

Abstract
Problem:
Although early environmental influences are thought to influence the development of inflammatory bowel disease (IBD), little is known about the role of the in utero environment on subsequent IBD risk. We hypothesized that prenatal exposure to bacterial lipopolysaccharide (LPS) could modify the subsequent development of dextran sulfate sodium (DSS)-induced ulcerative colitis in adulthood by influencing the associated cellular and immune response.
Method of Study:
To test this hypothesis, we exposed developing mice in utero to LPS or saline (PBS) at E17.5, and then induced colitis at 5 weeks. We then assessed colitis severity and effects on the microbiome. In order to define the developmental impact of any potential LPS effect, we also exposed one-week old mice to either LPS or saline before inducing colitis at 5 weeks.
Results:
Mice that had been exposed to LPS but not saline in utero were protected from subsequent colitis development, and their intestinal barrier integrity and tight junction expression distribution were similar to that of control mice that were not exposed to DSS. By contrast, mice exposed to either LPS or saline at day 7 of life all developed severe colitis upon subsequent DSS exposure.
Conclusion:
These results identify an informative time window during fetal development during which exposure to an otherwise pro-inflammatory agent like LPS protects against an inflammatory disease in adulthood.
Keywords: Inflammatory bowel disease, fetal development, lipopolysaccharide
Introduction
Inflammatory bowel disease (IBD), which is comprised of the clinical subtypes ulcerative colitis and Crohn’s disease, is characterized by episodes of excessive inflammation of the gastrointestinal tract. In ulcerative colitis, the inflammation is limited to the colonic mucosa, whereas Crohn’s disease is characterized by transmural inflammation that can extend anywhere from the mouth to the anus (1–3). Although the precise mechanisms leading to IBD remain incompletely understood, current thinking suggests that the disease stems from dysregulation of the mucosal immune response in a genetically susceptible host, in association with ill-defined dietary factors (1–5). Whilst environmental factors are thought to be important in driving disease development (6,7), the potential role of the in utero environment on IBD pathogenesis remains largely unexplored. Previous authors have shown that maternal smoking during early pregnancy (8), and the occurrence of preterm birth itself (9), both reduce the development of ulcerative colitis, while early life infections increase the risk of developing ulcerative colitis in adulthood (10). These findings together suggest that factors present during development or in early childhood can influence the subsequent development of IBD.
We now seek to test the hypothesis that in utero exposure to inflammatory stimuli will modify responses to subsequent inflammatory triggers in adulthood by attenuating the subsequent inflammatory response to factors that lead to IBD. We tested this hypothesis using a mouse model of in utero endotoxin exposure followed by the subsequent exposure to an established model of colitis. Specifically, we exposed developing mice in utero at E17.5 to either LPS or saline, and then at 5 weeks of age we chemically induced colitis using 2% dextran sodium sulfate (DSS). In control experiments, 7 day old mice were exposed to LPS or saline and the impact on subsequent IBD severity was determined. We now show that in utero LPS exposure protects against the subsequent development of colitis, and this protection was not present when mice received LPS at seven days of age. These results identify an informative time window in which exposure to an otherwise harmful agent like LPS could, in fact, provide protection against an inflammatory disease well into adulthood.
Methods
Mice
CD-1 mice were purchased from Charles River Laboratories (Wilmington, MA). Animal protocols were approved by the Johns Hopkins University Animal Care and Use Committee (protocol M017M304). Mice were housed in a pathogen free environment on a 12-hour light/12-hour dark cycle and given free access to standard rodent chow (PicoLab mouse diet 20, 22% kcal % fat) and acidified tap water after weaning before beginning the dextran sulfate sodium model. Mice were bred beginning at nine weeks of age and removed from the breeding colony after six months. The experimental procedures including the exposure to LPS in utero and the subsequent induction of colitis are described below.
Intrauterine exposure to LPS
Pregnant CD-1 mice, which normally deliver pups at day 19–21 days of gestation, were used for all mouse experiments (29). The induction of in utero inflammation was performed at E17.5 (referred to as E17 in this paper) under isofluorane anesthesia, which was administered to the pregnant mouse in an induction chamber via a nose cone. Exposure at E17 was chosen based on previous relevant literature that utilized the intrauterine inflammation model outlined below (30–32), and note that this model mimics a sub-clinical infection withithout inducing preterm birth. After establishment of adequate anesthesia, and after sterile preparation of the skin using 10% povidone-iodine (Professional Disposables), a laparotomy incision was made on the pregnant mouse’s abdomen. The cervix was then located via the laparotomy, and the two gestational sacks to the left of the cervix were exposed. LPS (Sigma-Aldrich L3129) at 2 mg/kg based on maternal body weight was injected into the uterine wall between the gestational sacks. Sterile saline was used to wash the gestational sacks before they were returned to the abdomen. The fascia was then closed with Ethicon 3–0 Braided Vicryl Sutures and the skin was closed with staples (Fine Science Tools, 9mm AutoClip). The mice were closely monitored for preterm birth of pups. 12 pregnant mice underwent the above LPS injection procedure, with eight litters surviving and each litter producing 4–12 live pups. Eight control mice underwent injection of phosphate buffered saline (PBS), with all litters surviving and each litter producing 10–16 live pups. There was an equal distribution of males and females in each experiment, premature delivery was not observed, and all pups were weaned at three weeks of age and separated by sex.
Endotoxemia at P7
At P7, CD-1 pups that were delivered spontaneously were given an intraperitoneal injection of LPS (L3129) at 2 mg/kg based on pup weight or PBS (0.9% Sodium Chloride, Hospira Inc.). Of the 63 pups injected with LPS, 51 survived the injection and all mice injected with PBS survived. The pups were then returned to their mother and weaned at three weeks of age and separated by sex. After this point, they were given free access to standard rodent chow (PicoLab mouse diet 20, 22% kcal % fat) and acidified tap water until beginning the dextran sulfate sodium model.
Dextran Sulfate Sodium-induced Colitis
All mice, regardless of the LPS or saline exposure they received at E17 or P7, underwent a six day DSS model as follows: On day 1 of the model, the mice were weighed and given either 2% DSS (MW 40,000–50,000 kDa, Thermo Scientific) in 0.5% sucrose drinking water; control mice received 0.5% sucrose drinking water alone. Mice were weighed on each day of the model. On model day three, fresh 2% DSS/0.5% sucrose or 0.5% sucrose water was offered. On day six, all mice were offered regular water and euthanized on day seven. Before euthanasia, colonoscopy (see below) was performed using isoflurane anesthesia administered via nose cone. Colon length was measured, and then colons were flash-frozen in liquid nitrogen and stored at −80 °C. Cages were separated by sex and exposure throughout the model. All mice were studied at 5–6 weeks after initial injection of LPS (either in utero or on day 7).
Immunofluorescence and histology
Colon samples were collected immediately after euthanasia and fixed in 4% paraformaldehyde (Electron Microscope Services, Cat #RT15700) in PBS overnight. The tissue was then processed using a Microm STP 120 Spin Tissue Processor (Thermo Scientific), embedded in paraffin, and 5-μm thick sections were cut using a CUT 6062 microtome (SLEE). The paraffin-embedded samples were then probed with primary antibodies in a 1:200 dilution in TBST overnight at 40 C and probed with 4’,6-diamidino-2-phenylindole (DAPI), anti-ZO-1 (ThermoFisher, Cat# 61–7300), and Alexa Fluor 555 donkey anti-mouse at 1:1000 for two hours at RT then washed and mounted in gelvatol mounting media. For Alcian blue staining, the collection, fixation, and section cutting processes outlined above were used. The paraffin-embedded samples were probed with Alcian blue (Sigma-Aldrich) for half an hour and nuclear fast red (Sigma-Aldrich) for one hour at room temperature then washed and mounted in Permount mounting media (Fischer Scientific).
Microscopy and fluorescence quantification
All immunofluorescence images were acquired using a Nikon Eclipse Ti microscope with a Nikon A1 confocal laser microscope system. Images were captured using a 20X objective (Nikon Plan Apo 20x/0.75 DIC N2) and maximum intensity z-projections of 3–6 3 μm optical sections. The images were then analyzed using Fiji, quantified using Excel, and visual representations were created using GraphPad Prism 7. All Alcian blue images were acquired using a Leica DMi8 inverted microscope. Images were captured using a 20X objective (Leica HC PLAN 20X/0.70). The images were analyzed using Fiji and quantified using Excel. Five unique mucosal thickness measurements and 15 unique crypt measurements were recorded for each sample and respective averages was calculated.
Mouse Colonoscopy
Colonoscopy was performed on six-week-old mice upon conclusion of the six day 2% DSS model before euthanasia. Isoflurane anesthesia was administered via a nose cone to anesthetize mice. Colonoscopy was then performed using a Karl Storz endoscope (Cat #61029D) with an attached air syringe. The endoscope was coated with surgical lubricant (Puralube) and inserted into the anus. Air was slowly pushed from the syringe to allow a field of view and pictures were taken using a StarTech HDMI Video Capture device. Mice were euthanized after the procedure was completed.
The degree of DSS-induced colitis was measured using a scoring system adapted from previously published literature. Thickening of the colon (0=transparent, 1=moderate, 2=marked, 3=intra-transparent), changes in the vascular pattern (0=normal, 1=moderate, 2=marked, 3=bleeding), and stool consistency (0=normal/solid, 1=shaped, 2=unshaped, 3=spread) were all measured. Total scores were added across all measurements, for a total possible score of nine.
16S rRNA sequencing of microbes in mouse stool
Stool from mice that underwent in utero injection of PBS or LPS and subsequent 2% DSS exposure or 0.5% sucrose control exposure was isolated from the colon at euthanasia by extraction from the tissue with forceps and flash-frozen using liquid nitrogen then stored at −80 °C. Microbial genomic DNA was isolated using a DNeasy PowerSoil Kit (Cat #12888–50, Qiagen), and processed at the Johns Hopkins Transcriptomics and Deep Sequencing Core for library preparation and 16S rRNA sequencing (MiSeq 2×150, V3V4 sequencing). Quality trimming (target error rate <0.5%), PhiX removal, chimera detection, host genome removal, and unknown contaminant assessment for non-16S sequences were performed. Data were normalized to 10,000 high-quality 16S rRNA sequences. High-resolution taxonomic assignment was performed by Resphera Insight (Baltimore, Maryland, United States), functional inference of gene content was found using PICRUSt, alpha and beta-diversity calculations were made using QIIME, and principal coordinates, differential abundance of alpha diversity, and taxonomic categories analyses were performed using R.
Quantitative Real-Time PCR
RNA was extracted from colon tissue using an Rneasy Mini Kit (Cat #74106, Qiagen) and converted into complementary DNA using a QuantiTect Reverse Transcription Kit (Cat #205314, Qiagen). Quantitative real-time PCR was performed on the Bio-Rad CFX96 Real-Time System using primers outlined in Table 1. All values were normalized relative to the housekeeping gene Ribosomal protein large P0 (Rplp0).
Table 1.
Primers used for qPCR experiments.
| Primer | Species | Forward Sequence | Reverse Sequence | Amplicon Size (bp) |
|---|---|---|---|---|
| RPL0 | Mouse/Rat/Human | GGCGACCTGGAAGTCCAACT | CCATCAGCACCACAGCCTTC | 143 |
| IL-6 | Mouse/Rat | CCAATTTCCAATGCTCTCCT | ACCACAGTGAGGAATGTCCA | 182 |
| IL1β | Mouse/Rat | AGTGTGGATCCCAAGCAATACCCA | TGTCCTGACCACTGTTGTTTCCCA | 175 |
| TNFα | Mouse/Rat | TTCCGAATTCACTGGAGCCTCGAA | TGCACCTCAGGGAAGAATCTGGAA | 144 |
| Tlr4 | Mouse | TTTATTCAGAGCCGTTGGTG | CAGAGGATTGTCCTCCCATT | 186 |
| Muc2 | Mouse | TAGTGGAGATTGTGCCGCTGAAGT | AGAGCCCATCGAAGGTGACAAAGT | 168 |
| Hes1 | Mouse | CAGCTCCGGGAAAGCAAGCCC | GCCACCTTTCTCTGAGTCACCGC | 141 |
Statistical Analysis
Data were analyzed using GraphPad Prism 7 software. All data were analyzed for significance using a two-tailed t-t test or one-way ANOVA and post hoc Bonferroni correction for multiple comparisons. Statistical significance was determined by a p-value of <0.05. All PCR was performed in triplicate. Data was assembled and graphed using GraphPad Prism 7. All E17 experiments were repeated three times. Careful consideration was taken to ensure that each exposure group (i.e. E17 LPS plus 2% DSS) did not come from a single litter but instead, after weaning, each litter was split into either DSS exposure or control. Data was pooled across experiments to generate each figure.
Results
Prenatal LPS exposure protects mice from subsequent development of experimental colitis in adulthood.
To determine the effects of prenatal endotoxin exposure on colitis severity in adulthood, we first analyzed colon tissue from six-week old CD-1 mice that had been exposed in utero to either LPS (2 mg/kg) or PBS, and which were subsequently exposed to a 6 day DSS-colitis model. Colonoscopy was performed on the 6-week-old mice while they were under general anesthesia in order to initially assess the severity of colonic inflammation. As shown in Figure 1, control mice that were injected with either saline or LPS in utero (Figure 1 A–B, I) or administered saline or LPS at day 7 of life (Figure 1 C–D, J), revealed healthy colonic mucosa with no evidence of inflammation. By contrast, in mice that were exposed to the DSS colitis model at 5 weeks of age, we observed significant inflammation, mucosal friability, and bleeding in the colon in mice that had received PBS at E17 (Figure 1E, I–J, p<0.0001). Strikingly, the in utero exposure at E17 to LPS significantly reduced the degree of inflammation, and the colonic mucosa appeared to resemble untreated mice (Figure 1F, I, p<0.0001). Colonoscopies were also performed in the mice that were induced to develop colitis after prior exposure to either LPS or PBS on day 7 of life. As shown in Figure 1 G–H and J, both of these DSS-exposed groups displayed significant inflammation and bleeding within the colon (p<0.0001). Taken together, these findings reveal that the administration of LPS in utero, but not in the early post-natal period, provides protection against the subsequent development of colitis. We next sought to quantify the extent of this effect.
Figure 1. Mouse colonoscopy reveals that prenatal LPS exposure protects mice from subsequent development of experimental colitis in adulthood.
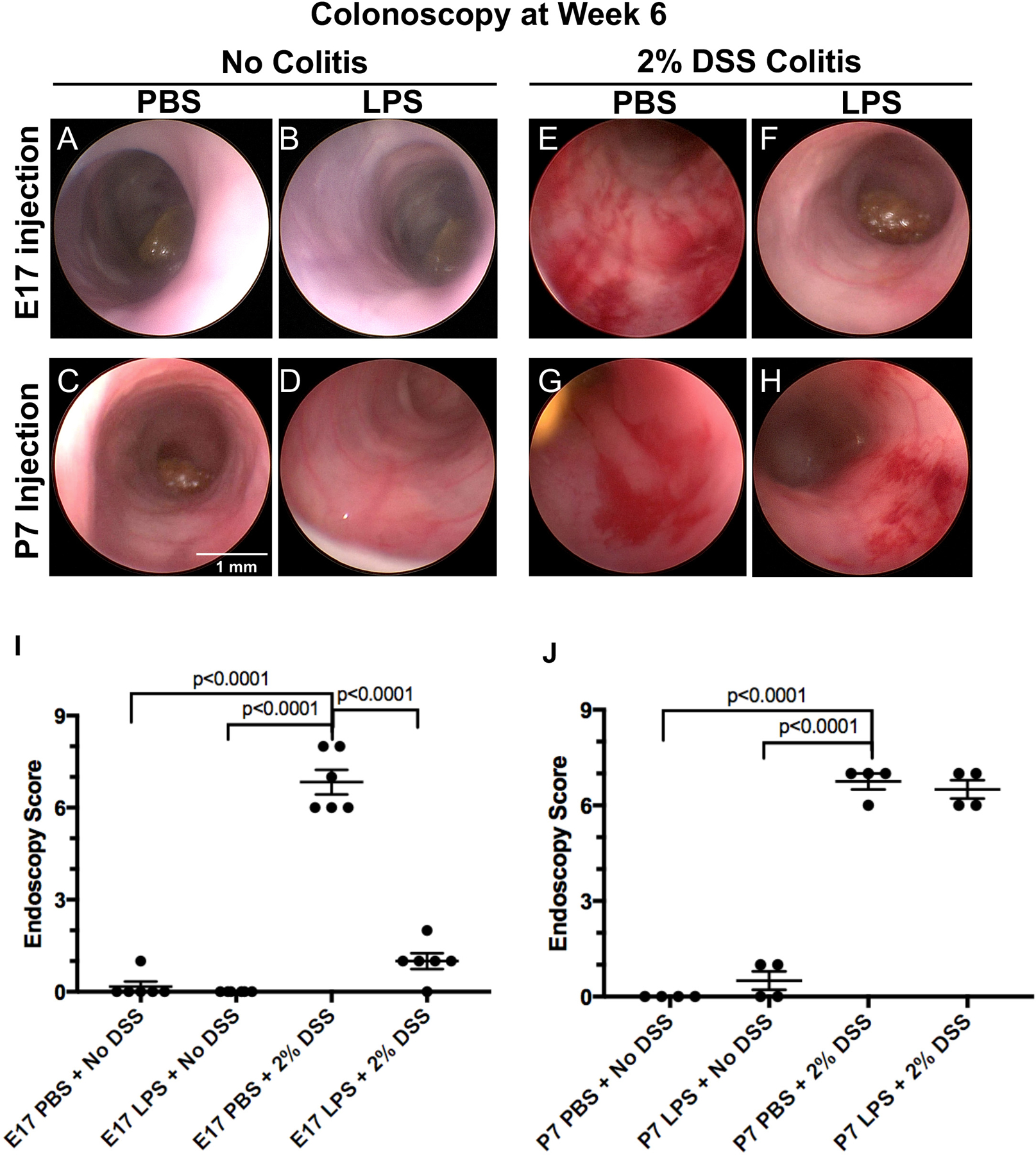
Representative colonoscopy images from 6 week old mice that either did not undergo 2% DSS model (A-D), or which were exposed to 2% DSS colitis (E-H) after exposure to either saline (PBS) or lipopolysaccharide (LPS, 2 mg/kg) at either E17 in utero or day 7 of life (P7) as indicated. I-J: Endoscopy scoring of colon thickness, vascular pattern, and stool consistency of 6 week old mice that were injected at E17 in utero with either PBS or LPS or injected at day 7 with either PBS or LPS. Each colonoscopy image and graph point is representative of a single mouse for both E17 (n=6 for each exposure group) and P7 (n=4 for each exposure group) experiments. One-way ANOVA tests were performed to determine significance.
Prenatal exposure to LPS reduces the effects of colitis on weight loss and colon length.
To further assess the effects of the in utero exposure to LPS on the subsequent development of colitis, we next assessed the degree of weight loss and colonic shortening, two important markers of colitis severity in this model (11,12). Mice were weighed on each day of the 6-day model, and their weight change over the six days is displayed in Figure 2A. The sex of the mice did not have an effect on colitis severity, as revealed by a lack of significant difference between male and female mice within each exposure group, after adjusting for initial weight differences between sexes (p=0.902 for non-DSS exposed mice, p=0.731 for DSS exposed mice). Control mice that were injected with PBS in utero that were not exposed to DSS at five weeks did not lose weight over the six days of the DSS model (mean weight gain= 2.1 g ± 0.9) as expected (Figure 2A, grey and black lines). There was also no weight loss in control mice injected with LPS at E17 that were not exposed to DSS at five weeks (mean weight gain=2.0 g ± 0.3, Figure 2B, grey and black lines), confirming that the initial LPS injection did not have major independent effects on growth. By contrast, mice that were exposed to PBS at E17 and then subsequently to DSS at five weeks showed significant colitis, as manifest by weight loss, losing an average of 0.6 ± 0.4 g over the six day model (Figure 2A blue and red bars). Strikingly, mice that were exposed to LPS in utero at E17 and then induced to develop colitis at five weeks showed significantly less weight loss than these control groups (mean weight gain=1.6 g ± 0.2, Figure 2B, blue and red lines, p<0.001 vs. PBS injection). These findings reveal that in utero LPS exposure protects against the weight loss that is associated with DSS colitis, and instead actually gained weight during the model.
Figure 2. Prenatal exposure to LPS reduces the effects of colitis on weight loss and colon length.
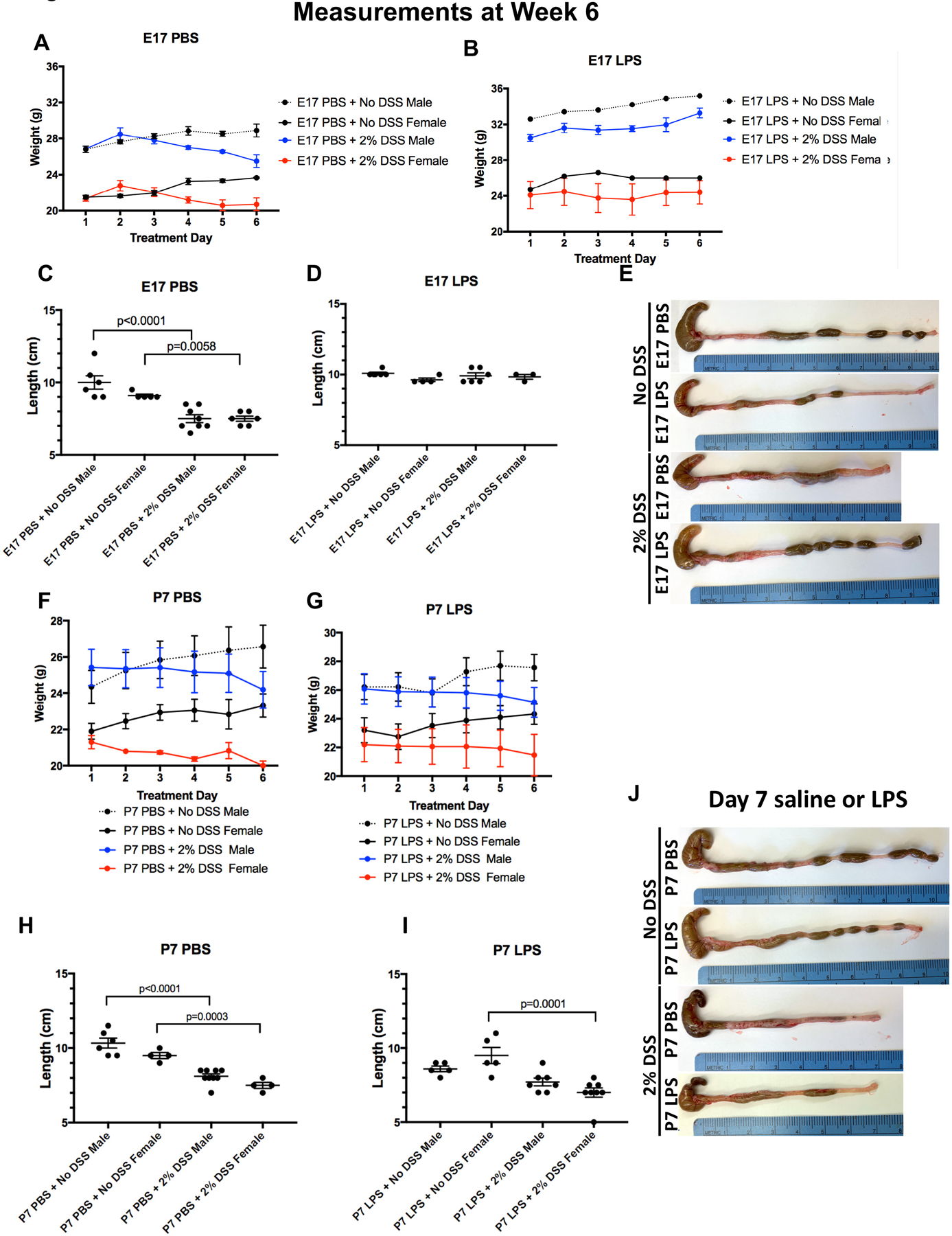
A-E: Daily weights (± SD, A-B) and colon lengths (C-E) of 6 week old mice (n ≥ 5 for males, n ≥ 3 for females) that were injected at E17 in utero with either saline (PBS, A, C) or LPS (B, D); A-B: Each point represents group average; E: Representative images of colons removed from in utero injected mice of the indicated group at the end of the colitis model. F-J: Daily weights (± SD F-G) and colon lengths (H-I) of 6 week old mice (n ≥ 5 for males, n ≥ 4 for females) that were injected at day 7 with either PBS (F, H) or LPS (G, I). F-G: Each point represents group average; J: Representative images of colons from the indicated group. All data is pooled across three separate experiments for both E17 and P7 (2 mg/kg) exposures, respectively, and t-tests were performed to determine significance.
Further validation of the protective effects of in utero exposure to LPS on the subsequent development of colitis is shown in Figure 2C–D, in which colon length measurements are shown and which stratified by sex. Consistent with measures of weight change, colon length was not significantly different between male and female mice within each exposure group. Unlike control mice which had average colon lengths between 9.1–9.9 cm, mice who were exposed to colitis after in utero exposure to PBS revealed a significantly shorter average colon length of 7.6 cm (Figure 2C), a finding consistent with the development of colonic inflammation. By contrast, the exposure in utero to LPS markedly reduced the degree of colonic length shortening when they were subsequently induced to develop colitis, and the lengths were between 9.1–9.9 cm, similar to control mice (Figure 2D). Representative images of the colon from each group with an attached ruler is provided in Figure 2E. Taken together, these results support the notion that the in utero exposure to LPS significantly protects mice from colon shortening associated with the onset of experimental colitis.
In seeking the potential mechanisms that could explain the protective effects of in utero LPS exposure, it was critical for us to next define whether the observed protective effects of LPS were attributable to LPS exposure in the in utero environment, or whether the protective effects of LPS occurred independent of the window of exposure. To test for this possibility directly, we next repeated the above experiments by exposing mice to PBS or LPS (2 mg/kg) at postnatal day 7 without prior in utero exposure. As shown in Figure 2 F–J, in contrast to the protective effect of in utero LPS injection, mice that received LPS at 7 days of age followed by the induction of DSS colitis at five weeks experienced no protection from LPS exposure in terms of weight loss (Figure 2F–G) or colonic shortening (Figure 2H–I, representative raw images of colons are in J). Taken together, these results indicate that in utero exposure of LPS at E17 significantly protects against the subsequent development of colitis, and that the protection is not seen when LPS is given at later time-points postnatally. We next sought to determine the mechanisms involved, and evaluated first the inflammatory gene expression in epithelial cells between groups.
Prenatal LPS exposure leads to reduced inflammation and increased Muc2 expression in the adult mouse colon.
In seeking to determine the mechanisms of in utero LPS exposure to subsequent colitis development, we next evaluated whether in utero exposure to LPS could alter the degree of mucosal pro-inflammatory cytokine expression. As shown in Figure 3A, there was a significant increase in expression of the pro-inflammatory cytokines IL1β, Tnfα, and IL-6 within the colons of mice that were exposed to DSS after prior in utero exposure to PBS at E17, consistent with the induction of colitis, and the expression of these cytokines was each significantly reduced after in utero exposure to LPS at E17. Further, the expression of the mucus producing goblet cell gene Muc2 was significantly reduced in mice exposed to the colitis model, while the expression of Muc2 was found to be at control levels in the colitis mice that had received LPS in utero at E17 (Figure 3B). The induction of colitis in control mice that received saline at E17 resulted in significant upregulation of the expression of the LPS receptor gene Tlr4, while mice that received LPS in utero at E17 showed significantly reduced expression of TLR4 upon the subsequent induction of colitis consistent with reduced colitis severity (Figure 3C). As expected, mice that were not exposed to colitis did not reveal any significant increase in Tlr4 (Figure 3C). Additional studies revealed that the protection achieved from LPS exposure required in utero administration, as mice with colitis that had been exposed to LPS day 7 showed no protection from the colitis-induced increased expression of IL1β, IL-6, Tnfα, and Tlr4 (Figures 3D and 3G). Surprisingly, Muc2 expression was not reduced in mice exposed to DSS compared to those that were not (Figure 3E). However, expression of Hes1, a transcriptional repressor that inhibits goblet cell formation, was significantly increased among P7 PBS plus DSS exposed mice compared to P7 PBS or LPS exposed mice not exposed to DSS (p=0.0003, p=0.0082, Figure 3F). Notably, there was an increase in baseline expression across all E17 exposed groups, regardless of exposure, compared to P7 exposed groups. This indicates a potential lasting effect of the in utero exposure level itself, though it should be noted that there still remains a significant difference between E17 exposed groups for all markers, thus making it unlikely that maternal laparotomy is the cause for DSS-related expression changes between E17 exposed and P7 exposed mice. Overall, these findings reveal that exposure to LPS in utero leads to reduced inflammation and Hes1 expression and increased Muc2 expression in mice with colitis, while such protection was not observed in mice that were treated with LPS or PBS at P7. To further investigate the mechanisms mediating this effect, we next evaluated whether epithelial cell barrier function was affected by in utero exposure to LPS.
Figure 3. Prenatal LPS exposure leads to reduced inflammation and increased Muc2 expression in the adult mouse colon.
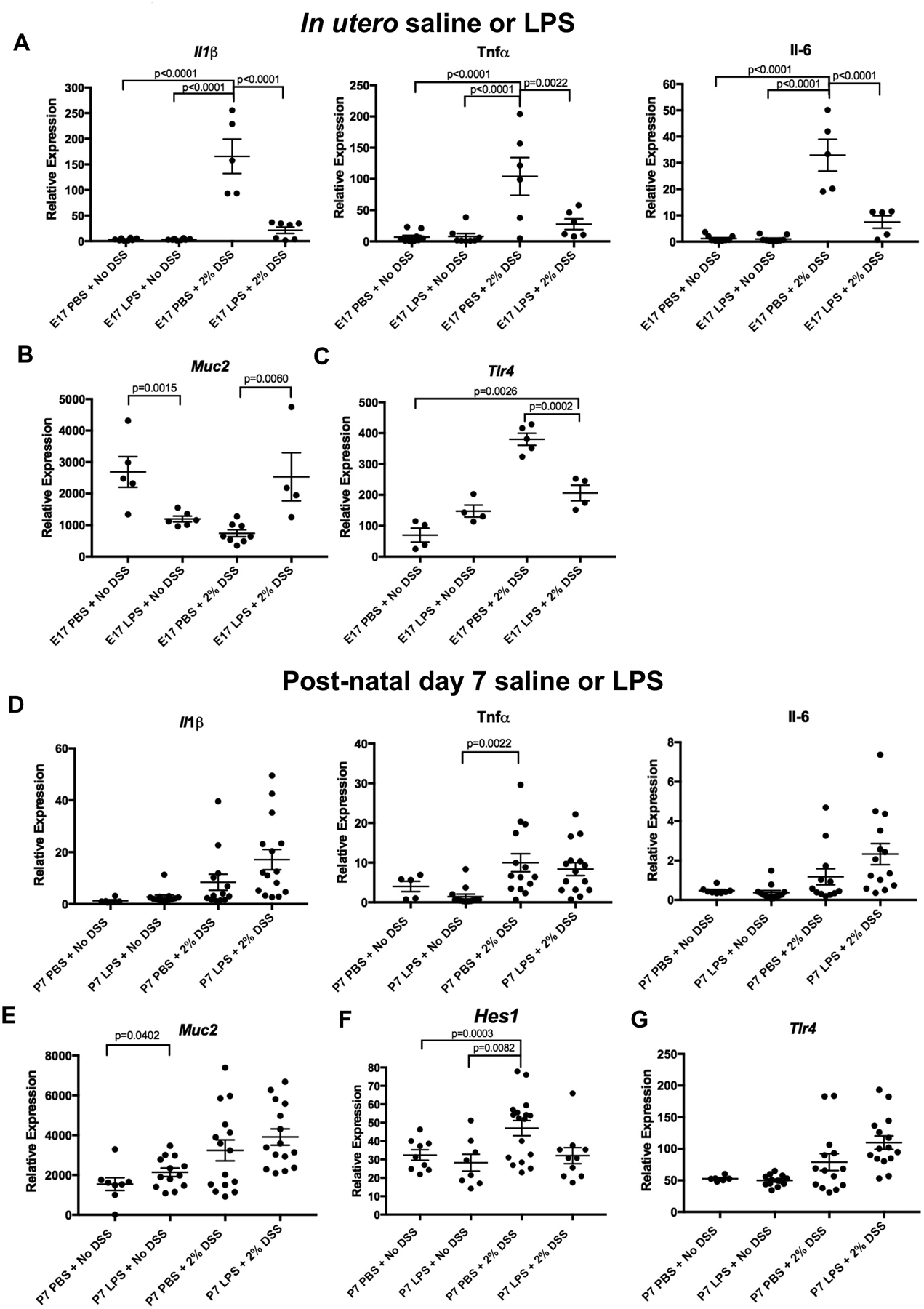
qRT-PCR measurement of the indicated gene at 6 weeks of age in mice with and without colitis that had been exposed to either saline (PBS) or LPS at E17 (A-C) or at day 7 (D-G). Each point represents an individual mouse and all expression levels are relative to RPL0 expression. n ≥ 4 animals analyzed per group for E17 experiments and n ≥ 5 for P7 experiments. All data is an aggregate of three separate experiments for both E17 and P7 exposures and one-way ANOVA tests were performed to determine significance.
Prenatal LPS exposure minimizes the loss of goblet cells and tight junctions in adult mice with experimental colitis.
LPS exposure in utero could protect against the subsequent development of colitis by enhancing the protective mucosal layer of the colonic epithelium, which is developmentally regulated through the Math1 pathway, and which we have shown to be influenced by activation of TLR4 (13). We therefore next investigated whether in utero exposure to LPS could alter the degree of mucin-producing cells within the gut. As shown in Figures 4A–B, the induction of colitis led to a significant loss of goblet cells in the intestinal epithelium in mice examined at 6 weeks of age that were exposed to PBS in utero compared to all other groups (p<0.0001). Importantly, this loss of goblet cells was not observed in mice exposed to LPS in utero, again reflective of in utero protection by LPS. To quantify the extent of mucus production, mucosal layer thickness was measured for each sample, as a validated readout of mucin production in the DSS model (11). As shown in Figure 4C, the mucosal thickness of mice with colitis that had been exposed to LPS in utero was similar to control mice that did not receive DSS, confirming that the protection extends towards mucous protection in adulthood. Control mice with colitis that were exposed to PBS at E17 displayed reduced goblet cells (Figure 4A) and a significantly reduced mucosal layer on the intestinal epithelium (Figure 4C) compared to all other exposure groups, as expected. Further evidence that in utero administration of LPS prevented epithelial layer injury was determined by evaluation of tight junction staining via ZO-1 staining. As shown in Figures 4D–E, there was a significant loss of tight junctions in mice with colitis that were injected in utero with PBS that was not seen in mice exposed to LPS in utero. Importantly, the injection of either LPS or PBS at day 7 did not influence the degree of colitis-induced reduction in goblet cells (Figure 5A–B), loss of mucosal thickness (Figure 5C), or tight junction loss (Figure 5D–E) when evaluated at week 6. Taken together, these findings reveal that in utero LPS exposure limits the loss of the structural barrier in the intestinal epithelium in the setting of subsequent experimental colitis. We next sought to assess whether these results were associated with any changes in the stool microbiome.
Figure 4. Prenatal LPS exposure minimizes the loss of goblet cells and tight junctions in adult mice with experimental colitis.
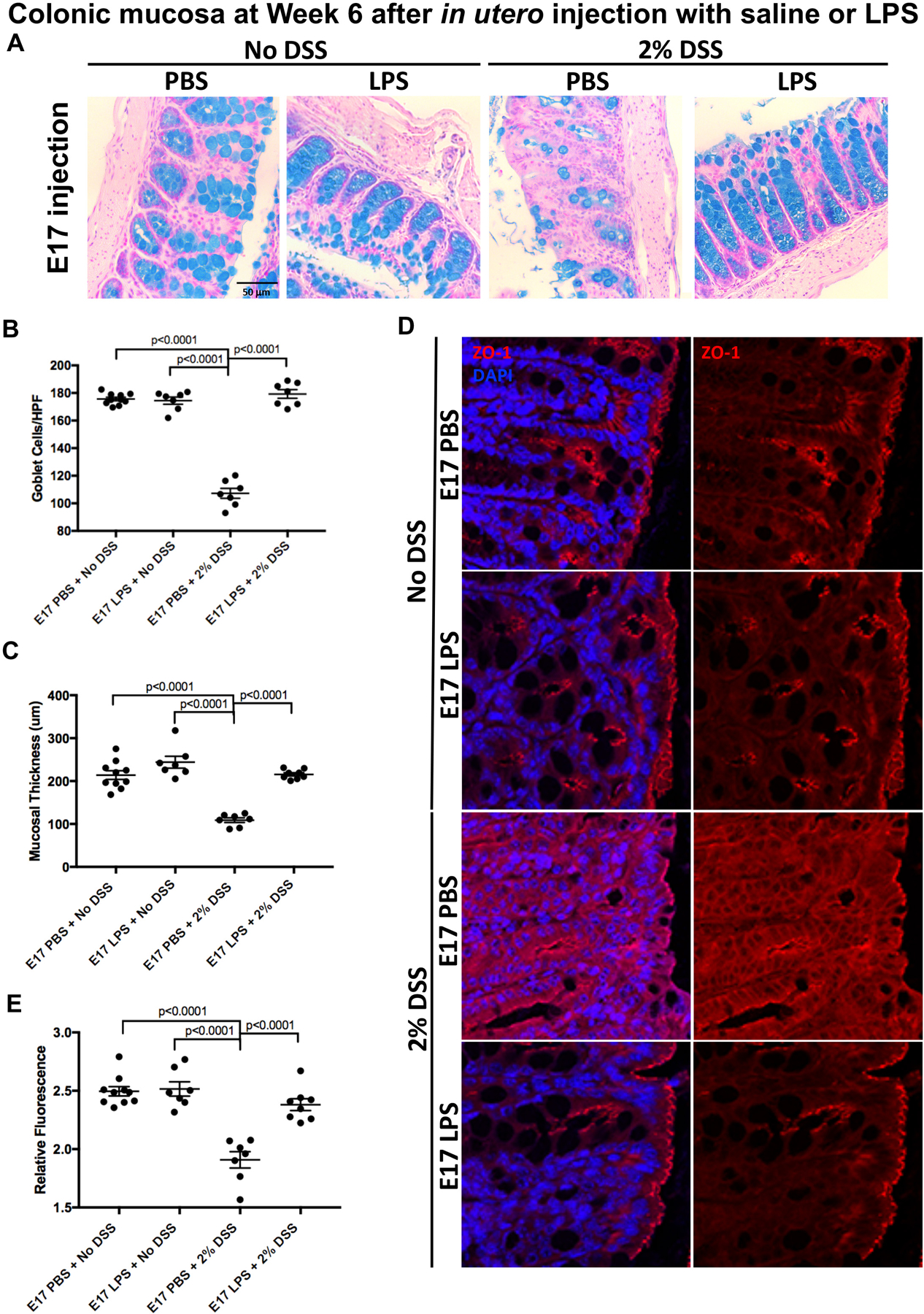
A: Representative micrograph of Alcian blue staining in the colonic mucosa of 6 week old mice that were exposed to either saline (PBS) or LPS (2mg/kg) in utero at E17, and then either not exposed to colitis (left two panels) or exposed to 2% DSS colitis for 6 days (right two panels); B: Alcian blue staining quantification using the average of 15 measurements from crypts within each sample (n ≥ 7 per group) as described in Methods, HPF=high powered field; C: Mucosal thickness quantification measured using the average of five measurements from each sample (n ≥ 7 per group) as described in Methods; D: Representative confocal micrographs of the colonic epithelium of 6 week old mice that were exposed in utero to LPS or saline and subsequently exposed to no colitis or to 2% DSS colitis as indicated; E: Quantification of relative fluorescence measurements of ZO-1 staining compared against DAPI staining (n ≥ 7 per group). Data was pooled across three separate experiments and one-way ANOVA tests were performed to determine significance.
Figure 5. LPS exposure at day 7 of age does not impact the colitis-induced loss of goblet cells and tight junctions in adult mice.
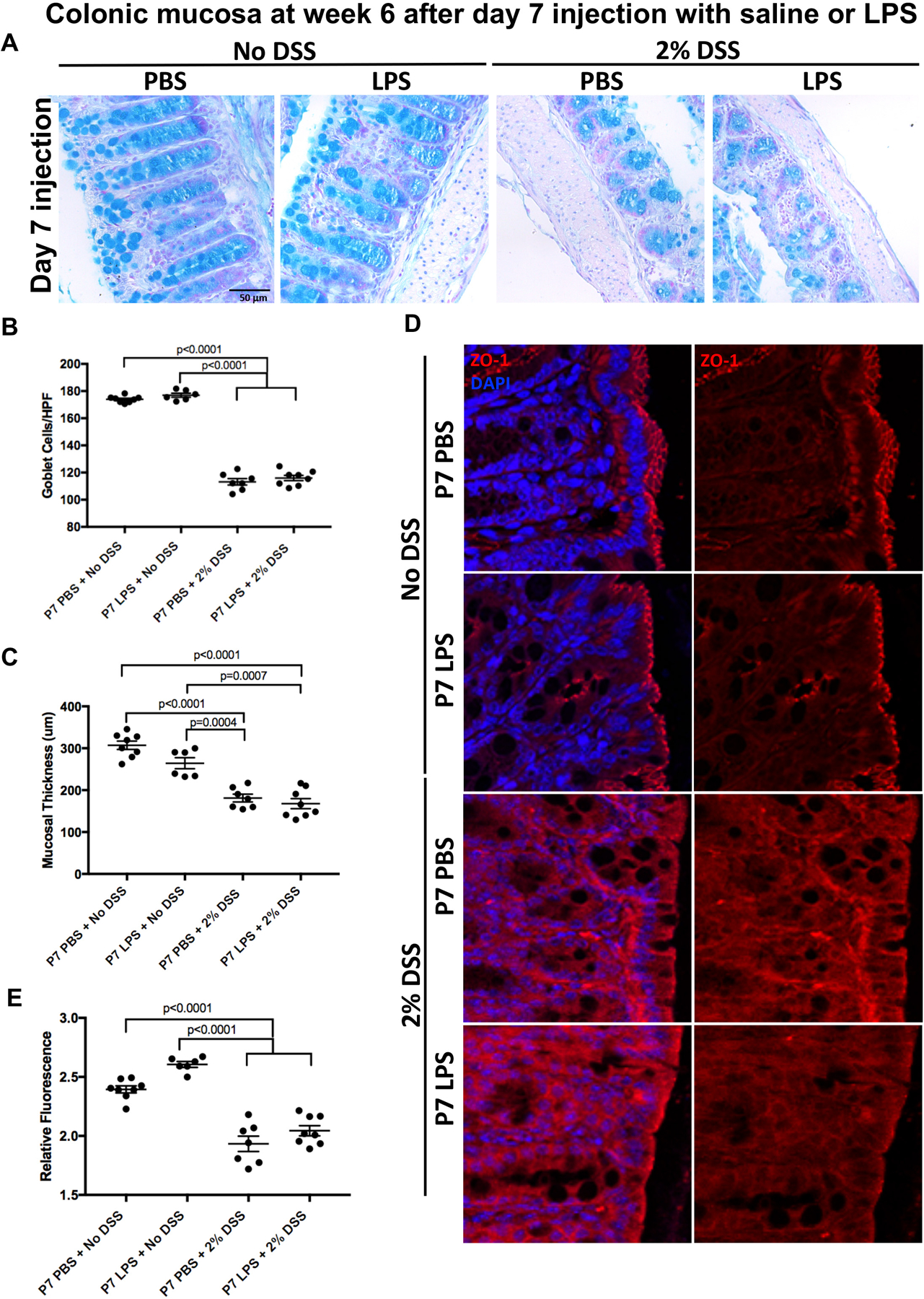
A: Representative micrograph of Alcian blue staining in the colonic mucosa of 6 week old mice that were exposed to either saline (PBS) or LPS (2mg/kg) at day 7, and then either not exposed to colitis (left two panels) or exposed to 2% DSS colitis for 6 days (right two panels); B: Alcian blue staining quantification using the average of 15 measurements from crypts within each sample (n ≥ 6 per group) as described in Methods, HPF=high powered field; C: Mucosal thickness quantification measured using the average of five measurements from each sample (n ≥ 6 per group) as described in Methods; D: Representative confocal micrographs of the colonic epithelium of 6 week old mice that were exposed day 7 to LPS (2mg/kg) or saline and subsequently exposed to no colitis or to 2% DSS colitis as indicated; E: Quantification of relative fluorescence measurements of ZO-1 staining compared against DAPI staining (n ≥ 6 per group). Data was pooled across three separate experiments and one-way ANOVA tests were performed to determine significance.
Effects of in utero exposure to LPS on 16S rRNA sequencing of the microbiome.
In the final series of experiments, we assessed whether the in utero exposure to LPS altered the stool microbiome during the subsequent development of colitis in adulthood. To do so, we performed 16S rRNA sequencing of stool samples from adult mice that were induced to develop colitis and which had been exposed to LPS in comparison with two mice that had not been exposed to LPS. Almost all of the top 50 taxonomic assignments were from the Bacteriodetes and Firmicutes phyla, consistent with the known makeup of the mouse microbiome. Diversity analysis using unweighted UniFrac and Bray-Curtis dissimilarity analyses found that axis 1 defined the in utero exposure (i.e. PBS or LPS) while axis 2 defined the DSS exposure (Figure 6A). Consistent with the known effects of DSS (14,15), we observed a significant reduction in alpha diversity amongst mice exposed to DSS at five weeks compared to those not exposed to DSS, regardless of their in utero exposure (p=0.05, Figure 6B). We also observed a significant increase in Firmicutes among E17 PBS exposed mice compared to E17 LPS exposed mice (Figure 6C). Interestingly, we also observed a significant increase in Flavobacteria, a genus within the Bacteroides phylum, amongst mice exposed to LPS at E17, regardless of their DSS exposure status (Figure 6D), consistent with a lasting effect of early LPS exposure. Overall, control mice that did not received DSS exposure had significantly more diverse microbiomes while mice that received E17 PBS had a significantly greater abundance of Firmicutes and a significantly reduced abundance of Flavobacteria than those that received E17 LPS. It is surprising that only mice exposed to LPS in utero at E17 showed persistent effects on Firmicutes bacteria, and suggest an enduring impact of the developmental exposure to LPS. These findings suggest an insight into protection as previous work has shown that ulcerative colitis severity is associated with an increase Firmicutes bacteria (16), that persists through adulthood. Taken together, these findings suggest the importance of in utero exposure to LPS in the protection against subsequent colitis development, and illustrate the long-term effects on barrier injury, gene expression, and the microbiome.
Figure 6. Effects of in utero exposure to LPS on 16S rRNA sequencing of the microbiome.
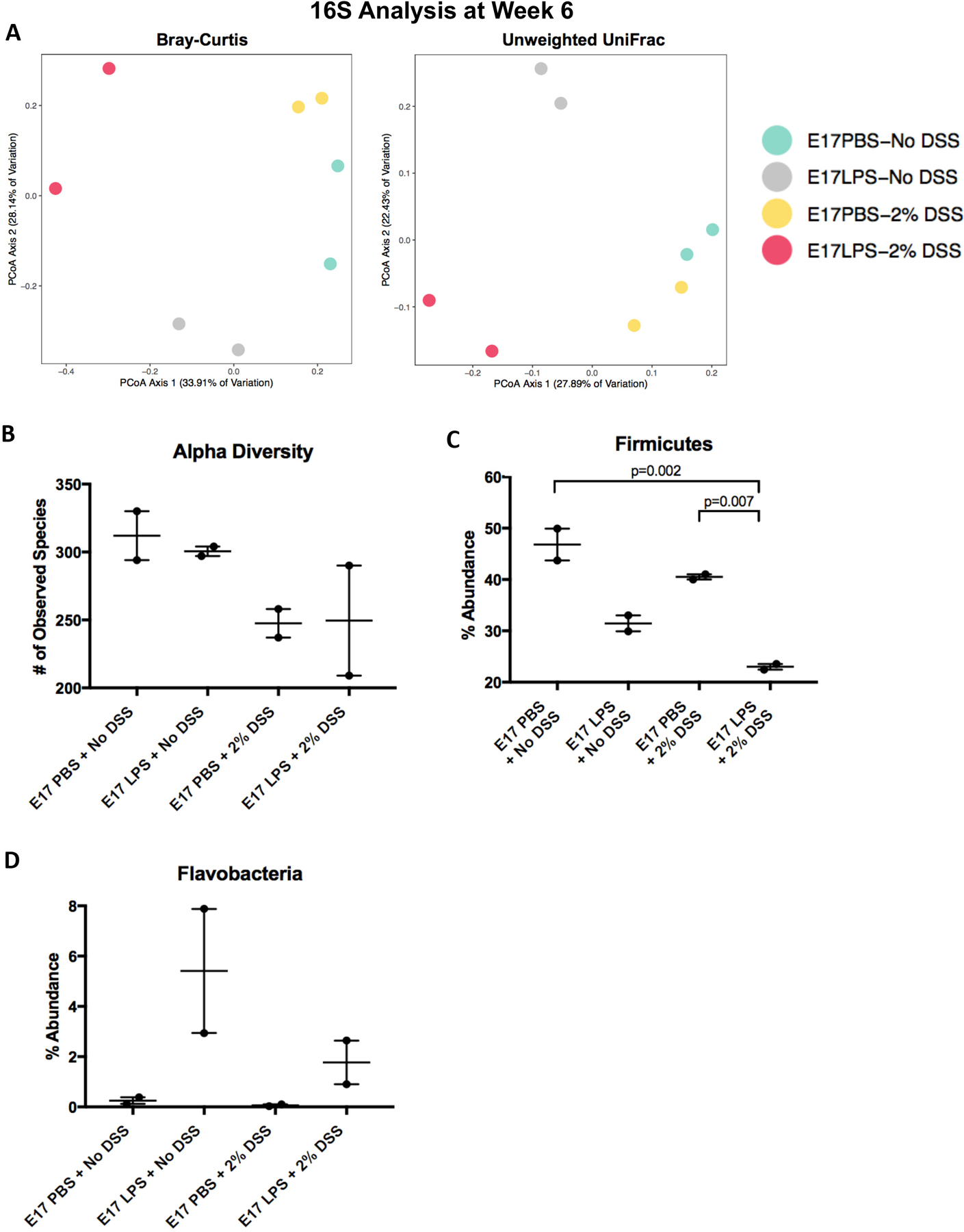
A: Diversity analysis using unweighted UniFrac and Bray-Curtis dissimilarity analyses of 6 week old mice in the indicated group, with and without colitis, that were exposed to saline (PBS) or LPS at E17; B: Analysis of alpha diversity amongst mice exposed to DSS at five weeks compared to those not exposed to DSS with LPS or PBS in utero exposure; C-D: Percent abundance of Firmicutes (C) and Flavobacteria in 6 week old mice exposed to PBS or LPS (2mg/kg) in utero. 16S rRNA sequencing of stool collected from each exposure group. N=2 for all groups and t-tests were performed to determine significance.
Discussion
The current findings support our initial hypothesis that in utero exposure to LPS influences the subsequent development of colitis in the adult. Specifically, we have now shown that mice exposed in utero at E17 to LPS and later DSS have increased mucosal thickness, decreased Tlr4, and decreased IL1β, Tnfα, and IL-6 expression compared to E17 PBS plus DSS exposed mice. Mice exposed to DSS that were exposed to LPS at E17 reveal minimal goblet cell and tight junction loss and showed no signs of colonic injury at colonoscopy.
Taken together, these findings reveal an informative window of LPS exposure in utero for protection against the subsequent development of colitis. We also note that the current study includes distinct comparisons of mice of both gender. In human studies, while there are reports of a male predisposition to ulcerative colitis development, these are largely in populations over the age of 45. Our model at 5-weeks in the adult mouse better mimics an earlier onset of disease in humans, where sex-specific difference in measures of weight loss and colon length are less consistently observed (17,18). These results provide insights into how the in utero immune environment can influence postnatal susceptibility to disease, and in particular to the development of ulcerative colitis, a condition which has been linked in the past to changes in the maternal environment, but for which there was prior little evidence to support this possibility. It is tempting to speculate that, given our prior description that the in utero activation of TLR4 regulates goblet cell differentiation through Math1 signaling, the early exposure to LPS could have long lasting effects on goblet cell production which we now identify (13).
The results suggest that in utero environment represents a key period of vulnerability in the development of the intestinal immune system. Ulcerative colitis has been shown experimentally to be caused by a variety of factors, one of which is dysregulation of the immune system (1,5). In the healthy intestine, the intestinal epithelium serves as a selectively permeable barrier, protecting tissue from pathogens that populate the lumen upon ingestion, absorbing nutrients from ingested food, and allowing the transfer of fluids and electrolytes between the tissue and lumen (2,19,20). Barrier integrity is achieved via the function of the intestinal epithelial cells, which surround the lumen of the colon and secrete mucin via MUC2 that acts as both a lubricant and is home to the intestinal microbiota, and tight junctions, which allow for selective passage of nutrients through the intestinal barrier (19–21). However, in ulcerative colitis, the intestine loses its selective permeability, instead allowing microbial pathogens to impact the colon and subsequently cause inflammation. Epithelial breakdown is also due, in part, to a loss of tight junctions due to a decrease in the number of ZO-1 proteins present in the tissue and reduced goblet cell function via decreased Muc2 protein levels (19,22). Previous studies have shown that reduced Muc2 protein levels had a strong association with ulcerative colitis, indicating an interruption of the necessary post-transcriptional modification of Muc2 in the breakdown of the epithelium seen in ulcerative colitis (23,24). Our experiments have shown that in utero LPS exposure protects the colon from this breakdown, in part due to a muted immune response, in the setting of subsequent chemically-induced colitis exposure.
Though usually a symbiotic relationship, alterations in microbiotic diversity can lead to intestinal inflammation. Overall, ulcerative colitis is characterized by a decrease in alpha diversity, a measure of species richness and diversity, as well as an increase in populations of beneficial bacteria and a decrease in populations of harmful bacteria in the colon (1,25–27). This reduction in alpha diversity was seen in our preliminary experiments among mice that received DSS exposure, regardless of their in utero exposure. We also saw a significant decrease in Firmicutes populations in both groups that received E17 LPS exposure. This is consistent with previous literature that found that increased Firmicutes levels are associated with reduced GLP-2. Reduced GLP-2 has been shown to cause worse colonic injury in a DSS mouse model due to a breakdown of the intestinal barrier. Interestingly, we also saw a significant increase in the abundance of amongst all mice exposed to E17 LPS. In 2016, Jiae, Hwang, and Cho demonstrated that a member of the genus Flavobacterium, Chryseobacterium sp., induces immune tolerance when injected into the gut, though there is little evidence that this genus has an effect on organisms other than freshwater fish (28). In addition to a decrease in Muc2 and increase in Hes 1 expression, epithelial breakdown also occurs when levels of glucagon-like peptide 2 (GLP-2), an intestinal epithelium growth factor responsible for barrier maintenance and prevention of LPS accumulation, are decreased. Changes in the number of Bacteroides and Firmicutes, two of the four major phyla within the gut microbiome, have been linked to reduced GLP-2 levels. Exposure to LPS in utero could alter these populations and cause a cascading effect in which Bacteroides increases in abundance and Firmicutes decreases in abundance. This would then increase GLP-2 levels and reduce intestinal permeability and overall LPS concentration, as LPS would not be able to cross the epithelial barrier. Increased barrier function would reduce any potential inflammatory response via TLR4 and, subsequently, NF-kB expression. These findings speak to the possibility that prenatal LPS exposure has a direct effect on the makeup of the microbiome, which impacts the protective effects described above. Though these results provide potential novel insights into the makeup of the microbiome after prenatal LPS exposure and the subsequent protective effects described, further confirmatory studies using greater numbers of mice, or even in different species including human, are required before definitive conclusions can be drawn.
In aggregate, the current findings support the notion that in utero LPS exposure protects mice from developing experimental ulcerative colitis. The protective effect may be due to changes in the microbiome that lead to tissue protection via immune tolerance, or to direct effects on the mucosal epithelium. Despite the confines of the animal model, the approach used in this study contributes to the growing body of work surrounding the effect of prenatal bacterial exposure on postnatal immune function. It is hoped that through a focus on maternal-fetal interactions during a vulnerable window of mucosal development, we can gain insights into the processes leading to the development of inflammatory diseases including IBD, and thus develop novel therapeutic approaches against these conditions.
Supplementary Material
Acknowledgements:
DJH is supported by R01GM078238 and R01DK083752 from the National Institutes of Health and the Robert James Garrett Fund for the Surgical Treatment of Children from the Johns Hopkins University.
Footnotes
Conflict of interest statement: None of the authors have any conflict of interest with the current work.
References
- 1.Shapiro JM, Subedi S, LeLeiko NS. Inflammatory Bowel Disease. Pediatr Rev 2016;37:337–47. [DOI] [PubMed] [Google Scholar]
- 2.Guan Q A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. 2019;7247238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brant SR. Update on the heritability of inflammatory bowel disease: The importance of twin studies. Inflamm Bowel Dis. 2011;17:1–5. [DOI] [PubMed] [Google Scholar]
- 5.Sairenji T, Collins KL, Evans DV. An Update on Inflammatory Bowel Disease. Prim Care Clin Off Pract. 2017;44:673–92. [DOI] [PubMed] [Google Scholar]
- 6.Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015; 12:205–217. [DOI] [PubMed] [Google Scholar]
- 7.Abegunde AT, Muhammad BH, Bhatti O, Ali T. Environmental risk factors for inflammatory bowel diseases: Evidence based literature review. World J Gastroenterol. 2016;22:6296–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aspberg S, Dahlquist G, Kahan T, Kallen B. Fetal and perinatal risk factors for inflammatory bowel disease. 2006;95:1001–4. [DOI] [PubMed] [Google Scholar]
- 9.Sonntag B, Stolze B, Heinecke A, Luegering A, Heidemann J, Lebiedz P, et al. Preterm birth but not mode of delivery is associated with an increased risk of developing inflammatory bowel disease later in life. Inflamm Bowel Dis. 2007;13:1385–90. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein CN, Burchill C, Targownik LE, Singh H, Roos LL. Events within the first year of life, but not the neonatal period, affect risk for later development of inflammatory bowel diseases. Gastroenterology 2019;156:2190–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim YE, Lee M, Gu H, Kim J, Jeong S, Yeo S, et al. HIF-1α activation in myeloid cells accelerates dextran sodium sulfate-induced colitis progression in mice. Dis Model Mech. 2018. 11:dmm033241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park YH, Kim N, Shim YK, Choi YJ, Nam RH, Choi YJ, et al. Adequate dextran sodium sulfate-induced colitis model in mice and effective outcome measurement Method. J Cancer Prev. 2015. 20:260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sodhi CP, Neal MD, Siggers RS, Sho SS, Ma C, et al. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012. 143:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Håkansson, Tormo-Badia N, Baridi A, Xu J, Molin G, Hagslätt ML, et al. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin Exp Med. 2014;15:107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagalingam N a, Kao JY, Young VB. Microbial ecology of the murine gut associated with the development of DSS-colitis. Imflammatory Bowel Dis. 2012;17:917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alam MT, Amos GCA, Murphy ARJ, Murch S, Wellington EMH, Arasaradnam RP. Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog. 2020;12:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greuter T, Manser C, Pittet V, Vavricka SR, Biedermann L. Gender Differences in Inflammatory Bowel Disease. Digestion. 2020. 1–7. doi: 10.1159/000504701. [DOI] [PubMed] [Google Scholar]
- 18.Shah SC, Khalili H, Gower-Rousseau C, Olen O, Benchimol EI, Lynge E, et al. Sex-based differences in incidence of inflammatory bowel diseases—pooled analysis of population-based studies from western countries. Gastroenterology 2018. 155:1079–1089 [DOI] [PubMed] [Google Scholar]
- 19.Henderson P, Van Limbergen JE, Schwarze J, Wilson DC. Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm Bowel Dis. 2011. 17:382–395. [DOI] [PubMed] [Google Scholar]
- 20.Okumura R, Takeda K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med. 2017;49:e338–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson LW, Artis D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. [DOI] [PubMed] [Google Scholar]
- 22.Johansson MEV, Holmén Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A. 2011;108:4659–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanski C, Born M, Foss HD, Marowski B, Mansmann U, Arastéh K, et al. Defective post-transcriptional processing of MUC2 mucin in ulcerative colitis and in Crohn’s disease increases detectability of the MUC2 protein core. J Pathol. 1999;188:304–11. [DOI] [PubMed] [Google Scholar]
- 24.Ma X, Dai Z, Sun K, Zhang Y, Chen J, Yang Y, et al. Intestinal epithelial cell endoplasmic reticulum stress and inflammatory bowel disease pathogenesis: An update review. Front Immunol. 2017;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog 2017;106:171–181. [DOI] [PubMed] [Google Scholar]
- 26.Earley H, Lennon G, Balfe Á, Coffey JC, Winter DC, O’Connell PR. The abundance of Akkermansia muciniphila and its relationship with sulphated colonic mucins in health and ulcerative colitis. Sci Rep. 2019;9:15683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen ZH, Zhu CX, Quan YS, Yang ZY, Wu S, Luo WW, et al. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J Gastroenterol. 2018;24:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J, Hwang S, Cho S. Immune tolerance to an intestine-adapted bacteria, Chryseobacterium sp., injected into the hemocoel of Protaetia brevitarsis seulensis. Sci Rep. 2016. 6;31722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bramanti P, Hybrid mice. Charles River Laboratories Technical Bulletin 1999. 1:13–16 [Google Scholar]
- 30.Burd I, Brown A, Gonzalez JM, Chai J, Elovitz MA. A mouse model of term chorioamnionitis: Unraveling causes of adverse neurological outcomes. Reprod Sci. 2011;18:900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burd I, Balakrishnan B, Kannan S. Models of Fetal Brain Injury, Intrauterine Inflammation, and Preterm Birth. Am J Reprod Immunol. 2012;67:287–294. [DOI] [PubMed] [Google Scholar]
- 32.Burd I, Chai J, Gonzalez J, Ofori E, Monnerie H, Le Roux PD, et al. Beyond white matter damage: fetal neuronal injury in a mouse model of preterm birth. Am J Obstet Gynecol. 2009;201:279.e1–e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.