

Summary
Most genome-wide association studies (GWAS) and transcriptome-wide association studies (TWAS) focus on European populations; however, these results cannot always be accurately applied to non-European populations due to genetic architecture differences. Using GWAS summary statistics in the Population Architecture using Genomics and Epidemiology study, which comprises ∼50,000 Hispanic/Latinos, African Americans, Asians, Native Hawaiians, and Native Americans, we perform TWAS to determine gene-trait associations. We compared results using three transcriptome prediction models derived from Multi-Ethnic Study of Atherosclerosis populations: the African American and Hispanic/Latino (AFHI) model, the European (EUR) model, and the African American, Hispanic/Latino, and European (ALL) model. We identified 240 unique significant trait-associated genes. We found more significant, colocalized genes that replicate in larger cohorts when applying the AFHI model than the EUR or ALL model. Thus, TWAS with population-matched transcriptome models have more power for discovery and replication, demonstrating the need for more transcriptome studies in diverse populations.
Subject Areas: Population, Genetics, Genomics, Human Genetics
Graphical Abstract
Highlights
-
•
TWAS mechanistically extends GWAS findings in diverse populations
-
•
Population-matched transcriptome models detect more replicable associations
-
•
Colocalization shows GWAS variants likely act through gene expression regulation
-
•
More GWAS and transcriptome modeling in diverse populations are needed
Population; Genetics; Genomics; Human Genetics
Introduction
Genome-wide association studies (GWAS) test single-nucleotide polymorphisms (SNPs) across the genome for association with diseases and other complex traits. GWAS have identified thousands of SNP-trait associations with complex traits; however, the majority of the studies exclusively include individuals of European ancestries (Buniello et al., 2019). As of 2017, within 4655 GWAS, 78% of individuals come from European ancestries (Morales et al., 2018), creating a significant gap of knowledge for those of non-European descent. Even when present in large scale biobanks, non-European populations are often excluded from genetic analyses (Peterson et al., 2019; Ben-Eghan et al., 2020), which further worsens under-representation of diverse populations in research. As those of European ancestries only make up a small fraction of the human population, expanding the number of non-European individuals in genomic research benefits all populations by more fully incorporating global genetic diversity in association studies. Since populations were isolated from each other by geography throughout large spans of human history, allele frequencies and effect sizes differ across populations, making current GWAS results poor genetic predictors for non-European populations (Mogil et al., 2018; Martin et al., 2019; Keys et al., 2020). To start to address this problem, the Population Architecture using Genomics and Epidemiology (PAGE) study performed 28 GWAS on clinical and behavioral phenotypes in a multi-ancestries cohort that included Hispanic/Latinos, African Americans, Asians, Native Hawaiians, and Native Americans (Wojcik et al., 2019). The PAGE study is the largest collection of GWAS conducted in non-Europeans.
Meanwhile, transcriptome-wide association studies (TWAS) incorporate transcriptome data along with genotype and phenotype data to make gene-trait associations (Gamazon et al., 2015; Gusev et al., 2016). In TWAS, expression quantitative trait loci (eQTL) data are used to build models that predict gene expression levels from genotypes. The models are integrated with GWAS data to test genes, rather than SNPs, for association with complex traits. Gene-trait associations identified through TWAS provide evidence that gene regulatory mechanisms underlie the trait's biology. TWAS have not yet been applied to the PAGE GWAS results.
Here, we perform TWAS with S-PrediXcan (Barbeira et al., 2018) in PAGE using GWAS summary statistics and three transcriptome prediction models built in the Multi-Ethnic Study of Atherosclerosis (MESA) (Bild et al., 2002, Liu et al., 2013, Mogil et al., 2018). We compared performance and replication of each transcriptome prediction model to determine whether population ancestry matching or sample size is more important in TWAS. We use one transcriptome model built in the MESA African American and Hispanic/Latino (AFHI) populations, one built in the MESA European population (EUR), and another built in the MESA African American, Hispanic/Latino, and European (ALL) populations combined. From there, we colocalize our S-PrediXcan results using COLOC software (Giambartolomei et al., 2014; Hormozdiari et al., 2016; Barbeira et al., 2018; Pividori et al., 2020; Barbeira et al., 2019) to provide more evidence the SNPs in discovered genes are acting through gene expression regulation to affect the associated phenotypes. We then tested discovered associations for replication using the PhenomeXcan database, which includes S-PrediXcan results from large, predominantly European GWAS (Pividori et al., 2020). We find a higher proportion of gene-trait pairs identified in PAGE replicate when we use the population-matched AFHI transcriptome prediction model than either the EUR or ALL transcriptome prediction models. All scripts used for analyses are available at https://github.com/WheelerLab/MESA_expression_prediction.
Results
We sought to perform TWAS in the PAGE study (Wojcik et al., 2019) to reveal new associations or show that previously discovered GWAS loci likely act through transcription regulation to affect the trait. We also sought to compare TWAS results in the diverse PAGE cohort using two different transcriptome prediction models, one built in populations that more closely match the genetic ancestries of PAGE and one that is composed of individuals of European genetic ancestries. In addition, we compared these results to a third transcriptome model that included all available populations. In the PAGE study, 28 GWAS on clinical and behavioral phenotypes (Table 1) were performed (Wojcik et al., 2019). Individuals in PAGE self-identified as Hispanic/Latino (n = 22,216), African American (n = 17,299), Asian (n = 4,680), Native Hawaiian (n = 3,940), Native American (n = 652), or Other (n = 1,052) (Wojcik et al., 2019). In comparison to any other GWAS, this study includes the most phenotypes tested in a single study, the most trait associations, and the highest number of non-European individuals (Wojcik et al., 2019). TWAS integrate genetically regulated gene expression into complex trait mapping studies, but like GWAS, most are performed in European populations (Gamazon et al., 2015; Gusev et al., 2016). We compared S-PrediXcan results using transcriptome prediction models trained with genotype and monocyte gene expression data from three populations in MESA to find genes associated with traits in PAGE. Two MESA models (Mogil et al., 2018) were built in populations of similar size: EUR (n = 578), which comprises individuals of European ancestries and reflects transcriptome data more readily available, and AFHI (n = 585), which comprises individuals of African American and Hispanic/Latino ancestries and more closely resembles the ancestries of individuals in PAGE. However, we also use ALL (n = 1,163), which includes both EUR and AFHI individuals, to see if increased sample size with increased population diversity improves our ability to discover and replicate TWAS associations.
Table 1.
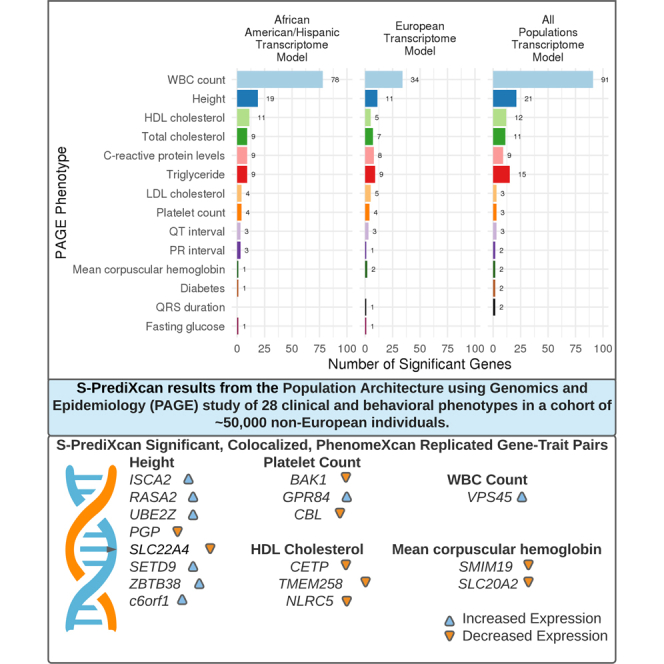
Population Architecture Using Genomics and Epidemiology (PAGE) Phenotypes Tested in TWAS and the Significant Gene Counts for Each Phenotype and Transcriptome Prediction Model
| Trait | Total N or N Cases/N Controls | Mean or % Cases | SD of Mean | TWAS with AFHI Count | TWAS with EUR Count | TWAS with all Count |
|---|---|---|---|---|---|---|
| Inflammatory traits | ||||||
| C-reactive protein (CRP) (mg/L) | 28,520 | 4.114 | 4.836 | 9 | 8 | 9 |
| White blood cell (WBC) count (109 cells/L) | 28,608 | 6.253 | 1.943 | 78 | 34 | 91 |
| Mean corpuscular hemoglobin concentration (MCHC) (g/dL) | 19,803 | 32.909 | 1.249 | 1 | 2 | 2 |
| Platelets (per mcL) | 29,328 | 246.783 | 64.273 | 4 | 4 | 3 |
| Lipid traits | ||||||
| HDL cholesterol (mg/dL)a | 33,063 | 50.738 | 15.372 | 11 | 5 | 12 |
| LDL cholesterol (mg/dL)a | 32,221 | 137.777 | 40.945 | 4 | 5 | 3 |
| Triglycerides (mg/dL)a | 33,096 | 137.830 | 92.125 | 9 | 9 | 15 |
| Total Cholesterol (mg/dL)a | 33,185 | 214.864 | 46.452 | 9 | 7 | 11 |
| Lifestyle traits | ||||||
| Cigarettes/day exclude nonsmokers | 15,862 | 12.507 | 9.088 | 0 | 0 | 0 |
| Coffee (cups/day) | 35,902 | 0.893 | 1.130 | 0 | 0 | 0 |
| Glycemic traits | ||||||
| HbA1c (mmol/mol)b | 11,178 | 36.823 | 4.520 | 0 | 0 | 0 |
| Fasting insulin (pmol/L)b | 21,551 | 10.233 | 7.979 | 0 | 0 | 0 |
| Fasting glucose (mmol/L)b | 23,911 | 5.050 | 0.633 | 1 | 1 | 0 |
| Type 2 diabetes (cases/controls) | 14,042/31,683 | 30.7% | 1 | 0 | 2 | |
| Electrocardiogram traits | ||||||
| QT interval (ms) | 17,348 | 410.678 | 30.580 | 3 | 3 | 3 |
| QRS interval (ms) | 17,046 | 89.023 | 9.596 | 0 | 1 | 2 |
| PR interval (ms) | 17,422 | 158.909 | 22.364 | 3 | 1 | 2 |
| Blood Pressure traits | ||||||
| Systolic blood pressure (mm Hg)a | 35,433 | 132.150 | 22.243 | 0 | 0 | 0 |
| Diastolic blood pressure (mm Hg)a | 35,433 | 80.681 | 13.827 | 0 | 0 | 0 |
| Hypertension (cases/controls) | 27,123/22,018 | 55.2% | 0 | 0 | 0 | |
| Anthropometric traits | ||||||
| WHR-femalesb | 24,838 | 0.855 | 0.082 | 0 | 0 | 0 |
| WHR-malesb | 9,066 | 0.952 | 0.066 | 0 | 0 | 0 |
| WHR | 33,904 | NA | NA | 0 | 0 | 0 |
| Height (cm) | 49,796 | 163.893 | 9.568 | 19 | 11 | 21 |
| BMI (kg/m2) | 49,335 | 29.333 | 6.285 | 0 | 0 | 0 |
| Kidney traits | ||||||
| Chronic kidney disease (cases/controls) | 4,154/41,573 | 10.0% | 0 | 0 | 0 | |
| End-stage renal disease (cases/controls) | 602/32,459 | 1.9% | 0 | 0 | 0 | |
| eGFR (mL/min)c | 27,900 | 90.548 | 21.880 | 0 | 0 | 0 |
Phenotype information and GWAS sample sizes were taken from Table S1 in Wojcik et al., 2019. Wojcik et al., 2019 had a combined Nmax = 49,839.
SD = standard deviation; WHR = waist-to-hip ratio; HbA1c = hemoglobin A1c; eGFR = estimated glomerular filtration rate; CRP = c-reactive protein; MCHC = mean corpuscular hemoglobin concentration; BMI = body mass index; AFHI = African American and Hispanic/Latino transcriptome prediction model; EUR = European transcriptome model; ALL = African American, Hispanic/Latino, and European transcriptome model; MESA = Multi-Ethnic Study of Atherosclerosis; PAGE = Population Architecture using Genomics and Epidemiology study.
Traits have been adjusted for medications by adding a constant.
Traits have been adjusted for BMI.
Estimated glomerular filtration rate (eGFR) was calculated using the CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) formula from Levey et al., 2009. See Wojcik et al., 2019 for details.
TWAS Identifies More Significant Genes when Using Larger and Population-Matched Gene Expression Prediction Models
We used S-PrediXcan with the summary statistics from the 28 PAGE GWAS and either the AFHI, EUR, or ALL MESA transcriptome prediction models to perform TWAS. We found 14 of the 28 different PAGE phenotypes returned significant gene-trait associations (Table 1). We identified 152 significant gene-trait pairs with the AFHI transcriptome prediction model, 91 significant gene-trait pairs with the EUR transcriptome prediction model, and 176 significant gene-trait pairs with the ALL transcriptome prediction model (Table S1, P < 0.05/n, where n is the number of genes tested for association with each trait). In total, we identified 206 unique genes and 240 unique gene-trait pairs. Of the 240 unique gene-trait pairs, we found 50 using all three MESA models, 53 using both AFHI and EUR MESA models, 63 using AFHI and ALL MESA models, 13 using EUR and ALL MESA models, and 57 overlapped with gene-trait pairs previously mapped as a nearby gene to SNPs discovered in the original PAGE GWAS (Table S1) (Wojcik et al., 2019). The Z-scores of the AFHI and EUR identified genes are highly correlated (R = 0.63), indicating that most genes have similar effects across population models and just miss reaching the significance threshold in one population or the other (Figure 1). This Z score correlation remains when all tested genes, not just those that reached significance with one population model, are compared (R = 0.69, Figure S1). If we are more conservative in our TWAS multiple testing adjustment and correct for all tests performed, not just tests within a trait, 95 gene-trait pairs remain significant with AFHI, 46 gene-trait pairs with EUR, and 121 gene-trait pairs with ALL (P < 1.1 × 10−7, Figure 2, Table S1).
Figure 1.

Z score Comparison of TWAS Significant Genes Identified by AFHI and EUR MESA Transcriptome Prediction Models in PAGE
Gene-trait pairs that were identified as significant (P < 0.05/n, n = the number of genes in the transcriptome model tested in S-PrediXcan) by either model are displayed. The Pearson correlation of displayed gene-trait pairs is shown in the upper left corner (R = 0.63). AFHI = African American and Hispanic/Latino transcriptome prediction model; EUR = European transcriptome prediction model; MESA = Multi-Ethnic Study of Atherosclerosis; PAGE = Population Architecture using Genomics and Epidemiology study.
Figure 2.

Manhattan Plot of the 14 of 28 PAGE Phenotypes Tested that Returned Significant TWAS Gene-Trait Pairs Using the AFHI, EUR, and ALL MESA Gene Expression Prediction Models
Each point represents the -log10(p) of a gene association test and gene chromosomal position colored by phenotype. Only significant gene-trait pairs are shown (P < 0.05/n, n = the number of genes in the transcriptome model tested in S-PrediXcan). The dotted line is at the more conservative significance threshold calculated using all tests (P < 1.1 × 10−7). 11 phenotypes have gene associations that meet this more stringent threshold. Using the AFHI, EUR, and ALL models, we identified 95, 46, and 121 significant gene-trait pairs, respectively, at this threshold. Gene-trait pairs with P < 1e-50 are displayed at P = 1e-50 for readability. AFHI = African American and Hispanic/Latino transcriptome prediction model; EUR = European transcriptome model; ALL = African American, Hispanic/Latino, and European transcriptome model; MCHC = mean corpuscular hemoglobin concentration; CRP levels = c-reactive protein levels; WBC count = white blood cell count; MESA = Multi-Ethnic Study of Atherosclerosis; PAGE = Population Architecture using Genomics and Epidemiology study.
Colocalization of TWAS Results Identifies SNPs Most Likely to Act through Gene Expression Regulation
Across all TWAS phenotypes, white blood cell (WBC) count had the highest number of significant genes for each transcriptome model. We identified 34 genes (91% on chromosome 1) significantly associated with WBC count using EUR models, 78 genes (96% on chromosome 1) using AFHI models, and 91 genes (99% on chromosome 1) using ALL models. Because linkage disequilibrium and gene co-regulation are potential confounders of TWAS results (Giambartolomei et al., 2014; Hormozdiari et al., 2016; Barbeira et al., 2018; Pividori et al., 2020; Gamazon et al., 2015; Wainberg et al., 2019), we further investigated whether the TWAS gene associations had colocalized signals with known eQTLs. Colocalization provides additional evidence that the SNPs in a given expression model are functioning via gene expression regulation to affect the associated trait (Giambartolomei et al., 2014; Hormozdiari et al., 2016; Barbeira et al., 2018; Pividori et al., 2020).
We applied COLOC (Giambartolomei et al., 2014) with the PAGE GWAS summary statistics and the AFHI, EUR, and ALL MESA eQTL data (Mogil et al., 2018). Only the SNPs that were included in the MESA model and the GWAS summary statistics were tested. This allows us to determine if eQTLs are shared between the gene expression prediction models and the GWAS results. In our S-PrediXcan analyses, we identified 152, 91, and 176 genome-wide significant gene-trait pairs using the AFHI, EUR, and ALL models, respectively. Of these gene-trait pairs, 32 AFHI gene-trait pairs, 20 EUR gene-trait pairs, and 37 ALL gene-trait pairs had a colocalization probability P4 > 0.5, suggesting the eQTL and GWAS signals are colocalized. Six of the gene-trait pairs were significant in all three (AFHI, EUR, and ALL) analyses. 13 gene-trait pairs were significant in only the AFHI and ALL analyses while another three gene-trait pairs were significant in the EUR and ALL analyses. 228 gene-trait pairs between AFHI, EUR, and ALL (70, 60, and 98 gene-trait pairs, respectively) were found to be independent (P3 > 0.5). However, COLOC could not confirm 50, 11, and 41 gene-trait pairs as either colocalized or independent signals (P3 < 0.5 and P4 < 0.5) in the AFHI, EUR, and ALL models, respectively. Whether these genes are contributing to their respective traits through gene expression regulation is unknown with current data and colocalization models.
More AFHI-Discovered Gene-Trait Pairs Replicate in PhenomeXcan Than EUR- or ALL-Discovered Gene-Trait Pairs
To determine if the gene associations we identified in PAGE replicated in TWAS studies of larger European populations, we used PhenomeXcan, a gene-trait association resource (Pividori et al., 2020). PhenomeXcan is a gene-based resource with the S-MultiXcan cross-tissue gene-trait association results from UK BioBank GWAS Summary Statistics, other accessible large-scale GWAS, and the Genotype-Tissue Expression Project (GTEx) version 8 models (Pividori et al., 2020; GTEx Consortium, 2020).
We tested the 62 unique colocalized gene-trait pairs for replication in the PhenomeXcan database, which includes results from larger European TWAS. We considered PhenomeXcan genes with P < 0.0008 (Bonferroni correction for 62 tests) and the same direction of effect with the same or similar trait as the discovery in PAGE to have replicated. Of the 32 AFHI colocalized discoveries, 11 (0.34) replicated in PhenomeXcan, of the 20 EUR discoveries, 5 (0.25) replicated in PhenomeXcan, and of the 37 ALL colocalized discoveries, 10 (0.27) replicated in PhenomeXcan with the same direction of effect (P < 0.0008 Table S2). Two of the PhenomeXcan replicated gene-trait pairs, BAK1 with platelet count and SLC22A4 with height, were significant in the AFHI, EUR, and ALL TWAS.
PhenomeXcan also reports the FASTENLOC calculated regional colocalization probabilities (RCPs) that are greater than 0.1. Given the conservative nature of colocalization approaches, this threshold limits reporting of false negatives (Pividori et al., 2020). When looking at the gene-trait pairs that replicated in PhenomeXcan, all gene-trait pairs had at least one study with an RCP >0.5, which provides strong evidence that these genes are colocalized and contributing to the trait through gene expression regulation (Table 2). These genes are ZBTB38, SLC22A4, SLC20A2, SMIM19, SETD9, CBL, and BAK1.
Table 2.
S-PrediXcan Significant Genes in PAGE with Colocalization Probability (P4) > 0.5 that Replicated in Independent Studies in PhenomeXcan
| Gene Name | Z Score | Effect Size | P | CHR | P3 | P4 | Model | Phenotype | Best PhenomeXcan P | RCP |
|---|---|---|---|---|---|---|---|---|---|---|
| CETP | −18 | −12 | 4.2 × 10−73 | 16 | 2.3 × 10−3 | 1 | AFHI | HDL cholesterol | 6.1 × 10−97 | NA |
| TMEM258 | −4.8 | −17 | 1.7 × 10−6 | 11 | 7.1 × 10−3 | 0.95 | AFHI | HDL cholesterol | 1.6 × 10−6 | NA |
| SETD9 | 4.7 | −9.7 | 2.3 × 10−6 | 5 | 0.19 | 0.80 | AFHI | Height | 9.6 × 10−17 | 0.57 |
| RASA2 | 4.5 | −7.7 | 5.7 × 10−6 | 3 | 6.5 × 10−2 | 0.92 | AFHI | Height | 2.1 × 10−105 | NA |
| UBE2Z | 5.4 | 9.4 | 2.7 × 10−8 | 17 | 0.23 | 0.77 | AFHI | Height | 4.5 × 10−48 | NA |
| ISCA2 | 4.8 | 0.09 | 1.3 × 10−6 | 14 | 0.03 | 0.97 | AFHI | Height | 5.8 × 10−25 | NA |
| SLC22A4 | −5.0 | −0.05 | 5.3 × 10−7 | 5 | 0.17 | 0.81 | AFHI | Height | 6.2 × 10−47 | NA |
| SMIM19 | −6.6 | 0.16 | 3.1 × 10−11 | 8 | 0.10 | 0.90 | AFHI | MCHC | 2.8 × 10−23 | 0.58 |
| BAK1 | −11 | 0.02 | 2.6E-30 | 6 | 4.4 × 10−3 | 1 | AFHI | Platelet count | 2.6 × 10−149 | 0.97 |
| CBL | −4.5 | −0.06 | 6.0 × 10−6 | 11 | 1.8 × 10−2 | 0.98 | AFHI | Platelet count | 6.9 × 10−60 | 0.81 |
| VPS45 | 9.7 | −0.05 | 3.9 × 10−22 | 1 | 2.2 × 10−2 | 0.95 | AFHI | WBC count | 5.8 × 10−6 | NA |
| ZBTB38 | 4.9 | −0.11 | 1.2 × 10−6 | 3 | 1.7 × 10−2 | 0.98 | EUR | Height | 9.5 × 10−150 | 0.58 |
| PGP | −4.9 | −2.6 | 8.0 × 10−7 | 16 | 6.7 × 10−3 | 0.99 | EUR | Height | 1.9 × 10−32 | NA |
| SLC22A4 | −4.4 | 0.08 | 9.8 × 10−6 | 5 | 4.8 × 10−2 | 0.95 | EUR | Height | 6.2 × 10−47 | NA |
| BAK1 | −12 | 0.08 | 2.8 × 10−32 | 6 | 2.5 × 10−3 | 1 | EUR | Platelet count | 2.6 × 10−149 | 0.97 |
| GPR84 | −5.7 | 0.11 | 1.4 × 10−6 | 12 | 3.3 × 10−3 | 1 | EUR | Platelet count | 3.9 × 10−47 | NA |
| BAK1 | −12 | −13 | 7.0 × 10−34 | 6 | 3.9 × 10−3 | 1 | ALL | Platelet count | 2.6 × 10−149 | 0.97 |
| c6orf1 | 7.5 | 0.74 | 6.7 × 10−14 | 6 | 0.21 | 0.54 | ALL | Height | 9.0 × 10−132 | NA |
| CETP | −20 | −7.7 | 4.2 × 10−73 | 16 | 2.3 × 10−3 | 1 | ALL | HDL cholesterol | 6.1 × 10−97 | NA |
| NLRC5 | −7.1 | −3.7 | 1.4 × 10−12 | 16 | 0.31 | 0.66 | ALL | HDL cholesterol | 2.0 × 10−65 | NA |
| PGP | −4.5 | −0.04 | 5.6 × 10−6 | 16 | 1.3 × 10−2 | 0.95 | ALL | Height | 1.9 × 10−32 | NA |
| SETD9 | 4.6 | 0.02 | 4.3 × 10−6 | 5 | 0.19 | 0.80 | ALL | Height | 9.6 × 10−17 | 0.57 |
| SLC20A2 | −4.5 | −0.25 | 7.9 × 10−6 | 8 | 0.32 | 0.68 | ALL | MCHC | 7.3 × 10−21 | 0.51 |
| SLC22A4 | −4.7 | −0.05 | 2.4 × 10−6 | 5 | 0.10 | 0.89 | ALL | Height | 6.2 × 10−47 | NA |
| VPS45 | 8.8 | 0.08 | 1.2 × 10−18 | 1 | 0.27 | 0.69 | ALL | WBC count | 5.8 × 10−6 | NA |
| ZBTB38 | 6.7 | 0.18 | 2.6 × 10−11 | 3 | 8.3 × 10−3 | 0.99 | ALL | Height | 9.5 × 10−150 | 0.58 |
Details of the studies used in PhenomeXcan are in Table S2.
P3 = COLOC probability eQTL and GWAS signals are independent; P4 = COLOC probability eQTL and GWAS signals are colocalized; AFHI = African American and Hispanic/Latino transcriptome prediction model; EUR = European transcriptome model; ALL = African American, Hispanic/Latino, and European transcriptome model; MESA = Multi-Ethnic Study of Atherosclerosis; PAGE = Population Architecture using Genomics and Epidemiology study; RCP = PhenomeXcan regional colocalization probability.
One gene that was identified as significantly associated with mean corpuscular hemoglobin concentration (MCHC) in both AFHI and EUR at the stringent threshold of 1.1 × 10−7 was SMIM19. In the PAGE GWAS, SNPs near SMIM19 were found to be associated with MCHC (Wojcik et al., 2019). In our analysis, SMIM19 was only found to have colocalized GWAS and eQTL signals with AFHI eQTLs (P4 = 0.90), but not with EUR (P4 = 0.047) or ALL (P4 = 0.052) eQTLs (Figure 3, Table S1). SMIM19 is also significantly associated with MCHC (P = 2.81 × 10−23, RCP = 0.578) in PhenomeXcan with GWAS summary statistics from the UKBioBank. A gene located next to SMIM19 on chromosome 8, SLC20A2, associated with MCHC and had colocalized signal with the ALL MESA eQTLs (P4 = 0.68). SLC20A2 is also significantly associated with MCHC (P = 7.28 × 10−21, RCP = 0.507) in PhenomeXcan with GWAS summary statistics from the UK BioBank. While both genes may be involved in MCHC, in our study, SMIM19 has stronger evidence of acting through gene expression regulation to affect MCHC than SLC20A2 as indicated by higher P4 in PAGE using AFHI, higher cross-validated prediction performance in all populations, and higher RCP in PhenomeXcan (Tables S1 and S2).
Figure 3.
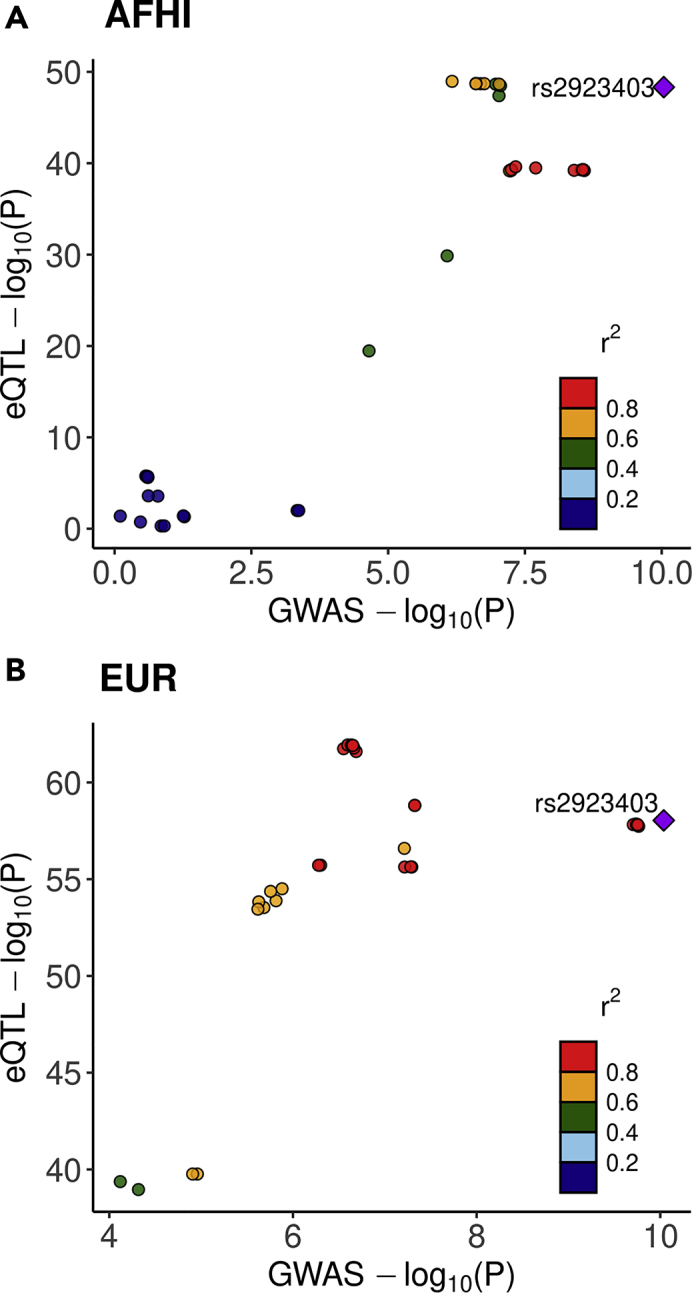
SMIM19 GWAS and eQTL Signals are Colocalized in AFHI, but not EUR
LocusCompare (Liu et al., 2019) plots for mean corpuscular hemoglobin concentration (MCHC) PAGE GWAS p values compared to (A) AFHI MESA eQTL p values and (B) EUR MESA eQTL p values of SNPs in the SMIM19 prediction models. When most points are located on the diagonal, it indicates the GWAS and eQTL signals are likely colocalized. The lead SNP in the AFHI eQTL and PAGE GWAS, rs2923403, is located among the top signals and in the upper right corner, supporting the COLOC evidence for colocalization AFHI (P4 = 0.90). When using EUR eQTL data in COLOC, the GWAS and eQTL signals did not colocalize (EUR P4 = 0.047). Points are colored according to the pairwise LD r2 with rs2923403 in (A) AMR and (B) EUR 1000 Genomes populations.
Of the 17 unique gene-trait pairs that replicated in PhenomeXcan, 5 of these gene-trait pairs do not appear in the GWAS Catalog and thus may represent new biology discovered through TWAS. These include ISCA2, SETD9, and SLC22A4, associated with height; VPS45 associated with WBC count; and GPR84 associated with platelet count. ISCA2, SETD9, SLC22A4, and VPS45 were significant in AFHI S-PrediXcan while only SLC22A4 and GPR84 were significant in EUR S-PrediXcan. SETD9, SLC22A4, and VPS45 were significant in ALL S-PrediXcan.
The other 12 gene-trait pairs that replicated in PhenomeXcan were found significant in at least one other GWAS of the same or similar phenotype. In the original PAGE GWAS, BAK1 in relation to platelet count, CETP in relation to HDL cholesterol, c6orf1 in relation to height, ZBTB38 in relation to height, and SMIM19 in relation to MCHC were all mapped as genes nearest to the significantly associated SNP (Table S3).
Discussion
We applied S-PrediXcan to GWAS results of 28 traits from the PAGE study and found a higher proportion of genes with colocalized GWAS and eQTL signals that replicated in PhenomeXcan using the AFHI transcriptome models than with using EUR or ALL models. This suggests that through using population-matched gene expression prediction models, we find more significant gene-trait pairs that replicate in larger, independent studies. We found that S-PrediXcan Z-scores are consistent between AFHI and EUR transcriptome models (R = 0.63), even if a particular gene was only found significant using one or the other population (Figure 1). As has been shown in SNP effect size comparisons (Stranger et al., 2012; Marigorta and Navarro, 2013; Wojcik et al., 2019; Shang et al., 2020), this strong gene effect size correlation indicates the underlying biological pathways affecting each complex trait do not differ between populations. Instead, our power to detect the associations differs and subsequently, predictive power between populations is reduced (Mogil et al., 2018; Martin et al., 2019; Keys et al., 2020). We have more power to detect associations in PAGE that replicate in independent cohorts using the AFHI transcriptome prediction model because the minor allele frequency and LD structure of AFHI more closely resembles that of PAGE than does the structure of either EUR or ALL (Mogil et al., 2018; Wojcik et al., 2019).
Four gene-trait pairs that replicated in PhenomeXcan mapped as the nearest gene to an associated SNP locus in the original PAGE study (Wojcik et al., 2019). These include BAK1, where here we found increased predicted BAK1 associated with decreased platelet count using all three transcriptome models. We identified CETP using the ALL and AFHI models, SMIM19 using the AFHI transcriptome model, and ZBTB38 using the EUR and ALL transcriptome models. Increased predicted CETP associated with decreased HDL cholesterol levels, supporting previous findings (Barter et al., 2003; Thompson et al., 2003; de Grooth et al., 2004; Kosmas et al., 2016; Andaleon et al., 2019). Increased predicted SMIM19 expression associated with decreased MCHC. In addition to associating in the original PAGE GWAS, SNPs near SMIM19 associated with MCHC in two independent GWAS (Hodonsky et al., 2017; Astle et al., 2016). Meanwhile, we found increased predicted ZBTB38 expression associated with increased height. This association is supported by 17 other independent GWAS (Gudbjartsson et al., 2008; Lettre et al., 2008; Sanna et al., 2008; Weedon et al., 2008; Cho et al., 2009; Soranzo et al., 2009; Kamatani et al., 2010; Kim et al., 2010; Lango Allen et al., 2010; N'Diaye et al., 2011; Bernt et al., 2013; Wood et al., 2014; He et al., 2015; Nagy et al., 2017; Tachmazidou et al., 2017; Kichaev, 2018; Akiyama et al., 2019; Wojcik et al., 2019).
Although not identified in the original PAGE GWAS (Wojcik et al., 2019), SNPs near PGP associated with height in European and Japanese GWASs (Tachmazidou et al., 2017; Akiyama et al., 2019). We found increased PGP predicted expression associated with decreased height, thus providing more evidence PGP affects height through gene expression regulation. Similar to PGP, SLC20A2 was not identified in the original PAGE GWAS but replicated in PhenomeXcan. We found SNPs near SLC20A2 associated with MCHC in independent GWAS (Kanai et al., 2018), and SNPs near SLC20A2 were also associated with mean corpuscular hemoglobin volume, a related phenotype to MCHC, in three other independent GWAS (Astle et al., 2016; Kanai et al., 2018; Chen et al., 2020). Here, we found increased SLC20A2 predicted expression associated with decreased MCHC. More work is needed to disentangle whether SMIM19 or SLC20A2, which are located next to each other on chromosome 8, is causal for MCHC. In our study, SMIM19 has stronger evidence of acting through gene expression regulation to affect MCHC, but both genes may be involved.
We discovered several gene-trait associations that replicated in PhenomeXcan but were not previously included in the GWAS Catalog and thus may represent new biological mechanisms underlying the traits. These include ISCA2, SETD9, SLC22A4, VPS45, and GPR84. Neither ISCA2 nor SETD9 were previously identified in GWAS as associated with height; we found increased expression of these genes associated with increased height. SLC22A4 was not previously identified as associated with height despite our findings demonstrating increased SLC22A4 expression is associated with decreased height. Similarly, no previous GWAS have linked increased GPR84 expression to increased platelet count. Mutations in VPS45 are known to cause neutrophil defect syndrome (Vilboux et al., 2013; Stepensky et al., 2013), and we found significant associations between predicted VPS45 expression and WBC count.
There are significantly more genes with no evidence of colocalization nor evidence of independence when analyzing the AFHI S-PrediXcan output. These 50 genes could be functioning through gene expression regulation. Better methods, specifically colocalization methods for recently admixed populations, are needed to determine whether these genes are likely functional.
In summary, we found more gene-trait pairs discovered in PAGE with AFHI transcriptome models replicated in PhenomeXcan (11/32, 34%) compared to the gene-trait pairs discovered with EUR models (5/20, 25%) and, to a smaller extent, ALL models (10/37, 27%). Since the largest populations in PAGE are of Hispanic/Latino and African American ancestries, TWAS with population-matched transcriptome models, i.e. AFHI rather than EUR, have more power for discovery and discovered genes are more likely to replicate. Transcriptome prediction models trained in a cohort with similar ancestries to the original GWAS should be used and thus more transcriptome studies in diverse populations are needed.
Limitations of the Study
Here we identified gene-trait pairs using MESA transcriptome models in conjunction with the PAGE GWAS summary statistics in a TWAS analysis. The MESA models were trained using monocyte transcriptomes, and other tissues are likely more relevant to the phenotypes studied. Better complex trait methods for handling linkage disequilibrium and local ancestry in admixed populations like PAGE and MESA are needed. While the GWAS summary statistics from the combined PAGE populations are currently available in the GWAS Catalog, making within population summary statistics publicly available in future studies will encourage meta-analyses and promote development of more sophisticated models to help narrow the diversity gap in genomics (Peterson et al., 2019; Ben-Eghan et al., 2020). More genomes and transcriptomes in more tissues in admixed populations are needed to enhance model development and to better understand the genetics of complex traits in all populations.
Resource Availability
Lead Contact
Further information and questions should be directed to and will be fulfilled by the Lead Contact, Heather Wheeler (hwheeler1@luc.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All scripts used for analyses are available at https://github.com/WheelerLab/MESA_expression_prediction. TWAS summary statistics, colocalization results, and MESA models from this study can be found at Mendeley Data: https://doi.org/10.17632/p8cgvyz4sz. PAGE GWAS summary statistics are available in the GWAS Catalog at https://www.ebi.ac.uk/gwas/publications/31217584.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Kathleen Delany for aiding in the early colocalization pipeline development and Ryan Schubert for providing manuscript feedback. This work is supported by the NIH National Human Genome Research Institute Academic Research Enhancement Award R15 HG009569 (H.E.W.), the Loyola Mulcahy Scholarship (E.G., I.G.), and the Loyola MS Bioinformatics Fellowship (E.G.).
Author Contributions
E.G. and H.E.W. conceived and designed the experiments. E.G. performed S-PrediXcan, colocalization analyses, and GWAS Catalog replication searches. I.G. performed the PhenomeXcan search and performed PubMed searches to identify replication studies. E.G. and H.E.W. wrote the paper. All authors read, provided feedback, and approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101850.
Supplemental Information
References
- Akiyama M., Ishigaki K., Sakaue S., Momozawa Y., Horikoshi M., Hirata M., Matsuda K., Ikegawa S., Takahashi A., Kanai M. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun. 2019;10:4393. doi: 10.1038/s41467-019-12276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andaleon A., Mogil L.S., Wheeler H.E. Genetically regulated gene expression underlies lipid traits in Hispanic cohorts. PLoS One. 2019;14:e0220827. doi: 10.1371/journal.pone.0220827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle W.J., Elding H., Jiang T., Allen D., Ruklisa D., Mann A.L., Mead D., Bouman H., Riveros-Mckay F., Kostadima M.A. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167 doi: 10.1016/j.cell.2016.10.042. 1415–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira A.N., Dickinson S.P., Bonazzola R., Zheng J., Wheeler H.E., Torres J.M., Torstenson E.S., Shah K.P., Garcia T., Edwards T.L. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeira A.N., Pividori M., Zheng J., Wheeler H.E., Nicolae D.L., Im H.K. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019;15:e1007889. doi: 10.1371/journal.pgen.1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barter P.J., Brewer H.B., Chapman M.J., Hennekens C.H., Rader D.J., Tall A.R. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003;23:160–167. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- Ben-Eghan C., Sun R., Hleap J.S., Diaz-Papkovich A., Munter H.M., Grant A.V., Dupras C., Gravel S. ‘Don’t ignore genetic data from minority populations’. Nature. 2020;585:184–186. doi: 10.1038/d41586-020-02547-3. [DOI] [PubMed] [Google Scholar]
- Bernt M., Braband A., Schierwater B., Stadler P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenet. Evol. 2013;69:328–338. doi: 10.1016/j.ympev.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Bild D.E., Bluemke D.A., Burke G.L., Detrano R., Diez Roux A.V., Folsom A.R., Greenland P., Jacob D.R., Kronmal R., Liu K. Multi-ethnic study of atherosclerosis: objectives and design. Am. J. Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.H., Raffield L.M., Mousas A., Sakaue S., Huffman J.E., Moscati A., Trivedi B., Jiang T., Akbari P., Vuckovic D. Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell. 2020;182 doi: 10.1016/j.cell.2020.06.045. 1198–e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y.S., Go M.J., Kim Y.J., Heo J.Y., Oh J.H., Ban H.J., Yoon D., Lee M.H., Kim D.J., Park M. A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat. Genet. 2009;41:527–534. doi: 10.1038/ng.357. [DOI] [PubMed] [Google Scholar]
- Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., Nicolae D.L. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Grooth G.J., Klerkx A.H., Stroes E.S., Stalenhoef A.F., Kastelein J.J., Kuivenhoven J.A. A review of CETP and its relation to atherosclerosis. J. Lipid Res. 2004;45:1967–1974. doi: 10.1194/jlr.R400007-JLR200. [DOI] [PubMed] [Google Scholar]
- GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson D.F., Walters G.B., Thorleifsson G., Stefansson H., Halldorsson B.V., Zusmanovich P., Sulem P., Thorlacius S., Gylfason A., Steinberg S. Many sequence variants affecting diversity of adult human height. Nat. Genet. 2008;40:609–615. doi: 10.1038/ng.122. [DOI] [PubMed] [Google Scholar]
- Gusev A., Ko A., Shi H., Bhatia G., Chung W., Penninx B.W., Jansen R., de Geus E.J., Boomsma D.I., Wright F.A. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016;48:245–252. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M., Xu M., Zhang B., Liang J., Chen P., Lee J.Y., Johnson T.A., Li H., Yang X. Meta-analysis of genome-wide association studies of adult height in East Asians identifies 17 novel loci. Hum. Mol. Genet. 2015;24:1791–1800. doi: 10.1093/hmg/ddu583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodonsky C.J., Jain D., Schick U.M., Morrison J.V., Brown L., McHugh C.P., Schurmann C., Chen D.D., Liu Y.M., Auer P.L. Genome-wide association study of red blood cell traits in Hispanics/Latinos: the Hispanic Community Health Study/Study of Latinos. PLoS Genet. 2017;13:e1006760. doi: 10.1371/journal.pgen.1006760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hormozdiari F., van de Bunt M., Segrè A.V., Li X., Joo J.W.J., Bilow M., Sul J.H., Sankararaman S., Pasaniuc B., Eskin E. Colocalization of GWAS and eQTL signals detects target genes. Am. J. Hum. Genet. 2016;99:1245–1260. doi: 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamatani Y., Matsuda K., Okada Y., Kubo M., Hosono N., Daigo Y., Nakamura Y., Kamatani N. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat. Genet. 2010;42:210–215. doi: 10.1038/ng.531. [DOI] [PubMed] [Google Scholar]
- Kanai M., Akiyama M., Takahashi A., Matoba N., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- Keys K.L., Mak A.C.Y., White M.J., Eckalbar W.L., Dahl A.W., Mefford J., Mikhaylova A.V., Contreras M.G., Elhawary J.R., Eng C. On the cross-population generalizability of gene expression prediction models. PLoS Genet. 2020;16:e1008927. doi: 10.1371/journal.pgen.1008927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichaev G. Integrative statistical methods to understand the genetic basis of complex trait. UCLA. 2018. https://escholarship.org/uc/item/3w07g23z
- Kim J.J., Lee H.I., Park T., Kim K., Lee J.E., Cho N.H., Shin C., Cho Y.S., Lee J.Y., Han B.G. Identification of 15 loci influencing height in a Korean population. J. Hum. Genet. 2010;55:27–31. doi: 10.1038/jhg.2009.116. [DOI] [PubMed] [Google Scholar]
- Kosmas C.E., DeJesus E., Rosario D., Vittorio T.J. CETP Inhibition: Past Failures and Future Hopes. Clin Med Insights Cardiol. 2016;10:37–42. doi: 10.4137/CMC.S32667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lango Allen H. Hundreds of variants influence human height and cluster within genomic loci and biological pathways. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G., Jackson A.U., Gieger C., Schumacher F.R., Berndt S.I., Sanna S., Eyheramendy S., Voight B.F., Butler J.L., Guiducci C. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey A.S., Stevens L.A., Schmid C.H., Zhang Y.L., Castro A.F., Feldman H.I., Kusek J.W., Eggers P., Van Lente F., Greene T., Coresh J. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Gloudemans M.J., Rao A.S., Ingelsson E., Montgomery S.B. Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet. 2019;51:768–769. doi: 10.1038/s41588-019-0404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Ding J., Reynolds L.M., Lohman K., Register T.C., De La Fuente A., Howard T.D., Hawkins G.A., Cui W., Morris J. Methylomics of gene expression in human monocytes. Hum. Mol. Genet. 2013;22:5065–5074. doi: 10.1093/hmg/ddt356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigorta U.M., Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9:e1003566. doi: 10.1371/journal.pgen.1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil L.S., Andaleon A., Badalamenti A., Dickinson S.P., Guo X., Rotter J.I., Johnson W.C., Im H.K., Liu Y., Wheeler H.E. Genetic architecture of gene expression traits across diverse populations. PLoS Genet. 2018;14:e1007586. doi: 10.1371/journal.pgen.1007586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 2018;19:21. doi: 10.1186/s13059-018-1396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy R., Boutin T.S., Marten J., Huffman J.E., Kerr S.M., Campbell A., Evenden L., Gibson J., Amador C., Howard D.M. Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017;9:23. doi: 10.1186/s13073-017-0414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Diaye A. Identification, replication, and fine-mapping of Loci associated with adult height in individuals of african ancestry. PLoS Genet. 2011;7:e1002298. doi: 10.1371/journal.pgen.1002298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson R.E., Kuchenbaecker K., Walters R.K., Chen C.Y., Popejoy A.B., Periyasamy S., Lam M., Iyegbe C., Strawbridge R.J., Brick L. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179:589–603. doi: 10.1016/j.cell.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pividori M. PhenomeXcan: mapping the genome to the phenome through the transcriptome. Sci. Adv. 2020 doi: 10.1126/sciadv.aba2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna S., Jackson A.U., Nagaraja R., Willer C.J., Chen W.M., Bonnycastle L.L., Shen H., Timpson N., Lettre G., Usala G. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat. Genet. 2008;40:198–203. doi: 10.1038/ng.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L., Smith J.A., Zhao W., Kho M., Turner S.T., Mosley T.H., Kardia S.L.R., Zhou X. Genetic architecture of gene expression in European and African Americans: an eQTL mapping study in GENOA. Am. J. Hum. Genet. 2020;106:496–512. doi: 10.1016/j.ajhg.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soranzo N., Rivadeneira F., Chinappen-Horsley U., Malkina I., Richards J.B., Hammond N., Stolk L., Nica A., Inouye M., Hofman A., Stephens J. Meta-analysis of genome-wide scans for human adult stature identifies novel Loci and associations with measures of skeletal frame size. PLoS Genet. 2009;5:e1000445. doi: 10.1371/journal.pgen.1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepensky P., Saada A., Cowan M., Tabib A., Fischer U., Berkun Y., Saleh H., Simanovsky N., Kogot-Levin A., Weintraub M. The Thr224Asn mutation in the VPS45 gene is associated with the congenital neutropenia and primary myelofibrosis of infancy. Blood. 2013;121:5078–5087. doi: 10.1182/blood-2012-12-475566. [DOI] [PubMed] [Google Scholar]
- Stranger B.E., Montgomery S.B., Dimas A.S., Parts L., Stegle O., Ingle C.E., Sekowska M., Smith G.D., Evans D., Gutierrez-Arcelus M. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachmazidou I., Süveges D., Min J.L., Ritchie G.R.S., Steinberg J., Walter K., Iotchkova V., Schwartzentruber J., Huang J., Memari Y. Whole-genome sequencing coupled to imputation discovers genetic signals for anthropometric traits. Am. J. Hum. Genet. 2017;100:865–884. doi: 10.1016/j.ajhg.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.F., Lira M.E., Durham L.K., Clark R.W., Bamberger M.J., Milos P.M. Polymorphisms in the CETP gene and association with CETP mass and HDL levels. Atherosclerosis. 2003;167:195–204. doi: 10.1016/s0021-9150(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Vilboux T., Lev A., Malicdan M.C., Simon A.J., Järvinen P., Racek T., Puchalka J., Sood R., Carrington B., Bishop K. A congenital neutrophil defect syndrome associated with mutations in VPS45. N. Engl. J. Med. 2013;369:54–65. doi: 10.1056/NEJMoa1301296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg M., Sinnott-Armstrong N., Mancuso N., Barbeira A.N., Knowles D.A., Golan D., Ermel R., Ruusalepp A., Quertermous T., Hao K. Opportunities and challenges for transcriptome-wide association studies. Nat. Genet. 2019;51:592–599. doi: 10.1038/s41588-019-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon M.N., Lango H., Lindgren C.M., Wallace C., Evans D.M., Mangino M., Freathy R.M., Perry J.R., Stevens S., Hall A.S. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik G.L., Graff M., Nishimura K.K., Tao R., Haessler J., Gignoux C.R., Highland H.M., Patel Y.M., Sorokin E.P., Avery C.L. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A.R., Esko T., Yang J., Vedantam S., Pers T.H., Gustafsson S., Chu A.Y., Estrada K., Luan J., Kutalik Z. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 2014;46:1173–1186. doi: 10.1038/ng.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scripts used for analyses are available at https://github.com/WheelerLab/MESA_expression_prediction. TWAS summary statistics, colocalization results, and MESA models from this study can be found at Mendeley Data: https://doi.org/10.17632/p8cgvyz4sz. PAGE GWAS summary statistics are available in the GWAS Catalog at https://www.ebi.ac.uk/gwas/publications/31217584.