

Abstract
Purpose of review
Degenerative ataxias are rare and currently untreatable movement disorders, primarily characterized by neurodegeneration in the cerebellum and brainstem. We highlight MRI studies with the most potential for utility in pending ataxia trials, and underscore advances in disease characterization and diagnostics in the field.
Recent findings
With availability of advanced MRI acquisition methods and specialized software dedicated to the analysis of MRI of the cerebellum, patterns of cerebellar atrophy in different degenerative ataxias are increasingly well-defined. The field further embraced rigorous multi-modal investigations to study network level microstructural and functional brain changes, and their neurochemical correlates. MRI and MRS were shown to be more sensitive to disease progression than clinical scales and to detect abnormalities in premanifest mutation-carriers.
Summary
MR techniques are increasingly well-placed for characterizing the expression and progression of degenerative ataxias. The most impactful work has arguably come through multi-institutional studies that monitor relatively large cohorts, multi-modal investigations that assess the sensitivity of different measures and their inter-relationships, and novel imaging approaches that are targeted to known pathophysiology (e.g., iron and spinal imaging in Friedreich ataxia). These multi-modal, multi-institutional studies are paving the way to clinical trial readiness and enhanced understanding of pathology in degenerative ataxias.
Keywords: Friedreich ataxia, spinocerebellar ataxia, cerebellar multiple system atrophy, magnetic resonance imaging, cerebellum
INTRODUCTION
Ataxia refers to deficits in the coordination of movement and balance. While ataxia can present as a symptom of other neurological diseases and occur due to acquired causes (e.g. alcoholic cerebellar degeneration, vitamin deficiency), here we focus on hereditary and sporadic degenerative ataxias that are characterized by neurodegeneration in the cerebellum and its afferent and efferent connections, and frequently also in other brain regions [1] (Table 1). Conventional MR imaging is part of the diagnostic work-up in ataxias to confirm cerebellar atrophy (Figure 1). In addition, quantitative MR technologies have been used to evaluate structure, connectivity, function and biochemistry in ataxias for over two decades [2]. Rigorous quantitative MRI studies were in particular facilitated by the discovery of genetic mutations for many degenerative ataxias, which allowed studies of genetically defined cohorts. More recently, with exciting developments toward viable disease modifying therapies [3], a critical need for non-invasive, validated biomarkers of cerebral and cerebellar pathology has emerged to facilitate upcoming clinical trials. To present a meaningful picture of the recent progress in cerebellar ataxia imaging, we will highlight the important contributions from the last three years. Progress in this field has been particularly facilitated by the wider availability of high field MR scanners (3 tesla and above), development and dissemination of advanced MRI and MR spectroscopy (MRS) acquisition methods [4,5], and development of specialized software and templates dedicated to the analysis of MR images from the human cerebellum and brainstem [6,7].
Table 1.
Genetic, clinical, and imaging features in degenerative ataxias
| Disease | Gene/Protein (locus) | Typical Age at Onset | Key-symptoms in addition to Cerebellar Ataxia | Morphometric Changes (MRI) | Neurochemistry (MRS), Function and Connectivity (fMRI, DWI) |
|---|---|---|---|---|---|
| AUTOSOMAL RECESSIVE ATAXIAS | |||||
| FRDA | FXN/frataxin (9q13) | Adolescence (around puberty) | Neuromuscular deformities, cardiac hypertrophy, abnormal eye movements | Spinal cord, brainstem, cerebellum; thalamus, and cortical motor areas (later stages) | ↓tNAA*1(/tCr)*1; ↑tCr*1; ↑mI*1; ↓cerebro-cerebellar and ↑cerebro-cerebral connectivity; cerebral compensation/ cerebellar dysfunction |
| AT | AT-mutated (ATM)/ATM protein (11q22–23) | Infancy - early childhood (2–4 years) | Oculomotor apraxia, cutaneous telangiectasias, dystonia chorea, immune-deficiency, malignancy | Cerebellum | ↓tNAA/tCho*1; ↓tCho/tCr*1 |
| AOA (1/2) | APTX/aprataxin (1) SETX/senataxin (2) | Early childhood (1), later childhood (2) | Oculomotor apraxia, movement disorders, pyramidal signs, intellectual disability | Cerebellum | (2): ↓tNAA*1; ↑mI*1,*2 |
| ARSACS | SACS/sacsin (13q12.12) | Infancy (12 – 18 month) | Spasticity, peripheral neuropathy | Cerebellum, parietal lobe (bilateral); linear pontine hypointensities | NA |
| AUTOSOMAL DOMINANT ATAXIAS | |||||
| SCA1 | ATXN1/ataxin-1 (6p22.3) | Early to mid-adulthood | Hypermetric saccades, pyramidal signs | Brainstem, cerebellum (preclinical), and basal ganglia | ↓tNAA*1,*2(/tCr)*1; ↓tCho/tCr*1; ↑tCr*1; ↑mI*2 |
| SCA2 | ATXN2/ataxin-2 (12q24.12) | Early child- to late adulthood | Slow saccades, peripheral neuropathy, motor neuron signs, autonomic dysfunction, cognitive impairment | Pons; cerebellum and brainstem (preclinical) | Altered inter-nodal cerebellar-cerebrum connectivity; ↓tNAA*1,*2(/tCr)*1; ↓tCho*2(/tCr)*1; ↑tCho*1; ↑mI*1,*2(/tCr)*1; ↑tCr*1 |
| SCA3 (Machado– Joseph disease) | ATXN3/ataxin-3 (14q32.12) | Childhood to mid-adulthood | Dystonia, sensory deficits, parkinsonism | Brainstem, cerebellum, basal ganglia; spinal cord, midbrain, substantia nigra (preclinical); cerebrum (later stages) | Microstructural abnormalities in cerebellar and cerebral peduncles (premanifest); ↓tNAA*1,*2(/tCr)*1; ↓tCho/tCr*1; ↑tCr*1; ↑mI*2 |
| SCA6 | CACNA1A (19p13.2) | Early to late adulthood | Almost purely ataxic | Cerebellum | ↓RS FC in attention network; impaired functional activity in SC and SMA; ↓tNAA*1(/tCr)*1; ↑mI*1 |
| SCA7 | ATNX7/ataxin-7 (3p14.1) | Infancy to adulthood | Visual loss, mitochondrial dysfunction | Cerebellum, brainstem | ↓tNAA*1,*2(/tCr)*1; ↓Pi(/PCr) (31P-MRS) in visual cortex during visual task |
| X-LINKED ATAXIAS | |||||
| FXTAS | FMR1 | Late adulthood (> 50 years) | Intention tremor, dementia, and parkinsonism | Midbrain, brainstem, and cerebellum | NA |
| SPORADIC DEGENERATIVE ATAXIAS | |||||
| MSA-C | - | Adulthood | Autonomic dysfunction, dysarthria, oculomotor dysfunction, cognitive impairment | Cerebellum (GM+WM), brainstem (GM+WM), pons; “hot cross bun” sign, middle cerebellar peduncle hyperintensity, putaminal hypointensity, and the hyperintense putaminal rim sign | ↓tNAA*1,*2(/tCr)*1,*2; ↑mI*1,*2; ↑tCr*1; microstructural white matter involvement (DTI); ↓cerebellar fiber density, and impairment of frontal and occipital white matter connectivity (DTI); ↓connectivity of motor networks |
| SAOA | - | Adulthood | sporadic adult progressive cerebellar ataxia of unknown etiology | Cerebellum (GM+WM), brainstem (GM) | Abnormal intra-cerebellar FC |
FRDA = Friedreich Ataxia; AT = Ataxia Telangiectasia; AOA = Ataxia with Oculomotor Apraxia; ARSACS = Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay; SCA = Spinocerebellar Ataxia; FXTAS = Fragile X-Associated Tremor/Ataxia Syndrome; MSA-C = Cerebellar Multiple System Atrophy; SAOA = Sporadic Adult Onset Ataxia; DTI = diffusion tensor imaging; tNAA = total N-acetylaspartate; tCr = total Creatine (creatine + phosphocreatine); tCho = total Choline; mI = myo-Inositol; *1 = in cerebellum (vermis and/or cerebellar hemispheres); *2 = in brainstem (pons); ↓ reduced compared to controls; ↑ increased compared to controls; RS = resting-state; FC = functional connectivity; SC = sensorimotor cortex; SMA = supplementary motor area; Pi/PCr = ratio of inorganic phosphate to phosphocreatine; 31P-MRS = 31-phosphorus magnetic resonance spectroscopy. The listed findings are primarily based on review articles and meta-analyses; therefore, mostly metabolite ratios are displayed for MRS findings, and the respective metabolic reference is indicated in brackets, i.e. (/tCr). The respective “numerator” simultaneously represents the metabolite concentration from individual findings, i.e. tNAA in tNAA(/tCr). In those cases where individual metabolite concentrations are listed, only few findings are available, and meta-analyses are missing (i.e. AOA2) or findings are mixed (i.e. ↑mI in SCA6).
1. Typical MRI findings in the cerebellum and brainstem in degenerative ataxias.
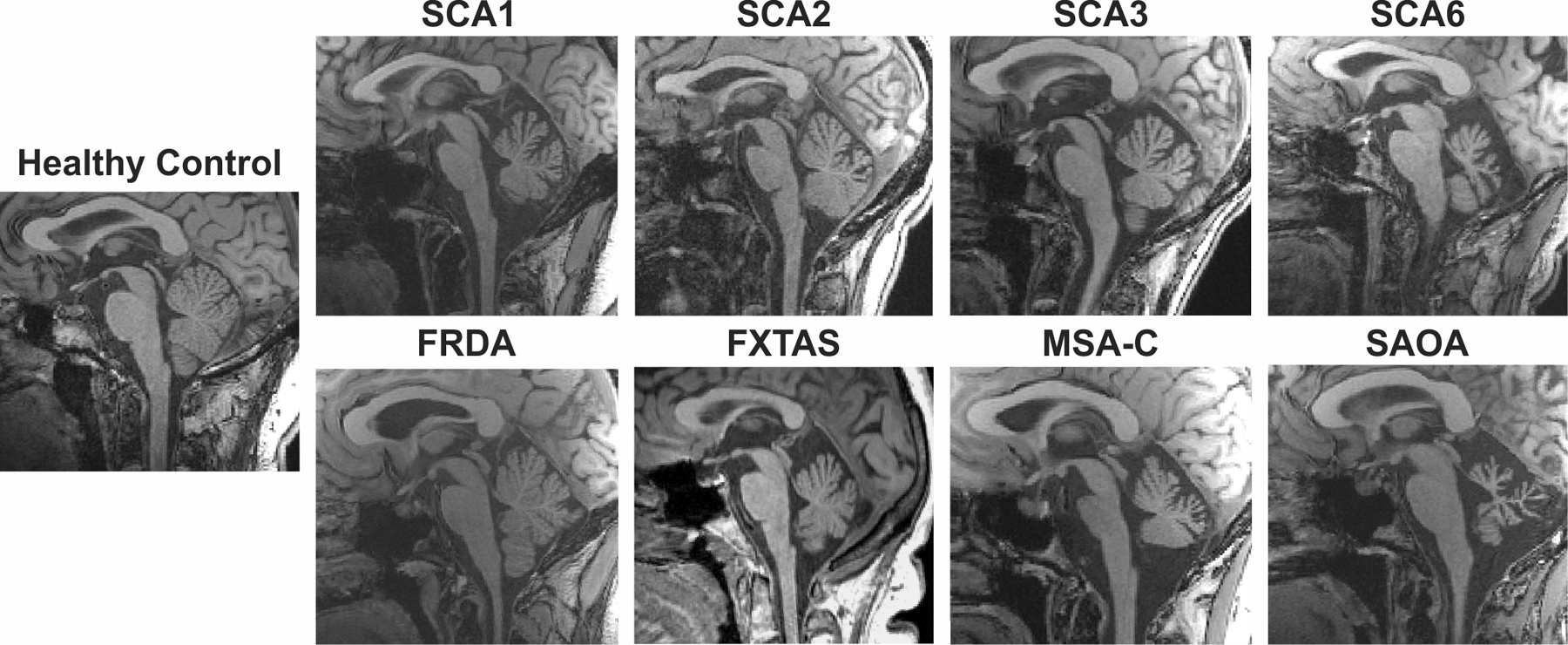
Varying degrees of cerebellar and pontine atrophy are shown on T1-weighted mid-sagittal images. SCA: Spinocerebellar ataxia; FRDA: Friedreich ataxia; FXTAS: fragile X-associated tremor/ataxia syndrome; MSA-C: Cerebellar multiple system atrophy; SAOA: Sporadic adult onset ataxia.
AUTOSOMAL RECESSIVE ATAXIAS
Quantitative neuroimaging research in autosomal recessive ataxias has largely focused on Friedreich ataxia (FRDA), the most common ataxia (Table 1). Rarer disorders have most often been the subject of qualitative case reports, although several recent case-control studies are available. Overall, recent MRI studies have not only brought better delineation of atrophy patterns in autosomal recessive ataxias, but they have also increasingly moved beyond a myopic focus on cerebellar macrostructure. Studies characterizing network-level microstructural and functional brain changes, spinal morphology, as well as longitudinal studies of pathophysiology (e.g. iron accumulation) have been particularly impactful in understanding the evolution of these diseases. Efforts to define disease staging, to disambiguate developmental versus degenerative brain changes, and to uncover factors associated with inter-individual variability (i.e., age of onset) have also provided increasingly nuanced disease descriptions.
Friedreich Ataxia: Brain Morphometry
With the availability of cerebellum-focused image analysis tools, a pattern of grey matter atrophy weighted towards lobules IV to VI, and reductions in brainstem and cerebellar white matter volume adjacent to the dentate nuclei and within the cerebellar peduncles, is now well-established [8–12] (Figure 2). Atrophy of the dentate nuclei was further supported using quantitative susceptibility mapping (QSM), alongside cross-sectional increases in iron concentration and longitudinal iron accumulation in these structures [13]. Outside of the cerebellum and brainstem, reports of more subtle anatomical changes remain mixed, with atrophy of the thalamus and cortical motor areas most consistently implicated and thought to reflect later-stage disease changes [9,10].
2. Examples of the most common quantitative imaging approaches in the most prevalent degenerative ataxias among autosomal recessive, autosomal dominant and sporadic ataxias, namely Friedreich ataxia (FRDA), spinocerebellar ataxia type 3 (SCA3), and cerebellar multiple system atrophy (MSA-C).
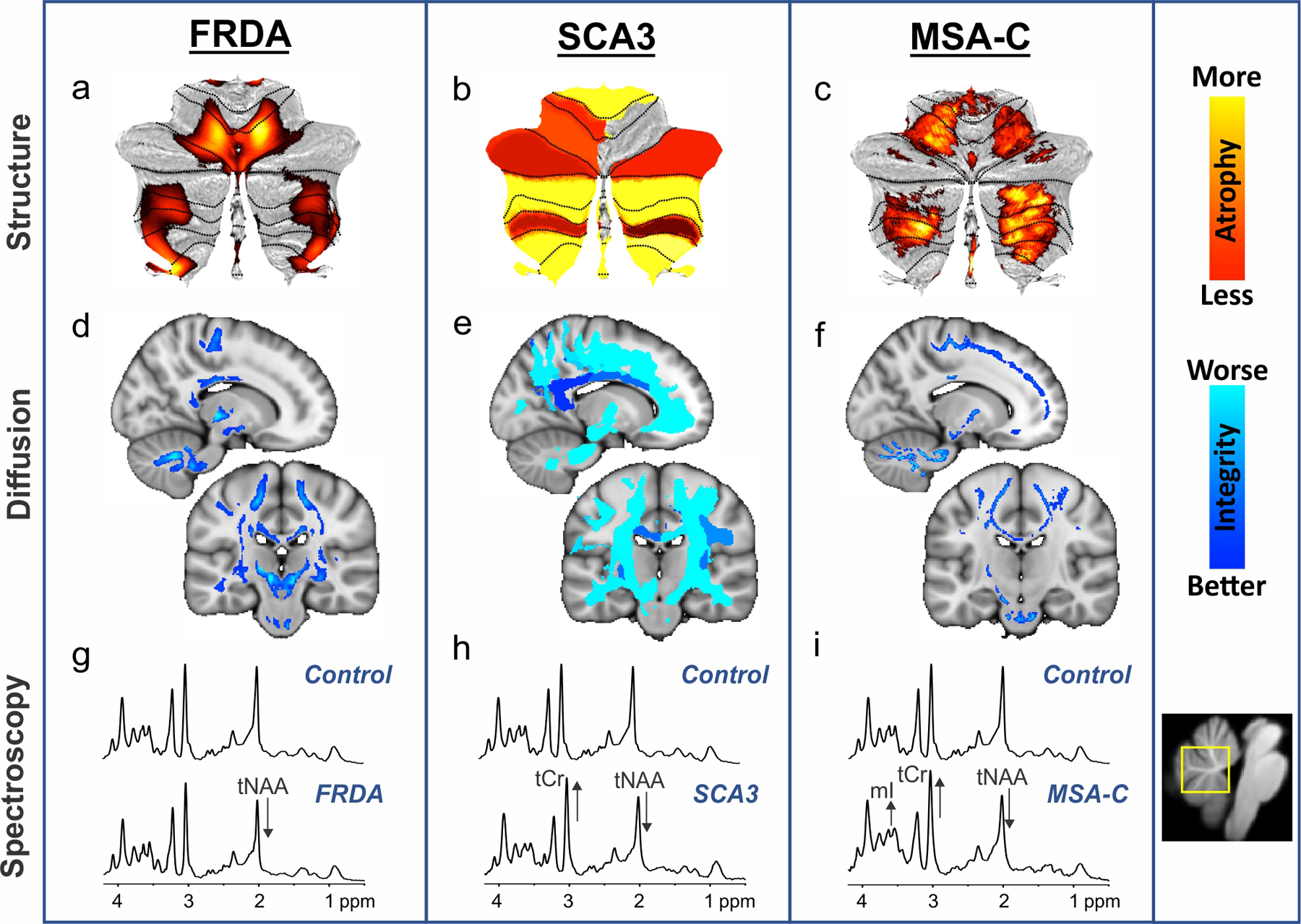
Profiles of cerebellar atrophy from voxel-wise (a, c) and region-of-interest (b) analyses are displayed in hot colors (top). Fractional anisotropy findings from diffusion-weighted imaging, reflecting white matter microstructural integrity, are shown in cool colors (middle) from voxel-wise (d), region-of-interest (e), and tract-based spatial statistics (TBSS) (f) analyses. These images reflect different analysis approaches, but exemplify their common utility in defining key disease characteristics. The bottom panels (g-i) display neurochemical abnormalities in MR spectra obtained at 7T from a vermis voxel (shown in yellow on the right) in individuals with ataxia relative to a control spectrum. The alterations visible in the spectra are marked with arrows. tNAA: total N-acetylaspartate; tCr: total Creatine; mI: myo-Inositol. The panels in this figure are based on data from prior publications [11, 25, 33, 45] or unpublished data.
Friedreich Ataxia: Connectivity, Neurochemistry and Function
Robust microstructural white matter abnormalities were detected not only in the cerebellar peduncles and in the brainstem, but more subtle changes also extend to the cerebrum, most notably in corticospinal, callosal, and long-range association tracts [10–12,14] (Figure 2). Microstructural impairments appear to manifest over-and-above volumetric atrophy [11] and correlate with biochemical markers of neuronal loss (N-acetyl-aspartate-to-creatine ratio) [14]. The patterns of abnormalities reported across different neuroimaging indices point to the potential for both myelin-related and degeneration-related white-matter pathology in FRDA.
Whole-brain functional MRI (fMRI) studies in FRDA have also revealed evidence of network-level functional changes. Reduced cerebro-cerebellar and increased cerebro-cerebral connectivity were found using resting-state fMRI [15], and a recent task-based study of finger-tapping function provided evidence of cerebral compensation in parallel with cerebellar dysfunction [16]. These studies indicate the potential for adaptive mechanisms to play a role in disease mitigation or expression.
Friedreich Ataxia: Spine
While neuroimaging studies have conventionally focused on brain changes in FRDA, spinal cord atrophy is also a clearly established primary site of pathology in this disease. Recent MRI evaluations of spinal cord morphometry have identified flattening and reduced cross-sectional area across the full length of the spinal cord, most markedly in cervical regions [9,10]. Spinal cord changes are proposed to be early, progressive, and clinically relevant features of FRDA.
Other Autosomal Recessive Ataxias
Available literature in other, often very rare, autosomal recessive inherited ataxias has largely consisted of qualitative case reports or retrospective case series. Aggregated case reviews, which in some recent cases include many tens of subjects, have provided generalizable clinical insights into common radiological features evident in disorders such as ataxia telangiectasia (AT) [17], ataxia with oculomotor apraxia (AOA) [18], and autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) [19]. However, these reports are based on qualitative clinical judgement and are restricted to large morphological features (e.g., “cerebellar atrophy” or “vermal hypoplasia”).
Several quantitative case-control assessments of brain integrity have also been undertaken in rare recessive ataxias. Using a multi-modal imaging approach in children with AT, global cerebellar volumetric and diffusion abnormalities were reported relative to healthy controls [20]. In the same study, the diffusion abnormalities correlated with spectroscopic indices of neuronal integrity and gliosis [20]. Similarly, although in a more limited sample of individuals with SYNE1 ataxia and matched controls, widespread grey and white matter impairments were identified throughout the brain [21].
AUTOSOMAL DOMINANT ATAXIAS
Autosomal dominantly inherited ataxias comprise spinocerebellar ataxias (SCAs) and episodic ataxias [1]. The majority of MRI literature available from the review period focusses on the most common SCAs, with only a few qualitative case reports available on MRI of episodic ataxias. Recent highlights in SCA imaging include the demonstration of the higher sensitivity of MRI and MRS to disease progression than clinical scales, detection of premanifest abnormalities in mutation-carriers, and developing a better understanding of the role of cerebellar degeneration in non-motor deficits using functional MR.
Spinocerebellar Ataxias: Morphometry
Genotype-specific atrophy patterns are increasingly well-mapped for the most common SCAs [22–26]. Atrophy is primarily found in the brainstem, cerebellum and basal ganglia in SCA1 and SCA3 [22,25] (Figure 2), the pons and cerebellum in SCA2 [24], and the cerebellum in SCA6 and SCA7 [22,26] (Figure 1). In addition, atrophy of the spinal cord is seen in SCA3 and cerebral atrophy is observed at late stages in this disease [25].
Such genotype-dependent volumetric measures were previously shown to correlate with the widely used and validated clinical scale, the Scale for the Assessment and Rating of Ataxia (SARA), and to be more sensitive to change than SARA [23]. This was confirmed by two other longitudinal studies in which volumetry outperformed clinical scores in the measurement of disease progression in SCA1, SCA2, SCA3 and SCA7 [27,28]. Together, these studies provide a strong rationale to supplement clinical outcome assessments with these objective, non-invasive MRI markers in clinical trials.
A large multisite study of individuals at risk of SCA has also shown brainstem and cerebellum atrophy prior to ataxia onset in SCA1 and SCA2 [29]. Similar results in preclinical and manifest SCA2 mutation carriers were recently obtained in a Cuban population [24]. Similarly, volume reductions were identified at the spinal cord, midbrain and substantia nigra at the preclinical stage in SCA3 [25].
Spinocerebellar Ataxias: Connectivity, Neurochemistry and Function
A number of reports prior to our review period had shown regional white matter abnormalities using diffusion tensor imaging (DTI) in the common SCAs [30,31]. Recent cross-sectional investigations further outlined differences in DTI metrics between patients with SCAs and controls in multiple brain regions, including the cerebellar peduncles and brainstem (pons) [25,27,32] (Figure 2). Most of these studies also showed correlations between DTI metrics and clinical severity in SCAs. Importantly, premanifest microstructural abnormalities were detected in the cerebellar and cerebral peduncles in SCA3 [25].
Similar to genotype-dependent morphometric findings, a recent MRS study showed genotype-dependent neurochemical abnormalities in common SCAs [33], which could be detected in presymptomatic mutation carriers up to 10 years prior to their estimated disease onset [33]. A longitudinal study in SCA1 also showed that both MRI volumetry and MRS were more sensitive to disease progression than SARA, and that MRS may have predictive value for disease progression [28]. In keeping with the move towards multimodal evaluations of disease, a novel statistical strategy to integrate multimodal biomarkers, including volumetry and MRS, was proposed to identify markers of disease progression in SCAs [34]. In addition to work directed towards biomarkers, an important contribution towards disease understanding was made when SCA7 was defined as a mitochondrial disease based in part on phosphorus (31P) MRS data obtained during a visual task [35].
Among the few recent functional connectivity studies, a network-based statistical approach showed altered inter-nodal cerebellar-cerebrum connectivity in SCA2, providing clinical and neural clues about cognitive and motor dysfunction [36]. In SCA6, decreased resting-state functional connectivity in the attention network [37] and impaired functional activity in the sensorimotor cortex and supplementary motor area was observed [38].
X-LINKED ATAXIAS
Fragile X-associated tremor/ataxia syndrome (FXTAS) is the only X-linked ataxia for which quantitative neuroimaging has been reported. Here, midbrain, brainstem, and cerebellar atrophy was shown, with a large retrospective cross-sectional study also indicating that cerebellar and brainstem atrophy is progressive in both unaffected premutation carriers and those with frank illness [39,40]. Further investigations have supported the critical involvement of the cerebellar peduncles. White matter lesions in infratentorial regions correlate with motor and cognitive dysfunction [41], and diffusion-based microstructural indices of middle and inferior cerebellar peduncle integrity are associated with methylation levels in the causative FMR1 gene and circulating FMR1 mRNA [42]. Longitudinal imaging also indicates that unaffected premutation carriers with smaller middle cerebellar peduncles may be at greater risk of transitioning to symptomatic states, perhaps reflecting a useful stratification biomarker [40].
SPORADIC DEGENERATIVE ATAXIAS
Sporadic degenerative ataxias comprise cerebellar multiple system atrophy (MSA-C) and the often more benign sporadic adult onset ataxia (SAOA) [1]. Recent MRI studies in sporadic ataxias focused on characterizing regional gray and white matter loss, as well as neurochemical and connectivity changes. A body of work has also attempted to use imaging to distinguish between MSA-C and SAOA, which may help with diagnosis early in the disease course.
Sporadic ataxias: Morphometry
Atrophy of the cerebellum and brainstem are common in both MSA-C and SAOA [43,44] (Figures 1, 2). This was recently further confirmed by a whole-brain morphometry study showing gray matter volume loss specifically in the cerebellar areas subserving sensorimotor functions in both diseases [45].
MRI can also help to distinguish between MSA-C and SAOA. For example, while white matter loss in the cerebellum was prominent in both SAOA and MSA-C, brainstem white matter was found to be reduced only in MSA-C [45,46]. Structural MRI features, including pons and/or middle cerebellar peduncle (MCP) atrophy, are even included in the second consensus statement on the diagnosis of MSA [47]. Other MRI features, such as the “hot cross bun” sign, MCP hyperintensity, putaminal hypointensity and the hyperintense putaminal rim sign have also been described in MSA-C [48].
Sporadic ataxias: Connectivity, Neurochemistry and Function
Using diffusion MRI, prominent microstructural white matter involvement has been observed in MSA-C (Figure 2), but not in SAOA [45]. In MSA-C, DTI studies have further revealed a reduction of cerebellar fiber density, and impairment of frontal and occipital white matter connectivity [49]. In addition, microstructural alterations of the motor subnetworks in the diencephalon, thalamus and cerebellar regions were observed in MSA-C, which correlated negatively with clinical features including the Unified Multiple System Atrophy Rating Scale (UMSARS) and duration of illness [50]. One MRS study also demonstrated that brainstem volume and N-acetyl-aspartate-to-creatine ratio in the cerebellum reliably distinguished patients with MSA-C from those with SAOA [51].
Functional resting-state connectivity has revealed diminished functional connectivity in the cerebellum, dentate nucleus, pons, basal ganglia, default mode network, temporo-parietal regions and limbic system in patients with MSA-C compared with healthy controls, which was again associated with clinical performance [52,53]. Among the few functional studies in SAOA, abnormal intra-cerebellar functional connectivity patterns were reported in areas with gray matter loss relative to intact cerebellar regions, suggesting that atrophy occurs in those cerebellar regions characterized by abnormal connectivity measures [54].
CONCLUSIONS
Recent ataxia imaging has been marked by a move towards multi-modal MR imaging, both in service of furthering the understanding of progressive disease pathology, and in preparation for upcoming clinical trials in the most common degenerative ataxias, in particular FRDA and common SCAs. Importantly, a number of studies have demonstrated detection of CNS abnormalities prior to ataxia onset, which can be used for patient stratification in clinical trials. In addition, MR metrics most sensitive to disease progression can be used for treatment monitoring at both premanifest and manifest stages. Namely, atrophic changes may be slowed down or stopped upon treatment, while microstructural, functional and neurochemical abnormalities may be reversible since they mark changes prior to, and in many cases independent of, cell loss. To improve the robustness of disease characterizations, and prepare for these upcoming trials through validation of longitudinal MRI and MRS markers, several prospective multi-site studies have been initiated (Figure 3), such as EUROSCA (http://www.eurosca.org/), READISCA (https://readisca.org/), ESMI (http://www.ataxia-study-group.net/html/studies/esmi), and retrospective data pooling platforms have been launched, such as ENIGMA-Ataxia (http://enigma.ini.usc.edu/ongoing/enigma-ataxia/). These efforts represent international collaborations, which are essential to gather sufficient trial-ready cohorts with these rare diseases, but also because of potential geographic differences in clinical characteristics of each disease entity. Similar multi-site, multi-modal, and longitudinal efforts are needed in recessive ataxias other than FRDA and in dominant ataxias other than the common SCAs. Harmonization of data collection methodologies is key for such multi-institutional efforts and is defined as a major goal for two recent initiatives, the SCA Global (http://ataxia-global-initiatives.net/sca-global/) and ARCA Global (http://ataxia-global-initiatives.net/arca-global/). Similar efforts are needed for sporadic ataxias, especially considering that MRI and MRS may have great utility in the clinic at early disease stages for diagnosis and prognosis in these conditions. Finally, while clinical case reports will continue to be a useful and necessary vehicle for characterizing very rare diseases, their utility would be greatly expanded through the use of quantitative measures where possible (e.g., Z-scores of cerebellar lobule volumes relative to a healthy control cohort).
3. Pipeline for validating MR imaging biomarkers for use in clinical practice or clinical trials and the status of different MR modalities in the pipeline for different degenerative ataxia categories.
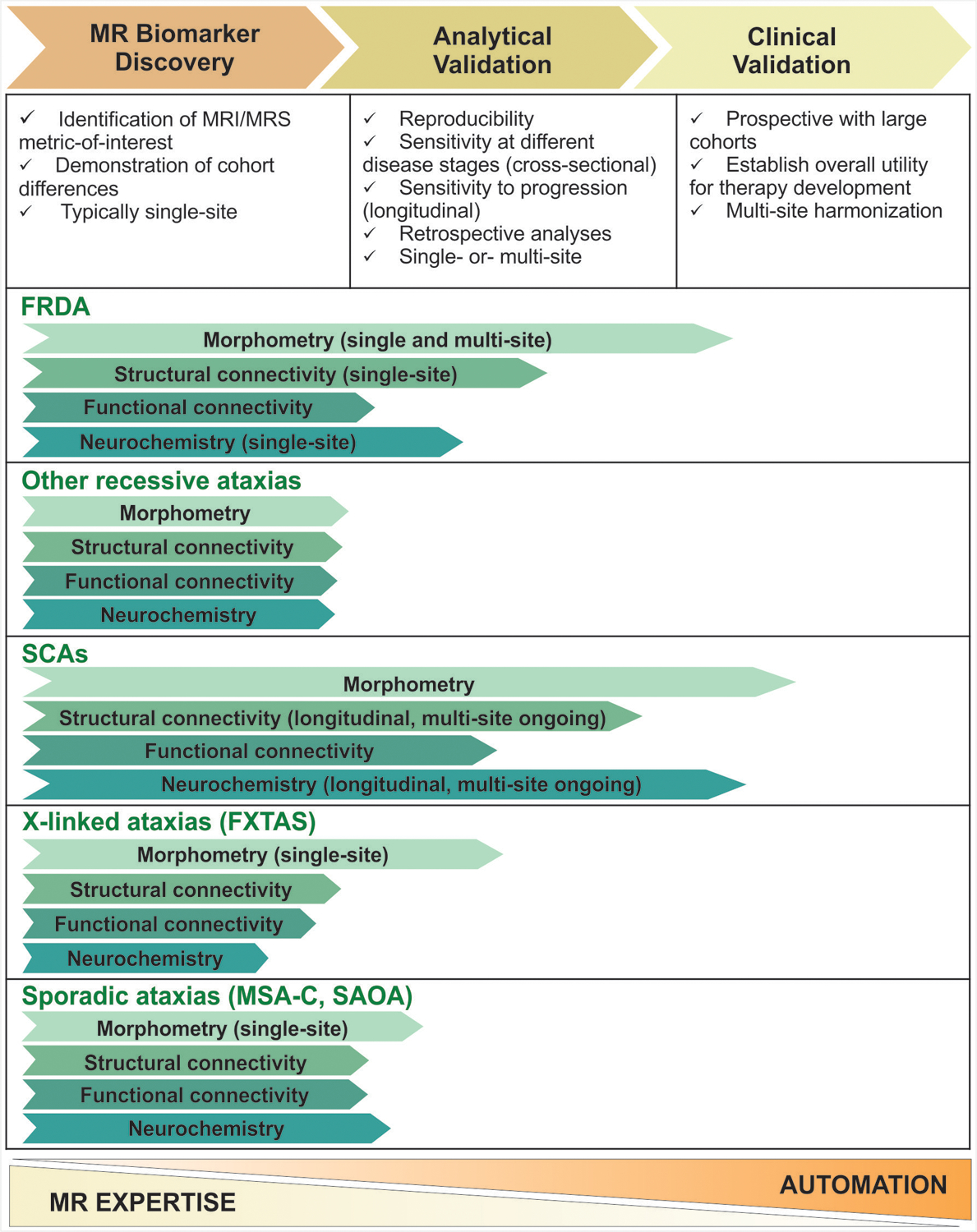
Note that the final phase, regulatory approval (“Qualification”) that follows the first three phases is not shown. The lengths of the arrows are meant to provide the reader an approximate idea of the place of the different markers in the pipeline relative to each other and relative to the phases defined on top. For example, prospective multi-site morphometry studies with large FRDA cohorts are in the planning stage, hence the arrow ends early in the third phase. Similarly, multi-site morphometry studies with relatively large SCA cohorts have been completed (e.g. EUROSCA), however sample sizes are still limited relative to natural history studies that have validated clinical scales. Typically, earlier stage biomarker discovery studies are conducted at sites with substantial MR expertise and as the identified biomarker moves through the pipeline, increased automation in data acquisition and analysis is necessary for clinical utility, as represented by the scale at the bottom.
KEY POINTS.
Quantitative MR imaging in degenerative ataxias has moved towards multi-modal, multi-institutional studies with large cohorts, both to understand progressive disease pathology and to prepare for testing of disease-modifying therapies in the pipeline, in particular for Friedreich ataxia (FRDA) and spinocerebellar ataxias (SCAs).
Similar efforts are needed in recessive ataxias other than FRDA, in dominant ataxias other than the common SCAs and in sporadic ataxias.
Patterns of cerebellar atrophy in different degenerative ataxias are increasingly well-defined thanks to availability of advanced MRI acquisition methods and specialized software dedicated to the analysis of MRI of the cerebellum.
Volumetric MRI and MR spectroscopy (MRS) are more sensitive to disease progression than validated clinical scales and cerebral and cerebellar abnormalities are detectable by structural and diffusion MRI and MRS at the premanifest stage in SCAs.
Acknowledgements
We acknowledge the commitment and generosity of patients with ataxias for participating in research. We would further like to thank our colleagues Drs. James Joers, Jennifer Faber, Thiago Rezende, Joanne Fielding, Scott Kolbe and Pierre-Gilles Henry for contributing data and images used in figure generation.
Financial support and sponsorship
G.Ö. is supported by the National Institutes of Health, National Ataxia Foundation and Biogen, Inc. I.H.H. is supported by the Australian National Health and Medical Research Council. K.R. and the position of J.K. are funded by the German Federal Ministry of Education and Research (BMBF 01GQ1402 to KR) and the German Research Foundation (IRTG 2150 269953372/GRK2150). K.R. is additionally supported by the German Federal Ministry of Education and Research (01DN18022), the German Research Foundation (ZUK32/1) and Alzheimer Forschung Initiative e.V (AFI 13812, NL-18002CB).
Funding: The preparation of this manuscript was supported by the National Institute of Neurological Disorders and Stroke (NINDS) grants R01 NS080816 and U01 NS104326, the Australian National Health and Medical Research Council (Fellowship 1106533 and Grant 1184403) and the German Federal Ministry of Education and Research (BMBF 01GQ1402). The Center for Magnetic Resonance Research is supported by the National Institute of Biomedical Imaging and Bioengineering (NIBIB) grant P41 EB015894 and the NINDS Institutional Center Cores for Advanced Neuroimaging award P30 NS076408.
Footnotes
Conflicts of interest
G.Ö. has received research grants from Takeda Pharmaceuticals, Inc. and Biogen, Inc. I.H.H. receives investigator-initiated research funding from Takeda Pharmaceuticals. K.R. has received honoraria for presentations or advisory boards from Lilly and Roche, as well as clinical trial grants from Pfizer, Merck, Minoryx, Biogen and Roche.
REFERENCES AND RECOMMENDED READING
- 1.Klockgether T, Paulson H. Milestones in ataxia. Mov Disord 2011; 26:1134–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Currie S, Hadjivassiliou M, Craven IJ et al. Magnetic resonance imaging biomarkers in patients with progressive ataxia: current status and future direction. Cerebellum 2013; 12:245–266. [DOI] [PubMed] [Google Scholar]
- 3.Ashizawa T, Öz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol 2018; 14:590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glasser MF, Smith SM, Marcus DS et al. The Human Connectome Project’s neuroimaging approach. Nat Neurosci 2016; 19:1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Öz G, Deelchand DK, Wijnen JP et al. Advanced single voxel 1H magnetic resonance spectroscopy techniques in humans: Experts’ consensus recommendations. NMR Biomed 2020; e4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diedrichsen J, Balsters JH, Flavell J et al. A probabilistic MR atlas of the human cerebellum. Neuroimage 2009; 46:39–46. [DOI] [PubMed] [Google Scholar]
- 7.Romero JE, Coupe P, Giraud R et al. CERES: A new cerebellum lobule segmentation method. Neuroimage 2017; 147:916–924. [DOI] [PubMed] [Google Scholar]
- 8.Lindig T, Bender B, Kumar VJ et al. Pattern of Cerebellar Atrophy in Friedreich’s Ataxia-Using the SUIT Template. Cerebellum 2019; 18:435–447. [DOI] [PubMed] [Google Scholar]
- 9.*.Dogan I, Romanzetti S, Didszun C et al. Structural characteristics of the central nervous system in Friedreich ataxia: an in vivo spinal cord and brain MRI study. J Neurol Neurosurg Psychiatry 2019; 90:615–617. [DOI] [PubMed] [Google Scholar]; The first MR investigation of whole-spine morphometry in Friedreich ataxia, revealing important insights into its global involvement in this disease and strongly motivating future work to investigate longitudinal changes.
- 10.*.Rezende TJR, Martinez ARM, Faber I et al. Developmental and neurodegenerative damage in Friedreich’s ataxia. Eur J Neurol 2019; 26:483–489. [DOI] [PubMed] [Google Scholar]; This study is the first to undertake a side-by-side comparison of children and adults with Friedreich ataxia to draw inferences regarding earlier (developmental) vs. later (degenerative) disease-related changes, making important contributions to defining disease staging.
- 11.*.Selvadurai LP, Corben LA, Delatycki MB et al. Multiple mechanisms underpin cerebral and cerebellar white matter deficits in Friedreich ataxia: The IMAGE-FRDA study. Hum Brain Mapp 2020; doi: 10.1002/hbm.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive study of white matter abnormalities in Friedreich ataxia, investigating a range of in vivo measures and their inter-relationships. This work suggests that there may be multiple pathways causing white matter damage, which must be considered in natural history studies and clinical trials.
- 12.Vavla M, Arrigoni F, Nordio A et al. Functional and Structural Brain Damage in Friedreich’s Ataxia. Front Neurol 2018; 9:747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.*.Ward PGD, Harding IH, Close TG et al. Longitudinal evaluation of iron concentration and atrophy in the dentate nuclei in friedreich ataxia. Mov Disord 2019; 34:335–343. [DOI] [PubMed] [Google Scholar]; The first use of quantitative susceptibility mapping (QSM) to investigate longitudinal changes in the dentate nuclei in Friedreich ataxia, highlighting its strong potential as a treatment monitoring biomarker.
- 14.*.Gramegna LL, Tonon C, Manners DN et al. Combined Cerebellar Proton MR Spectroscopy and DWI Study of Patients with Friedreich’s Ataxia. Cerebellum 2017; 16:82–88. [DOI] [PubMed] [Google Scholar]; Multi-modal integration of spectroscopic and diffusion measures, highlighting biochemical correlates of white matter changes to provide more in-depth and rebost biological interpretations.
- 15.*.Cocozza S, Costabile T, Tedeschi E et al. Cognitive and functional connectivity alterations in Friedreich’s ataxia. Ann Clin Transl Neurol 2018; 5:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first resting-state fMRI study in this disorder, supporting the emerging picture of complex whole-brain functional changes that may be either pathological or compensatory.
- 16.Harding IH, Corben LA, Delatycki MB et al. Cerebral compensation during motor function in Friedreich ataxia: The IMAGE-FRDA study. Mov Disord 2017; 32:1221–1229. [DOI] [PubMed] [Google Scholar]
- 17.Akturk H, Sutcu M, Somer A et al. Ataxia telangiectasia in Turkey: multisystem involvement of 91 patients. World J Pediatr 2017; 13:465–471. [DOI] [PubMed] [Google Scholar]
- 18.Renaud M, Moreira MC, Ben Monga B et al. Clinical, Biomarker, and Molecular Delineations and Genotype-Phenotype Correlations of Ataxia With Oculomotor Apraxia Type 1. JAMA Neurol 2018; 75:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rezende Filho FM, Parkinson MH, Pedroso JL et al. Clinical, ophthalmological, imaging and genetic features in Brazilian patients with ARSACS. Parkinsonism Relat Disord 2019; 62:148–155. [DOI] [PubMed] [Google Scholar]
- 20.**.Dineen RA, Raschke F, McGlashan HL et al. Multiparametric cerebellar imaging and clinical phenotype in childhood ataxia telangiectasia. Neuroimage Clin 2019; 25:102110. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive, multi-modal evaluation of cerebellar macrostructure, microstructure, and biochemistry in a large cohort of children with AT, highlighting relevant measures for longitudinal follow-up and biomarker validation.
- 21.Gama MTD, Piccinin CC, Rezende TJR et al. Multimodal neuroimaging analysis in patients with SYNE1 Ataxia. J Neurol Sci 2018; 390:227–230. [DOI] [PubMed] [Google Scholar]
- 22.Schulz JB, Borkert J, Wolf S et al. Visualization, quantification and correlation of brain atrophy with clinical symptoms in spinocerebellar ataxia types 1, 3 and 6. Neuroimage 2010; 49:158–168. [DOI] [PubMed] [Google Scholar]
- 23.Reetz K, Costa AS, Mirzazade S et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain 2013; 136:905–917. [DOI] [PubMed] [Google Scholar]
- 24.Reetz K, Rodriguez-Labrada R, Dogan I et al. Brain atrophy measures in preclinical and manifest spinocerebellar ataxia type 2. Ann Clin Transl Neurol 2018; 5:128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.**.Rezende TJR, de Paiva JLR, Martinez ARM et al. Structural signature of SCA3: From presymptomatic to late disease stages. Ann Neurol 2018; 84:401–408. [DOI] [PubMed] [Google Scholar]; This work reports about the structural damage in SCA3/MJD, beginning in the spinal cord, cerebellar peduncles, as well as substantia nigra and progresses to cerebral areas in the long term. These structural differences reveal some insights into the pathogenesis of SCA3/MJD and suggest a staging scheme to map the progression of the disease.
- 26.Chirino A, Hernandez-Castillo CR, Galvez V et al. Motor and cognitive impairments in spinocerebellar ataxia type 7 and its correlations with cortical volumes. Eur J Neurosci 2018; 48:3199–3211. [DOI] [PubMed] [Google Scholar]
- 27.Adanyeguh IM, Perlbarg V, Henry PG et al. Autosomal dominant cerebellar ataxias: Imaging biomarkers with high effect sizes. Neuroimage Clin 2018; 19:858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.*.Deelchand DK, Joers JM, Ravishankar A et al. Sensitivity of Volumetric Magnetic Resonance Imaging and Magnetic Resonance Spectroscopy to Progression of Spinocerebellar Ataxia Type 1. Mov Disord Clin Pract 2019; 6:549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]; First longitudinal SCA study where the sensitivity of MRS to disease progression was compared to that of both volumetry and the most widely used clinical scale in SCAs, SARA.
- 29.*.Jacobi H, Reetz K, du Montcel ST et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol 2013; 12:650–658. [DOI] [PubMed] [Google Scholar]; This is the first multi-centre European study showing clinical and imaging results in the most common preclinical spinocerebellar ataxias. It highlights the presence of mild coordination deficits and brain abnormalities in preclinical SCA1 and SCA2 mutation carriers.
- 30.Della Nave R, Ginestroni A, Tessa C et al. Brain white matter damage in SCA1 and SCA2. An in vivo study using voxel-based morphometry, histogram analysis of mean diffusivity and tract-based spatial statistics. Neuroimage 2008; 43:10–19. [DOI] [PubMed] [Google Scholar]
- 31.Falcon MI, Gomez CM, Chen EE et al. Early Cerebellar Network Shifting in Spinocerebellar Ataxia Type 6. Cereb Cortex 2016; 26:3205–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng H, Liang X, Long Z et al. Gene-Related Cerebellar Neurodegeneration in SCA3/MJD: A Case-Controlled Imaging-Genetic Study. Front Neurol 2019; 10:1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.*.Joers JM, Deelchand DK, Lyu T et al. Neurochemical abnormalities in premanifest and early spinocerebellar ataxias. Ann Neurol 2018; 83:816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of sensitivity of MRS to premanifest abnormalities in multiple common SCAs up to 10 years before predicted ataxia onset.
- 34.*.Garali I, Adanyeguh IM, Ichou F et al. A strategy for multimodal data integration: application to biomarkers identification in spinocerebellar ataxia. Brief Bioinform 2018; 19:1356–1369. [DOI] [PubMed] [Google Scholar]; A novel statistical strategy is proposed to combine brain volumetry, magnetic resonance spectroscopy, metabolomic and lipidomic analyses to obtain composite biomarkers of disease progression.
- 35.**.Ward JM, Stoyas CA, Switonski PM et al. Metabolic and Organelle Morphology Defects in Mice and Human Patients Define Spinocerebellar Ataxia Type 7 as a Mitochondrial Disease. Cell Rep 2019; 26:1189–1202 e1186. [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive study that combined data from patients, mice, and human stem cell derived neurons to categorize SCA7 as a mitochondrial disorder. Excellent example of utilizing 31P MRS during cerebral activation to reveal altered metabolism in a neurodegenerative disease.
- 36.Olivito G, Cercignani M, Lupo M et al. Neural substrates of motor and cognitive dysfunctions in SCA2 patients: A network based statistics analysis. Neuroimage Clin 2017; 14:719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira L, Airan RD, Fishman A et al. Resting-state functional connectivity and cognitive dysfunction correlations in spinocerebelellar ataxia type 6 (SCA6). Hum Brain Mapp 2017; 38:3001–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang N, Christou EA, Burciu RG et al. Sensory and motor cortex function contributes to symptom severity in spinocerebellar ataxia type 6. Brain Struct Funct 2017; 222:1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.*.Wang JY, Hessl D, Hagerman RJ et al. Abnormal trajectories in cerebellum and brainstem volumes in carriers of the fragile X premutation. Neurobiol Aging 2017; 55:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]; A large retrospective cross-sectional study providing insights into the rate of subtentorial atrophy in individuals with FXTAS.
- 40.**.Shelton AL, Wang JY, Fourie E et al. Middle Cerebellar Peduncle Width-A Novel MRI Biomarker for FXTAS? Front Neurosci 2018; 12:379. [DOI] [PMC free article] [PubMed] [Google Scholar]; Results from this study indicate that middle cerebellar peduncle structure may be useful for stratifying premutation carriers at higher-risk of converting to symptomatic FXTAS, allowing for sample enrichment in future observational studies or clinical trials.
- 41.Hocking DR, Loesch DZ, Trost N et al. Total and Regional White Matter Lesions Are Correlated With Motor and Cognitive Impairments in Carriers of the FMR1 Premutation. Front Neurol 2019; 10:832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.*.Shelton AL, Cornish KM, Godler D et al. White matter microstructure, cognition, and molecular markers in fragile X premutation females. Neurology 2017; 88:2080–2088. [DOI] [PubMed] [Google Scholar]; Innovative and comprehensive integration of disease-relevant neuroimaging, DNA markers, blood measures, and cognitive phenotyping.
- 43.Burk K, Globas C, Wahl T et al. MRI-based volumetric differentiation of sporadic cerebellar ataxia. Brain 2004; 127:175–181. [DOI] [PubMed] [Google Scholar]
- 44.Heim B, Krismer F, Seppi K. Structural Imaging in Atypical Parkinsonism. Int Rev Neurobiol 2018; 142:67–148. [DOI] [PubMed] [Google Scholar]
- 45.**.Faber J, Giordano I, Jiang X et al. Prominent White Matter Involvement in Multiple System Atrophy of Cerebellar Type. Mov Disord 2020; doi: 10.1002/mds.27987. [DOI] [PubMed] [Google Scholar]; This work describes imaging findings in sporadic degenerative ataxia such as multiple system atrophy with predominant cerebellar ataxia (MSA-C) and sporadic adult-onset ataxia (SAOA). It demonstrates the significance of microstructural white matter changes in the differentiation between the two conditions.
- 46.Dash SK, Stezin A, Takalkar T et al. Abnormalities of white and grey matter in early multiple system atrophy: comparison of parkinsonian and cerebellar variants. Eur Radiol 2019; 29:716–724. [DOI] [PubMed] [Google Scholar]
- 47.Gilman S, Wenning GK, Low PA et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carre G, Dietemann JL, Gebus O et al. Brain MRI of multiple system atrophy of cerebellar type: a prospective study with implications for diagnosis criteria. J Neurol 2020; doi: 10.1007/s00415-020-09702-w. [DOI] [PubMed] [Google Scholar]
- 49.Wang PS, Yeh CL, Lu CF et al. The involvement of supratentorial white matter in multiple system atrophy: a diffusion tensor imaging tractography study. Acta Neurol Belg 2017; 117:213–220. [DOI] [PubMed] [Google Scholar]
- 50.Shah A, Prasad S, Rastogi B et al. Altered structural connectivity of the motor subnetwork in multiple system atrophy with cerebellar features. Eur Radiol 2019; 29:2783–2791. [DOI] [PubMed] [Google Scholar]
- 51.Kadodwala VH, Hadjivassiliou M, Currie S et al. Is 1H-MR spectroscopy useful as a diagnostic aid in MSA-C? Cerebellum Ataxias 2019; 6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang H, Wang N, Luo X et al. Altered functional connectivity of dentate nucleus in parkinsonian and cerebellar variants of multiple system atrophy. Brain Imaging Behav 2019; 13:1733–1745. [DOI] [PubMed] [Google Scholar]
- 53.Ren S, Zhang H, Zheng W et al. Altered Functional Connectivity of Cerebello-Cortical Circuit in Multiple System Atrophy (Cerebellar-Type). Front Neurosci 2018; 12:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang X, Faber J, Giordano I et al. Characterization of Cerebellar Atrophy and Resting State Functional Connectivity Patterns in Sporadic Adult-Onset Ataxia of Unknown Etiology (SAOA). Cerebellum 2019; 18:873–881. [DOI] [PubMed] [Google Scholar]