

Abstract
G protein-coupled receptors (GPCRs) play critical roles in transmitting a variety of extracellular signals into the cells and regulate diverse physiological functions. Naturally occurring mutations that result in dysfunctions of GPCRs have been known as the causes of numerous diseases. Significant progresses have been made in elucidating the pathophysiology of diseases caused by mutations. The multiple intracellular signaling pathways, such as G protein-dependent and β-arrestin-dependent signaling, in conjunction with recent advances on biased agonism, have broadened the view on the molecular mechanism of disease pathogenesis. This review aims to briefly discuss biased agonism of GPCRs (biased ligands and biased receptors), summarize the naturally occurring GPCR mutations that cause biased signaling, and propose the potential pathophysiological relevance of biased mutant GPCRs associated with various endocrine diseases.
Keywords: G protein-coupled receptor, naturally occurring mutation, biased signaling, pathophysiology
1. Introduction
G protein-coupled receptors (GPCRs), with about 800 members identified in human genome, constitute the largest superfamily of cell surface receptors [1, 2]. Their topology consists of an extracellular N-terminus, a bundle of seven transmembrane domains (TMDs, TMD1 to TMD7) within the plasma membrane, connected by three extracellular loops (ECLs, ECL1 to ECL3) and three intracellular loops (ICLs, ICL1 to ICL3), and an intracellular C-terminus. GPCRs are divided into six subfamilies, based on sequence homology and functional similarity, including rhodopsin-like (Family A), secretin-like (Family B), metabotropic glutamate (Family C), pheromone (Family D), cAMP (Family E), and frizzled (Family F) GPCRs (information on the GPCR database: https://gpcrdb.org). Only four families (A, B, C, and F) among them are found in humans. Family A rhodopsin-like GPCRs, consisting of more than half of all GPCRs, are the largest family in this superfamily.
GPCRs transmit a variety of extracellular signals into the cells. These signals comprise chemical and sensory stimuli, such as hormones, neurotransmitters, chemokines, ions, light, and odorants [1], highlighting the significant roles of GPCRs in mediating diverse physiological functions. Therefore, GPCRs have been exploited as therapeutic targets with particular longstanding interest over the past several decades. Another reason of GPCRs being good drug targets is that ligands bind to GPCRs (non-olfactory GPCRs) in the orthosteric binding site, a deep cleft on the extracellular side of the receptor, making it easier to develop drugs since they do not need to be structurally and chemically modified to cross the plasma membrane [3], compared to drugs targeting intracellular kinases and proteases. Up to 2017, 481 of all FDA-approved drugs target GPCRs, accounting for approximately 34% of all approved drugs. They mediate the effects via 107 GPCRs, which constitutes only 27% of the non-olfactory human GPCRs [4].
GPCR-mediated signal transduction starts with ligand binding to receptors, followed by receptor conformational change and activation of signal mediators (Fig. 1). The conventional intracellular signaling is mediated by heterotrimeric G protein consisting of α, β, and γ subunits. Once receptor is activated, the α subunit dissociates from βγ heterodimer and then activates enzymes generating the second messengers and downstream mediators to transduce the signal. Four ubiquitously expressed subtypes of α subunit of G protein, including Gαs, Gαi/o, Gαq/11, and Gα12/13, have been discovered based on their different sequences and functions [5] (Fig. 1). Activated Gαs increases the activity of adenylyl cyclase (AC) (a membrane enzyme catalyzing the conversion of ATP to 3’,5’-cyclic adenosine monophosphate (cAMP) and pyrophosphate) to induce intracellular cAMP generation and subsequently activate protein kinase A (PKA). Conversely, activated Gαi/o decreases adenylyl cyclase activity to inhibit intracellular cAMP generation and PKA activity. Activated Gαq/11 triggers a distinct pathway by activating phospholipase C-β (PLCβ), which in turn induces diacylglycerol (DAG)-protein kinase C (PKC) pathway and inositol 1,4,5-trisphosphate (IP3)-Ca2+ mobilization. Activated Gα12/13 primarily affect Rho-specific guanine nucleotide exchange factor (RhoGEF), further activating small GTPase RhoA and increasing Rho-kinase activity [6]. In addition to α subunit, βγ heterodimer mediates signaling processes and modulates activation of ion channels, such as G protein-regulated inwardly rectifying K+ channels [7] and Ca2+ channels [8] (reviewed in [9]).
Figure 1.
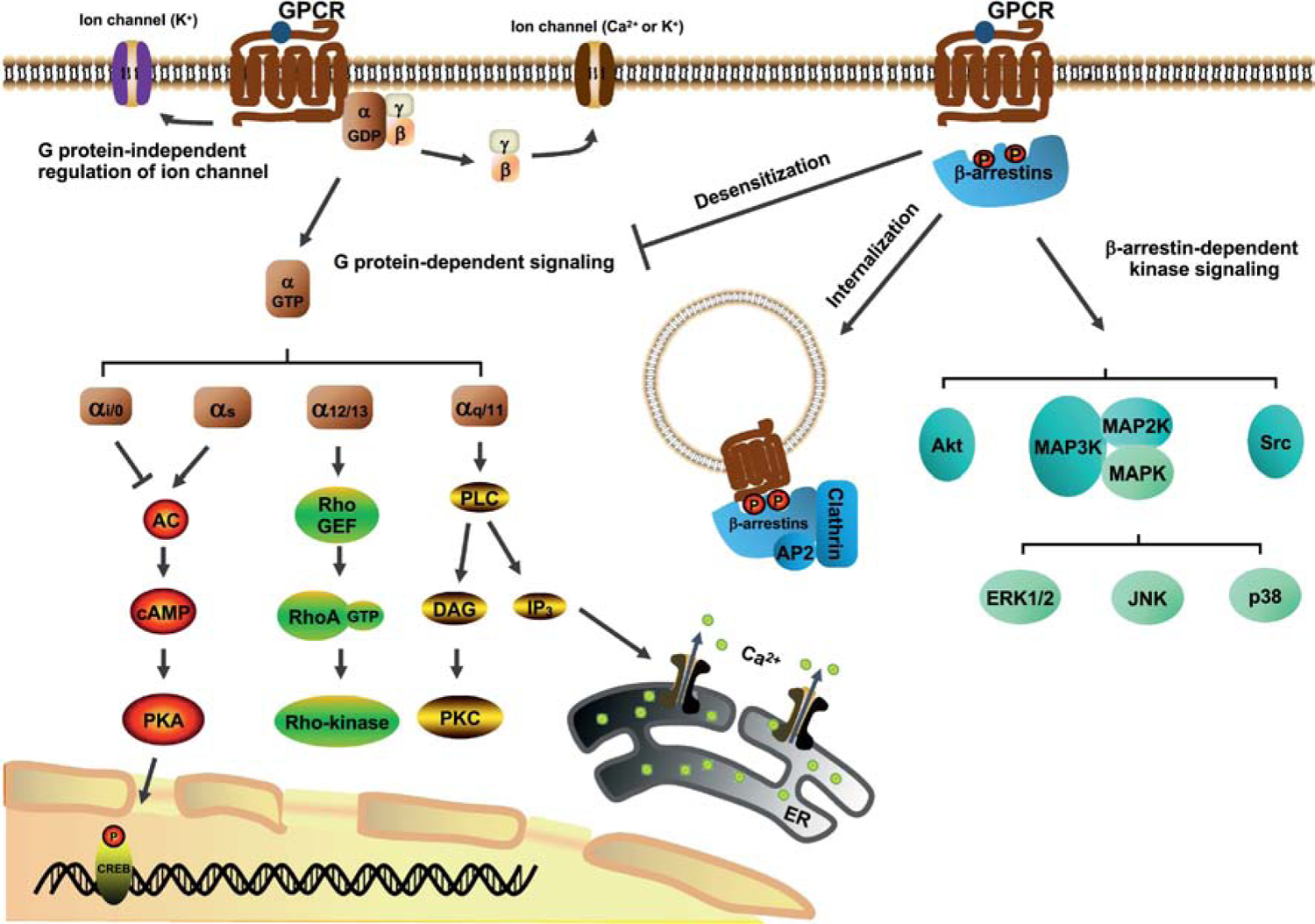
Intracellular signaling pathways triggered by GPCRs. Agonist binding to receptors triggers a variety of distinct downstream signaling pathways. Signal propagation can be mediated through G proteins (Gαs, Gαi/o, Gαq/11, Gα12/13, and Gβγ), thereby regulating activities of several kinases (PKA, PKC, and Rho-kinase) and Ca2+ mobilization. Recruitment of β-arrestins resulted from phosphorylation of GPCRs desensitizes G protein coupling, mediates receptor internalization, and initiates β-arrestin-dependent signaling and kinase activation.
Naturally occurring mutations that affect GPCR signal transduction can cause either impaired or enhanced protein function, which are classified as loss-of-function or gain-of-function mutations, respectively [10–14]. Loss-of-function mutations prevent signaling through several ways, such as defective receptor biosynthesis or trafficking, impairing ligand binding, and/or impairing basal or agonist-induced signaling [12, 14, 15]. For instance, rhodopsin is a light-sensitive GPCR essential for visual phototransduction. The mutant rhodopsin P23H is the first identified mutant in GPCRs that results in human diseases [16]. Biochemical and biophysical studies reveled that P23H is trapped intracellularly by quality control system in endosomal reticulum due to misfolding, thus exhibiting signaling deficiency and causing retinitis pigmentosa, a degenerative eye disease associated with severe vision impairment [17, 18]. Since the discovery of rhodopsin P23H mutation, the list of diseases resulting from GPCR loss-of-function mutations keeps expanding [12], including idiopathic hypogonadotropic hypogonadism (IHH), Kallmann syndrome, oligospermia and subfertility, ovarian dysgenesis and other human diseases (Table 1).
Table 1.
Loss-of-function GPCR mutations associated with diverse human diseases.
| Receptor | Disease | Clinical features | References |
|---|---|---|---|
| GPR54 | Idiopathic hypogonadotropic hypogonadism | Attenuated secretion of GnRH, sexual development disruption | [215] |
| PROKR2 | Kallmann syndrome | Impaired development of the olfactory system and reproductive axis | [271] |
| FSHR | Oligospermia and subfertility Ovarian dysgenesis |
Small testes with impaired spermatogenesis in males Absence of follicular maturation in females |
[272] |
| TSHR | Euthyroid hyperthyrotropinemia Congenital hypothyroidism |
TSH resistance with normal biological effects of TH TSH resistance with reduced biological effects of TH |
[234, 273] |
| CaSR | Familial hypocalciuric hypercalcemia | Lifelong moderate elevation of serum calcium concentration and inappropriately normal PTH levels | [274, 275] |
| Neonatal severe hyperparathyroidism | Neonatal marked elevation of serum calcium and PTH levels | ||
| AVPR2 | Nephrogenic diabetes insipidus | Polyuria, polydipsia, hypernatremia, low urine osmolality, and AVP resistance | [236] |
| MC1R | Pigmentation defect | Red hair color phenotype: red hair, fair skin, poor ability to tan, and increased risk of melanoma | [276] |
| MC2R | Familial glucocorticoid deficiency | Low or undetectable cortisol levels and elevated ACTH levels | [277] |
| MC4R | Obesity | Hyperphagia, increased fat and lean mass, and increased bone mass | [278] |
| PTHR1 | Blomstrand chondrodysplasia | Advanced endochondral bone maturation associated with breast development and tooth impaction | [279, 280] |
| Rhodopsin | Retinitis pigmentosa | Loss of vision, insensitivity in dim light | [16] |
Gain-of-function mutations can be due to decreased specificity, enhanced sensitivity in response to ligands, increased basal activity (constitutive activation), or increased receptor expression, thereby causing hyper-responsive phenotypes [11, 14]. The rhodopsin K296E is the first reported naturally occurring constitutively active mutation in GPCRs that is associated with human diseases [19]. Robinson et al. (1992) showed that the inverse agonist of rhodopsin, 11-cis-retinal, fails to suppress the constitutive activity of K296E since the retinal binding site is mutated, potentially explaining the mechanism that K296E causes retinitis pigmentosa with a persistent stimulation of phototransduction pathway [20]. Since then, numerous cases and studies have been reported, showing the gain-of-function mutations of GPCRs result in a wide range of human diseases [14, 21], including precocious puberty, spontaneous ovarian hyperstimulation, familial gestational hyperthyroidism and other diseases (Table 2).
Table 2.
Gain-of-function GPCR mutations associated with diverse human diseases.
| Receptor | Disease | Clinical features | References |
|---|---|---|---|
| GPR54 | Central precocious puberty | Premature secretion of GnRH, early pubertal development | [223] |
| FSHR | Spontaneous ovarian hyperstimulation | Hydrothorax, ascites, and ovarian enlargement | [281, 282] |
| TSHR | Familial gestational hyperthyroidism | Severe nausea and vomiting, weight loss, persistent tachycardia, and sweating during gestation | [283] |
| CaSR | Autosomal dominant hypocalcemia | Decreased serum calcium concentrations and inappropriately low PTH levels, hyperphosphatemia, hypercalciuria | [284, 285] |
| Bartter syndrome type V | Defective renal reabsorption of sodium and chloride, hypokalemia, and increased blood pH | ||
| AVPR2 | Nephrogenic syndrome of inappropriate antidiuresis | Hyponatremia, concentrated urine in adults and seizures in infants | [235] |
| MC2R | Cushing’s syndrome | Hypercortisolism | [269, 286] |
| PTHR1 | Jansen metaphyseal chondrodysplasia | Short-limbed dwarfism, disorganized metaphyseal regions | [287] |
| Rhodopsin | Congenital night blindness | Insensitivity in dim light without progressive loss of day vision | [288] |
Polymorphisms at GPCRs, generally defined as occurring in at least 1% of the population, can also be associated with various diseases [22]. Polymorphisms identified in coding regions usually result in dysfunctional receptors (e.g. rhodopsin, μ-opioid receptor (μ-OR), adrenergic receptors (ARs), follicle-stimulating hormone (FSH) receptor (FSHR), thyroid-stimulating hormone (TSH) receptor (TSHR), and vasopressin type 2 receptor (AVPR2)), while those identified in noncoding regions are believed to affect promoter activity or transcription (e.g. ARs, bradykinin B2 receptor, chemokine receptors, and dopamine receptors) (reviewed in [22]). Examination of these GPCR mutations and polymorphisms has a significant impact on our understanding of the relationship between genetic variants and various human diseases.
2. Multiple GPCR signaling pathways
For over two decades, biochemical and pharmacological studies demonstrated that a large number of GPCRs are pleiotropic receptors that can couple to multiple G proteins simultaneously [23]. For example, TSHR couples to all four G protein subtypes with similar potencies in thyroid gland [24, 25]. Other examples of GPCRs coupling to more than one G protein subtype include galanin receptor type 2 (Gαi, Gαq, and Gα12) [26], parathyroid hormone (PTH) receptor (PTHR) (Gαs, Gαi, and Gαq/11) [27], luteinizing hormone (LH) receptor (LHR) (Gαs, Gαi and Gαq) [28], melatonin receptor (Gαi/o and Gαq) [29], and melanin-concentrating hormone receptor (Gαi/o and Gαq) [30] (reviewed in [23]). Thus, the multiplicity of G protein coupling known for a variety of receptors provides one mechanism for the activation of multiple intracellular signaling pathways by a single receptor.
In the classical view, G protein signaling is typically terminated by desensitization, resulting from phosphorylation of the cytoplasmic tail and/or ICLs by G protein-coupled receptor kinases (GRKs) and recruitment of β-arrestins (β-arrestin-1 or β-arrestin-2) by phosphorylated receptor, with β-arrestins sterically competing with G-protein for binding to the GPCRs [31, 32]. Further, specific GRK-mediated phosphorylation pattern directs distinct β-arrestin conformations (phosphorylation “bar code” theory), thereby controlling the interaction partners and related functions of β-arrestins [33]. β-arrestins can act as the adaptors for clathrin, transporting phosphorylated receptors to clathrin-coated pits for receptor internalization. The internalized receptors can be further transported into endosomes where receptors can either recycle to the plasma membrane for resensitization or move into lysosomes for degradation. In addition, β-arrestins can serve as scaffolds to mediate signaling to mitogen-activated protein kinases (MAPKs) activation, such as extracellular signal-regulated kinases (ERKs), c-Jun-N-terminal kinases (JNKs), and p38 kinases, as well as other protein kinases [34–37] (reviewed in [32, 38, 39]) (Fig. 1). Although ERK1/2 activation can be caused by activated G protein, the ERK1/2 pathway mediated by β-arrestins has been shown to be segregated from G protein activation [40, 41]. In the case of type 1 angiotensin II receptor (AT1R), β-arrestins mediate slower and persistent ERK1/2 activation, whereas G protein mediates rapid and transient ERK1/2 activation [41].
In addition to G protein signaling from plasma membrane, the signaling originating from intracellular compartments, such as early endosomes and Golgi apparatus, also represents an important aspect of GPCR signaling and challenges the longstanding concept that GPCR only signals from plasma membrane and activation is silenced by receptor internalization [42, 43]. Up to now, several GPCRs have been reported to trigger sustained G protein signaling from intracellular compartments after internalization. Examples include PTHR, β2-AR, TSHR and AVPR2 for Gαs signaling [44–47] and calcium-sensing receptor (CaSR) for Gαq/11 signaling [48] from endosomes, and sphingolipid S1P receptor (S1P1R) for Gαi signaling from Golgi [49]. Although binding of G proteins and β-arrestins to prototypical GPCRs was thought to be mutually exclusive, structural analyses have revealed that in some GPCRs, internalized receptor could form a super-complex with both β-arrestin and G protein, leading to this sustained endosomal G protein signaling [47, 50]. In this complex, the receptor couples to G protein in canonical fashion and also allows β-arrestin binding to function as a regulator for G protein signaling, along with β-arrestin-mediated signaling [42, 50].
The diverse intracellular signaling pathways initiated by GPCRs have been shown to cause different cellular and physiological consequences and defects in these signaling pathways will lead to many diseases. Traditionally, functional characterizations of mutant receptors associated with diseases have primarily employed the canonical signaling pathway or preferential G protein, such as the Gαs/i-cAMP or Gαq-IP3/Ca2+ pathway. Thus, mutations are classified as loss-of-function or gain-of-function mutations according to the properties of this conventional pathway. However, with recent expansion of our understanding of the molecular pharmacology of GPCRs, this approach seems to be oversimplified, since the other signaling pathways may also be indispensable in mediating the physiological responses and contributing to pathophysiology when disrupted. Therefore, it is necessary to determine whether the mutation affects multiple signaling pathways and whether the mutation differentially influences these signaling pathways, exhibiting biased signaling.
The traditional methods of measuring GPCR activation is to directly detect the GTP/GDP exchange ([35S]-GTPγS binding assay widely used for Gαs and Gαq signaling) or second messenger levels (e.g. cAMP assay for Gαs and Gαi signaling, and inositol phosphate (IP) accumulation and intracellular calcium assays for Gαq/11 signaling). Recent advances on biochemical and biophysical techniques added more dimensions to GPCR signaling assays and facilitated us to investigate multiple signaling pathways rather than only conventional one. Cell-based reporter gene assay provides a popular homogenous assay system for assessment of GPCR signaling owing to its high sensitivity and wide dynamic range. In the reporter gene assay, GPCR-mediated second messenger regulates specific transcription factor, thereby activating or inhibiting a responsive element located in the promoter of reporter gene, which in turn regulates the reporter protein expression and this process can be quantitatively measured [51]. The Gαs-mediated cAMP production leads to activation of cAMP response element binding protein (CREB) [52]; Gαq/11-mediated intracellular Ca2+ mobilization can activate nuclear factor of activated T cells (NFAT) [53]; Gα12/13-mediated formation of filamentous actins activate serum response factor (SRF) [54]; ERK1/2 activation also activates SRF and downstream serum response element (SRE) [55]. The reporter can be β-galactosidase, luciferase, green fluorescent protein, or β-lactamase [51]. Of them, the luciferase reporter gene assay is the most commonly used for measurement of GPCR signaling.
Recently developed GPCR biosensors can monitor real-time GPCR activation in living cells under physiological conditions. For example, resonance energy transfer (RET) assay is based on the transfer of energy from an energy donor (fluorescent or bioluminescent molecule) attached to GPCR to an energy acceptor (another fluorescent molecule) attached to G protein or β-arrestin. The energy transfer can be changed and monitored when the proximity of GPCR to G protein or β-arrestin changes [56]. In the early studies of GPCR biased agonism, characterization of β-arrestin recruitment has significantly evolved with the development of FRET/BRET assays [56–58].
It is possible that differences may appear when different signaling assays are used to access GPCR activation [59]. For example, in one study of C-C chemokine receptor 7 (CCR7),both endogenous ligands (CCL19 and CCL21) activate G protein signaling (measured by [35S]-GTPγS binding assay), but only CCL19 results in an increased β-arrestin recruitment (measured by translocation to cell membrane) and β-arrestin-dependent ERK1/2 activation, suggesting ligand bias of CCL21 at CCR7 [60]. However, in one later study, inconsistent result was observed when BRET biosensors were used for measurement of both G protein coupling and β-arrestin-recruitment. CCL21 is shown to be less potent in activating G proteins and recruiting β-arrestin, refuting the previous notion of ligand bias [61]. Therefore, the reliability of signaling properties of receptor is likely related to the signaling assays used. It is more reliable to directly detect G protein coupling or β-arrestin recruitment using FRET/BRET biosensors, compared to using different types of signaling assays. Future studies involving multiple GPCR signaling assays should take this into consideration.
3. Biased agonism of GPCRs
Biased signaling is the preferential activation of one signaling pathway over another or others, different from the classical view on GPCR signal transduction that multiple downstream signaling pathways are activated with similar efficacies [32, 62]. The phenomenon of biased agonism can be explained by the multi-state model of receptor activation. The theory states that receptor activation is a highly dynamic process in which multiple active conformations can be induced by different ligands to mediate distinct signaling pathways [32, 63, 64]. Biased agonism, in most cases, is a property of the ligand-receptor complex. Therefore, biased signaling initiated by the receptor may be due to either ligand effect (biased ligand) or receptor itself (biased receptor) (Fig. 2A). Additionally, for many GPCRs with constitutive activities, biased signaling can also be induced by receptor in the absence of ligand stimulation, known as biased constitutive signaling.
Figure 2.
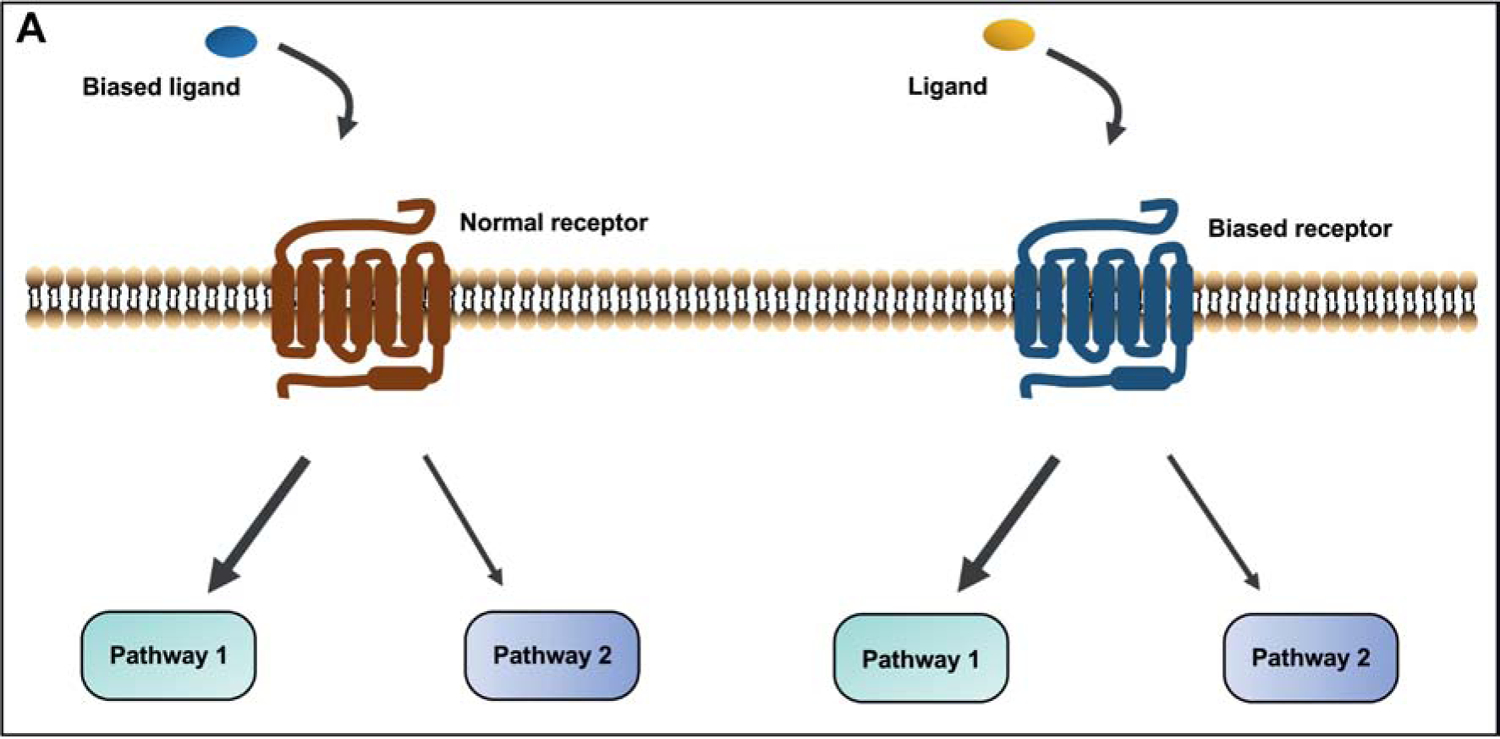
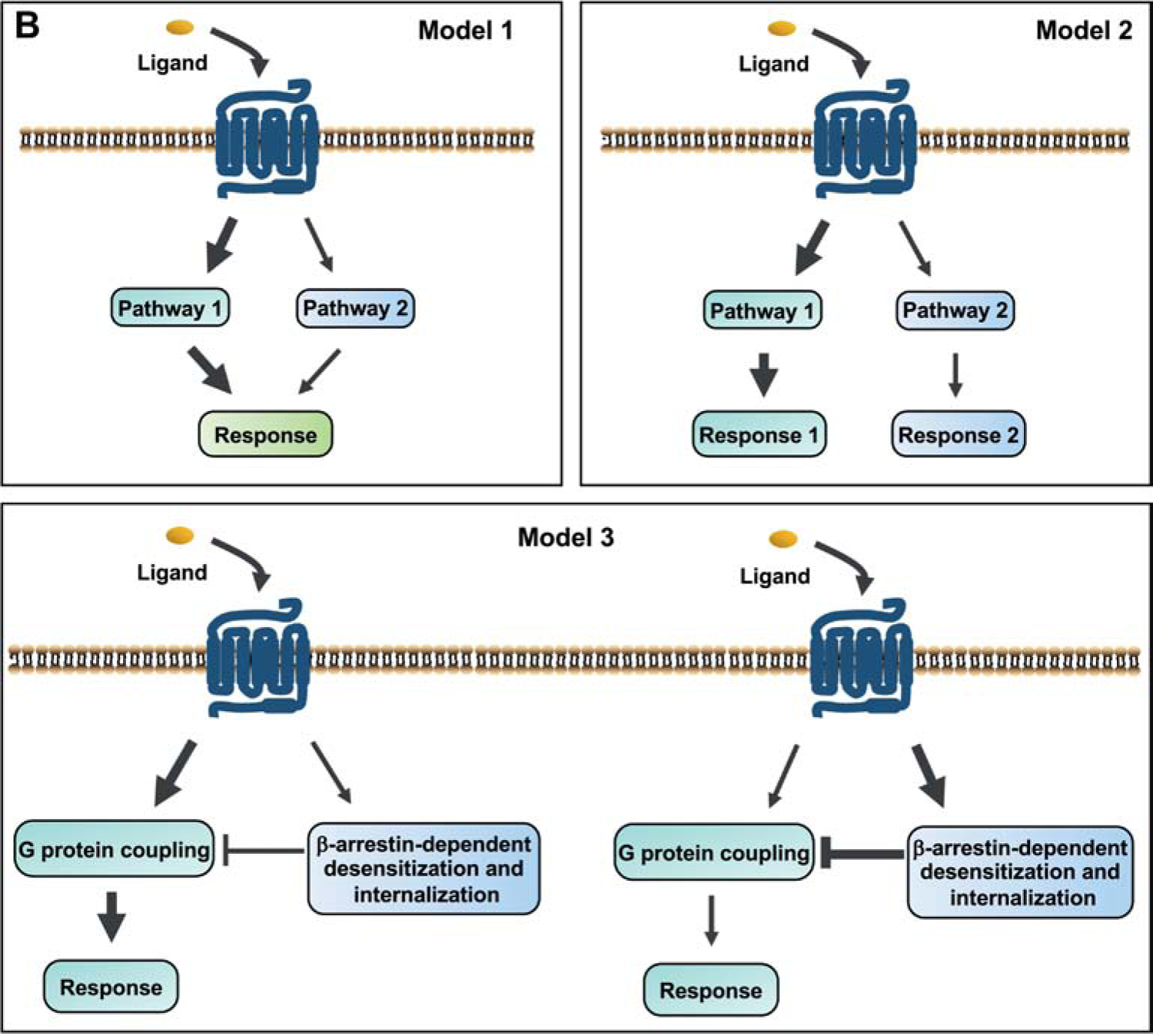
Models for biased signaling and pathophysiological relevance of biased mutants in GPCRs. (A) Two cases of biased agonism: the biased response is initiated by binding of biased ligand to the GPCR or a ligand to biased receptor. (B) Four types of biased mutant models that are proposed to be pathophysiologically relevant. Biased signaling can be initiated by mutant receptor between G protein and β-arrestins, or different G protein subtypes (models 1 and 2). The β-arrestin mediated receptor desensitization and internalization are considered as a counteractive pathway for G protein-dependent signaling (model 3). Mutant receptors can also trigger biased constitutive signaling in the absence of ligand. Sustained endosomal G protein signaling also represents an important pathway mediating physiological function of receptor (model 4).
3.1. Biased ligands
Biased ligands selectively stabilize GPCRs to distinct conformations, stimulating or inhibiting one (or some) of the possible signaling pathways [65, 66]. Some biased ligands preferentially stimulate the signaling associated with beneficial responses, providing the potential to develop new therapeutics with fewer side effects, greater efficacy and higher safety (for excellent reviews, see [67–70] for examples). Here, we highlight several biased ligands, selectively activating either G protein or β-arrestin signaling, or signaling of different G protein subtypes.
G protein biased ligands exert therapeutic benefits via inducing G protein-dependent signaling with little or no β-arrestin signaling. Biased ligands targeting G protein-dependent signaling to opioid receptors (ORs) provide striking examples. μ-Opioid agonists like morphine, fentanyl, and oxycodone exhibit powerful analgesic effect by activating μ-OR. However, their clinical utility is greatly limited due to the adverse events including nausea and vomiting, respiratory depression, constipation, and sedation [69, 71, 72]. Studies with β-arrestin-2 knockout mice showed that the Gαi signaling is associated with powerful opioid analgesia, while β-arrestin-2 signaling is involved in adverse events in respiratory and gastrointestinal systems [73–75]. Therefore, identification of G protein-biased ligands devoid of β-arrestin activation may provide powerful analgesics with increased safety and tolerability [72]. TRV130, PZM21, and SR-17018, are three novel μ-OR ligands identified by high-throughput screening, structure-based drug design, and synthesis-driven structure-activity relationship, respectively [76–78]. These ligands stimulate Gαi signaling that is associated with analgesia without β-arrestin-induced nausea or vomiting [69, 72]. Compared to μ-OR agonists, κ-opioid receptor (KOR) agonists do not activate the reward pathway, thus exhibiting nonaddictive analgesic effects [79]. Likewise, KOR exerts analgesic properties by activating Gαi signaling, while β-arrestin signaling is associated with notable dysphoria [79]. 6’GNTI is a partial agonist of KOR that stimulates G protein signaling with potent analgesic effect but not β-arrestin signaling [79, 80].
In other GPCRs, β-arrestin-biased ligands are preferred therapeutic agents. Due to the important roles in regulating cardiovascular and pulmonary functions, the ARs are the most common therapeutic targets of therapeutic agents. However, activation of the ARs shows biphasic effects in cardiovascular and pulmonary functions [81]. In β1-AR, β-arrestin mediates the transactivation of epidermal growth factor receptor, exerting cardioprotective effect, whereas G protein signaling is thought to be cardiotoxic [82–84]. Therefore, identifying biased ligands to selectively target β-arrestin signaling in ARs shows therapeutic benefits [85]. Carvedilol is a β-arrestin-biased ligand of β1-AR with improved cardioprotective effect [86, 87]. Rockman and colleagues revealed a previously unrecognized signaling mechanism of biased agonism resulting from carvedilol: 1). carvedilol results in the transition of the β1-AR from Gαs coupling to Gαi coupling; 2). the recruitment of Gαi leads to a unique receptor conformation, initiating β-arrestin-biased signaling [85]. The β2-AR biased ligands, propranolol, ICI 118,551, and BI-167107, selectively stimulate β-arrestin signaling, which is beneficial for hypertension and heart failure treatment [78, 88–91].
Some biased ligands activate receptors to couple with particular G protein subtype(s). For example, in α2-AR, catecholamine analogues are more potent in activating Gαq signaling while imidazoline or ox-/thiazoloazepine analogues selectively activate Gαi/o signaling [92]. Ligand with different modification or concentration selectively activates different G protein signaling pathways. The acidic (sialylated) isoforms of gonadotropin activate coupling to Gαs, whereas neutral/basic gonadotropin isoforms interact with Gαi signaling in gonadotropin receptors [93]. Different glycoforms of FSH also exert functional selectivity in Gs protein versus β-arrestin mediated signaling [94]. Thiazolidinone analogs activate Gαs signaling through FSHR at lower concentrations and Gαi signaling at higher concentrations, similar to the biased signaling induced by differentially glycosylated gonadotropins [93, 95, 96].
In addition to the G protein or β-arrestin biased ligands, ligands that activate the receptors with temporal or spatial preferences could be considered as a new type of biased ligands [42, 66]. The endosomal cAMP signaling potentially provides a mechanism to explain that the ligands exert temporal or spatial bias in signal generation at different subcellular localizations [97]. For example, analyses of FRET biosensors showed that PTHR, which plays an important role in regulating mineral ion homeostasis, exerts endosomal cAMP signaling [45]. PTH analogues and PTH-related peptide (PTHrP) bind to PTHR with different conformations, resulting in bias in signal generation [45, 97, 98]. The PTHrP preferentially triggers the transient cAMP signaling which are derived from ligand-receptor complexes at the plasma membrane, while the PTH analogues preferentially trigger the prolonged endosomal cAMP signaling [97, 99, 100]. The emerging endosomal PTHR cAMP signaling provides new insights into therapy for patients with hypoparathyroidism [42]. Due to the short half-life of the injected PTH (1–34), patients with hypoparathyroidism are currently treated by vitamin D and calcium supplements [101]. The LA-PTH is a PTH analog that preferentially trigger the prolonged endosomal cAMP signaling. Mouse studies showed that LA-PTH results in prolonged hypercalcemic and LA-PTH is now in preclinical development [42, 100].
Five melanocortin receptors (MCRs), members of Family A GPCRs, are unique in that there are two endogenous antagonists, Agouti-signaling protein (ASIP) for MC1R and Agouti-related protein (AgRP) for MC3R and MC4R. In the grey squirrel, ASIP is an inverse agonist at the wild-type (WT) MC1R but an agonist at a melanic variant [102]. We showed that although AgRP is an inverse agonist (negative antagonist) at the Gαs-cAMP pathway [103], it is an agonist of ERK1/2 activation at both MC3R and MC4R [104, 105]. Several additional small molecule inverse agonists also serve as biased ligands at the MC4R [104] (reviewed in [106]). A small molecule, AP1189, has also been shown to be a biased agonist at the MC1R and MC3R, activating the ERK1/2 pathway but not the Gαs-cAMP pathway [107].
3.2. Biased allosteric modulators
Allosterism has been shown to exist widely in various types of proteins since it was first described in the hemoglobin [108]. GPCRs are thought as natural allosteric machines because agonist binding on the extracellular portion of the receptor causes conformational change in the receptor, resulting in the binding of intracellular signal transducers such as G proteins or β-arrestins on the intracellular portion of the receptor [109]. Allosteric modulation of GPCRs, in recent years, has been emerging as a promising novel approach to develop therapeutic agents for treatment of various disorders, conferring advantages over orthosteric ligands such as potential enhanced selectivity for different subtypes of the same family of receptors and limited maximal signaling [67, 109, 110]. Allosteric modulators are a group of molecules that can change receptor response to its orthosteric ligand by allosterically binding to the receptor. They are generally classified as positive allosteric modulators (PAMs), which increase the receptor response to orthosteric ligand, negative allosteric modulators (NAMs), which decrease receptor responsiveness, and neutral allosteric ligands, which have no effects on the receptor responsiveness although allosterically bind to receptor [111].
Biased allosteric modulators are ligands that bind to allosteric sites of receptors and preferentially modulate particular signaling pathways triggered by the orthosteric ligands. This type of modulators is effective in modulation of GPCR signaling with therapeutic purposes. The allosteric modulators of the CaSR (cinacalcet, NPS-R568 and NPS-2143), promote biased signaling by exerting greater allosteric modulation of intracellular Ca2+ mobilization relative to ERK1/2 activation in response to extracellular Ca2+ [112]. The small molecule allosteric modulator, Org27569, displays biased allosteric effects on the CB1 cannabinoid receptor by blocking cAMP inhibition triggered by Gαi/o but having little or no effect on ERK1/2 activation with the presence of cannabinoid ligands [113].
One non-steroidal anti-inflammatory drug, fenoprofen, was shown to have positive allosteric modulator activity at some MCRs, with pro-resolving properties by promoting macrophage phagocytosis and efferocytosis [114]. Our recent study further showed that fenoprofen is a biased allosteric enhancer as it selectively induces ERK1/2 activation rather than cAMP production at both WT and naturally occurring mutant MC3Rs [115]. A variant of canine β-defensin 103 (a high-affinity agonist for MC1R [116]), cBD103ΔG23, is a weak allosteric agonist at the canine MC4R [117]. Although an orthosteric ligand for human MC4R [118], THIQ is an allosteric ligand at a fish MC4R [119].
3.3. Biased receptors
Biased receptor is a mutant receptor capable of selectively adopting particular active conformation upon ligand stimulation to induce specific signaling pathway, different from the WT receptor. Compared with extensive studies on identifying biased ligands, biased receptors are not widely investigated. However, the importance of characterization of biased receptors should also be highlighted since investigation of them provides an effective way to elucidate the relationship between receptor conformational change and downstream signaling. According to the multi-state model, it is possible that the signaling pathways can be selectively activated with specific amino acid modifications in GPCRs.
Indeed, mutagenesis studies have suggested that some residues are critical in maintaining the specific conformations of GPCRs, and the switches of different intracellular signaling pathways through mutant receptors are the consequences of mutations on the key residues. For instance, mutating the highly conserved DRY motif of AT1R to AAY leads to unbalanced activation of Gαq/11 signaling and β-arrestin-dependent ERK1/2 signaling [120]. Novel mutations of β2-AR designed based on evolutionary trace analysis, including T68F, Y132G, and Y219A, are shown to lead the receptor to adopt conformation incapable of activating Gαs but capable of recruiting β-arrestins to initiate receptor internalization and β-arrestin-dependent ERK1/2 signaling [121]. By predicting and mutating the key restudies potentially interacting with G proteins or β-arrestins, several engineered biased mutant D2 dopamine receptors, including β-arrestin-biased (A135R and M140D) and Gαi/o protein-biased (L125N and Y133L) mutants, have been characterized [122]. Biased signaling caused by mutating key residues were also reported in ghrelin receptor [123], apelin receptor [124, 125], neuropeptide Y receptors [126], and CB1 cannabinoid receptor [127] (reviewed in [128]).
We have used alanine scanning mutagenesis to systematically study the structure-function relationship of the neural melanocortin receptors, MC3R and MC4R. By comparing Gαs-cAMP and ERK1/2 signaling, we have identified numerous biased mutant receptors [129–132]. For example, in our studies of L140 in human MC4R (numbered as 3.43 according to the Ballesteros and Weinstein numbering scheme [133]), a highly conserved amino acid in Family A GPCRs and shown to be important in constraining the receptor in inactive conformation (reviewed in Ref. [14]), in addition to confirming that this residue is also important in constraining the MC4R in inactive conformation (some mutants are constitutively active), we showed that some mutants have biased constitutive signaling and some mutants exhibit biased ligand-stimulated signaling [130], similar to a previous study [134].
Recently, biophysical and structural studies have started identifying the structural elements dictating biased signaling in different GPCRs. For example, using fluorine-19 nuclear magnetic resonance at the β2-AR, it was shown that agonists and biased ligands induce different conformations, primarily at TMD6 and TMD7, respectively [135], with the biased ligands stabilizing the active conformation in TMD7 longer than balanced agonists [136]. Using double electron-electron resonance spectroscopy and molecular dynamic simulations at the AT1R, different conformational ensembles are observed with balanced and biased ligands [137, 138] (see these recent reviews [59, 139, 140] for additional examples in other GPCRs). These studies will promote rational in silico design of biased ligands with better therapeutic potential (see [138] for example).
4. Biased signaling in naturally occurring GPCR mutations and endocrine diseases
Besides lab-generated biased mutant receptors, naturally occurring GPCR mutations that cause biased signaling have been identified and characterized based on different signaling assays. The majority of these mutations are associated with various disorders (Table 3). We will mainly focus on review of mutations in MC4R, CaSR, and PROKR2 since there are relatively more literature reporting biased mutations of these receptors. Mutations in other GPCRs will also be briefly discussed.
Table 3.
Summary of naturally occurring GPCR mutations resulting in biased signaling.
| Receptor | Mutation | Signaling properties | ligand | Cell system | References |
|---|---|---|---|---|---|
| GPR54 | L148S | Strongly impairs Gαq-IP accumulation but retains β-arrestin-dependent ERK1/2 activation | Kisspeptin-54 | HEK293 | [219] |
| R386P | Normal IP accumulation but defective receptor internalization | Kisspeptin-10 | COS-7 | [223] | |
|
PROKR2 |
A51T, R85C, R85H, R164Q, R268C, M323I, and V331M | Impair at least one G protein-dependent signaling (Gαq-Ca2+, Gαs-cAMP, and Gαi/o-ERK1/2) but not β-arrestin recruitment (A51T: defective in Gαs and Gαi/o; R85C: defective in Gαq; R85H, R164Q, and V331M: defective in Gαq and Gαi/o; R268C and M323I: defective in Gαi/o) |
PROK2 |
HEK293 |
[203, 205, 289] |
| R80C | Impairs Gαs-cAMP signaling and β-arrestin recruitment but not Gαq-Ca2+ or Gαi/o-ERK1/2 signaling | ||||
| L173R and V334M | Preferentially impair Gαs-cAMP but not Gαq-IP signaling | ||||
| L218S, R270H, R268C and V331M | Preferentially impair Gαq (IP or Ca2+) but not Gαs-cAMP signaling | ||||
| FSHR | A189V | Undetectable cAMP production, PKA activation, and cAMP/PKA transcriptional-associated response but inducible β-arrestin-dependent ERK1/2 activation | FSH | HEK293N | [229] |
| N431I | Increases basal and ligand-induced cAMP production but impairs β-arrestin-dependent desensitization and internalization (normalized to cell surface expression) | FSH | HEK293 | [230] | |
| TSHR | L653V | Normal maximal cAMP production but impaired IP accumulation | TSH | COS-7 | [234] |
| CaSR | L727Q and V836L | Reduced efficacy on intracellular Ca2+ mobilization but enhanced potency or efficacy on ERK1/2 activation | Ca2 | HEK293 | [188, 189] |
| R680G | Normal intracellular Ca2+ mobilization but enhanced β-arrestin-dependent ERK1/2 activation | ||||
| S657C | Increased intracellular Ca2+ mobilization but decreased ERK1/2 activation | ||||
| G778D, N178D, and S820A | Not detectable intracellular Ca2+ mobilization but inducible ERK1/2 activation (G778D); Impaired intracellular Ca2+ mobilization without altering ERK1/2 activation (N178D and S820A) |
||||
| G778D | Not detectable Ca2+ mobilization but inducible ERK1/2 activation | ||||
| AVPR2 | L83Q | Gαs-biased signaling after rescue by pharmacoperone | AVP | HeLa | [239] |
| Y128S | Constitutive Gαs-biased signaling after rescue by pharmacoperone | ||||
| R137H | Inducible β-arrestin recruitment and endocytosis but resistance to AVP-stimulated cAMP production and ERK1/2 activation (constitutive β-arrestin recruitment and endocytosis) | AVP | HEK293 | [245] | |
| R181C | Gαs-biased receptor incapable of signaling to Gαq/11-IP accumulation or recruiting β-arrestin | AVP | PC12 | [246] | |
| R137C/L | Inducible β-arrestin recruitment and endocytosis but resistance to AVP-stimulated cAMP production and ERK1/2 activation (high basal cAMP levels and constitutive β-arrestin recruitment and endocytosis) | AVP | HEK293 | [245] | |
| I130N, F229V, and L312S | Higher basal cAMP production and reduced constitutive β-arrestin recruitment and/or Gαq/11-IP accumulation | HEK293T HEK293FT |
[247–249] | ||
|
MT1R MT2R |
G166E and I212T (MT1R) | Bias towards ERK1/2 activation | Melatonin | COSM1 HEK293 |
[255, 256] |
| V124I (MT2R) | Bias towards Gαi activation | ||||
| W22L, A52T, A74T, R138H, R138L, L166I, R222H, R330W, and I353T (MT2R) | Bias towards ERK1/2 activation without Gαi activation | ||||
| MC1R | R151C, R160W, and D294H | Defective cAMP production but normal ERK1/2 activation | [Nle4,D-phe7]-α-MSH | PC12 | [263] |
| MC2R | F278C | Normal cAMP production but defective receptor internalization | ACTH | Mouse Y6 | [269] |
| MC3R | I87T, N128S, M134I, and L297V | Biased cAMP production without significant activation of ERK1/2 | α-MSH | HEK293T | [153] |
| F82S, I183N, and I335S | Biased activation of ERK1/2 without increasing cAMP production | ||||
| MC4R | D90N | Biased coupling to Gαi/o protein rather than Gαs protein | α-MSH | HEK293 GT1–7 | [158] |
| V50M, H76R, and I170V | Biased cAMP production and defective β-arrestin recruitment | α- and/or β-MSH | CHO- K1 | [156] | |
| V103I and H158R | Biased Gq/11 activation | α- and β-MSHs | HEK293 | [163] | |
| G55V, I69T, M79I, N97D, G98R, I125K, W174C, A175T, I194T, A219V, P260Q, F261S, C271Y, Q307X, and C326R | Biased activation of ERK1/2 without increasing cAMP production | [155, 162] | |||
| R165G, R165W, and C172R | Biased ERK1/2 constitutive signaling | ||||
| CCR6 | A89T, R155W, and A369V | Decreased basal and ligand-induced Gαi signaling but unaffected β-arrestin recruitment | Chemokine (C-C motif) ligand 20 | HEK293 | [290] |
| β3-AR | S165P and S257P | Impaired Gαs-cAMP production but normal Gαi-ERK1/2 activation | CL316,243 | HEK293 CHO-K1 |
[291] |
| GPR56 | R565W and L640R | Altered Gα12/13 signaling but no effects on Gαq-independent Ca2+ channel | HEK293T/17 | [292] |
4.1. Neural MCR (MC3R and MC4R) mutations and biased signaling
Neural MCRs, including MC3R and MC4R, with high expression in the central nervous system, are two important regulators of energy homeostasis [141–143]. Targeted deletion of Mc3r or Mc4r in mice leads to obesity [144–146]. These two receptors have been extensively studied in obesity pathogenesis. Many naturally occurring mutations in MC3R and MC4R have been identified from obese patients, providing further evidence for their function in regulating energy homeostasis ( for reviews, see Refs. [147–150]).
MC3R couples to Gαs protein stimulating cAMP-PKA pathway, and Gαi/o protein inducing ERK1/2 pathway through activation of phosphoinositide 3-kinase (PI3K) [151, 152]. β-arrestin-dependent internalization has also been suggested as an intracellular event after MC3R activation. Several MC3R mutants have been characterized in both cAMP and ERK1/2 pathway and shown to be biased receptors [153]. We showed that upon α-melanocyte-stimulating hormone (α-MSH) stimulation, I87T, N128S, M134I, and L297V mutants display biased activation of Gαs-cAMP pathway, while F82S, I183N, and I335S mutants show biased activation of ERK1/2 pathway [153].
MC4R couples to multiple G proteins including Gαs, Gαi/o, and Gαq/11, and G protein-independent pathways such as regulation of potassium channel (Kir 7.1) and β-arrestin-mediated internalization [151, 152, 154–156]. Until now, based on investigations of signaling profiles of naturally occurring MC4R mutations, a number of mutations resulting in biased signaling have been identified. These mutations are mostly distributed in the regions of TMDs, ICLs, and C-terminus, which potentially affect interaction of receptor and G protein or other signaling regulators/mediators (e.g. β-arrestins).
The first biased MC4R mutant associated with severe early-onset obesity is D90N [157, 158]. This mutant, without initiation of Gαs-cAMP pathway despite normal α-MSH binding, is able to couple to Gαi/o protein thereby inhibiting cAMP production in pertussis toxin-sensitive manner [158]. D90 (2.50) is involved in interaction with NPxxY motif of TMD7 and plays a role in switch between inactive and active receptor conformation in other GPCRs, such as rhodopsin and TSHR [159, 160], possibly through changing the inter-helical space of receptors [135, 161]. This may suggest that D90 of MC4R is also important in inter-helical space change and thus critical for G protein coupling. We also investigated biased signaling between Gαs-cAMP and ERK1/2 pathways in 73 naturally occurring MC4R mutations, revealing 27 mutants with biased activation of either Gαs-cAMP or ERK1/2 pathway [162] (Table 3). Some MC4R mutants with altered β-arrestin recruitment [155, 156] and biased Gαq/11 activation [163] have also been identified, although another study just published reported no significant biased signaling in seven Class V mutants (mutants with no measurable change in receptor cell surface expression, ligand binding, and receptor-mediated intracellular signaling [15]) and two signaling defective mutants with the exception of one mutant, E308K [164].
The physiological relevance of intracellular signaling initiated by neural MCRs have been shown by several studies (reviewed in [165]). The conventional Gαs-cAMP signaling pathway through MC4R mediates energy expenditure since Gαs deficiency in central nervous system leads to defect in energy expenditure without changing food intake [166, 167]. Food intake is mediated by several other intracellular mediators, such as Gαq/11-IP3/Ca2+, AMPK, ERK1/2, and potassium channel (Kir 7.1) [154, 165–171]. For example, Gαq deletion in the paraventricular nucleus causes hyperphagic obesity without affecting energy expenditure [171]; MC4R activation decreases AMPK phosphorylation thereby inhibiting food intake [168] and inhibitory effects of α-MSH on AMPK phosphorylation can be blocked by ERK1/2 inhibitors (U0126 and PD184352) [170]; MC4R coupling to closure of potassium channel (Kir 7.1) results in depolarization of neurons within paraventricular nucleus of hypothalamus, generating an anorexigenic effect [154]. ERK1/2 activation through MC3R is also involved in regulation of feeding behavior [172]. In addition to regulation of energy homeostasis, MC4R-medtaied Gαs signaling results in increased blood pressure and heart rate through sympathetic activation, causing the cardiovascular side effects and potentially limiting the therapeutics targeting MC4R [171, 173–176].
The biased agonism at neural MCR mutants provides new understanding on obesity pathogenesis and its therapeutics. Mutant D90N MC4R with both inactivated Gαs and activated Gαi/o signaling, resulting in severe defect of cAMP production, potentially account for the obesity phenotype of patients carrying this mutation [177]. Biased defect of either Gαs-cAMP or ERK1/2 pathway of MC3R and MC4R mutants might also be one cause of disrupted energy homeostasis and obesity phenotype of patients with corresponding mutations. Indeed, among MC4R mutants, five Class V mutants including C40R, V50M, T112M, A154D, and S295P are defective in ERK1/2 activation despite normal cell surface expression, ligand binding, and ligand-stimulated Gαs-cAMP signaling, suggesting potentially important role of ERK1/2 activation in regulation of energy homeostasis other than Gαs-cAMP pathway [162]. This may suggest that different therapeutic strategies should be adopted for obese patients carrying either Gαs signaling defective or ERK1/2 defective mutations.
Two recent studies identified β-arrestin-biased MC4R mutations and proposed that β-arrestin-mediated signaling plays a significant role in MC4R-mediated body weight regulation [155, 156]. Gillyard et al. suggest that β-arrestin recruitment is a marker of normal MC4R function [156]. In their study, five of seven Class V MC4R mutants show altered activities in β-arrestin recruitment upon α-MSH and/or β-MSH stimulation. Another study on 61 naturally occurring gain-of-function (in terms of canonical Gαs-cAMP pathway) MC4R variants clarified that β-arrestin-mediated MC4R signaling is critical in human weight regulation and not associated with cardiovascular side effects. In their study, five MC4R gain-of-function variants, including T11S, T101N, F201L, G231S, and R236C, are shown to result in biased signaling of cAMP production, whereas four variants, including V103I, I251L, I289L, and I317V, exhibits signaling bias toward β-arrestin recruitment and enhanced ERK1/2 activation [155]. By performing a comprehensive genetic association analyses, they found that people carrying homozygous β-arrestin-biased MC4R alleles, compared to noncarriers, have approximately 50% lower risks of obesity, type 2 diabetes, and coronary artery disease. The risks of these diseases are intermediate even in carriers with one β-arrestin-biased MC4R allele. Carriers with β-arrestin biased variants also have normal blood pressure and heart rate. However, population carrying gain-of-function variants with biased cAMP production do not exhibit the lower risks of such diseases but are associated with increased blood pressure [155]. This supports that β-arrestin biased signaling is clearly associated with protection from obesity and its related cardio-metabolic diseases.
Characterization of MC4R biased mutations further dissects the molecular mechanisms underlying diseases caused by genetic variants, and thereby identify other clinically relevant pathways which could be preferentially modulated in obesity treatment to achieve more effective and safer therapeutic effects. Considering the protective effects of biased β-arrestin signaling, some biased ligands or biased allosteric modulators could be used to modulate this pathway with minimal alteration on conventional Gαs pathway to avoid cardiovascular complications.
4.2. CASR mutations and biased signaling
The CaSR is a Family C GPCR, expressed in parathyroid gland, kidney, bone, intestine, and brain [178]. By sensing the extracellular Ca2+, activated CaSR suppresses PTH secretion from parathyroid cells and Ca2+ reabsorption in the kidney tubules [178, 179], and stimulates calcitonin secretion from thyroid gland [180]. Activation of Gαq/11 and Gαi/o proteins through CaSR has been suggested as signaling pathways related to the inhibition of PTH release in response to extracellular Ca2+ stimulation [181, 182]. In addition, CaSR-mediated ERK1/2 activation, a convergent point of several signaling pathways mediated by Gαq/11 and Gαi/o, as well as other signal mediators, such as β-arrestins [183, 184], is also believed to be involved in inhibition of PTH secretion [185, 186]. Until now, more than 230 mutations have been identified in human CaSR [187]. Most of these mutations are clinically relevant and associated with human disorders related to dysregulation of extracellular Ca2+ level, such as familial hypocalciuric hypercalcemia (FHH) and autosomal dominant hypocalcemia (ADH). However, in many earlier studies, mutations were classified as loss-of-function or gain-of-function mutations simply based on Gαq/11 signaling pathway by measuring IP accumulation or intracellular Ca2+ mobilization, which may lead to neglect of other clinically important signaling pathway(s) and the therapeutic potential.
Recently, several groups have focused on biased agonism of CaSR and identified many CaSR mutations which can induce signaling bias between Gαq/11-Ca2+ mobilization and ERK1/2 activation [188–190]. Most of these biased mutations are located in the TMDs and extracellular domain or loops. In particular, some biased mutations appear at disease-switch residues (different mutations on the same residue cause opposite phenotypes, leading to either FHH or ADH) [190], suggesting that biased agonism of CaSR may play important role in disease pathogenesis. Investigation of biased signaling triggered by mutant CaSRs will provide us insights into developing new targets for therapies of disorders related to extracellular calcium regulation.
Leach and colleagues investigated the signaling properties of 21 mutant CaSRs on both Gαq/11-Ca2+ mobilization and ERK1/2 activation [188]. These naturally occurring mutations included several loss-of-function mutations associated with FHH and gain-of-function mutations associated with ADH. Functional studies demonstrated that some of these mutations lead to pathway-selective signaling [188]. Some FHH-causing mutations switch the CaSR from preferentially coupling to Gαq/11-Ca2+ mobilization to ERK1/2 activation. In contrast, some of the ADH-causing mutations (Q681H, E767K, L773R, S820F, and F821L) induce a signaling bias toward Gαq/11-Ca2+ mobilization with increased potencies but do not significantly change either potency or maximal response in ERK1/2 activation. Of particular interest is the fact that three mutations (L727Q, G778D, and V836L) show biased impact on maximal responses in these two pathways [188] (Table 3).
Gorvin et al. showed that an ADH-causing mutation, R680G, located at the extracellular end of TMD3, differentially impacts the intracellular signaling pathways of CaSR. Different from cysteine and histidine substitution on R680 (R680C and R680H) (which impair receptor cell surface expression, Gαq/11-Ca2+ mobilization, and ERK1/2 activation [188]), glycine substitution on arginine 680 (R680G) does not affect intracellular Gαq/11-Ca2+ signaling but enhances G protein-independent and β-arrestin-dependent ERK1/2 activation [190], leading to gain-of-function in MAPK signaling. The signaling bias towards β-arrestin-dependent ERK1/2 activation has been considered as a consequence of disruption of R680-E767 salt bridge, an important interaction between TMD3 and ECL2 of CaSR [189]. Based on structure model of metabotropic glutamate receptor 1, which also belongs to Family C GPCRs and may have similar structural topology to the CaSR TMDs, this disruption might lead to disruption of TMD3 and ECL2 interaction and outward shift of the TMD3, TMD4 and/or TMD5 of CaSR, thereby facilitating β-arrestin binding to the cytoplasmic side of receptor [189, 191]. The outward shift of TMDs and insertion of the β-arrestin finger loop was also observed in β2-AR [192]. The signaling bias of R680G provides further support for the role of CaSR-mediated MAPK signaling in inhibition of PTH synthesis and release, and also clear insights into structure-function relationship of CaSR.
Gorvin et al. further identified some disease-switch residues that may determine the CaSR signaling bias [190]. By using bias factor calculation formula [193, 194], in which both EC50s and maximal responses are calculated, several FHH- or ADH-causing mutations were shown to result in signaling bias towards either intracellular Ca2+ signaling or ERK1/2 signaling. It is possible that mutations at these disease-switch residues may switch receptor conformation between G protein coupling and β-arrestin coupling since many of these mutations are located in the critical regions (extracellular domain or TMDs) for ligand binding and receptor activation. For example, a disease-switch residue N178, located at hinge region of the extracellular domain, has been shown to appear at the interface of CaSR dimer upon receptor activation [195]. Different mutations on N178 showed distinct signaling bias between intracellular Ca2+ signaling and ERK1/2 signaling. FHH-causing mutation N178D has impaired Ca2+ signaling (increased EC50 and reduced maximal response) but normal ERK activation, thus typical biased Ca2+ signaling. However, ADH-causing mutation N178Y reinforces both Ca2+ and ERK signaling pathways. Two other disease-switch residues, S657 and S820, are located at the TMD2 and TMD6, respectively. ADH-causing mutation S657C causes biased intracellular Ca2+ signaling with decreased ERK1/2 activation, but FHH-causing mutation S820A impairs intracellular Ca2+ signaling without altering ERK1/2 activation [190].
CaSR can undergo clathrin-mediated endocytosis [196], indicating that signal transduction could be regulated by this process. Indeed, some mutations on σ subunit of adaptor protein-2 (AP2σ) (a critical protein recognizing clathrin and connecting it with receptor in coated vesicles) have impact on receptor activity and are linked with FHH [197]. These hypercalcemia-associated AP2σ mutations disrupt CaSR endocytosis, therefore increasing cell surface expression of CaSR. However, paradoxically, they impair CaSR-mediated intracellular signaling rather than generating signal amplification [198]. Hence, Gorvin et al. hypothesized that in addition to the signaling from plasma membrane, CaSR might also signal from endosomal compartment like some other GPCRs (such as AVPR2 and PTH1R), and disruption of receptor endocytosis might attenuate overall signal transduction [46, 48, 199]. They further found that endosomal CaSR could predominantly couple to Gαq/11 to generate a sustained intracellular signaling distinct from rapid signaling originated from plasma membrane and this pathway is independent of β-arrestins [48]. The discovery of endosomal CaSR signaling has enriched our knowledge on relationship between CaSR-related diseases and CaSR-mediated intracellular signaling pathways, providing a new scope for further investigation. Although there is no CaSR mutation so far reported to cause biased signaling through endosomal pathway, it is worthy to evaluate signaling properties of CaSR mutations in light of this endosomal pathway considering its novel roles in regulating receptor activity and therapeutic potential for CaSR-related diseases.
4.3. PROKR2 mutations and biased signaling
Prokineticin (PROK) receptor 2 (PROKR2), a Family A GPCR, is known as an important GPCR in olfactory bulb morphogenesis and sexual maturation. Dysfunctional PROKR2 signaling may cause Kallmann syndrome, a disorder associated with idiopathic hypogonadotropic hypogonadism (IHH) and olfactory abnormalities (anosmia or hyposmia) [200, 201]. The migration of embryonic GnRH neurons from nasal epithelium into forebrain is disrupted due to early impaired development of the peripheral olfactory system [202]. Naturally occurring mutations in the genes encoding PROKR2 and cognate ligand PROK2 have been identified in patients with this syndrome, implicating defective PROKR2 signaling in disease pathogenesis.
PROKR2 can trigger several different intracellular signaling pathways, including Gαq/11, Gαi/o, and Gαs-dependent pathways [203]. In addition, β-arrestin recruitment through PROKR2 was also observed upon PROK2 stimulation [203], although the interaction between PROKR2 and β-arrestins is not considered as an event for receptor internalization and ERK1/2 activation [204].
To date, several mutations in human PROKR2 associated with Kallmann syndrome have been characterized in multiple signaling pathways in order to evaluate whether other pathways, in addition to the conventional Gαq/11, are related to disease pathogenesis. Two groups reported that 10 missense mutations result in biased signaling through PROKR2 with respect to different G protein subtypes and β-arrestins (see details in Table 3) [203, 205]. Of these mutations, some impair at least one G protein-dependent signaling but not β-arrestin recruitment (A51T, R85C, R85H, R164Q, R268C, M323I, and V331M). One mutation (R80C) results in defect in β-arrestin recruitment and Gαs-dependent signaling without impacting Gαq/11 and Gαi/o pathways [203]. Two other mutations (L173R and V334M) have been shown to preferentially impair Gαs but not Gαq/11-dependent signaling [205]. A very recent functional study revealed that two IHH-associated mutations, L218P and R270H, impair Gαq/11-dependent signaling but maintain normal Gαs and ERK1/2 signaling [206].
Investigation of biased mutations of PROKR2 provides a distinctive view to better understand the molecular mechanisms of PROKR2 signaling and structure-function relationship of this receptor. For instance, defective β-arrestin recruitment of R80C could be explained by the unique location of this mutation. R80, highly conserved among PROKR2s of different species, is located at ICL1, the region that does not directly interact with G proteins but is close to TMD7 and the eighth α helix known for β-arrestin recruitment [135, 203, 207–209]. The ICL3 may be a critical region controlling biased coupling of G proteins (impairment of Gαq coupling). This intracellular loop is thought to interact with various G proteins [210, 211]. The structural model of PROKR2 shows that the residue R268 of ICL3 is located in the region that interacts with G proteins [203, 205]. Mutation R268C selectively impairs Gαq signaling (measured by IP accumulation), although no significant change when intracellular Ca2+ release was monitored [203]. Another biased mutation, R270H, is also presented in ICL3. R270 is only two residues distal to R268. Thus, it is possible that R270H selectively disrupt the interaction of ICL3 to Gαq protein, leading to a biased Gαs coupling and ERK1/2 signaling [206]. To demonstrate this, the glutathione S-transferase (GST)-tagged PROKR2-ICL3s (WT and R270H) were pulled down to detect the binding of Gαs and Gαq proteins [206]. Indeed, the R270H mutation only impaired Gαq binding to ICL3, with normal Gαs interaction. This biased signaling occurs only when substitution is from arginine to other basic amino acids, such as histidine and lysine, but not other non-basic amino acids [206].
The signaling properties of these biased PROKR2 mutants indicate that the conventional Gαq signaling is not the only pathway relevant for disease pathogenesis. Many biased mutations described above cause defects in other G protein signaling and/or β-arrestin recruitment, suggesting pathogenic involvement of these pathways. Further studies are needed to explore the relationship of these pathways to Kallmann syndrome. It is interesting to note that one of these mutations, R268C, resulting in severe impairment of Gαi/o pathway, is now considered to be nonpathogenic since this mutation is frequently present in African American population (even in homozygous state), indicating that Gαi/o-dependent pathway plays a minor role in Kallmann syndrome pathogenesis. R268C is speculated to be a positively selected mutation which is protective in infection process in which infectious agents, such as whooping cough bacterial agent and the human malaria parasite [212, 213], use Gαi/o pathway to exert detrimental effects [203].
4.4. Other GPCR mutations and biased signaling
4.4.1. KISS1R mutations
G protein-coupled receptor 54, GPR54 (encoded by KISS1R, also called kisspeptin receptor), and the cognate ligand, kisspeptin, are known as important regulators for gonadotropin-releasing hormone (GnRH) secretion [214, 215]. GnRH is a critical hormone in hypothalamic-pituitary-gonadal axis and regulator of reproduction. Kisspeptin activates various intracellular signaling pathways via GPR54, including Gαq/11-mediated IP3 accumulation, Ca2+ mobilization, PKC activation, and MAPK (ERK1/2 and p38) activation, important events in kisspeptin-dependent GnRH secretion [216, 217]. Recruitment of β-arrestin is involved in desensitization, internalization, and signal transduction of GPR54 [218]. Recently, Ahow et al. reported that kisspeptin-dependent luteinizing hormone secretion (downstream action of GnRH secretion) is impaired in mice lacking either β-arrestin-1 or β-arrestin-2 [219], further supporting the idea that β-arrestin-dependent signaling plays a significant role in reproductive function through GPR54.
Loss-of-function mutations in human KISS1R have been shown to be a cause of IHH [220], a disorder characterized by the absence of spontaneous sexual maturation due to deficiency in GnRH secretion. One KISS1R mutation associated with IHH and infertility, L148S, has been shown to impact on the receptor with strong uncoupling from Gαq/11-dependent IP accumulation (downstream metabolite of IP3) although it does not affect receptor cell surface expression and ligand binding [215, 221]. Further, Ahow et al. reported that L148S mutant retains β-arrestin-dependent ERK1/2 activation despite defective Gαq/11 signaling, suggesting that L148S is a biased receptor, potentially explaining why some patients with loss-of-function KISS1R mutations in terms of IP accumulation display variable gonadotropic deficiency [219]. Gαq/11-independent signaling through biased GPR54 mutants might partially account for GnRH secretion and prevent complete gonadotropin deficiency in those patients. For example, the loss-of-function mutation L102P delays pubertal maturation but does not impede it, which might be due to other Gαq/11-independent signaling through the mutant receptor, such as β-arrestin-biased ERK1/2 activation [219, 222].
The only gain-of-function KISS1R mutation is R386P identified from a girl with idiopathic central precocious puberty [223 932]. Functional studies showed that R386P mutant has comparable EC50 and maximal response in IP accumulation as WT receptor, but displays a prolonged activation of intracellular signaling pathways, which indicates that this mutation might preferentially disrupt β-arrestin-dependent receptor desensitization and increase the stimulatory effects of kisspeptin on GnRH synthesis and secretion, eventually leading to precocious puberty [223].
4.4.2. FSHR mutations
The FSHR is mainly expressed by the granulosa cells of the ovarian follicle and the Sertoli cells of the seminiferous tubules, playing critical roles in reproduction. The gonadotropin from pituitary gland, FSH, is the endogenous ligand for FSHR in the gonads. FSH action through FSHR is fundamental for ovarian follicle maturation, whereas it is not essential for males to develop fertility although necessary to achieve normal testis and sperm production [224, 225].
Several loss-of-function mutations in human FSHR associated with IHH have been described (reviewed in [226]). A189V was the first reported FSHR loss-of-function mutation [227]. Five Finnish men (the population where A189V is frequently present) carrying A189V mutation exhibit variable degrees of oligospermia and subfertility [225]. The mutation impairs FSH-stimulated cAMP production [227, 228]. One recent study demonstrated that although A189V mutant is defective in Gαs-cAMP pathway with undetectable cAMP production, PKA activation, and cAMP/PKA transcriptional-associated response, this mutant receptor can still induce normal β-arrestin-dependent ERK1/2 signaling at low cell surface expression level, suggesting that A189V FSHR is a biased receptor [229]. The biased signaling properties provide the plausible explanation for the mild abnormalities (oligospermia and subfertility) detected in men harboring A189V mutation. Compared with the complete inhibition of all potential FSH-dependent signaling pathways due to failure of mutant FSH binding and activating FSHR, the biased A189V FSHR mutant impairs Gαs-cAMP pathway but not β-arrestin-dependent ERK1/2 signaling, which is suspected to be also involved in regulating reproductive function in males [229].
N431I is a gain-of-function FSHR mutation detected in an asymptomatic man with normal spermatogenesis, suppressed serum FSH, and normal or increased levels of biomedical markers of FSH action [230]. The impaired agonist-stimulated desensitization and internalization caused by this mutation are thought to be one of the reasons for the apparent constitutive activity and increased agonist-induced cAMP production when receptor cell surface expression was normalized [230].
4.4.3. TSHR mutation
TSHR, expressed in thyroid gland, is essential for thyroid hormone (TH, including thyroxine and triiodothyronine) synthesis and secretion. Upon endogenous agonist TSH binding, TSHR primarily couples to Gαs-cAMP pathway. In addition, the receptor also couples to Gαq/11-IP3/Ca2+ pathway when higher TSH concentration is present [231]. These two intracellular G protein-dependent signaling pathways have been suggested to mediate distinct physiological processes. The Gαs-cAMP pathway is thought to mediate TSH-stimulated TH secretion, growth and differentiation of follicular cells, and iodide uptake through sodium iodide symporter, while the Gαq/11-IP3/Ca2+ pathway regulates TH synthesis by inducing production of H2O2 which is critical for activity of thyroid peroxidase, a key enzyme in iodination and production of TH [232, 233].
Loss-of-function mutations has been known to cause hypothyroidism and resistance to TSH [231]. However, one novel familial missense mutation, L653V, has been reported in individuals with euthyroid hyperthyrotropinemia [234]. Homozygous individuals carrying L653V TSHR mutation exhibit normal free and total TH levels but markedly increased serum TSH levels. In addition, these individuals have higher radio-iodide uptake in thyroid gland compared with heterozygous and normal family members, suggesting imbalanced iodide trapping and iodination [234]. Functional study revealed that in COS-7 cells, L653V mutant has comparable cell surface expression and TSH binding as WT receptor. The potencies of TSH for cAMP and IP production are affected equivalently by this mutation with same fold-change on EC50s for both pathways. However, reduced efficacy is observed in IP production with severely impaired maximal response in contrast to normal efficacy of TSH for cAMP production, indicating that L653V TSHR exhibits biased signaling [234].
Because Gαs-cAMP and Gαq/11-IP/Ca2+ mediate distinct physiological responses [232, 233], severely defective Gαq/11-IP3/Ca2+ pathway may be the reason for the development of clinical phenotype of individuals carrying this mutation as discussed above [234]. In those patients, serum concentration of TSH are increased to overstimulate the mutant TSHR and compensate for defective TH synthesis due to impaired Gαq/11-IP3/Ca2+ signaling (negative feedback regulation of hypothalamic-pituitary-thyroid axis). Elevated TSH, on the other hand, also overstimulates the Gαs-cAMP pathway to induce further increased iodide trapping [234]. Hence, it is assumed that patients with L653V mutation exhibiting normal TH levels but increased iodine uptake is due to biased signaling of the two G protein pathways.
4.4.4. AVPR2 mutations
The system of arginine vasopressin (AVP) and AVPR2 are important in the regulation of water homeostasis. After AVP binding, AVPR2 predominantly signals through coupling to Gαs and stimulating cAMP-PKA pathway to phosphorylate aquaporin 2 water channel (AQP2), thus promoting water reabsorption in the kidney distal collecting tube [235–237]. Moreover, ERK1/2 and Gαq/11 signaling through AVPR2 can also be activated after AVP binding [238, 239]. Mutations in AVPR2 gene may lead to disorders of water homeostasis, with either nephrogenic diabetes insipidus (NDI) (failure in water reabsorption) or nephrogenic syndrome of inappropriate antidiuresis (NSIAD) (excessive water reabsorption, hyponatremia, and seizures) [235, 236, 240].
L83Q, Y128S, R137H, and R180C, are four NDI-associated mutations. L83Q and Y128S mutants are retained in the endoplasmic reticulum or endoplasmic reticulum-Golgi intermediate compartment [241, 242]. Rescue of these two mutants with pharmacoperones causes AVP-induced biased Gαs signaling in L83Q and biased constitutive Gαs signaling in Y128S [239]. R137H cannot activates Gαs but can recruit β-arrestin and induce endocytosis [243]. Constitutive β-arrestin recruitment and receptor endocytosis cause loss of function by decreasing the number of receptors at the cell surface [243–245]. R181C mutant exhibits Gαs biased signaling incapable of signaling to Gαq/11-IP3 accumulation or recruiting β-arrestins [246].
Opposite to the clinical outcomes of R137H, two other mutations (R137C and R137L) on the same residue lead to NSIAD. R137C and R137L display higher basal cAMP accumulation compared to R137H, although they are also associated with defective AVP-stimulated cAMP production and constitutive β-arrestin recruitment as R137H. The balance between constitutive Gαs-cAMP signaling and β-arrestin-dependent endocytosis seems to determine the extent of basal cAMP levels, providing an explanation for the different disease phenotypes between NDI- and NSIAD-causing mutations [245].
Three other NSIAD-associated mutations, I130N, F229V, and L312S, exhibit similar biased signaling properties with constitutive cAMP production and defective constitutive β-arrestin recruitment [247–249]. This further confirms that constitutive Gαs signaling in mutant AVPR2 is the major determinant of NSIAD [249]. Tolvaptan, an aquaretic drug, is proposed to be used for treatment of hyponatremia in patients with NSIAD, due to its role of inverse agonist at constitutive cAMP production of I130N, F229V, and L312S mutants [247–249].
4.4.5. MTNR1A and MTNR1B mutations
Melatonin, the principal hormone of the pineal gland, is crucial in sleep induction, circadian rhythm regulation, and immune response [250]. Melatonin signaling is mediated by melatonin receptor 1 and 2 (MT1R and MT2R). RT-PCR or in situ hybridization showed that the transcripts of MTNR1A (encoding MT1R) are localized in the suprachiasmatic nuclei, cortex, hippocampus, thalamus and cerebellum, and transcripts of MTNR1B (encoding MT2R) are distributed in the retina, whole brain, and hippocampus (reviewed in [251]). MT1R and MT2R activation by melatonin modulates several signal transduction pathways, typically inducing Gαi signaling to inhibit cAMP-PKA activation, and stimulating ERK1/2 and/or JNK activation (reviewed in [252]). In some cells, these receptors also change intracellular diacylglycerol, IP3, and Ca2+ concentration as well as cGMP levels [253].
Alteration in melatonin signaling has been reported in autism spectrum disorders (ASD), circadian disorders, and diabetes. ASD is a developmental disability that affects communication, behavior, and sleep [254]. Chaste et al. [255] reported that in 295 ASD patients, six nonsynonymous mutations in MTNR1A and ten in MTNR1B are identified with altered receptor function. Particularly interesting mutants are G166E MT1R (no significant difference of sleep between patients and controls), I212T MT1R (identified from patients with severe mental retardation and autism), and V124I MT2R (identified from patient with seasonal affective disorder). The two MT1R mutants show reduced cell surface expression. They also display biased signaling by completely inhibiting Gαi signaling and retaining partial activity in ERK1/2 activation. V124I MT2R preferentially impairs ERK1/2 signaling and shows normal Gαi signaling in comparison to the WT receptor [255]. The biased signaling of these three mutants potentially accounts for the phenotypes of some patients.
Moreover, some MTNR1B mutations have been most extensively associated with increased fasting plasma glucose and increased type 2 diabetes risk. Functional analysis of 40 MT2R mutants revealed that nine mutants (W22L, A52T, A74T, R138H, R138L, L166I, R222H, R330W, and I353T) display ERK1/2 biased signaling without Gαi activation [256]. Identification of these mutants and validation of the biased signaling may provide further information for the development of precision therapies for ASD and type 2 diabetes patients.
4.4.6. MC1R mutations
Melanocortin-1 receptor (MC1R) is a Gαs-coupled receptor expressed in epidermal melanocytes. MC1R regulates production of melanin pigment, serving as a major determinant of skin pigmentation, skin phototype, ultraviolet radiation sensitivity and non-melanoma skin cancer risk [257, 258]. After endogenous ligand (α-MSH or other proopiomelanocortin-derived peptides) binding, MC1R activates cAMP-PKA and ERK1/2 signaling [259].
The MC1R is highly polymorphic with more than 100 nonsynonymous mutations reported so far [259, 260]. R151C, R160W, and D294H, are the commonest three loss-of-function MC1R mutations associated with the red hair color (RHC) phenotype (such as red hair, fair skin, freckling, poor tanning, and increased risk for melanoma and nonmelanoma skin cancers) [261, 262]. These three mutants display reduced cAMP signaling but activate ERK1/2 signaling as efficiently as WT MC1R, suggesting that they are biased receptors [263]. Impaired cAMP signaling in R151C, R160W and D294H may account for the loss-of-function in photoprotection [264, 265], and ERK1/2 activation plays a minor effect on MC1R-dependent pigmentation in human skin compared with cAMP pathway [259]. Biased constitutive signaling has also been reported for a naturally occurring mutation, E92K [266].
4.4.7. MC2R mutation
Adrenocorticotropic hormone (ACTH) and its cognate receptor, MC2R, play a pivotal role in adrenal development, carbohydrate metabolism, and stress response by regulating synthesis and secretion of aldosterone and glucocorticoid [267]}. ACTH induces Gαs-cAMP-PKA pathway through activating MC2R [267]. Dysfunctions of ACTH-MC2R system such as abnormal secretion of ACTH or MC2R mutations result in severe adrenal diseases. For example, pathological ACTH over-secretion results in Cushing’s syndrome (glucocorticoid over-secretion) and adrenocortical hyperplasia whereas loss-of-function mutations in MC2R cause familial glucocorticoid deficiency syndrome [267, 268].
A naturally occurring mutation in human MC2R (F278C) has been identified with ACTH-independent Cushing’s syndrome. F278C mutant displays similar ACTH-induced cAMP production but increased basal cAMP accumulation with impaired desensitization and internalization compared to WT receptor [269]. Desensitization of MC2R results from PKA-mediated phosphorylation [270]. Mutation of phosphorylation site S208 of MC2R also causes failure in desensitization. One possibility of the F278C-induced dysfunction in receptor desensitization is that F278C affects the phosphorylation sites, serine 280 and/or 294. These findings suggest that failure of desensitization in MC2R appears to be associated with enhanced basal receptor activity, causing Cushing’s syndrome [269].
5. Pathophysiological implications of biased mutant GPCRs in endocrine diseases
Over the past decade, biased ligands at GPCRs, with more beneficial effects and less detrimental side effects, have been suggested as novel therapeutic agents for disease treatments. Biased mutant GPCRs identified so far also provide us implications on pathogenesis and treatment of many diseases. Herein, we proposed four types of biased mutant receptor models relevant to disease pathogenesis (Fig. 2B).
The first model proposes that two intracellular signaling pathways are both required for mediating a certain physiological response. Impaired or enhanced activation on either one will induce lower or higher degree of such response, eventually leading to the abnormal phenotypes. This type of biased mutant receptors includes L148S GPR54, A189V FSHR, and some mutant PROKR2s, CaSRs, and neural MCRs [153, 158, 162, 188, 189, 203, 205, 219, 229]. In the second model, two intracellular signaling pathways are thought to be responsible for two distinct physiological responses, and defect in either one will lead to its distinct phenotype. Biased L653V TSHR belongs to this type [234].
In the third model, β-arrestin mediated receptor desensitization and internalization are considered as a counteractive pathway for G protein-dependent signaling cascades. Inactive or overactive β-arrestin coupling will lead to either hyperactivated or suppressed G protein-dependent signaling and then consequent abnormal phenotype. This type of biased receptor is represented by some mutant GPCRs, such as R386P GPR54, N431I FSHR, F278C MC2R, and some mutant AVPR2s [223, 230, 245–247, 269]. Considering the increasing evidence of endosomal signaling triggered by internalized GPCRs, we also present the model of biased endosomal signaling (model 4), in which mutant receptor may preferentially strengthen G protein activation through endosomal pathway, leading to a more sustained G protein signaling and relevant physiological response. Although there is no GPCR mutant characterized so far in this way, future research along this line will likely provide better understanding of the molecular and cellular basis of disease pathogenesis.
Comprehensive understanding of signaling properties of biased mutant GPCRs in pathophysiology facilitates us to select appropriate ligands that could specifically and precisely target the abnormal pathways without affecting the normal ones. Biased ligands and biased allosteric modulators described above might be better drug candidates considering their desirable effects on modulation of clinically relevant signaling pathways of normal receptors. However, to our knowledge, there is no report of such effective ligands for selective modification of the impaired pathways of biased mutant GPCRs. Hence, future studies are still required to identify or develop these ligands and to test their pharmacological abilities in terms of biased mutant GPCRs. The investigation of signaling properties of biased mutant GPCRs also provides a better perspective on structure-function relationship of these receptors and will be of benefit to develop drugs targeting some residues critical for receptor signaling and meet therapeutic effects in patients with loss-of-function or gain-of-function mutations.
6. Conclusions
Naturally occurring mutations in GPCRs have been known to cause various endocrine diseases. GPCR mutations that result in biased signaling have been identified and characterized in recent years, making significant progresses in elucidating the pathogenesis of diseases. The investigation of biased agonism of these GPCR mutations will strengthen our understanding of correlation between signaling properties and clinical phenotypes, thereby providing pathophysiological implications and novel therapies for patients with such mutations.
Highlights:
Naturally occurring mutations in GPCRs are the causes of numerous diseases.
Biased ligands and mutations can be found in GPCRs.
Some disease-causing GPCR mutations cause biased signaling.
Characterization of biased mutant GPCRs elucidates the pathophysiology of these diseases.
Acknowledgements
Our original studies on the neural MCRs were supported by following grants: National Institutes of Health grant R15DK077213, American Diabetes Association Grant 1-12-BS212, Intramural Grant Program of Auburn University, and Animal Health and Disease Research Program and Interdisciplinary Grant of Auburn University College of Veterinary Medicine. We thank former graduate students, whose work are cited in the manuscript, for their contributions to the original studies summarized in this review. Li-Kun Yang and Zhi-Shuai Hou received graduate fellowships from China Scholarship Council, People’s Republic of China.
Abbreviations:
- AC
adenylyl cyclase
- ACTH
adrenocorticotropic hormone
- ADH
autosomal dominant hypocalcemia
- AQP2
aquaporin 2
- AR
adrenergic receptor
- ASD
autism spectrum disorders
- AT1R
angiotensin II receptor type 1
- AVP
arginine vasopressin
- AVPR2
type 2 arginine vasopressin receptor
- cAMP
cyclic adenosine monophosphate
- CaSR
calcium-sensing receptor
- CCR
C-C chemokine receptor
- DAG
diacylglycerol
- ECL
extracellular loop
- ERK
extracellular signal-regulated kinase
- FHH
familial hypocalciuric hypercalcemia
- FSH (FSHR)
follicle-stimulating hormone (receptor)
- GnRH
gonadotropin-releasing hormone
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- hCG
human chorionic gonadotropin
- ICL
intracellular loop
- IHH
idiopathic hypogonadotropic hypogonadism
- IP
inositol phosphate
- IP3
inositol 1,4,5-trisphosphate
- JNK
c-Jun-N-terminal kinase
- LH (LHR)
luteinizing hormone (receptor)
- MAPK
mitogen-activated protein kinase
- MCR
melanocortin receptor
- MSH
melanocyte-stimulating hormone
- MT1/2R
melatonin receptor 1 and 2
- NDI
nephrogenic diabetes insipidus
- NSIAD
nephrogenic syndrome of inappropriate antidiuresis
- OR
opioid receptor
- PI3K
phosphoinositide 3-kinase
- PKA/C
protein kinase A/C
- PLC
phospholipase C
- PROK2 and PROKR2
prokineticin 2 and prokineticin receptor 2
- PTH (PTHR)
parathyroid hormone (receptor)
- RHC
red hair color
- RhoGEF
Rho-specific guanine nucleotide exchange factor
- TH
thyroid hormone
- TMD
transmembrane domain
- TSH (TSHR)
thyroid-stimulating hormone (receptor)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare that there is no conflict of interest with this manuscript.
References:
- [1].Bockaert J, Pin JP, Molecular tinkering of G protein-coupled receptors: an evolutionary success, EMBO J, 18 (1999) 1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tao YX, Molecular chaperones and G protein-coupled receptor maturation and pharmacology, Mol Cell Endocrinol, 511 (2020) 110862. [DOI] [PubMed] [Google Scholar]
- [3].Congreve M, de Graaf C, Swain NA, Tate CG, Impact of GPCR structures on drug discovery, Cell, 181 (2020) 81–91. [DOI] [PubMed] [Google Scholar]
- [4].Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE, Trends in GPCR drug discovery: new agents, targets and indications, Nat Rev Drug Discov, 16 (2017) 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Offermanns S, G-proteins as transducers in transmembrane signalling, Prog Biophys Mol Biol, 83 (2003) 101–130. [DOI] [PubMed] [Google Scholar]
- [6].Worzfeld T, Wettschureck N, Offermanns S, G12/G13-mediated signalling in mammalian physiology and disease, Trends Pharmacol Sci, 29 (2008) 582–589. [DOI] [PubMed] [Google Scholar]
- [7].Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE, The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart, Nature, 325 (1987) 321–326. [DOI] [PubMed] [Google Scholar]
- [8].Ikeda SR, Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits, Nature, 380 (1996) 255. [DOI] [PubMed] [Google Scholar]
- [9].Clapham DE, Neer EJ, G protein βγ subunits, Annu Rev Pharmacol Toxicol, 37 (1997) 167–203. [DOI] [PubMed] [Google Scholar]
- [10].Spiegel AM, Defects in G protein-coupled signal transduction in human disease, Annu Rev Physiol, 58 (1996) 143–170. [DOI] [PubMed] [Google Scholar]
- [11].Schoneberg T, Schulz A, Biebermann H, Hermsdorf T, Rompler H, Sangkuhl K, Mutant G-protein-coupled receptors as a cause of human diseases, Pharmacol Ther, 104 (2004) 173–206. [DOI] [PubMed] [Google Scholar]
- [12].Tao YX, Inactivating mutations of G protein-coupled receptors and diseases: Structure-function insights and therapeutic implications, Pharmacol Ther, 111 (2006) 949–973. [DOI] [PubMed] [Google Scholar]
- [13].Lania AG, Mantovani G, Spada A, Mechanisms of disease: Mutations of G proteins and G-protein-coupled receptors in endocrine diseases, Nat Clin Pract Endocrinol Metab, 2 (2006) 681–693. [DOI] [PubMed] [Google Scholar]
- [14].Tao YX, Constitutive activation of G protein-coupled receptors and diseases: Insights into mechanism of activation and therapeutics, Pharmacol Ther, 120 (2008) 129–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tao YX, Conn PM, Chaperoning G protein-coupled receptors: From cell biology to therapeutics, Endocr Rev, 35 (2014) 602–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dryja TP, McGee TL, Hahn LB, Cowley GS, Olsson JE, Reichel E, Sandberg MA, Berson EL, Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa, N Engl J Med, 323 (1990) 1302–1307. [DOI] [PubMed] [Google Scholar]
- [17].Sung CH, Davenport CM, Hennessey JC, Maumenee IH, Jacobson SG, Heckenlively JR, Nowakowski R, Fishman G, Gouras P, Nathans J, Rhodopsin mutations in autosomal dominant retinitis pigmentosa, Proc Natl Acad Sci U S A, 88 (1991) 6481–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sung CH, Davenport CM, Nathans J, Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain, J Biol Chem, 268 (1993) 26645–26649. [PubMed] [Google Scholar]
- [19].Keen TJ, Inglehearn CF, Lester DH, Bashir R, Jay M, Bird AC, Jay B, Bhattacharya SS, Autosomal dominant retinitis pigmentosa: four new mutations in rhodopsin, one of them in the retinal attachment site, Genomics, 11 (1991) 199–205. [DOI] [PubMed] [Google Scholar]
- [20].Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD, Constitutively active mutants of rhodopsin, Neuron, 9 (1992) 719–725. [DOI] [PubMed] [Google Scholar]
- [21].Vassart G, Costagliola S, G protein-coupled receptors: mutations and endocrine diseases, Nat Rev Endocrinol, 7 (2011) 362–372. [DOI] [PubMed] [Google Scholar]
- [22].Rana BK, Shiina T, Insel PA, Genetic variations and polymorphisms of G protein-coupled receptors: functional and therapeutic implications, Annu Rev Pharmacol Toxicol, 41 (2001) 593–624. [DOI] [PubMed] [Google Scholar]
- [23].Hermans E, Biochemical and pharmacological control of the multiplicity of coupling at G-protein-coupled receptors, Pharmacol Ther, 99 (2003) 25–44. [DOI] [PubMed] [Google Scholar]
- [24].Allgeier A, Offermanns S, Van Sande J, Spicher K, Schultz G, Dumont JE, The human thyrotropin receptor activates G-proteins Gs and Gq/11, J Biol Chem, 269 (1994) 13733–13735. [PubMed] [Google Scholar]
- [25].Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE, Schultz G, The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families, Proc Natl Acad Sci U S A, 93 (1996) 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wittau N, Grosse R, Kalkbrenner F, Gohla A, Schultz G, Gudermann T, The galanin receptor type 2 initiates multiple signaling pathways in small cell lung cancer cells by coupling to Gq, Gi and G12 proteins, Oncogene, 19 (2000) 4199. [DOI] [PubMed] [Google Scholar]
- [27].Schwindinger WF, Fredericks J, Watkins L, Robinson H, Bathon JM, Pines M, Suva LJ, Levine MA, Coupling of the PTH/PTHrP receptor to multiple G-proteins, Endocrine, 8 (1998) 201–209. [DOI] [PubMed] [Google Scholar]
- [28].Gudermann T, Nichols C, Levy F, Birnbaumer M, Birnbaumer L, Ca2+ mobilization by the LH receptor expressed in Xenopus oocytes independent of 3’,5’-cyclic adenosine monophosphate formation: evidence for parallel activation of two signalling pathways, Mol Endocrinol, 6 (1992) 272–278. [DOI] [PubMed] [Google Scholar]
- [29].Godson C, Reppert SM, The Mel1a melatonin receptor is coupled to parallel signal transduction pathways, Endocrinology, 138 (1997) 397–404. [DOI] [PubMed] [Google Scholar]
- [30].Hawes BE, Kil E, Green B, O’neill K, Fried S, Graziano MP, The melanin-concentrating hormone receptor couples to multiple G proteins to activate diverse intracellular signaling pathways, Endocrinology, 141 (2000) 4524–4532. [DOI] [PubMed] [Google Scholar]
- [31].Pitcher JA, Freedman NJ, Lefkowitz RJ, G protein-coupled receptor kinases, Annu Rev Biochem, 67 (1998) 653–692. [DOI] [PubMed] [Google Scholar]
- [32].Rajagopal S, Rajagopal K, Lefkowitz RJ, Teaching old receptors new tricks: biasing seven-transmembrane receptors, Nat Rev Drug Discov, 9 (2010) 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Reiter E, Ahn S, Shukla AK, Lefkowitz RJ, Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors, Annu Rev Pharmacol Toxicol, 52 (2012) 179–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ, β-Arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3, Science, 290 (2000) 1574–1577. [DOI] [PubMed] [Google Scholar]
- [35].Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ, Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds, Proc Natl Acad Sci U S A, 98 (2001) 2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sun Y, Cheng Z, Ma L, Pei G, β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation, J Biol Chem, 277 (2002) 49212–49219. [DOI] [PubMed] [Google Scholar]
- [37].Goel R, Phillips-Mason PJ, Raben DM, Baldassare JJ, α-Thrombin induces rapid and sustained Akt phosphorylation by β-arrestin1-dependent and-independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression, J Biol Chem, 277 (2002) 18640–18648. [DOI] [PubMed] [Google Scholar]
- [38].DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK, β-Arrestins and cell signaling, Annu Rev Physiol, 69 (2007) 483–510. [DOI] [PubMed] [Google Scholar]
- [39].Ma L, Pei G, β-arrestin signaling and regulation of transcription, J Cell Sci, 120 (2007) 213–218. [DOI] [PubMed] [Google Scholar]
- [40].Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM, β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation, J Biol Chem, 277 (2002) 9429–9436. [DOI] [PubMed] [Google Scholar]
- [41].Ahn S, Shenoy SK, Wei H, Lefkowitz RJ, Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor, J Biol Chem, 279 (2004) 35518–35525. [DOI] [PubMed] [Google Scholar]
- [42].Vilardaga JP, Jean-Alphonse FG, Gardella TJ, Endosomal generation of cAMP in GPCR signaling, Nat Chem Biol, 10 (2014) 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tsvetanova NG, Irannejad R, von Zastrow M, G protein-coupled receptor (GPCR) signaling via heterotrimeric G proteins from endosomes, J Biol Chem, 290 (2015) 6689–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ, Persistent cAMP-signals triggered by internalized G-protein-coupled receptors, PLoS Biol, 7 (2009) e1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP, Sustained cyclic AMP production by parathyroid hormone receptor endocytosis, Nat Chem Biol, 5 (2009) 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Feinstein TN, Yui N, Webber MJ, Wehbi VL, Stevenson HP, King JD, Hallows KR, Brown D, Bouley R, Vilardaga JP, Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin, J Biol Chem, 288 (2013) 27849–27860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Thomsen ARB, Plouffe B, Cahill TJ 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ, GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling, Cell, 166 (2016) 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gorvin CM, Insights into calcium-sensing receptor trafficking and biased signalling by studies of calcium homeostasis, J Mol Endocrinol, 61 (2018) R1–R12. [DOI] [PubMed] [Google Scholar]
- [49].Mullershausen F, Zecri F, Cetin C, Billich A, Guerini D, Seuwen K, Persistent signaling induced by FTY720-phosphate is mediated by internalized S1P1 receptors, Nat Chem Biol, 5 (2009) 428–434. [DOI] [PubMed] [Google Scholar]
- [50].Nguyen AH, Thomsen ARB, Cahill TJ 3rd, Huang R, Huang LY, Marcink T, Clarke OB, Heissel S, Masoudi A, Ben-Hail D, Samaan F, Dandey VP, Tan YZ, Hong C, Mahoney JP, Triest S, Little J.t., Chen X, Sunahara R, Steyaert J, Molina H, Yu Z, des Georges A, Lefkowitz RJ, Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex, Nat Struct Mol Biol, 26 (2019) 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thomsen W, Frazer J, Unett D, Functional assays for screening GPCR targets, Curr Opin Biotechnol, 16 (2005) 655–665. [DOI] [PubMed] [Google Scholar]
- [52].Montminy MR, Bilezikjian LM, Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene, Nature, 328 (1987) 175–178. [DOI] [PubMed] [Google Scholar]
- [53].Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR, Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression, Nature, 383 (1996) 837–840. [DOI] [PubMed] [Google Scholar]
- [54].Mao J, Yuan H, Xie W, Simon MI, Wu D, Specific involvement of G proteins in regulation of serum response factor-mediated gene transcription by different receptors, J Biol Chem, 273 (1998) 27118–27123. [DOI] [PubMed] [Google Scholar]
- [55].Galan B, Caboche J, Elk-1 a transcription factor with multiple facets in the brain, Front Neurosci, 5 (2011) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lohse MJ, Nuber S, Hoffmann C, Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling, Pharmacol Rev, 64 (2012) 299–336. [DOI] [PubMed] [Google Scholar]
- [57].Bertrand L, Parent S, Caron M, Legault M, Joly E, Angers S, Bouvier M, Brown M, Houle B, Menard L, The BRET2/arrestin assay in stable recombinant cells: a platform to screen for compounds that interact with G protein-coupled receptors (GPCRS), J Recep Signal Transduc Res, 22 (2002) 533–541. [DOI] [PubMed] [Google Scholar]
- [58].Vilardaga JP, Bünemann M, Krasel C, Castro M, Lohse MJ, Measurement of the millisecond activation switch of G protein–coupled receptors in living cells, Nature biotechnology, 21 (2003) 807–812. [DOI] [PubMed] [Google Scholar]
- [59].Seyedabadi M, Ghahremani MH, Albert PR, Biased signaling of G protein coupled receptors (GPCRs): Molecular determinants of GPCR/transducer selectivity and therapeutic potential, Pharmacol Ther, 200 (2019) 148–178. [DOI] [PubMed] [Google Scholar]
- [60].Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS, Differential desensitization, receptor phosphorylation, β-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7, J Biol Chem, 279 (2004) 23214–23222. [DOI] [PubMed] [Google Scholar]
- [61].Corbisier J, Galès C, Huszagh A, Parmentier M, Springael JY, Biased signaling at chemokine receptors, J Biol Chem, 290 (2015) 9542–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kenakin T, Inverse, protean, and ligand-selective agonism: matters of receptor conformation, FASEB J, 15 (2001) 598–611. [DOI] [PubMed] [Google Scholar]
- [63].Kenakin T, Ligand-selective receptor conformations revisited: the promise and the problem, Trends Pharmacol Sci, 24 (2003) 346–354. [DOI] [PubMed] [Google Scholar]
- [64].Perez DM, Karnik SS, Multiple signaling states of G-protein-coupled receptors, Pharmacol Rev, 57 (2005) 147–161. [DOI] [PubMed] [Google Scholar]
- [65].Violin JD, Lefkowitz RJ, β-Arrestin-biased ligands at seven-transmembrane receptors, Trends Pharmacol Sci, 28 (2007) 416–422. [DOI] [PubMed] [Google Scholar]
- [66].Pupo AS, Duarte DA, Lima V, Teixeira LB, Parreiras-e-Silva LT, Costa-Neto CM, Recent updates on GPCR biased agonism, Pharmacol Res, 112 (2016) 49–57. [DOI] [PubMed] [Google Scholar]
- [67].Kenakin T, Miller LJ, Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery, Pharmacol Rev, 62 (2010) 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kenakin T, Christopoulos A, Signalling bias in new drug discovery: detection, quantification and therapeutic impact, Nat Rev Drug Discov, 12 (2013) 205–216. [DOI] [PubMed] [Google Scholar]
- [69].Rominger DH, Cowan CL, Gowen-MacDonald W, Violin JD, Biased ligands: pathway validation for novel GPCR therapeutics, Curr Opin Pharmacol, 16 (2014) 108–115. [DOI] [PubMed] [Google Scholar]
- [70].Kenakin T, Biased receptor signaling in drug discovery, Pharmacol Rev, 71 (2019) 267–315. [DOI] [PubMed] [Google Scholar]
- [71].Kieffer BL, Opioids: first lessons from knockout mice, Trends Pharmacol Sci, 20 (1999) 19–26. [DOI] [PubMed] [Google Scholar]
- [72].Violin JD, Crombie AL, Soergel DG, Lark MW, Biased ligands at G-protein-coupled receptors: promise and progress, Trends Pharmacol Sci, 35 (2014) 308–316. [DOI] [PubMed] [Google Scholar]
- [73].Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT, Enhanced morphine analgesia in mice lacking β-arrestin 2, Science, 286 (1999) 2495–2498. [DOI] [PubMed] [Google Scholar]
- [74].Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG, μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence, Nature, 408 (2000) 720–723. [DOI] [PubMed] [Google Scholar]
- [75].Raehal KM, Walker JKL, Bohn LM, Morphine side effects in β-arrestin 2 knockout mice, J Pharmacol Exp Ther, 314 (2005) 1195–1201. [DOI] [PubMed] [Google Scholar]
- [76].Chen X, Pitis P, Liu G, Yuan C, Gotchev D, Cowan CL, Rominger DH, Koblish M, DeWire SM, Crombie AL, Structure-activity relationships and discovery of a G protein biased μ opioid receptor ligand, [(3-Methoxythiophen-2-yl) methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5] decan-9-yl] ethyl}) amine (TRV130), for the treatment of acute severe pain, J Med Chem, 56 (2013) 8019–8031. [DOI] [PubMed] [Google Scholar]
- [77].Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, Huang XP, Sassano MF, Giguere PM, Lober S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK, Structure-based discovery of opioid analgesics with reduced side effects, Nature, 537 (2016) 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bermudez M, Nguyen TN, Omieczynski C, Wolber G, Strategies for the discovery of biased GPCR ligands, Drug Discov Today, 24 (2019) 1031–1037. [DOI] [PubMed] [Google Scholar]
- [79].Rives ML, Rossillo M, Liu-Chen LY, Javitch JA, 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment, J Biol Chem, 287 (2012) 27050–27054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM, Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at κ-opioid receptors in striatal neurons, J Biol Chem, 288 (2013) 22387–22398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Woo AYH, Xiao RP, β-Adrenergic receptor subtype signaling in heart: from bench to bedside, Acta Pharmacol Sin, 33 (2012) 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Xiao RP, β-Adrenergic signaling in the heart: dual coupling of the β2-adrenergic receptor to Gs and Gi proteins, Sci Signal, 2001 (2001) re15. [DOI] [PubMed] [Google Scholar]
- [83].Lohse MJ, Engelhardt S, Eschenhagen T, What is the role of β-adrenergic signaling in heart failure?, Circ Res, 93 (2003) 896–906. [DOI] [PubMed] [Google Scholar]
- [84].Noma T, Lemaire A, Prasad SVN, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, β-Arrestin–mediated β1-adrenergic receptor transactivation of the EGFR confers cardioprotection, J Clin Invest, 117 (2007) 2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang JL, Hanada K, Staus DP, Makara MA, Dahal GR, Chen Q, Ahles A, Engelhardt S, Rockman HA, Gαi is required for carvedilol-induced β1 adrenergic receptor β-arrestin biased signaling, Nat Commun, 8 (2017) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ, A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling, Proc Natl Acad Sci U S A, 104 (2007) 16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, Rockman HA, β-Blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation, Proc Natl Acad Sci U S A, 105 (2008) 14555–14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G, β-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors, Proc Natl Acad Sci U S A, 100 (2003) 11406–11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Baker JG, Hall IP, Hill SJ, Agonist and inverse agonist actions of β-blockers at the human β2-adrenoceptor provide evidence for agonist-directed signaling, Mol Pharmacol, 64 (2003) 1357–1369. [DOI] [PubMed] [Google Scholar]
- [90].Violin JD, Ren XR, Lefkowitz RJ, G-protein-coupled receptor kinase specificity for β-arrestin recruitment to the β2-adrenergic receptor revealed by fluorescence resonance energy transfer, J Biol Chem, 281 (2006) 20577–20588. [DOI] [PubMed] [Google Scholar]
- [91].Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK, Structure of a nanobody-stabilized active state of the β2 adrenoceptor, Nature, 469 (2011) 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kukkonen JP, Jansson CC, Åkerman KE, Agonist trafficking of Gi/o-mediated α2A-adrenoceptor responses in HEL 92.1. 7 cells, Br J Pharmacol, 132 (2001) 1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Arey BJ, López FJ, Are circulating gonadotropin isoforms naturally occurring biased agonists? Basic and therapeutic implications, Rev Endocr Metab Disord, 12 (2011) 275–288. [DOI] [PubMed] [Google Scholar]
- [94].Zariñán T, Butnev VY, Gutiérrez-Sagal R, Maravillas-Montero JL, Martinez-Luis I, Mejía-Domínguez NR, Juárez-Vega G, Bousfield GR, Ulloa-Aguirre A, In vitro impact of FSH glycosylation variants on FSH receptor-stimulated signal transduction and functional selectivity, J Endocr Soc, 4 (2020) bvaa019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Arey BJ, Stevis PE, Deecher DC, Shen ES, Frail DE, Negro-Vilar A, López FJ, Induction of promiscuous G protein coupling of the follicle-stimulating hormone (FSH) receptor: a novel mechanism for transducing pleiotropic actions of FSH isoforms, Mol Endocrinol, 11 (1997) 517–526. [DOI] [PubMed] [Google Scholar]
- [96].Arey BJ, Yanofsky SD, Pérez MC, Holmes CP, Wrobel J, Gopalsamy A, Stevis PE, López FJ, Winneker RC, Differing pharmacological activities of thiazolidinone analogs at the FSH receptor, Biochem Biophys Res Commun, 368 (2008) 723–728. [DOI] [PubMed] [Google Scholar]
- [97].Cheloha RW, Gellman SH, Vilardaga JP, Gardella TJ, PTH receptor-1 signalling—mechanistic insights and therapeutic prospects, Nat Rev Endocrinol, 11 (2015) 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, Vilardaga JP, Retromer terminates the generation of cAMP by internalized PTH receptors, Nat Chem Biol, 7 (2011) 278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Okazaki M, Ferrandon S, Vilardaga JP, Bouxsein ML, Potts JT, Gardella TJ, Prolonged signaling at the parathyroid hormone receptor by peptide ligands targeted to a specific receptor conformation, Proc Natl Acad Sci U S A, 105 (2008) 16525–16530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Maeda A, Okazaki M, Baron DM, Dean T, Khatri A, Mahon M, Segawa H, Abou-Samra AB, Jüppner H, Bloch KD, Critical role of parathyroid hormone (PTH) receptor-1 phosphorylation in regulating acute responses to PTH, Proc Natl Acad Sci U S A, 110 (2013) 5864–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Winer KK, Zhang B, Shrader JA, Peterson D, Smith M, Albert PS, Cutler GB Jr, Synthetic human parathyroid hormone 1–34 replacement therapy: a randomized crossover trial comparing pump versus injections in the treatment of chronic hypoparathyroidism, J Clin Endocrinol Metab, 97 (2012) 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].McRobie HR, King LM, Fanutti C, Symmons MF, Coussons PJ, Agouti signalling protein is an inverse agonist to the wildtype and agonist to the melanic variant of the melanocortin-1 receptor in the grey squirrel (Sciurus carolinensis), FEBS Lett, 588 (2014) 2335–2343. [DOI] [PubMed] [Google Scholar]
- [103].Nijenhuis WA, Oosterom J, Adan RA, AgRP(83–132) acts as an inverse agonist on the human melanocortin-4 receptor, Mol Endocrinol, 15 (2001) 164–171. [DOI] [PubMed] [Google Scholar]
- [104].Mo XL, Tao YX, Activation of MAPK by inverse agonists in six naturally occurring constitutively active mutant human melanocortin-4 receptors, Biochim Biophys Acta, 1832 (2013) 1939–1948. [DOI] [PubMed] [Google Scholar]
- [105].Yang Z, Tao YX, Biased signaling initiated by agouti-related peptide through human melanocortin-3 and -4 receptors, Biochim Biophys Acta, 1862 (2016) 1485–1494. [DOI] [PubMed] [Google Scholar]
- [106].Tao YX, Constitutive activity in melanocortin-4 receptor: biased signaling of inverse agonists, Adv Pharmacol, 70 (2014) 135–154. [DOI] [PubMed] [Google Scholar]
- [107].Montero-Melendez T, Gobbetti T, Cooray SN, Jonassen TE, Perretti M, Biased agonism as a novel strategy to harness the proresolving properties of melanocortin receptors without eliciting melanogenic effects, J Immunol, 194 (2015) 3381–3388. [DOI] [PubMed] [Google Scholar]
- [108].Monod J, Wyman J, Changeux JP, On the nature of allosteric transitions: a plausible model, in: Selected Papers in Molecular Biology by Jacques Monod, Elsevier, 1978, pp. 593–623. [DOI] [PubMed]
- [109].Christopoulos A, Kenakin T, G protein-coupled receptor allosterism and complexing, Pharmacol Rev, 54 (2002) 323–374. [DOI] [PubMed] [Google Scholar]
- [110].Foster DJ, Conn PJ, Allosteric modulation of GPCRs: new insights and potential utility for treatment of schizophrenia and other CNS disorders, Neuron, 94 (2017) 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Conn PJ, Christopoulos A, Lindsley CW, Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders, Nat Rev Drug Discov, 8 (2009) 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A, Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor, Endocrinology, 153 (2012) 1232–1241. [DOI] [PubMed] [Google Scholar]
- [113].Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K, Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor, Mol Pharmacol, 88 (2015) 368–379. [DOI] [PubMed] [Google Scholar]
- [114].Montero-Melendez T, Forfar RA, Cook JM, Jerman JC, Taylor DL, Perretti M, Old drugs with new skills: fenoprofen as an allosteric enhancer at melanocortin receptor 3, Cell Mol Life Sci, 74 (2017) 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Yuan XC, Tao YX, Fenoprofen-An old drug rediscovered as a biased allosteric enhancer for melanocortin receptors, ACS Chem Neurosci, 10 (2019) 1066–1074. [DOI] [PubMed] [Google Scholar]
- [116].Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson DA, Nix MA, Kerns JA, Schmutz SM, Millhauser GL, Barsh GS, A β-defensin mutation causes black coat color in domestic dogs, Science, 318 (2007) 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Aono S, Dennis JC, He S, Wang W, Tao YX, Morrison EE, Exploring pleiotropic functions of canine β-defensin 103: Nasal cavity expression, antimicrobial activity, and melanocortin receptor activity, Anat Rec (Hoboken), (2020) 10.1002/ar.24300. [DOI] [PubMed]
- [118].Sebhat IK, Martin WJ, Ye Z, Barakat K, Mosley RT, Johnston DB, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre DE, van der Ploeg LH, Patchett AA, Nargund RP, Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium- 3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)- 2-[4-cyclohexyl-4-(1H-1,2,4-triazol- 1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist, J Med Chem, 45 (2002) 4589–4593. [DOI] [PubMed] [Google Scholar]
- [119].Yi TL, Yang LK, Ruan GL, Yang DQ, Tao YX, Melanocortin-4 receptor in swamp eel (Monopterus albus): Cloning, tissue distribution, and pharmacology, Gene, 678 (2018) 79–89. [DOI] [PubMed] [Google Scholar]
- [120].Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ, Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2, Proc Natl Acad Sci U S A, 100 (2003) 10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ, β-arrestin-dependent G protein-independent ERK1/2 activation by the β2 adrenergic receptor, J Biol Chem, 281 (2006) 1261–1273. [DOI] [PubMed] [Google Scholar]
- [122].Urs NM, Peterson SM, Caron MG, New concepts in dopamine D2 receptor biased signaling and implications for schizophrenia therapy, Biol Psychiatry, 81 (2017) 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Evron T, Peterson SM, Urs NM, Bai Y, Rochelle LK, Caron MG, Barak LS, G protein and β-arrestin signaling bias at the ghrelin receptor, J Biol Chem, 289 (2014) 33442–33455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Chen X, Bai B, Tian Y, Du H, Chen J, Identification of serine 348 on the apelin receptor as a novel regulatory phosphorylation site in apelin-13-induced G protein-independent biased signaling, J Biol Chem, 289 (2014) 31173–31187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ban T, Li X, Ma X, Yang H, Song Y, Sun Y, Shen M, Li N, Zhang MY, Ma Y, Zhong W, Zhang M, Hu LA, GPCR structure and function relationship: identification of a biased apelin receptor mutant, Biochem J, 475 (2018) 3813–3826. [DOI] [PubMed] [Google Scholar]
- [126].Ouedraogo M, Lecat S, Rochdi MD, Hachet-Haas M, Matthes H, Gicquiaux H, Verrier S, Gaire M, Glasser N, Mély Y, Distinct motifs of neuropeptide Y receptors differentially regulate trafficking and desensitization, Traffic, 9 (2008) 305–324. [DOI] [PubMed] [Google Scholar]
- [127].Gyombolai P, Tóth AD, Tímár D, Turu G, Hunyady L, Mutations in the ‘DRY’motif of the CB1 cannabinoid receptor result in biased receptor variants, J Mol Endocrinol, 54 (2015) 75–89. [DOI] [PubMed] [Google Scholar]
- [128].Reiter E, Ayoub MA, Pellissier LP, Landomiel F, Musnier A, Tréfier A, Gandia J, De Pascali F, Tahir S, Yvinec R, β-arrestin signalling and bias in hormone-responsive GPCRs, Mol Cell Endocrinol, 449 (2017) 28–41. [DOI] [PubMed] [Google Scholar]
- [129].Huang H, Tao YX, Pleiotropic functions of the transmembrane domain 6 of human melanocortin-4 receptor, J Mol Endocrinol, 49 (2012) 237–248. [DOI] [PubMed] [Google Scholar]
- [130].Mo XL, Yang R, Tao YX, Functions of transmembrane domain 3 of human melanocortin-4 receptor, J Mol Endocrinol, 49 (2012) 221–235. [DOI] [PubMed] [Google Scholar]
- [131].Huang H, Tao YX, Functions of the DRY motif and intracellular loop 2 of human melanocortin 3 receptor, J Mol Endocrinol, 53 (2014) 319–330. [DOI] [PubMed] [Google Scholar]
- [132].Yang Z, Huang ZL, Tao YX, Functions of DPLIY motif and helix 8 of human melanocortin-3 receptor, J Mol Endocrinol, 55 (2015) 107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Ballesteros JA, Weinstein H, Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors., Methods Neurosci, 25 (1995) 366–428. [Google Scholar]
- [134].Patten CS, Daniels D, Suzuki A, Fluharty SJ, Yee DK, Structural and signaling requirements of the human melanocortin 4 receptor for MAP kinase activation, Regul Pept, 142 (2007) 111–122. [DOI] [PubMed] [Google Scholar]
- [135].Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K, Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR, Science, 335 (2012) 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Lamichhane R, Liu JJ, White KL, Katritch V, Stevens RC, Wüthrich K, Millar DP, Biased signaling of the G-protein-coupled receptor β2AR is governed by conformational exchange kinetics, Structure, 28 (2020) 371–377 e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Wingler LM, Elgeti M, Hilger D, Latorraca NR, Lerch MT, Staus DP, Dror RO, Kobilka BK, Hubbell WL, Lefkowitz RJ, Angiotensin analogs with dvergent bias stabilize distinct receptor conformations, Cell, 176 (2019) 468–478 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Suomivuori CM, Latorraca NR, Wingler LM, Eismann S, King MC, Kleinhenz ALW, Skiba MA, Staus DP, Kruse AC, Lefkowitz RJ, Dror RO, Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor, Science, 367 (2020) 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Shukla AK, Singh G, Ghosh E, Emerging structural insights into biased GPCR signaling, Trends Biochem Sci, 39 (2014) 594–602. [DOI] [PubMed] [Google Scholar]
- [140].Al-Zoubi R, Morales P, Reggio PH, Structural insights into CB1 receptor biased signaling, Int J Mol Sci, 20 (2019) 1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Cone RD, Anatomy and regulation of the central melanocortin system, Nat Neurosci, 8 (2005) 571–578. [DOI] [PubMed] [Google Scholar]
- [142].Tao YX, Molecular mechanisms of the neural melanocortin receptor dysfunction in severe early onset obesity, Mol Cell Endocrinol, 239 (2005) 1–14. [DOI] [PubMed] [Google Scholar]
- [143].Tao YX, The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology, Endocr Rev, 31 (2010) 506–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F, Targeted disruption of the melanocortin-4 receptor results in obesity in mice, Cell, 88 (1997) 131–141. [DOI] [PubMed] [Google Scholar]
- [145].Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH, Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass, Nat Genet, 26 (2000) 97–102. [DOI] [PubMed] [Google Scholar]
- [146].Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD, A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse, Endocrinology, 141 (2000) 3518–3521. [DOI] [PubMed] [Google Scholar]
- [147].Tao YX, Mutations in melanocortin-4 receptor and human obesity, Prog Mol Biol Transl Sci, 88 (2009) 173–204. [DOI] [PubMed] [Google Scholar]
- [148].Tao YX, Mutations in the melanocortin-3 receptor (MC3R) gene: Impact on human obesity or adiposity, Curr Opin Investig Drugs, 11 (2010) 1092–1096. [PubMed] [Google Scholar]
- [149].Hinney A, Volckmar AL, Knoll N, Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis, Prog Mol Biol Transl Sci, 114 (2013) 147–191. [DOI] [PubMed] [Google Scholar]
- [150].Yang Z, Tao YX, Mutations in melanocortin-3 receptor gene and human obesity, Prog Mol Biol Transl Sci, 140 (2016) 97–129. [DOI] [PubMed] [Google Scholar]
- [151].Breit A, Wolff K, Kalwa H, Jarry H, Buch T, Gudermann T, The natural inverse agonist agouti-related protein induces arrestin-mediated endocytosis of melanocortin-3 and -4 receptors, J Biol Chem, 281 (2006) 37447–37456. [DOI] [PubMed] [Google Scholar]
- [152].Rodrigues AR, Almeida H, Gouveia AM, Intracellular signaling mechanisms of the melanocortin receptors: current state of the art, Cell Mol Life Sci, 72 (2015) 1331–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Yang F, Huang H, Tao YX, Biased signaling in naturally occurring mutations in human melanocortin-3 receptor gene, Int J Biol Sci, 11 (2015) 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ghamari-Langroudi M, Digby GJ, Sebag JA, Millhauser GL, Palomino R, Matthews R, Gillyard T, Panaro BL, Tough IR, Cox HM, Denton JS, Cone RD, G-protein-independent coupling of MC4R to Kir7.1 in hypothalamic neurons, Nature, 520 (2015) 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Lotta LA, Mokrosiński J, de Oliveira EM, Li C, Sharp SJ, Luan J, Brouwers B, Ayinampudi V, Bowker N, Kerrison N, Human gain-of-function MC4R variants show signaling bias and protect against obesity, Cell, 177 (2019) 597–607. e599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gillyard T, Fowler K, Williams SY, Cone RD, Obesity-associated mutant melanocortin-4 receptors with normal Gαs coupling frequently exhibit other discoverable pharmacological and biochemical defects, J Neuroendocrinol, 31 (2019) e12795. [DOI] [PubMed] [Google Scholar]
- [157].Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A, Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization, Diabetes, 52 (2003) 2984–2988. [DOI] [PubMed] [Google Scholar]
- [158].Büch TR, Heling D, Damm E, Gudermann T, Breit A, Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1–7 cells defines agouti-related protein as a biased agonist, J Biol Chem, 284 (2009) 26411–26420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Govaerts C, Lefort A, Costagliola S, Wodak SJ, Ballesteros JA, Van Sande J, Pardo L, Vassart G, A conserved Asn in transmembrane helix 7 is an on/off switch in the activation of the thyrotropin receptor, J Biol Chem, 276 (2001) 22991–22999. [DOI] [PubMed] [Google Scholar]
- [160].Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP, Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation, Proc Natl Acad Sci U S A, 100 (2003) 2290–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Kohlhoff KJ, Shukla D, Lawrenz M, Bowman GR, Konerding DE, Belov D, Altman RB, Pande VS, Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways, Nature chemistry, 6 (2014) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].He S, Tao YX, Defect in MAPK signaling as a cause for monogenic obesity caused by inactivating mutations in the melanocortin-4 receptor gene, Int J Biol Sci, 10 (2014) 1128–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Paisdzior S, Dimitriou IM, Schöpe PC, Annibale P, Scheerer P, Krude H, Lohse MJ, Biebermann H, Kühnen P, Differential signaling profiles of MC4R mutations with three different ligands, Int J Mol Sci, 21 (2020) 1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Sharma S, Thibodeau S, Lytton J, Signal pathway analysis of selected obesity-associated melanocortin-4 receptor class V mutants, Biochim Biophys Acta Mol Basis Dis, 1866 (2020) 165835. [DOI] [PubMed] [Google Scholar]
- [165].Yang LK, Tao YX, Biased signaling at neural melanocortin receptors in regulation of energy homeostasis, Biochim Biophys Acta Mol Basis Dis, 1863 (2017) 2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Chen M, Wang J, Dickerson KE, Kelleher J, Xie T, Gupta D, Lai EW, Pacak K, Gavrilova O, Weinstein LS, Central nervous system imprinting of the G protein Gsα and its role in metabolic regulation, Cell Metab, 9 (2009) 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Chen M, Berger A, Kablan A, Zhang J, Gavrilova O, Weinstein LS, Gsα deficiency in the paraventricular nucleus of the hypothalamus partially contributes to obesity associated with Gsα mutations, Endocrinology, 153 (2012) 4256–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB, AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus, Nature, 428 (2004) 569–574. [DOI] [PubMed] [Google Scholar]
- [169].Sutton GM, Duos B, Patterson LM, Berthoud HR, Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus, Endocrinology, 146 (2005) 3739–3747. [DOI] [PubMed] [Google Scholar]
- [170].Damm E, Buech TR, Gudermann T, Breit A, Melanocortin-induced PKA activation inhibits AMPK activity via ERK-1/2 and LKB-1 in hypothalamic GT1–7 cells, Mol Endocrinol, 26 (2012) 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Li YQ, Shrestha Y, Pandey M, Chen M, Kablan A, Gavrilova O, Offermanns S, Weinstein LS, Gq/11α and Gsα mediate distinct physiological responses to central melanocortins, J Clin Invest, 126 (2016) 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Begriche K, Marston OJ, Rossi J, Burke LK, McDonald P, Heisler LK, Butler AA, Melanocortin-3 receptors are involved in adaptation to restricted feeding, Genes Brain Behav, 11 (2012) 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].da Silva AA, Kuo JJ, Hall JE, Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin, Hypertension, 43 (2004) 1312–1317. [DOI] [PubMed] [Google Scholar]
- [174].Ni XP, Butler AA, Cone RD, Humphreys MH, Central receptors mediating the cardiovascular actions of melanocyte stimulating hormones, J Hypertens, 24 (2006) 2239–2246. [DOI] [PubMed] [Google Scholar]
- [175].Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, Balthasar N, Williams KW, Elmquist JK, Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons, Cell, 152 (2013) 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O’Rahilly S, Farooqi IS, Modulation of blood pressure by central melanocortinergic pathways, N Engl J Med, 360 (2009) 44–52. [DOI] [PubMed] [Google Scholar]
- [177].Breit A, Büch TR, Boekhoff I, Solinski HJ, Damm E, Gudermann T, Alternative G protein coupling and biased agonism: new insights into melanocortin-4 receptor signalling, Mol Cell Endocrinol, 331 (2011) 232–240. [DOI] [PubMed] [Google Scholar]
- [178].Brown EM, MacLeod RJ, Extracellular calcium sensing and extracellular calcium signaling, Physiol Rev, 81 (2001) 239–297. [DOI] [PubMed] [Google Scholar]
- [179].Brown EM, Role of the calcium-sensing receptor in extracellular calcium homeostasis, Best Pract Res Clin Endocrinol Metab, 27 (2013) 333–343. [DOI] [PubMed] [Google Scholar]
- [180].Fox J, Lowe SH, Conklin RL, Petty BA, Nemeth EF, Calcimimetic compound NPS R-568 stimulates calcitonin secretion but selectively targets parathyroid gland Ca2+ receptor in rats, J Pharmacol Exp Ther, 290 (1999) 480–486. [PubMed] [Google Scholar]
- [181].Quitterer U, Hoffmann M, Freichel M, Lohse MJ, Paradoxical block of parathormone secretion is mediated by increased activity of Gα subunits, J Biol Chem, 276 (2001) 6763–6769. [DOI] [PubMed] [Google Scholar]
- [182].Wettschureck N, Lee E, Libutti SK, Offermanns S, Robey PG, Spiegel AM, Parathyroid-specific double knockout of Gq and G11 α-subunits leads to a phenotype resembling germline knockout of the extracellular Ca2+-sensing receptor, Mol Endocrinol, 21 (2007) 274–280. [DOI] [PubMed] [Google Scholar]
- [183].Kifor O, MacLeod RJ, Diaz R, Bai M, Yamaguchi T, Yao T, Kifor I, Brown EM, Regulation of MAP kinase by calcium-sensing receptor in bovine parathyroid and CaR-transfected HEK293 cells, Am J Physiol-Renal Physiol, 280 (2001) F291–F302. [DOI] [PubMed] [Google Scholar]
- [184].Thomsen ARB, Hvidtfeldt M, Bräuner-Osborne H, Biased agonism of the calcium-sensing receptor, Cell Calcium, 51 (2012) 107–116. [DOI] [PubMed] [Google Scholar]
- [185].Almadén Y, Canalejo A, Ballesteros E, Añón G, Cañadillas S, Rodríguez M, Regulation of arachidonic acid production by intracellular calcium in parathyroid cells: effect of extracellular phosphate, J Am Soc Nephrol, 13 (2002) 693–698. [DOI] [PubMed] [Google Scholar]
- [186].Corbetta S, Lania A, Filopanti M, Vicentini L, Ballare E, Spada A, Mitogen-activated protein kinase cascade in human normal and tumoral parathyroid cells, J Clin Endocrinol Metab, 87 (2002) 2201–2205. [DOI] [PubMed] [Google Scholar]
- [187].Hannan FM, Thakker RV, Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism, Best Pract Res Clin Endocrinol Metab, 27 (2013) 359–371. [DOI] [PubMed] [Google Scholar]
- [188].Leach K, Wen A, Davey AE, Sexton PM, Conigrave AD, Christopoulos A, Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor, Endocrinology, 153 (2012) 4304–4316. [DOI] [PubMed] [Google Scholar]
- [189].Gorvin CM, Babinsky VN, Malinauskas T, Nissen PH, Schou AJ, Hanyaloglu AC, Siebold C, Jones EY, Hannan FM, Thakker RV, A calcium-sensing receptor mutation causing hypocalcemia disrupts a transmembrane salt bridge to activate β-arrestin–biased signaling, Sci Signal, 11 (2018) eaan3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Gorvin CM, Frost M, Malinauskas T, Cranston T, Boon H, Siebold C, Jones EY, Hannan FM, Thakker RV, Calcium-sensing receptor residues with loss- and gain-of-function mutations are located in regions of conformational change and cause signalling bias, Hum Mol Genet, 27 (2018) 3720–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V, Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator, Science, 344 (2014) 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang CR, Gu LL, Shan JM, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL Jr., Kobilka BK, Skiniotis G, Lefkowitz RJ, Visualization of arrestin recruitment by a G-protein-coupled receptor, Nature, 512 (2014) 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Luttrell LM, Maudsley S, Bohn LM, Fulfilling the promise of “biased” G protein-coupled receptor agonism, Mol Pharmacol, 88 (2015) 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Gundry J, Glenn R, Alagesan P, Rajagopal S, A practical guide to approaching biased agonism at G protein coupled receptors, Front Neurosci, 11 (2017) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Geng Y, Mosyak L, Kurinov I, Zuo H, Sturchler E, Cheng TC, Subramanyam P, Brown AP, Brennan SC, Mun HC, Structural mechanism of ligand activation in human calcium-sensing receptor, eLife, 5 (2016) e13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Holstein DM, Berg KA, Leeb-Lundberg LMF, Olson MS, Saunders C, Calcium-sensing receptor-mediated ERK1/2 activation requires Gαi2 coupling and dynamin-independent receptor internalization, J Biol Chem, 279 (2004) 10060–10069. [DOI] [PubMed] [Google Scholar]
- [197].Nesbit MA, Hannan FM, Howles SA, Reed AAC, Cranston T, Thakker CE, Gregory L, Rimmer AJ, Rust N, Graham U, Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3, Nat Genet, 45 (2013) 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Gorvin CM, Rogers A, Hastoy B, Tarasov AI, Frost M, Sposini S, Inoue A, Whyte MP, Rorsman P, Hanyaloglu AC, AP2σ mutations impair calcium-sensing receptor trafficking and signaling, and show an endosomal pathway to spatially direct G-protein selectivity, Cell reports, 22 (2018) 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Wehbi VL, Stevenson HP, Feinstein TN, Calero G, Romero G, Vilardaga JP, Noncanonical GPCR signaling arising from a PTH receptor–arrestin–Gβγ complex, Proc Natl Acad Sci U S A, 110 (2013) 1530–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Seminara SB, Hayes FJ, Crowley WF Jr, Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations, Endocr Rev, 19 (1998) 521–539. [DOI] [PubMed] [Google Scholar]
- [201].Zhao Y, Wu J, Wang X, Jia H, Chen DN, Li JD, Prokineticins and their G protein-coupled receptors in health and disease, Prog Mol Biol Transl Sci, 161 (2019) 149–179. [DOI] [PubMed] [Google Scholar]
- [202].Teixeira L, Guimiot F, Dodé C, Fallet-Bianco C, Millar RP, Delezoide AL, Hardelin JP, Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions, J Clin Invest, 120 (2010) 3668–3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Sbai O, Monnier C, Dode C, Pin JP, Hardelin JP, Rondard P, Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations, FASEB J, 28 (2014) 3734–3744. [DOI] [PubMed] [Google Scholar]
- [204].Yin W, Liu H, Peng Z, Chen D, Li J, Li JD, Mechanisms that underlie the internalization and extracellular signal regulated kinase 1/2 activation by PKR2 receptor, Cel Signal, 26 (2014) 1118–1124. [DOI] [PubMed] [Google Scholar]
- [205].Libri DV, Kleinau G, Vezzoli V, Busnelli M, Guizzardi F, Sinisi AA, Pincelli AI, Mancini A, Russo G, Beck-Peccoz P, Germline prokineticin receptor 2 (PROKR2) variants associated with central hypogonadism cause differental modulation of distinct intracellular pathways, J Clin Endocrinol Metab, 99 (2014) E458–E463. [DOI] [PubMed] [Google Scholar]
- [206].Zhao Y, Wu J, Jia H, Wang X, Zheng R, Jiang F, Chen DN, Chen Z, Li JD, PROKR2 mutations in idiopathic hypogonadotropic hypogonadism: selective disruption of the binding to a Gα-protein leads to biased signaling, FASEB J, 33 (2019) 4538–4546. [DOI] [PubMed] [Google Scholar]
- [207].Abreu AP, Noel SD, Xu S, Carroll RS, Latronico AC, Kaiser UB, Evidence of the importance of the first intracellular loop of prokineticin receptor 2 in receptor function, Mol Endocrinol, 26 (2012) 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy, Proc Natl Acad Sci U S A, 109 (2012) 6733–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Magnan R, Escrieut C, Gigoux V, De K, Clerc P, Niu F, Azema J, Masri B, Cordomi A, Baltas M, Distinct CCK-2 receptor conformations associated with β-arrestin-2 recruitment or phospholipase-C activation revealed by a biased antagonist, J Am Chem Soc, 135 (2013) 2560–2573. [DOI] [PubMed] [Google Scholar]
- [210].Hill SJ, G-protein-coupled receptors: past, present and future, Br J Pharmacol, 147 (2006) S27–S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Zhou XT, Chen DN, Xie ZQ, Peng Z, Xia KD, Liu HD, Liu W, Su B, Li JD, Functional analysis of the distal region of the third intracellular loop of PROKR2, Biochem Biophys Res Commun, 439 (2013) 12–17. [DOI] [PubMed] [Google Scholar]
- [212].Bokoch GM, Katada T, Northup JK, Ui M, Gilman AG, Purification and properties of the inhibitory guanine nucleotide-binding regulatory component of adenylate cyclase, J Biol Chem, 259 (1984) 3560–3567. [PubMed] [Google Scholar]
- [213].Harrison T, Samuel BU, Akompong T, Hamm H, Mohandas N, Lomasney JW, Haldar K, Erythrocyte G protein-coupled receptor signaling in malarial infection, Science, 301 (2003) 1734–1736. [DOI] [PubMed] [Google Scholar]
- [214].de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E, Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54, Proc Natl Acad Sci U S A, 100 (2003) 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS Jr., Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O’Rahilly S, Carlton MB, Crowley WF Jr., Aparicio SA, Colledge WH, The GPR54 gene as a regulator of puberty, N Engl J Med, 349 (2003) 1614–1627. [DOI] [PubMed] [Google Scholar]
- [216].Castellano JM, Navarro VM, Fernandez-Fernandez R, Castano JP, Malagon MM, Aguilar E, Dieguez C, Magni P, Pinilla L, Tena-Sempere M, Ontogeny and mechanisms of action for the stimulatory effect of kisspeptin on gonadotropin-releasing hormone system of the rat, Mol Cell Endocrinol, 257 (2006) 75–83. [DOI] [PubMed] [Google Scholar]
- [217].Castaño JP, Martínez-Fuentes AJ, Gutiérrez-Pascual E, Vaudry H, Tena-Sempere M, Malagón MM, Intracellular signaling pathways activated by kisspeptins through GPR54: do multiple signals underlie function diversity?, Peptides, 30 (2009) 10–15. [DOI] [PubMed] [Google Scholar]
- [218].Pampillo M, Camuso N, Taylor JE, Szereszewski JM, Ahow MR, Zajac M, Millar RP, Bhattacharya M, Babwah AV, Regulation of GPR54 signaling by GRK2 and β-arrestin, Mol Endocrinol, 23 (2009) 2060–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Ahow M, Min L, Pampillo M, Nash C, Wen J, Soltis K, Carroll RS, Glidewell-Kenney CA, Mellon PL, Bhattacharya M, Kiss1r signals independently of Gαq/11 and triggers LH secretion via the β-arrestin pathway in the male mouse, Endocrinology, 155 (2014) 4433–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Semple RK, Achermann JC, Ellery J, Farooqi IS, Karet FE, Stanhope RG, O’rahilly S, Aparicio SA, Two novel missense mutations in G protein-coupled receptor 54 in a patient with hypogonadotropic hypogonadism, J Clin Endocrinol Metab, 90 (2005) 1849–1855. [DOI] [PubMed] [Google Scholar]
- [221].Wacker JL, Feller DB, Tang XB, Defino MC, Namkung Y, Lyssand JS, Mhyre AJ, Tan X, Jensen JB, Hague C, Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function, J Biol Chem, 283 (2008) 31068–31078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Tenenbaum-Rakover Y, Commenges-Ducos M, Iovane A, Aumas C, Admoni O, de Roux N, Neuroendocrine phenotype analysis in five patients with isolated hypogonadotropic hypogonadism due to a L102P inactivating mutation of GPR54, J Clin Endocrinol Metab, 92 (2007) 1137–1144. [DOI] [PubMed] [Google Scholar]
- [223].Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, Seminara SB, Mendonca BB, Kaiser UB, Latronico AC, A GPR54-activating mutation in a patient with central precocious puberty, N Engl J Med, 358 (2008) 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Kumar TR, Wang Y, Lu N, Matzuk MM, Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility, Nat Genet, 15 (1997) 201–204. [DOI] [PubMed] [Google Scholar]
- [225].Tapanainen JS, Aittomaki K, Min J, Vaskivuo T, Huhtaniemi IT, Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility, Nat Genet, 15 (1997) 205–206. [DOI] [PubMed] [Google Scholar]
- [226].Tao YX, Segaloff DL, Follicle stimulating hormone receptor mutations and reproductive disorders, Prog Mol Biol Transl Sci, 89 (2009) 115–131. [DOI] [PubMed] [Google Scholar]
- [227].Aittomaki K, Lucena JLD, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, Kaskikari R, Sankila E-M, Lehfaslaiho H, Engel AR, Nieshlag E, Huhtaniemi I, de la Chapelle A, Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure, Cell, 82 (1995) 959–968. [DOI] [PubMed] [Google Scholar]
- [228].Rannikko A, Pakarinen P, Manna PR, Beau I, Misrahi M, Aittomaki K, Huhtaniemi I, Functional characterization of the human FSH receptor with an inactivating Ala189Val mutation, Mol Human Reprod, 8 (2002) 311–317. [DOI] [PubMed] [Google Scholar]
- [229].Tranchant T, Durand G, Gauthier C, Crepieux P, Ulloa-Aguirre A, Royere D, Reiter E, Preferential β-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189V mutation, Mol Cell Endocrinol, 331 (2011) 109–118. [DOI] [PubMed] [Google Scholar]
- [230].Casas-Gonzalez P, Scaglia HE, Perez-Solis MA, Durand G, Scaglia J, Zarinan T, Dias JA, Reiter E, Ulloa-Aguirre A, Normal testicular function without detectable follicle-stimulating hormone. A novel mutation in the follicle-stimulating hormone receptor gene leading to apparent constitutive activity and impaired agonist-induced desensitization and internalization, Mol Cell Endocrinol, 364 (2012) 71–82. [DOI] [PubMed] [Google Scholar]
- [231].Kopp P, The TSH receptor and its role in thyroid disease, Cell Mol Life Sci, 58 (2001) 1301–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M, Marians RC, Davies TF, Zannini MS, De Felice M, Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland, Proc Natl Acad Sci U S A, 99 (2002) 15462–15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF, Defining thyrotropin-dependent and-independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice, Proc Natl Acad Sci U S A, 99 (2002) 15776–15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Grasberger H, Van Sande J, Hag-Dahood Mahameed A, Tenenbaum-Rakover Y, Refetoff S, A familial thyrotropin (TSH) receptor mutation provides in vivo evidence that the inositol phosphates/Ca2+ cascade mediates TSH action on thyroid hormone synthesis, J Clin Endocrinol Metab, 92 (2007) 2816–2820. [DOI] [PubMed] [Google Scholar]
- [235].Feldman BJ, Rosenthal SM, Vargas GA, Fenwick RG, Huang EA, Matsuda-Abedini M, Lustig RH, Mathias RS, Portale AA, Miller WL, Nephrogenic syndrome of inappropriate antidiuresis, N Engl J Med, 352 (2005) 1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Fujiwara TM, Bichet DG, Molecular biology of hereditary diabetes insipidus, J Am Soc Nephrol, 16 (2005) 2836–2846. [DOI] [PubMed] [Google Scholar]
- [237].Spanakis E, Milord E, Gragnoli C, AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance, J Cell Physiol, 217 (2008) 605–617. [DOI] [PubMed] [Google Scholar]
- [238].Charest PG, Bouvier M, Palmitoylation of the V2 vasopressin receptor carboxyl tail enhances β-arrestin recruitment leading to efficient receptor endocytosis and ERK1/2 activation, J Biol Chem, 278 (2003) 41541–41551. [DOI] [PubMed] [Google Scholar]
- [239].Janovick JA, Spicer TP, Bannister TD, Scampavia L, Conn PM, Pharmacoperone rescue of vasopressin 2 receptor mutants reveals unexpected constitutive activity and coupling bias, PloS One, 12 (2017) e0181830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Bichet DG, V2R mutations and nephrogenic diabetes insipidus, Prog Mol Biol Transl Sci, 89 (2009) 15–29. [DOI] [PubMed] [Google Scholar]
- [241].Ala Y, Morin D, Mouillac B, Sabatier N, Vargas R, Cotte N, Dechaux M, Antignac C, Arthus MF, Lonergan M, Turner MS, Balestre MN, Alonso G, Hibert M, Barberis C, Hendy GN, Bichet DG, Jard S, Functional studies of twelve mutant V2 vasopressin receptors related to nephrogenic diabetes insipidus: molecular basis of a mild clinical phenotype, J Am Soc Nephrol, 9 (1998) 1861–1872. [DOI] [PubMed] [Google Scholar]
- [242].Jean-Alphonse F, Perkovska S, Frantz MC, Durroux T, Mejean C, Morin D, Loison S, Bonnet D, Hibert M, Mouillac B, Mendre C, Biased agonist pharmacochaperones of the AVP V2 receptor may treat congenital nephrogenic diabetes insipidus, J Am Soc Nephrol, 20 (2009) 2190–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Barak LS, Oakley RH, Laporte SA, Caron MG, Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus, Proc Natl Acad Sci U S A, 98 (2001) 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Bernier V, Lagace M, Lonergan M, Arthus MF, Bichet DG, Bouvier M, Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059, Mol Endocrinol, 18 (2004) 2074–2084. [DOI] [PubMed] [Google Scholar]
- [245].Rochdi MD, Vargas GA, Carpentier E, Oligny-Longpre G, Chen S, Kovoor A, Gitelman SE, Rosenthal SM, von Zastrow M, Bouvier M, Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: implications for treatments, Mol Pharmacol, 77 (2010) 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Armstrong SP, Seeber RM, Ayoub MA, Feldman BJ, Pfleger KD, Characterization of three vasopressin receptor 2 variants: an apparent polymorphism (V266A) and two loss-of-function mutations (R181C and M311V), PloS One, 8 (2013) e65885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Carpentier E, Greenbaum LA, Rochdi D, Abrol R, Goddard WA, Bichet DG, Bouvier M, Identification and characterization of an activating F229V substitution in the V2 vasopressin receptor in an infant with NSIAD, J Am Soc Nephrol, 23 (2012) 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Erdélyi LS, Mann WA, Morris-Rosendahl DJ, Groß U, Nagel M, Várnai P, Balla A, Hunyady L, Mutation in the V2 vasopressin receptor gene, AVPR2, causes nephrogenic syndrome of inappropriate diuresis, Kidney Int, 88 (2015) 1070–1078. [DOI] [PubMed] [Google Scholar]
- [249].Tiulpakov A, White CW, Abhayawardana RS, See HB, Chan AS, Seeber RM, Heng JI, Dedov I, Pavlos NJ, Pfleger KD, Mutations of vasopressin receptor 2 including novel L312S have differential effects on trafficking, Mol Endocrinol, 30 (2016) 889–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Brzezinski A, Melatonin in humans, N Engl J Med, 336 (1997) 186–195. [DOI] [PubMed] [Google Scholar]
- [251].Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, Cecon E, Zlotos DP, Update on melatonin receptors: IUPHAR Review 20, Br J Pharmacol, 173 (2016) 2702–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Jockers R, Maurice P, Boutin JA, Delagrange P, Melatonin receptors, heterodimerization, signal transduction and binding sites: what’s new?, Br J Pharmacol, 154 (2008) 1182–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Vanecek J, Cellular mechanisms of melatonin action, Physiol Rev, 78 (1998) 687–721. [DOI] [PubMed] [Google Scholar]
- [254].Richdale AL, Schreck KA, Sleep problems in autism spectrum disorders: prevalence, nature, & possible biopsychosocial aetiologies, Sleep Med Rev, 13 (2009) 403–411. [DOI] [PubMed] [Google Scholar]
- [255].Chaste P, Clement N, Mercati O, Guillaume JL, Delorme R, Botros HG, Pagan C, Périvier S, Scheid I, Nygren G, Identification of pathway-biased and deleterious melatonin receptor mutants in autism spectrum disorders and in the general population, PloS One, 5 (2010) e11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Bonnefond A, Clément N, Fawcett K, Yengo L, Vaillant E, Guillaume JL, Dechaume A, Payne F, Roussel R, Czernichow S, Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes, Nat Genet, 44 (2012) 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Sturm RA, Skin colour and skin cancer–MC1R, the genetic link, Melanoma Res, 12 (2002) 405–416. [DOI] [PubMed] [Google Scholar]
- [258].Rees JL, The genetics of sun sensitivity in humans, Am J Human Genet, 75 (2004) 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Herraiz C, Garcia-Borron JC, Jimenez-Cervantes C, Olivares C, MC1R signaling. Intracellular partners and pathophysiological implications, Biochim Biophys Acta, 1863 (2017) 2448–2461. [DOI] [PubMed] [Google Scholar]
- [260].Pérez Oliva AB, Fernéndez LP, DeTorre C, Herráiz C, Martínez-Escribano JA, Benítez J, Lozano Teruel JA, García-Borrón JC, Jiménez-Cervantes C, Ribas G, Identification and functional analysis of novel variants of the human melanocortin 1 receptor found in melanoma patients, Hum Mutat, 30 (2009) 811–822. [DOI] [PubMed] [Google Scholar]
- [261].Sturm RA, Duffy DL, Box NF, Chen W, Smit DJ, Brown DL, Stow JL, Leonard JH, Martin NG, The role of melanocortin-1 receptor polymorphism in skin cancer risk phenotypes, Pigment Cell Melanoma Res, 16 (2003) 266–272. [DOI] [PubMed] [Google Scholar]
- [262].Beaumont KA, Liu YY, Sturm RA, The melanocortin-1 receptor gene polymorphism and association with human skin cancer, Prog Mol Biol Transl Sci, 88 (2009) 85–153. [DOI] [PubMed] [Google Scholar]
- [263].Herraiz C, Jimenez-Cervantes C, Zanna P, Garcia-Borron JC, Melanocortin 1 receptor mutations impact differentially on signalling to the cAMP and the ERK mitogen-activated protein kinase pathways, FEBS Lett, 583 (2009) 3269–3274. [DOI] [PubMed] [Google Scholar]
- [264].Busca R, Ballotti R, Cyclic AMP a key messenger in the regulation of skin pigmentation, Pigment Cell Melanoma Res, 13 (2000) 60–69. [DOI] [PubMed] [Google Scholar]
- [265].Slominski A, Tobin DJ, Shibahara S, Wortsman J, Melanin pigmentation in mammalian skin and its hormonal regulation, Physiol Rev, 84 (2004) 1155–1228. [DOI] [PubMed] [Google Scholar]
- [266].Benned-Jensen T, Mokrosinski J, Rosenkilde MM, The E92K melanocortin 1 receptor mutant induces cAMP production and arrestin recruitment but not ERK activity indicating biased constitutive signaling, PLoS One, 6 (2011) e24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Clark AJ, Weber A, Adrenocorticotropin insensitivity syndromes, Endocr Rev, 19 (1998) 828–843. [DOI] [PubMed] [Google Scholar]
- [268].Cooray SN, Chan L, Metherell L, Storr H, Clark AJL, Adrenocorticotropin resistance syndromes, Endocr Dev, 13 (2008) 99–116. [DOI] [PubMed] [Google Scholar]
- [269].Swords FM, Baig A, Malchoff DM, Malchoff CD, Thorner MO, King PJ, Hunyady L, Clark AJ, Impaired desensitization of a mutant adrenocorticotropin receptor associated with apparent constitutive activity, Mol Endocrinol, 16 (2002) 2746–2753. [DOI] [PubMed] [Google Scholar]
- [270].Baig AH, Swords FM, Noon LA, King PJ, Hunyady L, Clark AJ, Desensitization of the Y1 cell adrenocorticotropin receptor: evidence for a restricted heterologous mechanism implying a role for receptor- effector complexes, J Biol Chem, 276 (2001) 44792–44797. [DOI] [PubMed] [Google Scholar]
- [271].Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, Lespinasse J, Lienhardt-Roussie A, Mathieu M, Moerman A, Morgan G, Murat A, Toublanc JE, Wolczynski S, Delpech M, Petit C, Young J, Hardelin JP, Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2, PLoS Genet, 2 (2006) e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Themmen APN, Huhtaniemi IT, Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function, Endocr Rev, 21 (2000) 551–583. [DOI] [PubMed] [Google Scholar]
- [273].Biebermann H, Schoneberg T, Krude H, Schultz G, Gudermann T, Gruters A, Mutations of the human thyrotropin receptor gene causing thyroid hypoplasia and persistent congenital hypothyroidism, J Clin Endocrinol Metab, 82 (1997) 3471–3480. [DOI] [PubMed] [Google Scholar]
- [274].Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG, Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism, Cell, 75 (1993) 1297–1303. [DOI] [PubMed] [Google Scholar]
- [275].Pollak MR, Chou YH, Marx SJ, Steinmann B, Cole DE, Brandi ML, Papapoulos SE, Menko FH, Hendy GN, Brown EM, Seidman CE, Seidman JG, Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype, J Clin Invest, 93 (1994) 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Valverde P, Healy E, Jackson I, Rees JL, Thody AJ, Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans, Nat Genet, 11 (1995) 328–330. [DOI] [PubMed] [Google Scholar]
- [277].Clark AJ, McLoughlin L, Grossman A, Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor, Lancet, 341 (1993) 461–462. [DOI] [PubMed] [Google Scholar]
- [278].Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S, Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene, N Engl J Med, 348 (2003) 1085–1095. [DOI] [PubMed] [Google Scholar]
- [279].Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, Le Merrer M, Silve C, Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia, J Clin Invest, 102 (1998) 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Wysolmerski JJ, Cormier S, Philbrick WM, Dann P, Zhang JP, Roume J, Delezoide AL, Silve C, Absence of functional type 1 parathyroid hormone (PTH)/PTH-related protein receptors in humans is associated with abnormal breast development and tooth impaction, J Clin Endocrinol Metab, 86 (2001) 1788–1794. [DOI] [PubMed] [Google Scholar]
- [281].Smits G, Olatunbosun O, Delbaere A, Pierson R, Vassart G, Costagliola S, Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor, N Engl J Med, 349 (2003) 760–766. [DOI] [PubMed] [Google Scholar]
- [282].Vasseur C, Rodien P, Beau I, Desroches A, Gerard C, de Poncheville L, Chaplot S, Savagner F, Croue A, Mathieu E, Lahlou N, Descamps P, Misrahi M, A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome, N Engl J Med, 349 (2003) 753–759. [DOI] [PubMed] [Google Scholar]
- [283].Rodien P, Bremont C, Sanson ML, Parma J, Van Sande J, Costagliola S, Luton JP, Vassart G, Duprez L, Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin, N Engl J Med, 339 (1998) 1823–1826. [DOI] [PubMed] [Google Scholar]
- [284].Hendy GN, D’Souza-Li L, Yang B, Canaff L, Cole DE, Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia, Hum Mutat, 16 (2000) 281–296. [DOI] [PubMed] [Google Scholar]
- [285].Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, Chikatsu N, Fujita T, Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome, Lancet, 360 (2002) 692–694. [DOI] [PubMed] [Google Scholar]
- [286].Swords FM, Noon LA, King PJ, Clark AJ, Constitutive activation of the human ACTH receptor resulting from a synergistic interaction between two naturally occurring missense mutations in the MC2R gene, Mol Cell Endocrinol, 213 (2004) 149–154. [DOI] [PubMed] [Google Scholar]
- [287].Schipani E, Langman CB, Parfitt AM, Jensen GS, Kikuchi S, Kooh SW, Cole WG, Jüppner H, Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen’s metaphyseal chondrodysplasia, N Engl J Med, 335 (1996) 708–714. [DOI] [PubMed] [Google Scholar]
- [288].Sieving PA, Fowler ML, Bush RA, Machida S, Calvert PD, Green DG, Makino CL, McHenry CL, Constitutive “light” adaptation in rods from G90D rhodopsin: a mechanism for human congenital nightblindness without rod cell loss, J Neurosci, 21 (2001) 5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Zhao YG, Wu JY, Jia H, Wang XY, Zheng RZ, Jiang F, Chen DN, Chen ZH, Li JD, PROKR2 mutations in idiopathic hypogonadotropic hypogonadism: selective disruption of the binding to a Gα-protein leads to biased signaling, FASEB J, 33 (2019) 4538–4546. [DOI] [PubMed] [Google Scholar]
- [290].Julian B, Gao K, Harwood BN, Beinborn M, Kopin AS, Mutation-induced functional alterations of CCR6, J Pharmacol Exp Ther, 360 (2017) 106–116. [DOI] [PubMed] [Google Scholar]
- [291].Huang Q, Zhang LF, Cheng Y, Zhao YC, Si L, Gao Y, Wei W, Trp64Arg (rs4994) polymorphism of β3-adrenergic receptor gene is associated with hyperuricemia in a Chinese male population, Clinical chemistry and laboratory medicine, 51 (2013) 1755–1760. [DOI] [PubMed] [Google Scholar]
- [292].Kishore A, Hall RA, Disease-associated extracellular loop mutations in the adhesion G protein-coupled receptor G1 (ADGRG1; GPR56) differentially regulate downstream signaling, J Biol Chem, 292 (2017) 9711–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]