

Abstract
Humoral immunity in mammals relies on the function of two developmentally and functionally distinct B cell subsets - B1 and B2 cells. While B2 cells are responsible for the adaptive response to environmental antigens, B1 cells regulate the production of polyreactive and low affinity antibodies for innate humoral immunity. The molecular mechanism of B cell specification into different subsets is understudied. In this study, we identified lysine methyltransferase NSD2 (MMSET/WHSC1) as a critical regulator of B1 cell development. In contrast to its minor impact on B2 cells, deletion of the catalytic domain of NSD2 in primary B cells impairs the generation of B1 lineage. Thus, NSD2, a histone H3 K36 dimethylase, is the first-in-class epigenetic regulator of a B cell lineage in mice.
Keywords: B1 cells, NSD2, MMSET, histone methylation
Introduction
Two developmentally and functionally distinct B cell populations support the humoral immunity in mice and man [1]. Most peripheral B cells, which are defined as B2 cells, are generated in the bone marrow and have an immensely diverse, mostly non self-directed antibody repertoire [2, 3]. B2 cells can expand rapidly upon infection or antigen stimulation, followed either by immediate differentiation into antibody producing plasma cells or by the germinal center reaction and ensuing generation of cells expressing high affinity antibody [4].
In contrast to B2 cells, the majority of B1 cells are generated by late fetal and/or neonatal definitive hematopoiesis and reside predominately within well-defined anatomical compartments, such as peritoneal cavity and pleural cavity [5, 6]. They do not proliferate in response to antigen stimulation, but divide in a seemingly autonomous fashion at a low rate [7]. The antibody repertoire of B1 cells is represented largely by self-reactive or poly-reactive low affinity antibodies, mostly of the IgM or IgG3 (and IgA in mucosal surfaces) isotypes [8-10]. In addition to the markedly different developmental and functional features, B1 cells display a distinct pattern of surface proteins, including CD5 or CD11b that are expressed normally on T cells or macrophages, respectively [6, 11, 12]. Accordingly, the CD5-positive B1 cells are defined as B1a cells and the CD5-negative B1 cells as B1b cells. The ontogeny of B1 cells is not well understood and opposing ideas have been posited. Some findings suggested these cells develop from a distinctive fetal lineage [13, 14], while others indicated that B1 differentiation is “instructed” by signals downstream of their surface antigen receptors [15]. These concepts may not be mutually exclusive, since B1 cells expresses poly-reactive antigen receptors [16] that could be particularly amenable to stimulation from self or environmental antigens, leading to the surface expression of characteristic markers. The discovery of lin28b as a key regulator of B1 cell development [17] support the existence of a separate lineage for these cells.
NSD2 (nuclear receptor SET domain-containing protein 2, also known as MMSET, multiple myeloma SET-domain containing protein or WHSC1, Wolf–Hirschhorn syndrome candidate 1) is one of three members of the NSD family of histone lysine methyltransferases [18] that contains, in addition to the catalytic SET domain, PHD (plant homeodomain) fingers, PWWP (Pro-Trp-Trp-Pro) domains, and a NSD specific Cys-His-rich C5HCH domain. Hereafter, we will refer to this protein as NSD2.
The substrate specificity of NSD2, while most likely being the Lys36 of histone H3 in vivo, remains somewhat controversial and in vitro depends on the nature of a substrate [19, 20]. The methylation of Lys36 of histone H3 has been implicated in the process of RNA elongation during transcription, thus suggesting that NSD2 contributes to the generation of full-length transcripts [21]. NSD2 function is essential for normal development in mice and humans, and NSD2 deficiency in mice leads to neonatal death due to severe growth retardation [22]. NSD2 is often deleted in Wolf-Hirschhorn syndrome [23] and a great deal of attention for NSD2 stems from its link to aggressive multiple myeloma in humans [24], whereby the t(4;14) translocation places the Nsd2 gene, which encodes NSD2, under the control of the IgH Eμ-enhancer and leads to NSD2 over-expression [25]. This molecular signature is linked to aggressive myeloma and poor prognosis [26]. The mechanism of NSD2 contribution to myelomagenesis and/or tumor progression is not well understood.
Here, we present data on the essential and selective role of the NSD2 histone methyltransferase in mouse B cells, as it is required for generation of the B1 lineage.
Methods
Ethical statement.
Nsd2loxSET/ loxSET and Nsd2ΔSET/ ΔSET mice on C57/BL6 background were generated in our laboratory. Nsd2loxSET/ loxSET littermates (not crossed to the mice expressing Cre recombinase) were used as controls. Mice were housed under specific pathogen-free conditions and experimental protocols were approved by the Rockefeller University Institutional Animal Care and Use Committee. All studies were conducted in accordance with the GlaxoSmithKline plc (GSK) Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the Institution, where the work was performed.
Generation of Nsd2-flox-SET mice.
To create the targeting vector pBSmmsetflox, a single loxP site [27], a BsoBI (Ava I) restriction site, and a NeoR selection marker cassette flanked by FRT sites [28], were introduced into a HindIII site in intron 19 and an additional loxP site was inserted in a BglII site in intron 17 of the mouse Nsd2 locus (Fig. 1A). ES cells at embryonic day 14.1 were transfected and selected by standard techniques. Successful recombinants were identified by Southern blot analysis (BsoBI digested total DNA with 5’ or 3’ probe; NcoI digested total DNA with 3’ probe). Targeted ES cells were used to generate mice. The FRT site-flanked NeoR cassette was removed by breeding to FLP deleter mice [29]. The resulting mice were designated Nsd2loxSET/ loxSET (Fig. 1A).
Fig. 1: Expression of Nsd2 and generation of mice with B cell-specific expression of catalytically inactive NSD2.
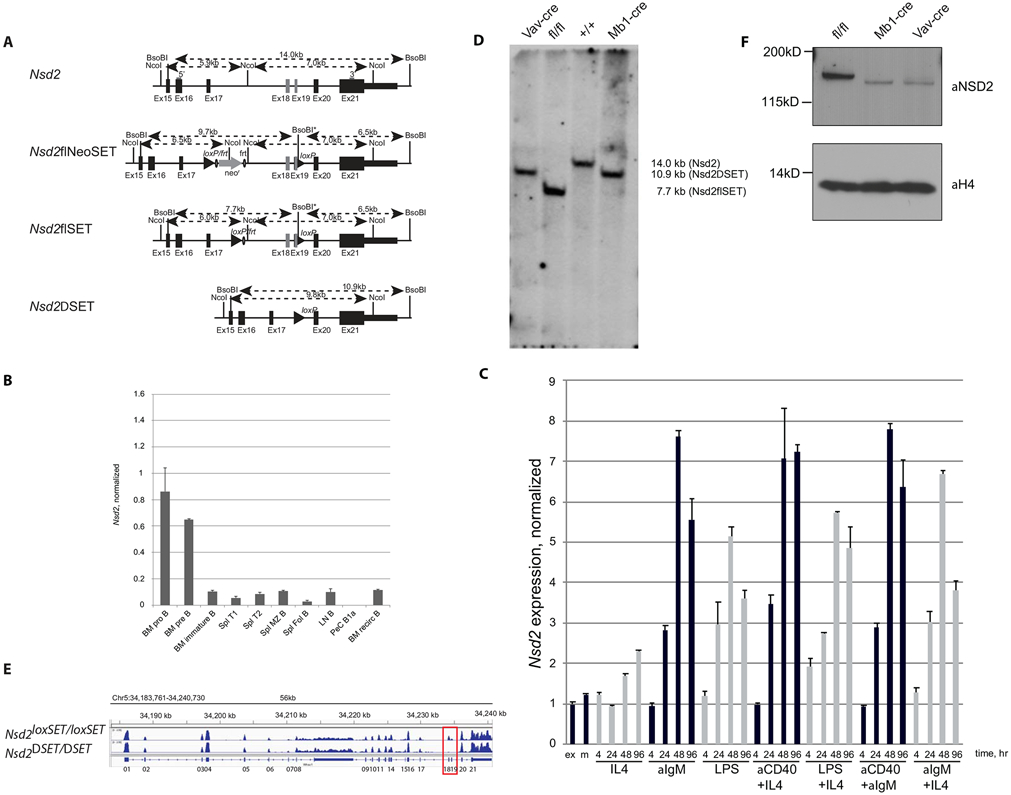
A) Structure of the wild type Nsd2 locus, the targeted locus (Nsd2loxSET/FRT), the targeted locus after FLP-recombination mediated deletion of the NeoR-cassette (Nsd2loxSET/loxSET), and the SET-domain deleted locus after Cre-recombination (Nsd2ΔSET/ΔSET). Numbered rectangles depict exons, filled triangles and circles represent loxP and FRT sites, respectively. Restriction sites and distances are indicated above each locus. The 5’ and 3’ probes used in Southern blots are shown as grey bars. B) Nsd2 expression profile in different B cell populations purified from 6-12 weeks old mice was determined by qPCR and normalized to Tbp using primer pairs tcatgggaaacacaattcagca/ aagtagcttcaaagggtgtcg and gctctggaattgtaccgcag/ctggctcatagctcttggctc for Nsd2 and Tbp, respectively. Bone marrow (BM) pro-B cells (pro B), pre-B cells (pre B), immature B cells (immature B), splenic (Spl) transitional 1 B cells (T1), transitional 2 B cells (T2), marginal zone B cells (MZ B), follicular B cells (Fol B), lymph node (LN) B cells (B), peritoneal cavity (PeC) B1a cells (Bia), recirculating B cells (recirc B). A representative experiment out of 3 performed (3 biological and 3 technical replicates each). C) Nsd2 expression is upregulated upon B cell activation in vitro. Purified splenic B cells were stimulated with different agents for up to 96 hours and RNA level of the gene of interest was measured at 4, 24, 48, and 96 hours. A representative experiment out of 3 performed (3 biological and 3 technical replicates each). D) Mb1Cre- and Vav1Cre- mediated deletion of the SET-domain encoding exons 18 and 19 of the Nsd2 gene in splenic B cells. Southern blot analysis of DNA isolated from purified B cell with the 5’ probe after BsoBI digest is shown. The 7.7 kb band corresponds to the targeted locus (fl/fl) and the 14 kb and the 10.9 kb bands correspond to the wild type (+/+) and Cre modified Nsd2 gene, respectively. A representative experiment out of 2 performed (no technical replicates). E) Whole-transcriptome profile of Nsd2 gene in splenic B cells isolated from Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice. IGV tracks show the relative RNA expression level. Exons 18 and 19 encoding the SET domain are boxed. A representative experiment out of 3 performed (no technical replicates). F) Truncated NSD2ΔSET protein is less stable compared to its full-length counterpart in purified splenic B cells from Nsd2loxSET/loxSET (fl/fl), Mb1creNsd2loxSET/loxSET (Mb1cre) and Vav1creNsd2loxSET/loxSET (Vavcre) mice. Exons 18 and 19 encode for 83 amino acids, which are missing in NSD2ΔSET. A representative experiment out of 2 performed (no technical replicates).
Generation of mice with germline- or B cell-specific ablation of the NSD2 SET domain.
To ablate NSD2 SET domain in germline, the Nsd2loxSET/ loxSET mice were crossed with the B6.C-Tg(CMV-cre)1Cgn/J [30]. For B-cell-specific ablation, the following deletor mice were used: B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ (Mb1-Cre) [31], B6.Cg-Commd10Tg(Vav1-icre)A2Kio/J (Vav1-Cre) [32], and B6.129P2(C)-Cd19tm1(cre)Cgn/J (CD19-Cre) [33]. All Cre deletor mice were from the Jackson Laboratory (Bar Harbor, USA). All experiments were performed with 8-16 week old mice.
Antibodies.
The following antibodies were purchased from either BD Biosciences (San Jose, USA), eBioscience/Thermo Fisher Scientific (Waltham, USA) or the Jackson Laboratory: B220 (RA3-6B2), IAb (AF6-120.1), IgM (115-116-075), IgD (11-26c.2a), CD5 (53-7-3), CD11b (M1/70), CD21/35 (7G6), CD19 (B3B4), CD23 (1D3), CD43 (S7), CD86 (GL1), CD90 (53-2.1). The B cell receptor specific antibodies PE-3H7 (anti-VH11id), and APC-13B5 (anti-Vk9id) were kindly provided by Kyoko Hayakawa (Fox Chase Cancer Center, Philadelphia, USA) and the NSD2 antibody (29D1) was purchased from Abcam (Cambridge, UK).
Definition of cell types by cell surface phenotype.
Bone marrow pro-B cells (IgM−B220+CD43+), bone marrow pre-B cells (IgM−B220+CD43−), bone marrow immature B cells (IgM+B220+), bone marrow recirculating B cells (IgM+B220hi), splenic T1 (IgM+CD21/35−), splenic T2 (IgM+CD21/35hi), splenic marginal zone (MZ) B cells (CD19+CD21highCD23low/-), splenic follicular B cells (CD19+CD21medCD23hi), splenic T cells (CD3ε+), lymph nodes B cells (CD19+), Peyer’s Patch IgA expressing B cells (B220+IgM−IgA+), Peyer’s Patch T cells (CD3ε+), splenic germinal center (GC) B cells (B220+ CD95+CD38dull), splenic GC B cells in the light zone (LZ) (B220+CD95+CD38dull CD86+CXCR4lo) and dark zone (B220+CD95+CD38dullCD86−CXCR4+), peritoneal cavity (PeC) T cells (CD5+IgM−), PeC B1a cells (IgMhiCD11b+CD5+), PeC B1b cells (IgMhiCD11b+ CD5−) PeC B2 cells (IgM+CD11b−CD5−).
ChIP-seq.
ChIP was performed as previously described [34, 35]. In brief, 107 cells were cross-linked with 0.5% formaldehyde at room temperature for 10 min. Chromatin was sonicated to 300–500 bp in RIPA buffer with 0.3 M NaCl. 5–10 μg antibodies were pre-incubated with Dynabead Protein A/G (Invitrogen/ Thermo Fisher Scientific) for at least 8 h before incubating with sonicated chromatin overnight. After that, beads were washed in modified RIPA wash buffer (100 μM LiCl) and 1× in TE. After overnight cross-link reversal at 65°C, RNase digestion, and proteinase K digestion, ChIP DNA and input DNA were purified using the QuiaQuick PCR purification kit (Qiagen, Hilden, Germany). For validation of ChIP-Seq, ChIP DNA was analyzed via qPCR using SYBR Green PCR Master Mix and the LightCycler 480 (Roche, Basel, Switzerland). Primer sequences are available upon request.
For ChIP-Seq, 30 μl of ChIP DNA were used to generate blunt-ended DNA using reagents supplied with the Epicenter DNA EndRepair kit (Epicentre Biotechnologies, Madison, USA) according to the manufacturer’s instructions. The end-repaired DNA was purified using the QuiaQuick PCR purification kit. Using Klenow Fragment (3′ to 5′ exo-, New England Biolabs, Ipswich, USA), the “A” bases were added to the DNA. The DNA was purified using the MinElute PCR purification kit (Qiagen). The T4 DNA ligase (New England Biolabs) was used for ligation of Illumina/Solexa adapters to the DNA fragments. The adaptor-ligated DNA was purified with the MinElute PCR purification kit (Qiagen). The DNA fragments were subjected to 18 cycles of PCR using the Illumina/Solexa primers 1.0 and 2.0 to generate the ChIP-Seq libraries. The ChIP-Seq libraries were purified with the MinElute PCR purification kit (Qiagen).
Samples were sequenced on the Illumina Hiseq2000 platform for 50 cycles, and raw sequencing data were processed using the CASAVA_v1.8.2 software to generating fastq files. Sequencing reads were aligned to the mouse genome (mm9) using Bowtie v0.12.7 [36]. Reads were kept if they aligned with two errors or fewer and did not align to more than one location in the genome. A 25-bp density coverage map was created by extending each read for 100 bp to account for mean library fragment length and mapping the number of reads per 25 bp bin using IGVtools [37]. Values in each sample were normalized to fpkm values by calculating the fraction of mapped reads per bin in one million total reads.
For comparative analysis of promoter regions, the number of aligned reads in the area surrounding the transcriptional start site (±3 kb) of each gene was used.
Preparation of libraries for RNA sequencing.
2 μg total RNA were used per sample, ribosomal RNA was removed with the Ribo-Zero Magnetic kit (Epicentre Biotechnologies), and libraries were prepared with the ScripSeq v2 RNA-Seq Library Prepapration kit (Epicentre Biotechnologies) following the manufacturer’s instructions. Samples were sequenced in the same manner as the ChIP-seq samples, but using 100 cycles instead of 50. Fastq reads were aligned to the mouse reference genome mm9 using Tophat [38] to account for splicing and alternative promoter usage as well as insertions and deletions. Subsequently, the cufflinks RNA-seq analysis tool cuffdiff [39] was used to assess differential gene expression, alternative promoter usage, and splicing variation between experimental datasets. The resulting fpkm values were used for further data analysis and visualization.
Quantitative PCR.
Total RNA was extracted from freshly isolated cells using an RNase minikit (Qiagen) according to the manufacturer’s protocol. RNA was DNase treated using an RNase free DNase set (Qiagen) and cDNA was synthesized using reagents supplied with a first strand cDNA synthesis kit (Roche). Quantitative real-time PCR was performed using SYBR Green (Roche) on a Roche Lightcycler 480. Primers were designed with the Primer3 program. MMSET-F: TCATGGGAAACACAATTCAGCA; MMSET-R: AAGTAGCTTCAAAGGGTGTCG; TBP-F: GCTCTGGAATTGTACCGCAG; TBP-R: CTGGCTCATAGCTCTTGGCTC
Analysis of V(D)J junctions.
Analysis of immunoglobulin gene rearrangements was conducted as described previously [40] in 3-fold dilutions using Thy1.2 as a loading control.
Protein expression and radioactive methyltransferase assay.
His6-NSD2 (cloned into pET19b) was expressed in E. coli and purified over Ni-NTA spin columns (Qiagen). Methyltransferase assay was performed as described previously with 10 mM of Adenosyl-L-methionine, S- [methyl-3H] (GE Healthcare, Chicago, USA) for 45 min at 30°C [41]. Protein gels were incubated with EN3HANCE (PerkinElmer, Waltham, USA), dried, and exposed to Kodak Biomax XAR film (Sigma-Aldrich, St. Louis, USA) for 2 – 10 days at −80°C.
Flow cytometry.
Single-cell suspensions from indicated tissues were prepared. All antibodies were used at dilutions ranging from 1:100-1:3,000 and incubated for 30 min at 4°C. Flow cytometric analysis and cell sorting were performed using a FACS LSR II or Aria (Becton Dickinson, Franklin Lakes, USA) and data were analyzed with FlowJo software (Becton Dickinson).
B cell purification, in vitro activation and proliferation.
Splenic B cells were purified by depleting CD43+ cells, using anti-CD43 beads and magnetic columns (Miltenyi Biotec, Bergisch Gladbach, Germany) and stimulated in vitro with 10 μg/ml F(ab’)2 fragment of goat anti-mouse IgM (Jackson ImmunoResearch, West Grove, USA) in combination with 25U/ml of recombinant mouse IL-4 (R&D Systems, Minneapolis, USA), 5μg/ml bacterial LPS (Sigma-Aldrich), or 5 μg/ml of bacterial LPS in combination with 25 U/ml of recombinant mouse IL-4. Labeling of cells with 5-(6-) carboxyfluorescein diacetate, succinimidyl ester (CFSE, Molecular Probes, Eugene, USA) for analysis of proliferation was performed following the manufacturer’s instructions. The decline in CFSE fluorescence as a measure of B cell proliferation was determined by FACS analysis.
Histone post translational modification analysis.
Histones were extracted in acid and chemically derivatized twice, digested with trypsin, followed two more rounds of derivatization and the peptides were desalted by using C18 stage-tips, as described earlier [42]. Samples were analyzed using an EASY-nLC nanoHPLC (Thermo Fisher Scientific) in a gradient of 0-35% solvent B (A = 0.1% formic acid; B = 95% MeCN, 0.1% formic acid) over 30 min and from 34% to 100% solvent B in 20 minutes at a flow-rate of 250 nL/min. Nano-liquid chromatography was coupled with a Q-Exactive mass spectrometer (Thermo Fisher Scientific). Full scan MS spectrum (m/z 290−1650) was performed in the Orbitrap (Thermo Fisher Scientific) with a resolution of 30,000 (at 400 m/z) with an AGC target of 1×10e6. The MS/MS events included both data-dependent acquisition and target, the latter for isobaric peptides to enable MS/MS-based quantification. The relative abundance of histone H3 and H4 peptides were calculated by using EpiProfile [43].
Cell survival assay.
Purified B cells were cultured either in medium alone or in the presence of 1.56-25ng ml− of recombinant BAFF (R&D Systems) for the indicated time and stained with Annexin V (Roche) and 7-aminoactinomycin D (7-AAD; Sigma-Aldrich).
Statistical analysis
was performed in Prism (GraphPad Software, San Diego, USA) with the unpaired t-test.
Results
The SET domain of NSD2 is required for postnatal survival
To determine whether the catalytic (SET) domain of NSD2 is essential for survival, we generated mutant mice by gene targeting in ES cells. The loxP sites were introduced in introns 17 and 19 of the NSD2-encoding gene (Nsd2) and Nsd2loxSET/ loxSET mice were produced (Fig. 1A). These mice were bred to the CMVcre germline deleter mice and then intercrossed to produce the Nsd2ΔSET/ ΔSET offspring. Similar to NSD2−/− [22], homozygous Nsd2ΔSET/ ΔSET mice die early after birth (data not shown). Therefore, the SET domain of NSD2 is required for postnatal survival.
B cell-specific ablation of the NSD2 SET domain
To measure Nsd2 expression in the B cell compartment, we purified RNA from the wild type B cells of different developmental stages and measured its RNA level by qRT-PCR (Fig. 1B). Nsd2 is expressed in early B2 cell progenitors (pre- and pro-B cells) and its transcription is decreased at later stages. Of note, Nsd2 expression in B1 cells was below the level of detection (Fig 1B). In vitro activation of B2 cells by a variety of stimuli upregulated Nsd2 transcripts (Fig. 1C). We conclude that Nsd2 expression levels vary throughout B cell development.
To define the contribution of the NSD2 SET domain to B and plasma cell development, we conditionally deleted it in the B cell lineage. Nsd2loxSET/loxSET mice were crossed to Mb1cre mice for conditional deletion in pro-B cells [31] or to CD19cre mice for conditional deletion in pre-B cells [33]. They were also crossed to Vav1cre mice for conditional ablation in multiple lineages during the early stages of definitive hematopoiesis [32]. The deletion of the NSD2 SET domain in B cells was incomplete in CD19creNsd2loxSET/loxSET (Suppl. Fig. 1) and complete in Mb1creNsd2loxSET/loxSET and Vav1creNsd2loxSET/loxSET (Fig. 1D). We therefore chose Mb1creNsd2loxSET/loxSET for all subsequent experiments and Nsd2loxSET/loxSET littermates were used as control.
Splenic B cells were present in mutant mice, and successful deletion of Nsd2 exons 18 and 19 was confirmed by RNA-seq analysis of purified B cells (Fig. 1E). By Western blot, the NSD2ΔSET protein displayed the expected shift in size and was less stable compared to the wild type NSD2 (Fig. 1F). We conclude that the NSD2 SET domain is largely deleted from B2 cells.
Histone methylation changes in NSD2ΔSET B2 cells
In vitro, NSD2 is a histone methyltransferase with broad specificity [19, 20]. In contrast, on intact nucleosomes NSD2 predominantly methylates histone H3 with only some residual activity toward histone H4 (Suppl. Fig. 2). To evaluate the catalytic function of NSD2 in B2 cells, we compared the pattern of histone modifications in NSD2 versus NSD2ΔSET expressing cells by mass spectrometry. Selective changes were noted in the pattern of histone H3 modification. In agreement with the previously reported specificity of NSD2 towards dimethylation of lysine 36 of histone H3 (H3K36me2) [19], overall H3K36me2 and H3K36me3 levels were reduced in NSD2ΔSET B cells, correlating with a corresponding increase in the level of unmodified H3K36 (Fig. 2A-C). The reduction in H3K36me2/3 methylation affected over 50% of all modified histones and the loss of H3K36me2/3 appears to be independent of other modifications on the same histone. We conclude that in B2 cells H3K36me2 is the main substrate of NSD2.
Fig. 2: Selective methylation of histone H3 at Lys36 by NSD2.
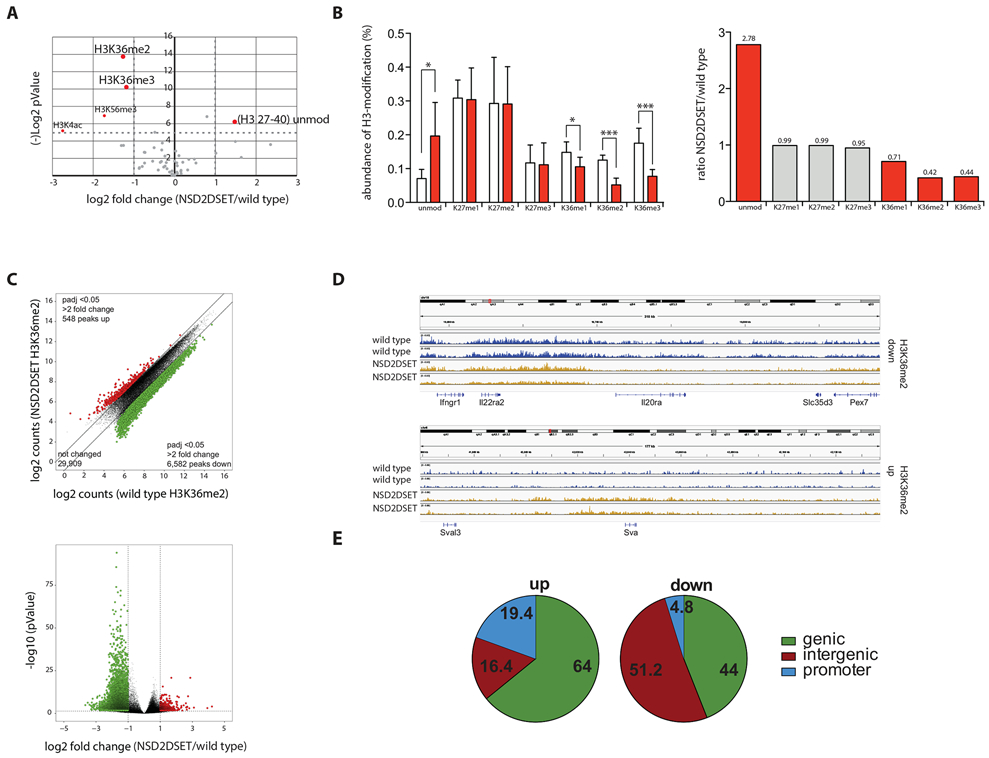
A) Volcano plot shows changes in relative abundance of distinct modifications of histones extracted from splenic B cells purified from Nsd2loxSET/loxSET (wild type) or Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice. Values over 4.32 in the Y-axis [(−)log2 pValue] corresponding to −log2 (0.05) are significant and highlighted in red. The data represent 2 biological replicates and 3 independent measurements. Significance was determined by unpaired Student’s T-test. B) The fraction of histone H3 modified at Lys36 was determined by combining the frequencies for all histone H3 peptides carrying this modification, independent of other modifications present on the same peptide. Peptides of controls (from splenic B cells isolated from Nsd2loxSET/loxSET mice) are represented by white bars, peptides from splenic B cells isolated from Mb1creNsd2loxSET/loxSET mice are in red (left graph). The significant changes in abundance of H3-peptides carrying the indicated modifications are depicted in red bars, not significant changes in grey bars. Numbers above the bars indicate the fold change of Mb1creNsd2loxSET/loxSET (NSD2ΔSET) over Nsd2loxSET/loxSET (wild type) (right graph). The data represent 2 biological replicates and 3 independent measurements. C) Scatter plot of H3K36me2 peaks of splenic B cells Nsd2loxSET/loxSET (wild type) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice. Significant increase (red) or decrease (green) in specific peaks are indicated (top). Volcano plot illustrating differential H3K36me2 levels in splenic B cells from Nsd2loxSET/loxSET (wild type) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice (bottom). Significant increase (red) or decrease (green) in specific peaks are indicated. Data plotted are average normalized H3K36me2 values of called peaks from 3 biological replicates. D) Sample IGV tracks of H3K36me2 confirming the locus specific changes in Nsd2loxSET/loxSET (wild type) vs. Mb1creNsd2loxSET/loxSET (NSD2ΔSET) splenic B cells. Data represents 2 biological replicates and 3 independent measurements. E) The gain (left) and loss (right) of H3K36me2 in Mb1creNsd2loxSET/loxSET vs. Nsd2loxSET/loxSET splenic B cells in genic, intergenic, and promoter regions. The data represent 2 biological replicates and 3 independent measurements.
To evaluate changes in the distribution of H3K36me2 across the genome, we performed ChIP-seq on wild type and NSD2ΔSET mutant B2 cells (Fig. 2D). This analysis showed significant (p-adjusted <0.05) locus-specific changes: signal reduction of over two-fold was observed at 6,582 peaks and increase of over two-fold at 548 peaks. The gain of H3K36me2 in NSD2ΔSET B cells occurred mainly in genic (64%) and promoter regions (19.4%) while the loss of H3K36me2 was observed mainly in intergenic regions (51.2%; Figure 2E). We conclude that changes in H3K36me2 occur at specific sites in the genome.
Data availability
Sequencing data generated for this study are available through the GEO database: H3K36me2 ChIP-sequencing and RNA-Seq (accession no. GSE155689).
The NSD2 SET domain is dispensable for B2 cell development
Flow cytometric analysis of bone marrows showed a similar distribution of B cell progenitors in mice with NSD2 versus NSD2ΔSET expressing B cells (Suppl. Fig. 3A), while the number of recirculating B cell progenitors and of follicular B cells in the spleen were mildly reduced in the mutant (Suppl. Fig. 3B). We tested the impact of NSD2ΔSET on proliferation in response to antigen receptor- or polyclonaly-triggered B cell proliferation in vitro and found no defect (Suppl. Fig. 3C). The ability of B2 cells to respond to pro-survival signals such as BAFF was not affected by NSD2ΔSET (Suppl. Fig. 3D). Thus, the catalytic function of NSD2 has only mild effects on the steady-state size and composition of the B2 cell compartment.
The lack of NSD2 SET domain controls isotype class switching, splenic germinal center formation and the humoral immune response
Previous studies reported that NSD2 is required for B cell class switch recombination [44, 45]. In agreement with these studies, we found reduced serum levels of IgM, IgG3, and IgA in Mb1creNsd2loxSET/loxSET mice compared to their Nsd2loxSET/loxSET littermate controls (Fig. 3A) and a reduced percentage of IgA positive B cells in the Peyer’s Patches (Fig. 3B). To determine whether NSD2ΔSET B cells have impaired switching to IgA in vitro, we stimulated B2 cells in the presence of TGF-β or all trans-retinoic acid (ATRA) and measured class switch recombination by flow cytometry (Fig. 3C). IgA switching was consistently decreased in NSD2ΔSET B cells, while their ability to upregulate the integrin α4β7 – a known target of RA-signaling [46] – was maintained. Switching to IgG3 in response to LPS was also significantly impaired, while switching to IgG1 in response to LPS+IL4 showed no defect (Fig. 3D).
Fig. 3: NSD2 controls selective class switch recombination in B cell.
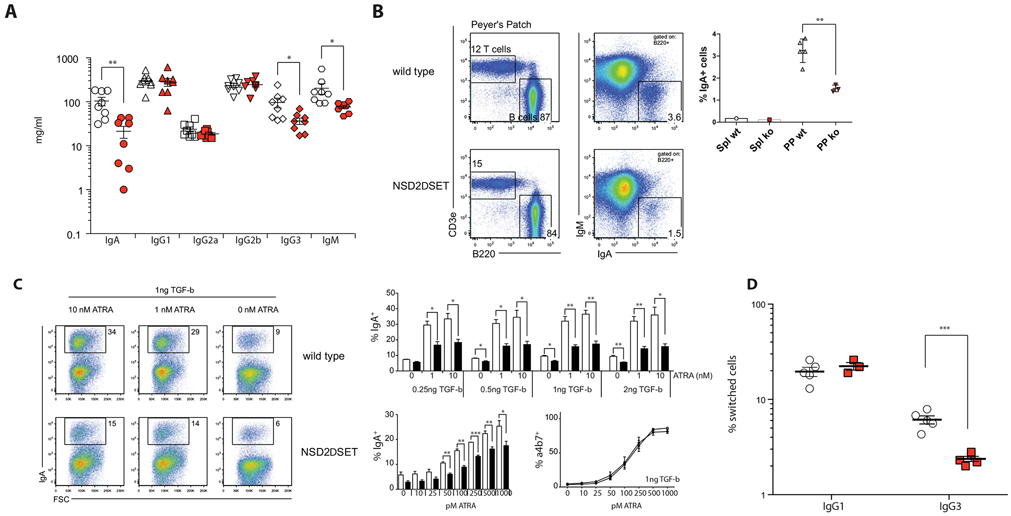
A) Concentration of serum immunoglobulins in Nsd2loxSET/loxSET (white symbols) and Mb1creNsd2loxSET/loxSET (red symbols) mice were quantified by ELISA. Each symbol represents one mouse (8 mice per genotype, 2 independent experiments). Significance was determined by unpaired t-test *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001. B) FACS plots show relative abundance of T cells (T cells, CD3e+) and B cells (B cells, B220+); and the relative abundance of surface IgA positive (B220+IgA+) B cells in Peyer’s Patches of Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET; ko) mice. The percent of IgA+ B2 cells in spleen (Spl) and Peyer’s Patches (PP) is indicated on the right. A representative experiment out of 3 performed (3 mice each). C) NSD2ΔSET B2 cells have a defect in switching to IgA in vitro. Splenic B2 cells isolated from Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice were stimulated in vitro in the presence of LPS, TGF-β and all trans-retinoic acid (ATRA). The percentage of IgA positive B cells is indicated. Representative plots of more than 3 independent experiments with 3 or more mice per group are shown. Bar diagrams indicate the percent of IgA positive B2 cells after 3 days in various culture conditions. Control Nsd2loxSET/loxSET cells (white bars) and cells from Mb1creNsd2loxSET/loxSET (black bars) mice (top) are shown. NSD2ΔSET B2 cells have a defect in switching to IgA, but not in the induction of the integrin α4β7 in response to ATRA. The percentage of IgA positive B cells is response to LPS and a stable amount of TGF-β (1ng) with increasing amounts of ATRA (0-1000pM) in Nsd2loxSET/loxSET (white bars) and Mb1creNsd2loxSET/loxSET (black bars) B2 cells is indicated (bottom left). The percentage of integrin α4β7-positive B cells is response to LPS and a stable amount of TGF-β (1ng) with increasing amounts of ATRA (0-1000pM) in Nsd2loxSET/loxSET (open symbols) and Mb1creNsd2loxSET/loxSET (closed symbols) B2 cells is indicated (bottom right). D) NSD2ΔSET B2 cells have a defect in switching to IgG3, but not IgG1 in vitro. B2 cells isolated from Nsd2loxSET/loxSET (open symbols) and Mb1creNsd2loxSET/loxSET (red symbols) mice were stimulated in vitro in the presence of LPS or LPS+IL4. The percentages of switched B cells after 3 days in vitro culture are indicated. Each symbol represents one mouse (3-5 mice per genotype, 2 independent experiments). Significance was determined by unpaired t-test *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001.
In response to infection or vaccination B cells form germinal centers (GC) in lymphoid organs [47]. To evaluate the role of the NSD2 SET domain in GC formation, we immunized mice with sheep red blood (SRB) cells and analyzed splenic GCs by flow cytometry. NSD2ΔSET GCs were smaller both under steady state conditions and upon immunization, while the distribution of GC light versus dark zone was similar (Fig. 4A). To evaluate the antigen-specific humoral response, mice were immunized with model antigens. The T-independent antigen NP-Ficoll induced the typical antigen-specific IgM, IgG3, and λ-chain response [48], with a slight reduction in the number of IgM specific antibodies at day 14 post immunization in mice with NSD2ΔSET B cells (Fig. 4B). In response to immunization with the T-cell dependent antigen NP22-CGG we found a mild antigen-specific IgG1 defect and more pronounced IgG3 defect in the recall response to secondary immunization (Fig. 4C). We conclude that NSD2ΔSET in B cells mildly alters isotype class switching, splenic germinal center formation and the humoral immune response.
Fig. 4. NSD2 controls splenic germinal center response.
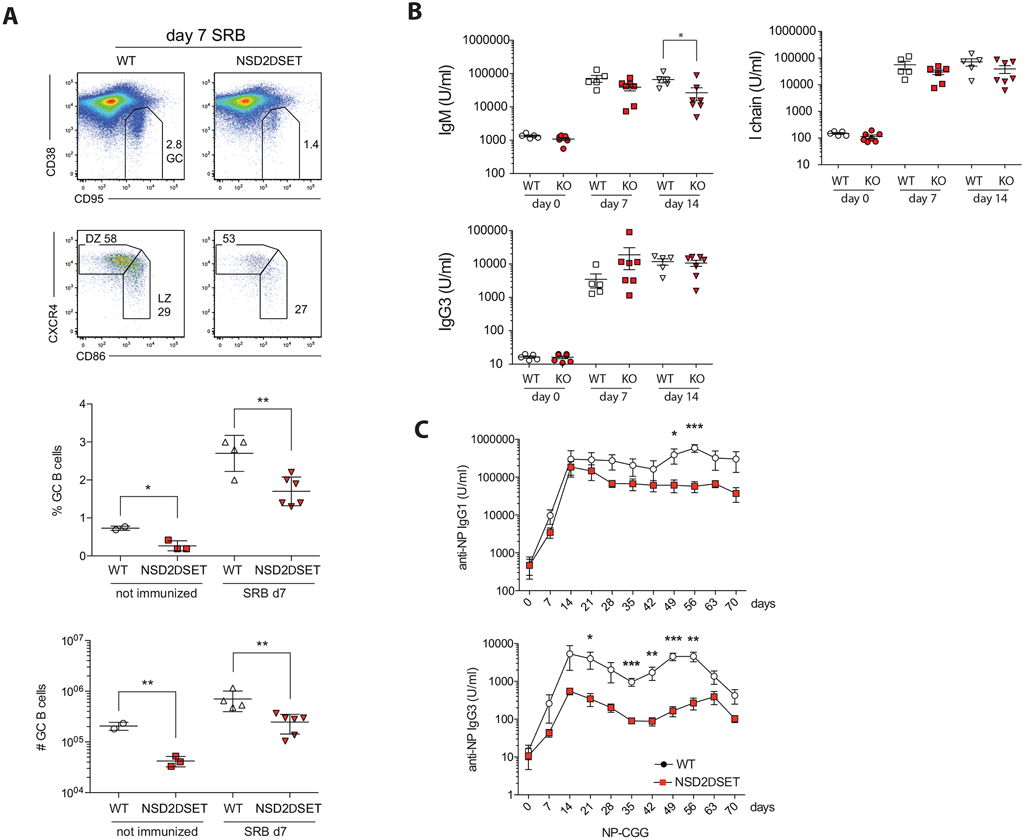
A) Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice were injected IP with sheep red blood cells and the percent of germinal center (GC) cells (GC; CD95+CD38dull) and the percent of GC B cells in the light zone (LZ; CD86+CXCR4dull) and dark zone (DZ; CD86−CXCR4+) of the spleen are indicated in the representative FACS plots. More than 3 independent experiments with 3 or more mice per group were performed. The percent and total number of GC B cells in Nsd2loxSET/loxSET (WT, open symbols) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET, red symbols) mice was plotted below. Each symbol represents one mouse (4 wild type and 6 NSD2ΔSET mice). Significance was determined by unpaired t-test *: p ≤ 0.05, **: p ≤ 0.01. B) T-independent immunization. Nsd2loxSET/loxSET (WT, open symbols) and Mb1creNsd2loxSET/loxSET (KO, red symbols) mice were immunized with the T independent model antigen (NP-Ficoll) and titers of the antigen specific serum antibodies were determined by ELISA. Each symbol represents one mouse (5 wild type and 7 NSD2ΔSET mice in 2 independent experiments). C) T-dependent immunization. Nsd2loxSET/loxSET (open symbols) and Mb1creNsd2loxSET/loxSET (red symbols) mice were immunized (day 0) and boosted (day 35) with the T-dependent model antigen NP22-CGG and titers of the antigen specific serum antibodies were determined by ELISA. Data summarized for 5 mice in each group in 2 independent experiments. Significance was determined by unpaired t-test *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001. The experiment was performed once with 5-8 mice per experimental group.
NSD2 is required for the generation of B1 cells
Contrary to the mild effects on B2 cells, peritoneal B1 cells were strongly reduced in Mb1creNsd2loxSET/loxSET mice. FACS analysis of peritoneal cells derived from these animals revealed an over 12-fold reduction in the number of IgMhiCD5hiCD11bhi B1a cells and a nearly 3-fold reduction of the IgMhiCD5loCD11bhi B1b cells (Fig. 5A, B). Phosphotidylcholine-specific antibodies in B1 cells are enriched for Vh11 or Vh12 heavy chains paired with Vκ4 or Vκ9 light chains [49]. Peritoneal B cells expressing Vh11/Vκ9 antibodies were nearly absent in Mb1creNsd2loxSET/loxSET mice (Fig. 5C). In agreement with this finding, Vh12 – Jh rearrangements revealed a defect in Vh12-Jh1/Jh3 junctions in NSD2ΔSET B1 cells (Fig. 5D). We conclude that intact NSD2 is required for the generation of B1 cells.
Fig. 5: NSD2 is essential for B1 cell generation.
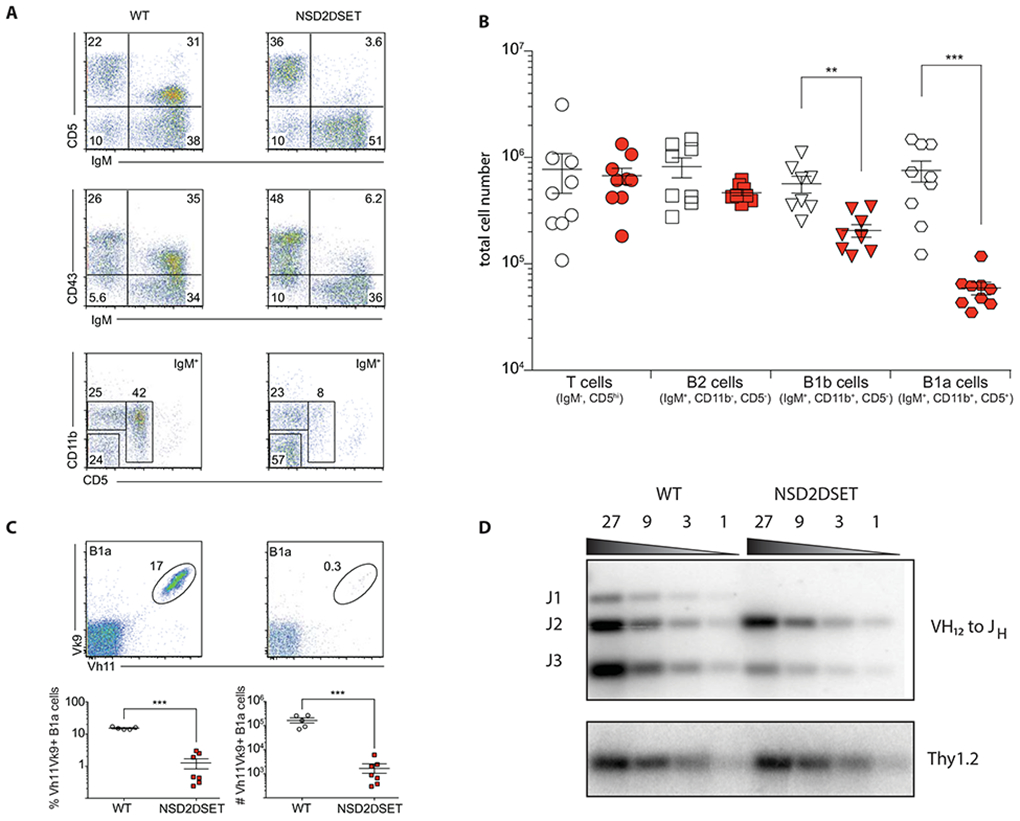
A) FACS plots show relative abundance of T cells (CD5+IgM−), B1a (IgMhiCD11b+CD5+), and B1b (IgMhiCD11b+ CD5−), and B2 (IgM+CD11b−CD5−) cells in the peritoneal cavity. The gating for the top and the middle plots in on live cells, for the bottom plots on live and IgM+ cells. Representative plots of more than 3 independent experiments with 3 or more mice per group are shown. B) Absolute numbers of distinct lymphoid cells in the peritoneal cavity. Open symbols (Nsd2loxSET/loxSET) and red symbols (Mb1creNsd2loxSET/loxSET) mice. Each symbol represents one mouse (9 mice per genotype in 3 independent experiments). C) The frequency (bottom left) and absolute number (bottom right) of peritoneal cavity B1a cells (gated on live/IgMhi and CD5+) expressing phosphatidylcholine specific B cells receptor (Vκ9 Vh11) in Nsd2loxSET/loxSET (WT, open symbols) and Mb1creNsd2loxSET/loxSET (red symbols) are shown. Representative plots of 2 independent experiments with six mice in total are depicted. Significance was determined by unpaired t-test *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. D) Southern blotting analysis of Vh12 to Jh rearrangements in splenic B cells isolated from Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice. Thy1.2 was probed as loading control. A representative experiment out of 2 performed is shown (no technical replicates).
To address the question whether NSD2 is essential for the generation or the maintenance of B1 cells, we analyzed 3-week old mice. While littermate control mice (Nsd2loxSET/loxSET) had a large percentage of B1a and B1b cells, young Nsd2ΔSET/ΔSET mice displayed a significant reduction in the percent and number of these populations, indicating that the development, and not the maintenance of B1 cells, is impaired (Suppl. Fig. 4).
Discussion
In this work we demonstrate that NSD2 (MMSET), a SET domain-containing histone lysine methyltransferase, dimethylates lysine 36 of histone H3 (H3K36me2) in vivo, affecting majority of all modified histones. The selectivity of H3K36me2 down-regulation only at some gene targets suggest a locus-specific mechanism of NSD2 targeting to the chromatin in B2 cells. How such specificity is achieved remains to be investigated further. A likely scenario is that NSD2 is recruited to chromatin with the help of its non-catalytic domains, which differ between distinct NSD family members [20].
While being dispensable for B2 cell development sensu stricto, this enzyme appears to contribute to the control of isotype class switching, splenic germinal center formation and the humoral immune response. We propose that NSD2 might play a role in the regulation of peripheral B2 cell maintenance. One of the important conclusions of this study is related to the obvious lack of NSD2 contribution to the B cell division. This particular finding argues against the current view on NSD2 as an important regulator of cell proliferation [50-53]. The unaltered pro-B to pre-B cell transition in Nsd2ΔSET/ΔSET mice implies that NSD2 does not play a critical role in IgH gene rearrangement and expression, as well as division of B cell progenitors. The wild-type like pattern of immature B2 cell generation in the bone marrow of Nsd2ΔSET/ΔSET mice also suggests that NSD2 is not crucial for surface expression of IgM and signaling, required for the generation of immature B cells.
In contrast to the fairly minor effects on B2 cells, peritoneal B1 cells are strongly affected when the NSD2-encoding gene, Nsd2, was ablated in B cell-specific manner. NSD2 mRNA is expressed at lower levels in adult B1 cells as compared to B2 cells. This observation, combined with the selective reduction of the B1 cells following NSD2 gene ablation suggests that NSD2 is likely to contribute to fetal B lymphopoiesis rather than to B1 cell maintenance during adulthood The major reduction of B1 cells numbers is mirrored by a decrease in serum antibody titers. It is well established that B1 cells are the largest contributors to the overall serum levels of IgM and IgG3 [54, 55], which, in addition to the switching defect in B2 cells, can explain the reduction in serum levels of these immunoglobulins.
Understanding the exact mechanisms of NSD2 contribution to B1 cell differentiation will require the development of approaches that allow for efficient inactivation of NSD2 in B1 cells during embryonic development, as well as after establishment of the mature B1 cell compartment. However, already at this point, our highly unexpected findings revealed NSD2 as the first-in-class epigenetic master regulator of a major B cell compartment in mice. This observation, while currently limited to studies in experimental animals, may help to understand the contribution of NSD2 malfunction to lymphoid tumor development in human.
Supplementary Material
Suppl. Fig. 2. NSD2 methylates histones H3 and H4 only in the context of nucleosomes.
NSD2SET or NSD2SET Y1092A (NSD2SETmut) were incubated with purified histone H3 (H3), intact nucleosomes (N), octamers of histones (O) or mixed histones, not assembled into nucleosomes (H) in the presence of Adenosyl-L-methionine, S-[methyl-3H]. Coomassie (top) and autoradiograph (methyl-3H, bottom) are shown.
Suppl. Fig. 1. CD19Cre- mediated deletion of the SET-domain encoding exons 18 and 19 of the Nsd2 gene in splenic B cells. Southern blot analysis of DNA isolated from purified splenic B cell with the 5’ probe after BsoBI digest is shown. The 7.7 kb band corresponds to the targeted locus (floxed) and the 14 kb and the 10.9 kb bands correspond to the wild type (WT) and CD19-Cre modified Nsd2 gene (Δ), respectively. The genotype of the purified B cells is indicated above each lane. Nsd2+/+ (wild type), Nsd2loxSET/loxSET (floxed SET), CD19creNsd2loxSET/loxSET (ΔSET; CD19-cre).
Suppl. Fig. 4. B1 cells do not develop in young Nsd2ΔSET/ΔSET mice. FACS plots show relative abundance of B1a (IgMhiCD11b+CD5+) and B1b (IgMhiCD11b+ CD5−) cells in the peritoneal cavity of 3 weeks old Nsd2loxSET/loxSET control (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice. Absolute numbers of distinct lymphoid cells in the peritoneal cavity (bottom left) and percent (bottom right) are indicated. Nsd2loxSET/loxSET (WT, open symbols) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET, red symbols). Representative plots of one experiment with 4-5 mice per group are shown. Significance was determined by unpaired t-test *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Suppl. Fig. 3: NSD2 is not essential for the development and maturation of B2 cells. A) FACS dot plots show distribution of developing and mature peripheral B2 cells. The percentage of cells in Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice in bone marrow (BM) and spleen (SPL) is indicated. Abbreviations used in the figure: pre/pro-B cells (pre/pro), immature B cells (imm), recirculating B cells (reci), pro-B cells (pro), T cells (T), B cells (B), follicular B cells (Fol), marginal zone B cells (MZ), transitional 1 B cells (T1), and transitional 2 B cells (T2). Representative plots of more than 3 independent experiments with 3 or more mice per group are shown. B) The absolute numbers of developing and mature B2 cells in the bone marrow (top) and spleen (bottom) of Nsd2loxSET/loxSET control (white symbols) and Mb1creNsd2loxSET/loxSET (red symbols) mice are shown. Each symbol represents one mouse. The experiments were performed 3-4 times with 2-3 mice per genotype. Significance was determined by unpaired t-test *: p ≤ 0.05, **: p ≤ 0.01. C) NSD2 catalytic function is dispensable for B cell proliferation. Expression of CD86 and IAb on freshly isolated Nsd2loxSET/loxSET control (grey) and Mb1creNsd2loxSET/loxSET (red) splenic B cells in the absence (filled histograms) or presence of stimuli (open histograms). Proliferation of freshly isolated Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) B cells in the absence (filled histograms) or presence of stimuli (open histograms) was measured by CFSE dilution. The experiments were performed 3-4 times with 2-3 mice per genotype. D) Dispensable role of NSD2 catalytic function in B cell survival. Frequencies of viable Nsd2loxSET/loxSET control (open symbols) and Mb1creNsd2loxSET/loxSET (red symbols) splenic B cells in the presence of 0 – 25 ng of recombinant BAFF. Data are means ± s.d. from 3 independent experiments.
Acknowledgements
We thank Dr. Davide Robbiani (Rockefeller University), Dr. Brad Rosenberg (Icahn School of Medicine at Mount Sinai) for critical reading of the manuscript. This work was supported by the NIH grants AI118891 and CA196539, and a Leukemia and Lymphoma Robert Arceci Scholar Award to B.A.G.; International Myeloma Foundation (Brian D. Novis Award) and Wendy Will Case Cancer Fund to V.Y.; and Open Philanthropy Project/Good Ventures Fund (project Histone mimicry by viruses) and GlaxoSmithKline (project Histone mimicry by pathogens) to A.T. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations:
- BCR
B cell receptor
- H3K36me2
histone 3 lysine 36 di-methylation
- NSD2
nuclear receptor SET domain-containing protein 2
- MMSET, WHSC1
methyltransferase NSD2
Footnotes
Conflicts of Interest
Authors declare no conflict of interest.
Disclaimer
Part of this work has been published as a pre-print [56]
References
- 1.McHeyzer-Williams MG (2003) B cells as effectors, Curr Opin Immunol. 15, 354–61. [DOI] [PubMed] [Google Scholar]
- 2.Yurasov S & Nussenzweig MC (2007) Regulation of autoreactive antibodies, Curr Opin Rheumatol. 19, 421–6. [DOI] [PubMed] [Google Scholar]
- 3.LeBien TW & Tedder TF (2008) B lymphocytes: how they develop and function, Blood. 112, 1570–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nutt SL, Hodgkin PD, Tarlinton DM & Corcoran LM (2015) The generation of antibody-secreting plasma cells, Nat Rev Immunol. 15, 160–71. [DOI] [PubMed] [Google Scholar]
- 5.Martin F & Kearney JF (2001) B1 cells: similarities and differences with other B cell subsets, Curr Opin Immunol. 13, 195–201. [DOI] [PubMed] [Google Scholar]
- 6.Su I & Tarakhovsky A (2000) B-1 cells: orthodox or conformist?, Curr Opin Immunol. 12, 191–4. [DOI] [PubMed] [Google Scholar]
- 7.Deenen GJ & Kroese FG (1992) Murine peritoneal Ly-1 B cells do not turn over rapidly, Ann N Y Acad Sci. 651, 70–1. [DOI] [PubMed] [Google Scholar]
- 8.Baumgarth N (2011) The double life of a B-1 cell: self-reactivity selects for protective effector functions, Nat Rev Immunol. 11, 34–46. [DOI] [PubMed] [Google Scholar]
- 9.Montecino-Rodriguez E & Dorshkind K (2006) New perspectives in B-1 B cell development and function, Trends Immunol. 27, 428–33. [DOI] [PubMed] [Google Scholar]
- 10.Rothstein TL (1990) Polyreactive low-affinity IgM antibodies produced by CD5+ B cells, Immunol Today. 11, 152. [DOI] [PubMed] [Google Scholar]
- 11.Ghosn EE, Yang Y, Tung J, Herzenberg LA & Herzenberg LA (2008) CD11b expression distinguishes sequential stages of peritoneal B-1 development, Proc Natl Acad Sci U S A. 105, 5195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forster I, Gu H, Muller W, Schmitt M, Tarlinton D & Rajewsky K (1991) CD5 B cells in the mouse, Curr Top Microbiol Immunol. 173, 247–51. [DOI] [PubMed] [Google Scholar]
- 13.Hardy RR & Hayakawa K (1991) A developmental switch in B lymphopoiesis, Proc Natl Acad Sci U S A. 88, 11550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayakawa K, Hardy RR, Herzenberg LA & Herzenberg LA (1985) Progenitors for Ly-1 B cells are distinct from progenitors for other B cells, J Exp Med. 161, 1554–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haughton G, Arnold LW, Whitmore AC & Clarke SH (1993) B-1 cells are made, not born, Immunol Today. 14, 84–7; discussion 87-91. [DOI] [PubMed] [Google Scholar]
- 16.Kantor AB, Merrill CE, Herzenberg LA & Hillson JL (1997) An unbiased analysis of V(H)-D-J(H) sequences from B-1a, B-1b, and conventional B cells, J Immunol. 158, 1175–86. [PubMed] [Google Scholar]
- 17.Yuan J, Nguyen CK, Liu X, Kanellopoulou C & Muljo SA (2012) Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis, Science. 335, 1195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett RL, Swaroop A, Troche C & Licht JD (2017) The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer, Cold Spring Harb Perspect Med. 7, a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ 3rd, Qiao Q, Neubert TA, Xu RM, Gozani O & Reinberg D (2009) The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate, J Biol Chem. 284, 34283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morishita M, Mevius D & di Luccio E (2014) In vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and NSD3/WHSC1L, BMC Struct Biol. 14, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greer EL & Shi Y (2012) Histone methylation: a dynamic mark in health, disease and inheritance, Nat Rev Genet. 13, 343–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ & Kaneda Y (2009) A histone H3 lysine 36 trimethyltransferase links Nkx2–5 to Wolf-Hirschhorn syndrome, Nature. 460, 287–91. [DOI] [PubMed] [Google Scholar]
- 23.Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P & South ST (2014) Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn Syndrome, Eur J Hum Genet. 22, 464–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirabella F, Wu P, Wardell CP, Kaiser MF, Walker BA, Johnson DC & Morgan GJ (2013) MMSET is the key molecular target in t(4;14) myeloma, Blood Cancer J. 3, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chesi M, Nardini E, Lim RS, Smith KD, Kuehl WM & Bergsagel PL (1998) The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts, Blood. 92, 3025–34. [PubMed] [Google Scholar]
- 26.Keats JJ, Maxwell CA, Taylor BJ, Hendzel MJ, Chesi M, Bergsagel PL, Larratt LM, Mant MJ, Reiman T, Belch AR & Pilarski LM (2005) Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16;q32)-positive multiple myeloma patients, Blood. 105, 4060–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sauer B & Henderson N (1988) Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1, Proc Natl Acad Sci U S A. 85, 5166–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlake T & Bode J (1994) Use of mutated FLP recognition target (FRT) sites for the exchange of expression cassettes at defined chromosomal loci, Biochemistry. 33, 12746–51. [DOI] [PubMed] [Google Scholar]
- 29.Dymecki SM (1996) Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice, Proc Natl Acad Sci U S A. 93, 6191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwenk F, Baron U & Rajewsky K (1995) A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells, Nucleic Acids Res. 23, 5080–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R & Reth M (2006) Testing gene function early in the B cell lineage in mb1-cre mice, Proc Natl Acad Sci U S A. 103, 13789–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ & Kioussis D (2003) Transgenic mice with hematopoietic and lymphoid specific expression of Cre, Eur J Immunol. 33, 314–25. [DOI] [PubMed] [Google Scholar]
- 33.Rickert RC, Roes J & Rajewsky K (1997) B lymphocyte-specific, Cre-mediated mutagenesis in mice, Nucleic Acids Res. 25, 1317–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee TI, Johnstone SE & Young RA (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location, Nat Protoc. 1, 729–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, Wen D, Chapgier A, DeKelver RC, Miller JC, Lee YL, Boydston EA, Holmes MC, Gregory PD, Greally JM, Rafii S, Yang C, Scambler PJ, Garrick D, Gibbons RJ, Higgs DR, Cristea IM, Urnov FD, Zheng D & Allis CD (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions, Cell. 140, 678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Trapnell C, Pop M & Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome, Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorvaldsdóttir H, Robinson JT & Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration, Brief Bioinform. 14, 178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R & Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL & Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq, Nat Biotechnol. 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torres RM & Kühn R (1997) Laboratory protocols for conditional gene targeting, Oxford Uninversity Press, Oxford, New York. [Google Scholar]
- 41.Donlin LT, Andresen C, Just S, Rudensky E, Pappas CT, Kruger M, Jacobs EY, Unger A, Zieseniss A, Dobenecker MW, Voelkel T, Chait BT, Gregorio CC, Rottbauer W, Tarakhovsky A & Linke WA (2012) Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization, Genes Dev. 26, 114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhanu NV, Sidoli S & Garcia BA (2016) Histone modification profiling reveals differential signatures associated with human embryonic stem cell self-renewal and differentiation, Proteomics. 16, 448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan ZF, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR & Garcia BA (2018) EpiProfile 2.0: a computational platform for processing epi-proteomics mass spectrometry data, J Proteome Res. 17, 2533–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen HV, Dong J, Panchakshari RA, Kumar V, Alt FW & Bories JC (2017) Histone methyltransferase MMSET promotes AID-mediated DNA breaks at the donor switch region during class switch recombination, Proc Natl Acad Sci U S A. 114, E10560–E10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Li N, Yin Y, Zheng N, Min M, Lin B, Zhang L, Long X, Zhang Y, Cai Z, Zhai S, Qin J & Wang X (2018) Methyltransferase NSD2 ensures germinal center selection by promoting adhesive interactions between B cells and follicular dendritic cells, Cell Rep. 25, 3393–3404 e6. [DOI] [PubMed] [Google Scholar]
- 46.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C & Song SY (2004) Retinoic acid imprints gut-homing specificity on T cells, Immunity. 21, 527–38. [DOI] [PubMed] [Google Scholar]
- 47.Victora GD & Nussenzweig MC (2012) Germinal centers, Annu Rev Immunol. 30, 429–57. [DOI] [PubMed] [Google Scholar]
- 48.Swanson CL, Wilson TJ, Strauch P, Colonna M, Pelanda R & Torres RM (2010) Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response, J Exp Med. 207, 1485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clarke SH & McCray SK (1993) VH CDR3-dependent positive selection of murine VH12-expressing B cells in the neonate, Eur J Immunol. 23, 3327–34. [DOI] [PubMed] [Google Scholar]
- 50.Brito JL, Walker B, Jenner M, Dickens NJ, Brown NJ, Ross FM, Avramidou A, Irving JA, Gonzalez D, Davies FE & Morgan GJ (2009) MMSET deregulation affects cell cycle progression and adhesion regulons in t(4;14) myeloma plasma cells, Haematologica. 94, 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kojima M, Sone K, Oda K, Hamamoto R, Kaneko S, Oki S, Kukita A, Machino H, Honjoh H, Kawata Y, Kashiyama T, Asada K, Tanikawa M, Mori-Uchino M, Tsuruga T, Nagasaka K, Matsumoto Y, Wada-Hiraike O, Osuga Y & Fujii T (2019) The histone methyltransferase WHSC1 is regulated by EZH2 and is important for ovarian clear cell carcinoma cell proliferation, BMC Cancer. 19, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J, Yin C, Okamoto H, Mushlin H, Balgley BM, Lee CS, Yuan K, Ikejiri B, Glasker S, Vortmeyer AO, Oldfield EH, Weil RJ & Zhuang Z (2008) Identification of a novel proliferation-related protein, WHSC1 4a, in human gliomas, Neuro Oncol. 10, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu C, Jiang YH, Zhao ZL, Wu HW, Zhang L, Yang Z, Hoffman RM & Zheng JW (2019) Knockdown of histone methyltransferase WHSC1 induces apoptosis and inhibits cell proliferation and tumorigenesis in salivary adenoid cystic carcinoma, Anticancer Res. 39, 2729–2737. [DOI] [PubMed] [Google Scholar]
- 54.Baumgarth N (2016) B-1 cell heterogeneity and the regulation of natural and antigen-induced IgM production, Front Immunol. 7, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffman W, Lakkis FG & Chalasani G (2016) B cells, antibodies, and more, Clin J Am Soc Nephrol. 11, 137–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobenecker MW, Yurchenko V, Marcello J, Becker A, Rudensky E, Bahnu NV, Carrol T, Garcia BA, Rosenberg BR, Prinjha R & Tarakhovsky A (2019) Histone methyltransferase MMSET/NSD2 is essential for generation of B1 cell compartment in mice, bioRxiv, 687806. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 2. NSD2 methylates histones H3 and H4 only in the context of nucleosomes.
NSD2SET or NSD2SET Y1092A (NSD2SETmut) were incubated with purified histone H3 (H3), intact nucleosomes (N), octamers of histones (O) or mixed histones, not assembled into nucleosomes (H) in the presence of Adenosyl-L-methionine, S-[methyl-3H]. Coomassie (top) and autoradiograph (methyl-3H, bottom) are shown.
Suppl. Fig. 1. CD19Cre- mediated deletion of the SET-domain encoding exons 18 and 19 of the Nsd2 gene in splenic B cells. Southern blot analysis of DNA isolated from purified splenic B cell with the 5’ probe after BsoBI digest is shown. The 7.7 kb band corresponds to the targeted locus (floxed) and the 14 kb and the 10.9 kb bands correspond to the wild type (WT) and CD19-Cre modified Nsd2 gene (Δ), respectively. The genotype of the purified B cells is indicated above each lane. Nsd2+/+ (wild type), Nsd2loxSET/loxSET (floxed SET), CD19creNsd2loxSET/loxSET (ΔSET; CD19-cre).
Suppl. Fig. 4. B1 cells do not develop in young Nsd2ΔSET/ΔSET mice. FACS plots show relative abundance of B1a (IgMhiCD11b+CD5+) and B1b (IgMhiCD11b+ CD5−) cells in the peritoneal cavity of 3 weeks old Nsd2loxSET/loxSET control (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice. Absolute numbers of distinct lymphoid cells in the peritoneal cavity (bottom left) and percent (bottom right) are indicated. Nsd2loxSET/loxSET (WT, open symbols) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET, red symbols). Representative plots of one experiment with 4-5 mice per group are shown. Significance was determined by unpaired t-test *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Suppl. Fig. 3: NSD2 is not essential for the development and maturation of B2 cells. A) FACS dot plots show distribution of developing and mature peripheral B2 cells. The percentage of cells in Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) mice in bone marrow (BM) and spleen (SPL) is indicated. Abbreviations used in the figure: pre/pro-B cells (pre/pro), immature B cells (imm), recirculating B cells (reci), pro-B cells (pro), T cells (T), B cells (B), follicular B cells (Fol), marginal zone B cells (MZ), transitional 1 B cells (T1), and transitional 2 B cells (T2). Representative plots of more than 3 independent experiments with 3 or more mice per group are shown. B) The absolute numbers of developing and mature B2 cells in the bone marrow (top) and spleen (bottom) of Nsd2loxSET/loxSET control (white symbols) and Mb1creNsd2loxSET/loxSET (red symbols) mice are shown. Each symbol represents one mouse. The experiments were performed 3-4 times with 2-3 mice per genotype. Significance was determined by unpaired t-test *: p ≤ 0.05, **: p ≤ 0.01. C) NSD2 catalytic function is dispensable for B cell proliferation. Expression of CD86 and IAb on freshly isolated Nsd2loxSET/loxSET control (grey) and Mb1creNsd2loxSET/loxSET (red) splenic B cells in the absence (filled histograms) or presence of stimuli (open histograms). Proliferation of freshly isolated Nsd2loxSET/loxSET (WT) and Mb1creNsd2loxSET/loxSET (NSD2ΔSET) B cells in the absence (filled histograms) or presence of stimuli (open histograms) was measured by CFSE dilution. The experiments were performed 3-4 times with 2-3 mice per genotype. D) Dispensable role of NSD2 catalytic function in B cell survival. Frequencies of viable Nsd2loxSET/loxSET control (open symbols) and Mb1creNsd2loxSET/loxSET (red symbols) splenic B cells in the presence of 0 – 25 ng of recombinant BAFF. Data are means ± s.d. from 3 independent experiments.
Data Availability Statement
Sequencing data generated for this study are available through the GEO database: H3K36me2 ChIP-sequencing and RNA-Seq (accession no. GSE155689).