

Hybrids optimize metabolic abundance through diurnal nonadditive expression of key enzymes in photosynthetic and photorespiratory pathways to enhance photosynthetic efficiency and plant growth.
Abstract
Heterosis or hybrid vigor is widespread in plants and animals. Although the molecular basis for heterosis has been extensively studied, metabolic and proteomic contributions to heterosis remain elusive. Here we report an integrative analysis of time-series metabolome and proteome data in maize (Zea mays) hybrids and their inbred parents. Many maize metabolites and proteins are diurnally regulated, and many of these show nonadditive abundance in the hybrids, including key enzymes and metabolites involved in carbon assimilation. Compared with robust trait heterosis, metabolic heterosis is relatively mild. Interestingly, most amino acids display negative mid-parent heterosis (MPH), i.e., having lower values than the average of the parents, while sugars, alcohols, and nucleoside metabolites show positive MPH. From the network perspective, metabolites in the photosynthetic pathway show positive MPH, whereas metabolites in the photorespiratory pathway show negative MPH, which corresponds to nonadditive protein abundance and enzyme activities of key enzymes in the respective pathways in the hybrids. Moreover, diurnally expressed proteins that are upregulated in the hybrids are enriched in photosynthesis-related gene-ontology terms. Hybrids may more effectively remove toxic metabolites generated during photorespiration, and thus maintain higher photosynthetic efficiency. These metabolic and proteomic resources provide unique insight into heterosis and its utilization for high yielding maize and other crop plants.
INTRODUCTION
Heterosis or hybrid vigor refers to superior growth or fitness in the hybrid progeny compared with one or both parents (Birchler et al., 2010; Chen, 2013; Schnable and Springer, 2013; Hochholdinger and Baldauf, 2018). The use of heterosis has successfully improved yield and quality in maize (Zea mays), which is also an excellent model plant for studying heterosis (Duvick, 2001). Maize hybrids exhibit extraordinary heterosis in many traits from sugar content to plant height, biomass, and grain yield (Springer and Stupar, 2007; Paschold et al., 2010; Ko et al., 2016; Li et al., 2018). Biomass heterosis in seedlings is established shortly after germination (Ko et al., 2016). In over a century of research, genetic models such as “dominance” and “overdominance” have been extensively studied but cannot fully explain heterosis; it might be better to jump outside these theoretical dogmas to investigate heterosis (Birchler et al., 2010).

Genome-wide gene expression studies have revealed possible roles of transcription-level regulation in heterosis, including additive gene expression (Guo et al., 2006; Springer and Stupar, 2007; Thiemann et al., 2014), nonadditive gene expression (Swanson-Wagner et al., 2009; Jahnke et al., 2010; Baldauf and Marcon, 2016), and single-parent expression (Baldauf et al., 2018). These observations of different gene expression modes may reflect complexity of heterosis and could be related to low correlations between mRNA and protein abundance levels (de Sousa Abreu et al., 2009; Vogel and Marcotte, 2012), which could be as low as ∼5% of the ∼2,700 examined protein and mRNA levels in maize (Jiang et al., 2019). As the end products of gene expression regulatory processes, proteins and small molecules (metabolites) play a direct role in plant cell structure and metabolism, which can shape plant phenotypes including heterosis (Goff and Zhang, 2013; Kosová et al., 2015).
Metabolomic analysis shows large natural variation across different genotypes in maize (Riedelsheimer et al., 2012; Xu et al., 2019; Zhou and Kremling, 2019), and metabolite variation is implicated in heterosis. For example, differential accumulation of metabolites has been observed between hybrids and parents in maize (Römisch-Margl et al., 2010; Wang et al., 2014), Arabidopsis (Arabidopsis thaliana; Korn et al., 2010; Meyer et al., 2012), and Brassica juncea (Bajpai and Reichelt, 2019). Compared with inbreds, maize hybrids show lower metabolite variability in root tissue with all possible inheritance patterns including additive and dominance (Lisec et al., 2011). A recent simulation study shows that it is possible to explain most of the biomass heterosis in Arabidopsis F1 hybrids by additive effects coupled with epistasis through a large metabolic network (Vacher and Small, 2019). Interestingly, many metabolites in Arabidopsis show diel rhythms (24-h) and are under circadian regulation, especially for those involved in photosynthetic pathway (Kim et al., 2017).
Compared with gene expression, protein studies are limited in maize leaves (Facette et al., 2013) and roots (Marcon et al., 2015). Using isobaric tags for a relative and absolute quantitation-based approach, ∼2,700 proteins identified concurrently have reliable mRNA abundance measurements in 98 maize inbreds (Jiang et al., 2019). Comparative proteomics has inferred heterosis in maize (reviewed in Xing et al., 2016). For example, of the 970 proteins examined in maize seminal roots, 85 display nonadditive accumulation in reciprocal hybrids between B73 and Mo17 (Marcon et al., 2013). Moreover, changes in the total and the mitochondrial proteome can also contribute to heterosis in maize hybrids (Dahal et al., 2012). However, the relationship between proteome and metabolome, and their roles in heterosis, remain largely unknown.
The circadian clock controls growth and development in plants and animals (Bendix et al., 2015; Chen and Mas, 2019) through rhythmic transcriptome changes (Hazen et al., 2009; Khan et al., 2010; Staiger et al., 2013), which are transduced into protein rhythms and further metabolite oscillations in a variety of signaling pathways (Chen and Mas, 2019). The circadian clock plays a role in heterosis (Chen, 2013). In Arabidopsis allotetraploids and Arabidopsis F1 hybrids, expression of key circadian clock genes in the central clock feedback loop is altered during the day by chromatin modifications to promote expression of clock output genes involved in chlorophyll, photosynthesis, and starch metabolism (Ni et al., 2009). As a result, more chlorophyll, sugar, and starch are produced during the day, and more starch can be degraded and used at night to promote growth. As a trade-off for growth, circadian-mediated temporal repression of stress-related genes (Miller et al., 2015) and ethylene biosynthesis and signaling genes (Song et al., 2018) also contributes to biomass heterosis. In maize, the binding activities of CIRCADIAN CLOCK ASSOCIATED1 (ZmCCA1) in the hybrids relative to inbred parents is temporally shifted toward early morning, which can activate morning-phased genes to promote photosynthesis and growth (Ko et al., 2016). However, the role for the circadian clock in regulating protein and metabolic rhythms remains to be tested.
In this study, we investigated temporal regulation of metabolome and proteome in maize hybrids relative to their parental inbreds. Notably, a large fraction of the maize metabolome and proteome is under diel (and possibly diurnal) regulation and displays distinct patterns between the F1 hybrids and inbred parents. These differences in metabolic and proteomic levels may contribute to heterosis. Particularly, by diurnally regulating the nonadditive expression of key enzymes involved in photosynthetic and photorespiratory pathways, hybrids may optimize the balance of metabolite abundance to promote carbon assimilation and plant growth.
RESULTS
Diel Regulation and Distinct Patterns of the Metabolome in Maize Hybrids and Their Parents
Metabolite levels were examined in the seedlings 9 d after planting, DAP) of maize inbreds B73 and Mo17, their F1 hybrid (B73XMo17, BM), and the reciprocal hybrid Mo17XB73 (MB) with three biological replicates every 3 h within one diel cycle. Using nontargeted gas chromatography time-of-flight mass spectrometry (GC-TOF-MS; Morgenthal et al., 2005), we identified 447 nonredundant metabolites including 191 annotated metabolites for each genotype at six day-time and three night-time points (Supplemental Data Set 1). Metabolome data generated in this study were generally of high quality, with coefficient of variation across biological replicates <0.1 for 90% of the metabolites (Supplemental Figure 1).
We performed a principal component analysis (PCA) of the identified metabolites across the nine time points (Figure 1A). Notably, metabolites in inbreds and hybrids were clearly separated from each other (corresponding to PC2), demonstrating the existence of major metabolomic differences between the hybrids and their parents. Although the data were limited to one diel cycle, the separation of metabolites between day- and night time points in inbreds and hybrids (corresponding to PC1) indicates potential diurnal regulation of metabolites in maize. The metabolomes between the two inbred parents were more different than between the two hybrids, which were in the middle, between those of the parents.
Figure 1.
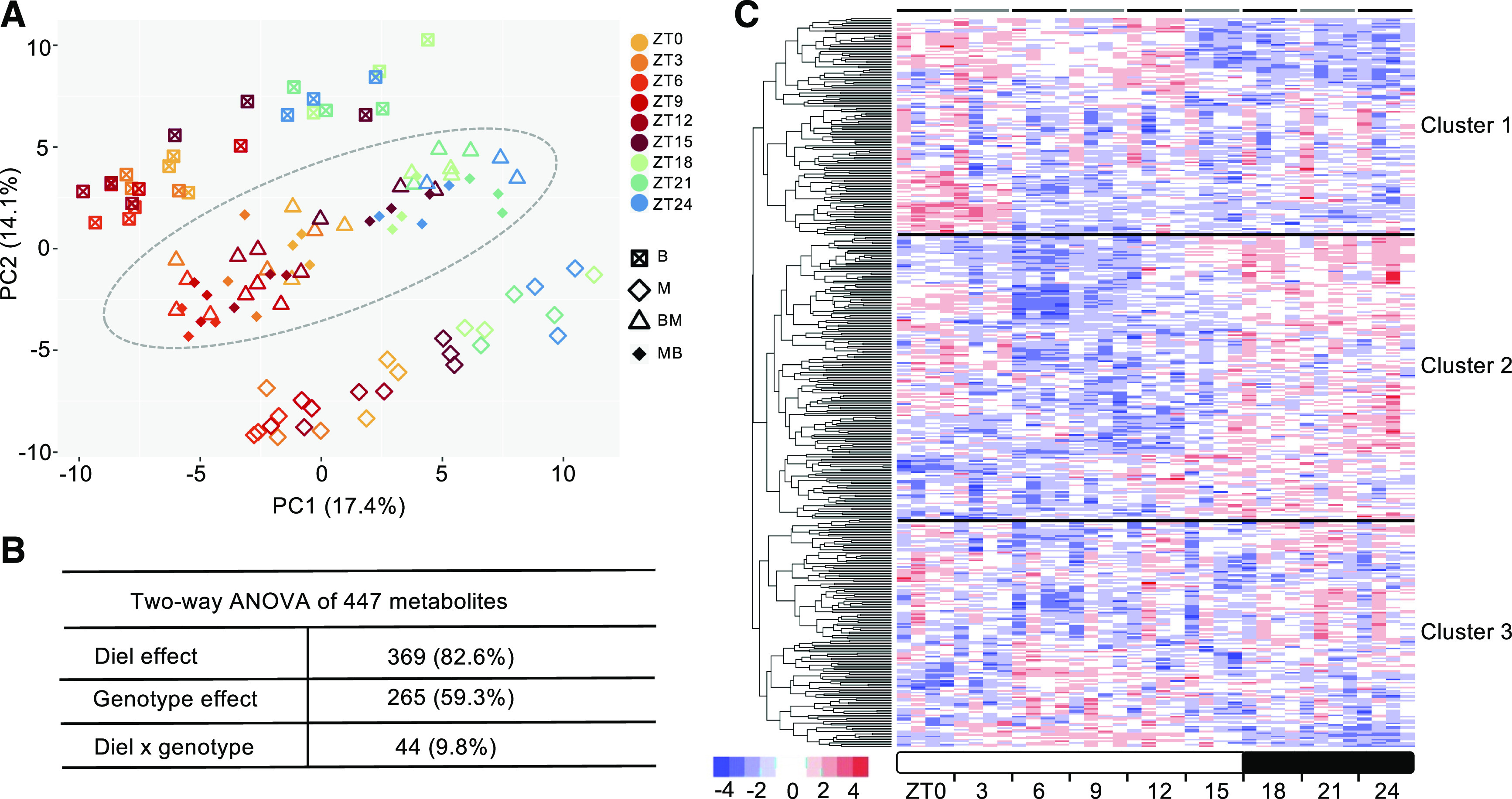
Patterns of Temporal and Genotypic Effects on Maize Metabolomes.
(A) PCA of maize metabolomes across nine time points in four genotypes (B73, Mo17, and two reciprocal hybrids, F1BM and F1MB) at 9 DAP. Biological replicates were shown as individuals. Hybrid samples were indicated by the ellipse; day- and night-time points are ZT0 to ZT15 and ZT18 to ZT24, respectively.
(B) Two-way ANOVA of 447 metabolites, showing diel or genotype effect at a statistically significant level (P < 0.05).
(C) Hierarchical clustering analysis of metabolic abundance profiles throughout the diel cycle. Metabolic abundance was averaged, and z-score transformed. Each bar (time point) consists of four columns representing B73, Mo17, F1BM, and F1MB (left to right). GO enrichment of metabolic pathways in each cluster was summarized in the Supplemental Table. The horizontal lines indicate the boundary between clusters.
ANOVA further confirmed both genotypic and diel effects on those metabolites (false discovery rate [FDR]-adjusted P < 0.05). Of the 447 metabolites, 369 (82.6%) were subjected to diel effects, and 265 (59.3%) showed genotypic effects, while 44 (9.8%) showed an interaction between genotypic and diel effects (Figure 1B; Supplemental Data Set 2). Some important components or intermediates of key metabolic processes were among the top metabolites, including pyruvate (pyruvic acid) and Gal-6-phosphate (diel effect) as CO2 shuttle and intermediate in C4 photosynthesis, Fru and Man-6-phosphate (diel effect) as energy source or signaling molecules in plant development, and Rib, Ade and Thr (genotypic effect) as building blocks of macromolecules (Table 1).
Table 1. ANOVA Identifies Top Metabolites Subjected to Diel (15) or Genotypic Effects (10).
| Metabolites | P Values | Effects | Categories |
|---|---|---|---|
| Pyruvic acid | 3.00E-40 | Diel | Acids |
| Methanol phosphate | 1.00E-35 | Diel | Sugar phosphates |
| Alpha-ketoglutarate | 2.00E-30 | Diel | Acids |
| Ala | 1.00E-24 | Diel | Amino acids |
| 2-Hydroxyglutaric acid | 1.00E-24 | Diel | Acids |
| 2-Isopropylmalic acid | 1.00E-22 | Diel | Acids |
| Erythronic acid lactone | 6.00E-21 | Diel | Others |
| Linoleic acid methyl ester | 1.00E-20 | Diel | Lipids |
| Fru | 3.00E-20 | Diel | Sugars |
| Gal-6-phosphate | 1.00E-19 | Diel | Sugar phosphates |
| Man-6-phosphate | 3.00E-19 | Diel | Sugar phosphates |
| Guanosine | 2.00E-18 | Diel | Nucleosides |
| Xylulose | 2.00E-16 | Diel | Sugars |
| Xylonolactone | 2.00E-16 | Diel | Others |
| Gly | 2.00E-15 | Diel | Amino acids |
| Tocopherol gamma | 1.00E-07 | Genotypic | Alcohols |
| Adenine | 4.00E-06 | Genotypic | Nucleosides |
| Adenosine | 6.00E-06 | Genotypic | Nucleosides |
| Phenylethylamine | 5.00E-05 | Genotypic | Amine |
| Rib | 5.00E-05 | Genotypic | Sugars |
| Citramalic acid | 8.00E-05 | Genotypic | Acids |
| Thr | 2.00E-04 | Genotypic | Amino acids |
| Lyxitol | 3.00E-04 | Genotypic | Alcohols |
| Xylonolactone | 5.00E-04 | Genotypic | Others |
| 5-Hydroxynorvaline | 5.00E-04 | Genotypic | Amino acids |
Hierarchical clustering analysis of metabolic abundance demonstrated the diel regulation of the maize metabolome (Figure 1C). Three major clusters were identified: cluster 1, consisting of 132 metabolites, showed higher abundance during the early day than at night; cluster 2, the largest cluster, consisting of 174 metabolites, peaked at night; cluster 3, consisting of 141 metabolites, showed mixed patterns of abundance among the genotypes at different time points. Analysis of metabolic pathways (FDR-adjusted P < 0.05) indicated an enrichment in clusters 1 and 3 of metabolites, which are mainly involved in the citrate cycle, carbon fixation, and amino acid metabolism (Supplemental Table).
Prevalent Nonadditivity and Mild Heterosis Level of Metabolites in the Hybrids
We next investigated how many of the identified metabolites in the hybrids show nonadditive patterns of accumulation deviating from the mid-parent value (MPV, average of the two parents; Figure 2A). Among 447 metabolites identified, 356 (79.6%) were identified as nonadditively accumulated or nonadditive metabolites (P < 0.05, Student’s t test) in the reciprocal hybrids in one or more time points (with 265 and 266 for F1BM and F1MB, respectively), and ∼49.2% (175/356) metabolites were shared by both hybrids (Figure 2B). Most of them (133, 76%) showed changes in the same direction, either higher or lower than the MPV in both BM and MB. Numbers of nonadditive metabolites during the day and at night were 291 and 232, respectively, and 57.4% (167/291) of the daytime nonadditive metabolites overlapped with nighttime ones (Figure 2B). The average number of nonadditive metabolites per time point was slightly higher during the day than at night (103 versus 88; P = 0.08, Welch’s t test). The number of nonadditive metabolites above or below the MPV was comparable across day time points, while the fraction above MPV in the early night (Zeitgeber Time [ZT]18) was significantly lower than the later time points (ZT21, ZT24; P < 0.005, Fisher’s exact test; Figure 2A).
Figure 2.
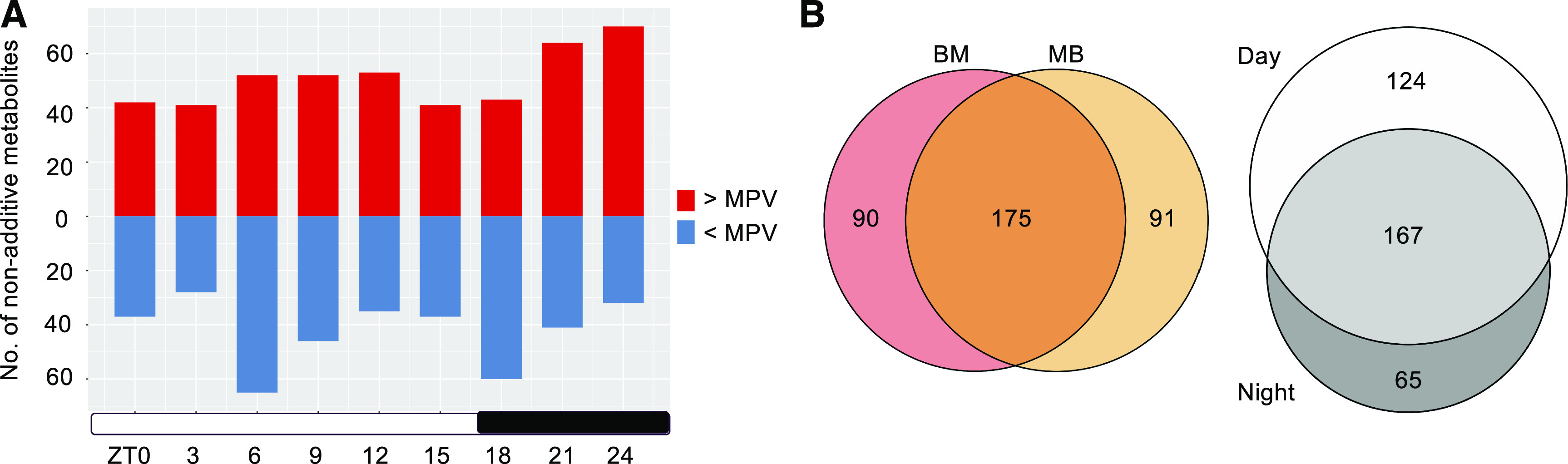
Analysis of Nonadditively Expressed Metabolites in Hybrids during the Day and Night.
(A) Number of nonadditive metabolites at each ZT were identified using Student’s t test (P < 0.05). Red and blue bars indicate the levels of metabolites in the hybrid that were higher or lower than the MPV, respectively. The double asterisks indicate the statistically significant level of P < 0.005 (Fisher’s exact test).
(B) Overlap of nonadditive metabolites between the reciprocal hybrids (F1BM and F1MB, left) and between day and night time points (right).
To quantitatively study metabolic heterosis, we calculated percent mid-parent heterosis (MPH) for each of the 191 annotated metabolites as MPH = 100*D/A, where “D” is the deviation between the hybrid and the average of the two parents (“A”). We found that metabolites such as amines and amino acids show significant negative MPH, while sugars, lipids, and nucleosides tend to show positive MPH (Figures 3A and 3B). Surprisingly, the median of absolute MPH for day- and night time points was 2.7% and 2.6%, respectively, and the maximum MPH observed was 33.4%. Among them, 119 (62.3%) and 108 (56.5%) metabolites had relatively “small” absolute MPH, from 2.0% to 5.0% in day- and night time points, respectively (Figures 3C and 3D), while 42 (22.0%) and 37 (19.4%) of the annotated metabolites in day- and night time points, respectively, had “large” absolute mean MPH, >5.0% across all time points. This scale of heterosis for metabolites is mild for maize hybrids and is in sharp contrast to the much higher phenotypic heterosis reported previously (Springer and Stupar, 2007; Paschold et al., 2010).
Figure 3.
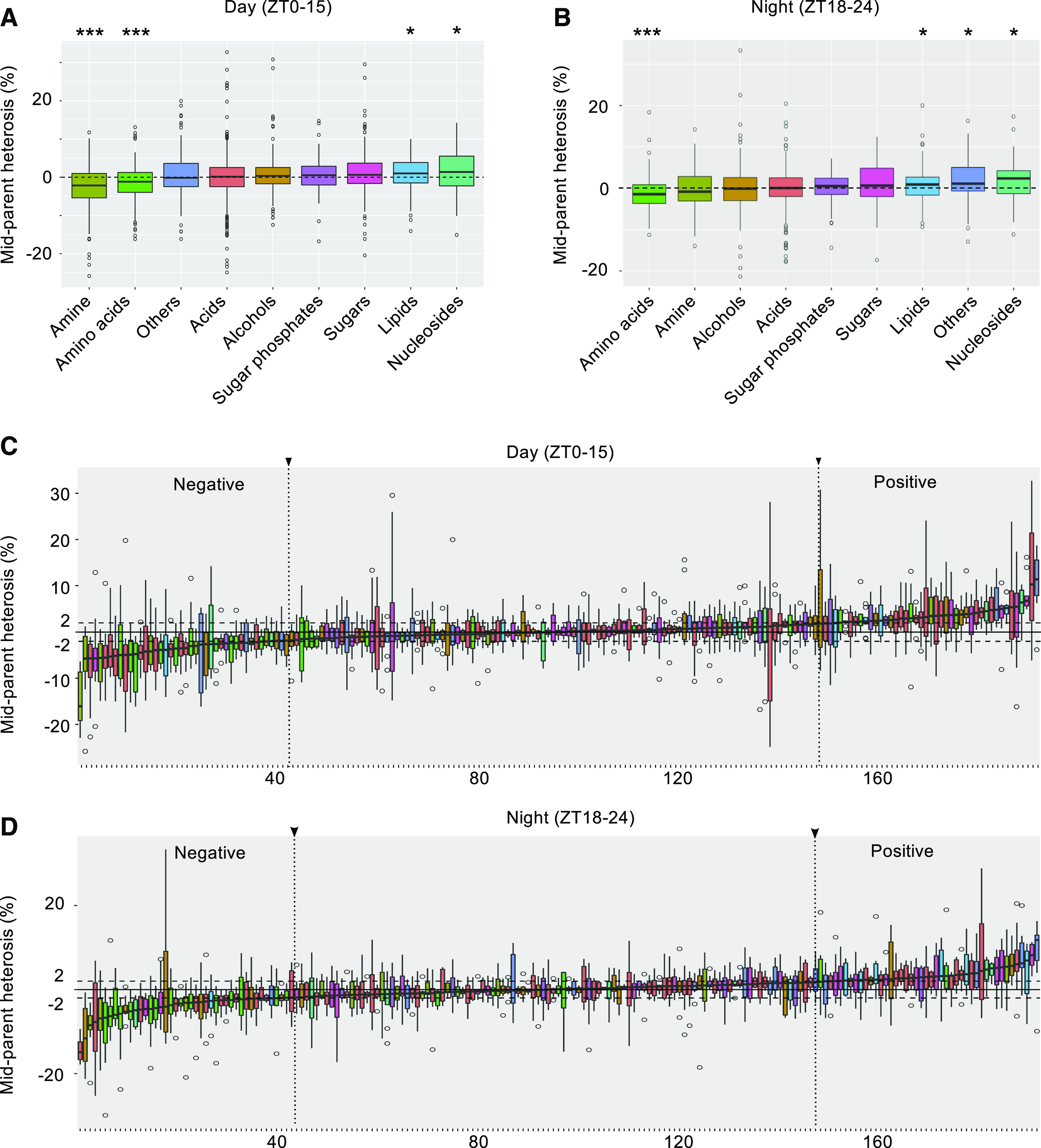
Diel Regulation of Metabolic Heterosis in Maize.
(A) and (B) Boxplot showing the distribution of relative MPH in each metabolic category during the day (ZT0, ZT3, ZT6, ZT9, ZT12, and ZT15) and at night (ZT18, ZT21,and ZT24). One and three asterisks indicate the mean value of MPH at statistically significant levels of P < 0.05 and P < 0.0005, respectively (one-sample Student’s t test).
(C) and (D) Boxplots showing the distribution of MPH for each known metabolite during day and night time points. Negative heterosis (median MPH value < −2%) and positive heterosis (median MPH value > 2%) are indicated by dashed lines. The color code in Figures 3C and 3D is the same as in Figures 3A and 3B. Vertical dotted lines indicate the cut-off for metabolites with negative or positive MPH.
Rhythmic Metabolites Tend to be Nonadditive in the Hybrids
The metabolic data from nine time points throughout the diel cycle enable us to investigate the rhythmicity of the maize metabolome. Diurnal rhythmicity was identified according to the JTK_CYCLE method (Hughes et al., 2010), in which 266 (ranging from 145 to 158 among genotypes) of 447 (59.5%) metabolites were identified as rhythmic metabolites (FDR-adjusted P < 0.05; Figure 4A). Over half of the metabolites in each major metabolic category showed rhythmicity, especially the amino acids and alcohols; >70% of these were rhythmic. Most amino acids (86%) peaked during the day, and nearly half (47%) peaked at ZT9 (Figure 4B). At night, over half of acids, alcohols, and sugars displayed peaks for each category with the largest fraction peaking at ZT21. Importantly, all sugars peaked from afternoon (ZT9) to the midnight (ZT21), and no sugars peaked at the end of the night (ZT24).
Figure 4.
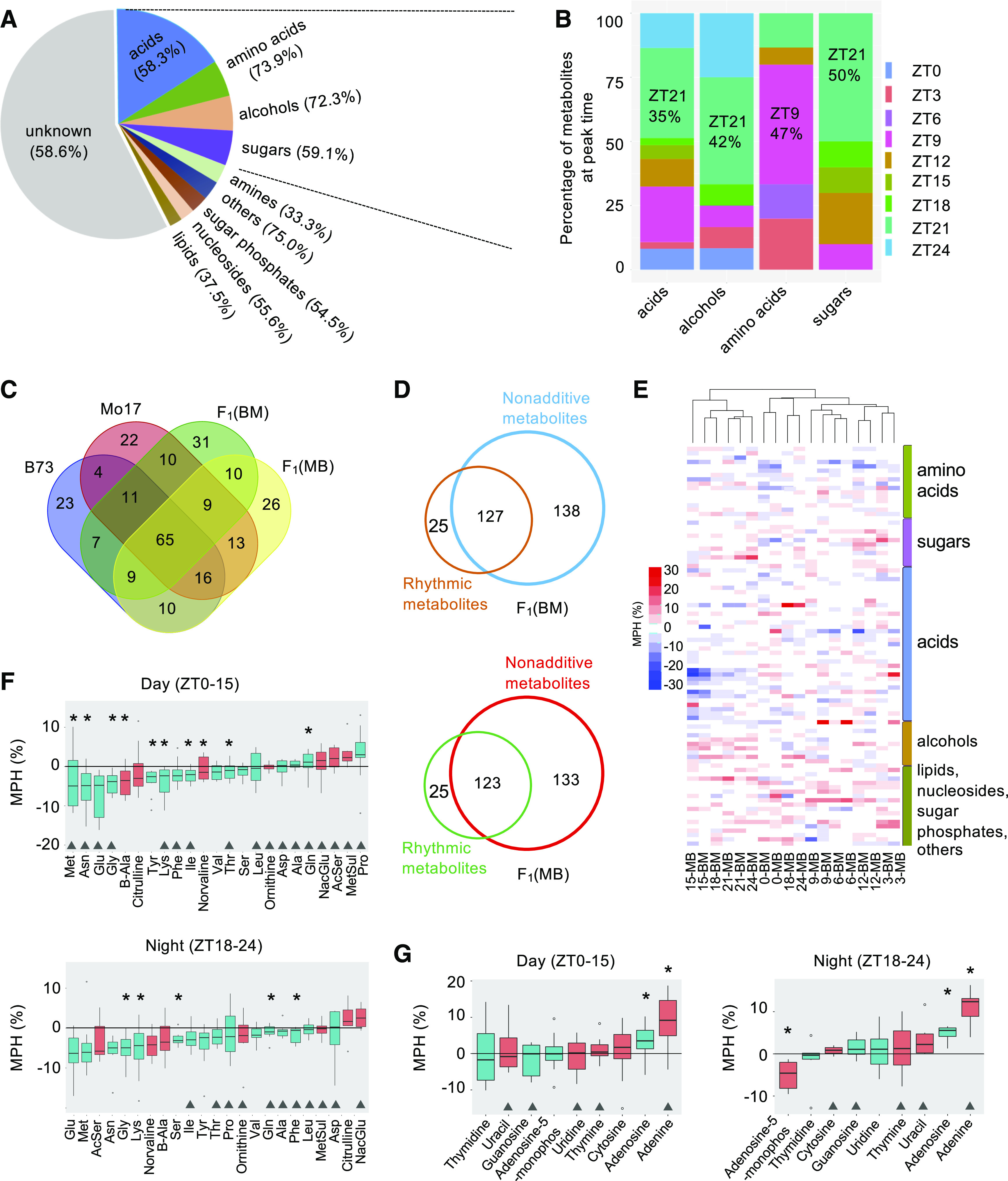
Diel and Nonadditive Regulation of Metabolome in Maize Hybrids and Their Inbreds.
(A) Pie chart showing the relative composition of oscillated metabolites in different categories. The percentage of rhythmic metabolites in each category is shown in parentheses.
(B) Distribution of oscillated metabolites in the four major categories at the peak time.
(C) Comparison of rhythmic metabolites in the F1 hybrids and their inbred parents.
(D) Comparison of rhythmic and nonadditive metabolites in the reciprocal F1 hybrids (BM and MB).
(E) Heatmap showing relative MPH of all annotated rhythmic metabolites.
(F) Changes of MPH in individual amino acids; triangles mark the rhythmic metabolites; green and red boxplots indicate standard and modified amino acids, respectively. An asterisk in (F) indicates mean value of the MPH at significant level of P < 0.05 (one-sample Student’s t test).
(G) Changes of MPH in individual nucleosides; triangles mark the rhythmic metabolites; green and red boxplots indicate nucleotide and nucleotide base, respectively. An asterisk in (G) indicates mean value of the MPH at significant level of P < 0.05 (one-sample Student’s t test).
For the 266 rhythmic metabolites, 65 (24.4%) were shared across all four genotypes, and 96 (48.2%) and 93 (42.9%) were shared by the two inbred parents and by the reciprocal hybrids, respectively (Figure 4C). This result indicates that rhythmic metabolites are variable between the inbred parents and/or between the inbreds and F1 hybrids. Some variation could also be attributed to technical limitations in measuring the metabolites. Interestingly, most rhythmic metabolites for F1BM (127/152) and F1MB (133/158) also showed nonadditive patterns in the hybrids (Figure 4D). This fraction of rhythmically nonadditive metabolites (83.6% for F1BM, P = 2.3E-08 or 84.2% for F1MB P = 5.8E-09) was significantly higher than that (55.6%, 265/447) of nonadditive ones of all metabolites examined (Fisher’s exact test), indicating that rhythmic metabolites tend to show nonadditive patterns in the hybrids.
Furthermore, most amino acids display negative MPH in the hybrids, while sugars, alcohols, lipids, nucleosides, sugar phosphates, and other metabolites tend to show positive MPH with variation across time points (Figure 4E). Acids showed large variation of MPH across time points and in the reciprocal hybrids and were more likely to exhibit negative MPH during dawn and at night (Figure 4E). Fifteen and 19 of all 23 amino acids had median MPH below zero among day- and night time points, respectively, 14 of which were statistically significant (Figure 4F). Some amino acids, such as Pro, Cit, and Gln, changed their directions of MPH during the day and at night. There were more nucleosides having positive MPH at night than during the day (Figure 4G). Adenosine and Ade consistently showed significant positive MPH in both day and night. Interestingly, adenosine-5-monophosphate switched its MPH direction from no significance during the day to significantly negative at night.
Positive MPH in the Photosynthetic Pathway and Negative MPH in the Photorespiratory Pathway
These data of nonadditive accumulation of rhythmic metabolites between day and night and between parents and the hybrids may indicate temporal regulation of metabolic pathways in metabolic heterosis. Further analysis showed that the metabolites involved in photosynthesis, the Calvin cycle, and sugar metabolism (Figure 5A) tended to exhibit positive MPH (Figures 5C and 5D), including pyruvate and malate (important CO2 shuttles in photosynthesis), ribulose 1,5-bisphosphate (RuBP; CO2 acceptor in the Calvin cycle), and Suc and Mal (products of photosynthesis). The metabolites involved in the photorespiratory pathway (Figure 5B), such as Gln, Gly, and Ser, displayed negative MPH at a statistically significant level (Figures 5C and 5D). Moreover, many of these metabolites were rhythmic and possessed different degrees of MPH in the hybrids between day and night (Figure 5E). For example, pyruvate only shows significantly positive MPH during the day (P = 0.001), but not at night (P = 0.84), which is consistent with its role in photosynthesis. These results indicate that higher photosynthetic efficiency in the hybrids, compared with the parents, may result from elevation of key CO2 transporters for photosynthesis and from suppression of photorespiration. Collectively, these changes may allow hybrids to enhance net CO2 assimilation by increasing photosynthetic production while reducing photorespiratory consumption.
Figure 5.
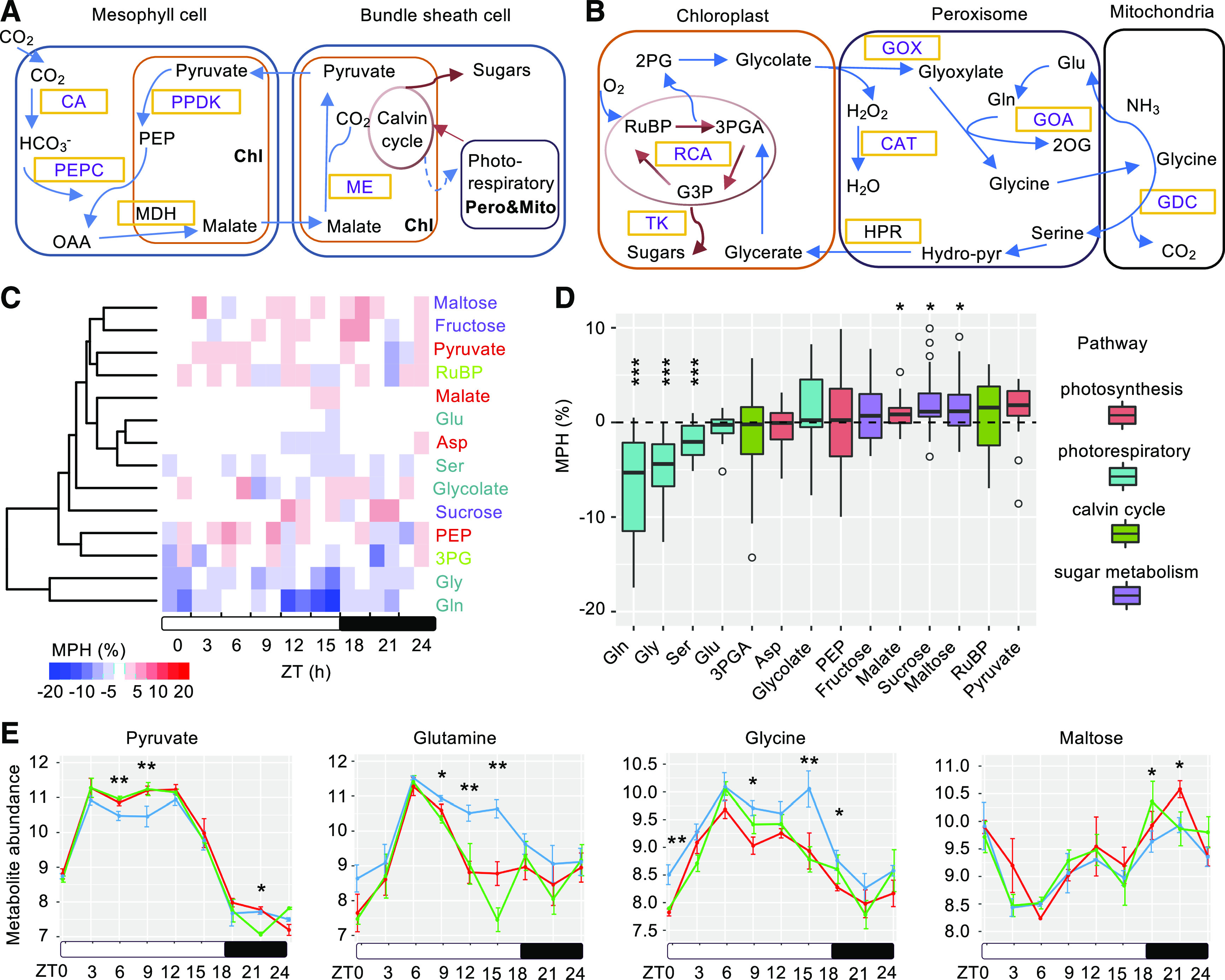
Diel Regulation of Metabolic Heterosis in C4 Photosynthetic and Photorespiratory Pathways.
(A) Schematic diagram showing key enzymes and metabolites in C4 photosynthesis. Chl, chloroplast; Pero, peroxisome; Mito, mitochondrion. CO2 enters the leaf mesophyll cell and is converted to bicarbonate (HCO3−) by CA. HCO3− is used to produce PEP by PEPC. The produced oxaloacetate (OAA) is converted into malate by malate dehydrogenase (MDH) in mesophyll chloroplasts, and then diffused into the bundle-sheath cell. Malate is decarboxylated by ME in the bundle-sheath chloroplast to release CO2, which finally enters the Calvin cycle and is fixed to produce sugars.
(B) Schematic diagram showing key enzymes and metabolites in photorespiratory pathway. In chloroplast, RuBP is converted into 3PGA and 2PG by Rubisco. 3PGA is then converted back to RuBP in the Calvin cycle, and 2PG is converted to glycolate, which is oxidized to glyoxylate and H2O2 by GOX in the peroxisome. Along with glutamate, glyoxylate is used to produce Gly, which is then converted to Ser with liberation of CO2 and ammonia by GDC in mitochondria. In peroxisome, hydroxypyruvate (Hydro-pyr) is made from Ser and then reduced to glycerate by hydroxypyruvate reductase (HPR). Photorespiration is completed when glycerate is eventually converted back to 3PGA in chloroplast, which can reenter the Calvin cycle to produce carbohydrates. 2OG, 2-oxoglutarate; G3P, glyceraldehyde 3-phosphate.
(C) Cluster analysis of metabolites identified in C4 photosynthetic, photorespiratory, Calvin cycle, and sugar metabolism pathways; the two columns in each time point indicate two F1 (BM and MB) hybrids; the heat map color scale indicates MPH (%); text colors for metabolites indicate different pathways as in (D). Asp, asparagine; Glu, glutamic acid.
(D) Metabolites in different pathways show distinct patterns. One and three asterisks indicate mean value of the metabolite MPH at significant levels of P < 0.05 and P < 0.0005, respectively (one-sample Student’s t test).
(E) Examples of rhythmic regulation in metabolic profiles in maize C4 photosynthetic and photorespiratory pathways. Line colors indicate MPV (blue), F1 (BM, red), and F1 (MB, green). The y axis indicates metabolite abundance of the normalized raw spectra with log2-transformation. Single and double asterisks above time points indicate statistically significant levels of P < 0.05 and P < 0.01, respectively, between MPV and one of the hybrids or both (Student’s t test).
The Maize Proteome Exhibits Diel Regulation and Differences between Inbreds and Hybrids
Various metabolites in plants are intermediates or end products of metabolic activities, which are catalyzed by numerous enzymes (proteins). To trace back the cause of metabolite differences between inbred parents and their hybrids, we analyzed the proteome of maize seedlings (stage 9 DAP) from four different time points (ZT3, ZT9, ZT15, and ZT21) in B73 and Mo17 and their reciprocal hybrids (BM and MB). Proteins that were present in all three replicates of each sample were subjected to subsequent analyses. Using a label-free liquid chromatography-tandem mass spectrometry (LC-MS/MS) method (Theodoridis et al., 2008), 4,792 nonredundant proteins were detected as expressed in at least one sample, ranging from 2,312 to 3,290 proteins across genotypes and time points (Supplemental Figure 2A). PCA results showed clear separation of maize proteome between inbreds and hybrids and across different time points (Supplemental Figure 2B).
Generally, the plants expressed more proteins in the morning and early afternoon (ZT3 and ZT9, n = 3,114 and 3,036) than at dusk (ZT15, n = 2,522) or at night (ZT21, n = 2,662). While inbreds and hybrids expressed a comparable number of proteins in the morning (ZT3; 3,155 versus 3,072) and midday (ZT9; 3,054 versus 3,018), significantly more proteins were expressed in inbred parents than hybrids at dusk (2,654 versus 2,391, P < 0.0005) and at night (2,902 versus 2,423, P < 0.0001, Supplemental Figure 2A, and see “Methods”). On average, approximately two-thirds of proteins are shared among all four genotypes across time points (Supplemental Figure 3). Of all proteins identified (4,792), 743 (15.5%), and 1,392 (29%) represent significant genotype and time effects, respectively, and 306 (6.4%) proteins represent significant interaction effects between genotype and time (P < 0.05, two-way ANOVA). Approximately half of the proteins that showed genotype effects also showed a significant time-of-day effect (Supplemental Figures 4A and 4B).
In the hybrids, 1,025 proteins were identified as nonadditively expressed in at least one time point (P < 0.05, Student’s t test), with relatively more nonadditive proteins detected in the early morning (ZT3, 362 proteins) and at night (ZT21, 503 proteins) than other time points (Supplemental Figure 4C). Between the reciprocal hybrids, 205 of 592 (34.6%) nonadditive proteins in F1BM overlapped with the nonadditive proteins in F1MB (Supplemental Figure 4D). Across different time points, the overlapping fraction of nonadditive proteins was small (Supplemental Figure 4E), indicating rapid temporal regulation of nonadditively expressed proteins in the hybrids. There were 380 and 691 nonadditive proteins above and below MPV, respectively (Supplemental Figure 4F), with significantly more nonadditive proteins below MPV (110) than above MPV (62) across time points (P = 0.014, Student’s t test).
Across time points, 2,540 proteins were identified as rhythmic by JTK_CYCLE (FDR-adjusted P < 0.05), which accounted for over half (53%) of all detectable proteins (Supplemental Figure 5A). Among the rhythmic proteins, ∼60% were specific to individual genotypes (Supplemental Figure 5B). A phase distribution analysis of the rhythmic proteins showed similar phases between the hybrids and inbred parents, but compared with inbred parents, hybrids showed a larger fraction of rhythmic proteins in early morning (ZT3) and a smaller fraction at night (ZT21; Supplemental Figure 6). This data supports our previous findings for the temporal shift of ZmCCA1 binding targets to the early morning in the hybrids as a head-start for photosynthetic activities (Ko et al., 2016).
Approximately 30% of nonadditive proteins overlapped with rhythmic proteins (Supplemental Figures 5C and 5D). Interestingly, for those proteins showing both nonadditivity and rhythmicity, upregulated proteins in the hybrids were exclusively enriched in gene ontology (GO) biological process terms associated with photosynthesis (GO:0015979, P = 1.00E-12), photosynthesis-light reaction (GO:0019684, P = 7.70E-11), photosynthetic electron transport in PSI (GO:0009773, P = 3.80E-10), and cellular components such as photosynthetic membrane (GO:0034357, P = 7.00E-15; Supplemental Data Set 3). This enrichment is similar to the GO terms enrichment for metabolite abundance in the hybrids.
Concerted Changes in Proteins (Enzymes) and Metabolites in the Hybrids
The above data suggest that concerted changes between enzymes and metabolites in photosynthetic and photorespiratory pathways may occur in maize hybrids. We examined the abundance of key enzymes in the C4 photosynthetic pathway (Figure 5A) that plays an important role in CO2 transportation and fixation. In the F1MB hybrid, the abundance of carboxylate phosphoenolpyruvate carboxylase (PEPC) and malic enzyme (ME) were both significantly upregulated (P < 0.05, Student’s t test) in the morning (ZT3), at midday (ZT9), and at night (ZT21; Figure 6). In the F1BM hybrid, PEPC abundance was upregulated at ZT9 and ZT21, and ME at ZT21. Pyruvate orthophosphate dikinase (PPDK) was upregulated in the morning (ZT3) for both reciprocal hybrids and at night (ZT21) for F1MB. Carbonic anhydrase (CA) was upregulated only at ZT21 for both hybrids. Moreover, all these enzymes have relatively higher abundance in the morning and midday (ZT3 and ZT9) than at dusk (ZT15) and at night (ZT21). These results are consistent with photosynthetic rate changes in the seedlings at the same stage (Ko et al., 2016). The upregulation of these key enzymes during the day may confer higher photosynthetic efficiency to the hybrids and can also explain the positive MPH as well as the nonadditively higher abundance observed for related metabolites like carboxylate phosphoenolpyruvate (PEP), malate, pyruvate, RuBP, Suc, and maltose (Figures 5C to 5E).
Figure 6.

Temporal Abundance of Key Enzymes in the C4 Photosynthesis Pathway.
Protein abundance is shown as log2-transformed and normalized spectral counts (y axis). An asterisk indicates the significant higher level (P < 0.05) of protein abundance in the hybrids than the MPV (Student’s t test). CA, GRMZM2G121878; PEPC, GRMZM2G083841; ME, GRMZM2G085019; PPDK, GRMZM2G097457.
In the photorespiratory pathway (Figure 5B), several enzymes were highly abundant in the hybrids relative to MPV (Figure 7A). Glycolate oxidase (GOX) had higher abundance than MPV across four time points, with statistically significant abundance at ZT9. We propose that nonadditive upregulation of GOX abundance in the hybrids could generate higher H2O2 concentrations. To test this, we measured the H2O2 concentration in leaves of the same stage seedlings for each genotype (Supplemental Figure 7; see Methods). H2O2 concentration was higher in hybrids than the MPV at most of the time points, but the difference was only statistically significant in the morning (ZT3, P < 0.05). After analysis where the day time points (ZT3, ZT9, and ZT15) were combined and compared with the night (ZT21) time point, we found the H2O2 concentration in both reciprocal hybrids was significantly higher than the MPV during the day, but not the night.
Figure 7.

Temporal Abundance of Key Enzymes in Photorespiratory Pathway and Calvin Cycle.
(A) Examples of protein abundance changes in the photorespiratory pathway. Protein abundance is shown as log2-transformed and normalized spectral counts (y axis).
An asterisk in (A) and (B) indicates the significantly higher level (P < 0.05) of protein abundance in the hybrids than the MPV (Student’s t test), with one exception (below the MPV as indicated by the arrow). GOX, GRMZM2G129246; CAT, GRMZM2G090568; GDC, GRMZM2G104310; GOA (glutamine oxoglutarate aminotransferase), GRMZM2G036609.
(B) Examples of protein abundance changes in the Calvin cycle. RCA, GRMZM2G162200; TK, GRMZM2G033208.
This trend observed for H2O2 is consistent with catalase (CAT) protein abundance, which was upregulated at night (ZT21) for both hybrids (Figure 7A); CAT is responsible for scavenging H2O2 produced during glycolate oxidation. We further measured the enzyme activities of GOX and CAT, which were consistent with their protein levels and showed nonadditive increases during the midday and at night, respectively (Supplemental Figure 8), supporting their respective roles in H2O2 generation and scavenging.
For some key metabolites in the hybrids, PEP and pyruvate (photosynthesis) showed higher abundance in the morning or early afternoon, Gly and Gln (photorespiration) with lower abundance in the afternoon, and Suc and Fru (Calvin cycle) with higher abundance in the afternoon and night for some time points (Supplemental Figure 9). The enzyme glycine decarboxylase (GDC) was upregulated at night (ZT21) and downregulated at midday (ZT9). Glutamine oxoglutarate aminotransferase was upregulated in the F1BM hybrid at ZT3, ZT9, and ZT21 (Supplemental Figure 8). As a result, these enzymes may accelerate rapid turnover of metabolites such as Gln, Gly, and Ser during midday to early afternoon, leading to lower abundance at night (Supplemental Figure 9).
Rubisco activase (RCA) and transketolase (TK), two key enzymes in the Calvin cycle, both had relatively high abundance in the morning and at midday (Figure 7B). RCA, which activates Rubisco, was upregulated at ZT15 in the reciprocal hybrids. Notably, it is the abundance of the activated Rubisco sites, not the Rubisco abundance per se, that correlates positively with photosynthesis rate and fresh weight in maize Rubisco-overexpressing lines (Salesse-Smith et al., 2018). Moreover, RCA gene expression is positively associated with maize grain yield (Zhang et al., 2019). TK, which is a key enzyme in the Calvin cycle and related to sugar synthesis, was upregulated in ZT9 and ZT21 for both hybrids. A small decrease in TK activity was found to have dramatic inhibitory effects on RuBP regeneration and photosynthesis in tobacco (Nicotiana tabacum; Henkes et al., 2001). Consistent with the results of protein abundance, RCA and TK also showed significantly higher enzyme activities compared with MPV at different time points (Supplemental Figure 8), which may lead to the nonadditively higher abundance of photosynthetic products such as Suc, Fru, and Mal in the afternoon and night (Figure 5E; Supplemental Figure 9).
Metabolites such as 2-phosphoglycolate (2PG), glycolate, and glyoxylate are toxic to Calvin-cycle and Rubisco activity (Dellero et al., 2016b). Thus, it is important for plants to effectively detoxify those metabolites (Dellero et al., 2016b). We suggest that the upregulation of the aforementioned key enzymes in the photorespiratory pathway may help hybrids maintain a relatively more efficient photosynthesis system by effectively removing inhibitory metabolites. This hypothesis is supported by the finding that photorespiratory-related metabolites such as Gly, Ser, and Gln show negative MPH in hybrids (Figures 5C and 5D). In addition, downregulation of these metabolites in the hybrids may also indicate their faster conversion to 3-phosphoglycerate (3PGA), which can be used in the Calvin cycle for sugar biosynthesis.
DISCUSSION
In this study, integrative analysis of time-series data showed the diel regulation of the maize metabolome and proteome with distinct and nonadditive patterns between F1 hybrids and their inbred parents. These differences in metabolite and protein levels may contribute to hybrid vigor. Specifically, by diurnally regulating nonadditive expression of key enzymes involved in photosynthetic and photorespiratory pathways, hybrids may optimize the abundance of corresponding metabolites to improve carbon assimilation and detoxify toxic metabolites, which ultimately promotes plant growth.
Metabolic and Proteomic Rhythmicity in Maize Hybrids and Inbreds
Diurnal regulation of gene expression in maize occurs for an estimated 10% to 30% of expressed transcripts (Hayes et al., 2010; Khan et al., 2010; Jończyk et al., 2011). This regulation has been shown to affect heterosis in maize (Ko et al., 2016) and other plant species (Ni et al., 2009). However, little is known about diurnal regulation of metabolites and proteins, the end products of gene expression, and their roles in heterosis in maize. Previous studies using different methods identified 632 (36%) and 354 (12%) rhythmic proteins in maize leaves (Feng et al., 2017) and rice seedlings (Hwang et al., 2011), respectively. In this study, although the available data sets of metabolome and proteome in one diel cycle with limited time points may hinder JTK_CYCLE to capture certain subtle rhythmicity resides, our analysis has identified several general trends, providing some directions for future studies.
We found that ∼82% (369/447) of metabolites and 29% (1,392/4,792) of proteins detected had rhythmic accumulation, indicating extensive diel regulation of the metabolome and proteome of both hybrids and inbreds. For instance, all sugars had peak accumulation beginning at midday (ZT9) and continuing through to the middle of night (ZT21), and no sugars peaked at the end of the night (ZT24; Figure 4B). This observation coincides with the net photosynthesis rate changes, which increase from early morning and peak at midday, and then decrease until the end of the day (Ko et al., 2016). The concentration of carbohydrates such as Suc, peaks at midday, and remains at a high level until the middle of night, until it is depleted by the end of night (Kalt-Torres and Huber, 1987; Kalt-Torres et al., 1987).
Consistent with our previous findings for the temporal shift of ZmCCA1 binding targets to the early morning in the hybrids (Ko et al., 2016), the hybrids displayed more rhythmic proteins than the parents in the early morning (ZT3; Supplemental Figure 6). This result indicates that hybrids may have the advantage of head-starting in gene and protein expression and consequently in physiological processes such as photosynthesis. Although the overall amplitudes among all rhythmic proteins are similar between the inbreds and hybrids, some of the nonadditive proteins show increased levels of amplitudes, suggesting a potential mechanism to maintain circadian robustness (Chen, 2013; Chen and Mas, 2019). Changes in circadian clock amplitudes can be achieved by chromatin and other epigenetic modifications on circadian clock genes as demonstrated in Arabidopsis (Ni et al., 2009; Ng et al., 2014; Song et al., 2019).
Nonadditive Metabolites and Proteins in the Hybrids
Most metabolites examined in the study show nonadditive values in the hybrids at different time points. Similarly, over half of the examined metabolites exhibited nonadditivity in different Arabidopsis hybrids (Rodriguez Cubillos et al., 2018). The fraction of proteins showing nonadditivity (∼21%) in this study of seedlings is higher than that (∼9%) found in previous work in root (Marcon et al., 2013). This discrepancy could result from several factors such as proteomic assays, plant tissues, or sampling time points. We found 30% of the nonadditive proteins that show rhythmicity are enriched in photosynthesis GO terms and may contribute to heterosis. It should be noted that the other nonadditive proteins that are not identified as rhythmic, such as heat shock proteins (Fu et al., 2011) and ribosomal proteins (Marcon et al., 2013), can also play a role in heterosis.
It is also notable that the overlap of nonadditive proteins (Supplemental Figure 4D) or metabolites (30%, Figure 2B) between the reciprocal hybrids is relatively low. Similar low-overlap results between the reciprocal hybrids were reported in proteomic data (∼9%) from seminal roots (Marcon et al., 2013) and gene expression data (∼10%) in the endosperm (Springer and Stupar, 2007). This could be related to a parent-of-origin effect, as observed in the hybrids of Arabidopsis (Ng et al., 2014) or maize endosperm (Springer and Stupar, 2007). In our study, we revealed a temporal effect on the proteomic data. Based on the PCA plot (Supplemental Figure 2B), the variation between the reciprocal hybrids is largely separated by different time points, despite their being clustered in a given time point. Although the cause for this difference needs further investigation, we predict that the difference between the reciprocal hybrids is biologically relevant.
The mild MPH for metabolites observed in this study is comparable to a previous finding (Lisec et al., 2011). Similar mild MPH for metabolites was also reported for B. juncea (Bajpai and Reichelt, 2019) and Arabidopsis (Lisec et al., 2009). Although the data from this work were obtained from seedlings, it is likely that the multitude and amplification of mild increases in metabolic processes may result in a much higher scale of heterosis for phenotypic traits such as the increased biomass, plant height, root length, leaf length, and carbon fixation observed in maize hybrids (Springer and Stupar, 2007; Paschold et al., 2010; Ko et al., 2016). Moreover, positive heterosis in photosynthesis and negative heterosis in photorespiration may interactively produce greater phenotypic heterosis in an accelerated cascade manner.
Temporal Positive and Negative Regulation of Key Enzymes and Metabolites for Heterosis
Our observation of positive MPH of metabolites in photosynthetic, Calvin-cycle, and sugar metabolism pathways, and of negative MPH in the photorespiratory pathway in the hybrids, is consistent with the abundance of key enzymes involved in these pathways. For example, the positive MPH of sugars (Figure 3) is supported by upregulated proteins that are enriched in GO terms in photosynthetic-related processes such as the pyruvate (GO:0006090, P = 2.00E-06), sugar and starch (GO:0019252, P = 6.80E-06), disaccharide (GO:0005984, P = 0.0003), and monosaccharide catabolic metabolisms (GO:0046365, P = 0.0011; Supplemental Data Set 3).
The negative MPH of metabolites in the photorespiratory pathway may be associated with the nonadditive accumulation of GOX proteins in the hybrids (Figure 7A). As the oxygenase activity of Rubisco is ubiquitous and unavoidable (Jordan and Ogren, 1981), the synthesis of glycolate is inevitable, which can be mediated through the photorespiratory process to avoid toxic levels of glycolate accumulation (Zelitch et al., 2009). In C4 plants such as maize, photorespiration is suppressed due to the increased CO2 concentration in bundle-sheath chloroplasts by the carbon shuttle, which results in higher photosynthetic efficiency compared with C3 plants. However, photorespiration is not only essential for C4 plants, but also plays an important role in the evolution of C4 photosynthesis (Sage et al., 2012).
In photorespiration, GOX is responsible for the catalyzation of the oxidation of glycolate to glyoxylate in higher plants (Figure 7B). Effective purge of glycolate is essentially important to maintain photosynthetic efficiency. For example, low GOX activity can severely impair CO2 assimilation rates in leaves of rice (Oryza sativa; Xu et al., 2009; Lu et al., 2014), Arabidopsis (Dellero et al., 2016a), and maize (Zelitch et al., 2009), possibly through inhibiting Rubisco activity (Dellero et al., 2016b). The higher activity of GOX in the maize hybrids (Figure 7A) may enable the more rapid removal of the toxic glycolate. Indeed, glycolate shows negative MPH at ZT9 (−7.7% for F1BM and −2.4% for F1MB), which remains negative until early night (ZT15; Figure 5C). Furthermore, nonadditive upregulation of GOX activity at ZT9 in the hybrids is consistent with the maximum net photosynthesis rate at the same time (Kalt-Torres et al., 1987; Ko et al., 2016), which may suggest that high GOX activities are required for the maintenance of high levels of photosynthesis to prevent photoinhibition (Yamaguchi and Nishimura, 2000). As a result, photosynthetic assimilates such as Suc start peaking from ZT9.
H2O2 is concomitantly produced as a byproduct during photosynthesis from the catalyzation of glycolate to glyoxylate by GOX (Figure 5B). The role of reactive oxygen species in plant defense has been well documented (Camejo et al., 2016). For instance, the H2O2 produced by GOX is associated with both gene-for-gene and non-host–resistance response in plants (Rojas et al., 2012). There is increasing evidence to support the idea that reactive oxygen species play an integral role as signaling molecules in regulating numerous biological processes such as plant growth and development (Mittler et al., 2011; Baxter et al., 2014). The nonadditive accumulation of H2O2 during the day (Supplemental Figure 7) may enhance photosynthetic efficiency in the hybrids as a signaling molecule and was consequently detoxified at night by increased CAT activity.
Roles of Photosynthetic and Photorespiratory Pathways in Maize Seedling Heterosis
Based on our observation and available data, we propose that at dawn or early morning, hybrids may sense the light more effectively than inbred parents through the circadian clock protein-binding activities (Ko et al., 2016) and/or other signaling molecules such as H2O2 (Baxter et al., 2014), which can enhance an acclimation to the increasing light condition. At midday, when light intensity reaches its highest level, hybrids can tolerate the photoinhibition stress more effectively and maintain a higher level of the photosynthesis rate without excessively elevating its photorespiratory machinery compared with the inbreds, which is possibly achieved by the rapid detoxification of glycolate through upregulated GOX activity.
This is consistent with the findings that the proteins nonadditively upregulated in the hybrids are exclusively enriched in photosynthesis-related GO terms, and the upregulation of abundance and activities of key enzymes in both photosynthetic (PEPC, ME, and PPDK) and Calvin-cycle pathways (RCA and TK). As a result, metabolites involved in the photosynthetic pathway, such as malate, pyruvate, and RuBP, show positive MPH, while the metabolites involved in the photorespiratory pathway such as Gly, Gln, and Ser, display negative MPH. Consequently, hybrids can accumulate more photosynthetic assimilates such as Suc and Mal. At night, when the circadian clock and/or signaling molecule level shifts, increased CAT activity can reduce H2O2 abundance to the MPV level to protect plants from the excess oxidative stress. Overall, these new insights into metabolic and proteomic processes of photosynthesis and photorespiration will provide a conceptual advance for breeding and biotechnological improvement of hybrid crops.
METHODS
Plant Materials
Maize (Zea mays) inbred lines B73 and Mo17 and their F1 hybrid (BM) and the reciprocal hybrids (MBs) were generously provided by James A. Birchler at the University of Missouri. All plants were grown at 600 μmol m−2 s−1 under 16 h of light at 8 h of dark cycle supplemented by model no. Pro 325 LED lights (LumiGrow) with a temperature 28°C (light) and 23°C (dark) in the growth facility at The University of Texas at Austin and Nanjing Agricultural University (for enzyme activity assays). Experiment was in a randomized design, and plants were rotated daily to minimize positional effects. Seeds were soaked to maximize germination consistency across genotypes before planting.
The above-ground tissues of seedlings at 9 DAP (V2 stage) from four genotypes were used for the study at a 3-h interval or nine time points (ZT0, ZT3, ZT6, ZT9, ZT12, ZT15, ZT18, ZT21, and ZT24) for metabolome assays and at a 6-h interval or four time points (ZT3, ZT9, ZT15, and ZT21) for proteome analysis. Above-ground seedling tissues were harvested and frozen immediately in liquid nitrogen and stored at −80°C until further processing. Three biological replicates were used for each time point of each genotype.
Untargeted Metabolomics Methods
Metabolite Extraction
Metabolites were extracted for three biological replicates for each genotype (B73, Mo17, BM, and MB) at each time point. For each biological replicate of each sample, frozen tissue was ground in liquid nitrogen to fine powder, and an aliquot (20 mg) of tissue powder was extracted with 1 mL of degassed extraction solvent (5:2:2 methanol/chloroform/water, v/v/v). The mixture was vortexed for 15 min at 4°C and then centrifuged at 13,500g for 3 min at 4°C. The supernatant was collected and dried under vacuum at 40°C overnight (Fiehn et al., 2000). Derivatization was performed as previously described by Fiehn et al. (2008). In brief, 10 μL of methyloxamine hydrochloride (40 mg/mL in pyridine) was added to each sample and then the samples were incubated with shaking at 30°C for 1.5 h. A mixture (91 μL) of N-methyl-N-(trimethylsilyl) trifluoroacetamide containing fatty acid methyl ester markers was added into the sample and incubated at 37°C for 30 min.
GC-TOF-MS
Samples were assayed by GC-TOF-MS as previously described by Fiehn et al. (2008). In brief, derivatized sample injections of 0.5 μL were made in splitless mode with a purge time of 25 s, and the temperature program was set as follows: from 50°C to 275°C at a linear rate of 12°C/s and held for 3 min. A model no. 6890 Gas Chromatograph (Agilent) was used with a 30-m–long, 0.25-mm i.d., Rtx5Sil-MS column with a 0.25-μm 5% diphenyl film; then an additional 10-m integrated guard column was used. A constant flow of 1 mL/min was used to perform chromatography, and the oven temperature was ramped from 50°C to 330°C over 22 min at a linear rate. A Pegasus IV TOF Mass Spectrometer (Leco) with 280°C transfer line temperature, electron ionization at −70 V, and an ion source temperature of 250°C was used to perform mass spectrometry. Mass spectra were acquired from m/z 85 to 500 at 20 spectra/s and 1,750-V detector voltage.
Metabolite Database Search and Data Processing
The exported result files were processed by the metabolomics BinBase database (https://fiehnlab.ucdavis.edu/projects/binbase-setup; Kind et al., 2009). Retention index and mass spectrum information or the NIST11 commercial library (https://www.nist.gov/system/files/documents/srd/NIST1a11Ver2-0Man.pdf) were used to match all database entries in BinBase against the Fiehn mass spectral library of 1,200 authentic metabolite spectra. Identified metabolites were reported if present with at least 50% of the samples per study design group as defined in the MiniX database (https://fiehnlab.ucdavis.edu/projects/binbase-setup). Sample data was first exported to the netCDF format and then evaluated with BinBase. In brief, the output results were exported to the BinBase database. Noisy and inconsistent peaks were filtered out according to multiple parameters. Quantitation of each metabolite was reported as peak height using the unique ion as default. Based on the underlying assumption that the total amounts of ionized metabolites that reach the detector are similar for different samples, the raw data was first log2-transformed to conform to normal distribution for inter-sample comparisons and then normalized by the median of the sum of all peak heights of all metabolites in each sample. To define cluster borders, we used heatmap.2 in the R package (R Core Team, 2018), which calculates the Euclidean distance between measurements to obtain the distance matrix and employs complete agglomeration method for clustering.
Label-Free Proteomics Methods
Protein Extraction and Preparation
Proteins were extracted for three biological replicates for each genotype at each time point. For each biological replicate, frozen tissue (∼2 g) was ground in liquid nitrogen to fine powder. Proteins were then precipitated and washed with methanol (at −20°C) with 0.2 mM of Na3VO4 three times, followed by washing with acetone (−20°C) three times. Protein pellets were then dried at 4°C in a vacuum concentrator. Subsequently, proteins were resuspended in 1 mL of extraction buffer (0.1% [w/v] SDS, 1 mM of EDTA, and 50 mM of HEPES buffer at pH 7). Cysteines were reduced by 1 mM of Tris (2-carboxyethyl) phosphine at 95°C for 5 min and then alkylated by 2.5 mM of iodoacetamide at 37°C in the dark for 15 min. Proteins were digested with trypsin (enzyme/substrate w:w ratio = 1:100; Roche) overnight. A second trypsin digestion (enzyme/substrate w:w ratio = 1:100) was performed the next day for 4 h. Digested peptides were purified on an Oasis mixed-mode, strong cation-exchange cartridge (MCX; Waters) to remove SDS. Peptides were eluted from the MCX column with 1 mL of 50% (v/v) isopropyl alcohol and 400 mM of NH4HCO3 at pH 9.5 and then dried in a vacuum concentrator at 4°C. A Pierce Protein Assay Kit (Pierce) was used to quantify the peptide amount after using the MCX column. Peptides were then resuspended in 1% (v/v) formic acid to a final pH of 3.0 and used for MS analysis.
Mass Spectrum and Peptide Identification
A model no. 1100 HPLC System (Agilent Technologies) delivered a flow rate of 600 nL min−1 to a three-phase capillary chromatography column through a splitter. Then, 5 μm of Zorbax SB-C18 (Agilent) was packed into fused silica capillary tubing (250-μm ID; 360-μm OD; 30-cm long) using a custom pressure cell to form the first dimension reverse phase section of the column (RP1). A 5-cm–long, strong cation exchange (SCX) section of the column packed with 5 μm of PolySulfoethyl (PolyLC) was connected to RP1 using a zero dead volume 1-μm filter (model no. M548; Upchurch) attached to the exit of the RP1 column. At the analytical section of the column (RP2), a fused silica capillary (200-μm ID, 360-μm OD, 20-cm long) packed with 5 μm of Zorbax SB-C18 (Agilent), was connected to the SCX section. The electrospray tip of the fused silica tubing was pulled to a sharp tip with the inner diameter smaller than 1 μm using a laser puller (model no. P-2000; Sutter). Using the custom pressure cell, the peptide mixtures were loaded onto the RP1 column section. Then the three sections were joined and mounted on a custom electrospray adapter for on-line nested elution. For each LC-MS/MS analysis, a new set of columns was used. A 0% to 80% (v/v) acetonitrile gradient was first used to elute the peptides from the RP1 column section to the SCX column section for 150 min. The peptides were then fractionated by the SCX column section using a series of 29 step salt gradients (0 mM, 5 mM, 10 mM, 15 mM, 20 mM, 22.5 mM, 25 mM, 27.5 mM, 30 mM, 32.5 mM, 35 mM, 37.5 mM, 40 mM, 42.5 mM, 45 mM, 47.5 mM, 50 mM, 52.5 mM, 55 mM, 57.5 mM, 60 mM, 65 mM, 70 mM, 75 mM, 80 mM, 85 mM, 90 mM, 150 mM, and 1 M of ammonium acetate, for 20 min each), followed by high-resolution reverse phase separation on the RP2 section of the column using an acetonitrile gradient of 0% to 80% for 120 min.
An LTQ Velos Linear Ion Trap MS/MS Spectrometer (Thermo Electron), which employed automated, data-dependent acquisition, was used to analyze the spectra. The mass spectrometer was operated in positive ion mode with a source temperature of 250°C. As a final fractionation step, gas phase separation in the ion trap was employed to separate the peptides into three mass classes before scanning (300 to 800, 800 to 1,100, and 1,100 to 2,000 D). Each MS scan was followed by five MS/MS scans of the most intense ions from the parent MS scan. To improve the duty cycle, a dynamic exclusion of 1 min was used.
Protein Database Search
The raw data was extracted and searched using Spectrum Mill v3.03 (Agilent Technologies). MS/MS spectra with a sequence tag length of 1 or less were discarded. The remaining MS/MS spectra were searched against the maize B73 RefGen_v2 5a working gene set downloaded from http://ensembl.gramene.org/Zea_mays/Info/Index. The enzyme parameter was limited to full tryptic peptides, allowing a maximum miscleavage of 1. All other search parameters were set to Spectrum Mill’s default settings (carbamidomethylation of cysteines, ±2.5 D for precursor ions, ±0.7 D for fragment ions, and a minimum matched peak intensity of 50%). A 1:1 concatenated forward/reverse database was constructed to calculate the FDR. The tryptic peptides in the reverse database were compared with the forward database and were shuffled if they matched to any tryptic peptides from the forward database. Cutoff scores were dynamically assigned to each data set to maintain the FDR < 0.05% for spectra (608 decoy hits among 1,269,618 spectra), 0.13% for unique peptide (71 decoy hits among 55,676 unique peptide), 0.41% for proteins (81 decoy hits among 19,651 proteins), and 0.68% for protein groups (58 decoy hits among 8,625 protein groups). To address the protein database redundancy, proteins that share common peptides were grouped using principles of parsimony. Thus, proteins within the same group shared the same set or subset of unique peptides. The group leader of each protein group was defined as the protein presented by the highest number of unique peptide identifications or showed the highest value of protein coverage. Protein abundance levels were quantified by spectral counting. Spectral counts for each protein represent the total number of peptide spectral matches to that protein.
Normalization and Filtering
Proteins entered into subsequent analysis must fulfill the following criteria: three or more unique peptides mapped to the protein or two unique peptides mapped to the protein but with at least 30% coverage. Raw spectrum counts of each protein in each sample were first normalized by dividing the calculated size factor for each sample (calculated from dividing spectrum counts of each sample by the median total spectrum counts of all samples) to account for library size variations across samples and then log2-transformed. A total of 4,792 proteins that were detectable by the two criteria noted above in at least one genotype or time point were classified as expressed proteins. Raw and normalized data set of spectrum counts for each sample can be downloaded from https://de.cyverse.org/dl/d/DB13CC69-0F5F-4889-9CFC-D84C78EC5A68/Maize_proteome_9DAP_BM_upload.xlsx.
H2O2 Measurement
The H2O2 concentration was measured following the protocol by Chakraborty et al. (2016) with minor modifications. An Amplex Red Assay Kit (Amplex Red, DMSO, Horseradish peroxidase, and phosphate buffer, 5×) was obtained from Invitrogen (Life Technologies). Amplex Red was prepared as per manufacturer’s protocol. For each genotype at ZT3, ZT9, ZT15, and ZT21, the second leaf tissue at 9 DAP was harvested and ground in liquid nitrogen using a Qiagen TissueLyser (Qiagen). Then, phosphate buffer (500 μL) was added into powdered plant sample (∼50 mg). The suspended tissue was vortexed for 10 s and shaken continuously at room temperature for 60 min. Samples were centrifuged at room temperature for 5 min at 12,000g. The supernatant (200 μL) was transferred to a new pre-labeled tube and stored on ice until use. We then added 5 μL of supernatant into 95 μL of premixed Amplex red + Horseradish peroxidase cocktail. Assaying H2O2 concentration was performed in a fluorometer plate reader, using 560-nm excitation and 590-nm emission filters (Omega Optical). Three biological replicates were assayed for each genotype at each time point. A standard calibration curve was prepared using 5 μL of the stock H2O2 standards to yield H2O2 concentrations of 0, 0.25, 0.5, 1, 1.5, 2, 3, and 4 μM with two biological replicates. Background noise values, as the value at 0 μM, were subtracted from the other standard concentrations. The fitted regression line is y = 0.001482x – 0.01372, adjusted R2 = 0.9997. The intensity of fluorescent signal was then converted to H2O2 concentration as μmol/g (fresh weight) according to the calibration curve.
Enzyme Activity Measurement
For each genotype at ZT3, ZT9, ZT15, and ZT21, above-ground seedling tissues were harvested with three biological replicates and frozen immediately in liquid nitrogen and stored at −80°C until further processing. Activities of CAT (CAT-1-Y) and GOX (DLO-1-G) were measured using commercial kits (Keming Biotechnology) following the manufacturer’s protocol. Activities of RCA and TK were measured using commercial kits (RCA Enzyme Kit and TK Enzyme Kit; Solarbio) following the manufacturer’s instructions.
Bioinformatics
Kyoto Encyclopedia of Genes and Genomes Enrichment
Metabolites were functionally annotated using the Kyoto Encyclopedia of Genes and Genomes database (http://www.kegg.jp/) and were mapped to Kyoto Encyclopedia of Genes and Genomes pathways. Enrichment analysis for metabolites in each cluster in Figure 1C was conducted. Pathways with P < 0.05 were considered as significantly enriched.
GO Enrichment
GO enrichment of proteins was conducted using the web-based tool agriGO v2.0 (http://systemsbiology.cau.edu.cn/agriGOv2/) with singular enrichment analysis (Tian et al., 2017). Only the FDR-adjusted P < 0.01 was reported in the main text. The filtered working gene list together with the gene annotation of maize was obtained from http://ensembl.gramene.org/Zea_mays/Info/Index?db=core.
Putative enzyme annotation: putative functional annotation of key enzymes was obtained from the Gramene database (http://ensembl.gramene.org/Zea_mays/Info/Index?db=core).
Statistical Analysis
Three biological replicates were used to calculate mean values of each metabolite or protein for each genotype in a given time point. Two-way ANOVA was used to evaluate the significant effect of genotype, circadian regulation, and the interaction of metabolites and proteins (P < 0.05). The FDR-corrected P-value was used to account for possible excessive false-positives generated by multiple tests. Nonadditivity of metabolites/proteins in hybrids was detected by comparing the abundance of hybrids and MPVs using Student’s t test (P < 0.05). Significantly deviated values of MPH from zero were detected by a one-sample t test (P < 0.05). The difference of the average number of expressed proteins between inbreds and hybrids at each time point was tested by calculating the standardized normal deviate parameter z (Pocock, 2006). Here, z = (inbred – hybrid)/square root (inbred + hybrid), which estimates the difference of the two values (hybrid and hybrid) divided by the square root of their sum. The “inbred” and “hybrid” in the formula indicate the numbers of expressed proteins for inbred parents (the average between B73 and Mo17) and hybrids (the average between BM and MB), respectively, at each time point. Hierarchical clustering was performed using the heatmap.2 function in the R package (gplots, v3.0.1.1; https://cran.r-project.org/web/packages/gplots/index.html). PCA was conducted using the prcomp function in R with data centered and scaled (https://www.rdocumentation.org/packages/stats/versions/3.6.2/topics/prcomp).
Rhythmicity Test
Rhythmicity of metabolites and proteins were identified according to the JTK_CYCLE method (Hughes et al., 2010). Period was set as 24 h and the amplitude and the phase as free parameters. FDR-adjusted P < 0.05 was used as the cut-off to determine rhythmic metabolites and proteins.
Accession Numbers
Sequence data for genes reported and discussed in this article can be found in the GenBank library/MaizeGDB gene records under the following accession numbers: NC_050098.1 (220987608..220997146)/ GRMZM2G121878; NC_050104.1 (70927112..70932455)/GRMZM2G083841; NC_050098.1 (7184775..7189972)/GRMZM2G085019; NC_050103.1 (109319267..109324789)/GRMZM2G097457; NC_050102.1 (5745473..5748512)/ GRMZM2G129246; NC_050096.1 (7097456..7099732)/GRMZM2G090568; NC_050105.1 (5519993..5526181)/GRMZM2G104310; NC_050102.1 (180078786..180096656)/GRMZM2G036609; NC_050099.1 (1225439..1227877)/GRMZM2G162200; and NC_050104.1 (24660295..24664743)/GRMZM2G033208.
Supplemental Data
Supplemental Figure 1. Quality assessment of maize metabolomes.
Supplemental Figure 2. Patterns of proteomic changes between maize inbred parents and hybrids.
Supplemental Figure 3. Overlap of expressed proteins across genotypes at each time point.
Supplemental Figure 4. Summary of nonadditively expressed proteins in hybrids.
Supplemental Figure 5. Number of rhythmic proteins.
Supplemental Figure 6. Phase distribution of rhythmic proteins among four genotypes.
Supplemental Figure 7. H2O2 concentration in each genotype across time points.
Supplemental Figure 8. Temporal activities of key enzymes in the photorespiratory pathway and Calvin cycle.
Supplemental Figure 9. Temporal abundance of key metabolites in the C4 photosynthesis pathway, photorespiratory pathway, and Calvin cycle.
Supplemental Table. Enrichment of metabolic pathways (P < 0.05) for metabolites in different clusters according to hierarchical clustering analysis.
Supplemental Data Set 1. Log2-transformed metabolites for four genotypes at nine time points with three replicates.
Supplemental Data Set 2. Details of two-way ANOVA for metabolites.
Supplemental Data Set 3. GO enrichment of nonadditively up- and downregulated proteins with rhythmicity in the hybrids.
DIVE Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
We are indebted to James A. Birchler, Curators’ Professor of Biological Sciences at the University of Missouri, for his generous support and gift of maize hybrid and inbred materials for this study. We thank the Texas Advanced Computing Center for computational support, the Westcoast Metabolomics Center at the University of California, Davis for metabolomic assays, and JadeBio (La Jolla, California) for protein MS analysis. We also thank the State Key Laboratory of Crop Genetics and Germplasm Enhancement at Nanjing Agricultural University for providing support and assistance for Z.L. to perform enzyme assays during article revision, when Z.L. could not return to the United States due to travel bans during the COVID-19 pandemic. This work was funded by the National Science Foundation (grant IOS1238048 to Z.J.C. and F.G.H.) and the National Institute of General Biomedical Sciences (grant GM109076 to Z.J.C.). Z.J.C. is the D. J. Sibley Centennial Professor of Plant Molecular Genetics.
AUTHOR CONTRIBUTIONS
Z.J.C. conceived and designed the experiments; H.Y.C. and A.Z. performed the experiments; Z.L. and A.Z. performed data analysis; Q.X.S. and F.G.H. contributed reagents and materials; Z.L. and Z.J.C. wrote the article.
Footnotes
Articles can be viewed without a subscription.
References
- Bajpai P.K., Reichelt M.(2019). Heterotic patterns of primary and secondary metabolites in the oilseed crop Brassica juncea. Heredity (Edinb) 123: 318–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf J.A., Marcon C.(2016). Nonsyntenic genes drive tissue-specific dynamics of differential, nonadditive, and allelic expression patterns in maize hybrids. Plant Physiol. 171: 1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf J.A., Marcon C., Lithio A., Vedder L., Altrogge L., Piepho H.P., Schoof H., Nettleton D., Hochholdinger F.(2018). Single-parent expression is a general mechanism driving extensive complementation of non-syntenic genes in maize hybrids. Curr. Biol. 28: 431–437 e434. [DOI] [PubMed] [Google Scholar]
- Baxter A., Mittler R., Suzuki N.(2014). ROS as key players in plant stress signalling. J. Exp. Bot. 65: 1229–1240. [DOI] [PubMed] [Google Scholar]
- Bendix C., Marshall C.M., Harmon F.G.(2015). Circadian clock genes universally control key agricultural traits. Mol. Plant 8: 1135–1152. [DOI] [PubMed] [Google Scholar]
- Birchler J.A., Yao H., Chudalayandi S., Vaiman D., Veitia R.A.(2010). Heterosis. Plant Cell 22: 2105–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camejo D., Guzmán-Cedeño Á., Moreno A.(2016). Reactive oxygen species, essential molecules, during plant–pathogen interactions. Plant Physiol. Biochem. 103: 10–23. [DOI] [PubMed] [Google Scholar]
- Chakraborty S., Hill A.L., Shirsekar G., Afzal A.J., Wang G.L., Mackey D., Bonello P.(2016). Quantification of hydrogen peroxide in plant tissues using Amplex Red. Methods 109: 105–113. [DOI] [PubMed] [Google Scholar]
- Chen Z.J.(2013). Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 14: 471–482. [DOI] [PubMed] [Google Scholar]
- Chen Z.J., Mas P.(2019). Interactive roles of chromatin regulation and circadian clock function in plants. Genome Biol. 20: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahal D., Mooney B.P., Newton K.J.(2012). Specific changes in total and mitochondrial proteomes are associated with higher levels of heterosis in maize hybrids. Plant J. 72: 70–83. [DOI] [PubMed] [Google Scholar]
- de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C.(2009). Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5: 1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellero Y., Jossier M., Glab N., Oury C., Tcherkez G., Hodges M.(2016b). Decreased glycolate oxidase activity leads to altered carbon allocation and leaf senescence after a transfer from high CO2 to ambient air in Arabidopsis thaliana. J. Exp. Bot. 67: 3149–3163. [DOI] [PubMed] [Google Scholar]
- Dellero Y., Jossier M., Schmitz J., Maurino V.G., Hodges M.(2016a). Photorespiratory glycolate-glyoxylate metabolism. J. Exp. Bot. 67: 3041–3052. [DOI] [PubMed] [Google Scholar]
- Duvick D.N.(2001). Biotechnology in the 1930s: The development of hybrid maize. Nat. Rev. Genet. 2: 69–74. [DOI] [PubMed] [Google Scholar]
- Facette M.R., Shen Z., Björnsdóttir F.R., Briggs S.P., Smith L.G.(2013). Parallel proteomic and phosphoproteomic analyses of successive stages of maize leaf development. Plant Cell 25: 2798–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng D., Wang Y., Lu T., Zhang Z., Han X.(2017). Proteomics analysis reveals a dynamic diurnal pattern of photosynthesis-related pathways in maize leaves. PLoS ONE. 12: e0180670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O., Kopka J., Dörmann P., Altmann T., Trethewey R.N., Willmitzer L.(2000). Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18: 1157–1161. [DOI] [PubMed] [Google Scholar]
- Fiehn O., Wohlgemuth G., Scholz M., Kind T., Lee D.Y., Lu Y., Moon S., Nikolau B.(2008). Quality control for plant metabolomics: Reporting MSI-compliant studies. Plant J. 53: 691–704. [DOI] [PubMed] [Google Scholar]
- Fu Z., Jin X., Ding D., Li Y., Fu Z., Tang J.(2011). Proteomic analysis of heterosis during maize seed germination. Proteomics 11: 1462–1472. [DOI] [PubMed] [Google Scholar]
- Goff S.A., Zhang Q.(2013). Heterosis in elite hybrid rice: Speculation on the genetic and biochemical mechanisms. Curr. Opin. Plant Biol. 16: 221–227. [DOI] [PubMed] [Google Scholar]
- Guo M., Rupe M.A., Yang X., Crasta O., Zinselmeier C., Smith O.S., Bowen B.(2006). Genome-wide transcript analysis of maize hybrids: Allelic additive gene expression and yield heterosis. Theor. Appl. Genet. 113: 831–845. [DOI] [PubMed] [Google Scholar]
- Hayes K.R., Beatty M., Meng X., Simmons C.R., Habben J.E., Danilevskaya O.N.(2010). Maize global transcriptomics reveals pervasive leaf diurnal rhythms but rhythms in developing ears are largely limited to the core oscillator. PLoS One 5: e12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen S.P., Naef F., Quisel T., Gendron J.M., Chen H., Ecker J.R., Borevitz J.O., Kay S.A.(2009). Exploring the transcriptional landscape of plant circadian rhythms using genome tiling arrays. Genome Biol. 10: R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkes S., Sonnewald U., Badur R., Flachmann R., Stitt M.(2001). A small decrease of plastid transketolase activity in antisense tobacco transformants has dramatic effects on photosynthesis and phenylpropanoid metabolism. Plant Cell 13: 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochholdinger F., Baldauf J.A.(2018). Heterosis in plants. Curr. Biol. 28: R1089–R1092. [DOI] [PubMed] [Google Scholar]
- Hughes M.E., Hogenesch J.B., Kornacker K.(2010). JTK_CYCLE: An efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25: 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H., Cho M.H., Hahn B.S., Lim H., Kwon Y.K., Hahn T.R., Bhoo S.H.(2011). Proteomic identification of rhythmic proteins in rice seedlings. Biochim. Biophys. Acta 1814: 470–479. [DOI] [PubMed] [Google Scholar]
- Jahnke S., Sarholz B., Thiemann A., Kühr V., Gutiérrez-Marcos J.F., Geiger H.H., Piepho H.P., Scholten S.(2010). Heterosis in early seed development: A comparative study of F1 embryo and endosperm tissues 6 days after fertilization. Theor. Appl. Genet. 120: 389–400. [DOI] [PubMed] [Google Scholar]
- Jiang L.G., Li B., Liu S.X.(2019). Characterization of proteome variation during modern maize breeding. Mol. Cell. Proteomics 18: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jończyk M., Sobkowiak A., Siedlecki P., Biecek P., Trzcinska-Danielewicz J., Tiuryn J., Fronk J., Sowiński P.(2011). Rhythmic diel pattern of gene expression in juvenile maize leaf. PLoS One 6: e23628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan D.B., Ogren W.L.(1981). Species variation in the specificity of ribulose biphosphate carboxylase/oxygenase. Nature 291: 513–515. [Google Scholar]
- Kalt-Torres W., Huber S.C.(1987). Diurnal changes in maize leaf photosynthesis: III. Leaf elongation rate in relation to carbohydrates and activities of sucrose metabolizing enzymes in elongating leaf tissue. Plant Physiol. 83: 294–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalt-Torres W., Kerr P.S., Usuda H., Huber S.C.(1987). Diurnal changes in maize leaf photosynthesis: I. Carbon exchange rate, assimilate export rate, and enzyme activities. Plant Physiol. 83: 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S., Rowe S.C., Harmon F.G.(2010). Coordination of the maize transcriptome by a conserved circadian clock. BMC Plant Biol. 10: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.A., Kim H.S., Choi S.H., Jang J.Y., Jeong M.J., Lee S.I.(2017). The importance of the circadian clock in regulating plant metabolism. Int. J. Mol. Sci. 18: 2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T., Wohlgemuth G., Lee D.Y., Lu Y., Palazoglu M., Shahbaz S., Fiehn O.(2009). FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 81: 10038–10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko D.K., Rohozinski D., Song Q., Taylor S.H., Juenger T.E., Harmon F.G., Chen Z.J.(2016). Temporal shift of circadian-mediated gene expression and carbon fixation contributes to biomass heterosis in maize hybrids. PLoS Genet. 12: e1006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn M., Gärtner T., Erban A., Kopka J., Selbig J., Hincha D.K.(2010). Predicting Arabidopsis freezing tolerance and heterosis in freezing tolerance from metabolite composition. Mol. Plant 3: 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosová K., Vítámvás P., Urban M.O., Klíma M., Roy A., Prášil I.T.(2015). Biological networks underlying abiotic stress tolerance in temperate crops—a proteomic perspective. Int. J. Mol. Sci. 16: 20913–20942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Coffey L., Garfin J., Miller N.D., White M.R., Spalding E.P., de Leon N., Kaeppler S.M., Schnable P.S., Springer N.M., Hirsch C.N.(2018). Genotype-by-environment interactions affecting heterosis in maize. PLOS One 13: e0191321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisec J., Römisch-Margl L., Nikoloski Z., Piepho H.P., Giavalisco P., Selbig J., Gierl A., Willmitzer L.(2011). Corn hybrids display lower metabolite variability and complex metabolite inheritance patterns. Plant J. 68: 326–336. [DOI] [PubMed] [Google Scholar]
- Lisec J., Steinfath M., Meyer R.C., Selbig J., Melchinger A.E., Willmitzer L., Altmann T.(2009). Identification of heterotic metabolite QTL in Arabidopsis thaliana RIL and IL populations. Plant J. 59: 777–788. [DOI] [PubMed] [Google Scholar]
- Lu Y., Li Y., Yang Q., Zhang Z., Chen Y., Zhang S., Peng X.X.(2014). Suppression of glycolate oxidase causes glyoxylate accumulation that inhibits photosynthesis through deactivating Rubisco in rice. Physiol. Plant. 150: 463–476. [DOI] [PubMed] [Google Scholar]
- Marcon C., Lamkemeyer T., Malik W.A., Ungrue D., Piepho H.P., Hochholdinger F.(2013). Heterosis-associated proteome analyses of maize (Zea mays L.) seminal roots by quantitative label-free LC-MS. J. Proteomics 93: 295–302. [DOI] [PubMed] [Google Scholar]
- Marcon C., Malik W.A., Walley J.W.(2015). A high-resolution tissue-specific proteome and phosphoproteome atlas of maize primary roots reveals functional gradients along the root axes. Plant Phys 168: 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer R.C., et al. (2012). Heterosis manifestation during early Arabidopsis seedling development is characterized by intermediate gene expression and enhanced metabolic activity in the hybrids. Plant J. 71: 669–683. [DOI] [PubMed] [Google Scholar]
- Miller M., Song Q., Shi X., Juenger T.E., Chen Z.J.(2015). Natural variation in timing of stress-responsive gene expression predicts heterosis in intraspecific hybrids of Arabidopsis. Nat. Commun. 6: 7453. [DOI] [PubMed] [Google Scholar]
- Mittler R., Vanderauwera S., Suzuki N., Miller G., Tognetti V.B., Vandepoele K., Gollery M., Shulaev V., Van Breusegem F.(2011). ROS signaling: The new wave? Trends Plant Sci. 16: 300–309. [DOI] [PubMed] [Google Scholar]
- Morgenthal K., Scholz M., Selbig J., Weckwerth W.(2005). Correlative GC-TOF-MS-based metabolite profiling and LC-MS-based protein profiling reveal time-related systemic regulation of metabolite--protein networks and improve pattern recognition for multiple biomarker selection. Metabolomics 1: 109–121. [Google Scholar]
- Ng D.W., Miller M., Yu H.H., Huang T.Y., Kim E.D., Lu J., Xie Q., McClung C.R., Chen Z.J.(2014). A role for CHH methylation in the parent-of-origin effect on altered circadian rhythms and biomass heterosis in Arabidopsis intraspecific hybrids. Plant Cell 26: 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z., Kim E.D., Ha M., Lackey E., Liu J., Zhang Y., Sun Q., Chen Z.J.(2009). Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457: 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschold A., Marcon C., Hoecker N., Hochholdinger F.(2010). Molecular dissection of heterosis manifestation during early maize root development. Theor. Appl. Genet. 120: 383–388. [DOI] [PubMed] [Google Scholar]
- Pocock S.J.(2006). The simplest statistical test: How to check for a difference between treatments. BMJ 332: 1256–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A Language and Environment for Statistical Computing. (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Riedelsheimer C., Lisec J., Czedik-Eysenberg A., Sulpice R., Flis A., Grieder C., Altmann T., Stitt M., Willmitzer L., Melchinger A.E.(2012). Genome-wide association mapping of leaf metabolic profiles for dissecting complex traits in maize. Proc. Natl. Acad. Sci. USA 109: 8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez Cubillos A.E., Tong H., Alseekh S., de Abreu E.L.F.(2018). Inheritance patterns in metabolism and growth in diallel crosses of Arabidopsis thaliana from a single growth habitat. Heredity 120: 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas C.M., Senthil-Kumar M., Wang K., Ryu C.M., Kaundal A., Mysore K.S.(2012). Glycolate oxidase modulates reactive oxygen species-mediated signal transduction during nonhost resistance in Nicotiana benthamiana and Arabidopsis. Plant Cell 24: 336–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römisch-Margl L., Spielbauer G., Schützenmeister A., Schwab W., Piepho H.P., Genschel U., Gierl A.(2010). Heterotic patterns of sugar and amino acid components in developing maize kernels. Theor. Appl. Genet. 120: 369–381. [DOI] [PubMed] [Google Scholar]
- Sage R.F., Sage T.L., Kocacinar F.(2012). Photorespiration and the evolution of C4 photosynthesis. Annu. Rev. Plant Biol. 63: 19–47. [DOI] [PubMed] [Google Scholar]
- Salesse-Smith C.E., Sharwood R.E., Busch F.A.(2018). Overexpression of Rubisco subunits with RAF1 increases Rubisco content in maize. Nat. Plants 4: 802–810. [DOI] [PubMed] [Google Scholar]
- Schnable P.S., Springer N.M.(2013). Progress toward understanding heterosis in crop plants. Annu. Rev. Plant Biol. 64: 71–88. [DOI] [PubMed] [Google Scholar]
- Song Q., Ando A., Xu D., Fang L., Zhang T., Huq E., Qiao H., Deng X.W., Chen Z.J.(2018). Diurnal down-regulation of ethylene biosynthesis mediates biomass heterosis. Proc. Natl. Acad. Sci. USA 115: 5606–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q., Huang T.Y., Yu H.H., Ando A., Mas P., Ha M., Chen Z.J.(2019). Diurnal regulation of SDG2 and JMJ14 by circadian clock oscillators orchestrates histone modification rhythms in Arabidopsis. Genome Biol. 20: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer N.M., Stupar R.M.(2007). Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res. 17: 264–275. [DOI] [PubMed] [Google Scholar]
- Staiger D., Shin J., Johansson M., Davis S.J.(2013). The circadian clock goes genomic. Genome Biol. 14: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson-Wagner R.A., DeCook R., Jia Y., Bancroft T., Ji T., Zhao X., Nettleton D., Schnable P.S.(2009). Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 326: 1118–1120. [DOI] [PubMed] [Google Scholar]
- Theodoridis G., Gika H.G., Wilson I.D.(2008). LC-MS-based methodology for global metabolite profiling in metabonome. TrAC Trends Analyt. Chem. 27: 251–260. [Google Scholar]
- Thiemann A., Fu J., Seifert F., Grant-Downton R.T., Schrag T.A., Pospisil H., Frisch M., Melchinger A.E., Scholten S.(2014). Genome-wide meta-analysis of maize heterosis reveals the potential role of additive gene expression at pericentromeric loci. BMC Plant Biol. 14: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian T., Liu Y., Yan H., You Q., Yi X., Du Z., Xu W., Su Z.(2017). agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45 (W1): W122–W129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher M., Small I.(2019). Simulation of heterosis in a genome-scale metabolic network provides mechanistic explanations for increased biomass production rates in hybrid plants. Nature 5: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C., Marcotte E.M.(2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Xue Z., Wang T.(2014). Differential analysis of proteomes and metabolomes reveals additively balanced networking for metabolism in maize heterosis. J. Proteome Res. 13: 3987–4001. [DOI] [PubMed] [Google Scholar]
- Xing J., Sun Q., Ni Z.(2016). Proteomic patterns associated with heterosis. Biochim. Biophys. Acta 1864: 908–915. [DOI] [PubMed] [Google Scholar]
- Xu G., Cao J., Wang X., Chen Q., Jin W., Li Z., Tian F.(2019). Evolutionary metabolomics identifies substantial metabolic divergence between maize and its wild ancestor, teosinte. Plant Cell 31: 1990–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Zhang J., Zeng J., Jiang L., Liu E., Peng C., He Z., Peng X.(2009). Inducible antisense suppression of glycolate oxidase reveals its strong regulation over photosynthesis in rice. J. Exp. Bot. 60: 1799–1809. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K., Nishimura M.(2000). Reduction to below threshold levels of glycolate oxidase activities in transgenic tobacco enhances photoinhibition during irradiation. Plant Cell Physiol. 41: 1397–1406. [DOI] [PubMed] [Google Scholar]
- Zelitch I., Schultes N.P., Peterson R.B., Brown P., Brutnell T.P.(2009). High glycolate oxidase activity is required for survival of maize in normal air. Plant Physiol. 149: 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhou Y., Sun Q., Deng D., Liu H., Chen S., Yin Z.(2019). Genetic determinants controlling maize Rubisco activase gene expression and a comparison with rice counterparts. BMC Plant Biol 19: 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Kremling K.A.(2019). Metabolome-scale genome-wide association studies reveal chemical diversity and genetic control of maize specialized metabolites. Plant Cell 31: 937–955. [DOI] [PMC free article] [PubMed] [Google Scholar]