAM file | 155 |
| into RNA–DNA mates in interleaved FASTQ format | |||
| -l specifies minimum length; RNA or DNA with length less than specified will be omitted | |||
| -n renames the prefix of each read | |||
| -o outputs to file in HDF5 format | |||
| evaluate | GridTools.py evaluate calculates quality and quantity of each pair of RNA–DNA | 157 | |
| mates from the BAM file mapped to the genome. | |||
| -g specifies gene annotation in GTF format | |||
| -k specifies bin size (kb) of the genome (default: 10 kb) | |||
| -m specifies moving window for smoothing in bins (default: 10) | |||
| -o outputs mapping information to the HDF5 file | |||
| stats | GridTools.py stats calculates statistics of GRID-seq data | 158 | |
| -p specifies prefix of output file names | |||
| -b outputs the summary of base-position information for RNA, Linker and DNA | |||
| -c outputs the summary of mapping information in read counts | |||
| -l outputs the distribution of sequence length for RNA, Linker and DNA | |||
| -r outputs the resolution information of the library | |||
| RNA | GridTools.py RNA identifies chromatin-enriched RNAs and evaluates the gene | 159 | |
| expression levels as well as interaction scopes | |||
| -e specifies output file for the gene expression | |||
| -s specifies output file for the RNA interaction scope | |||
| DNA | GridTools.py DNA identifies RNA-enriched chromatin regions in background (trans) and foreground (cis) | 160 | |
| matrix | GridTools.py matrix evaluates the RNA–chromatin interaction matrix | 161 | |
| -k specifies cutoff of RNA reads per kilobase in the gene body | |||
| -x specifies cutoff of DNA reads per kilobase at the maximum bin | |||
| model | GridTools.py model builds a network model to deduce the enhancer–promoter proximity | Box 3 | |
| -e specifies BED file of regulatory elements (e.g., enhancers and promoters) | |||
| -k specifies cutoff of RNA reads per kilobase in the gene body | |||
| -x specifies cutoff of DNA reads per kilobase at the maximum bin size | |||
| -z specifies z score used to filter for significant proximity | |||
| bgzip | Block compression/decompression utility | 157 | |
| tabix | Generic indexer for TAB-delimited genome position files | 157 | |
| -p specifies input file in GFF or GTF format |
Materials
Biological materials
C57BL/6 mouse ES cells (B6 mESCs; ref. 8), a gift from B. Ren (University of California, San Diego).
F1 Mus musculus castaneus × S129/SvJae mouse ES cells (F123 mESCs), a gift from B. Ren (University of California, San Diego). ! CAUTION Before use, cells should always be checked for authenticity and for contamination by mycoplasma. ▲ CRITICAL We demonstrate how to perform the GRID-seq procedure and present the results generated with the B6 and F123 mESC lines described above. However, we also used Drosophila S2, human MDA-MB-231, MM.1S, MCF7, HeLa S3, K562 and HepG2 cells for successful library construction.
Reagents
▲ CRITICAL Extreme caution should be taken to avoid nuclease contamination. Use nuclease-free reagents and materials. Change gloves regularly throughout the Procedure.
2-Mercaptoethanol (50 mM; Thermo Fisher Scientific, cat. no. 31350010)
5′ DNA Adenylation Kit (New England Biolabs, cat. no. E2610S)
Acetic acid (100% (vol/vol); EMD Millipore, cat. no. 100056)
Acrylamide/bis solution (19:1, 40% (wt/vol); Bio-Rad, cat. no. 1610144) ! CAUTION Acrylamide/bis solution is toxic and may be carcinogenic. Wear gloves and safety glasses.
Agarose (Seakem LE Agarose; Lonza, cat. no. 50004)
AluI (Thermo Fisher Scientific, cat. no. ER0012)
Ammonium persulfate (APS; 10% (wt/vol); Bio-Rad, cat. no. 161–0700)
ATP (100 mM; Thermo Fisher Scientific, cat. no. R0441)
BSA (20 mg/ml; New England Biolabs, cat. no. B9000S)
CHIR99021 (Stemgent Stemolecule, cat. no. 04–0004)
Distilled water (DNase/RNase-free; Thermo Fisher Scientific, cat. no. 10977015)
Disuccinimidyl glutarate (DSG; Thermo Fisher Scientific, cat. no. 20593)
DTT (100 mM; Thermo Fisher Scientific, cat. no. 707265ML)
DMEM (no glucose, no glutamine, no phenol red; Thermo Fisher Scientific, cat. no. A1443001)
DNA ladder (10 bp; Thermo Fisher Scientific, cat. no. 10821–015) Alternatively, 20-bp DNA ladders can be used
DNA ladder (100 bp; Thermo Fisher Scientific, cat. no. 15628–019)
dNTP mix (10 mM each; Thermo Fisher Scientific, cat. no. R0192)
EDTA (0.5 M, pH 8.0; Thermo Fisher Scientific, cat. no. AM9260G)
Ethanol (100% (vol/vol); Pharmco-AAPER, cat. no. 111000200) ! CAUTION Ethanol is highly flammable.
Formaldehyde solution (37% (wt/vol) in H2O; Sigma-Aldrich, cat. no. 252549) ! CAUTION Formaldehyde is toxic; always use it in a fume hood.
Gel-loading dye (New England Biolabs, cat. no. B7022S)
Glycine (Sigma-Aldrich, cat. no. 357002–1KG)
GlycoBlue coprecipitant (Thermo Fisher Scientific, cat. no. AM9515)
GlutaMax Supplement (Gibco, cat. no. 34050061)
Hydrochloric acid (HCl; Sigma-Aldrich, cat. no. 258148)
Isopropanol (100% (vol/vol); Pharmco-AAPER, cat. no. 111000190) ! CAUTION Isopropanol is highly flammable and harmful; contact with skin or eyes and inhalation should be avoided. Use it inside a fume hood.
Klenow fragment (3′–5′ exo-; New England Biolabs, cat. no. M0212S)
Knockout DMEM (Thermo Fisher Scientific, cat. no. 10829018)
Knockout serum replacement (Thermo Fisher Scientific, cat. no. 10828028)
L-Glutamine (200 mM solution; Thermo Fisher Scientific, cat. no. 25030081)
Leukemia inhibitory factor (LIF; final concentration is 1,000 units/ml; Millipore, cat. no. ESG1107)
MEM non-essential amino acid solution (100×; Gibco, cat. no. 11140076)
MmeI (New England Biolabs, cat. no. R0637S)
Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific, cat. no. 65001)
New England Biolabs Buffer 2 (10×; New England Biolabs, cat. no. B7002S)
Nonidet P-40 substitute (NP-40; Sigma-Aldrich, cat. no. 11332473001)
Oligonucleotides (Integrated DNA Technologies; see Table 1 for sequences) ▲ CRITICAL Oligonucleotides L1 and L2 should be RNase-free HPLC-purified; A1 and A2, Primer1 and Primer2 should be HPLC or PAGE-purified. Linker and adaptor oligonucleotides can be purchased in bulk and dissolved in nuclease-free water to make 100 µM solutions. These stocks can be divided into aliquots and stored at −80 °C indefinitely.
PD0325901 (Stemgent Stemolecule, cat. no. 04–0006)
Penicillin–streptomycin (liquid; Gibco, cat. no. 15070063)
Phenol/chloroform/isoamyl alcohol (pH 8.0; Thermo Fisher Scientific, cat. no. 15593049) ! CAUTION Phenol, chloroform and isoamyl alcohol are harmful; contact with skin or eyes and inhalation should be avoided. Use it inside a fume hood.
PBS (1×; Thermo Fisher Scientific, cat. no. 10010023)
Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific, cat. no. F530S). ▲ CRITICAL We recommend using polymerase from this supplier, as using DNA polymerase from other manufacturers results in higher background during PCR amplification of the library.
Potassium chloride (KCl; 2 M; Thermo Fisher Scientific, cat. no. AM9640G)
Protease inhibitor cocktail tablets (Sigma-Aldrich, cat. no. S8830–2TAB)
Proteinase K (Thermo Fisher Scientific, cat. no. AM2546)
RiboLock RNase inhibitor (40 U/µl; Thermo Fisher Scientific, cat. no. EO0382)
RNase A, DNase and protease free (10 mg/ml; Thermo Fisher Scientific, cat. no. EN0531)
RNaseZap (Thermo Fisher Scientific, cat. no. AM9782)
Shrimp alkaline phosphatase (rSAP; New England Biolabs, cat. no. M0371S)
Sodium acetate (NaOAc; 3 M (pH 5.5); Thermo Fisher Scientific, cat. no. AM9740)
Sodium chloride (NaCl; 5 M; Thermo Fisher Scientific, cat. no. AM9759)
SDS (Sigma-Aldrich, cat. no. L3771–100G)
Sodium hydroxide NaOH; 10 M; Sigma-Aldrich, cat. no. 72068–100ML)
SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, cat. no. 18080051)
SuperScript III Reverse Transcriptase (Thermo Fisher Scientific, cat. no. 18080044)
SYBR Gold nucleic acid gel stain (Thermo Fisher Scientific, cat. no. S11494)
T4 DNA ligase (Thermo Fisher Scientific, cat. no. 15224025) ▲ CRITICAL Use ligase only from this supplier in in situ DNA ligation because of the compatible buffer system at related steps. T4 DNA ligase from an alternative supplier can be used if the reagent contains an equivalent enzyme concentration and is compatible with the buffer system.
T4 DNA ligase, high concentration (New England Biolabs, cat. no. M0202T) ▲ CRITICAL Use ligase from this specific supplier in adaptor ligation because of the compatible buffer system at related steps. T4 DNA ligase from an alternative supplier can be used if the reagent contains equivalent enzyme concentration and is compatible with the buffer system.
T4 polynucleotide kinase (PNK; New England Biolabs, cat. no. M0201)
T4 polynucleotide kinase (PNK; Thermo Fisher Scientific, cat. no. EK0032)
T4 RNA ligase 2 (truncated KQ; New England Biolabs, cat. no. M0373S)
TBE (10×; Thermo Fisher Scientific, cat. no. AM9865)
Tetramethylethylenediamine (TEMED; Bio-Rad, cat. no. 161–0800) ! CAUTION TEMED is toxic; contact with skin or eyes and inhalation should be avoided. Use it inside a fume hood.
Tris-HCl buffer (1 M, pH 7.5; Thermo Fisher Scientific, cat. no. 15567027)
Tris-HCl buffer (1 M, pH 8.0; Thermo Fisher Scientific, cat. no. 15568025)
Triton X-100 (Sigma-Aldrich, cat. no. T9284)
Tween 20 (Sigma-Aldrich, cat. no. P9416)
Dimethyl sulfoxide (DMSO; Sigma-Aldrich, cat. no. D8418)
Trypan blue (Sigma-Aldrich, cat. no. T8154)
6× loading buffer (New England Biolabs, cat. no. B7024S)
10% and 12% native polyacrylamide gels (home-made; see ‘Reagent setup’)
Equipment
▲ CRITICAL Nuclease-free equipment is required: spray pipettes, pipette aids and bench with RNaseZap.
Freezer (–20 °C; Panasonic, model. no. BZ10145190)
Freezer (–80 °C; Thermo Fisher Scientific, model. no. 989)
Filter unit (0.22 μm; Olympus, cat. no. 25–244)
Centrifuge tubes (50 ml; Genesee, cat. no. 21–106)
Agarose electrophoresis cell (Bio-Rad, model. no.1704469)
PCR tubes (Axygen, 0.2 ml; Corning, cat. no. PCR-02-C)
Barnstead Labquake Rotator (Thermo Fisher Scientific, cat. no. 415110)
Bright-Line counting chamber (Hausser Scientific, cat. no. 3200)
Cell culture incubator (Thermo Fisher Scientific, model. no. Heracell VIOS 250i), set to 37 °C and 5% carbon dioxide
Cell scraper (Sigma-Aldrich, cat. no. SIAL0008–150EA)
Centrifuge tube filters (Costar Spin-X; 45-µm; Thermo Fisher Scientific, cat. no. 7200387)
Culture dishes (Nest, cat. no. 704001)
Dark Reader transilluminator (Clare Chemical Research, cat. no. DR89X)
Fisherbrand electric pipette controller (Thermo Fisher Scientific, cat. no. 14–955-202)
Inverted microscope (Olympus, model. no. CKX53)
LoBind microcentrifuge tubes (1.5 ml; Eppendorf, cat. no. 022431021 ▲ CRITICAL We strongly recommend using Eppendorf LoBind microcentrifuge tubes throughout the protocol to minimize loss of nucleic acids.
MagnaRack magnetic separation rack (Thermo Fisher Scientific, cat. no. CS15000)
MaXtract high-density tube Qiagen, cat. no. 129046)
Spectrophotometers (NanoDrop 2000; Thermo Fisher Scientific, model no. ND-2000)
Universal power supply (PowerPac; Bio-Rad, cat. no. 1645070)
Qubit fluorometer (Thermo Fisher Scientific, model. no. Q32857)
Razor blades (0.009 inch; Thermo Fisher Scientific, cat. no. 940115)
Refrigerated centrifuge (Eppendorf, model no. 5804R)
Refrigerated microcentrifuge (Eppendorf, model no. 5425R)
Thermal cycler (SimpliAmp, 96-well; Thermo Fisher Scientific, model no. A24811)
Single-channel manual pipettes (0.5–10 μl, 20–200 μl and 100–1,000 μl; Rainin, cat. nos. 17014388, 17014391 and 17014382)
SpeedVac dryer (Thermo Fisher Scientific, cat. no. 20–548-134)
Manual homogenizer (Squisher-Single; Zymo, cat. no. H1001–50)
Tetra vertical electrophoresis cell (for 1-mm-thick hand-cast gels; Bio-Rad, cat. no. 1658001FC)
Thermal mixer C (Thermomixer C; Eppendorf, cat. no. Z605271)
Vortex mixer (Vortex-Genie 2; Scientific Industries, model no. G560/SI-0236)
Sequencer (Illumina, model no. HiSeq 2500)
Computer, programs and source code
A computer with 16 or more 64-bit processors and 64 GB or more of RAM is recommended
A Ubuntu Linux distro, such as Ubuntu server 18.04 LTS, is recommended (https://www.ubuntu.com/download/server)
BWA software package (v.0.7.12-r1039; http://www.bio-bwa.sourceforge.net)
Cutadapt package (v.1.22; https://cutadapt.readthedocs.io/en/stable/)
Python 3 (v.3.5.2; https://www.python.org)
GridTools.py (v.1.1; https://github.com/biz007/gridtools)
SAMtools (v.1.3; http://www.samtools.sourceforge.net)
Cytoscape (v.3.5; https://cytoscape.org)
Example datasets
F123 mESC: http://fugenome.ucsd.edu/gridseq/datasets/gridseq.test10M.raw.fq.gz
mm10.enhancers.bed: http://fugenome.ucsd.edu/gridseq/datasets/mm10.enhancers.bed
Reagent setup
▲ CRITICAL All reagents, solutions and buffers should be made with nuclease-free distilled water.
2i/LIF medium
Prepare this medium by supplementing Knockout DMEM with 15% Knockout serum replacement, 2 mM l-glutamine, 1× penicillin–streptomycin, 1× MEM non-essential amino acid solution and 0.1 mM 2-mercaptoethanol. Prepared medium can be stored at 4 °C for up to 1 month. 1,000 U/ml LIF, 3 μM CHIR99021 and 1 μM PD0325901 are added to the medium immediately before feeding to the cells.
LIF medium without 2i
Prepare this medium by supplementing Knockout DMEM with 15% Knockout serum replacement, 1× penicillin–streptomycin, 1× MEM non-essential amino acid solution, 1× GlutaMax, 1,000 U/ml LIF and 0.4 mM 2-mercaptoethanol. Prepared medium can be stored at 4 °C for up to 1 month.
150 mM NaOH
Prepare 1 ml of 150 mM NaOH by diluting 15 µl of 10 M NaOH with 985 µl of nuclease-free water. ! CAUTION NaOH is corrosive; contact with skin or eyes should be avoided. ▲ CRITICAL 150 mM NaOH can be prepared in advance and stored in 100-µl aliquots at −80 °C for up to a year. Use a freshly thawed aliquot each time.
500 mM DSG
Prepare a 500 mM DSG stock solution by dissolving 50 mg of DSG in 306.5 µl of DMSO. ! CAUTION DSG is a strong irritant. Use it inside a fume hood. ▲ CRITICAL Store at −20 °C for up to a year.
10% (vol/vol) Triton X-100
Prepare 50 ml of 10% (vol/vol) Triton X-100 by diluting 5 ml of Triton X-100 with 45 ml of nuclease-free water. Filter-sterilize the solution with a 0.22-μm filter. Store at room temperature (20–25 °C) for up to a year.
10% (vol/vol) Tween 20
Prepare 50 ml of 10% (vol/vol) Tween 20 by diluting 5 ml of Tween 20 in 45 ml of nuclease-free water. Filter-sterilize the solution with a 0.22-μm filter. Store at room temperature for up to a year.
10% (vol/vol) NP-40
Prepare 50 ml of 10% (vol/vol) NP-40 by diluting 5 ml of NP-40 with 45 ml of nuclease-free water. Filter-sterilize the solution with a 0.22-μm filter. Store at room temperature for up to a year.
1.2 M Glycine
Prepare 250 ml of 1.2 M glycine by dissolving 22.5 g of glycine in 220 ml of nuclease-free water. Adjust the volume to 250 ml and filter-sterilize with a 0.22-μm filter. Store at 4 °C for up to 6 months.
10% (wt/vol) SDS
Prepare 50 ml of 10% (wt/vol) SDS by dissolving 5 g of SDS in 40 ml of nuclease-free water. Heat the solution to 60 °C to facilitate dissolving. Adjust the volume to 50 ml with nuclease-free water. This solution can be stored at room temperature for up to 3 months. ! CAUTION SDS is a strong irritant. Wear a mask when weighing SDS and cleaning the area, as it disperses easily.
100× protease inhibitor cocktail
Dissolve one tablet of protease inhibitor cocktail in 1 ml of nuclease-free distilled water. ▲ CRITICAL 100× protease inhibitor can be prepared in advance and stored in 50-µl aliquots at −80 °C for up to a year. Use a freshly thawed aliquot each time.
1× PBST
Prepare 100 ml of 1× PBST by adding 1 ml of 10% (vol/vol) Tween 20 to 99 ml of 1× PBS. Store at −4 °C for up to a year.
Equilibrium buffer
Prepare 1 ml of equilibrium buffer by mixing 469 μl of nuclease-free distilled water, 5 μl of 1 M Tris-HCl, pH 7.5, 1 μl of 5 M NaCl, 10 μl of 10% (vol/vol) NP-40, 10 μl of RiboLock RNase inhibitor and 5 μl of 100× protease inhibitor cocktail. ▲ CRITICAL The buffer without the RNase inhibitor and the protease inhibitor can be prepared in advance and stored at 4 °C for up to 6 months.
Annealing buffer
Prepare 1 ml of annealing buffer by adding 100 µl of 1 mM Tris-HCl, pH 8.0, and 40 µl of 5 M NaCl to 860 µl of nuclease-free distilled water. Store at −20 °C for up to a year.
3% (wt/vol) Formaldehyde solution
To prepare this solution, mix 270 μl of 37% (wt/vol) formaldehyde solution with 3 ml of 1× PBS. ▲ CRITICAL The 3% (wt/vol) formaldehyde solution should always be prepared immediately before use.
2× proteinase K buffer
Prepare 10 ml of proteinase K buffer by mixing 200 µl of 1 M Tris-HCl, pH 7.5, 400 µl of 5 M NaCl, 40 µl of 0.5 M EDTA and 2 ml of 10% (wt/vol) SDS. Store at 4 °C for up to 6 months.
Cell washing buffer
Prepare 200 µl of cell washing buffer by mixing 176 µl of nuclease-free distilled water, 20 µl of 10× Tango Buffer (supplied with AluI), 2 µl of 10% (vol/vol) Tween 20 and 2 µl of RiboLock RNase inhibitor. ▲ CRITICAL The buffer without the RNase inhibitor can be prepared in advance and stored at 4 °C for up to 6 months.
Gel elution buffer
Prepare 10 ml of gel elution buffer by mixing 9.2 ml of nuclease-free distilled water, 100 µl of 1 M Tris-HCl, pH 8.0, 20 µl of 0.5 M EDTA, 100 µl of 10% (vol/vol) Tween 20 and 600 µl of 5 M NaCl. Store at 4 °C for up to 6 months.
2× Binding and washing buffer (2× B&W buffer)
Prepare 10 ml of 2× B&W buffer by mixing 5.84 ml of nuclease-free distilled water, 100 µl of 1 M Tris-HCl, pH 7.5, 20 µl of 0.5 M EDTA, 40 µl of 10% (vol/vol) Tween 20 and 4 ml of 5 M NaCl. Store at 4 °C for up to 6 months.
1× Binding and washing buffer (1× B&W buffer)
Prepare 10 ml of 1× B&W buffer by diluting 5 ml of 2× B&W buffer with 5 ml of nuclease-free distilled water. Store at 4 °C for up to 6 months.
50× SAM
Prepare 12.8 µl of 50× SAM by diluting 1 μl of 640× SAM (supplied with MmeI) with 11.8 μl of nuclease-free distilled water. ▲ CRITICAL SAM is unstable at room temperature. Avoid repeated freeze–thaw cycles of the 640× stock. The 50× solution should always be made immediately before use.
75% (vol/vol) ethanol
Prepare 50 ml of 75% (vol/vol) ethanol by diluting 37.5 ml of 100% (vol/vol) ethanol with 12.5 ml of nuclease-free distilled water. Store at 4 °C for up to a year.
10× TE buffer
Prepare 50 ml of 10× TE buffer by mixing 5 ml of 1 M Tris-HCl, pH 7.5, and 1 ml of 0.5 M EDTA, pH 8.0, with 44 ml of nuclease-free distilled water. Filter-sterilize the solution using a 0.22-μm filter unit. Store at 4 °C for up to a year.
1× TBE buffer
Prepare 50 ml of 1× TBE buffer by diluting 5 ml of 10× TBE with 45 ml of nuclease-free distilled water. Filter-sterilize the solution using a 0.22-μm filter unit. Store at 4 °C for up to a year.
10% (wt/vol) APS
Prepare 10 ml of 10% (wt/vol) APS by dissolving 1 g of APS in 10 ml of nuclease-free distilled water. Filter-sterilize the solution with a 0.22-μm filter. Store at −20 °C for up to a year.
10% (wt/vol) separating gel
Prepare 8 ml of 10% (wt/vol) separating gel by mixing 2 ml of 40% (wt/vol) acrylamide/bis solution, 5.51 ml of nuclease-free distilled water, 400 µl of 10× TBE, 80 µl of 10% (wt/vol) APS and 8 µl of TEMED. ▲ CRITICAL APS and TEMED should be added immediately before pouring the gel.
12% (wt/vol) separating gel
Prepare 8 ml of 12% (wt/vol) separating gel by mixing 2.4 ml of 40% (wt/vol) acrylamide/bis solution, 5.11 ml of nuclease-free distilled water, 400 µl of 10× TBE, 80 µl of 10% (wt/vol) APS and 8 µl of TEMED. ▲ CRITICAL APS and TEMED should be added immediately before pouring the gel.
18% (wt/vol) separating gel
Prepare 8 ml of 18% (wt/vol) separating gel by mixing 3.6 ml of 40% (wt/vol) acrylamide/bis solution, 3.91 ml of nuclease-free distilled water, 400 µl of 10× TBE, 80 µl of 10% (wt/vol) APS and 8 µl of TEMED. ▲ CRITICAL APS and TEMED should be added immediately before pouring the gel.
4% (wt/vol) stacking gel
Prepare 3 ml of 4% (wt/vol) stacking gel by mixing 0.3 ml of 40% (wt/vol) acrylamide/bis solution, 2.52 ml of nuclease-free distilled water, 150 µl of 10× TBE, 30 µl of 10% (wt/vol) APS and 3 µl of TEMED. ▲ CRITICAL APS and TEMED should be added immediately before pouring the gel. ▲ CRITICAL All separating gels used in this protocol should be prepared with a stacking gel portion on top.
Procedure
Linker preparation ● Timing 24 h
▲ CRITICAL Annealed linker and adaptor solution should be prepared in advance of the GRID-seq experiment, which begins with ‘Cell harvest and dual crosslinking’ (Step 13).
-
1Set up a 40-µl adenylation reaction in a nuclease-free 0.2-ml PCR tube, as shown in the table below. Mix and incubate the reaction in a thermal cycler for 1 h at 65 °C.
Reagent Volume (µl) per reaction Final concentration Oligo L1 (100 µM; Table 1) 6 15 µM Mth RNA Ligase (included in the 5′ DNA Adenylation Kit) 8 10 µM 10× 5′ DNA Adenylation Reaction Buffer (from the 5′ DNA Adenylation Kit) 4 1× ATP (1 mM; included in the 5′ DNA Adenylation Kit) 20 500 µM RiboLock RNase inhibitor 1 1 U/µl Nuclease-free distilled water 1 -
2
Transfer the mixture to a MaXtract high-density tube. Dilute the mixture with 200 μl of nuclease-free distilled water and add 240 μl of phenol/chloroform/isoamyl alcohol. Mix well by shaking for 1 min.
! CAUTION Phenol/chloroform/isoamyl alcohol is highly toxic. Always handle it in a fume hood.
▲ CRITICAL STEP Insufficient mixing may lead to incomplete phase separation and loss of the supernatant.
-
3
Centrifuge the tube for 5 min at 16,000g at 4 °C. Transfer the supernatant to a 1.5-ml LoBind tube.
-
4
Add 1.5 μl of GlycoBlue coprecipitant and 24 μl of 3 M NaOAc to the supernatant and mix well by pipetting.
-
5
Add 720 μl of 100% (vol/vol) ethanol and vortex thoroughly. Allow the solution to precipitate for 1 h at −80 °C.
■ PAUSE POINT The solution can be left overnight and stored for up to 3 d at −80 °C.
-
6
Centrifuge the solution for 20 min at 16,000g at 4 °C. Remove all the supernatant and air-dry the pellet for 3 min in a SpeedVac dryer.
▲ CRITICAL STEP Do not to let the pellet dry completely, as this will greatly decrease its solubility.
-
7
Re-dissolve the pellet in 68 μl of annealing buffer, and add 1 μl of RiboLock RNase inhibitor and 6 μl of 100 µM oligo L2 (Table 1). Mix the solution thoroughly by pipetting.
-
8
Allow the oligos to anneal by incubating the 75-µl solution in a preheated thermal cycler that gradually decreases the temperature from 80 °C to 25 °C.
▲ CRITICAL STEP We typically set the thermal cycler ramping to 0.1 °C/s for annealing. Fast ramping may yield more unannealed oligos.
-
9
Take 1 µl of the annealed linker and measure the concentration with a spectrophotometer. The concentration is usually within the range of 6–10 mM (150–250 ng/µl).
-
10
Take 100 ng (~4 pmol) of the annealed linker for QC1, which evaluates the integrity of the linker (Box 1).
▲ CRITICAL STEP Storing the annealed linker at a temperature higher than −80 °C may affect the integrity of the ssRNA portion of the linker.
? TROUBLESHOOTING
■ PAUSE POINT The annealed linker can be stored for at least 6 months at −80 °C.
- 11
-
12
Allow the oligos to anneal by incubating the solution in a preheated thermal cycler that gradually decreases the temperature from 95 °C to 25 °C.
▲ CRITICAL STEP We typically set the thermal cycler ramping to 0.1 °C/s for annealing. Fast ramping may yield more unannealed oligos.
■ PAUSE POINT The annealed adaptor can be stored indefinitely at −80 °C.
Cell harvest and dual crosslinking ● Timing 3 h
-
13
Seed and culture cells under recommended conditions to reach ~80% confluency in 24 h. A typical GRID-seq experiment requires ~2 × 106 mammalian cells or ~1 × 107 Drosophila cells. However, we recommend starting with more cells and performing 2–3 biological repeats started from different cell stock vials in parallel.
▲ CRITICAL STEP Check cells for Mycoplasma contamination before setting up the experiment.
▲ CRITICAL STEP Adherent cells can be harvested by scraping or trypsinization.
-
14
Resuspend the cells with 10 ml of 1× PBS, and then transfer the suspension to a 50-ml tube.
-
15
Centrifuge the cells for 5 min at 500g at 4 °C. Remove the supernatant from the pellet.
-
16
Wash the cells twice more by repeating Steps 14 and 15.
-
17
Resuspend the cells in 5 ml of 1× PBS. Fix the cells by mixing with 20 μl of 500 mM DSG.
-
18
Rock the tube gently for 45 min at room temperature.
-
19
Centrifuge the cells for 5 min at 500g at 4 °C. Remove the supernatant and resuspend cells in 5 ml of 1× PBS.
▲ CRITICAL STEP After crosslinking by DSG, some cell types, such as Drosophila S2 cells and human MCF7 cells, tend to stick to the wall of the tube and are difficult to spin down. In such cases, addition of Tween 20 at a final concentration of 0.1% (vol/vol) is recommended for all further steps involving washing and resuspending the cells.
-
20
Wash the cells once more by repeating Steps 14 and 15.
-
21
Resuspend the cell pellet with 3 ml of 3% (wt/vol) formaldehyde and rock gently for 10 min at room temperature.
-
22
Quench the formaldehyde by mixing the suspension with 1.4 ml of 1.2 M glycine. Incubate the tube for 5 min at room temperature.
-
23
Centrifuge the cells for 5 min at 1,000g at 4 °C.
-
24
Remove the supernatant and wash the cells by repeating Steps 14 and 15.
-
25
Resuspend the cell pellet with 500 μl of equilibrium buffer. Transfer the suspension to a new 1.5-ml tube and leave it on ice for 15 min.
-
26
During the incubation, take 5 μl of the cell suspension for cell counting. Adjust the total number of mammalian cells to 1.5 × 106–2.5 × 106 for each sample using equilibrium buffer.
▲ CRITICAL STEP More than 2.5 × 106 mammalian cells could lead to inefficient cell lysis and poor quality of nuclei isolation. This limit can be increased fivefold for Drosophila S2 cells.
? TROUBLESHOOTING
■ PAUSE POINT The cell suspension can be left on ice for 1 h without affecting the library quality.
-
27
Centrifuge the cells for 3 min at 1,500g at 4 °C.
-
28
Discard the supernatant and resuspend the cells in 200 µl of ice-cold cell washing buffer.
-
29
Centrifuge the cells for 3 min at 1,500g at 4 °C.
-
30Prepare 300 μl of SDS buffer for each sample as shown in the table below.
Reagent Volume (μl) per reaction Final concentration Nuclease-free distilled water 261.6 10× Tango Buffer (supplied with AluI) 32 1× 10% (wt/vol) SDS 6.4 0.2% ▲ CRITICAL STEP The concentration of SDS in this buffer affects the efficiency of nuclei isolation. The concentration can be adjusted within a range from 0.1% to 0.5%, depending on the cell type used. A final concentration of 0.2% was used for the two mES cell lines used as examples and it is recommended as a starting concentration for condition optimization for a new cell type. Higher SDS concentration could de-crosslink and dissociate the fixed nuclear matrix.
-
31
Remove the supernatant and resuspend cells in 300 µl of SDS buffer.
-
32
Incubate the suspension for 10 min at 62 °C in a thermal mixer without any mixing.
▲ CRITICAL STEP Shaking of the suspension at this step usually leads to clumping of the cells, which may be difficult to disassociate.
-
33
Quench the SDS with 50 μl of 10% (vol/vol) Triton X-100 and incubate the samples on ice.
■ PAUSE POINT The nuclei suspension can be left on ice for 1 h without affecting the library quality.
-
34
Take 5 μl of the nuclei suspension for counting, using a hemocytometer under a microscope. High-quality nuclei isolation generally yields >80% cytoplasm-free nuclei. A typical GRID-seq experiment requires no less than ~1 × 106 cytoplasm-free nuclei in mammals or ~1 × 107 in Drosophila. Trypan blue can be used to assist visualization.
? TROUBLESHOOTING
Chromatin and RNA fragmentation ● Timing 4 h
-
35Centrifuge the nuclei for 3 min at 2,500g at 4 °C. Prepare 500 μl of the digestion reaction mixture for each sample as shown in the table below.
Reagent Volume (µl) per reaction Final concentration Nuclease-free distilled water 360 10× Tango Buffer (supplied with AluI) 50 1× 10% (vol/vol) Triton X-100 50 1% RiboLock RNase inhibitor 10 0.8 U/µl 100× protease inhibitor 5 1× AluI 25 0.5 U/µl Total 500 -
36
Remove the supernatant and resuspend the nuclei pellet in the reaction mixture.
-
37
Incubate the tube for 2 h at 37 °C in a thermal mixer with intermittent mixing at 800 r.p.m.
-
38
Take 20 µl of the digested nuclei for QC2, which evaluates the efficiency of chromatin fragmentation (Box 2).
-
39
Centrifuge the 480-µl sample for 3 min at 2,500g at 4 °C.
-
40During centrifugation of digested nuclei, prepare 400 μl of the end-repairing reaction mixture for each sample, as shown in the table below.
Reagent Volume (μl) per reaction Final concentration Nuclease-free distilled water 341 10× Tango Buffer (supplied with AluI) 40 1× ATP (100 mM) 4 1 mM RiboLock RNase inhibitor 10 1 U/µl T4 PNK (Thermo Fisher Scientific) 5 125,000 U/l Total 400 -
41
Remove and discard the supernatant and resuspend the nuclei pellet in the reaction mixture.
-
42
Incubate the tube for 1.5 h at 37 °C in a thermal mixer with intermittent mixing at 800 r.p.m.
In situ ligation of RNA and DNA ● Timing ~8 h
-
43
Centrifuge the tube for 3 min at 2,500g at 4 °C, and discard the supernatant.
-
44Prepare 400 μl of RNA pre-ligation buffer for each sample as shown in the table below. Always prepare this buffer immediately before use.
Reagent Volume (µl) per reaction Final concentration Nuclease-free distilled water 352 10× RNA Ligase Buffer (supplied with T4 RNA ligase
2, truncated KQ)40 1× 10% (vol/vol) Tween 20 4 0.1% RiboLock RNase inhibitor 4 0.4 U/µl Total 400 -
45
Resuspend the pelleted nuclei in 200 µl of the RNA pre-ligation buffer and wash by repeated pipetting of the suspension with a 200-µl pipette tip for about 20 times.
-
46
(Optional) Incubate the nuclei suspension for 5 min at 70 °C, and then cool the tube immediately on ice.
▲ CRITICAL STEP This step uniformly fragments RNA in situ. This step should be skipped if quality check and final results indicate strong RNA degradation.
-
47
Centrifuge the nuclei for 3 min at 2,500g at 4 °C. Discard the supernatant.
-
48
Wash the nuclei once more by repeating Steps 45 and 47, using another 200 µl of the RNA pre-ligation buffer.
-
49Prepare 330 µl of the ssRNA ligation mixture for each sample as shown in the table below.
Reagent Volume (μl) per reaction Final concentration Nuclease-free distilled water 252.5 10× RNA Ligase Buffer (supplied with T4 RNA ligase 2, truncated KQ) 50 1× Annealed linker (from Step 10) 25 ~0.4 µM RiboLock RNase inhibitor 12.5 1 U/µl Total 330 -
50
Resuspend the nuclei in the mixture and let it sit on ice for 5 min.
-
51
Mix the suspension with 10 µl of T4 RNA Ligase 2, truncated KQ.
-
52
Use a clean blade to cut the first 5 mm from the tip of a 200-µl pipette tip. Use the cut tip to transfer 150 µl of 50% (wt/vol) PEG-8000 (supplied with T4 RNA ligase 2, truncated KQ) to the suspension and mix by pipetting.
▲ CRITICAL STEP The 50% (wt/vol) PEG-8000 is viscous. Take care to gently homogenize the suspension after adding PEG. Do not vortex.
▲ CRITICAL STEP It is essential to add linker, ligase and PEG in the exact order specified. Pre-mixing these reagents may substantially decrease the linker ligation efficiency on the RNA side.
-
53
Incubate the reaction for 2 h at 25 °C in a thermal mixer with intermittent mixing at 1,000 r.p.m.
-
54Sequentially add the following reagents for primer extension to the reaction from Step 53 and mix by pipetting.
Reagent Volume (μl) per reaction Nuclease-free distilled water 29 2 M KCl 18 10 mM dNTP mix 32 5× RT First Strand Buffer (included in the SuperScript III Reverse Transcriptase) 28 100 mM DTT 28 SuperScript III RT (included in the SuperScript III First-Strand Synthesis System and the SuperScript III Reverse Transcriptase) 5 -
55
Incubate the reaction for 45 min at 50 °C in a thermal mixer with intermittent mixing (i.e., 10 s of mixing followed by 20 s of still incubation) at 1,000 r.p.m.
-
56
Centrifuge the tube for 5 min at 2,500g at 4 °C. Discard the supernatant.
-
57Prepare 400 µl of DNA pre-ligation buffer for each sample as shown in the table below.
Reagent Volume (μl) per reaction Final concentration Nuclease-free distilled water 352 10× DNA Ligation Buffer (supplied with T4 DNA ligase from New England Biolabs, without PEG) 40 1× RiboLock RNase inhibitor 4 0.4 U/µl 10% (vol/vol) Tween 20 4 0.1% Total 400 -
58
Resuspend the nuclei in 200 µl of the DNA pre-ligation buffer and wash by repeated pipetting of the suspension with a 200-µl pipette tip for about 20 times.
-
59
Centrifuge the tube for 3 min at 2,500g at 4 °C. Discard the supernatant.
-
60
Wash the nuclei once more by repeating Steps 58 and 59.
-
61Prepare 1.2 ml of dsDNA ligation mixture for each sample as shown in the table below.
Reagent Volume (μl) per reaction Final concentration Nuclease-free distilled water 924 10× DNA Ligation Buffer (supplied with T4 DNA ligase from New England Biolabs, without PEG) 120 1× BSA (20 mg/ml) 6 0.1 mg/ml 10% (vol/vol) Triton X-100 120 1% RiboLock RNase inhibitor 6 0.2 U/μl T4 DNA ligase (Thermo Fisher Scientific) 24 0.02 U/μl Total 1200 ▲ CRITICAL STEP Do not use the 10× DNA Ligation Buffer from Thermo Fisher Scientific in this reaction, as it contains PEG, which increases superfluous DNA–DNA ligation.
-
62
Resuspend the nuclei and incubate for at least 4 h at 25 °C in a thermal mixer with intermittent mixing at 800 r.p.m.
■ PAUSE POINT Alternatively, the reaction can be incubated at 16 °C overnight.
Reverse crosslinking and purification ● Timing ~6 h
-
63
Centrifuge the nuclei for 5 min at 2,500g at 4 °C. Remove the supernatant.
-
64
Resuspend the nuclei in 400 μl of 1× PBST and transfer the suspension to a fresh 1.5-ml tube.
-
65
Wash the nuclei by pipetting and then centrifuge for 5 min at 2,500g at 4 °C. Remove the supernatant.
-
66
Resuspend the nuclei in 133 μl of nuclease-free distilled water and 133 μl of 2× proteinase K buffer. Add 13.3 μl of proteinase K.
-
67
Incubate the reaction for 30 min at 65 °C in a thermal mixer with intermittent mixing at 800 r.p.m.
-
68
Add 20 μl of 5 M NaCl to the reaction and incubate for another 1.5 h.
-
69
Allow the tube to naturally cool to room temperature.
-
70
Transfer all the solution to a MaXtract high-density tube and mix with 300 μl of phenol/chloroform/isoamyl alcohol by shaking.
! CAUTION Phenol/chloroform/isoamyl alcohol is highly toxic. Always handle it in a fume hood.
▲ CRITICAL STEP Insufficient mixing may lead to incomplete phase separation and loss of the supernatant.
-
71
Centrifuge the tube for 5 min at 16,000g at 4 °C.
-
72
Transfer the supernatant to a new 1.5-ml tube. Add 2 μl of GlycoBlue and 30 μl of 3 M NaOAc, and mix well by pipetting.
-
73
Add 900 μl of 100% (vol/vol) ethanol and vortex thoroughly.
-
74
Allow the solution to precipitate for at least 30 min at −80 °C.
■ PAUSE POINT The solution can be left overnight and stored indefinitely at −80 °C.
-
75
Centrifuge the solution for 30 min at 16,000g at 4 °C. Remove the supernatant and air-dry the pellet for 3 min in a SpeedVac dryer.
-
76
Dissolve the pellet in 51 μl of nuclease-free distilled water. Take 1 μl of the nucleic acid solution to measure the concentration with a spectrophotometer. Expect a total yield of nucleic acid in the range of 5–20 μg.
▲ CRITICAL STEP If the yield exceeds 15 μg, the sample should be divided into two or more aliquots, and one of these should be stored at −20 °C. Start the following procedure with samples containing no more than 15 μg of nucleic acid.
-
77
Add the 50 µl of 2× B&W buffer to the sample solution and mix well. Hold the sample solution at room temperature.
-
78
Take 30 μl of Dynabeads MyOne Streptavidin C1 and mix it with 100 μl of 1× B&W buffer. Wash the beads by repeated pipetting of the suspension with a 200-µl pipette tip for about 20 times.
-
79
Isolate the beads with a magnetic separator and repeat the washing one more time. Equilibrate the beads in 200 μl of 1× B&W buffer at 37 °C in a thermal mixer with intermittent mixing at 800 r.p.m.
-
80
Combine the sample solution from Step 77 with the equilibrated bead suspension.
-
81
Incubate the suspension for 30 min at 37 °C in a thermal mixer with intermittent mixing at 800 r.p.m.
-
82
Magnetically isolate the beads for 1 min and discard the supernatant. Take the tube off the magnet and thoroughly wash the beads by pipetting with 500 μl of 1× B&W buffer.
-
83
Repeat the washing (Step 82) four times.
-
84
After the final wash, resuspend the beads in 100 μl of freshly prepared 150 mM NaOH and incubate for 10 min at 25 °C in a thermal mixer.
-
85
Isolate the beads and transfer the supernatant to a new 1.5-ml tube. Neutralize the supernatant with 11 μl of 10× TE buffer and 6.5 μl of 100% (vol/vol) acetic acid.
▲ CRITICAL STEP Take care not to include the beads in the supernatant.
-
86
Add 2 μl of GlycoBlue and 12 μl of 3 M NaOAc. Mix well by pipetting.
-
87
Add 120 μl of 100% (vol/vol) isopropanol and vortex thoroughly.
-
88
Allow the solution to precipitate for 1h at −20 °C.
▲ CRITICAL STEP A precipitation time of 1 h is usually sufficient. No significant increase in yield was observed for longer precipitation periods.
■ PAUSE POINT The precipitates in isopropanol can be stored indefinitely at −80 °C.
Paired-end tag generation by MmeI ● Timing ~6 h
-
89
Pellet the DNA by centrifugation for 30 min at 16,000g at 4 °C. Remove the supernatant.
-
90
Wash the DNA pellet with 75% (vol/vol) ethanol for 5 min and centrifuge for 10 min at 16,000g at 4 °C.
-
91
Remove all supernatant and air-dry the pellet for 3 min in a SpeedVac dryer.
-
92
Dissolve the pellet in 31 μl of nuclease-free distilled water.
-
93
Take 1 μl of the DNA solution to measure the concentration with a spectrophotometer. Expect a total yield of DNA in the range of 200 ng–1 μg.
▲ CRITICAL STEP Use the ‘ssDNA mode’ of the spectrophotometer to measure the DNA concentration. If the amount exceeds 1 μg, the sample should be divided into two or more aliquots and one of them should be stored at −20 °C. Start the following procedure with samples containing no more than 1 µg of ssDNA.
-
94
Transfer the solution to a 0.2-ml tube. Add 5 μl of the random hexamer primers (included in the SuperScript III First-Strand Synthesis System) and 5 μl of 10× CutSmart Buffer (supplied with rSAP) to the tube. Adjust the total volume to 48.5 µl with nuclease-free distilled water. Mix well by pipetting.
-
95
Incubate the tube for 5 min at 98 °C in a thermal cycler, and then immediately snap-chill the tube on ice.
-
96
Add 0.5 μl of 10 mM dNTP mix and 1 μl of Klenow fragment, 3′–5′ exo-, to the cold solution and mix by pipetting.
-
97
Incubate the reaction at 37 °C for 1 h in a thermal cycler.
-
98
Inactivate the enzyme by heating the tube for 10 min to 70 °C. Allow the tube to cool to room temperature naturally.
-
99
Freshly prepare 50× SAM by diluting 2 μl of 640× SAM stock (supplied with MmeI) with 23.6 μl of ice-cold nuclease-free water.
-
100
Add 2 μl of 50× SAM and 0.5 μl of MmeI to the cooled solution. Mix well by pipetting.
▲ CRITICAL STEP SAM is unstable at room temperature and should be diluted immediately before starting the reaction. We recommend using SAM that has been produced within <3 months.
-
101
Incubate the digestion reaction for 30 min at 37 °C in a thermal cycler.
-
102
Add another 2 μl of MmeI to the reaction and incubate it for 30 min.
-
103
Mix the reaction with 2 μl of proteinase K.
-
104
Incubate the reaction for 20 min at 65 °C in a thermal cycler. Allow the tube to cool to room temperature naturally.
-
105
Dilute the solution with 240 μl of nuclease-free water to a total volume of ~300 µl.
-
106
Repeat Steps 70–72.
-
107
Add 300 μl of 100% (vol/vol) isopropanol and vortex thoroughly. Allow the solution to precipitate for 30 min at −20 °C.
■ PAUSE POINT The precipitates in isopropanol can be stored indefinitely at −80 °C.
-
108
Centrifuge the solution for 30 min at 16,000g at 4 °C. Remove the supernatant and air-dry the pellet for 3 min in a SpeedVac dryer.
-
109
Dissolve the pellet in 10 μl of nuclease-free distilled water.
-
110
Add 2 μl of the 6× loading buffer and load all the solution into a 12% (wt/vol) native polyacrylamide gel. Load 2 μl each of 100-bp DNA ladder and 10-bp DNA ladder in parallel.
-
111
Perform gel electrophoresis in 1× TBE buffer at 100 V for 15 min and then at 120 V for ~45 min.
▲ CRITICAL STEP The 6× loading buffer contains purple dye and blue dye as size indicators. End the electrophoresis before the blue dye reaches the end of the gel.
-
112
Place the gel in 30 ml of 1× TBE buffer containing 1 μl of SYBR Gold, and stain for 10 min on a shaker set to 10 r.p.m. in a dark room.
-
113
Visually check the DNA bands resolved on the gel with a Dark Reader transilluminator. See Fig. 2a for an example gel image. This is QC3.
? TROUBLESHOOTING
-
114
Cut the 85-bp band with a new blade and cut another piece of similar size from the same lane at an ~100-bp location as the mock sample. Collect the slices into new 1.5-ml tubes.
▲ CRITICAL STEP Use a new blade to cut each gel piece if multiple samples are processed in parallel to avoid cross-contamination.
▲ CRITICAL STEP The mock sample serves as a critical indicator of quality control for the following adaptor ligation steps. Typically, the gel slice that is cut at ~100 bp as a mock sample contains only smeared background. However, the amount of background DNA it contains is adequate to serve the purpose of a control sample. Process the mock sample in parallel during the following steps (Steps 115–145). Only one mock sample is needed for an experiment with multiple samples.
-
115
Use a Squisher to pulverize the gel slices in the tubes and add 500 μl of gel elution buffer to resuspend the gel debris. Set the tubes on a rotator for at least 2 h at room temperature.
▲ CRITICAL STEP Incomplete pulverization of the gel leads to a decreased yield of DNA.
■ PAUSE POINT The elution can be left on the rotator overnight.
-
116
Transfer the eluent and gel debris to a 0.45-µm Spin-X tube filter and centrifuge it for 2 min at 12,000g at 4 °C.
-
117
Transfer the flow-through to a MaXtract high-density tube and mix it with 500 μl of phenol/chloroform/isoamyl alcohol by shaking.
! CAUTION Phenol/chloroform/isoamyl alcohol is highly toxic. Always handle it in a fume hood.
▲ CRITICAL STEP Insufficient mixing may lead to incomplete phase separation and loss of the supernatant.
-
118
Centrifuge the tube for 5 min at 16,000g at 4 °C.
-
119
Transfer the supernatant to a new 1.5-ml tube. Add 2 μl of GlycoBlue, 50 μl of 3 M NaOAc and mix well by pipetting.
-
120
Add 500 μl of 100% (vol/vol) isopropanol and vortex thoroughly. Allow the solution to precipitate for at least 30 min at −20 °C.
■ PAUSE POINT The solution can be left overnight and stored indefinitely at −80 °C.
Adaptor ligation and PCR ● Timing ~8 h
-
121
Pellet the DNA precipitates and wash the pellet by repeating Steps 89–91.
-
122
Dissolve the pellet in 10 μl of nuclease-free distilled water and transfer the solution to a 0.2-ml tube.
-
123
Mix the solution with 1.1 µl of 10× CutSmart Buffer (supplied with rSAP) and 0.5 μl of rSAP.
-
124
Incubate the de-phosphorylation reaction for 30 min at 37 °C in a thermal cycler.
-
125
Inactivate the enzyme by heating the solution for 5 min at 65 °C. Allow the tube to cool to room temperature naturally.
-
126Add and mix the following reagents sequentially with each of the samples.
Reagent Volume (μl) per reaction 10× CutSmart Buffer (supplied with rSAP) 2 ATP (100 mM) 0.5 Annealed adaptor (A1/A2; Step 12; 25 µM) 2.4 T4 DNA ligase (New England Biolabs, high concentration) 2 24% (wt/vol) PEG-6000 (supplied with T4 PNK from Thermo Fisher Scientific) 9.6 ▲ CRITICAL STEP Thoroughly mix the MmeI-digested DNA and the adaptors before adding the ligase and PEG. Adding ligase directly to the digested DNA or premixing ligase and adaptors results in substantially decreased yield of the desired product.
-
127
Incubate the reaction for 1 h at 25 °C in a thermal cycler.
-
128Add the following reagents sequentially to each of the samples. A supplement of T4 PNK at this step improves the efficiency of ligation and nick-sealing.
Reagent Volume (μl) per reaction ATP (100 mM) 0.5 DTT (100 mM) 2 T4 PNK (New England Biolabs) 2 -
129
Incubate the reaction for 1 h at 25 °C in a thermal cycler.
-
130Add the following reagents sequentially to each of the samples. A supplement of T4 DNA ligase at this step improves the efficiency of ligation and nick-sealing.
Reagent Volume (μl) per reaction Nuclease-free distilled water 2.1 10× CutSmart Buffer (supplied with rSAP) 1 ATP (100 mM) 0.4 T4 DNA ligase (New England Biolabs, high concentration) 1.5 -
131
Incubate the reaction for 1 h at 25 °C in a thermal cycler.
-
132
Dilute the solution with 240 μl of nuclease-free distilled water to a total volume of about 300 µl.
-
133
Perform DNA extraction and resuspension by repeating Steps 70–74 and 108 and 109.
▲ CRITICAL STEP GlycoBlue coprecipitant may form a precipitate with PEG; this does not affect the yield of nucleic acids.
■ PAUSE POINT The precipitation reaction can be left at −20 °C overnight. The DNA in isopropanol can be stored indefinitely at −80 °C.
-
134
Add 2 μl of the 6× loading buffer to the sample solution and load all into a 10% (wt/vol) native polyacrylamide gel. Load 2 μl each of 100-bp DNA ladder and 10-bp DNA ladder in parallel.
▲ CRITICAL STEP The mock sample solution should be resolved in parallel in the same gel.
-
135
Perform gel electrophoresis in 1× TBE buffer at 100 V for 15 min and then at 120 V for ~1 h.
▲ CRITICAL STEP The 6× loading buffer contains purple dye and blue dye as size indicators. End the electrophoresis before the blue dye reaches the end of the gel.
-
136
Place the gel in 30 ml of 1× TBE buffer containing 1 μl of SYBR Gold, and stain for 10 min on a shaker in a dark room.
-
137
Visually check the DNA bands resolved on the gel with a Dark Reader transilluminator. See Fig. 2b for an example gel image. This is QC4.
-
138
Cut the single band typically located within the range of 175–190 bp. Collect the gel slices in 1.5-ml tubes. Cut a gel slice of similar size at the same position on the mock sample lane that lacks the desired band; this will be the new mock sample.
▲ CRITICAL STEP Use a new blade to cut each band if multiple samples are processed in parallel to avoid cross-contamination.
▲ CRITICAL STEP Due to the secondary structures formed at the unannealed side of the Y-shaped adaptors, the actual size of the molecules may differ from the observed molecular weight indicated by the DNA ladder on a native gel. Depending on the composition of the gel and buffer, the resolved location of the desired band may vary. Thus, it is essential to check that the desired band is missing at the corresponding position in the lane resolving the mock sample (Fig. 2b).
? TROUBLESHOOTING
-
139
Extract DNA from the gel fragments and precipitate the DNA by repeating Steps 115–120.
■ PAUSE POINT The solution can be left overnight and stored indefinitely at −80 °C.
-
140
Pellet the DNA precipitates and wash the pellet by repeating Steps 89–91.
-
141
Dissolve the pellet in 20 μl of nuclease-free distilled water.
■ PAUSE POINT This DNA solution is the pre-PCR library, which is enough for four sets of PCR amplifications. The leftover should be well labeled and can be stored at −80 °C indefinitely.
-
142Prepare the 20-µl PCR amplification mixture as shown in the table below and add to each sample to be amplified. The mock sample should be included for PCR in parallel if amplifying the libraries for the first time.
Reagent Volume (μl) Per reaction Final concentration Nuclease-free distilled water 8.4 Pre-PCR library 5 5× Phusion HF Buffer (supplied with Phusion High-Fidelity DNA polymerase) 4 1× Phusion High-Fidelity DNA polymerase 0.2 0.4 U/µl 10 mM dNTP mix 0.4 0.2 mM Primer1 (10 µM; Table 1) 1 0.5 µM Primer2 (10 µM; Table 1) 1 0.5 µM Total 20 -
143Use the following thermal cycling to perform PCR amplification.
Cycle number Denature (98 °C) Anneal (65 °C) Extend (72 °C) 1 30 s 2–15 10 s 30 s 15 s 16 5 min ▲ CRITICAL STEP We routinely amplify libraries for 16 cycles. Total cycle number can be increased up to 20 to increase the yield. However, doing so may give rise to duplicates and substantially decrease the library complexity.
-
144
Prepare the sample and perform electrophoresis by repeating Steps 134–136.
-
145
Visually check the DNA bands resolved on the gel with a Dark Reader transilluminator. See Fig. 2c for an example gel image. This is QC5.
▲ CRITICAL STEP The mock sample may show a distinct band at ~100 bp, which is the amplified adaptor-dimer, but no band should be visible above this size (Fig. 2c).
-
146
Cut the 188-bp PCR product. Collect the gel slices in 1.5-ml tubes.
▲ CRITICAL STEP Use a new blade to cut each band if multiple samples are processed in parallel to avoid cross-contamination.
-
147
Extract the DNA from the gel fragments and precipitate the DNA by repeating Steps 115–120.
■ PAUSE POINT The solution can be left overnight and stored indefinitely at −80 °C.
-
148
Pellet the DNA precipitates and wash the pellet by repeating Steps 89–91.
-
149
Dissolve the pellet in 21 μl of nuclease-free distilled water.
-
150
Take 1 μl of the barcoded library and measure the concentration by a Qubit fluorometer, following the manufacturer’s instructions. The typical yield for each sample is in the range of 10–80 ng.
? TROUBLESHOOTING
■ PAUSE POINT This solution is the final barcoded library ready for Illumina sequencing. It can be stored indefinitely at −80 °C.
Deep sequencing ● Timing ~1 d
-
151
Sequence the pooled GRID-seq libraries with an Illumina HiSeq 2500 sequencer. We sequence for 100 bp through the linker, using single-end mode. A library from human or mouse cells typically requires a sequencing depth of ~200 million reads.
Processing of raw reads ● Timing ~12 h
▲ CRITICAL We use ‘test’ as a placeholder name for GRID-seq files and the prefix of resultant file names. Users should replace it with their real file name. Demo data with the name ‘gridseq.test10M.raw. fq.gz’ can also be downloaded from the link in the ‘Equipment’ section. A list of the programs and parameters used in the analysis and explanations of parameter functions can be found in Table 2.
-
152Trim empty bases and the adaptor sequences in FASTQ files, using the Cutadapt package18 as follows:
cutadapt -l 86 --max-n 5 -o test.trimmed.fq.gz test.fq.gz
▲ CRITICAL STEP Length of the input reads must be kept to ≥86 bp, including empty bases denoted as ‘N’.
-
153Construct the BWA index file of the linker sequence as GRIDv1.fa (sequence in RNA–- linker–DNA orientation) using BWA19.
bwa index -p GRIDv1 GRIDv1.fa
-
154Align reads to the linker, using the BWA mem module as follows:
bwa mem -k 5 -L 4 -B 2 -O 4 -p GRIDv1 test.trimmed.fq.gz | / samtools view -ub | samtools sort -m 4G -o test.GRIDv1.bam samtools index test.GRIDv1.bam
-
155Separate the RNA and DNA read pairs, using GridTools.py matefq, generating an interleaved FASTQ file that records each RNA read followed by its paired DNA read in a single file.
GridTools.py matefq -l 19 -n Test -o test.h5 test.GRIDv1.bam | / gzip –c > test.mate.fq.gz
Identification of chromatin-associated RNAs and background signals ● Timing ~12 h
-
156Align read pairs to the genome (GRCm38 build for mouse used as an example), using BWA and use SAMtools20 to filter unambiguously mapped reads and mark duplicated read mates.
bwa mem -k 17 -w 1 -T 1 -p mm10_index test.mate.fq.gz | / samtools sort -n -m 1G | / samtools fixmate -pm - - | / samtools view -bf 0×1 | // samtools sort -m 1G | // samtools markdup - test.mate.srt.bam samtools index test.mate.srt.bam samtools sort -n -m 1G -o test.mate.mrk.bam test.mate.srt.bam
▲ CRITICAL STEP The genome index files prefixed with mm10_index should be prepared with reference genome files in FASTA format by BWA before Step 156, using the command bwa index -p mm10_index mm10.fa, according to the BWA software manual.
-
157Quality-check the alignment and generate metadata using GridTools.py evaluate:
GridTools.py evaluate -k 1 -m 10 -g mm10.gtf.gz -o test.h5 test.mate. mrk.bam
▲ CRITICAL STEP The gene annotation file mm10.gtf.gz needs to be indexed by tabix before Step 157, using the command grep -v ^”#” mm10.gtf | sort -k1,1 -k4,4n | bgzip > mm10.gtf.gz && tabix -p gff mm10.gtf.gz, according to the tabix software manual.
-
158Generate metadata of the GRID-seq library, including read count; length distribution of RNA, DNA and linker sequences; nucleotide frequency; sequencing quality distribution; and genomic bin-wise signal correlation at various resolutions by using GridTools.py stats. Some of these statistics are illustrated in Fig. 4a–c,f.
GridTools.py stats -bclr -p test.stats test.h5
? TROUBLESHOOTING
-
159Calculate the gene expression and features of RNAs using GridTools.py RNA. It identifies enriched genes on the basis of RNA quantification and generates distribution of RNA interaction range and strand distributions of RNA and the paired DNA reads.
GridTools.py RNA -e test.gene_exprs.txt -s test.gene_scope.txt test.h5
? TROUBLESHOOTING
-
160Quantify genome accessibility for DNA using GridTools.py DNA. Background and specific interaction signals (foreground) are reported as log2 transformed B values, indicating read density in the genomic bins normalized by total number of reads and sizes of chromosomes.
GridTools.py DNA test.h5 | gzip -c > test.dna.txt.gz
Construction of RNA–chromatin contact matrix ● Timing ~6 h
-
161Calculate the interaction density (V values) of all chromatin-associated RNA and genomic bins and generate the contact matrix using GridTools.py matrix as follows:
GridTools.py matrix -k 100 -x 10 test.h5 | gzip -c > test.matrix.gz
Further downstream analysis can be performed based on the contact matrix, including modeling an enhancer–promoter proximity network (Box 3).
Troubleshooting
Troubleshooting advice can be found in Table 3.
Table 3 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 10, Box 1 | Low yield of annealed linker | Oligonucleotide was incompletely dissolved or problems in oligo synthesis | Check the quality of the single-stranded oligonucleotide by measuring concentrations and/or resolve a 5-pmol sample on a denaturing polyacrylamide gel |
| Incomplete annealing | Double-check the sequences of oligonucleotides, salt concentration of annealing reaction and the ramping speed of the thermal cycler; HPLC-purify oligos | ||
| Incomplete precipitation | All precipitations require at least 30 min incubation at <4 °C. Longer time of incubation may improve yield for short DNA molecules | ||
| 26 | Loss of cells (>50% loss, visually estimated by cell pellet size) | Incomplete pelleting of cells | Increase the starting number of cultured cells; use a swinging-bucket rotor for centrifuging cells instead of a fixed-angle rotor; centrifugation time and speed can be increased up to 5 min at 2,300g |
| Fixed cells stick to the tube during centrifugation | Try different types of conical tubes; use PBST instead of PBS | ||
| 34 | Loss of nuclei (>50% loss, visually estimated by nuclei pellet size) | Incomplete pelleting of nuclei | Increase the starting number of cultured cells; put the 1.5-ml LoBind tube inside a 50-ml conical tube and then use swinging-bucket rotors for centrifuging; centrifugation time and speed can be increased up to 5 min at 2,300g |
| SDS concentration was too high | Incubating fixed cells with SDS at 62 °C may dissolve the cell membrane as well as the fixed nuclear matrix, leaving only debris in the solution. The SDS concentration should be decreased in such cases | ||
| >50% of cells with cytoplasm intact around the nuclei | Cells are not in a single-cell suspension | Separate the cells by pipetting with smaller pipette tips (e.g., 20- to 200-µl tips) or by passing them through a syringe and checking under a microscope before adding SDS | |
| The SDS concentration is too low to efficiently lyse cell membranes | SDS concentration can be increased up to 0.5% (wt/vol) | ||
| 113 | No band visible and/or band obscured by a strong background | DNA impurity | Perform DNA extraction and use ethanol to wash and precipitate the DNA again at −20 °C overnight after Step 107 |
| Too much input DNA | DNA input can be further decreased to 500 ng after Step 93 | ||
| Incomplete ssDNA release from Dynabeads MyOne Streptavidin C1 | Use fresh Dynabeads MyOne Streptavidin C1 and 150 mM NaOH; pool samples if the concentration in Step 93 is <200 ng | ||
| Insufficient washing of Dynabeads MyOne Streptavidin C1 | Increase the washing time and strength by vortex or rotation | ||
| Low efficiency of MmeI digestion | Add more MmeI by repeating Step 102 before adding proteinase K in Step 103; it is recommended to estimate the molar amount of restriction sites in the DNA sample and adjust MmeI amount to close to a 1:1 molar ratio; replace and use fresh SAM | ||
| DNA ligation and/or RNA ligation failed | Replace relevant reagents; check linker pre-adenylation efficiency | ||
| No 85-bp band, but the 65-bp band is present | DNA ligation failed | Replace relevant reagents. Note that DTT may be degraded in old ligase buffer | |
| Nothing on gel | Low amount of input DNA | Pool samples if concentration in Step 93 is <200 ng | |
| 138 | Specific band in 175- to 190-bp range is not missing in the mock sample | Oligo A1 and A2 were not annealed | Check annealed adaptors in an 18% native polyacrylamide gel |
| Incomplete precipitation of input DNA | Increase isopropanol precipitation time to 3 h; detergent sometimes affects low-quantity DNA precipitation steps after gel extraction. Remove the detergent by phenol–chloroform extraction before precipitation, if necessary | ||
| Desired band migrates out of the 175- to 190-bp range | This can happen if the electrophoresis conditions are different. Any band that is clearly missing in the mock control should be purified for PCR | ||
| Strong background smear and/or multiple undesired bands | Impurity of the paired-end tags | Cut the 85-bp band as close as possible in Step 114 to avoid gDNA contamination | |
| Low paired-end tag input | Multiple 85-bp bands in Step 114 from the same sample could be pooled to increase input DNA amount for adaptor ligation | ||
| 150 | Low yield of the amplified 188-bp DNA | Incomplete precipitation of input DNA | Increase isopropanol precipitation time to 3 h |
| Low amount of input DNA | Pool parallel adaptor-ligated samples to increase the input template amount for PCR | ||
| Strong background smear and/or multiple unspecific bands | Impurity of the adaptor-ligated library | Cut the adaptor-ligated band as close as possible in Step 138 to avoid adaptor-ligated gDNA contamination | |
| Usage of a different polymerase | Use Phusion polymerase from Thermo Fisher Scientific (cat. no. F530S). Phusion polymerase from other manufactures generated a much higher background with the same pre-PCR library | ||
| 158 | High level of duplicated mate-pair reads (>10%) | Low complexity of the library due to excessive PCR amplification | Reduce the number of PCR cycles |
| Low PCC at 1 kb/bin (<0.5) | Low complexity of the library | Pool parallel adaptor-ligated samples to increase the input template amount for PCR | |
| 159 | Low number of chromatin-associated RNA species (<500) | Low complexity of the library | Pool parallel adaptor-ligated samples to increase the input template amount for PCR |
| Box 1, step 6 | The RNA stretch is missing on the polyacrylamide gel | RNA degradation | The RNA stretch of oligo L1 is labile. Repeated freeze–thaw cycles and nuclease contamination should be avoided |
| Nuclease contamination in the polyacrylamide gel | Use fresh reagents and nuclease-free water to make gels and staining solutions | ||
| Box 2, step 15 | Too many DNA fragments >1 kb | Insufficient permeabilization of the nuclei | Increase the SDS concentration in Step 30 up to 0.5% (wt/vol) |
| Low AluI activity | Use no more than 2.5 × 106 mammalian or 1.3 × 107 Drosophila nuclei for each digestion reaction; use fresh enzymes; increase the time for AluI digestion |
Timing
Steps 1–12, preparation of linker, adaptors and cell culture: 1 d Steps 13–34, cell harvest and dual crosslinking: 3 h
Steps 35–42, chromatin fragmentation: 4 h
Steps 43–62, in situ ligation of RNA and DNA: ~8 h Steps 63–88, reverse crosslinking and purification: ~6 h Steps 89–120, paired-end tag generation by MmeI: ~6 h Steps 121–150, adaptor ligation and PCR: ~8 h
Step 151, deep sequencing: ~1 d
Steps 152–155, processing of raw reads: ~12 h
Steps 156–160, identification of chromatin-associated RNAs and background signals: ~12 h Step 161, construction of interactome matrices: ~6 h
Box 1, quality-control checkpoint 1 for linker integrity: 3 h
Box 2, quality-control checkpoint 2 for AluI digestion efficiency: 3 h Box 3, modeling the network of enhancer–promoter proximity: 3 h
Anticipated results
A successful mammalian GRID-seq dataset typically consists of ~50 million uniquely mapped read pairs, filtered from 250–400 million raw reads (Fig. 4a). More than 90% of RNA and DNA reads should be 20 or 21 bp in length (Fig. 4b). Nucleotides should be uniformly distributed in RNA and DNA, except for the first two bases of DNA, which correspond to half of the AluI recognition motif (Fig. 4c). Generally, at least 500 genes are detected that transcribe chromatin-associated RNAs, ~90% of which are protein-coding genes (Fig. 4e), with RNA reads mapped to both exons and introns. However, as shown in the example, the number of detected RNAs is highly dependent on the library complexity and sequencing depth. RNA reads should map only to the sense strand of the reference genome that produces the transcripts, but DNA reads should map to the reference genome without strand specificity (Fig. 4d). The final product of the method is a genome-wide interaction map of RNA on chromatin, in the format of a 2D matrix in which each gene is one row and each genomic bin is one column. A typical replicate-combined interactome matrix should have a median PCC value >0.8 at a 1 kb/bin resolution (Fig. 4f). Values in the matrix represent the background-corrected interaction density (V values) (Fig. 5b). Such a matrix can be used for generating global and local GRID-seq interaction maps, as well as for analyzing detailed binding events of specific RNAs (Fig. 5b–d).
Additional evaluation of GRID-seq experiments can be performed to analyze the agreement between biological replicates. Direct comparison of uniquely mapped reads between replicated datasets should have a high concordance (R2 ≥ 0.90) of RNA reads for each gene (Supplementary Fig. 2a) and DNA reads aligned to binned genomes (Supplementary Fig. 2b). Similarly, the datasets should score a higher agreement (R2 ≥ 0.98) of background signals (Supplementary Fig. 2c) and foreground signals (Supplementary Fig. 2d) between replicates, as compared to that of DNA or RNA reads. The sets of detected chromatin-associated RNA species can be different between replicates due to the different read volume and complexity of each replicate. Due to the increased read depth in the combined data, more RNAs pass the threshold shown in Supplementary Fig. 1 and are identified as chromatin-associated RNAs as compared with those in each of the replicates (Supplementary Fig. 2e, f). The replicated interactome matrix can also be visualized to assess robustness at different resolutions (Supplementary Fig. 2g,h).
In most cases, cis-acting RNAs display the highest interaction density on their genic regions of origin. The density declines gradually along the linear genome, but specific binding peaks can still be detected away from their transcribing loci. Such peaks of cis-acting RNAs reflect the spatial proximity of underlying chromatin loci to the site of transcription for such RNAs. Based on this principle (Box 3a) and annotation of enhancers and active promoters, a network of enhancer–promoter proximity can be inferred (Box 3b). It is common that a single RNA species can link multiple promoters to enhancers, forming a transcription hub (an example is shown in Box 3c). In some cases, linked enhancers and promoters are mega-bases away in linear genomic distance.
Supplementary Material
Acknowledgements
We thank B. Ren (University of California, San Diego) for the generous gifts of C57BL/6 mouse ES cells and F1 Mus musculus castaneus × S129/SvJae mouse ES cells. This work was supported by NIH grants (HG004659, HG007005, GM049369 and DK098808) to X.-D.F.; a grant from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16010113) to B.Z.; grants from the National Science Foundation of China (31872191, 31472263) and the China Scholarship Council (201606275037) to D.L.; and a grant from the National Natural Science Foundation of China (91640115) to Y.Z.
Footnotes
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
The in-house software, GridTools, is publicly available in GitHub at https://github.com/biz007/gridtools under an MIT license. It also houses a demo code for the F123 mESC test dataset. All analyses of the results and the performance evaluation of the pipeline in the current study were based on GridTools v.1.1 and are also available as Supplementary Software.
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41596-019-0172-4.
Journal peer review information: Nature Protocols thanks Xiaohua Shen and other anonymous reviewer(s) for their contribution to the peer review of this work.
Data availability
Data obtained with B6 mESCs have been deposited in the Gene Expression Omnibus under accession no. GSE82312, and a test dataset on the F123 mESCs is accessible at http://fugenome.ucsd.edu/gridseq/datasets/gridseq.test10M.raw.fq.gz.
References
- 1.Djebali S et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu XD Non-coding RNA: a new frontier in regulatory biology. Natl. Sci. Rev 1, 190–204 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rinn JL & Chang HY Genome regulation by long noncoding RNAs. Annu. Rev. Biochem 81, 145–166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen TC, Zaleta-Rivera K, Huang X, Dai X & Zhong S RNA, action through interactions. Trends Genet 34, 867–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu C, Qu K, Zhong FL, Artandi SE & Chang HY Genomic maps of long noncoding RNA occupancy reveal principles of RNA–chromatin interactions. Mol. Cell 44, 667–678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simon MD et al. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 108, 20497–20502 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engreitz JM et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X et al. GRID-seq reveals the global RNA–chromatin interactome. Nat. Biotechnol 35, 940–950 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sridhar B et al. Systematic mapping of RNA–chromatin interactions in vivo. Curr. Biol 27, 602–609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell JC et al. Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. Elife 7, e27024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang R et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 26, 1345–1348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saha S et al. Using the transcriptome to annotate the genome. Nat. Biotechnol 20, 508–512 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Fullwood MJ et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462, 58–64 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 3–12 (2011). [Google Scholar]
- 19.Li H & Durbin R Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data obtained with B6 mESCs have been deposited in the Gene Expression Omnibus under accession no. GSE82312, and a test dataset on the F123 mESCs is accessible at http://fugenome.ucsd.edu/gridseq/datasets/gridseq.test10M.raw.fq.gz.