

Abstract
NADPH oxidase 4 (NOX4) is the most abundant NOX isoform in the kidney; however, its importance for renal function has only recently emerged. The NOX4-dependent pathway regulates many factors essential for proper sodium handling in the distal nephron. However, the functional significance of this pathway in the control of sodium reabsorption in the initiation of chronic kidney disease is not established. We show that genetic ablation of Nox4 in Dahl salt-sensitive (SS) rat attenuates a high-salt (HS)-induced increase in epithelial Na+ channel (ENaC) activity in the cortical collecting duct. We also found that H2O2 upregulated ENaC activity, and H2O2 production was reduced in both the renal cortex and medulla in SSNox4−/− rats fed an HS diet. NaCl cotransporter expression was increased in the streptozotocin model of hyperglycemia-induced renal injury compared to healthy controls, while expression values between SS and SSNox4−/− groups were similar. ENaC activity in hyperglycemic animals was elevated in SS but not SSNox4−/− rats. These data emphasize a critical contribution of the NOX4-mediated pathway in maladaptive upregulation of ENaC-mediated sodium reabsorption in distal nephron in the conditions of HS- and hyperglycemia-induced kidney injury.
Keywords: chronic kidney disease, salt-sensitive hypertension, NOX4, ENaC, diabetic nephropathy, H2O2
INTRODUCTION
Chronic kidney disease (CKD) is a progressive disorder characterized by the impaired ability of the kidney to control fluid and electrolyte balance and to excrete metabolic wastes. CKD afflicts millions of Americans, accounts for significant morbidity and kidney failure. Although a variety of pathological conditions can cause CKD, specifically hypertension and diabetes mellitus are responsible for the majority of renal injury and progressive deterioration of kidney function and structure (1). Identification of the underlying molecular mechanisms will help to develop appropriate preventive therapies.
Evidence from clinical observations and basic science studies suggests that the progression of renal dysfunction in CKD is associated with an increase in oxidative stress as a consequence of reactive oxygen species (ROS) overproduction, an impaired antioxidant system, and mitochondrial function (2-4). Although many endogenous sources of ROS were described in the kidney, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, and specifically the most abundant isoform in the kidney NADPH oxidase 4 (NOX4), are generally accepted as their major producers (5, 6). NOX4 is expressed in several compartments of the kidney, and it was reported that its abundance could be altered by multiple factors that play an essential role in the initiation and progression of kidney injury, such as hyperglycemia, high salt (HS) intake in salt-sensitive rats (SS) rats, as well as other factors important for kidney disease progression (5, 7, 8). Nox4 deletion or pharmacological targeting of this NADPH oxidase was shown to reduce oxidative stress and renal injury in many renal and cardiovascular pathological conditions (9). Global Nox4 knockout (10, 11) and apocynin treatment (12, 13) decrease SS hypertension by reducing ROS levels (14). In streptozotocin (STZ)-induced diabetic ApoE−/− mice, administration of the most specific Nox1/4 inhibitor, GKT137831, replicated the renoprotective effects of Nox4 deletion (15). The novel pan-NOX inhibitor APX-115 also demonstrated similar protective effects in diabetic nephropathy (16). Together these studies emphasize the significance of the NOX4-dependent pathways in the development of CKD pathophysiology.
NOX4 is highly expressed in renal tubular segments, including the aldosterone-sensitive distal nephron (ASDN), which consists of the late distal convoluted tubule (DCT) and collecting duct (CD) (17, 18). Although sodium is reabsorbed along the whole nephron, the ASDN has a pivotal role in final urinary Na+ excretion. Na+ reabsorption in the CD is mainly mediated by the epithelium sodium channel (ENaC) (17). Upregulation of ENaC expression and activity is observed in experimental models of HS-induced hypertension (Dahl SS rats), and in an STZ model of type 1 diabetes, that might be a physiological compensation for the water and electrolyte wasting that is associated with these disease states. However, this also contributes significantly to the progression of hypertension (19). Numerous studies have shown that ENaC regulation by endocrine factors, such as angiotensin II (Ang II), prorenin, and insulin, increase epithelial sodium transport in the cortical collecting duct (CCD) through ROS-dependent mechanisms (20). Lu and colleagues reported that activation of prorenin receptors increases ENaC activity in mpkCCD immortalized cell culture, and both siRNA-mediated NOX4 knockdown and the dual NOX1/4 inhibitor GKT137892 blunts the effect of prorenin (21). ENaC activity might also be directly upregulated by the application of H2O2 in A6 distal nephron cells (22). Furthermore, our recent studies revealed that Dahl SS rats lacking Nox4 have lower blood pressure and renal injury when placed on an HS diet than wild type controls (10). Finally, Nox4 deletion protects against Ang II-mediated arterial and pulse pressure increase in an experimental model of hypertension in mice (23). These data suggest that ENaC upregulation in the context of renal injury might result from overstimulation of the NOX4-dependent pathway and increased ROS generation.
In contrast to NOX1-3 and NOX5, which are activated by specific agonists or Ca2+,and depend on cytosolic subunits, NOX4 is constitutively active and H202 production modulated by NOX4 expression levels (24). According to Rajaram and colleagues, the constitutive character of NOX4 activity may represent a “double-edge sword” for the kidneys and the heart. The authors summarize existing literature and conclude that although targeting NOX4 is beneficial in some cardiorenal conditions (e.g. in diabetic glomerulopathy, cardiac remodeling and hypertension), a number of detrimental effects such as tubule-interstitial injuries, atherosclerosis and cardiac ischemia may occur. This duality requires a careful evaluation of the potential adverse effects of NOX4 inhibition (9). The present study aimed to investigate the influence of Nox4 deletion in Dahl SS rats on the production of renal H2O2 and epithelial Na+ transport in the ASDN in two different models of CKD: salt-induced hypertension and STZ-induced type 1 diabetes.
MATERIALS AND METHODS
Experimental protocol and animals
Animal use and welfare procedures adhered to the ARRIVE guidelines and National Institutes of Health Guide for the Care and Use of Laboratory Animals. The following protocols were reviewed and approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. Experiments were performed on male Dahl salt-sensitive (SS; SS/JrHsdMcwi) and SSNox4−/− rats bred at the Medical College of Wisconsin. SSNox4−/− rats were created using zinc-finger nucleases technology. Genetic manipulations resulted in an 8 bp deletion in the Nox4 exon 7 and appearance of an early stop codon that caused missing most of the C-terminal (10). Rats were provided with normal salt food (0.4% NaCl AIN-76 purified rodent chow; Dyets #113755, Bethlehem, PA) and water ad libitum. The study was performed using two main protocols, as described below. Tissues for biochemical and electrophysiological experiments described in Protocol 2 were collected from the same animals as recently used for glomerular analysis (25).
Protocol 1.
For the study of the role of NOX4-dependent pathway in the model of HS-induced hypertension, eight weeks old SS and SSNox4−/− rats were switched to an HS diet (4%, Dyets #113756) for either three days or three weeks for the electrophysiological measurements. To collect urine, rats were placed in metabolic cages for 24 hrs periods on days 0, 3, and 21 of HS diet supplementation. Urine electrolytes and creatinine were measured with a blood gas and electrolyte analyzer (ABL system 800 Flex; Radiometer, Copenhagen, Denmark). At the end of the experiments, rats were euthanized and their kidneys were flushed and collected for electrophysiological analysis.
Protocol 2.
For the study of the role of Nox4-dependent pathway in the model of type 1 diabetes, 7 week old SS and SSNox4−/− rats received one intraperitoneal (i.p.) injection of streptozotocin (STZ, 75 mg/kg; Sigma-Aldrich, St Louis, MO) or vehicle (50 mM sodium citrate, pH 4.5) for controls as previously described (25-27). Briefly, seven days after STZ injection, both SS and SSNox4−/− groups of rats underwent surgery for subcutaneous implantation of slow-release insulin pellets (LinShin, Canada) to maintain moderate hyperglycemia (approximately 300 mg/dl) throughout the protocol timeline. Control groups were implanted with blank pellets. Rats were fed a normal salt (0.4% NaCl) diet throughout the protocol. At the end of the experiments, rats were euthanized, and kidneys were flushed and collected for electrophysiological measurements or Western blot analysis.
Measurement of H2O2 level in vivo
The detailed application of the biosensor amperometry technique to assess H2O2 levels in the kidney was previously described (28, 29). Briefly, animals were prepared using an anesthetic mixture of ketamine (20 mg/kg, intramuscularly) and inactin (50 mg/kg, i.p.) and placed on a temperature-controlled surgical table. Supplementary anesthetic (inactin) was administered i.p., as required. A midline incision was made to expose and isolate the left kidney from the surrounding fat before placing it in a stainless-steel kidney cup to reduce breathing artifacts (Fig. 4A). The exposed kidneys were not denervated. Following surgery and a 20-min equilibrium period, control baseline measurements were recorded. A H2O2-sensitive biosensor (tip − 0.5 mm; Sarissa Biomedical, UK) was inserted into the kidney cortex or medulla with a micromanipulator (MN-153, Narishige International USA, Inc). Interstitial infusion of catalase (2 μg/ml; #C40, ≥10,000 units/mg protein, Sigma-Aldrich) was performed directly to the kidney via an implanted catheter connected to a peristaltic pump to scavenge interstitial H2O2 and block the signal detected by the biosensors. The obtained levels of the catalase sensitive current were calculated as interstitial H2O2 concentrations corresponding to a linear calibration curve performed before each study as previously described (28, 29).
Electrophysiology
Electrophysiological recordings were performed using the cell-attached patch-clamp technique in a voltage-clamp configuration. ENaC activity was measured in split-open freshly isolated CCDs. Renal tubules were isolated manually or using a vibrodissociation technique described previously (30, 31). Recordings have been made using extracellular solution (in mM): 150 NaCl, 2 MgCl2, 10 HEPES (pH 7.4). Patch pipettes were filled with a solution of the following composition (in mM): 140 LiCl, 2 MgCl2, and 10 HEPES (pH 7.35). Resistances of patch pipettes ranged from 7 to 12 MΩ. After a high resistance seal was obtained, the cell-attached recordings were performed immediately and analyzed as described previously (30, 31).
Western blotting
Kidney cortical lysates were prepared as follows: the kidney cortex was excised immediately after perfusion and snap-frozen in liquid nitrogen. Cortical sections were cut, weighed and dissolved in Laemmli with protease inhibitors cocktail (Roche) at 20 mg/ml with pulse sonication for 5-10 sec. Samples were subjected to PAGE, transferred onto a nitrocellulose membrane (Millipore) for probing with antibodies against total and phosphorylated NCC kindly provided by Dr. David H. Ellison (Oregon Health & Science University).
Statistical analysis
Data presented as mean±SEM. To test for a significant difference among means, a two-factor ANOVA was applied. For two mean comparisons, unpaired, two-tailed, Student’s t-tests or non-parametric Mann-Whitney U test was performed. For paired experiment data, a Wilcoxon signed-rank test was used. For all hypothesis testing, a significance threshold of p<0.05 was used.
RESULTS
Nox4 deletion increases HS-induced diuresis and natriuresis, and prevents ENaC activation in SS rat
SSNox4−/− rats fed an HS diet was recently demonstrated to develop significantly lower blood pressure and lesser levels of albumin excretion, tubular necrosis and glomerular injury than SS controls that implicates a role of NOX4-dependent signaling in salt-induced hypertension and associated renal injury (10). Since impairment of pressure natriuresis/diuresis dependency is considered a major contributor to blood pressure elevation in this model of hypertension, our first aim was to test whether Nox4 deletion in SS rats improves sodium/water handling under HS intake. In the model of HS-induced hypertension, blood pressure increase is well established to occur in two phases: early reversible phase, which lasts 3-7 days, followed by the second phase that lasts weeks and is associated with irreversible kidney damage (32, 33). Figure 1A shows a schematic protocol of our experiment, where SS and SSNox4−/− male rats fed a normal salt diet (0.4% NaCl) were switched to an HS (4% NaCl) chow for either 3 days or 3 weeks with subsequent 24-hrs urine collections and analysis. Figure 1 illustrates that the HS diet increases urinary volume and sodium excretion similarly to earlier reports (34, 35). HS diet and associated hypertension led to renal damage, which can be detected by elevated albuminuria. As shown in Figs. 1B,C SSNox4−/− rats demonstrated increased daily diuresis and natriuresis on day 3 of dietary intervention indicating improved pressure-natriuretic response at an early stage. We also confirmed earlier findings (10) that SSNox4−/− animals have lower albuminuria (Fig. 1D). Urinary excretion of potassium and creatinine exhibited similar trends throughout the experiment, and no statistical difference is found between genotypes (Figs. 1E,F), which is indicative of equal food intake in both groups.
Figure 1.

Nox4 deletion increases diuresis in SS rats. A) Experimental protocol of the studies with high salt diet. Wild type male SS and SSNox4−/− rats were kept on a normal (0.4% NaCl) salt diet before switched to a high salt (4% NaCl) diet for three weeks. Urine samples were collected before and after (3 days and 3 weeks) switch to a high salt diet. Patch clamp analysis was performed at the same time points. B) 24-hr urine volumes collected in metabolic cages. Daily urinary excretion of sodium (C), albumin, (D) potassium (E) and creatinine (F) normalized to 100 g body weight. * p<0.05, ** p<0.01, *** p<0.001.
Previous studies demonstrated that HS diet induces increased ENaC activity in CCDs, which may contribute to the development of HS-induced hypertension (19, 34). To test the involvement of NOX4 in the upregulation of ENaC in our model, channel activity was probed with the patch-clamp technique in freshly isolated split opened CCDs. Fig. 2 illustrates representative examples of ENaC single-channel current, recorded in the cell-attached mode at different time points of the experiment and corresponding summary graphs of ENaC activity (NPo). Our data revealed that a significant increase in ENaC NPo in CCDs from SS controls could be detected after three days on HS diet, and is sustained with the HS intake. In contrast, the activity of ENaC in SSNox4−/− rats did not increase during the HS challenge (Fig. 2). Po remained stable in all groups and NPo upregulation in SS control rats was mediated by the higher number of active channels (N) − 1.2±0.2, 2.8±0.8 and 2.0±0.2 on Days 0, 3 and 21 respectively, whereas in SSNox4−/− rats N did not exceed 1.1±0.1. These data indicate that the NOX4-dependent pathway is involved in the upregulation of ENaC activity in Dahl SS rats.
Figure 2.
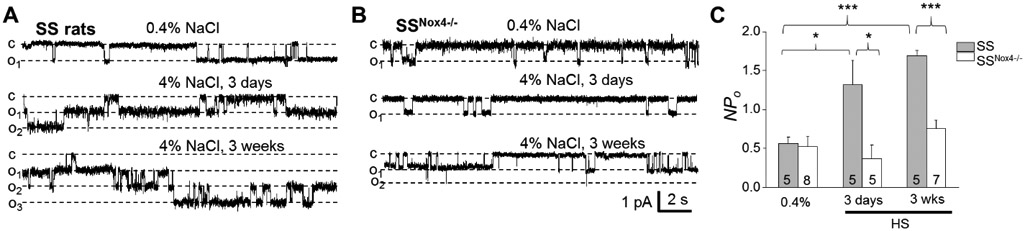
Nox4 deletion prevents HS-induced ENaC activation caused by a HS diet. A, B) Representative current traces from cell-attached patches containing ENaC channels and recorded from the apical membrane of split-open cortical collecting duct tubules. Shown are representative traces from SS (A) and SSNox4−/− (B) rats fed a normal (0.4% NaCl) and high salt (4% NaCl; 3 days and 3 weeks). “c” and “oi” denote closed and opened states of the channel, respectively; scale bars for entire and expanded traces are shown; holding potential is −40 mV; channel openings are downward traces. C) Summary graphs of ENaC activity (NPo) in SS and SSNox4−/− rats fed corresponding diets. Number of experiments is shown. * p<0.05.
NOX4 modulates ENaC activity via control of H2O2 level in the kidney
A group of studies reviewed in Cowley et al. (3) indicated that ROS’s excessive renal production increases sodium retention. It was previously demonstrated that elevated H2O2 level in the renal medulla of SS rats contributes to substantial differences in blood pressure between SS and control animals. While changes in H2O2 levels in the renal cortex were also observed (36), whether oxidative stress, and particularly H2O2, modulates ENaC properties in cortical segments of SS rat distal nephron has not been determined. To address this question, we evaluated the effect of H2O2 applications on ENaC activity in CCDs freshly isolated from adult SS rats. Fig. 3A shows a representative example of single-channel ENaC activity recorded in cell-attach mode in the principal CCD cell apical membrane before and after the addition of 0.5 mM H2O2 into the bath. Summary data reveals that the application of H2O2 to the bath solution results in increased ENaC NPo from 0.92 ± 0.40 to 1.84±0.4 (p=0.03; Fig. 3B).
Figure 3.
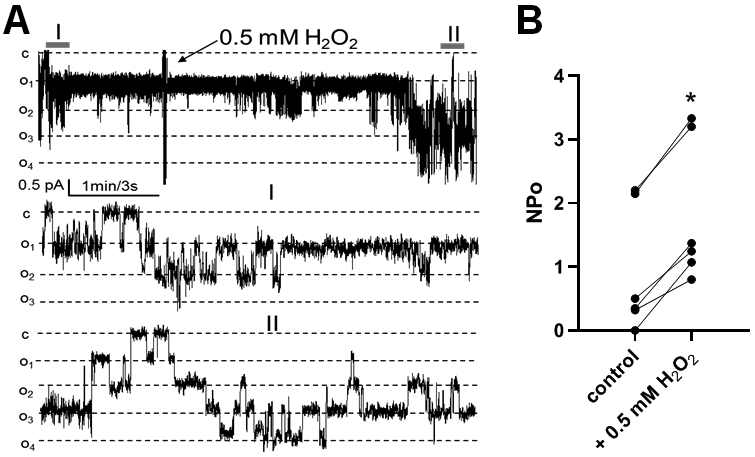
Effect of H2O2 application on ENaC activity in CCDs of SS rat. A) Representative current trace of ENaC activity measured at test potential of −60 mV before (I) and after (II) the addition of 0.5 mM H2O2 to bath solution. Dashed lines show respective current levels with “c” and “oi” denoting closed and opened states, respectively. B) Summary graph of ENaC activity (NPo) recorded before and 3-5 min after application of 0.5 mM H2O2. *p < 0.05.
Next, we examined Nox4 contribution to the interstitial levels of H2O2 levels in renal medulla and cortex of hypertensive SS rats (HS diet for 3 weeks). Figs. 4A,B represent the procedure and representative experiment for detecting renal interstitial H2O2 levels in anesthetized SS and SSNox4−/− rats using in vivo biosensors amperometry. The H2O2 signal's specificity was confirmed with an interstitial application of catalase enzyme, known for the high efficiency for converting H2O2 to water and oxygen. Our data revealed that SSNox4−/− rats exhibit a lower H2O2 concentration in both medullar and cortical renal tissues compared to SS control rats (Fig. 4C). Taken together, these data suggest that decreased H2O2 levels may underlie increased natriuresis and the absence of increased ENaC activity in SSNox4−/− rats fed with an HS diet.
Figure 4.

SSNox4−/− rats exhibit lower renal H2O2 levels on a HS diet. A) An example of the preparation for the measurement of H2O2 levels in vivo by the biosensors amperometry. The kidney was exposed and mounted in a kidney cup. The biosensor was inserted into the kidney cortical or medullar layer and connected to a dual-channel potentiostat for amperometry recordings. The reference electrode was placed onto the kidney surface and attached to the potentiostat to ensure reduced electrical noise. Interstitial perfusion of catalase was simultaneously performed. B) Representative recording of interstitial H2O2 levels. Perfusion of catalase (2 μg/ml) is shown. C) Summary graphs of the renal cortex and medulla interstitial H2O2 in SS and SSNox4−/− rats fed a HS diet. *p < 0.05.
Nox4 deletion prevents ENaC activation in the STZ model of diabetic nephropathy in Dahl SS rats
Production of ROS plays an essential role in the pathogenesis of diabetic nephropathy. The ameliorative effect of Nox4 deletion or pharmacological inhibition of the NOX4-dependent pathway on hyperglycemia-induced kidney injury was reported in rodent models of type 1 diabetes (25, 37-39). Also, the higher expression of membrane proteins responsible for sodium reabsorption in distal nephron was reported previously in STZ-treated Sprague-Dawley rats (40). Dahl SS rats were recently reported to be more susceptible to the development of STZ-induced renal injury than other strains (27, 41, 42); however, the extent of the sodium reabsorption machinery affected in this strain under the conditions of hyperglycemia has yet to be determined. In our recent studies, we reported that upon induction of type 1 diabetes with STZ, SSNox4−/− rats exhibited less kidney injury than control STZ-treated SS rats (25). We used the same groups of animals to test the contribution of ENaC and NCC in SSNox4−/− rats injected with STZ. We found that ENaC activity (NPo) in the chronically hyperglycemic SS rats was elevated compared to control SS rats (0.71±0.10 in sham controls and 1.27±0.2 in STZ treated conditions; p<0.05) and this effect was attributed to the changes in single-channel open probability (Po was 0.43±0.06 in control vs. 0.86±0.07 in hyperglycemic rats). In SSNox4−/− rats, hyperglycemia only slightly, but not significantly increased ENaC activity with no effect on Po (0.51±0.06 in sham control and 0.51±0.07 in STZ-treated group, respectively), which delineates the importance of NOX4 activation for the regulation of ENaC open probability in this model of diabetic nephropathy (Fig. 5).
Figure 5.
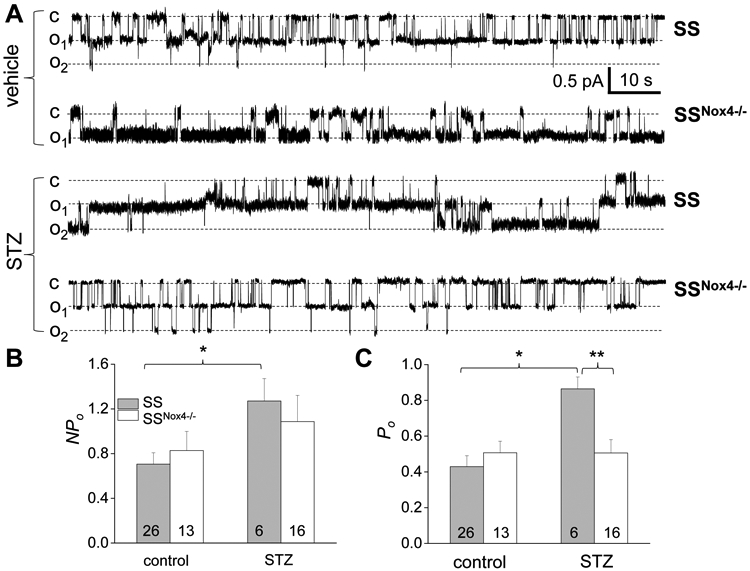
NOX4 deficiency prevents upregulation of ENaC activity in CCDs of STZ-injected SS rats. A) Representative current traces demonstrating ENaC activity in split-open CCDs isolated from vehicle- and STZ-treated SS and SSNox4−/− rats. “c” and “oi” denote closed and opened states of the channel, respectively; test potential is −40 mV. B, C) Summary graphs of total ENaC activity (NPo; B) and the open probability of individual channels (Po; C). Number of experiments is shown. * p<0.05, ** p<0.01, *** p<0.001.
Finally, using Western blot analysis, we found a significantly increased expression of total NCC in kidney tissue of SS rats treated with STZ compared to the vehicle-treated group. The abundance of the phosphorylated form of NCC from renal cortical tissue was also elevated demonstrating a significant activation of this cotransporter under hyperglycemia conditions (Fig. 6). However, in contrast to experiments with ENaC, Nox4 deletion does not affect hyperglycemia-induced upregulation of NCC expression.
Figure 6.
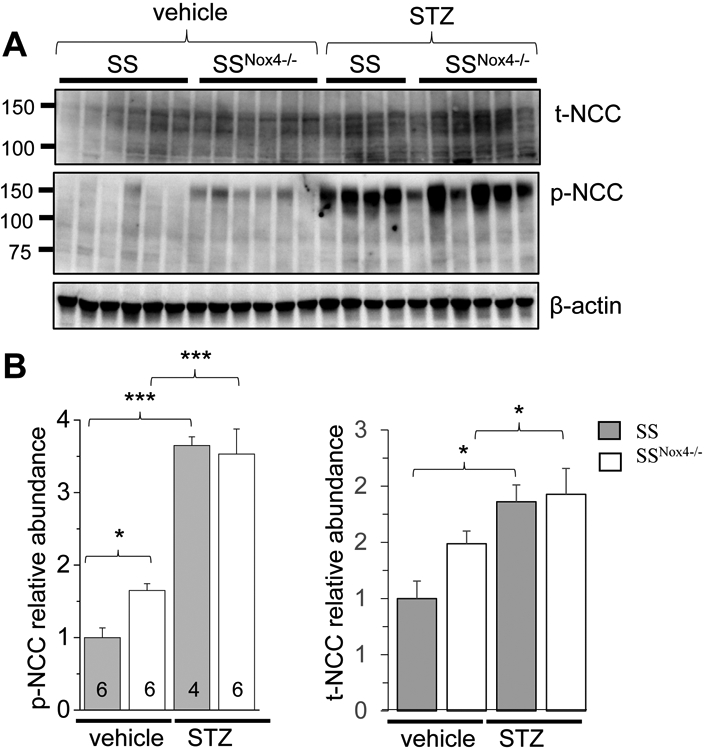
Nox4 deletion does not prevents upregulation of NCC expression in CCDs of SS rat in STZ model of type 1 diabetic nephropathy. A) Western blot analysis of total NCC (t-NCC) and phosphorylated form of NCC (p-NCC) in the renal cortex from vehicle- and STZ-treated SS and SSNox4−/− rats. β-actin was used as a loading control. B) Summary graphs of p-NCC and t-NCC representing the average relative density of the bands (normalized to β-actin) in the groups shown in A. * p<0.05, ** p<0.01, *** p<0.001.
DISCUSSION
Diabetes and hypertension, either alone or synergistically, are recognized as major causes of developing CKD (1, 43). A well-documented feature for both pathologies, which is the overproduction of ROS in the kidney, has been mainly attributed to the upregulation of NOX4-dependent signaling. Increased Nox4 expression was previously shown in the SS hypertension model by Cowley et al.; high salt diet significantly elevated Nox4 mRNA level in the cortex of Dahl SS rats (10). In an 8-week extended diabetic model induced by STZ injection in Wistar rats, Etoh and colleagues demonstrated an increase of Nox4 mRNA by ~35% in the cortex and twice in medulla (37), which may have contributed to a 2/3 increase in renal H2O2 levels observed in the Wistar Furth rat background (44). A 75% cortical Nox4 mRNA increase was also shown in STZ-induced type 1 diabetes in Sprague-Dawley rats (45). Nox4 deletion or its inhibition ameliorated the high salt or high glucose-induced kidney injury, underscoring the important role of NOX4-dependent pathways in the progression of kidney dysfunction. However, the underlying mechanisms are largely unidentified. In the present study we show that: a) Nox4 deletion in Dahl salt-sensitive (SS) rat challenged with an HS diet attenuates HS-induced upregulation of ENaC activity in CCDs; b) H2O2 upregulated ENaC activity in freshly isolated CCDs; c) H2O2 levels in the kidney of SSNox4−/− fed with HS was significantly lower than that of similarly treated SS controls; d) upregulation of renal NCC expression was similar in SS and SSNox4−/− groups in the conditions of STZ-induced hyperglycemia; and e) ENaC activity in hyperglycemic animals was elevated in SS controls but not SSNox4−/− rats.
Ample experimental data obtained in Dahl SS rats support the use of this strain as a model of salt-induced hypertension. Increased NADPH oxidase activity causing ROS hyperproduction was identified as a significant contributor to the development of hypertension (12). SS rats have an initial rapid and reversible increase in blood pressure observed within a few days on the HS diet, followed by a slow aggravation of hypertension associated with an increasingly irreversible component (33). The development of SS hypertension in these rats is mainly attributed to the reduction in renal excretory function, which leads to the progressive rightward shift of chronic pressure-natriuresis relationship and, thus, promoting maladaptive retention of salt and water, and the expansion of extracellular fluid volume, which ultimately results in the increase of blood pressure (33, 46). Cowley and colleagues previously identified H202 as a regulator of the pressure natriuresis response (47, 48). Here we observed a robust natriuretic response to an HS in SSNox4−/− rats on Day 3 of the dietary challenge. However, by the end experiment, urinary volume and sodium excretion became similar in both groups. Therefore, NOX4-deficient rats might appear to more robustly able to compensate to a HS load with a greater natriurestic response compared to SS rats thus hindering the renal damage. Longer high salt administration in SSNox4−/− rats leads to renal injury, which is detected by a significant increase of albuminuria (Fig. 1D), and sodium retention at Day 21. It was shown previously that in salt-resistant animal strains, ENaC is downregulated in response to an HS challenge (19). In contrast, an HS diet for several weeks induced in Dahl SS rats a substantial increased ENaC expression and activity, which significantly exacerbates the ramifications of HS excess (19).
The important role of ENaC in the development of HS-induced hypertension was confirmed in previous studies, where treatment with benzamil and amiloride (ENaC inhibitors) during HS consumption attenuated blood pressure elevation (34, 49). Interestingly, in our hands, a significant ENaC NPo increase was observed in SS rats as early as the third day of the HS diet (Fig. 2C) and during the later course of the HS challenge. This observation suggests that ENaC upregulation contributes to both the initial and late stages of hypertension progression in this model. However, in our previous studies with benzamil pretreatment, we did not observe a significant effect of ENaC inhibition on blood pressure during the early stage of HS-induced hypertension development (34). Moreover, although ENaC activity was low in the knockout rats, NOX4 deficiency did not improve natriuresis on the late stage of hypertension, indicating that in the developed renal damage, other parts of the nephron provide a major impact to natriuresis. The most probable candidate is the medullary thick ascending limb (mTAL) of the loop of Henle, where ROS increases sodium reabsorption via NKCC2 (50).
In SSNox4−/− rats fed a standard diet (0.4 % NaCl) we did not observe differences in either daily urine output or in urinary sodium levels compared to the SS control group. When experimental groups were challenged with HS, an increase in fluid and salt levels were visible at the beginning of HS intake in both groups; however, in SSNox4−/− rats, this increase was more striking than in SS controls. Early increase in diuresis and natriuresis in SSNox4−/− rats in response to HS is accompanied by reduced ENaC activity in the distal nephron. As ENaC is a well-recognized contributor to sodium and water retention, we suggest that the difference in urine and salt output in SSNox4−/− and SS control rats challenged with HS could be partly attributed to the elimination of renal sodium reabsorption that emerged from an overproduction of ENaC.
NOX4-dependent pathways are upregulated in the model of HS-induced hypertension and involved in the regulation of many factors important for the control of ENaC activity. In our study application of H2O2 increased ENaC NPo in CCD of SS rats, which complements other studies showing the positive regulation of ENaC activity by ROS. H2O2 application was shown to increase ENaC Po in amphibian cell cultures (22). SS rats on a HS diet exhibited elevated ROS levels in both the renal medulla and cortex, which was shown to contribute to a reduction in medullary perfusion, Na+ excretion, glomerular sclerosis, tubular injury and interstitial fibrosis (2). In our study, SSNox4−/− rats on HS diet exhibit a lower interstitial H2O2 interstitial concentration in both medullar and cortical renal tissues compared to SS control rats, corroborating the important role of NOX4 in the generation of H2O2 in the kidney. Taken together, our data suggest that in the model of HS-induced hypertension, upregulation of NOX4 leads to increased H2O2 production and subsequent ENaC activity. Increased NOX4 activity and NADPH-dependent ROS production was earlier demonstrated in the cortical tissues isolated from STZ-treated rats (51). In diabetes, sodium retention leads to fluid imbalance, edema, and eventually, hypertension (52, 53). Culshaw et al. reported a substantial impairment of pressure natriuresis in STZ-treated Sprague Dawley rats: acute elevation in blood pressure did not increase renal medullary blood flow, tubular sodium reabsorption was not downregulated, and proximal tubule sodium reabsorption, measured by lithium clearance, was unaffected (54). These data suggest that distal tubule sodium reabsorption was not downregulated with increased blood pressure in STZ-treated Sprague Dawley rats, and the tubular defect responsible for an impaired pressure-natriuretic response in early type 1 diabetes mellitus is located in the distal nephron (54). Wild type and Nox4 knockout Dahl SS rats subjected to the STZ treatment in our previous study demonstrated similar sodium excretion in both genotypes (25). Although we did not test pressure-natriuresis in acute settings in the present study, we suggest that pressure-natriuresis in Dahl rats in the setting of STZ-induced diabetes can potentially be impaired due to NOX4-driven ENaC activation, but this speculation requires further investigation.
A plethora of studies reported that ENaC function could be upregulated by a physiologically high glucose level. These high levels increase ENaC mRNA and protein expression in cell cultures (55). Reports on the effect of high blood glucose on ENaC function in vivo significantly vary depending on selected models and genetic backgrounds. However, biochemical analysis shows increased ENaC subunit expression in hyperglycemic animals (40, 56, 57) or following long exposure to media enriched with oligosaccharides (58). However, the effect of systemic hyperglycemia on ENaC activity was not well characterized in STZ-treated Sprague-Dawley or SS rats. This is different from hyperglycemic STZ-treated SD rats due to the genetic background, prehypertension state and other conditions, which enable the SS rat to better mimic clinical nephropathy. Importantly, our observations were performed in STZ-treated SS rats, which develop both hyperglycemia and strong signs of diabetic nephropathy including glomerular damage and proteinuria (25) that is not typical for STZ-induced type 1 diabetes in Sprague-Dawley rats (27). We confirmed that hyperglycemia results in significant activation of ENaC and found that Nox4 deficiency precluded ENaC activation. Our experiments also revealed a dramatically increased NCC expression in response to hyperglycemia, which is in agreement with earlier data from other groups (40, 59). However, in contrast to its effects on ENaC, Nox4 deficiency did not affect NCC levels in STZ-treated animals.
Earlier studies established an interaction between pressure-natriuresis and H2O2 production. In Sprague Dawley rats, increased Na+ and fluid delivery in mTAL perfused in vitro leads to the generation of superoxide, which can be reduced to H2O2 by superoxide dismutase (2, 60, 61). Interestingly, mitochondrial ROS production in mTAL was not mediated by NADPH oxidases (14). Involvement of H2O2 on the cortical collecting ducts has not as understood. Biosensor amperometry revealed increased interstitial H2O2 levels on a 4% NaCl diet, whereas Nox4 deficiency contributes to H2O2 production. As NOX4 is a membrane protein, we suggest that it can serve as both basolateral (or interstitial) and luminal source of H2O2 in CCD. Figure 7 provides a summary scheme illustrating that in pathological conditions, overexpression of NOX4 causes excessive H2O2 production in the cortical interstitium. H2O2 increases ENaC expression in the apical membrane (observed on a HS diet), whereas in the absence of NOX4 ENaC activity remained stable (Fig. 2). Factors capable of mediating the effect of basolateral H2O2 on ENaC are still not completely clear; experiments on immortalized CCD cell cultures showed that H2O2 implicates prostaglandin E2 (PGE2) in the regulation of ENaC (62). Severe oxidative stress in the kidney inhibits the EGF-ERK pathway (63); in normal conditions ERK promotes ENaC subunits phosphorylation and ubiquitination by Nedd4 (64). We can speculate that our data reported here and in earlier studies (34, 65) reflect the following mechanism: HS diet reduces EGF tissue level in Dahl SS rats cortex and increases the number of active ENaC on the plasma membrane, potentially resulting from impaired ubiquitination. The stimulatory effect of H2O2 on ENaC was shown in both basolateral and apical applications to amphibian A6 monolayer (66). Patch-clamp experiments revealed that H2O2 increases ENaC open probability Po in both apical and basolateral applications but did not affect ENaC if applied from the cytosolic side. Interestingly, inhibition of Phosphoinositide 3-kinase (PI3-kinase) with LY290002 prevents the effect of apical (not basolateral) H2O2 application. Data in Fig. 2 indicates that ENaC upregulation on the HS diet was mediated by a higher number of active channels on the membrane without increasing Po of individual channels. We suggest that in Dahl SS rats on a HS diet, basolateral NOX4 and auto/paracrine H2O2 production contribute to ENaC upregulation by recruiting intracellular mechanisms disturbing ENaC recycling from the plasma membrane. An elevated ENaC expression and/or increased trafficking to the membrane also cannot be excluded. Stable Po may reflect low involvement of apical NOX4 and lack of machinery mediating H2O2 signal across the isolated mammalian principal cells (e.g., the effect on cytoskeleton capable of modulating Po). This assumption is supported by the recent data by Kumar et al., indicating equal PI3-kinase and AMPK pathways activity in SS compared to SSNox4−/− rats on a 4% NaCl diet (11).
Figure 7.

Schematic illustration of ENaC regulation by NOX4 in the Dahl SS rats. Elevated expression of NOX4 in hypertension and diabetic conditions increases H2O2 level in the interstitium and on the luminal side. Oxidative stress acts via intracellular pathways (such as modulation of expression and activity of EGF Receptor, PGE2, small GTPase Rac1, and other signaling pathways) to elevate expression of ENaC on the apical membrane (19). Apical H2O2 production in diabetes facilitates ENaC open probability presumably by activation of PI3-kinase signaling.
In the diabetic conditions, NOX4 upregulation increases ENaC Po that indirectly may indicate a strong influence of apical NOX4 derived H2O2 and involvement of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) production in the membrane. Such a phenomenon is in accordance with the literature reporting that diabetes induced by STZ does not increase ENaC mRNA (54) in Sprague-Dawley rats. An increase of total renal ENaC was shown by Ecelbarger et al (40), and the group later dissected this effect and reported that it occurs in the medullary layers, whereas cortical ENaC level did not change (67). The authors also suggested that increased ENaC protein level in diabetes could be mediated by the presence of vasopressin, since diabetes is associated with elevated arginine vasopressin (AVP) (67). Although the AVP increase is a compensatory reaction against the natriuretic loss of sodium in acute diabetes (68), it contributes to renal injury (69, 70). Details on how vasopressin release is stimulated primarily by increases in plasma osmolality and regulates ENaC can be found in reviews by Bankir (71) and Stockand (72). We conclude that in diabetic conditions hyperglycemia increases luminal NOX4-dependent production of H2O2, which stimulates PIP3 production and ENaC Po (Fig. 7)
It was also reported that expression of total cortical NCC was significantly increased in STZ-treated Sprague-Dawley rats (40, 67), which is consistent with our data in the Dahl SS rat background (Fig. 6). The role of NCC in salt sensitive hypertension was reported but findings are less straightforward. In patients with Pseudohypoaldosteronism type II, NCC is constitutively active and not properly suppressed by a high salt diet, leading to abnormally increased salt reabsorption and salt-sensitive hypertension. NCC is upregulated by the WNK -SPAK/OSR1 signaling pathway, which is activated by RAAS, insulin and low potassium diet (73). We also demonstrated that knockout of renin in the Dahl SS background reduces NCC expression (31). However, there is a number of evidence summarized by Zicha et al, indicating that salt-induced hypertension in Dahl SS rats is not dependent on NCC as it depends on ENaC and NKCC2, although some aspects of NCC regulation can activate ENaC (74).
A synergistic effect of hypertension and diabetes is recognized as a driving force causing kidney damage, and an increasing number of studies have addressed the interaction between all factors in the development of CKD (75, 76). ENaC contributes to salt and water imbalance in both conditions, however the channel activity (visible as differential subunits regulation or electrophysiological characteristics) is determined by local ROS levels which depend on the endocrine profile of major hormonal factors such as as RAAS, vasopressin and atrial natriuretic peptides (19, 71). ENaC regulation in the context of superposition of hypertensive and diabetic conditions requires additional investigations. Conway et al. reported that STZ-induced diabetes does not lead to renal injury if the animals remain normotensive. The development of hypertension induced with the genetic renin-2 gene activation dramatically increased kidney damage during hyperglycemia (77). It was further reported that hyperglycemic Dahl SS rats display moderate hypertension and sodium loss (25, 27). Williams’ group generated an obese leptin and inulin insensitive model in the Dahl SS background which exhibits hypertension, euglycemia, hyperlipidemia and massive renal damage on a 1% NaCl diet (78, 79). Further comparison of pressure-natriuresis sensitivity and sodium transporter activity in these models would be helpful to explore the differential hormonal regulation of sodium handling and water-electrolyte balance in polygenic cardiorenal conditions such as metabolic syndrome.
ACKNOWLEDGMENT
This research was supported by the National Heart, Lung, and Blood Institute grants: R35 HL135749, P01 HL116264 (A.W.C. and A.S.), R01 HL122662 (A.W.C. and A.S.) R00 HL116603 (T.S.P.), R00 DK105160 (D.V.I.), American Heart Association grants: 16EIA26720006 (A.S.) and 17SDG33660149 (O. P.), American Society of Nephrology Carl W. Gottschalk Grant (T.S.P), and Department of Veteran Affairs grant I01 BX004024 (to A.S.). The authors declare that they have no conflicts of interest.
Nonstandard Abbreviations:
- Ang II
angiotensin II
- AVP
arginine vasopressin
- ASDN
aldosterone-sensitive distal nephron
- CCD
cortical collecting duct
- DCT
distal convoluted tubule
- DKD
diabetic kidney disease
- EGF
epidermal growth factor
- ENaC
epithelial Na+ channel
- HS
high salt
- mTAL
medullary thick ascending limb
- NCC
Na+-Cl− cotransporter
- NOX4
NADPH oxidase 4
- PGE2
prostaglandin E2
- PI3-kinase
Phosphoinositide 3-kinase
- PIP3
phosphatidylinositol (3,4,5)-trisphosphate
- ROS
reactive oxygen species
- SS
salt-sensitive
- SSNox4−/−
Nox4 knock out in Dahl SS rat background
- STZ
streptozotocin
REFERENCES
- 1.Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, and Rodrigues-Diez RR (2020) Targeting the progression of chronic kidney disease. Nat Rev Nephrol 16, 269–288 [DOI] [PubMed] [Google Scholar]
- 2.Cowley AW Jr., Abe M, Mori T, O'Connor PM, Ohsaki Y, and Zheleznova NN (2015) Reactive oxygen species as important determinants of medullary flow, sodium excretion, and hypertension. Am J Physiol Renal Physiol 308, F179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishimoto Y, Tanaka T, Yoshida Y, and Inagi R (2018) Physiological and pathophysiological role of reactive oxygen species and reactive nitrogen species in the kidney. Clin Exp Pharmacol Physiol 45, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jha JC, Ho F, Dan C, and Jandeleit-Dahm K (2018) A causal link between oxidative stress and inflammation in cardiovascular and renal complications of diabetes. Clin Sci (Lond) 132, 1811–1836 [DOI] [PubMed] [Google Scholar]
- 5.Sedeek M, Nasrallah R, Touyz RM, and Hebert RL (2013) NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 24, 1512–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheleznova NN, Yang C, and Cowley AW Jr. (2016) Role of Nox4 and p67phox subunit of Nox2 in ROS production in response to increased tubular flow in the mTAL of Dahl salt-sensitive (SS) rats. Am J Physiol Renal Physiol, 311, F450–F458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thallas-Bonke V, Jandeleit-Dahm KA, and Cooper ME (2015) Nox-4 and progressive kidney disease. Curr Opin Nephrol Hypertens 24, 74–80 [DOI] [PubMed] [Google Scholar]
- 8.Yang Q, Wu FR, Wang JN, Gao L, Jiang L, Li HD, Ma Q, Liu XQ, Wei B, Zhou L, Wen J, Ma TT, Li J, and Meng XM (2018) Nox4 in renal diseases: An update. Free Radic Biol Med 124, 466–472 [DOI] [PubMed] [Google Scholar]
- 9.Rajaram RD, Dissard R, Jaquet V, and de Seigneux S (2018) Potential benefits and harms of NADPH oxidase type 4 in the kidneys and cardiovascular system. Nephrol Dial Transpl 34, 567–576 [DOI] [PubMed] [Google Scholar]
- 10.Cowley AW, Yang C, Zheleznova NN, Staruschenko A, Kurth T, Rein L, Kumar V, Sadovnikov K, Dayton A, Hoffman M, Ryan RP, Skelton MM, Salehpour F, Ranji M, and Geurts A (2016) Evidence of the Importance of Nox4 in Production of Hypertension in Dahl Salt-Sensitive Rats. Hypertension 67, 440–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar V, Kurth T, Zheleznova NN, Yang C, and Cowley AW (2020) NOX4/H2O2/mTORC1 Pathway in Salt-Induced Hypertension and Kidney Injury. Hypertension 76, 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor NE, Glocka P, Liang M, and Cowley AW Jr. (2006) NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension 47, 692–698 [DOI] [PubMed] [Google Scholar]
- 13.Tian N, Moore RS, Phillips WE, Lin L, Braddy S, Pryor JS, Stockstill RL, Hughson MD, and Manning RD Jr. (2008) NADPH oxidase contributes to renal damage and dysfunction in Dahl salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol 295, R1858–R1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohsaki Y, O'Connor P, Mori T, Ryan RP, Dickinson BC, Chang CJ, Lu Y, Ito S, and Cowley AW Jr. (2012) Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am J Physiol Renal Physiol 302, F95–F102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HHHW, and Jandeleit-Dahm KA (2014) Genetic Targeting or Pharmacologic Inhibition of NADPH Oxidase Nox4 Provides Renoprotection in Long-Term Diabetic Nephropathy. J Am Soc Nephrol 25, 1237–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urner S, Ho F, Jha JC, Ziegler D, and Jandeleit-Dahm K (2020) NADPH oxidase inhibition: preclinical and clinical studies in diabetic complications. Antioxid Redox Signal doi: 10.1089/ars.2020.8047 [DOI] [PubMed] [Google Scholar]
- 17.Staruschenko A (2012) Regulation of transport in the connecting tubule and cortical collecting duct. Compreh Physiol 2, 1541–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feraille E, Dizin E, Roth I, Derouette JP, Szanto I, Martin PY, de Seigneux S, and Hasler U (2014) NADPH oxidase 4 deficiency reduces aquaporin-2 mRNA expression in cultured renal collecting duct principal cells via increased PDE3 and PDE4 activity. PLoS One 9, e87239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavlov TS, and Staruschenko A (2017) Involvement of ENaC in the development of salt-sensitive hypertension. Am J Physiol Renal Physiol 313, F135–F140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilatovskaya DV, Pavlov TS, Levchenko V, and Staruschenko A (2013) ROS production as a common mechanism of ENaC regulation by EGF, insulin, and IGF-1. Am J Physiol Cell Physiol 304, C102–C111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu X, Wang F, Liu M, Yang KT, Nau A, Kohan DE, Reese V, Richardson RS, and Yang T (2016) Activation of ENaC in collecting duct cells by prorenin and its receptor PRR: involvement of Nox4-derived hydrogen peroxide. Am J Physiol Renal Physiol 310, F1243–F1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma HP (2011) Hydrogen Peroxide Stimulates the Epithelial Sodium Channel through a Phosphatidylinositide 3-Kinase-dependent Pathway. J Biol Chem 286, 32444–32453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouabout G, Ayme-Dietrich E, Jacob H, Champy MF, Birling MC, Pavlovic G, Madeira L, Fertak LE, Petit-Demouliere B, Sorg T, Herault Y, Mudgett J, and Monassier L (2018) Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch Cardiov Dis 111, 41–52 [DOI] [PubMed] [Google Scholar]
- 24.Schröder K (2020) NADPH oxidases: Current aspects and tools. Redox Biology 34, 101512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilatovskaya DV, Blass G, Palygin O, Levchenko V, Pavlov TS, Grzybowski MN, Winsor K, Shuyskiy LS, Geurts AM, Cowley AW Jr., Birnbaumer L, and Staruschenko A (2018) A NOX4/TRPC6 Pathway in Podocyte Calcium Regulation and Renal Damage in Diabetic Kidney Disease. J Am Soc Nephrol 29, 1917–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spires D, Ilatovskaya DV, Levchenko V, North PE, Geurts AM, Palygin O, and Staruschenko A (2018) Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am J Physiol Renal Physiol 315, F1091–F1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slaughter TN, Paige A, Spires D, Kojima N, Kyle PB, Garrett MR, Roman RJ, and Williams JM (2013) Characterization of the development of renal injury in Type-1 diabetic Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 305, R727–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palygin O, Evans LC, Cowley AW Jr., and Staruschenko A (2017) Acute In Vivo Analysis of ATP Release in Rat Kidneys in Response to Changes of Renal Perfusion Pressure. J Am Heart Assoc 6, e006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palygin O, Levchenko V, Evans LC, Blass G, Cowley AW Jr., and Staruschenko A (2015) Use of enzymatic biosensors to quantify endogenous ATP or H2O2 in the kidney. J Vis Exp 104, e53059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isaeva E, Fedoriuk M, Bohovyk R, Klemens CA, Khedr S, Golosova D, Levchenko V, El-Meanawy A, Palygin O, and Staruschenko A (2019) Vibrodissociation method for isolation of defined nephron segments from human and rodent kidneys. Am J Physiol Renal Physiol 317, F1398–F1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavlov TS, Levchenko V, Ilatovskaya DV, Moreno C, and Staruschenko A (2016) Renal sodium transport in renin deficient Dahl salt-sensitive rats. J Renin Angiotensin Aldosterone Syst 17, 1470320316653858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abais-Battad JM, Alsheikh AJ, Pan X, Fehrenbach DJ, Dasinger JH, Lund H, Roberts ML, Kriegel AJ, Cowley AW Jr., Kidambi S, Kotchen TA, Liu P, Liang M, and Mattson DL (2019) Dietary Effects on Dahl Salt-Sensitive Hypertension, Renal Damage, and the T Lymphocyte Transcriptome. Hypertension 74, 854–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Vliet BN, Chafe LL, Halfyard SJ, and Leonard AM (2006) Distinct rapid and slow phases of salt-induced hypertension in Dahl salt-sensitive rats. J Hypertens 24, 1599–1606 [DOI] [PubMed] [Google Scholar]
- 34.Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, Mattson DL, Sorokin A, Lombard JH, Cowley AW Jr., and Staruschenko A (2013) Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol 24, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polina I, Domondon M, Fox R, Sudarikova A, Troncoso M, Vasileva V, Kashyrina Y, Gooz MB, Schibalski R, DeLeon-Pennell KY, Fitzgibbon WR, and Ilatovskaya DV (2020) The differential effects of low dose sacubitril and/or valsartan on renal disease in salt-sensitive hypertension. Am J Physiol Renal Physiol 319, F63–F75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Ryan RP, Cowley AW Jr., and Staruschenko A (2013) Real-time electrochemical detection of ATP and H2O2 release in freshly isolated kidneys. Am J Physiol Renal Physiol 305, F134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H, and Nawata H (2003) Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia 46, 1428–1437 [DOI] [PubMed] [Google Scholar]
- 38.Thallas-Bonke V, Jha JC, Gray SP, Barit D, Haller H, Schmidt HH, Coughlan MT, Cooper ME, Forbes JM, and Jandeleit-Dahm KA (2014) Nox-4 deletion reduces oxidative stress and injury by PKC-alpha-associated mechanisms in diabetic nephropathy. Physiol Rep 2, e12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Q, Lee D-Y, Féliers D, Abboud HE, Bhat MA, and Gorin Y (2020) Interplay between RNA-binding protein HuR and Nox4 as a novel therapeutic target in diabetic kidney disease. Mol Metab 36, 100968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song J, Knepper MA, Verbalis JG, and Ecelbarger CA (2003) Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am J Physiol Renal Physiol 285, F1125–1137 [DOI] [PubMed] [Google Scholar]
- 41.Spires D, Poudel B, Shields CA, Pennington A, Fizer B, Taylor L, McPherson KC, Cornelius DC, and Williams JM (2018) Prevention of the progression of renal injury in diabetic rodent models with preexisting renal disease with chronic endothelin A receptor blockade. Am J Physiol Renal Physiol 315, F977–f985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, and Staruschenko A (2015) Podocyte injury in diabetic nephropathy: implications of angiotensin II-dependent activation of TRPC channels. Sci Rep 5, 17637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staruschenko A (2017) Hypertension and diabetes mellitus: the chicken and egg problem. Hypertension 69, 787–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bell RC, Carlson JC, Storr KC, Herbert K, and Sivak J (2000) High-fructose feeding of streptozotocin-diabetic rats is associated with increased cataract formation and increased oxidative stress in the kidney. Br J Nutr 84, 575–582 [PubMed] [Google Scholar]
- 45.Gojo A, Utsunomiya K, Taniguchi K, Yokota T, Ishizawa S, Kanazawa Y, Kurata H, and Tajima N (2007) The Rho-kinase inhibitor, fasudil, attenuates diabetic nephropathy in streptozotocin-induced diabetic rats. Eur J Pharmacol 568, 242–247 [DOI] [PubMed] [Google Scholar]
- 46.Roman RJ, and Kaldunski M (1991) Pressure natriuresis and cortical and papillary blood flow in inbred Dahl rats. Am J Physiol 261, R595–602 [DOI] [PubMed] [Google Scholar]
- 47.O'Connor PM, and Cowley AW Jr. (2010) Modulation of pressure-natriuresis by renal medullary reactive oxygen species and nitric oxide. Curr Hypertens Rep 12, 86–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin C, Hu C, Polichnowski A, Mori T, Skelton M, Ito S, and Cowley AW Jr. (2009) Effects of renal perfusion pressure on renal medullary hydrogen peroxide and nitric oxide production. Hypertension 53, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S, and Tomita K (2009) Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens 27, 1679–1689 [DOI] [PubMed] [Google Scholar]
- 50.Haque MZ, Ares GR, Caceres PS, and Ortiz PA (2011) High salt differentially regulates surface NKCC2 expression in thick ascending limbs of Dahl salt-sensitive and salt-resistant rats. Am. J Physiol Renal Physiol 300, F1096–F1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, and Abboud HE (2005) Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280, 39616–39626 [DOI] [PubMed] [Google Scholar]
- 52.Ray EC, Rondon-Berrios H, Boyd CR, and Kleyman TR (2015) Sodium retention and volume expansion in nephrotic syndrome: implications for hypertension. Adv Chronic Kidney Dis 22, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spires D, Manis AD, and Staruschenko A (2019) Ion channels and transporters in diabetic kidney disease. Curr Top Membr 83, 353–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Culshaw GJ, Costello HM, Binnie D, Stewart KR, Czopek A, Dhaun N, Hadoke PWF, Webb DJ, and Bailey MA (2019) Impaired pressure natriuresis and non-dipping blood pressure in rats with early type 1 diabetes mellitus. J Physiol 597, 767–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hills CE, Bland R, Bennett J, Ronco PM, and Squires PE (2006) High glucose up-regulates ENaC and SGK1 expression in HCD-cells. Cell Physiol Biochem 18, 337–346 [DOI] [PubMed] [Google Scholar]
- 56.Riazi S, Khan O, Tiwari S, Hu X, and Ecelbarger CA (2006) Rosiglitazone regulates ENaC and Na-K-2Cl cotransporter (NKCC2) abundance in the obese Zucker rat. Am J Nephrol 26, 245–257 [DOI] [PubMed] [Google Scholar]
- 57.Oh YK, Joo KW, Lee JW, Jeon US, Lim CS, Han JS, Knepper MA, and Na KY (2007) Altered renal sodium transporter expression in an animal model of type 2 diabetes mellitus. J Korean Med Sci 22, 1034–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khedr S, Palygin O, Pavlov TS, Blass G, Levchenko V, Alsheikh A, Brands MW, El-Meanawy A, and Staruschenko A (2019) Increased ENaC activity during kidney preservation in Wisconsin solution. BMC Nephrol 20, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wyse B, Ali N, and Ellison DH (2002) Interaction with grp58 increases activity of the thiazide-sensitive Na-Cl cotransporter. Am. J Physiol Renal Physiol 282, F424–F430 [DOI] [PubMed] [Google Scholar]
- 60.Abe M, O'Connor P, Kaldunski M, Liang M, Roman RJ, and Cowley AW Jr. (2006) Effect of sodium delivery on superoxide and nitric oxide in the medullary thick ascending limb. Am. J Physiol Renal Physiol 291, F350–F357 [DOI] [PubMed] [Google Scholar]
- 61.Silva GB, Ortiz PA, Hong NJ, and Garvin JL (2006) Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension 48, 467–472 [DOI] [PubMed] [Google Scholar]
- 62.Soodvilai S, Jia Z, and Yang T (2007) Hydrogen peroxide stimulates chloride secretion in primary inner medullary collecting duct cells via mPGES-1-derived PGE2. Am. J Physiol Renal Physiol 293, F1571–F1576 [DOI] [PubMed] [Google Scholar]
- 63.Arany I, Faisal A, Nagamine Y, and Safirstein RL (2008) p66shc Inhibits Pro-survival Epidermal Growth Factor Receptor/ERK Signaling during Severe Oxidative Stress in Mouse Renal Proximal Tubule Cells. J Biol Chem 283, 6110–6117 [DOI] [PubMed] [Google Scholar]
- 64.Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, and Garty H (2002) Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem 277, 13539–13547 [DOI] [PubMed] [Google Scholar]
- 65.Staruschenko A, Palygin O, Ilatovskaya DV, and Pavlov TS (2013) Epidermal growth factors in the kidney and relationship to hypertension. Am. J Physiol Renal Physiol 305, F12–F20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Markadieu N, Crutzen R, Blero D, Erneux C, and Beauwens R (2005) Hydrogen peroxide and epidermal growth factor activate phosphatidylinositol 3-kinase and increase sodium transport in A6 cell monolayers. Am J Physiol Renal Physiol 288, F1201–F1212 [DOI] [PubMed] [Google Scholar]
- 67.Klein JD, Rash A, Sands JM, Ecelbarger CM, and Tiwari S (2009) Candesartan Differentially Regulates Epithelial Sodium Channel in Cortex Versus Medulla of Streptozotocin-Induced Diabetic Rats. J Epithel Biol Pharmacol 2, 23–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riazi S, Maric C, and Ecelbarger CA (2006) 17-β Estradiol attenuates streptozotocin-induced diabetes and regulates the expression of renal sodium transporters. Kidney Int 69, 471–480 [DOI] [PubMed] [Google Scholar]
- 69.Bardoux P, Bruneval P, Heudes D, Bouby N, and Bankir L (2003) Diabetes-induced albuminuria: role of antidiuretic hormone as revealed by chronic V2 receptor antagonism in rats. Nephrol Dial Transpl 18, 1755–1763 [DOI] [PubMed] [Google Scholar]
- 70.Bardoux P, Martin H, Ahloulay M, Schmitt F, Bouby N, Trinh-Trang-Tan MM, and Bankir L (1999) Vasopressin contributes to hyperfiltration, albuminuria, and renal hypertrophy in diabetes mellitus: study in vasopressin-deficient Brattleboro rats. Proc Natl Acad Sci U S A 96, 10397–10402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bankir L, Bichet DG, and Bouby N (2010) Vasopressin V2 receptors, ENaC, and sodium reabsorption: a risk factor for hypertension? Am J Physiol Renal Physiol 299, F917–F928 [DOI] [PubMed] [Google Scholar]
- 72.Stockand JD (2012) The role of the epithelial Na(+) channel (ENaC) in high AVP but low aldosterone states. Front Physiol 3, 304–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Furusho T, Uchida S, and Sohara E (2020) The WNK signaling pathway and salt-sensitive hypertension. Hypertens Res, doi: 10.1038/s41440-020-0437-x [DOI] [PubMed] [Google Scholar]
- 74.Zicha J, Hojná S, Vaňourková Z, Kopkan L, and Vaněčková I (2019) Is renal ß-adrenergic-WNK4-NCC pathway important in salt hypertension of Dahl rats? Physiol Res 68, 873–882 [DOI] [PubMed] [Google Scholar]
- 75.Wang Z, Carmo J. M. d., Aberdein N, Zhou X, Williams JM, Silva A. A. d., and Hall JE (2017) Synergistic Interaction of Hypertension and Diabetes in Promoting Kidney Injury and the Role of Endoplasmic Reticulum Stress. Hypertension 69, 879–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tanaka M, and Itoh H (2019) Hypertension as a Metabolic Disorder and the Novel Role of the Gut. Curr Hypert Rep 21, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conway BR, Rennie J, Bailey MA, Dunbar DR, Manning JR, Bellamy CO, Hughes J, and Mullins JJ (2012) Hyperglycemia and Renin-Dependent Hypertension Synergize to Model Diabetic Nephropathy. J Am Soc Nephrol 23, 405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McPherson KC, Taylor L, Johnson AC, Didion SP, Geurts AM, Garrett MR, and Williams JM (2016) Early development of podocyte injury independently of hyperglycemia and elevations in arterial pressure in nondiabetic obese Dahl SS leptin receptor mutant rats. Am J Physiol Renal Physiol 311, F793–F804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McPherson KC, Shields CA, Poudel B, Fizer B, Pennington A, Szabo-Johnson A, Thompson WL, Cornelius DC, and Williams JM (2019) Impact of obesity as an independent risk factor for the development of renal injury: implications from rat models of obesity. Am J Physiol Renal Physiol 316, F316–f327 [DOI] [PMC free article] [PubMed] [Google Scholar]