

Summary
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is a single-stranded, enveloped RNA virus and the etiological agent of the current coronavirus disease 2019 pandemic. Efficient replication of the virus relies on the activity of nonstructural protein 1 (Nsp1), a major virulence factor shown to facilitate suppression of host gene expression through promotion of host mRNA degradation and interaction with the 40S ribosomal subunit. Here, we report the crystal structure of the globular domain of SARS-CoV-2 Nsp1, encompassing residues 13 to 127, at a resolution of 1.65 Å. Our structure features a six-stranded, capped β-barrel motif similar to Nsp1 from SARS-CoV and reveals how variations in amino acid sequence manifest as distinct structural features. Combining our high-resolution crystal structure with existing data on the C-terminus of Nsp1 from SARS-CoV-2, we propose a model of the full-length protein. Our results provide insight into the molecular structure of a major pathogenic determinant of SARS-CoV-2.
Subject areas: Biochemistry, Virology, Structural Biology
Graphical Abstract
Highlights
-
•
SARS-CoV-2 Nsp1 features a capped β-barrel structure, similar to that of SARS-CoV
-
•
Distinct structural features distinguish SARS-CoV-2 Nsp1 from its SARS-CoV ortholog
-
•
The plasticity of the Nsp1 protein fold is evident through comparison with homologs
Biochemistry; Virology; Structural Biology
Introduction
In March of 2020, the World Health Organization declared coronavirus disease 2019 (COVID-19) a global pandemic. As of October 2020, there have been more than 35,400,000 cases of infection reported globally and approximately 1,040,000 deaths attributed to COVID-19 (Dong et al., 2020). The etiological agent of this pandemic has been identified as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), a member of the Betacoronavirus genus and closely related to the severe acute respiratory syndrome coronavirus (SARS-CoV) that caused the severe acute respiratory syndrome outbreak of 2002–2004 (Zhou et al., 2020; Chan et al., 2020).
Coronaviruses infect a diverse array of vertebrates, with infection typically resulting in respiratory disease or gastroenteritis (Weiss and Navas-Martin, 2005). Their broad host range allows for a substantial reservoir for human infection and mutation facilitates cross-species transmission (Yang et al., 2015). There is mounting evidence that SARS-CoV-2 originated via mutation and cross-species transmission of a pangolin coronavirus with which it shares ∼97% sequence identity (Zhang et al., 2020a). This highlights the dramatic impact that relatively few amino acid substitutions can have in coronaviruses and underscores the urgent need for characterization of these infectious agents at a molecular level.
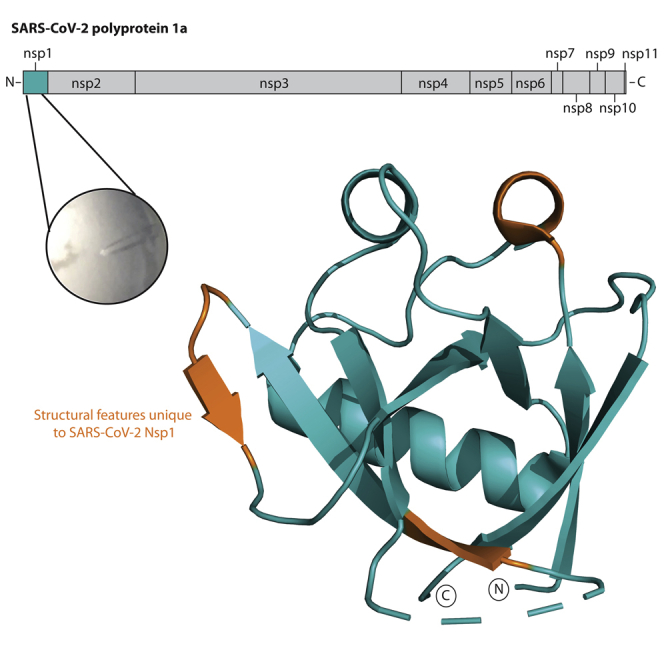
SARS-CoV-2 is an enveloped, positive-sense RNA virus with a single-stranded genome approximately 30 kb in size. The genome is 5′-capped and 3′poly-adenylated and the first ∼2/3 of the genome encode two overlapping reading frames that produce polyprotein 1a and 1ab (Masters, 2006). Downstream of this region, the remaining 1/3 of the genome encodes for structural proteins and a number of open reading frames (ORFs) that produce small accessory proteins. Polyprotein 1a and 1ab are large polypeptides that are processed post-translationally by viral-encoded proteases to produce non-structural proteins (Nsp) 1–16 (Narayanan et al., 2015). The genome also contains 5′ and -3′ untranslated regions (UTRs), the former of which plays a critical role in self-recognition that allows for SARS-CoV-2 protein production to occur unabated while host gene expression is suppressed (Tanaka et al., 2012). At the core of this mechanism is Nsp1, a 180 amino acid (AA) protein produced via processing of polypeptide 1a and 1ab by the Papain-like protease domain of Nsp3 (Figure 1A) (Harcourt et al., 2004).
Figure 1.

The structure of SARS-CoV-2 Nsp113-127
(A) Schematic of Orf1ab from SARS-CoV-2. The gray shading encompasses the non-structural proteins produced by Orf1a. Note, nsp11 is not produced by Orf1ab due to ribosomal frameshift (Naqvi et al., 2020). (B) Topological arrangement of SARS-CoV-2 Nsp113-127.
(C) Cartoon representation of the SARS-CoV-2 Nsp113-127 structure colored from N-terminus (blue) to C-terminus (red). Secondary structure elements and the N- and C-termini are labeled.
(D) Ribbon depiction of the structure, with side chains that contribute to the hydrophobic core of the beta-barrel shown in stick representation. The three layers of side chains (shown in red, blue, and green), plus the charged residues (shown in pink) at the bottom are colored and the residues within the layers are labeled.
(E) Electrostatic surface potential of the likely RNA-binding interface of SARS-CoV-2 Nsp113-127. R124 which is critical for the interaction with SL1 of the 5′UTR is labeled.
Nsp1 mediates a two-pronged approach to suppression of host gene expression. Firstly, it inhibits translation of host proteins during the initiation stage through interaction with the 40S ribosomal subunit. Secondly, Nsp1 promotes the degradation of host mRNA by endonucleolytic cleavage within the 5′UTR, which in turn leads to accelerated Xrn1-mediated mRNA decay (Kamitani et al., 2006, 2009). Viral mRNAs are able to avoid the fate of host mRNAs through interaction between Nsp1 and the stem-loop 1 (SL1) motif found in the viral 5′ UTR (Tanaka et al., 2012). Nsp1 from SARS-CoV has been shown to inhibit the type I interferon response in infected cells, allowing the virus to circumvent the innate immune response (Narayanan et al., 2008). Expression of SARS-CoV Nsp1 has also been shown to induce the production of chemokines, suggesting this protein may play a role in the “cytokine storm”, a maladaptive release of cytokines in response to infection, associated with a number of COVID-19 infections (Law et al., 2007; Ye et al., 2020). Thus, Nsp1 has emerged as a major pathogenicity factor that plays a critical role in the coronavirus infection cycle (Zust et al., 2007). Deletion or mutation of Nsp1 results in viral attenuation in infection models and restores the innate immune response in infected cells (Wathelet et al., 2007). Based on its central role in suppression of the host immune response and essentiality to infection, Nsp1 has been proposed as a therapeutic target for the treatment of COVID-19 (Zust et al., 2007). A prerequisite to any investigation into possible interventions targeting Nsp1 is high-resolution structural data that can facilitate robust in silico screening. A partial structure corresponding to the N-terminal fragment of Nsp1 from SARS-CoV was resolved by nuclear magentic resonance (NMR) spectroscopy and revealed a unique β-barrel motif. The corresponding domain from SARS-CoV-2 shares 86% sequence identity with its SARS-CoV ortholog, which is a considerable level of sequence diversity compared to many other non-structural proteins encoded by the SARS-CoV-2 genome (e.g. Nsp12 shares 96% identity with SARS-CoV ortholog (Zhang et al., 2020b)). Recent structural and biochemical studies involving SARS-CoV-2 non-structural proteins have revealed a level of functional divergence that would not have otherwise been predicted based on the high degree of sequence conservation with SARS-CoV orthologs. Biochemical characterization of the Nsp12-Nsp7-Nsp8 replicase complex showed that minor changes in AA sequence compared to SARS-CoV resulted in reduced enzymatic activity (Peng et al., 2020). Structural characterization of Nsp9 identified a novel peptide binding site not predicted bioinformatically or reported for Nsp9 from SARS-CoV (Littler et al., 2020). These findings are illustrative of the need for continued pursuit of structural characterization of SARS-CoV-2 proteins.
Here, we report the high-resolution crystal structure of the globular N-terminal domain of Nsp1 from SARS-CoV-2 at 1.65 Å resolution (PDB: 7K3N). Our data reveal a high level of structural conservation between Nsp1 of SARS-CoV-2 and SARS-CoV but also some unique structural features that likely contribute to increased stability of the β-barrel fold in SARS-CoV-2 Nsp1. Comparative analysis reveals additional structural homologs in Nsp1 proteins from Alphacoronaviruses, despite low levels of shared sequence identity. These results highlight the critical role this unique protein fold plays in facilitating viral infection and suppression of host gene expression.
Results
Crystal structure of SARS-CoV-2 Nsp113-127
In pursuit of structural characterization of Nsp1 from SARS-CoV-2, we expressed a codon-optimized version of the ORF encoding this protein in E. coli. Using this expression system, we obtained purified full-length Nsp1 after a two-step purification protocol (see details in Methods). However, we were unable to crystallize the full-length Nsp1 protein, likely due to the presence of flexibly disordered regions at the N- and C-termini.
Previous studies of Nsp1 from SARS-CoV reported the presence of a distinct globular domain comprising residues 13 to 127 (Almeida et al., 2007). To structurally characterize the N-terminal fragment of Nsp1 from SARS-CoV-2 (Nsp113-127), we sub-cloned it and expressed it in E. coli. The expression level of Nsp113-127 was comparable to that of the full-length protein, and with this fragment, we obtained well-diffracting crystals in several conditions that enabled structural determination of this portion of Nsp1.
The crystal structure of SARS-CoV-2 Nsp113-127 was determined via molecular replacement using the SARS-CoV Nsp1(PDB: 2HSX) solution structure as the search model and refined to a resolution of 1.65 Å (Table 1). The asymmetric unit contained a single molecule of Nsp113-127. Analysis of the Nsp113-127 structure by PDBePISA revealed that the protein has a total solvent-exposed area of 6102 Å2 (Krissinel and Henrick, 2007).
Table 1.
X-ray crystallographic statistics
| SARS-Cov-2 Nsp113-127 (PDB: 7K3N) | |
|---|---|
| Data Collection | |
| Space group |
P43212 |
| Cell dimensions | |
| a, b, c (Å) | 30.0, 37.0, 144.8 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Wavelength | 0.97913 |
| Resolution (Å) | 50.0-1.65 |
| (1.68-1.65) | |
| Rmeasa | 0.058 (1.091) |
| Rpimb | 0.020 (0.387) |
| I/sI | 35.6 (2.0) |
| Completeness (%) | 99.8 (99.5) |
| Redundancy | 8.2 (7.4) |
| CC1/2 | 0.997 (0.690) |
| Refinement | |
| Resolution (Å) | 32.95-1.65 |
| No. reflections | 12,884 |
| Rwork/Rfreec | 0.203/0.248 |
| No. atoms | |
| Protein | 828 |
| Water | 115 |
| Average B-factors (Å2) | 20.7 |
| Protein | 19.2 |
| Water | 31.2 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 0.872 |
| Ramachandran (%) | |
| Favored | 97.1 |
| Allowed | 2.9 |
| Disallowed | 0 |
Values in brackets refer to highest resolution shells.
Rmeas = Σhkl√ (n/n − 1) Σnj=1|Ihkl.j − ⟨Ihkl⟩|/ΣhklΣjIhk,j.
Rpim = Σhkl√ (1/n − 1) Σnj= 1|Ihkl.j − ⟨Ihkl⟩|/ΣhklΣjIhk,j.
R = Σ|Fpobs − Fpcalc|/ΣFpobs, where Fpobs and Fpcalc are the observed and calculated structure factor amplitudes, respectively.
As in the case of SARS-CoV, the structure of SARS-CoV-2 Nsp113-127 features a unique topological arrangement resulting in the formation of a six-stranded (n = 6) β-barrel that is primarily antiparallel, with the exception of strands β1 (Q15 – V20) and β2 (C51 – V54) (Figure 1B). Additional major structural features include α1 (V35 – D48) helix, which is positioned as a cap along one opening of the β-barrel, two 310 helices that run parallel to each other, and the β5 strand (I95 – Y97) which is not part of the β-barrel but forms a β-sheet interaction with the β4 strand (V84 – L92) (Figure 1C).
The core of the β-barrel is highly hydrophobic and mainly comprises the side chains of thirteen AAs organized into three layers. The first layer, which is adjacent to α1 helix, is formed by side chains of residues L16, L18, V69, L88, L107, and L123 (Figure 1D). The opening of the β-barrel at this layer is obstructed by the α1 helix, which contributes the side chain of L46 to the center of this first layer of core hydrophobic residues (Figure 1D). The middle layer of the β-barrel comprises the side chains of residues V20, L53, I71, V86, and V121, while the bottom layer features residues V84 and L104. Adjacent to this bottom layer are four solvent-exposed residues: E55, R73, E102, and R119 whose side chains point inward toward the core of the β-barrel (Figure 1D). Consequently, the hydrophobic core at both ends of the β-barrel is occluded: by the α1 helix on one side and the charged residues (E54, R73, E102, R119) on the other.
A distinctive feature of Nsp1 that is evident in the crystal structure of Nsp113-127 is the large number of flexible loops. For one of these loops (A76 – H81), we were unable to resolve the structure due to a lack of interpretable electron density. This region has been previously shown to be highly flexible as evidenced by the multitude of backbone confirmations observed in the NMR structure of SARS-CoV Nsp1 (Almeida et al., 2007). The positions of other loops in the crystal structure were stabilized via interactions between specific secondary structure elements. Specifically, the two 310 helices are aligned in the crystal structure forming H-bonds between R24 and Q63. This interaction appears to stabilize the position of two of the largest loops in the crystal structure, the β1-α1 (L21 – S34) loop and the β2-β3 (E54 – P67) loop.
Electrostatic surface potential analysis of the SARS-CoV-2 Nsp113-127 structure revealed several regions of charged residue colocalization (Baker et al., 2001). The most electronegative and electropositive patches appear adjacent on the Nsp113-127 surface, separated by a small electroneutral region corresponding to the α1 helix. The electropositive patch continues around the surface of the protein, where the R124 residue is localized (Figure 1E). This residue has been shown to be essential for the interaction between Nsp1 and SL1 of the 5′ UTR, implicating this charged surface in RNA binding (Tanaka et al., 2012).
Comparison with SARS-CoV Nsp1
The overall fold of SARS-CoV-2 Nsp113-127 was highly similar to that of the corresponding SARS-CoV Nsp1 fragment in accordance with the significant primary sequence identity shared between the two proteins (Figure 2A). However, detailed comparative analysis revealed several notable differences between the two structures. The SARS-CoV-2 structure of Nsp113-127 features two 310 helices, compared to the single one found in SARS-CoV Nsp1. The primary sequence corresponding to the second 310 helix is completely conserved between SARS-CoV-2 and SARS-CoV Nsp1 proteins, suggesting that this secondary structure element may be transient in nature or influenced by differences elsewhere in SARS-CoV-2 Nsp113-127. The two 310 helices interact with each other through H-bonds in SARS-CoV-2 Nsp113-127, but this interaction is notably absent in the NMR structure of Nsp1 from SARS-CoV (Almeida et al., 2007).
Figure 2.

Comparison of SARS-CoV-2 Nsp113-127 to SARS-CoV Nsp1
(A) Alignment of residues 13–127 of Nsp1 with conserved residues highlighted in black.
(B) Ribbon representation of SARS-CoV-2 (PDB: 7K3N) and SARS-CoV (PDB: 2HSX) Nsp113-127. β4 (shown in cartoon representation) is extended in the SARS-CoV-2 structure allowing for H-bonding with and formation of β5, which is absent in the SARS-CoV Nsp1 structure. The loop region (and equivalent in SARS-CoV Nsp1) stabilized through interaction between β4 and β5 is colored in purple.
(C) Comparison of electrostatic surface potential of SARS-CoV-2 and SARS-CoV Nsp113-127.
Two of the most striking differences between the two Nsp1 structures are the extension of β4 and the presence of an additional β-strand in the case of SARS-CoV-2 Nsp113-127. AA changes at positions 84 (K to V) and 85 (V to M) between SARS-CoV and SARS-CoV-2 Nsp1 result in an extension of β4 on the N-terminus by three residues (Figure 2B). This extension results in an increased number of H-bonds formed between β4 and β7 in SARS-CoV-2 Nsp1 compared to the pairing between β4 and β6 that occurs in SARS-CoV Nsp1. This extension also facilitates the formation of the β5 strand which is absent in SARS-CoV Nsp1. The β5 and β4 strands are part of a small β-sheet that stabilizes the loop region between β5 and β6 (equivalent to β4 and β5 in SARS-CoV Nsp1) (Figure 2B).
Comparison of the electrostatic surface potential of Nsp113-127 from SARS-CoV-2 and SARS-CoV shows additional differences between the two proteins. The SARS-CoV structure features a single patch of electronegative surface potential opposite a separate patch of electropositive surface potential. These two charged surfaces are oriented 180° opposite one another, and there is a ring of electroneutral surface potential separating the two charged surfaces (Figure 2C). The SARS-CoV-2 Nsp113-127 structure features an equivalent patch of electronegativity, but the electropositive surface is divided into two by a patch of mild electronegative surface potential.
Mapping the non-conserved residues onto the SARS-CoV-2 Nsp113-127 structure revealed that most of the differences are in solvent-exposed residues. Distinct patches of variation are observed immediately adjacent to both ends of β4 and in the loop between β3 and β4 (S74 – H83), where 4 of 9 residues differ between SARS-CoV-2 and SARS-CoV (Figure 3A). The remaining sequence variation is broadly distributed across the Nsp113-127 structure. Residues that contribute to the hydrophobic core of the β-barrel are conserved between SARS-CoV-2 and SARS-CoV, suggesting that this particular organization is critical to the function of Nsp1.
Figure 3.

Comparison of the structure of SARS-CoV-2 Nsp113-127 to structural homologs
(A) Cartoon representation of the SARS-CoV-2 Nsp113-127 structure with non-conserved residues between SARS-CoV-2 and SARS-CoV highlighted in magenta.
(B) SARS-CoV-2 Nsp113-127 (PDB: 7K3N) is shown in green; SARS-CoV Nsp1 (PDB: 2HSX), TGEV Nsp1 (PDB: 6IVC), and PEDV Nsp1 (PDB: 5XBC) are shown in gray with noticeable structural differences (compared to SARS-CoV-2) highlighted in red. Distinctive secondary structure features from the SARS-CoV-2 Nsp113-127 structure are labeled.
(C) Homology model of β-barrel domain of Nsp1 from MERS-CoV. Conserved residues between MERS-CoV and SARS-CoV-2 Nsp1 are highlighted in magenta and the N- and C-termini are labeled.
Comparison with other structural homologs
AA sequence-based searches for homologs of Nsp1 reveal a high level of sequence conservation among representatives of lineage B of betacoronaviruses, which includes several animal viruses closely related to SARS-CoV and SARS-CoV-2. Despite the presence of functionally equivalent Nsp1 proteins, no sequence-based homologs of SARS-CoV-2 Nsp1 were detected in other lineages of betacoronaviruses. This lack of sequence conservation is observed even when comparing SARS-CoV-2 Nsp1 to other coronaviruses known to infect humans, including Middle East respiratory syndrome coronavirus (MERS-CoV), human coronavirus (HCoV)-OC43, and HCoV-HKU1.
A search for structural homologs using our high-resolution structure of SARS-Cov-2 Nsp113-127 identified three closely related structural homologs, all annotated as Nsp1 proteins (Table 2) (Holm and Laakso, 2016). Unsurprisingly, the Nsp1 structure from SARS-CoV was identified as the closest homolog, but the search also indicated strong similarity to the Nsp1 proteins from transmissible gastroenteritis virus (TGEV) and porcine epidemic diarrhea virus (PEDV). These Alphacoronavirus homologs superimposed with SARS-CoV-2 Nsp113-127 with root-mean-square deviation (RMSD) values of 2.8 Å and 2.9 Å over 114 and 118 Cα atoms, respectively (Table 2) (Shen et al., 2019). Such appreciable levels of structural conservation, despite the low levels (<15%) of shared sequence identity, between these proteins highlight the plasticity of the unique protein fold found in coronaviral Nsp1 proteins (Figure 3B).
Table 2.
Structural homologs of SARS-Cov-2 Nsp113-127
To further examine the plasticity of this fold, we examined the predicted secondary structure of Nsp1 proteins from other coronaviruses known to infect humans (MERS, OC43, HKU1, 229E, NL63). These Nsp1 proteins share minimal primary sequence identity with Nsp1 from SARS-CoV-2, and a BLASTP search failed to identify them as homologs (Table S1). Despite the low primary sequence identity, we were able to identify topological fingerprints, regions comprising 6–7 β-strands and at least one α-helix, in each of the Nsp1 sequences that are indicative of an arrangement capable of forming the capped β-barrel motif observed in the SARS-CoV-2 Nsp113-127 crystal structure (Figure S1). We then modeled the region of MERS-CoV Nsp1 identified through this approach using our crystal structure as the threading template. This resulted in a high-scoring (C-score = 0.48) homology model featuring the capped β-barrel motif characteristic of the structure of SARS-CoV-2 Nsp113-127 (Figure 3C). Mapping the modest primary sequence conservation between MERS-CoV and SARS-CoV-2 onto the homology model revealed it to be distributed throughout the structure (Figure 3C). The β-barrel in the MERS-CoV Nsp1 homology model retains the hydrophobic core observed in SARS-CoV-2, highlighting a level of functional conservation between the two proteins that may not have been predicted through sequence-based comparisons alone. At the C-terminus of the model is the R146/K147 dipeptide (R124/K125 in SARS-CoV-2) that has been shown to be involved in Nsp1-mediated RNA cleavage (Terada et al., 2017).
Nsp113-127 structure in context of the full-length SARS-CoV-2 Nsp1
Recent cryogenic electron microscopy (cryo-EM) data of SARS-CoV-2 Nsp1 in complex with the 40S ribosome were able to resolve the structure of the C-terminal portion of Nsp1 at a resolution of 2.6 Å (Thoms et al., 2020) and 2.8 Å (Schubert et al., 2020). These studies confirm the presence of a C-terminal α-helix spanning residues S166 to N178 and revealed a second helix spanning residues P153 to N160 that was not predicted bioinformatically. The resulting helix-turn-helix motif interacts with the ribosome and anchors Nsp1 to the pre-initiation complex.
To place the crystal structure of Nsp113-127 in context of full-length protein, we analyzed the AA sequence using PSIPRED and DISPRRED3 (Buchan and Jones, 2019; Jones and Cozzetto, 2015). These tools predicted the N-terminal 12 residues outside of our structure containing a disordered protein-binding region and a largely unstructured C-terminal domain with the exception of a single predicted α-helix. The presence of large disordered regions adjacent to the globular domain of Nsp1 provides strong rationale for why the full-length protein failed to crystallize.
Using our crystal structure data in combination with insights into the C-terminal domain of Nsp1, we generated a homology-based model of the full-length Nsp1 protein. This model maintains the capped β-barrel motif observed in our crystal structure and features a largely disordered linker between the end of the globular domain and the C-terminal helix-turn-helix. The C-terminal domain is shown as extending outward from the core of the structure at the bottom of the β-barrel, at the face opposite of where the α1 helix localizes; however, the disordered linker connecting this motif from the rest of the structure suggests that the position of this domain is likely to be highly dynamic and capable of multiple configurations (Figure 4). This predicted full-length structure aligns well with the partial density and model proposed by the recent cryo-EM structures of the SARS-CoV-2 Nsp1 C-terminus bound to the 40S ribosome (Thoms et al., 2020).
Figure 4.

Model of full-length SARS-CoV-2 Nsp1
Cartoon representation of the model of the full-length SARS-CoV-2 Nsp1 colored from N-terminus (blue) to C-terminus (red). Distinctive secondary structure features are labeled.
Discussion
SARS-CoV-2 Nsp1 arrests translation of host proteins through interaction with the 40S ribosome and in doing so suppresses the innate immune response to viral infection. Through interaction with SL1 in the viral 5′UTR, Nsp1 also distinguishes viral transcripts from host transcripts and allows for viral gene expression to continue unabated, highlighting the critical role this nonstructural protein plays in SARS-CoV-2 pathogenesis. Nsp1 proteins encoded by representatives of the Coronaviridae family feature significant AA sequence diversity, highlighted by the inability to identify homologs via BLAST of SARS-CoV-2 Nsp1 outside of the B lineage of betacoronaviruses. Leveraging our high-resolution structural data, we were able to identify additional structural homologs of SARS-CoV-2 Nsp1, Nsp1 of TGEV, and PEDV, with which it shares low levels of sequence identity. This result is in accordance with the recent data demonstrating that coronaviral Nsp1 proteins exhibit similar function in suppression of host translation (Terada et al., 2017). These observations suggest that the distinctive features of the SARS-CoV-2 Nsp1, highlighted by our structural data, may be critical to its function in self-recognition, via interaction with SL1, of viral RNAs and its role in promoting the degradation of host transcripts. This also provides insight into molecular features that could be expected in functionally similar proteins found in other important human pathogens, such as Nsp1 from MERS-CoV.
Our sequence and structural based queries using SARS-CoV-2 Nsp1 were only able to identify homologs within the Coronaviridae family. This raises the possibility that the specific fold and functions of Nsp1 are unique to coronaviruses; however, the failure of sequence-based searches to identify Nsp1 from TGEV and PEDV as homologs of SARS-CoV-2 Nsp1 highlights the need for continued structural characterization of proteins involved in viral pathogenesis.
Limitations of the study
The crystal structure presented lacks the C-terminal domain of Nsp1 that has been shown to be critical and sufficient for facilitating interaction with the ribosome and inhibition of translation. Experimentally determining the full-length structure of Nsp1, alone or with an interaction partner, remains an important yet unfulfilled advancement to furthering our understanding of this essential component of SARS-CoV-2 infection.
Resource availability
Lead contact
Further information and requests for resources should be directed to the corresponding author, Alexei Savchenko (alexei.savchenko@ucalgary.ca)
Materials availability
Plasmids used in this study are available upon request.
Data and code availability
The structure of SARS-CoV-2 Nsp113-127 and all associated data have been deposited to the protein databank (www.rcsb.org) and are available under the accession code PDB: 7K3N.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank Changsoo Chang and the Structural Biology Center Team at APS for data collection. The structure presented was solved as part of the Center for Structural Genomics of Infectious Diseases (CSGID). This project has been funded in whole or in part with U.S. Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract Nos. HHSN272200700058C, HHSN272201200026C, and HHSN272201700060C.
Author contributions
Conceptualization, C.S. and A.S.; Investigation, C.S.; Formal Analysis, C.S. and N.W.; Writing – Original Draft, C.S. and A.S.; Writing – Review and Editing, C.S., N.W., and A.S.; Funding Acquisition – A.S.
Declaration of interests
The authors declare that they have no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101903.
Supplemental information
References
- Almeida M.S., Johnson M.A., Herrmann T., Geralt M., Wuthrich K. Novel beta-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:3151–3161. doi: 10.1128/JVI.01939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N.A., Sept D., Joseph S., Holst M.J., Mccammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan D.W.A., Jones D.T. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019;47:W402–W407. doi: 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F., Kok K.H., Zhu Z., Chu H., To K.K., Yuan S., Yuen K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Laakso L.M. Dali server update. Nucleic Acids Res. 2016;44:W351–W355. doi: 10.1093/nar/gkw357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T., Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015;31:857–863. doi: 10.1093/bioinformatics/btu744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W., Narayanan K., Huang C., Lokugamage K., Ikegami T., Ito N., Kubo H., Makino S. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl. Acad. Sci. U S A. 2006;103:12885–12890. doi: 10.1073/pnas.0603144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Law A.H., Lee D.C., Cheung B.K., Yim H.C., Lau A.S. Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J. Virol. 2007;81:416–422. doi: 10.1128/JVI.02336-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler D.R., Gully B.S., Colson R.N., Rossjohn J. Crystal structure of the SARS-CoV-2 non-structural protein 9, Nsp9. iScience. 2020;23:101258. doi: 10.1016/j.isci.2020.101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi A.A.T., Fatima K., Mohammad T., Fatima U., Singh I.K., Singh A., Atif S.M., Hariprasad G., Hasan G.M., Hassan M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165878. doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Huang C., Lokugamage K., Kamitani W., Ikegami T., Tseng C.T., Makino S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008;82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Ramirez S.I., Lokugamage K.G., Makino S. Coronavirus nonstructural protein 1: common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015;202:89–100. doi: 10.1016/j.virusres.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X., Wang Q., Sun Y., Fan Z., Qi J. Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep. 2020;31:107774. doi: 10.1016/j.celrep.2020.107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert K., Karousis E.D., Jomaa A., Scaiola A., Echeverria B., Gurzeler L.A., Leibundgut M., Thiel V., Muhlemann O., Ban N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020;27:959–966. doi: 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- Shen Z., Wang G., Yang Y., Shi J., Fang L., LI F., Xiao S., Fu Z.F., Peng G. A conserved region of nonstructural protein 1 from alphacoronaviruses inhibits host gene expression and is critical for viral virulence. J. Biol. Chem. 2019;294:13606–13618. doi: 10.1074/jbc.RA119.009713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Kamitani W., Dediego M.L., Enjuanes L., Matsuura Y. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J. Virol. 2012;86:11128–11137. doi: 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada Y., Kawachi K., Matsuura Y., Kamitani W. MERS coronavirus nsp1 participates in an efficient propagation through a specific interaction with viral RNA. Virology. 2017;511:95–105. doi: 10.1016/j.virol.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., Kratzat H., Hayn M., Mackens-Kiani T., Cheng J. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S.R., Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005;69:635–664. doi: 10.1128/MMBR.69.4.635-664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liu C., Du L., Jiang S., Shi Z., Baric R.S., Li F. Two mutations were critical for bat-to-human transmission of Middle East respiratory syndrome coronavirus. J. Virol. 2015;89:9119–9123. doi: 10.1128/JVI.01279-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q., Wang B., Mao J. The pathogenesis and treatment of the `Cytokine Storm' in COVID-19. J. Infect. 2020;80:607–613. doi: 10.1016/j.jinf.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Wu Q., Zhang Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020;30:1346–1351.e2. doi: 10.1016/j.cub.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.F., Stephen P., Theriault J.F., Wang R., Lin S.X. Novel coronavirus polymerase and nucleotidyl-transferase structures: potential to target new outbreaks. J. Phys. Chem. Lett. 2020;11:4430–4435. doi: 10.1021/acs.jpclett.0c00571. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zust R., Cervantes-Barragan L., Kuri T., Blakqori G., Weber F., Ludewig B., Thiel V. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007;3:e109. doi: 10.1371/journal.ppat.0030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structure of SARS-CoV-2 Nsp113-127 and all associated data have been deposited to the protein databank (www.rcsb.org) and are available under the accession code PDB: 7K3N.