

Graphical abstract

Keywords: ATP synthase, Structure-activity relationships, Synthesis, Tetrahydroquinolines, Tuberculosis
Abbreviations: DCM, dichloromethane; DIPEA, N,N-diisopropylethylamine; Et2O, diethyl ether; EtOAc, ethyl acetate; THF, tetrahydrofuran; MeOH, methanol
Abstract
A series of 5,8-disubstituted tetrahydroisoquinolines were shown to be effective inhibitors of M. tb in culture and modest inhibitors of M. tb ATP synthase. There was a broad general trend of improved potency with higher lipophilicity. Large substituents (e.g., Bn) at the tetrahydroquinoline 5-position were well-tolerated, while N-methylpiperazine was the preferred 8-substituent. Structure-activity relationships for 7-linked side chains showed that the nature of the 7-linking group was important; –CO– and –COCH2– linkers were less effective than –CH2– or –CONH– ones. This suggests that the positioning of a terminal aromatic ring is important for target binding. Selected compounds showed much faster rates of microsomal clearance than did the clinical ATP synthase inhibitor bedaquiline, and modest inhibition of mycobacterial ATP synthase.
1. Introduction
The treatment of drug-resistant tuberculosis (TB) has in recent times become a major global health problem.1 Recent encouragement has come from the discovery2 of the drug bedaquiline (1) (Fig. 1), which is a selective inhibitor of the ATP synthase enzyme of the causal bacterium Mycobacterium tuberculosis (M.tb), and is clinically effective against drug-resistant strains.3 A “second-generation” analogue of bedaquiline (TBAJ-876) has recently began clinical trial.4, 5, 6, 7
Fig. 1.

Examples of bioactive N-substituted tetrahydroisoquinolines.
In a search for new drugs for the treatment of tuberculosis we report here, from a screening lead (compound 13 of Table 1), the synthesis and structure–activity relationships of a new class of N-substituted 5,8-disubstituted tetrahydroisoquinolines and develop initial structure–activity relationships for a series of amide and urea analogues of these. N-Substituted tetrahydroisoquinolines have previously been reported as antagonists of the 5-hydroxytryptamine 5HT1B receptor8 (e.g., 2, Fig. 1), as potential anti-depressive agents9 (e.g., 3) and as anti-hypertensive agents10 (e.g., 4), and as inhibitors of the excitatory neuropeptides orexin-A and orexin-B (e.g., 5),11 but have not been reported as anti-tubercular agents.
Table 1.
Structural and biological data for 5, N-disubstituted 8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinolines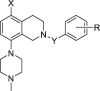
| MIC | IC50 | ||||||
|---|---|---|---|---|---|---|---|
| No | X | Y | R | MABAa | LORAb | VEROc | clogPd |
| 6 | Me | 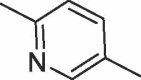 |
2-Me, 4-Cl | 1.9 | 2.4 | 22 | 6.03 |
| 7 | Me | 4-tBu | 1.4 | 3.4 | 18 | 7.12 | |
| 8 | OMe | 2-Me, 4-Cl | 1.9 | 3.6 | 12 | 5.43 | |
| 9 | OMe | 3,5-diCF3 | 7.9 | 12 | 19 | 6.31 | |
| 10 | H | 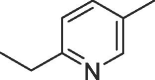 |
4-Cl | 1.8 | 16 | 16 | 5.17 |
| 11 | H | 2-Me, 4-Cl | 3.6 | 11 | 15 | 5.36 | |
| 12 | Me | 2-Me, 4-Cl | 0.79 | 5.2 | 14 | 5.86 | |
| 13 | OMe | 2-Me, 4-Cl | 1.8 | 6.1 | 12 | 5.27 | |
| 14 | SMe | 2-Me, 4-Cl | 3.4 | 6.0 | 11 | 5.81 | |
| 15 | F | 2-Me, 4-Cl | 3.8 | 6.1 | 24 | 5.68 | |
| 16 | F | 2,4-diF | 15 | 21 | 22 | 5.06 | |
| 17 | F | 2-CF3, 4-Cl | 2.3 | 5.4 | 11 | 5.65 | |
| 18 | H | 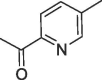 |
2-Me, 4-Cl | 11 | 13 | 20 | 4.80 |
| 19 | H | 4-Cl | 16 | 20 | 27 | 4.60 | |
| 20 | H | 3-Cl | 15 | 11.3 | 24 | 4.60 | |
| 21 | OMe | 4-Cl | 13 | 12 | 11 | 4.51 | |
| 22 | OMe | 2-Me, 4-Cl | 8.9 | 8.0 | 22 | 5.30 | |
| 23 | H | 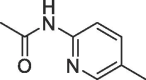 |
4-OCF3 | 3.6 | 3.9 | 14 | 5.02 |
| 24 | H | 2-Me, 4-Cl | 6.2 | 7.7 | 11 | 4.83 | |
| 25 | Me | 2,4-diCl | 1.95 | 1.99 | 12 | 5.60 | |
| 26 | Me | 3,5-diCF3 | 3.1 | 5.3 | >32 | 6.22 | |
| 27 | Et | 4-CF3 | 1.0 | 0.9 | 12 | 5.73 | |
| 28 | Et | 2-Me, 4-Cl | 2.9 | 1.8 | 12 | 5.74 | |
| 29 | Bn | 2-Me, 4-Cl | 1.8 | 1.5 | 11 | 7.08 | |
| 30 | Bn | 2,4-diMe | 2.0 | 1.9 | 11 | 6.66 | |
| 31 | OMe | 3,5-diaza | >32 | >32 | >32 | 2.59 | |
| 32 | OMe | 3-aza, 4-OMe | 15.5 | 20 | 14 | 3.30 | |
| 33 | OMe | 4-OMe | 7.1 | 7.4 | 16 | 3.82 | |
| 34 | OMe | 2-Cl, 4-CF3 | 3.6 | 3.2 | 12 | 5.19 | |
| 35 | OMe | 2-Me, 4-Cl | 1.9 | 3.1 | 12 | 4.73 | |
| 36 | OMe | 2,4-diCl | 1.9 | NDe | >32 | 5.01 | |
| 37 | OMe | 4-OCF3 | 3.1 | 3.0 | 12 | 4.93 | |
| 38 | OMe | 3,5-diCF3 | 1.3 | NDe | 10 | 6.67 | |
| 39 | SMe | 3,5-diCF3 | 3.6 | 4.0 | 20 | 6.17 | |
| 40 | SMe | 2-Me, 4-Cl | 1.9 | 2.0 | 13 | 5.28 | |
| 41 | F | 3-CF3, 4-Cl | 1.4 | 2.9 | 10 | 6.18 | |
| 42 | F | 2-Me, 4-Cl | 1.2 | 5.4 | 12 | 5.14 | |
| 43 | H | 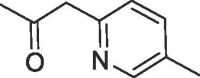 |
4-Cl | 14.1 | 20.3 | 19 | 4.05 |
| 44 | H | H | >32 | >32 | >32 | 3.32 | |
| 45 | H | 3-Cl | 15.0 | 21.7 | 25 | 4.05 | |
| 46 | H | 4-OCF3 | 13.7 | 12.4 | 20 | 4.45 | |
| 47 | H | 2-Me, 4-Cl | 14.0 | 19.4 | 11 | 4.25 | |
| 48 | Me | 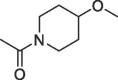 |
4-OCF3 | 6.4 | 3.9 | 19 | 5.71 |
| 49 | Me | 2-CF3 | 7.7 | 7.3 | 11 | 5.64 | |
| 50 | OMe | 4-OCF3 | 7.6 | 7.0 | 15 | 5.12 | |
| 51 | OMe | 2-CF3 | 12 | 10.0 | 24 | 5.05 | |
bMIC90 (µg/mL); minimum inhibitory concentration for inhibition of growth of M.tb strain H37Rv, determined under aerobic (replicating; MABA)12 or non-replicating (LORA)13 conditions, determined at the Institute for Tuberculosis Research, University of Illinois at Chicago. cCytotoxicity (IC50, µg/mL) in Vero green monkey-derived epithelial kidney cells;14dclogP calculated by ChemDraw Ultra v12.0.2. (CambridgeSoft); eNot done.
2. Results and discussion
2.1. Chemistry
The tetrahydroisoquinolines of Table 1 were prepared from the appropriate 5-substituted-8-bromoisoquinolines. Seven different 5-substituents were evaluated, and four of these (X = H, Me, OMe, F; compounds 56–59 respectively) were commercially available. The SMe-, Et- and Bn-substituted analogues (61–63) were prepared from the known isoquinoline 60 as shown in Scheme 1. Bromination of 60 with NBS gave the 5-Br compound 64, which was thiomethylated to give 65, then reduced to give 61. Lithiation of 64 and treatment with acetaldehyde gave 66, which was successively reduced with Et3SiH to give 67, then with NaCNBH3 to 62. Similar condensation of 64 with benzaldehyde gave 68, which was similarly reduced via 69 to give 63.
Scheme 1.

Syntheses of 5-substituted tetrahydroisoquinoline intermediates.
Reagents and conditions: (i) NBS, DMF, 20 °C, 3 days; (ii) nBuLi, THF, −78 °C, 5 min, then MeSSMe, −78 °C, 2 h; (iii) NaCNBH3, BF3.Et2O, MeOH, reflux, 22 h; (iv) nBuLi, THF, −78 °C, 5 min, then RCHO, −78 °C, 2 h; (v) Et3SiH, TFA, 75 °C, 1 h.
The compounds of Table 1 were then prepared by elaboration of the various side chains on these tetrahydroisoquinolines. Compounds 6, 7 and 9 (directly linked pyridyl analogues) were synthesized by selective reaction of tetrahydroisoquinolines 57 and 58 with 2,5-dibromopyridine (70) to give compounds (71, 72) that were Suzuki-coupled with the appropriate phenylboronic acids (73) or 81 (Scheme 2A). Compound 8 was synthesized by Buchwald coupling of 58 with the pre-formed sidechain bromide 75.
Scheme 2.


Syntheses of the tetrahydroisoquinolines 6–17 of Table 1.
Compounds 10, 11, 14–17 (CH2-linked analogues) were prepared by reaction of known tetrahydroisoquinolines 56, 59 and 61 with 5-bromo-2-(chloromethyl)pyridine (76) to give intermediates 77–79 (Scheme 2B). These were coupled with appropriate phenylboronic acids (73) or 81 to give 10, 11, 14–17 of Table 1. Compound 13 was prepared by the N-alkylation reaction of tetrahydroisoquinoline 58 with the fully pre-formed sidechain bromide 84. This was synthesised by Suzuki coupling of aldehyde 80 and boronic acid 81 to give aldehyde 82, which was reduced to alcohol 83 and brominated to give 84. Compound 12 was synthesised by Buchwald coupling of the sidechain-preinstalled tetrahydroisoquinoline-8-bromide 86 with N-methylpiperazine. 86 was prepared from tetrahydroisquinoline-8-bromide 85 by the N-alkylation with 84.
2A: directly-linked pyridyl side chains; compounds 6–9 of Table 1
Reagents and conditions: (i) Xantphos, Pd2(dba)3, NaOtBu, toluene, 100 °C, 4 h ; (ii) R-PhB(OH)2 (73) or 81, 2 M aq Na2CO3, PdCl2dppf, toluene, EtOH, 85 °C, 20 ~ 24 h; (iii) HBr, Br2, HCl, NaNO2, −5°C; (iv) 58, BINAP, Pd2(dba)3, NaOtBu, toluene, reflux, 1 h.
2B: –CH2-linked pyridyl side chains; compounds 10–17 of Table 1
Reagents and conditions: (i) K2CO3, DMF, 20 °C,15 h; (ii) R-PhB(OH)2 (73) or 81, aq Na2CO3, PdCl2dppf, toluene, EtOH, 80 °C, various hours; (iii) K3PO4·H2O, water, acetonitrile, dioxane, Pd(PPh3)4, reflux, 5 h; (iv) NaBH4, anhydrous MeOH, 4 h, 20 °C; (v) MsCl, Et3N, DCM, 20 °C, 15 min; then LiBr, anhydrous acetone, reflux, 1.5 h; (vi) 58, K2CO3, DMF, 20 °C, 5 h (vii) K2CO3, DMF, 20 °C, 5 h; (viii) N-methylpiperazine, BINAP, Pd2(dba)3, NaOtBu, toluene, reflux, 4 h.
The CO-linked compounds 18–22 of Table 1 were prepared as shown in Scheme 3, by HATU-mediated coupling of tetrahydroisoquinolines 56 or 58 with commercially-available phenyl-substituted 5-phenylpicolinic acids (87–89).
Scheme 3.

Synthesis of CO-linked compounds 18–22 of Table 1.
Reagents and conditions: (i) HATU, DIPEA, DMF, 20 °C, 16 h
Two general methods, chosen broadly on the availability of the starting materials, were used to prepare the amide-linked series of compounds 23–42 of Table 1 (Scheme 4). Reaction of tetrahydroisoquinolines (56–59, 61–63) with preformed nitrobenzylcarbamates 92 gave Table I compounds directly. Alternatively, reaction of the tetrahydroisoquinolines with the intermediate 4-nitrophenyl (5-bromopyridin-2-yl)carbamate) 93 gave the bromides 94, which were coupled with substituted phenylboronic acids (73) or 81.
Scheme 4.

Synthesis of CONH-linked compounds 23–42 of Table 1.
Reagents and conditions: (i) R-PhB(OH)2 (73) or 81, Pd(PPh3)4, 2 M Na2CO3, DME, reflux, 2.5 h; (ii) 4-nitrophenyl chloroformate, pyridine, DCM, 20 °C, 15 h; (iii) MeCN, 70 °C, overnight; (iv) 4-nitrophenylchloroformate, pyridine, DCM, 2–20 °C, overnight (v) DMF, 75 °C, 26 h; (vi) R-PhB(OH)2 (73) or 81, PdCl2dppf, 2 M Na2CO3, toluene/EtOH, 85 °C, 15 h.
Compounds 43–47 of Table 1 evaluated the utility of a more flexible- COCH2– linker. They were prepared as noted in Scheme 5, by reacting tetrahydroisoquinoline 56 with 2-(5-bromopyridin-2-yl)acetic acid 96 to give the bromide 97. This underwent Suzuki coupling with substituted phenylboronic acids (73) or 81 to provide the listed compounds.
Scheme 5.

Synthesis of –COCH2-linked compounds 43–47 of Table 1.
Reagents and conditions: (i) EDCI, DIPEA, HOBt, DMAP, DMF, 20 °C, 15 h; (ii) R-PhB(OH)2 (73) or 81, PdCl2dppf, 2 M Na2CO3, toluene/EtOH, 85 °C, 15 h.
Compounds 48–51 in Table 1, with carbamate-linked non-aromatic sidechain terminal units, were prepared by reacting 4-nitrophenylcarbonyl chloride 98 and amines 99, 100 to give the 4-nitrophenylcarbamate intermediates 101 and 102, which were in turn coupled with tetrahydroisoquinolines 57 or 58 (Scheme 6).
Scheme 6.

Synthesis of –CO-linked non-aromatic sidechain compounds 48–51 of Table 1.
Reagents and conditions: (i) NEt3, DCM, 20 °C, 2 h (90%); (ii) DMAP, toluene, 110 °C, 3 days.
Finally, the compounds of Table 2 explore variations in the 8-N-methylpiperazine unit, by replacing it with a range of related 6-membered heterocycles containing a fixed 2-pyridyl linker and a 2-methyl-4-chlorophenyl side chain (compounds 52–55 of Table 2). Similar to the synthesis of compound 12, treatment of 86 (see Scheme 2B) with various cyclic amines gave compounds 52–54. (Scheme 7). Compound 55 was synthesised by the same procedure as for compound 13 in Table 1 by the N-alkylation reaction of 106 with 84. 106 was prepared from 104 by Buchwald amination followed by reduction.
Table 2.
Structures of 8-substituted analogues and their in vitro assay data.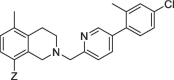
| MIC (µg/mL) | IC50 | ||||
|---|---|---|---|---|---|
| No | Z | MABAa | LORAb | VEROc | clogPd |
| 12 | 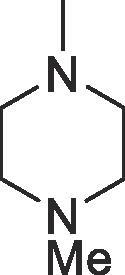 |
0.79 | 5.2 | 14 | 5.86 |
| 52 | 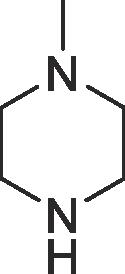 |
1.8 | 4.4 | 12 | 5.29 |
| 53 | 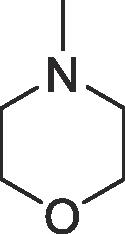 |
7.8 | 12 | >32 | 5.30 |
| 54 | 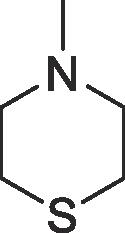 |
3.4 | 5.9 | >32 | 6.14 |
| 55 | 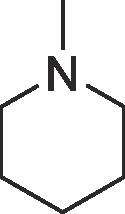 |
2.6 | NDe | >32 | 6.69 |
a–d As for Table 1; eNot done.
Scheme 7.

Syntheses of the tetrahydroisoquinolines 52–55 of Table 2.
Reagents and conditions: (i) Pd2(dba)3, NaOtBu, rac-BINAP, toluene, N-BOC-piperidine, 100 °C, 5 h; (ii) TFA, DCM; (iii) conditions as for i with morpholine or thiomorpholine; (iv) conditions as for i with piperidine; (v) NaBH3CN, MeOH, 0 °C for 10 min, then BF3.OEt2 0 °C for 1 h and reflux 3 h; (vi) 84, K2CO3, DMF, 20 °C, 5 h.
2.2. Structure-activity relationships
The compounds of Table 1 were evaluated for their minimum inhibitory concentrations (MICs; µg/mL) against Mycobacterium tuberculosis (strain H37Rv) cultured under both aerobic (MABA)12 and anerobic (LORA)13 conditions. The mammalian cytotoxicity (IC50s) of the compounds was assessed against a VERO cell line,14 and the results are recorded in Table 1.
The active compounds possess a wide range of lipophilicities, with calculated clogP values from 2.59 to 7.12. Previously, in several series of quite different classes of compounds that hit different M.tb targets, overall positive correlations are seen between antitubercular potency (measured as MICs) and compound lipophilicity (measured as clogP),15, 16, 17 and the same broad trend is shown here for the compounds with measurable MICs (equation 1).
| Log(MICMABA) = -0.31(±0.06)clogP + 2.25 (±0.30) | (1) |
n = 44, R = 0.65, F1,42 = 30.1
However, some structure–activity trends over and above this one can be seen. MIC potency falls off sharply at overall clogP values below about 4.5 (e.g., compounds 31, 33, 43–47), regardless of the molecular structure. Due to the diversity of structures in the set of compounds, it is more difficult to identify positive SAR contributions to potency. Compounds (28–30) with the most lipophilic X substituents Me, Et or Bn had an average MIC (MABA) for of 2.8 µg/mL, compared to an average MIC of 5.0 µg/mL for a larger group of 35 compounds where X was H or OMe, but this may be just the global lipophilicity effect.
For the vast majority of the compounds, the Y-component was a 2-pyridyl, attached to the tetrahydroisoquinoline by a variety of linkers of varying polarity and geometry. Since not all of these sets contained an identical set of R substituents on the terminal phenyl ring there is some noise in the data, but some conclusions can be drawn for the larger sets. Thus compounds 6–9 (direct link), 10–17 (CH2 link), and 23–42 (para CONH link) had average MICs (MABA) of 3.3, 4.1 and 3.7 µg/mL respectively, whereas compounds 18–22 CO linker) and 43–47 (COCH2 linker) had much poorer average MICs (12.2 and 17.7 µg/mL respectively). The aliphatic-linked compounds (48–51) showed potencies in between, with an average MIC of 8.3 µg/mL. Finally, the average MIC (MABA) for the 11 compounds bearing (more lipophilic) alkyl 5-substituents was 2.8 µg/mL, compared with average MICs of 4.8 µg/mL for the 17 5-F compounds and 5.4 µg/mL for the 18 5-OMe compounds of Table 1.
From these results, we selected the CH2-pyridyl linker and the 2-methyl-4-chlorophenyl terminal group as suitable for exploring the nature of the substituent space around the tetrahydroisoquinoline 8-position, by replacing the N-methylpiperazine with various alternates (Table 2).
The results show that a substituted piperazine functional group at the tetrahydroisoquinoline 4-position provides the best anti-tubercular activity (compounds 12 and 52). The more lipophilic N-piperidyl compound 55 maintains relatively good potency in the MABA assay.
Representative examples of the more potent compounds of Table 1 were evaluated in human and mouse liver microsomes to determine their primary microsomal clearance rates (Table 3). All of the compounds evaluated showed much faster clearance than bedaquiline (1), where the very slow human clearance of bedaquiline and its M2 metabolite may contribute to drug-related toxicities.18
Table 3.
Microsomal clearance data for representative compounds of Table 1.
| No | HLM Clinta | MLM Clinta |
|---|---|---|
| 1 | 3.0 | 7.0 |
| 6 | 22 | 35 |
| 7 | 41 | 56 |
| 8 | 17 | 42 |
| 10 | 121 | 238 |
| 12 | 92 | 239 |
| 27 | 107 | 810 |
| 36 | 48 | 584 |
| 42 | 50 | 65 |
| 48 | 48 | 40 |
aClearance (µL/min/kg protein). HLM (Human liver microsomes), MLM (Mouse liver microsomes).
Bedaquiline (1) and representative compounds of the tetrahydroisoquinolines of Table 1 were also evaluated for their ability against and selectivity for the mycobacterial (M. smegmatis) ATP synthase enzyme (Table 4). Bedaquiline (1) is known to be a potent) and very selective inhibitor of mycobacterial ATP synthase,19 with an IC50 of 0.55 µg/mL for the M. smegmatis enzyme. The most potent of the tetrahydroisoquinolines was compound 42 (IC50 of 1.8 µg/mL) and a lesser but still useful selectivity (9.4-fold) over the human enzyme. Other analogues (e.g., 10, 23) were less potent but more selective inhibitors (>70- and >80-fold respectively).
Table 4.
ATP synthase inhibition for representative compounds of Table 1.
| No | M. smeg ATPsyntha | Mamm. ATPsyntha |
|---|---|---|
| 1 | 0.55 | >1000 |
| 6 | 5.5 | 23 |
| 8 | 4.2 | 16 |
| 10 | 7.2 | >500 |
| 23 | 6.2 | >500 |
| 26 | 14 | 13 |
| 27 | 2.2 | 7.2 |
| 35 | 6.3 | 20 |
| 36 | 3.0 | 27 |
| 42 | 1.8 | 17 |
| 49 | 6.2 | 48 |
aIC50 values (µg/mL) for inhibition of M. smegmatis and human mitochondrial ATP synthase.22
3. Conclusions
A new class of tetrahydroquinoline compounds are effective inhibitors of M.tb in culture, with one-third of the compounds showing MICs of <2 µg/mL. As expected13-15 there was a broad general trend of improved potency with higher lipophilicity, with potency falling off sharply below clogP below 4.5. Large substituents (e.g., Bn) at the tetrahydroquinoline 5-position were well-tolerated. There were interesting group differences between sets of compounds with varying linker units Y; compounds 18–22 (–CO- linker) and compounds 43–47 (–COCH2- linker) were much less effective. This might be due to different positioning of the terminal aromatic ring for target binding, which does seem to be important, since compounds 48–51, with a non-aromatic terminal ring, were also relatively ineffective.
Pleasingly, all of the representative compounds evaluated in more detail for microsomal clearance showed much faster rates than 1, and were effective inhibitors of M. tb (M. smegmatis) ATP synthase, albeit with lesser differentials than 1 for M. tb over human enzyme.
4. Experimental
4.1. Chemistry
Final products were analysed by reverse-phase HPLC (Alltima C18 5 µm column, 15 × 3.2 mm; Alltech Associated, Inc., Deerfield, IL) using an Agilent HP1100 equipped with a diode-array detector. Mobile phases were gradients of 80% CH3CN/20% H2O (v/v) in 45 mM NH4HCO2 at pH 3.5 and 0.5 mL/min. Purity was determined by monitoring at 330 ± 50 nm and was ≥95% for all final products. Melting points were determined on an Electrothermal 9100 melting point apparatus. NMR spectra were obtained on a Bruker Avance 400 spectrometer at 400 MHz for 1H. Low-resolution atmospheric pressure chemical ionization (APCI) mass spectra were measured for methanol solutions on a ThermoFinnigan Surveyor MSQ mass spectrometer, connected to a Gilson autosampler.
4.1.1. Preparation of 5-substituted dihydroisoquinoline intermediates 61–63 (Scheme 1)
4.1.1.1. 8-(4-Methylpiperazin-1-yl)-5-(methylthio)isoquinoline (61)
A solution of 4- methylpiperazin-1-yl)isoquinoline (60) (1.76 g, 7.73 mmol) in DMF (20 mL) was cooled to 2 °C, N-bromosuccinimide (1.4 g, 8.11 mmol) was added, and the mixture was stirred at room temperature for 66 h. The mixture was diluted with ice water and the aqueous layer was extracted with DCM (5x). The combined extract was washed with water, brine, dried (MgSO4) and concentrated under reduced pressure. The crude product was purified by flash chromatography eluting with 3–9% MeOH/DCM to yield 5-bromo-8-(4-methylpiperazin-1-yl)isoquinoline (64) as a brown solid (1.69 g, 71%); mp 91–93 °C. 1H NMR (CDCl3) δ 9.56 (d, J = 0.6 Hz, 1H), 8.62 (d, J = 5.9 Hz, 1H), 8.62 (dd, J = 5.9, 0.8 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.01 (d, J = 8.1 Hz, 1H), 3.19 (br, 4H), 2.73 (br, 4H), 2.43 (s, 3H). MS (APCI): [M + H]+ 306.2 [79Br], 308.2 [81Br].
A solution of 64 (1.47 g, 4.80 mmol) in anhydrous THF (12 mL) at −78 °C under nitrogen was treated dropwise with n-butyllithium (2.6 mL, 5.28 mmol). The mixture turned black and was maintained at −78 °C for 5 min. Methyl disulfide (0.86 mL, 9.6 mmol) was added dropwise under nitrogen and the mixture was stirred at −78 °C for 2 h. The mixture was then quenched with water at −78 °C and then further diluted with water. The aqueous mixture was extracted with EtOAc (2x), and the combined extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to give a dark brown oil. Flash chromatography of the crude product using 4–8% MeOH/DCM as eluent gave 8-(4-methylpiperazin-1-yl)-5-(methylthio)isoquinoline (65) as a dark brown oil (95% pure). 1H NMR (CDCl3) δ 9.58 (d, J = 0.9 Hz, 1H), 8.58 (d, J = 5.9 Hz, 1H), 8.06 (dd, J = 5.9, 0.9 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 3.19 (br, 4H), 2.74 (br, 4H), 2.51 (s, 3H), 2.43 (s, 3H). MS (APCI): 274.1 [M + H]+.
A solution of 65 (0.934 g, 3.42 mmol) in MeOH (52 mL) at 2 °C under nitrogen was treated with NaCNBH3 (966 mg, 15.37 mmol) in three portions. After stirring at 2 °C for 10 min, BF3.OEt2 (1.9 mL, 15.37 mmol) was added dropwise. The orange mixture was maintained at the same temperature for another 30 min, then was refluxed under nitrogen for 22 h. The cooled mixture was then neutralised with 2 M Na2CO3 solution and extracted with EtOAc (3x). The combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to give a yellow solid. Flash chromatography on silica using 5–33% MeOH/DCM gave 61 as a yellow solid (0.643 g, 68%). mp 77–79 °C. 1H NMR (CDCl3) δ 7.06 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 4.00 (s, 2H), 3.17 (t, J = 6.2 Hz, 2H), 2.88 (t, J = 4.8 Hz, 4H), 2.73 (t, J = 6.2 Hz, 2H), 2.55 (br, 4H), 2.43 (s, 3H), 2.35 (s, 3H). MS (APCI): 274.4 [M + H]+
4.1.1.2. 5-Ethyl-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (62)
A solution of 64 (1.49 g, 4.15 mmol) in anhydrous THF (10 mL) at −78 °C under nitrogen was treated with n-butyllithium (2.3 mL, 4.56 mmol), then stirred at −78 °C for 5 min. Acetaldehyde (0.70 mL, 12.44 mmol) was added dropwise under nitrogen and the mixture was stirred at −60 °C for 30 min, then gradually warmed up to −10 °C over 30 min. The mixture was quenched with saturated NH4Cl solution, diluted with water and extracted with EtOAc (5x). This solution was washed with brine, dried (MgSO4) and concentrated under reduced pressure to give the crude product. Flash chromatography using 4–12% MeOH in DCM provided 1-(8-(4-methylpiperazin-1-yl)isoquinolin-5-yl)ethan-1-ol (66) as a brown gel (713 mg, contaminated with minor impurities), which was used in the next step without further purification. 1H NMR (CDCl3) δ 9.61 (d, J = 0.8 Hz, 1H), 8.54 (d, J = 6.0 Hz, 1H), 7.89 (dd, J = 6.0, 0.7 Hz, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.14 (d, J = 7.8 Hz, 1H), 5.54 (q, J = 6.4 Hz, 1H), 3.18 (br, 4H), 2.73 (br, 4H), 2.43 (s, 3H), 1.65 (d, J = 6.4 Hz, 3H). MS (APCI): 272.4 [M + H]+.
Triethylsilane (4.2 mL, 26.33 mmol) was added slowly to a solution of 66 (0.712 g, 2.63 mmol) in trifluoroacetic acid (30 mL) at room temperature, and then heated at 75 °C for 1 h. The mixture was poured into cold saturated NaHCO3 solution and 4 M NaOH solution was added to bring the mixture to pH 14. The aqueous mixture was extracted with DCM (4x), and the combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to give the crude product as a brown oil. Flash chromatography using 0–10% MeOH/DCM gave 5-ethyl-8-(4-methylpiperazin-1-yl)isoquinoline (67) as a dark brown oil (477 mg, 71%). 1H NMR (CDCl3) δ 9.61 (s, 1H), 8.53 (d, J = 5.9 Hz, 1H), 7.75 (d, J = 5.9 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.08 (d, J = 7.7 Hz, 1H), 3.17 (br, 4H), 3.00 (q, J = 7.5 Hz, 2H), 2.72 (br, 4H), 2.42 (s, 3H), 1.34 (t, J = 7.5 Hz, 3H). MS (APCI): 256.2 [M + H]+.
A solution of 67 (0.51 g, 2.0 mmol) in MeOH (32 mL) at 2 °C under nitrogen was treated with NaCNBH3 (0.564 g, 8.99 mmol) in three portions. The mixture was stirred at the same temperature for 10 min, then BF3.OEt2 (0.85 mL, 8.987 mmol) was added dropwise. The mixture was kept at 2 °C for 30 min, then heated to reflux under nitrogen overnight. The cooled mixture was neutralised with 2 M Na2CO3 and extracted with DCM (5x). The combined organic extracts were washed with brine, dried (MgSO4) and concentrated under reduced pressure to give a yellow solid. This solid was purified by flash chromatography on silica using 5–15% MeOH/DCM to give 62 as a yellow solid (0.333 g, 64%); mp 72–75 °C. 1H NMR (CDCl3) δ 7.05 (d, J = 8.1 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 4.04 (s, 2H), 3.17 (t, J = 6.2 Hz, 2H), 2.88 (t, J = 4.7 Hz, 4H), 2.73 (t, J = 6.2 Hz, 2H), 2.52–2.57 (m, 6H), 2.35 (s, 3H), 1.20 (t, J = 7.5 Hz, 3H). MS (APCI): 260.2 [M + H]+.
4.1.1.3. 5-Benzyl-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (63)
A solution of 5-bromo-8-(4-methylpiperazin-1-yl)isoquinoline (64) (0.975 g, 3.13 mmol) in anhydrous THF (8 mL) at −78 °C under nitrogen was treated dropwise with n-butyllithium (1.7 mL, 3.4 mmol). The mixture turned dark brown and was maintained at −78 °C for 2 min. Benzaldehyde (0.95 mL, 9.4 mmol) was added dropwise and the mixture was stirred at −78 °C for 2 h, then quenched with half-saturated NH4Cl solution and diluted with water. The aqueous mixture was extracted with EtOAc (4x), and the combined extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to give the crude product as a brown oil. Flash chromatography of the crude product (0–12% MeOH in DCM as eluent) provided (8-(4-methylpiperazin-1-yl)isoquinolin-5-yl)(phenyl)methanol (68) as a pale yellow semi-solid (568 mg, 55%). 1H NMR (CDCl3) δ 9.58 (d, J = 0.7 Hz, 1H), 8.44 (d, J = 6.0 Hz, 1H), 7.80 (dd, J = 6.0, 0.7 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.27–7.37 (m, 5H), 7.11 (d, J = 7.8 Hz, 1H), 6.41 (s, 1H), 3.17 (br, 4H), 2.73 (br, 4H), 2.43 (s, 3H). MS (APCI): 334.2 [M + H]+.
A solution of 68 (0.557 g, 1.67 mmol) in trifluoroacetic acid (18 mL) at room temperature was treated slowly with triethylsilane (2.70 mL, 16.7 mmol). The mixture was heated at 75 °C for 40 min, then left at room temperature overnight. Volatiles were removed under reduced pressure, and the residue was diluted in DCM and neutralised with saturated NaHCO3 solution. The aqueous mixture was extracted with DCM (3x), and the combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to yield the crude product as a brown liquid. The crude product was chromatographed on silica using 0–9% MeOH in DCM to give 5-benzyl-8-(4-methylpiperazin-1-yl)isoquinoline 69 as a yellow brown oil (400 mg, 75%). 1H NMR (CDCl3) δ 9.60 (d, J = 0.6 Hz, 1H), 8.47 (d, J = 6.0 Hz, 1H), 7.70 (dd, J = 6.0, 0.6 Hz, 1H), 7.38 (d, J = 7.7 Hz, 1H), 7.28–7.29 (m, 2H), 7.16–7.22 (m, 3H), 7.08 (d, J = 7.7 Hz, 1H), 4.34 (s, 2H), 3.19 (br, 4H), 2.73 (br, 4H), 2.43 (s, 3H). MS (APCI): 318.2 [M + H]+.
NaCNBH3 (0.350 g, 5.57 mmol) was added in three portions to a solution of 69 (0.393 g, 1.24 mmol) in MeOH (19 mL) at 2 °C under nitrogen. The mixture was stirred at 2 °C for 10 min, then BF3.OEt2 (0.52 mL, 5.6 mmol) was added dropwise. The mixture was maintained at the same temperature for another 30 min and subsequently refluxed under nitrogen for 3 h. The mixture was then cooled to room temperature and then neutralised with 2 M Na2CO3 solution, then extracted with DCM (3x). The combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to furnish 5-benzyl-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (63) as a yellow solid (0.383 g, 96%); mp 116–118 °C. 1H NMR (CDCl3) δ 7.24–7.28 (m, 2H), 7.16–7.20 (m, 1H), 7.11–7.13 (m, 2H), 6.96 (d, J = 8.1 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 4.02 (s, 2H), 3.90 (s, 2H), 3.10 (t, J = 6.2 Hz, 2H), 2.89 (t, J = 4.7 Hz, 4H), 2.63 (t, J = 6.2 Hz, 2H), 2.55 (m, 4H), 2.35 (s, 3H). MS (APCI): 322.2 [M + H]+.
4.1.2. Preparation of the directly-linked 2-pyridyl compounds 6–9 (Scheme 2A)
4.1.2.1. N-(5-(4-chloro-2-methylphenyl)pyridin-2-yl)-5-methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (6)
A mixture of 57 (300 mg, 1.22 mmol) and 70 (434 mg, 1.83 mmol) in toluene (10 mL) was purged with nitrogen before xantphos (35 mg, 0.06 mmol) and Pd2(dba)3 (28 mg, 0.03 mmol) were added. The mixture was heated in an oil bath at 80 °C, then NaOtBu (176 mg, 1.83 mmol) was added. The resulting mixture was heated at 100 °C for 4 h under nitrogen, then it was cooled to room temperature and ice was added to quench the reaction. The aqueous phase was extracted with EtOAc, the combined organic phase was washed with brine, dried over anhydrous Na2SO4 and filtered through a pad of alumina. The solvent was removed to give the crude product, this was purified by alumina chromatography, eluting with a mixture of EtOAc and petrol ether (1:4) to give 2-(5-bromopyridin-2-yl)-5-methyl-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (71) as a white solid (407 mg, 83%). 1H NMR (CDCl3) δ 8.23 (d, J = 2.4 Hz, 1H), 7.55 (dd, J = 9.0, 2.5 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.59 (d, J = 9.0 Hz, 1H), 4.52 (s, 2H), 3.75 (t, J = 5.7 Hz, 2H), 2.96 (t, J = 5.7 Hz, 2H), 2.92 (t, J = 4.7 Hz, 4H), 2.59 (br, 4H), 2.37 (s, 3H), 2.28 (s, 3H). MS (APCI): 490.2 [79Br], 492.2 [81Br] [M + H]+.
A mixture of 71 (100 mg, 0.25 mmol), (4-chloro-2-methylphenyl)boronic acid (81) (127 mg, 0.75 mmol) and aqueous Na2CO3 (2 M, 0.75 mL, 1.50 mmol) in toluene (2 mL) and EtOH (1 mL) was purged with nitrogen before (dppf)PdCl2.DCM (10 mg, 0.012 mmol) was added. The resulting mixture was heated in an oil bath at 85 °C for 24 h. After the solvent was removed, the residue was taken in EtOAc and washed with water and brine, dried over anhydrous Na2SO4 and filtered through a pad of alumina. The solvent was removed to give the crude product. Chromatography on alumina (4:1 hexanes:EtOAc), followed by trituration with Et2O, gave 6 as a white solid (72 mg, 65%); mp 169–172 °C. HPLC 96.5%. 1H NMR (CDCl3) δ 8.17 (d, J = 2.3 Hz, 1H), 7.47 (dd, J = 8.7, 2.4 Hz, 1H), 7.21 (dd, J = 8.2, 2.1 Hz, 1H), 7.13 (d, J = 8.2 Hz, 1H), 7.06 (d, J = 8.0 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.75 (d, J = 8.8 Hz, 1H), 4.61 (s, 2H), 3.84 (t, J = 5.7 Hz, 2H), 3.00 (t, J = 5.6 Hz, 2H), 2.93 (t, J = 4.7 Hz, 4H), 2.60 (br, 4H), 2.37 (s, 3H), 2.31 (s, 3H), 2.29 (s, 3H). HRMS calcd. for C27H31ClN4 (M + H+) m/z 447.2310, found 447.2296.
4.1.2.2. N-(5-(4-(tert-butyl)phenyl)pyridin-2-yl)-5-methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (7)
Using the same procedure as for compound 6, Reaction of 71 with 4-(tert-butyl)phenyl)boronic acid (73: R = 4-tBu) (133 mg, 0.75 mmol), gave 7 as white crystals (72 mg, 64%); HPLC 99.4%. mp 151–153 °C. 1H NMR (CDCl3) δ 8.50 (d, J = 2.3 Hz, 1H), 7.75 (dd, J = 8.8, 2.5 Hz, 1H), 7.50–7.44 (m, 4H), 7.05 (d, J = 8.0 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.77 (d, J = 8.8 Hz, 1H), 4.61 (s, 2H), 3.84 (t, J = 5.7 Hz, 2H), 3.00 (t, J = 5.6 Hz, 2H), 2.93 (t, J = 4.7 Hz, 4H), 2.59 (br, 4H), 2.37 (s, 3H), 2.31 (s, 3H), 1.36 (s, 9H). HRMS calcd. for C30H38N4 (M + H+) m/z 498.3169, found 498.3153.
4.1.2.3. N-(5-(4-Chloro-2-methylphenyl)pyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (8)
To a solution of 74 in aqueous HBr (48% wt, 6 mL) at −5 °C was added bromine (0.25 mL, 9.71 mmol) and aqueous HCl (37% wt, 1.13 mL, 11.3 mmol), followed by dropwise addition of a solution of NaNO2 (1.19 g, 17.4 mmol) in water (6 mL). The mixture was stirred at −5 °C for 1 h, then it was neutralized with 1 M NaOH. The aqueous layer was extracted with DCM, dried and evaporated. Column chromatography (49:1 hexanes:EtOAc) afforded 2-bromo-5-(4-chloro-2-methylphenyl)pyridine (75) as a white solid (0.36 g, 28%);, which was used directly. 1H NMR (CDCl3) δ 8.32 (dd, J = 2.3 Hz, J = 0.8 Hz, 1H), 7.55 (dd, J = 8.1 Hz, J = 0.8 Hz, 1H), 7.48 (dd, J = 8.1 Hz, J = 2.3 Hz, 1H), 7.30 (d, J = 2.2 Hz, 1H), 7.23–7.28 (m, 1H), 7.11 (d, J = 8.1 Hz, 1H), 2.24 (s, 3H).
A solution of 75 (0.108 g, 0.383 mmol) in toluene (5 mL) was purged with nitrogen for 1 min. BINAP (0.012 g, 0.019 mmol), Pd2(dba)3 (0.017 g, 0.019 mmol) and 5-methoxy-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (58, 0.100 g, 0.383 mmol) were then added, and the mixture stirred at 80 °C for 15 min. A bright orange solution formed, upon which NaOtBu (0.055 g, 0.574 mmol) was added, and the mixture heated at reflux for 1 h. The reaction was cooled and partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (19:1 DCM:MeOH) gave 8 as a light brown solid (0.085 g, 48%); mp 62–64 °C. 1H NMR (CDCl3) δ 8.15 (dd, J = 2.4 Hz, J = 0.6 Hz, 1H), 7.54 (dd, J = 8.7 Hz, J = 2.4 Hz, 1H), 7.26–7.28 (m, 1H), 7.20 (dd, J = 8.1 Hz, J = 1.9 Hz, 1H), 7.12 (d, J = 8.1 Hz, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.74 (dd, J = 8.7 Hz, J = 1.5 Hz, 2H), 4.72 (s, 2H), 3.90 (t, J = 6.0 Hz, 2H), 3.81 (s, 3H), 2.97 (t, J = 4.7 Hz, 4H), 2.91 (t, J = 6.0 Hz, 2H), 2.71 (s, 4H), 2.47 (s, 3H), 2.28 (s, 3H). Anal. (C27H31ClN4O) C, H, N.
4.1.2.4. N-(5-(3,5-bis(trifluoromethyl)phenyl)pyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (9)
Reaction of 58 (150 mg, 0.57 mmol), 70 (204 mg, 0.86 mmol), xantphos (17 mg, 0.03 mmol) and Pd2(dba)3 (13 mg, 0.014 mmol) and NaOBut (83 mg, 0.86 mmol) in toluene (5 mL) using conditions developed for the synthesis of 71 and purification by alumina chromatography (1:2 EtOAc: hexanes) gave 2-(5-bromopyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (72) as a pale solid (200 mg, 83%); 1H NMR (CDCl3) δ 8.21 (dd, J = 0.4, 2.5 Hz, 1H), 7.53 (dd, J = 9.0, 2.6 Hz, 1H), 7.00 (d, J = 8.7 Hz, 1H), 6.72 (d, J = 8.7 Hz, 1H), 6.58 (d, J = 9.0 Hz, 1H), 4.63 (s, 2H), 3.80 (s, 3H), 3.80 (t, J = 6.2 Hz, 2H), 2.91 (t, J = 4.7 Hz, 4H), 2.87 (t, J = 6.0 Hz, 4H), 2.63 (br, 4H), 2.40 (s, 3H); HRMS calcd. for C20H26BrN4O (M + H+) m/z 417.1285 [79Br], 419.1266 [81Br], found 417.1270, 419.1253.
A mixture of 72 (80 mg, 0.14 mmol), 3,5-bis(trifluoromethyl)phenyl)boronic acid (73: R = 3,5-bisCF3) (111 mg, 0.43 mmol) and aqueous Na2CO3 (2 M, 0.43 mL, 0.86 mmol) in toluene (2 mL) and EtOH (1 mL) was purged with nitrogen before (dppf)PdCl2-DCM (6 mg, 0.007 mmol) was added. The resulting mixture was heated in an oil bath at 85 °C for 20 h and then purified using conditions developed for the purification of 6, to give 9 as a white solid (58 mg, 73%); HPLC 93.7%. mp 145–146 °C. 1H NMR δ 8.50 (d, J = 2.2 Hz, 1H), 7.93 (s, 2H), 7.78–7.74 (m, 2H), 7.03 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 6.74 (d, J = 8.8 Hz, 1H), 4.77 (s, 2H), 3.91 (t, J = 6.1 Hz, 2H), 3.84 (s, 3H), 2.95–2.90 (m, 6H), 2.65 (br, 4H), 2.41 (s, 3H); HRMS calcd. for C28H28F6N4O (M + H+) m/z 551.2240, found 551.2227.
Note: The X = OMe compounds 8 and 9 were unstable in CDCl3 solution over 1–2 days.
4.1.3. Preparation of the –CH2-linked 2-pyridyl compounds 10–17 of Scheme 2B and Table 1
General procedure for further purification of methylamine linked compounds of Table 1: A minimal amount of EtOH (~1 mL) was added to the tetrahydroisoquinoline (~50 mg) in a vial, then approximately 3 equivalents of 1.25 M methanolic HCl were added. The resulting solution was stirred at room temperature for 5 min, then diisopropyl ether was added dropwise until the solution turned cloudy. The vial was left in the freezer overnight to allow precipitation of the HCl salt. The solution was carefully decanted (using a Pasteur pipette) and the solid carefully rinsed twice with Et2O (not filtered open to air, very hydroscopic) and then dried. The solid was then dissolved in water (~5 mL) and added to ~ 50 mL of 1 M aqueous HCl (50 mL). The aqueous layer was extracted twice with Et2O (2 × 50 mL), and then aqueous layer was basified with aqueous ammonia. The compound was then back extracted twice with Et2O (50 mL) and the organic layer was dried and evaporated to afford pure methylamine linked compounds if Table 1.
4.1.3.1. 2-((5-(4-Chlorophenyl)pyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (10)
A solution of 8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (56) (0.422 g, 1.82 mmol) and 5-bromo-2-(chloromethyl)pyridine (76) (0.414 g, 2.01 mmol) in anhydrous DMF was treated with K2CO3 (0.378 g, 2.74 mmol) at 20 °C. The mixture was stirred overnight, then diluted with water and extracted with DCM (3x). The combined extract was washed with water, brine, dried (MgSO4) and concentrated under reduced pressure. The crude product was chromatographed on silica (0–5% MeOH in DCM) to afford 2-((5-bromopyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (77) as a light yellow oil (0.525 g, 72%). 1H NMR (CDCl3) δ 8.63 (dd, J = 2.4, 0.6 Hz, 1H), 7.79 (dd, J = 8.3, 2.4 Hz, 1H), 7.44 (d, J = 8.2 Hz, 1H), 7.13 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 7.9 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 3.82 (s, 2H), 3.69 (s, 2H), 2.93–2.87 (m, 6H), 2.74 (t, J = 6.1 Hz, 2H), 2.51 (br, 4H), 2.34 (s, 3H). MS (APCI): 401.1 [79Br], 403.1 [81Br] [M + H]+.
A mixture of 77 (0.267 mmol), 4-chlorophenylboronic acid (73: R = 4-Cl) (0.293 mmol) and 2 M Na2CO3 solution (1.067 mmol) in toluene and EtOH (2:1) was purged with nitrogen. PdCl2dppf (5 mol%) was added and the mixture was purged again with nitrogen and heated at 80 °C for 2 h. The cooled mixture was diluted with water and extracted with DCM (3x). The combined organic extracts were washed with brine, dried (MgSO4) and evaporated to give the crude product. Chromatography on silica gel, eluting with MeOH/DCM mixtures, gave 10; mp 140–142 °C. HPLC 97.5%. 1H NMR (CDCl3) δ 8.77 (dd, J = 2.3, 0.6 Hz, 1H), 7.83 (dd, J = 8.1, 2.4 Hz, 1H), 7.59 (d, J = 8.1 Hz, 1H), 7.53 (AB br d, J = 8.7 Hz, 2H), 7.45 (AB br d, J = 8.7 Hz, 2H), 7.13 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 7.9 Hz, 1H), 6.89 (d, J = 7.9 Hz, 1H), 3.91 (s, 2H), 3.74 (s, 2H), 2.96–2.88 (m, 6H), 2.78 (t, J = 6.1 Hz, 2H), 2.50 (br, 4H), 2.32 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C26H30ClN4: 433.2154, found: 433.2146.
4.1.3.2. 2-((5-(2,4-Dichlorophenyl)pyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (11)
Similar reaction of 77 and 2-methyl-4-chlorophenylboronic acid (81) gave 11 as a brown gel. HPLC 95.5%. 1H NMR (CDCl3) δ 8.52 (dd, J = 2.1, 0.9 Hz, 1H), 7.62–7.56 (m, 2H), 7.30 (d, J = 2.1 Hz, 1H), 7.28–7.24 (m, 2H), 7.17–7.12 (m, 2H), 6.91 (d, J = 7.9 Hz, 1H), 6.88 (d, J = 7.9 Hz, 1H), 3.92 (s, 2H), 3.73 (s, 2H), 2.96 (t, J = 6.0 Hz, 2H), 2.89 (t, J = 4.7 Hz, 2H), 2.82 (t, J = 6.0 Hz, 2H), 2.50 (br, 4H), 2.32 (s, 3H), 2.27 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H32ClN4: 447.2310, found: 447.2293.
4.1.3.3. 2-((5-(4-Chlorophenyl)pyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-5-methyl-1,2,3,4-tetrahydroisoquinoline (12)
A mixture of 5-bromopicolinaldehyde (80) (5.00 g, 26.9 mmol), 2-methyl-4-chlorophenyl boronic acid (81) (4.16 g, 24.4 mmol) and K3PO4·H2O (11.23 g, 48.8 mmol) in water (25 mL), acetonitrile (75 mL) and dioxane (75 mL) was purged with nitrogen for 1 min. Pd(PPh3)4 (0.62 g, 0.54 mmol) was then added, and the mixture stirred at reflux for 5 h. The reaction was cooled and the solvent removed under reduced pressure. The residue was partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (19:1, x4:EtOAc) afforded 5-(4-chloro-2-methylphenyl)picolinaldehyde (82) as a tan solid (3.20 g, 57%), which was used directly. 1H NMR (CDCl3) δ 10.14 (s, 1H), 8.74 (s, 1H), 8.04 (d, J = 7.9 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.26–7.34 (m, 2H), 7.17 (d, J = 8.1 Hz, 1H), 2.28 (s, 3H).
A mixture of 82 (0.91 g, 3.93 mmol) and NaBH4 (0.30 g, 7.86 mmol) in MeOH (50 mL, anhydrous) was stirred at 20 °C for 1 h. The solvent was then removed and the residue partitioned between EtOAc and water. The organic layer was dried and evaporated to afford (5-(4-chloro-2-methylphenyl)pyridin-2-yl)methanol (83) as a white solid (0.90 g, 98%). 1H NMR (CDCl3) δ 8.50 (d, J = 1.6 Hz, 1H), 7.62 (dd, J = 8.0 Hz, J = 2.2 Hz, 1H), 7.24–7.33 (m, 3H), 7.13 (d, J = 8.0 Hz, 1H), 4.83 (s, 2H), 2.25 (s , 3H), which was used directly.
To a solution of 83 (0.26 g, 1.11 mmol) and triethylamine (0.23 mL, 1.67 mmol) in DCM (7.5 mL) at 20 °C was added mesyl chloride (0.10 mL, 1.34 mmol) dropwise. After 15 min, the reaction was diluted with DCM (20 mL) and the organic layer washed with sat. NaHCO3, dried and evaporated. The residue was redissolved in acetone (15 mL, anhydrous), lithium bromide (~2.5 g, excess) was added, and the mixture heated at reflux for 1.5 h. The solution was then cooled and the solvent evaporated to give a residue which was partitioned between EtOAc and water. The aqueous layer was extracted twice with EtOAc and the organic layer was dried and evaporated to afford 2-(bromomethyl)-5-(4-chloro-2-methylphenyl)pyridine (84) as a light brown solid (0.31 g, 94%), which was used directly. 1H NMR (CDCl3) δ 8.52 (d, J = 2.2 Hz, 1H), 7.64 (dd, J = 8.0 Hz, J = 2.2 Hz, 1H), 7.51 (dd, J = 8.0 Hz, J = 0.7 Hz, 1H), 7.23–7.33 (m, 3H), 7.13 (d, J = 8.0 Hz, 1H), 4.61 (s, 2H), 2.26 (s , 3H).
To a solution of 8-bromo-5-methyl-1,2,3,4-tetrahydroisoquinoline (85)20 (0.062 g, 0.274 mmol) and 84 (0.105 g, 0.356 mmol) in DMF (2.5 mL) was added K2CO3 (0.057 g, 0.411 mmol) and the resultant mixture stirred at room temperature for 5 h. The reaction was then diluted with EtOAc (50 mL) and washed with water (10 mL), and the organic layer was dried and evaporated. Column chromatography (4:1, X4:EtOAc) afforded 86 as a yellow oil (0.098 g, 81%). 1H NMR (CDCl3) δ 8.53 (dd, J = 2.2 Hz, J = 0.7 Hz, 1H), 7.61 (dd, J = 8.0 Hz, J = 2.2 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.23–7.33 (m, 3H), 7.17 (d, J = 8.0 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 3.95 (s, 2H), 3.72 (s, 2H), 2.84 (t, J = 5.3 Hz, 2H), 2.78 (t, J = 5.3 Hz, 2H), 2.28 (s, 3H), 2.19 (s, 3H), which was used directly.
A solution of 86 (0.100 g, 0.226 mmol) in toluene (2.5 mL) was purged with nitrogen for 1 min. BINAP (0.007 g, 0.011 mmol), Pd2(dba)3 (0.005 g, 0.0057 mmol) and N-methylpiperazine (0.038 mL, 0.34 mmol) were then added, and the mixture stirred at 80 °C for 15 min. A bright orange solution formed, upon which NaOtBu (0.105 g, 1.09 mmol) was added, and the mixture stirred at reflux under nitrogen for 4 h. The reaction was cooled and partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (19:1, DCM:MeOH) gave 12 as a yellow oil (0.037 g, 36%), which was further purified by the general salt formation/back extraction procedure as described above. 1H NMR (CDCl3) δ 8.52 (t, J = 1.0 Hz, 1H), 7.55–7.62 (m, 2H), 7.23–7.33 (m, 2H), 7.15 (d, J = 8.0 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.88 (d, J = 8.0 Hz, 1H), 3.92 (s, 2H), 3.75 (s, 2H), 2.87–2.96 (m, 8H), 2.57 (s, 4H), 2.39 (s, 3H), 2.29 (s, 3H), 2.19 (s, 3H); Anal. (C28H36Cl4N4) C, H, N (tri-HCl salt).
4.1.3.4. 2-((5-(4-Chlorophenyl)pyridin-2-yl)methyl)-8-(4-methoxypiperazin-1-yl)-5-methyl-1,2,3,4-tetrahydroisoquinoline (13)
To a solution of 58 (0.017 g, 0.065 mmol) and 84 (0.023 g, 0.078 mmol) in DMF (1 mL) was added K2CO3 (0.013 g, 0.098 mmol) and the resultant mixture stirred at r.t. for 5 h. The reaction was then diluted with EtOAc (50 mL) and washed with water (5 × 10 mL), and the organic layer was dried and evaporated. Column chromatography (9:1, DCM:MeOH) afforded 13 as a pale yellow oil (0.025 g, 80%), which was further purified by the general salt formation/back extraction procedure as described above: 1H NMR (CDCl3) δ 8.52 (s, 1H), 7.57–7.62 (m, 2H), 7.23–7.32 (m, 2H), 7.16 (d, J = 8.6 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.68 (d, J = 8.6 Hz, 1H), 3.91 (s, 2H), 3.80 (s, 3H), 3.75 (s, 2H), 2.90 (s, 4H), 2.81 (s, 3H), 2.59 (s, 4H), 2.39 (s, 3H), 2.27 (s, 3H); Anal. (C18H14N2O3) C, H, N.
4.1.3.5. 2-((5-(4-Chloro-2-methylphenyl)pyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-5-(methylthio)-1,2,3,4-tetrahydroisoquinoline (14)
Similar reaction of 8-(4-methylpiperazin-1-yl)-5-(methylthio)-1,2,3,4-tetrahydroisoquinoline (61) with 76, and chromatography on silica, eluting with 2–10% MeOH in DCM gave 2-((5-bromopyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-5-(methylthio)-1,2,3,4-tetrahydroisoquinoline (79) as a yellow gum (85%). 1H NMR (CDCl3) δ 8.64 (d, J = 1.9 Hz, 1H), 7.79 (dd, J = 8.3, 2.4 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 3.82 (2, 2H), 3.70 (s, 2H), 2.88–2.82 (m, 6H), 2.79–2.74 (m, 2H), 2.51 (br, 4H), 2.42 (s, 3H), 2.34 (s, 3H). MS (APCI): 447.1 [79Br], 449.1 [81Br] [M + H]+.
Reaction of 79 with boronic acid (81) as above gave 14 (76% yield); mp 65–68 °C. 1H NMR (CDCl3) δ 8.52 (dd, J = 2.2, 0.9 Hz, 1H), 7.62–7.55 (m, 2H), 7.30 (d, J = 2.1 Hz, 1H), 7.24 (d, J = 2.2 Hz, 1H), 7.16 (d, J = 8.2 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8.4 Hz, 1H), 3.91 (s, 2H), 3.74 (s, 2H), 2.90–2.85 (m, 8H), 2.50 (br, 4H), 2.42 (s, 3H), 2.32 (s, 3H), 2.27 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H34ClN4S: 493.2117, found: 493.2191
4.1.3.6. 2-((5-(4-Chloro-2-methylphenyl)pyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-5-fluoro-1,2,3,4-tetrahydroisoquinoline (15)
Similar reaction of 5-fluoro-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (59) with 76 gave 2-((5-bromopyridin-2-yl)methyl)-8-(4-methylpiperazin-1-yl)-5-fluoro-1,2,3,4-tetrahydroisoquinoline (78) (0.237 g, 64%); mp 93–95 °C. 1H NMR (CDCl3): δ 8.64 (d, J = 1.9 Hz, 1H), 7.80 (dd, J = 8.3, 2.4 Hz, 1H), 7.42 (d, J = 8.3 Hz, 1H), 6.91–6.82 (m, 2H), 3.82 (2, 2H), 3.69 (s, 2H), 2.86–2.82 (m, 6H), 2.74 (t, J = 6.0 Hz, 2H), 2.50 (br, 4H), 2.33 (s, 3H). MS (APCI): 419.1 [79Br], 421.1 [81Br] [M + H]+.
Reaction of 78 with boronic acid (81) as above gave 15 as an oil. HPLC 83.7%. 1H NMR (CDCl3) δ 8.54 (d, J = 1.6 Hz, 1H), 7.64 (dd, J = 7.9, 2.2 Hz, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 2.0 Hz, 1H), 7.27 (dd, J = 7.9, 1.9 Hz, 1H), 7.18 (d, J = 8.2 Hz, 1H), 6.93–6.84 (m, 2H), 3.93 (s, 2H), 3.74 (s, 2H), 2.92–2.82 (m, 8H), 2.50 (br, 4H), 2.33 (s, 3H), 2.28 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30ClFN4: 465.22158, found: 465.22036.
4.1.3.7. 2-((5-(2,4-Difluorophenyl)pyridin-2-yl)methyl)-5-fluoro-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (16)
Reaction of 78 with boronic acid 73 (R = 2,4-diF) as above gave 16 as an oil. HPLC 78.5%. 1H NMR (CDCl3) δ 8.71 (s, 1H), 7.83 (dt, J = 8.1, 1.9 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.48–7.40(m, 1H), 7.03–6.93 (m, 2H), 6.92–6.82 (m, 2H), 3.92 (s, 2H), 3.73 (s, 2H), 2.89–2.79 (m, 8H), 2.49 (br, 4H), 2.32 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C26H28F3N4: 453.22606, found: 453.22619.
4.1.3.8. 2-((5-(4-Chloro-2-(trifluoromethyl)phenyl)pyridin-2-yl)methyl)-5-fluoro-8-(4-methylpiperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (17)
Reaction of 78 with boronic acid 73 (R = 2-CF3, 4-Cl) as above gave 17 as a solid (56%); mp 102–104 °C. HPLC 93.9%. 1H NMR (CDCl3) δ 8.81 (d, J = 2.2 Hz, 1H), 7.88 (dd, J = 8.1, 2.4 Hz, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.73 (s, 1H), 7.63 (d, J = 8.2 Hz, 1H), 7.58 (d, J = 8.2 Hz, 1H), 6.93–6.83 (m, 2H), 3.94 (s, 2H), 3.75 (s, 2H), 2.88–2.83 (m, 6H), 2.79 (d, J = 5.7 Hz, 2H), 2.49 (br, 4H), 2.32 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H28ClF4N4: 519.19331, found: 519.19372.
4.1.4. Preparation of the –CO-linked pyridyl side compounds 18–22 of Scheme 3 and Table 1
General procedure: A suspension of the substituted phenylpicolinic acids (87–89) (0.319 mmol) in anhydrous DMF was treated at room temperature under nitrogen with diisopropylethylamine (0.319 mmol). The mixture was then treated with HATU (0.319 mmol) and was stirred for 5 min. Tetrahydroisoquinolines 56 or 58 (0.290 mmol) were added and the mixture was stirred at room temperature overnight. The mixture was diluted in water and the aqueous mixture was extracted with DCM (3x). The combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to furnish the crude product, which was chromatographed on silica using mixtures of MeOH and DCM to yield the clean products as yellow brown solids in 25–35% yields.
4.1.4.1. (5-(4-Chloro-2-methylphenyl)pyridin-2-yl)(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (18)
m.p. 132–135 °C. HPLC 96.0%. 1H NMR (CDCl3) δ (two rotamers, data on the major one is presented) 8.54 (s, 1H), 7.78–7.69 (m, 2H), 7.30–7.25 (m, 2H), 7.20–7.12 (m, 2H), 7.00–6.93 (m, 2H), 4.90 (s, 2H), 3.85 (br, 2H), 3.40 (br, 4H), 3.20 (br, 4H), 3.00 (br, 7H), 2.25 (br, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30ClN4O: 461.2103, found 461.2099.
4.1.4.2. (5-(4-Chlorophenyl)pyridin-2-yl)(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (19)
m.p. 144–147 °C. HPLC 97.7%. 1H NMR (CDCl3) δ (two rotamers, data on the major one is presented) 8.78 (d, J = 1.8 Hz, 1H), 7.95 (dd, J = 8.1, 1.8 Hz, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.58–7.45 (m, 4H), 7.18 (t, J = 7.7 Hz, 1H), 7.01–6.97 (m, 2H), 4.89 (s, 2H), 3.80 (t, J = 5.6 Hz, 2H), 3.34 (br, 4H), 3.17 (br, 4H), 2.97 (t, J = 5.6 Hz, 2H), 2.91 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C26H28ClN4O: 447.1946, found: 447.1934.
4.1.4.3. (5-(3-Chlorophenyl)pyridin-2-yl)(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (20)
m.p. 105–108 °C. HPLC 96.7%. 1H NMR (CDCl3) δ (two rotamers, data on the major one is presented) 8.81 (d, J = 1.8 Hz, 1H), 7.99 (dd, J = 8.2, 2.2 Hz, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.53–7.41 (m, 4H), 7.24–7.17 (m, 1H), 7.05 (t, J = 7.8 Hz, 1H), 7.02–6.96 (m, 1H), 4.91 (s, 2H), 3.84 (t, J = 5.9 Hz, 2H), 3.31 (br, 4H), 3.21 (br, 4H), 3.00 (t, J = 5.9 Hz, 2H), 2.90 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C26H28ClN4O: 447.1946, found: 447.1931.
4.1.4.4. (5-(4-Chlorophenyl)pyridin-2-yl)(5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (21)
21% yield; m.p. 124–127 °C. HPLC 99.8%. 1H NMR (CDCl3) δ (two rotamers, data on the major one is presented) 8.81 (apparent bd,1H), 7.97 (dd, J = 8.1, 2.1 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.55 (AB d, J = 8.1 Hz, 1H), 7.49 (AB d, J = 8.1 Hz, 1H), 7.06 (d, J = 7.7 Hz, 1H), 6.74 (d, J = 7.7 Hz, 1H), 4.92 (s, 2H), 3.99 (t, J = 5.6 Hz, 2H), 3.81 (s, 3H). 3.09 (br, 6H), 2.94–2.87 (m, 2H), 2.78 (br, 2H), 2.74 (s, 3H). HRMS (ESI + ): [M + H] + calculated for C27H30ClN4O2: 477.2052, found: 477.2031.
4.1.4.5. (5-(4-Chloro-2-methylphenyl)pyridin-2-yl)(5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (22)
29% yield; mp 125–128 °C. HPLC 98.5%. 1H NMR (CDCl3) δ (two rotamers, data on the major one is presented) 8.81 (apparent t,1H), 7.77–7.71 (m, 2H), 7.33 (apparent d,1H, 1H), 7.28 (dd, J = 8.2, 2.0 Hz, 1H), 7.17 (d, J = 8.1 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 4.91 (s, 2H), 3.85–3.82 (m, 5H). 3.26 (br, 2H), 3.16 (br, 4H), 2.96–2.80 (m, 7H), 2.28 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H32ClN4O2: 491.2208, found: 491.2202.
4.1.5. Preparation of the –CONH-linked pyridyl side compounds 23–42 of Scheme 4 and Table 1
Two general methods were used to prepare this class of compounds, the chosen method was dependant on the availability of the starting materials.
Method 1
4.1.5.1. N-(5-(4-Chloro-2-methylphenyl)pyridin-2-yl)-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (24).
A mixture of 2-amino-5-bromopyridine (90) (0.996 g, 5.76 mmol), 4-chloro-2-methylphenyl boronic acid (81) (0.771 g, 4.53 mmol) and 1 M Na2CO3 solution (9.5 mL, 9.465 mmol) in dimethoxyethane (18 mL) was flushed with nitrogen for 5 min. Tetrakis(triphenylphosphine)palladium(0) (47.6 mg, 0.0412 mmol) was added. The mixture was flushed again with nitrogen for 5 min, and then refluxed under nitrogen for 2.5 h. The mixture was cooled to room temperature and was diluted in water. The aqueous mixture was extracted with EtOAc (3x). The combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to furnish the crude product as a brown semi-solid. The crude product was chromatographed on silica using mixtures of hexane/EtOAc and finally 100% EtOAc to afford 4-nitrophenyl-(5-(4-chloro-2-methylphenyl)pyridine-2-yl)carbamate (91: R = 2-Me, 4-Cl) as a cream solid (0.905 g, 72%). 1HNMR (CDCl3) δ 8.01 (dd, J = 2.4, 0.6 Hz, 1H), δ 7.38 (dd, J = 8.4, 2.4 Hz, 1H), 7.26–7.25 (partially overlapped with residue of CHCl3, 1H), 7.20 (dd, J = 8.2, 2.0 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 6.56 (dd, J = 8.4, 0.7 Hz, 1H), 4.49 (br s, 2H, NH2), 2.25 (s, 3H). It was used directly.
Compound 91 (0.392 g, 1.793 mmol) was dissolved in anhydrous DCM (6 mL), anhydrous pyridine (0.17 mL, 2.151 mmol) was added, followed by 4-nitrophenyl chloroformate (443.6 mg, 2.151 mmol). The white slurry was stirred at room temperature under nitrogen overnight. The mixture was passed through a filter paper-lined Buchner funnel under vacuum, the white solid was washed with DCM and further dried to yield N-((5-(4-chloro-2-methylphenyl)pyridin-2-yl)carbamoyl)-2-(4-nitrophenyl)acetamide (92: R = 4-Cl, 2-Me) as a white solid (0.522 g, 76%); mp 201–203 °C, which was used directly. 1H NMR (DMSO‑d6) δ (3:1 mixture of rotamers) 11.05 (s, 1H), 10.8–10.4 (br s, 1H), 8.29 (t, J = 1.5 Hz, 1H), 8.14–8.09 (m, 2H), 7.86 (dd, J = 2.5, 0.6 Hz, 1H), 7.85–7.79 (br s, 2H), 7.43 (d, J = 2 Hz, 1H), 7.40–7.32 (m, 3H), 7.28 (d, J = 8.2 Hz, 1H), 7.28–7.24 (m, 1H), 6.96–6.90 (m, 2H), 2.27 (s, 3H), 2.24 (s, 3H), MS (APCI): 277.2 [35Cl] and 279.2 [37Cl] [M−p−NO2PhO + MeO]+.
A mixture of 56 (121 mg, 0.52 mmol) and 92 (R = 2-Me, 4-Cl, 301 mg, 0.78 mmol) in acetonitrile (3 mL) was stirred overnight at 70 °C. The mixture was directly adsorbed onto silica and chromatographed using 3–10% MeOH in DCM to afford 24 as pale yellow solid (57%); mp 86–88 °C. HPLC 96.7%. 1H NMR (CDCl3) δ 8.16–8.11 (m, 2H), 7.60 (d, J = 8.6, 2.4 Hz, 1H), 7.34 (s, 1H), 7.28 (d, J = 2.0 Hz, 1H), 7.24–7.21 (m, 2H), 7.13 (d, J = 8.1 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 7.4 Hz, 1H), 4.73 (s, 2H), 3.73 (t, J = 6.1 Hz, 2H), 2.99–2.94 (m, 6H), 2.66 (br, 4H), 2.39 (s, 3H), 2.26 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H31ClN5O 476.2212, found: 476.2199.
The following compounds were similarly prepared:
4.1.5.2. 8-(4-Methylpiperazin-1-yl)-N-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (23)
Intermediate 92 (R = 4-OCF3)21 was made by the above procedure and used without purification. Reaction of 56 and 92 (R = 4-OCF3) as above gave 23 (75% yield); mp 209–211 °C. HPLC 99.7%. 1H NMR (CDCl3) δ 8.42 (dd, J = 2.4, 0.6 Hz, 1H), 8.16 (dd, J = 8.7, 0.6 Hz, 1H), 7.85 (dd, J = 8.7, 2.4 Hz, 1H), 7.58–7.55 (m, 2H), 7.35 (s, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.22 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.96 (d, J = 7.4 Hz, 1H), 4.73 (s, 2H), 3.73 (t, J = 6.1 Hz, 2H), 3.00–2.94 (m, 6H), 2.66 (br, 4H), 2.40 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H29F3N5O2 512.2268, found: 512.2253.
4.1.5.3. 5-Methoxy-N-(6′-methoxy-[3,3′-bipyridin]-6-yl)-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (32)
Reaction of 58 and 92 (R = 3-aza, 4-OMe) as above gave 32; mp 93–96 °C. HPLC 95.0%. 1H NMR (CDCl3) δ 8.38 (dd, J = 2.4, 0.4 Hz, 1H), 8.35 (dd, J = 1.6, 0.6 Hz, 1H), 8.14 (dd, J = 8.7, 0.6 Hz, 1H), 7.80 (dd, J = 8.7, 2.5 Hz, 1H), 7.74 (dd, J = 8.6, 2.6 Hz, 1H), 7.42 (s, 1H, NH), 7.04 (d, J = 8.7 Hz, 1H), 6.83 (dd, J = 7.6, 0.6 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 4.72 (s, 2H), 3.98 (s, 3H), 3.81 (s, 3H), 3.73 (t, J = 6.1 Hz, 2H), 2.92–2.88 m, 6H), 2.67 (br, 4H), 2.41 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H33N6O3 489.2609, found: 489.2607.
4.1.5.4. N-(5-(4-chloro-2-methylphenyl)pyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (35)
From 58 and 92 (R = 2-Me, 4-Cl). 78% yield; mp 99–101 °C. HPLC 99.5%. 1H NMR (CDCl3) δ 8.14 (dd, J = 2.3, 0.7 Hz, 1H), 8.12 (dd, J = 8.6, 0.7 Hz, 1H), 7.60 (dd, J = 8.6, 2.4 Hz, 1H), 7.41 (s, 1H, NH), 7.28 (d, J = 2.1 Hz, 1H), 7.23 (dd, J = 8.2, 1.9 Hz, 1H), 7.13 (d, J = 8.1 Hz, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 4.72 (s, 2H), 3.82 (s, 3H), 3.73 (t, J = 6.1 Hz, 2H), 2.92–2.88 m, 6H), 2.65 (br, 4H), 2.39 (s, 3H), 2.25 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H33ClN5O2 506.2317, found: 506.2310.
4.1.5.5. 5-Methoxy-8-(4-methylpiperazin-1-yl)-N-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (37)
From 58 and 92 (R = 4-OCF3). 79% yield; mp 196–198 °C. HPLC 99.8%. 1H NMR (CDCl3) δ 8.41 (dd, J = 2.4, 0.5 Hz, 1H), 8.16 (dd, J = 8.7, 0.6 Hz, 1H), 7.84 (dd, J = 8.7, 2.5 Hz, 1H), 7.56 (AB br d, J = 8.8 Hz, 2H), 7.54 (s, 1H, NH), 7.30 (d, J = 8.0 Hz, 2H), 7.04 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 4.73 (s, 2H), 3.81 (s, 3H), 3.73 (t, J = 6.1 Hz, 2H), 2.91–2.88 m, 6H), 2.64 (br, 4H), 2.39 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H31F3N5O3 542.2374, found: 542.2368.
Method II
4.1.5.6. 5-Methoxy-8-(4-methylpiperazin-1-yl)-N-(5-(pyrimidin-5-yl)pyridin-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (31)
Pyridine (1.2 mL, 6.6 mmol) was added to a solution of 2-amino-5-bromopyridine (90, 1.035 g, 5.982 mmol) in acetonitrile (20 mL) at 2 °C, followed by 4-nitrophenylchloroformate (1.326 g, 6.581 mmol). The resulting white slurry was stirred overnight. The white solids were filtered off, washed with DCM, dried under ambient conditions to give 4-nitrophenyl (5-bromopyridin-2-yl)carbamate (93, ~1.5 g); mp 236–239 °C. No mass spectroscopic and NMR data could be obtained for this compound due to the low solubility in suitable solvents.
A typical procedure is given for the synthesis of 94: A mixture of 58 (0.13 g, 0.50 mmol) and 93 (0.254 g, 0.751 mmol) in DMF (3.5 mL) was stirred at 75 °C for 25 h. The mixture was diluted in water, then extracted with DCM (3x). The combined organic extract was washed with water, brine, dried (MgSO4) and concentrated to yield the crude product as a brown oil. The crude product was chromatographed using 0–15% MeOH in DCM to provide N-(5-bromopyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (94: X = OMe) as an off-white foam (166 mg, 72%); mp 76–79 °C. HPLC 92.8%. 1H NMR (CDCl3) δ 8.25 (dd, J = 2.4, 0.5 Hz, 1H), 8.02 (dd, J = 9.0, 0.6 Hz, 1H), 7.73 (dd, J = 9.0, 2.4 Hz, 1H), 7.28 (s, 1H), 7.03 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 4.69 (s, 2H), 3.81 (s, 3H), 3.69 (t, J = 6.1 Hz, 2H), 2.88 (apparent t, 6H), 2.63 (br, 4H), 2.38 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C21H27BrN5O2 [79Br] 460.1343, found: 460.1341.
A mixture of (94: X = OMe) (0.138 mmol), pyrimidin-5-ylboronic acid (73: R = 3,5-diaza) (0.413 mmol) and 2 M Na2CO3 solution (0.590 mmol) in a mixture of toluene and EtOH (2:1, 3 mL) was purged with nitrogen. PdCl2dppf (5 mol%) was added, the mixture was purged again with nitrogen and heated at 85 °C overnight. The mixture was then diluted with water and extracted with EtOAc (3x). The combined organic extract was washed with brine, dried (MgSO4) and concentrated under reduced pressure to afford the crude product. This was chromatographed on silica using mixtures of MeOH and DCM to yield clean 31 (55%); mp 201–204 °C. HPLC 93.7%. 1H NMR (CDCl3) δ 9.22 (s, 1H), 8.94 (s, 2H), 8.45 (dd, J = 2.5, 0.7 Hz, 1H), 8.25 (dd, J = 8.7, 0.7 Hz, 1H), 7.88 (dd, J = 8.8, 2.5 Hz, 1H), 7.44 (s, 1H, NH), 7.05 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 4.73 (s, 2H), 3.82 (s, 3H), 3.74 (t, J = 6.1 Hz, 2H), 2.93–2.90 m, 6H), 2.65 (br, 4H), 2.40 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C25H30N7O2 460.2461, found: 460.2444.
4.1.5.7. 5-Methoxy-N-(5-(4-methoxyphenyl)pyridin-2-yl)-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (33)
From 94 (X = OMe) and 73 (R = 4-OMe). 63% yield; mp 207–209 °C. HPLC 97.1%. 1H NMR (CDCl3) δ 8.39 (dd, J = 2.5, 0.6 Hz, 1H), 8.10 (dd, J = 8.7, 0.6 Hz, 1H), 7.82 (dd, J = 8.7, 2.5 Hz, 1H), 7.47 (AB br d, J = 8.8 Hz, 2H), 7.40 (s, 1H, NH), 7.03 (d, J = 8.7 Hz, 1H), 6.98 (AB br d, J = 8.8 Hz, 2H), 6.75 (d, J = 8.7 Hz, 1H), 4.72 (s, 2H), 3.85 (s, 3H), 3.81 (s, 3H), 3.72 (t, J = 6.1 Hz, 2H), 2.91–2.88 m, 6H), 2.64 (br, 4H), 2.39 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H34N5O3 488.2656, found: 488.2665.
4.1.5.8. N-(5-(2-Chloro-4-(trifluoromethyl)phenyl)pyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (34)
From 94 (X = OMe) and 73 (X = 2-Cl, 4-CF3). 15% yield; mp 61–64 °C. HPLC 92.6%. 1H NMR (CDCl3) δ 8.29 (dd, J = 2.4, 0.8 Hz, 1H), 8.19 (dd, J = 8.6, 0.6 Hz, 1H), 7.79–7.75 (m, 2H), 7.69 (s, 1H, NH), 7.58 (dd, J = 8.0, 0.8 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H), 6.74 (d, J = 8.8 Hz, 1H), 4.73 (s, 2H), 3.80 (s, 3H), 3.73 (t, J = 6.0 Hz, 2H), 2.91–2.88 m, 6H), 2.64 (br, 4H), 2.39 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H30ClF3N5O2 560.2035, found: 560.2036.
4.1.5.9. N-(5-(2,4-Dichlorophenyl)pyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (36)
From 94 (X = OMe) and 73 (X = 2,4-diCl). 55% yield; mp 108–111 °C. HPLC 91.8%. 1H NMR (CDCl3) δ 8.26 (dd, J = 2.4, 0.7 Hz, 1H), 8.14 (dd, J = 8.7, 0.7 Hz, 1H), 7.73 (dd, J = 8.7, 2.4 Hz, 1H), 7.50 (d, J = 2.0 Hz, 1H), 7.44 (s, 1H, NH), 7.32 (dd, J = 8.2, 2.0 Hz, 1H), 7.26 (d, J = 8.3 Hz, 1H), 7.04 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 4.72 (s, 2H), 3.81 (s, 3H), 3.72 (t, J = 6.1 Hz, 2H), 2.91–2.88 m, 6H), 2.65 (br, 4H), 2.40 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30Cl2N5O2 526.1771, found: 526.1766.
4.1.5.10. N-(5-(3,5-Bis(trifluoromethyl)phenyl)pyridin-2-yl)-5-methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (38)
From 94 (X = OMe) and 73 (R = 3,5-di-CF3). 54% yield; mp 106–109 °C. HPLC 97.7%. 1H NMR (CDCl3) δ 8.47 (d, J = 1.9 Hz, 1H), 8.22 (dd, J = 8.8, 0.5 Hz, 1H), 7.97 (s, 2H), 7.90 (dd, J = 8.8, 2.5 Hz, 1H), 7.87 (s, 1H), 7.45 (s, 1H, NH), 7.02 (d, J = 8.7 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 4.72 (s, 2H), 3.82 (s, 3H), 3.73 (t, J = 6.1 Hz, 2H), 2.96–2.90 m, 6H), 2.76 (br, 4H), 2.47 (s, 3H). (ESI+): [M + H]+ calculated for C29H30F6N5O2 594.2298, found: 594.2316.
The following compounds were similarly prepared:
4.1.5.11. N-(5-(2,4-Dichlorophenyl)pyridin-2-yl)-5-methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (25)
Following the typical procedure for 94 (X = OMe), N-(5-bromopyridin-2-yl)-5-methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (94: X = Me) was prepared from 57 and 93. It was purified by flash chromatography (2–15% MeOH in DCM) to give a yellow crystalline solid (71%); mp 117–120 °C. 1H NMR (CDCl3) δ 8.26 (dd, J = 2.5, 0.6 Hz, 1H), 8.05 (dd, J = 8.9, 0.6 Hz, 1H), 7.76 (dd, J = 9.0, 2.4 Hz, 1H), 7.28 (s, 1H, NH), 7.06 (d, J = 8.1 Hz, 1H), 6.93 (d, J = 8.4 Hz, 1H), 4.57 (s, 2H), 3.68 (t, J = 5.8 Hz, 2H), 2.95 (t, J = 5.3 Hz, 6H), 2.65 (br, 4H), 2.42 (s, 3H), 2.26 (s, 3H). MS (APCI): 444.1 [79Br], 446.1 [81Br] [M + H]+.
From 94 (X-Me) and 73 (R = 2,4-diCl) gave 25 (68% yield); mp 104–106 °C. HPLC 98.0%. 1H NMR (CDCl3) δ 8.27 (dd, J = 2.3, 0.7 Hz, 1H), 8.17 (dd, J = 8.7, 0.7 Hz, 1H), 7.76 (d, J = 8.7, 2.4 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.40 (s, 1H), 7.32 (dd, J = 8.3, 2.1 Hz, 1H), 7.28 (s, 1H, NH) 7.06 (d, J = 8.1 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 4.60 (s, 2H), 3.71 (t, J = 5.7 Hz, 2H), 2.97 (t, J = 5.7 Hz, 2H), 2.92 (t, J = 4.7 Hz, 4H), 2.59 (br, 4H), 2.38 (s, 3H), 2.26 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30Cl2N5O 510.18219, found: 510.18188.
4.1.5.12. N-(5-(3,5-Bis(trifluoromethyl)phenyl)pyridin-2-yl)-5-methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (26)
From 94 (X = Me) and 73 (R = 3,5-diCF3). 62% yield; mp 114–117 °C. HPLC 98.1%. 1H NMR (CDCl3) δ 8.48 (d, J = 1.9 Hz, 1H), 8.26 (dd, J = 7.9, 0.5 Hz, 1H), 7.98 (s, 2H), 7.92 (dd, J = 8.7, 2.5 Hz, 1H), 7.87 (s, 1H), 7.46 (s, 1H, NH), 7.06 (d, J = 8.1 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 4.61 (s, 2H), 3.72 (t, J = 5.8 Hz, 2H), 2.97 (t, J = 4.5 Hz, 6H), 2.72 (br, 4H), 2.46 (s, 3H), 2.28 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C29H30F6N5O 578.23545, found: 578.2345.
4.1.5.13. 5-Ethyl-8-(4-methylpiperazin-1-yl)-N-(5-(4-(trifluoromethyl)phenyl)pyridin-2-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (27)
Following the typical procedure for 94 (X = OMe), N-(5-bromopyridin-2-yl)-5-ethyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (94: X = Et) was prepared from 62 and 93. It was purified by flash chromatography (0–10% MeOH in DCM) to give a yellow crystalline solid (70%); mp 73–76 °C. 1H NMR (CDCl3) δ 8.25 (d, J = 2.5 Hz, 1H), 8.01 (dd, J = 8.9, 0.4 Hz, 1H), 7.73 (dd, J = 9.0, 2.5 Hz, 1H), 7.29 (s, 1H, NH), 7.11 (d, J = 8.2 Hz, 1H), 7.01 (d, J = 8.2 Hz, 1H), 4.70 (s, 2H), 3.71 (t, J = 6.1 Hz, 2H), 2.92 (t, J = 4.7 Hz, 6H), 2.64–2.55 (m, 6H), 2.38 (s, 3H), 1.19 (t, J = 7.6 Hz, 3H). MS (APCI): 458.2 [79Br], 460.2 [81Br] [M + H]+.
Reaction of 94 (X = Et) and 73 (R = 4-CF3) as above gave 27 (35% yield); mp 190–192 °C. HPLC 97.2%. 1H NMR (CDCl3) δ 8.46 (dd, J = 2.4, 0.6 Hz, 1H), 8.19 (dd, J = 8.7, 0.6 Hz, 1H), 7.92 (dd, J = 8.7, 2.5 Hz, 1H), 7.71 (AB br d, J = 8.4 Hz, 2H), 6.66 (AB br d, J = 8.4 Hz, 2H), 7.40 (s, 1H, NH), 7.12 (d, J = 8.2 Hz, 1H), 7.02 (d, J = 8.2 Hz, 1H), 4.74 (s, 2H), 3.75 (t, J = 6.1 Hz, 2H), 2.93 (apparent t, 6H), 2.65–2.59 (m, 6H), 2.39 (s, 3H), 1.20 (t, J = 7.5 Hz, 3H). HRMS (ESI + ): [M + H] + calculated for C29H33F3N5O 524.2610, found: 524.2632.
4.1.5.14. N-(5-(4-Chloro-2-methylphenyl)pyridin-2-yl)-5-ethyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (28)
From 94 (X = Et) and 81. 71% yield; mp 180–182 °C. HPLC 94.0%. 1H NMR (CDCl3) δ 8.14 (dd, J = 2.4, 0.6 Hz, 1H), 8.12 (dd, J = 8.6, 0.6 Hz, 1H), 7.60 (dd, J = 8.6, 2.4 Hz, 1H), 7.37(s, 1H, NH), 7.28 (d, J = 2.1 Hz, 1H), 7.23 (dd, J = 8.1, 2.0 Hz, 1H), 7.14–7.11 (apparent t, 2H), 7.02 (d, J = 8.2 Hz, 1H), 4.74 (s, 2H), 3.74 (t, J = 6.1 Hz, 2H), 2.93 (apparent t, 6H), 2.65–2.59 (m, 6H), 2.39 (s, 3H), 2.25 (s, 3H), 1.19 (t, J = 7.5 Hz, 3H). HRMS (ESI+): [M + H]+ calculated for C29H35ClN5O 504.2525, found: 504.2534.
4.1.5.15. 5-Benzyl-N-(5-(4-chloro-2-methylphenyl)pyridin-2-yl)-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (29)
Following the typical procedure for 94 (X = OMe), 5-benzyl-N-(5-bromopyridin-2-yl)-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (94: X = Bn) was prepared from 63 and 93. It was purified by flash chromatography (2–7% MeOH in DCM) to give a cream foamy solid (83%); mp 74–77 °C. 1H NMR (CDCl3) δ 8.24 (dd, J = 2.4, 0.6 Hz, 1H), 8.00 (dd, J = 8.9, 0.6 Hz, 1H), 7.73 (dd, J = 9.0, 2.4 Hz, 1H), 7.29–7.25 (m, 2H), 7.21–7.17 (m, 2H), 7.07–7.10 (m, 3H), 7.01 (d, J = 8.2 Hz, 1H), 4.69 (s, 2H), 3.97 (s, 2H), 3.59 (t, J = 6.1 Hz, 2H), 2.93 (t, J = 4.7 Hz, 4H), 2.81 (t, J = 6.1 Hz, 2H), 2.65 (br, 4H), 2.38 (s, 3H). MS (APCI): 520.2 [79Br], 522.1 [81Br] [M + H]+.
Reaction of 94 (X = Bn) and 81 as above gave 29 (49% yield); mp 107–110 °C. HPLC 96.5%. 1H NMR (CDCl3) δ 8.13 (dd, J = 2.3, 0.6 Hz, 1H), 8.09 (dd, J = 8.6, 0.6 Hz, 1H), 7.59 (dd, J = 8.6, 2.4 Hz, 1H), 7.29–7.27 (m, 3H), 7.24–7.18 (m, 2H), 7.14–7.07 (m, 4H), 7.02 (d, J = 8.2 Hz, 1H), 4.73 (s, 2H), 3.98 (s,2H), 3.63 (t, J = 6.1 Hz, 2H), 2.95 (t, J = 4.4 Hz, 4H), 2.83 (t, J = 6.1 Hz, 2H), 2.66 (br, 4H), 2.39 (s, 3H), 2.25 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C34H37ClN5O 566.2681, found: 566.2663.
4.1.5.16. 5-Benzyl-N-(5-(2,4-dimethylphenyl)pyridin-2-yl)-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (30)
From 94 (X = Bn) and 73 (R = 2,4-diMe). 35% yield; mp 94–97 °C. HPLC 97.2%. 1H NMR (CDCl3) δ 8.16 (dd, J = 2.3, 0.6 Hz, 1H), 8.07 (dd, J = 8.6, 0.6 Hz, 1H), 7.62 (dd, J = 8.6, 2.4 Hz, 1H), 7.29–7.27 (m, 3H), 7.21–7.18 (m, 1H), 7.11–7.05 (m, 6H), 7.02 (d, J = 8.2 Hz, 1H), 4.73 (s, 2H), 3.98 (s,2H), 3.63 (t, J = 6.1 Hz, 2H), 2.95 (t, J = 4.4 Hz, 4H), 2.83 (t, J = 6.1 Hz, 2H), 2.66 (br, 4H), 2.39 (s, 3H), 2.36 (s, 3H), 2.24 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C35H40N5O 546.3227, found: 546.3214.
The following compounds were similarly prepared:
4.1.5.17. N-(5-(3,5-Bis(trifluoromethyl)phenyl)pyridin-2-yl)-8-(4-methylpiperazin-1-yl)-5-(methylthio)-3,4-dihydroisoquinoline-2(1H)-carboxamide (39)
Following the typical procedure for 94 (X = OMe), N-(5-bromopyridin-2-yl)-8-(4-methylpiperazin-1-yl)-5-(methylthio)-3,4-dihydroisoquinoline-2(1H)-carboxamide (94: X = SMe) was prepared from 61 and 93. It was purified by flash chromatography (0–15% MeOH in DCM) to give an off-white solid (71%); mp 160–162 °C. 1H NMR (CDCl3) δ 8.25 (dd, J = 2.4, 0.5 Hz, 1H), 8.00 (dd, J = 8.9, 0.6 Hz, 1H), 7.74 (dd, J = 9.0, 2.4 Hz, 1H), 7.29 (s, 1H, NH), 7.18 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 4.69 (s, 2H), 3.71 (t, J = 6.2 Hz, 2H), 3.00 (t, J = 6.1 Hz, 2H), 2.94 (t, J = 4.6 Hz, 4H), 2.67 (br, 4H), 2.43 (s, 3H), 2.40 (s, 3H). MS (APCI): 476.1 [79Br], 478.1 [81Br], [M + H]+.
Reaction of 94 (X = SMe) and 73 (R = 3,5-diOCF3) gave 39 (58% yield); mp 106–109 °C. HPLC 94.5%. 1H NMR (CDCl3) δ 8.47 (dd, J = 2.5, 0.6 Hz, 1H), 8.22 (dd, J = 8.8, 0.6 Hz, 1H), 7.97 (s, 2H), 7.90 (dd, J = 8.8, 2.5 Hz, 1H), 7.87 (s, 1H), 7.45 (s, 1H, NH), 7.17 (d, J = 8.2 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 4.72 (s, 2H), 3.76 (t, J = 6.2 Hz, 2H), 3.03 (t, J = 6.2 Hz, 2H), 2.98 (t, J = 4.6 Hz, 4H), 2.76 (br, 4H), 2.46 (s, 3H), 2.44 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C29H30F6N5OS 610.2075, found: 610.2062.
4.1.5.18. N-(5-(4-Chloro-2-methylphenyl)pyridin-2-yl)-8-(4-methylpiperazin-1-yl)-5-(methylthio)-3,4-dihydroisoquinoline-2(1H)-carboxamide (40)
From 94 (X = SMe) and 81. 54% yield. mp 111–114 °C. HPLC 95.2%. 1H NMR (CDCl3) δ 8.15 (dd, J = 2.4, 0.7 Hz, 1H), 8.11 (dd, J = 8.6, 0.7 Hz, 1H), 7.60 (dd, J = 8.6, 2.4 Hz, 1H), 7.34 (s, 1H, NH), 7.28 (d, J = 2.1 Hz, 1H), 7.24–7.18 (m, 2H), 7.13 (d, J = 8.2 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 4.73 (s, 2H), 3.75 (t, J = 6.1 Hz, 2H), 3.02 (t, J = 6.1 Hz, 2H), 2.93 (t, J = 4.6 Hz, 4H), 2.65 (br, 4H), 2.44 (s, 3H), 2.39 (s, 3H), 2.25 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H33ClN5OS 522.2094, found: 522.2077.
4.1.5.19. N-(5-(4-Chloro-3-(trifluoromethyl)phenyl)pyridin-2-yl)-5-fluoro-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (41)
Following the typical procedure for 94 (X = OMe), N-(5-bromopyridin-2-yl)-5-fluoro-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (94: X = F) was prepared from 59 and 93. It was purified by flash chromatography (2–15% MeOH in DCM) to give a white foamy solid (71%); mp 160–162 °C. 1H NMR (CDCl3) δ 8.26 (dd, J = 2.4, 0.6 Hz, 1H), 8.01 (dd, J = 8.9, 0.6 Hz, 1H), 7.75 (dd, J = 9.0, 2.4 Hz, 1H), 7.30 (s, 1H, NH), 7.01 (dd, J = 8.7, 4.9 Hz, 1H), 6.94 (t, J = 8.8 Hz, 1H), 4.69 (s, 2H), 3.72 (t, J = 6.1 Hz, 2H), 2.94 (t, J = 6.1 Hz, 2H), 2.90 (t, J = 4.7 Hz, 4H), 2.63 (br, 4H), 2.38 (s, 3H). MS (APCI): 448.3 [79Br], 450.3 [81Br] [M + H]+.
Reaction of 94 (X = F) and 73 (R = 3-CF3, 4-Cl) gave 41 (30% yield); mp 97–100 °C. HPLC 93.5%. 1H NMR (CDCl3) δ 8.43 (dd, J = 2.5, 0.6 Hz, 1H), 8.18 (dd, J = 8.8, 0.6 Hz, 1H), 7.87–7.84 (m, 2H), 7.65 (dd, J = 8.3, 2.1 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.42 (s, 1H, NH), 7.04–7.00 (m, 1H), 6.95 (t, J = 8.7 Hz, 1H), 4.73 (s, 2H), 3.76 (t, J = 6.1 Hz, 2H), 2.96 (t, J = 6.1 Hz, 2H), 2.92 (t, J = 4.6 Hz, 4H), 2.66 (br, 4H), 2.40 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H27ClF4N5O 548.18348, found: 548.18230.
4.1.5.20. N-(5-(4-Chloro-2-methylphenyl)pyridin-2-yl)-5-fluoro-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinoline-2(1H)-carboxamide (42)
From 94 (X = F) and 81. 57% yield; mp 98–101 °C. HPLC 93.2%. 1H NMR (CDCl3) δ 8.15 (dd, J = 2.4, 0.7 Hz, 1H), 8.10 (dd, J = 8.6, 0.7 Hz, 1H), 7.61 (dd, J = 8.6, 2.3 Hz, 1H), 7.34 (s, 1H, NH), 7.28 (d, J = 2.1 Hz, 1H), 7.24 (dd, J = 8.4, 2.0 Hz, 1H), 7.14 (d, J = 8.2 Hz, 1H), 7.04–7.00 (m, 1H), 6.94 (t, J = 8.7 Hz, 1H), 4.73 (s, 2H), 3.76 (t, J = 6.1 Hz, 2H), 2.95 (t, J = 6.0 Hz, 2H), 2.91 (t, J = 4.4 Hz, 4H), 2.64 (br, 4H), 2.39 (s, 3H), 2.26 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30ClFN5O 494.21174, found: 494.21231.
4.1.6. Preparation of the –COCH2-linked pyridyl side compounds 43–47 of Scheme 5 and Table 1
A mixture of (4-methylpiperazin-1-yl)isoquinoline (56) (119.0 mg, 0.514 mmol), 2-(5-bromopyridin-2-yl)acetic acid (96) (111.1 mg, 0.514 mmol), dimethylamino-4-pyridine (62.8 mg, 0.514 mmol), N-hydroxybenzotriazole (69.5 mg, 0.514 mmol) and diisopropylethylamine (0.30 mL, 1.80 mmol) in anhydrous N,N-dimethylformamide (7 mL) was treated under nitrogen at room temperature with EDCI hydrochloride (167.5 mg, 0.874 mmol). The mixture was stirred at room temperature overnight, it was then diluted with water. The aqueous mixture was extracted with DCM (3x) and the combined extract was washed with water, brine, dried (MgSO4) and concentrated under reduced pressure. The crude product was chromatographed on silica using 3–9% MeOH in DCM to afford 2-(5-bromopyridin-2-yl)-1-(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (97) as a yellow oil (0.188 g, 85%). 1H NMR (CDCl3) δ (1:1 mixture of rotamers, two sets of data). 8.59 (d, J = 2.0 Hz, 1H), 8.56 (d, J = 2.1 Hz, 1H), 7.77 (dd, J = 8.3, 2.4 Hz, 1H), 7.72 (dd, J = 8.3, 2.4 Hz, 1H), 7.30 (d, J = 8.4 Hz, 1H), 7.23–7.16 (m, 3H), 6.97 (t, J = 7.8 Hz, 2H), 6.92–6.87 (m, 2H), 4.75 (s, 2H), 4.66 (s, 2H), 4.00 (s, 2H), 3.94 (s, 2H), 3.81–3.76 (m, 4H), 2.92 (t, J = 4.7 Hz, 4H), 2.88–2.82 (m, 8H), 2.61 (br, 8H), 2.38 (s, 3H), 2.35 (s, 3H). MS (APCI): 429.1 [79Br], 431.2 [81Br], [M + H]+.
Reaction of 97 with the appropriate boronic acids (73) or 81 using the reaction conditions and purification procedures described above for 31, gave the following compounds of Table 1:
4.1.6.1. 2-(5-(4-Chlorophenyl)pyridin-2-yl)-1-(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (43)
M.p. 49–52 °C. HPLC 93.4%. 1H NMR (CDCl3) δ (1:1 mixture of rotamers, two sets of data). 8.72 (d, J = 2.0 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 7.81 (dd, J = 8.1, 2.4 Hz, 1H), 7.76 (dd, J = 8.1, 2.4 Hz, 1H), 7.52–7.42 (m, 9H), 7.37 (d, J = 8.1 Hz, 1H), 7.21–7.15 (m, 2H), 7.01–6.96 (m, 2H), 6.93–6.87 (m, 2H), 4.77 (s, 2H), 4.71 (s, 2H), 4.10 (s, 2H), 4.03 (s, 2H), 3.85–3.81 (m, 4H), 2.93–2.84 (m, 12H), 2.60 (br, 8H), 2.36 (s, 3H), 2.35 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30ClN4O 461.2103, found: 461.2101.
4.1.6.2. 1-(8-(4-Methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)-2-(5-phenylpyridin-2-yl)ethan-1-one (44)
Semi-solid. HPLC 94.4%. 1H NMR (CDCl3) δ (1:1 mixture of rotamers, two sets of data). 8.76 (d, J = 2.0 Hz, 1H), 8.74 (d, J = 2.1 Hz, 1H), 7.85 (dd, J = 8.1, 2.4 Hz, 1H), 7.80 (dd, J = 8.1, 2.4 Hz, 1H), 7.58–7.52 (m, 4H), 7.49–7.44 (m, 5H), 7.42–7.35 (m, 3H), 7.19–7.15 (m, 2H), 7.01–6.96 (m, 2H), 6.93–6.87 (m, 2H), 4.78 (s, 2H), 4.71 (s, 2H), 4.10 (s, 2H), 4.04 (s, 2H), 3.85–3.81 (m, 4H), 2.93–2.90 (m, 6H), 2.90–2.83 (m, 6H), 2.60 (br, 8H), 2.36 (s, 3H), 2.34 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H31N4O 427.2492, found: 427.2486.
4.1.6.3. 2-(5-(3-Chlorophenyl)pyridin-2-yl)-1-(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (45)
Semi-solid. HPLC 94.7%. 1H NMR (CDCl3) δ (1:1 mixture of rotamers, two sets of data). 8.73 (d, J = 2.0 Hz, 1H), 8.70 (d, J = 2.1 Hz, 1H), 7.82 (dd, J = 8.1, 2.4 Hz, 1H), 7.77 (dd, J = 8.1, 2.4 Hz, 1H), 7.57–7.37 (m, 10H), 7.21–7.15 (m, 2H), 7.01–6.96 (m, 2H), 6.93–6.87 (m, 2H), 4.77 (s, 2H), 4.71 (s, 2H), 4.11 (s, 2H), 4.04 (s, 2H), 3.85–3.81 (m, 4H), 2.94–2.84 (m, 12H), 2.61 (br, 8H), 2.37 (s, 3H), 2.35 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C27H30ClN4O 461.2103, found: 461.2095.
4.1.6.4. 1-(8-(4-Methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)-2-(5-(4-(trifluoromethoxy)phenyl)pyridin-2-yl)ethan-1-one (46)
Semi-solid. HPLC 95.2%. 1H NMR (CDCl3) δ (1:1 mixture of rotamers, two sets of data). 8.73 (d, J = 2.2 Hz, 1H), 8.70 (d, J = 2.2 Hz, 1H), 7.82 (dd, J = 8.1, 2.4 Hz, 1H), 7.77 (dd, J = 8.1, 2.4 Hz, 1H), 7.59–7.52 (m, 4H), 7.37 (d, J = 8.1 Hz, 1H), 7.39 (d, J = 8.1 Hz, 1H), 7.32 (d, J = 8.5 Hz, 4H), 7.21–7.15 (m, 2H), 7.01–6.96 (m, 2H), 6.93–6.87 (m, 2H), 4.78 (s, 2H), 4.72 (s, 2H), 4.10 (s, 2H), 4.04 (s, 2H), 3.85–3.81 (m, 4H), 2.93–2.84 (m, 12H), 2.60 (br, 8H), 2.36 (s, 3H), 2.34 (s, 3H). HRMS (ESI+): [M + H]+ calculated for C28H30F3N4O2 511.2315, found: 511.2312.
4.1.6.5. (2-(5-(4-Chloro-2-methylphenyl)pyridin-2-yl)-1-(8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (47)
M.p. 45–48 °C. HPLC 95.9%. 1H NMR (CDCl3) δ (1:1 mixture of rotamers, two sets of data). 8.47 (d, J = 2.2 Hz, 1H), 8.45 (d, J = 2.2 Hz, 1H), 7.60–7.53 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.30–7.10 (m, 8H), 7.01–6.96 (m, 2H), 6.93–6.87 (m, 2H), 4.79 (s, 2H), 4.74 (s, 2H), 4.10 (s, 2H), 4.04 (s, 2H), 3.86–3.83 (m, 4H), 2.94–2.84 (m, 12H), 2.60 (br, 8H), 2.36 (s, 3H), 2.35 (s, 3H), 2.25 (s, 3H), 2.22 (s, 3H). HRMS (ESI+): [M+H]+ calculated for C28H32ClN4O 475.2259, found: 475.2244.
4.1.7. Preparation of the carboxypiperidine-linked compounds 48–51 of Scheme 6 and Table 1
4.1.7.1. (5-Methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)methanone (48)
Et3N (0.837 mL, 6.00 mmol) was added to a suspension of 4-(4-(trifluoromethoxy)phenoxy)piperidine hydrochloride (99-HCl) (523 mg, 2.00 mmol) in dry DCM (7.5 mL) at 0 °C, followed by the addition of 4-nitrophenyl carbonochloridate (98) (484 mg, 2.40 mmol). The resulting mixture was allowed to warm to room temperature and was stirred for 3 h. The solvent was evaporated and the residue was dissolved in EtOAc, washed with 5% ammonia five times, then brine and dried over anhydrous Na2SO4. Removal of solvent gave 4-nitrophenyl 4-(4-(trifluoromethoxy)phenoxy)piperidine-1-carboxylate (1 0 1) as an off-white solid (772 mg, 90%). 1H NMR (CDCl3) δ 8.28–8.24 (m, 2H), 7.33–7.29 (m, 2H), 7.16 (d, J = 8.5 Hz, 2H), 6.95–6.91 (m, 2H), 4.59–4.54 (m, 1H), 3.88–3.62 (m, 4H), 3.38–3.31 (m, 2H), 1.98–1.88 (m, 2H); HRMS calcd. for C19H17F3N2O6 (M + H+) m/z 427.1113, found 427.1116.
A mixture of 57 (80 mg, 0.33 mmol), 101 (200 mg, 0.47 mmol), DIPEA (0.115 mL, 0.66 mmol) and DMAP (4 mg, 0.033 mmol) in toluene (5 mL) was heated in an oil bath at 110 °C for 3 days. The solvent was evaporated and the residue was taken in EtOAc and washed with 5% ammonia five times (until the aqueous phase became colourless). The organic phase was washed with brine, dried over anhydrous Na2SO4 and filtered through a pad of alumina (Aluminium Oxide 90, Standardised, Merck). The solvent was removed to give the crude product, which was purified by column chromatography (alumina), using gradient mixtures of EtOAc and hexanes (1:2, then 2:3) as the eluent, to give 48 as a white solid (53 mg, 30%). A sample was further purified by preparative HPLC (Column: Synergi-Max RP 4, 250 × 21.20 mm; Mobile phase: A/B = from 90% to 2% (A: 0.1% THA in water pH 2.56, B: 90% acetonitrile in water); flow rate 9 mL/ min, gradient method; wave length: 254 nm, 288 nm). HPLC 90.4%. M.p. 91–94 °C. 1H NMR (CDCl3) δ 7.14 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.1 Hz, 1H), 6.93–6.89 (m, 3H), 4.50–4.45 (m, 1H), 4.35 (s, 2H), 3.60–3.54 (m, 2H), 3.46 (t, J = 5.6 Hz, 2H), 3.25–3.19 (m, 2H), 2.92–2.89 (m, 6H), 2.56 (br, 4H), 2.35 (s, 3H), 2.21 (s, 3H), 2.04–1.98 (m, 2H), 1.87–1.82 (m, 2H); HRMS calcd. for C28H35F3N4O3 (M + H+) m/z 533.2733, found 533.2743.
4.1.7.2. (5-Methyl-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)(4-(2-(trifluoromethyl)phenoxy)piperidin-1-yl)methanone (49)
A solution of 4-(2-(trifluoromethyl)phenoxy)piperidine (100) (500 mg, 2.04 mmol) in dry DCM (5 mL) was cooled in an ice bath, then treated with triethylamine (0.500 mL, 3.57 mmol), followed by 98 (493 mg, 2.45 mmol). The resulting mixture was allowed to warm up to room temperature and was stirred for 3 h. The solvent was evaporated and the residue was taken in EtOAc, and washed with 5% ammonia five times (until the aqueous phase became colourless). The solvent was removed to give the crude product, which was purified by column chromatography (silica), using a mixture of EtOAc and petrol hexanes (1:1) as the eluent, to give 4-nitrophenyl 4-(2-(trifluoromethyl)phenoxy)piperidine-1-carboxylate (102) as a colourless gum (618 mg, 74%). 1H NMR (CDCl3) δ 8.28–8.21 (m, 2H), 7.62–7.60 (m, 1H), 7.52–7.48 (m, 1H), 7.33–7.30 (m, 2H), 7.05–7.00 (m, 2H), 4.82–4.78 (m, 1H), 3.93–3.82 (m, 2H), 3.75–3.69 (m, 1H), 3.62–3.55 (m, 1H), 2.08–1.96 (m, 4H); HRMS calcd. for C19H17F3N2O5 (M + H+) m/z 411.1160, found 411.1168.
A mixture of 57 (80 mg, 0.33 mmol), 102 (200 mg, 0.49 mmol) and DIPEA (0.115 mL, 0.66 mmol) in toluene (5 mL) was heated in an oil bath at 100 °C for 48 h. The solvent was evaporated and the residue was taken in EtOAc and washed with 5% ammonia five times (until the aqueous phase became colourless). The organic phase was washed with brine, dried over anhydrous Na2SO4 and filtered through a pad of alumina. The solvent was removed to give the crude product, which was purified by column chromatography (alumina), using gradient mixtures of EtOAc and hexanes (1:1, then 2:1) as the eluent, followed by recrystallisation from ether and heptane to give 49 as a white solid (90 mg, 54%); mp 55–58 °C. HPLC 94.6%. 1H NMR (CDCl3) δ 7.58 (dd, J = 8.0, 1.1 Hz, 1H), 7.49–7.45 (m, 1H), 7.03–6.98 (m, 3H), 6.90 (d, J = 8.0 Hz, 1H), 4.72–4.68 (m, 1H), 4.34 (s, 2H), 3.55–3.50 (m, 2H), 3.45 (t, J = 5.6 Hz, 2H), 3.39–3.34 (m, 2H), 2.92–2.89 (m, 6H), 2.56 (br, 4H), 2.35 (s, 3H), 2.21 (s, 3H), 2.04–1.90 (m, 4H); HRMS calcd. for C28H35F3N4O2 (M + H+) m/z 517.2784, found 517.2795.
4.1.7.3. (5-Methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)methanone (50)
A mixture of 58 (80 mg, 0.33 mmol), 101 (200 mg, 0.47 mmol), DIPEA (0.115 mL, 0.66 mmol) and DMAP (4 mg, 0.033 mmol) in toluene (5 mL) was heated in an oil bath at 110 °C for 3 days. Workup as above gave the crude product, which was purified by column chromatography packed with alumina, using gradient mixtures of EtOAc and petrol ether (v/v = 1:2, then 1:1) as eluents, followed by recrystallisation from ether and heptane to give 50 as a white solid (76 mg, 43%); mp 49–51 °C. HPLC 90.3%. 1H NMR (CDCl3) δ 7.14 (dd, J = 9.0, 0.7 Hz, 2H), 7.00 (d, J = 8.6 Hz, 1H), 6.92–6.88 (m, 2H), 6.71 (d, J = 8.7 Hz, 1H), 4.48–4.44 (m, 3H), 3.80 (s, 3H), 3.60–3.53 (m, 2H), 3.49 (t, J = 6.0 Hz, 2H), 3.48–3.17 (m, 2H), 2.92–2.89 (m, 4H), 2.81 (t, J = 6.0 Hz, 2H), 2.61 (br, 4H), 2.38 (s, 3H), 2.05–1.97 (m, 2H), 1.86–1.78 (m, 2H); HRMS calcd. for C28H35F3N4O4 (M + H+) m/z 549.2694, found 549.2697
4.1.7.4. (5-Methoxy-8-(4-methylpiperazin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)(4-(2-(trifluoromethyl)phenoxy)piperidin-1-yl)methanone (51)
A mixture of 58 (85 mg, 0.32 mmol), 102 (200 mg, 0.49 mmol) and Et3N (0.090 mL, 0.64 mmol) in MeCN (5 mL) of was heated in an oil bath at 70 °C for 48 h. Workup and purification as above gave 51 as a white solid (98 mg, 57%); mp 74–77 °C. HPLC 97.0%. 1H NMR (CDCl3) δ 7.58 (d, J = 7.0 Hz, 1H), 7.49–7.45 (m, 1H), 7.01–6.98 (m, 3H), 6.71 (d, J = 8.7 Hz, 1H), 4.68–4.66 (m, 1H), 4.45 (s, 2H), 3.80 (s, 3H), 3.55–3.48 (m, 4H), 3.34–3.29 (m, 2H), 2.87 (t, J = 4.6 Hz, 4H), 2.81 (t, J = 6.0 Hz, 2H), 2.58 (br, 4H), 2.36 (s, 3H), 2.03–1.97 (m, 2H), 1.94–1.87 (m, 2H); HRMS calcd. for C28H35F3N4O3 (M + H+) m/z 533.2733, found 533.2740.
4.1.8. Preparation of the carbamate-linked compounds 52–55 of Scheme 7 and Table 2
4.1.8.1. 2-((5-(4-Chloro-2-methylphenyl)pyridin-2-yl)methyl)-5-methyl-8-(piperazin-1-yl)-1,2,3,4-tetrahydroisoquinoline (52)
A solution of 86 (0.230 g, 0.521 mmol) in PhMe (5 mL) was purged with nitrogen for 1 min. BINAP (0.017 g, 0.026 mmol), Pd2(dba)3 (0.007 g, 0.0076 mmol) and N-Bocpiperazine (0.146 g, 0.782 mmol) were then added, and the mixture stirred at 80 °C for 15 min. A bright orange solution formed, upon which NaOtBu (0.075 g, 0.782 mmol) was added, and the mixture stirred at reflux under nitrogen for 18 h. The reaction was cooled and partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (1:1, x4:EtOAc) afforded tert-butyl 4-(2-((5-(4-chloro-2-methylphenyl)pyridin-2-yl)methyl)-5-methyl-1,2,3,4-tetrahydroisoquinolin-8-yl)piperazine-1-carboxylate 103 (0.099 g, 35%) as a yellow solid; mp 76–79 °C. 1H NMR (CDCl3) δ 8.52 (d, J = 1.5 Hz, 1H), 7.52–7.63 (m, 2H), 7.22–7.33 (m, 2H), 7.15 (d, J = 7.8 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 3.91 (s, 2H), 3.78 (s, 2H), 3.43 (s, 4H), 2.88 (t, J = 5.4 Hz, 2H), 2.75–2.83 (m, 6H), 2.25 (s, 3H), 2.19 (s, 3H), 1.48 (s, 9H); Anal. (C32H39ClN4O2) C, H, N.
Trifluoroacetic acid (0.5 mL) was added to a solution of 103 (0.070 g, 0.128 mmol) in DCM (2.5 mL) at room temperature and the mixture stirred at this temperature for 2 h. Sat. NaHCO3 was added and the reaction was extracted with DCM. The organic phase was dried, filtered and evaporated to afford 52 as a yellow oil (0.025 g, 35%). 1H NMR (CDCl3) δ (tri-HCl salt) 8.69 (dd, J = 2.2 Hz, J = 0.8 Hz, 1H), 7.94 (dd, J = 8.0 Hz, J = 2.2 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.40 (d, J = 2.1 Hz, 1H), 7.32 (dd, J = 8.1 Hz, , J = 2.2 Hz, 1H), 7.23–7.39 (m, 2H), 7.14 (d, J = 8.1 Hz, 1H), 4.69 (s, 4H), 3.71 (s, 2H), 3.39 (s, 4H), 3.09–3.16 (m, 6H), 2.28 (s, 6H); Anal. (C27H34Cl4N4) C, H, N (tri-HCl salt).
4.1.8.2. 4-(2-((5-(4-Chloro-2-methylphenyl)pyridin-2-yl)methyl)-5-methyl-1,2,3,4-tetrahydroisoquinolin-8-yl)morpholine (53)
A solution of 86 (0.093 g, 0.21 mmol) in toluene (2 mL) was purged with nitrogen for 1 min. BINAP (0.007 g, 0.010 mmol), Pd2(dba)3 (0.005 g, 0.0053 mmol) and morpholine (0.027 mL, 0.316 mmol) were then added, and the mixture stirred at 80 °C for 15 min. A bright orange solution formed, upon which NaOtBu (0.030 g, 0.316 mmol) was added, and the mixture stirred at reflux under nitrogen for 4 h. The reaction was cooled and partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (2:1, x4:EtOAc) afforded 53 (0.024 g, 26%) as a yellow oil, which was further purified by the general salt formation/back extraction procedure as described above. 1H NMR (CDCl3) δ 8.52 (dd, J = 2.0 Hz, J = 1.0 Hz, 1H), 7.58–7.62 (m, 2H), 7.22–7.32 (m, 2H), 7.15 (d, J = 7.8 Hz, 1H), 7.02 (d, J = 8.0 Hz, 1H), 6.87 (d, J = 8.0 Hz, 1H), 3.91 (s, 2H), 3.74–3.76 (m, 6H), 2.78–2.89 (m, 8H), 2.29 (s, 3H), 2.19 (s, 3H); Anal. (C27H33Cl4N3O) C, H, N (tri-HCl salt).
4.1.8.3. 4-(2-((5-(4-Chloro-2-methylphenyl)pyridin-2-yl)methyl)-5-methyl-1,2,3,4-tetrahydroisoquinolin-8-yl)yhiomorpholine (54)
A solution of 86 (0.071 g, 0.161 mmol) in toluene (2.5 mL) was purged with nitrogen for 1 min, treated with BINAP (0.005 g, 0.008 mmol), Pd2(dba)3 (0.004 g, 0.004 mmol) and thiomorpholine (0.022 mL, 0.241 mmol) as above, to give 54 as a yellow solid (0.053 g, 71%); mp 133–135 °C. 1H NMR (CDCl3) δ 8.53 (dd, J = 2.0 Hz, J = 0.8 Hz, 1H), 7.55–7.63 (m, 2H), 7.25–7.30 (m, 2H), 7.16 (d, J = 7.8 Hz, 1H), 7.01 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 8.0 Hz, 1H), 3.90 (s, 2H), 3.72 (s, 2H), 3.06 (s, 4H), 2.85 (t, J = 5.5 Hz, 2H), 2.79 (t, J = 5.5 Hz, 2H), 2.27 (s, 3H), 2.15 (s, 3H); Anal. (C27H30ClN3S) C, H, N.
4.1.8.4. 2-((5-(4-Chloro-2-methylphenyl)pyridin-2-yl)methyl)-5-methyl-8-(piperidin-1-yl)-1,2,3,4-tetrahydroisoquinoline (55)
A solution of 104 (0.267 g, 1.18 mmol) in toluene (7.5 mL) was purged with nitrogen for 1 min. BINAP (0.037 g, 0.059 mmol), Pd2(dba)3 (0.054 g, 0.059 mmol) and piperidine (0.175 mL, 1.77 mmol) were then added, and the mixture stirred at 80 °C for 15 min. A bright orange solution formed, upon which NaOtBu (0.170 g, 1.77 mmol) was added, and the mixture stirred at reflux under nitrogen for 4 h. The reaction was cooled and partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (4:1, x4:EtOAc) afforded 5-methyl-8-(piperidin-1-yl)isoquinoline (105) as a brown oil (0.169 g, 62%), which was used directly. 1H NMR (CDCl3) δ 9.59 (s, 1H), 8.53 (d, J = 5.9 Hz, 1H), 7.68 (dd, J = 7.9 Hz, J = 0.8 Hz, 1H), 7.51 (dd, J = 7.9 Hz, J = 0.8 Hz, 1H), 7.00 (d, J = 7.9 Hz, 1H), 3.07 (s, 4H), 2.58 (s, 3H), 1.82–1.88 (m, 4H), 1.65–1.78 (m, 2H).
To a stirred solution of 105 (0.169 g, 1.25 mmol) in MeOH (7.5 mL) at 0 °C was added NaCNBH3 (0.235 g, 3.73 mmol), and the mixture stirred at this temperature for 10 min·BF3.OEt2 (0.46 mL, 3.73 mmol) was then added and the mixture stirred at 0 °C for 1 h, then at reflux for 3 h. The reaction was cooled and quenched with sat. NaHCO3, and the solvent removed under reduced pressure. The residue was partitioned between EtOAc and water, and the organic phase was dried, filtered and evaporated. Column chromatography (19:1, DCM:MeOH) afforded 5-methyl-8-(piperidin-1-yl)-1,2,3,4-tetrahydroisoquinoline (106) as a brown oil (0.171 g, 100%), which was used directly. 1H NMR (CDCl3) δ 7.01 (t, J = 8.0 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 4.09 (s, 2H), 3.24 (t, J = 6.2 Hz, 3H), 2.17 (s, 3H), 1.65–1.78 (m, 2H), 1.49 (s, 2H), 2.67–2.72 (m, 6H), 2.17 (1H).
To a solution of 106 (0.094 g, 0.408 mmol) and 2-(bromomethyl)-5-(4-chloro-2-methylphenyl)pyridine (84) (0.133 g, 0.449 mmol) in DMF (8 mL) was added K2CO3 (0.068 g, 0.492 mmol) and the resultant mixture stirred at room temperature for 5 h. The reaction was then diluted with EtOAc (50 mL) and washed with water (5 × 10 mL), and the organic layer was dried and evaporated. Column chromatography (2:1, x4:EtOAc) afforded compound 55 as a tan solid (0.059 g, 32%); mp 96–98 °C. 1H NMR (CDCl3) δ 8.52 (d, J = 1.5 Hz, 1H), 7.57–7.62 (m, 2H), 7.23–7.32 (m, 2H), 7.15 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 8.0 Hz, 1H), 3.92 (s, 2H), 3.78 (s, 2H), 2.86 (t, J = 5.4 Hz, 2H), 2.71–2.81 (m, 6H), 2.25 (s, 3H), 2.18 (s, 3H), 1.55–1.62 (m, 4H), 1.49 (s, 2H); Anal. (C28H32ClN3) C, H, N.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the Bill & Melinda Gates Foundation (#OPP1017459), the U.S. Agency for International Development (GHS-A-00-08-00012-00), the U.K. Department for International Development (DFID), and Irish Aid, 23-27 Henry Street, Limerick, Eire.
References
- 1.WHO Global Tuberculosis Report 2019 (https://www.who.int/tb/publications/global_report/en/): accessed 2nd July 2020.
- 2.Andries K., Verhasselt P., Guillemont J. Science. 2005;307:223–227. doi: 10.1517/13543784.14.7.917. [DOI] [PubMed] [Google Scholar]
- 3.Pym A.S., Diacon A.H., Tang S.J., Conradie F. On behalf of the TMC207-C209 study group. Eur. Respir. J. 2016;47:564–574. doi: 10.1183/13993003.00724-2015. [DOI] [PubMed] [Google Scholar]
- 4.Global Alliance for TB Drug Development website (https://www.tballiance.org/portfolio/compound/tbaj-876-diarylquinoline); accessed 2nd July 2020.
- 5.Sutherland H.S., Tong A.S.T., Choi P.J. 3,5-Dialkoxypyridine analogs of bedaquiline are potent antitubercular agents with minimal inhibition of the hERG channel. Bioorg Med Chem. 2019;27:1292–1307. doi: 10.1016/j.bmc.2019.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarathy JP, Ragunathan P, Shin J, Cooper CB, Upton AM, Grüber G, Dick T. TBAJ-876 retains bedaquiline’s activity against subunits c and ε of Mycobacterium tuberculosis F-ATP synthase. Antimicrob Agents Chemother 2019, 63, e01191-19. DOI: 10.1128/AAC.01191-19. [DOI] [PMC free article] [PubMed]
- 7.Sarathy J.P., Ragunathan P., Cooper C.B., Upton A.M., Grüber G., Dick T. TBAJ-876 displays bedaquiline-like Mycobactericidal potency without retaining the parental drug’s uncoupler activity. Antimicrob. Agents Chemother. 2020;64:e01540–e1619. doi: 10.1128/AAC.01540-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowe JA. Tetrahydroisoquinolines as antagonists of the 5HT1b receptor. WO 2004108682 A2.
- 9.Angst C, Haberlein M, Hill D, Jacobs R, Moore G, Pierson E, Ashokkumar BS, Shenvi AB. Preparation of isoquinolines as 5-HT antagonists for treatment of psychiatric disorders. WO 2003037887 A1.
- 10.Mathison I.W., Fowler K.C., Morgan P.H., Tidwell R.R. Synthesis and hypotensive properties of tetrahydroisoquinolines. J Med Chem. 1973;16(4):332–336. doi: 10.1021/jm00262a005. [DOI] [PubMed] [Google Scholar]
- 11.Perrey D.A., German N.A., Gilmour B.P. Substituted tetrahydroisoquinolines as selective antagonists for the orexin 1 receptor. J Med Chem. 2013;56(17):6901–6916. doi: 10.1021/jm400720h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins L.A., Franzblau S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cho S.H., Warit S., Wan B., Hwang C.H., Pauli G.F., Franzblau S.G. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falzari K., Zhu Z., Pan D., Liu H., Hongmanee P., Franzblau S.G. In vitro and in vivo activities of macrolide derivatives against Mycobacterium tuberculosis. AAC. 2005;49(4):1447–1454. doi: 10.1128/AAC.49.4.1447-1454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang D., Lu Y.u., Liu K., Liu B., Wang J., Zhang G., Zhang H., Liu Y., Wang B., Zheng M., Fu L., Hou Y., Gong N., Lv Y., Li C., Cooper C.B. Identification of less lipophilic riminophenazine derivatives for the treatment of drug-resistant tuberculosis. J Med Chem. 2012;55(19):8409–8417. doi: 10.1021/jm300828h. [DOI] [PubMed] [Google Scholar]
- 16.Blaser A., Sutherland H.S., Tong A.S.T., Choi P.J., Conole D., Franzblau S.G., Cooper C.B. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Bioorg Med Chem. 2019;27(7):1283–1291. doi: 10.1016/j.bmc.2019.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elkins S., Freundlich J.S., Choi I., Sarker M., Talcott C. Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends Microbiol. 2011;19:65–74. doi: 10.3389/fmicb.2018.01367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Svensson E.M., Murray S., Karlsson M.O., Dooley K.E. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti-TB drug. J Antimicrob Chemother. 2015;70:1106–1114. doi: 10.1093/jac/dku504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haagsma A.C., Abdillahi-Ibrahim R., Wagner M.J., Krab K., Vergauwen K., Guillemont J. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. AAC. 2009;53(3):1290–1292. doi: 10.1128/AAC.01393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aiaaoul H, Boss C, Richard-Bildstein, S. Heterocyclyl derivatives and their use as prostaglandin d2 receptor modulators. WO 2013093842.
- 21.Merkul E., Schäfer E., Müller T.J.J. Rapid synthesis of bis(hetero)aryls by one-pot Masuda borylation–Suzuki coupling sequence and its application to concise total syntheses of meridianins A and G. Org Biomol Chem. 2011;9(9):3139. doi: 10.1039/c1ob05310h. [DOI] [PubMed] [Google Scholar]