

Abstract
Malignant gliomas are the most lethal form of primary brain tumors. Despite advances in cancer therapy, the prognosis of glioma patients has remained poor. Cytochrome c (Cytc), an endogenous heme-based protein, holds tremendous potential to treat gliomas because of its innate capacity to trigger apoptosis. To this end, a hybrid cytochrome c-chlorotoxin (Cytc-CTX) protein was biosynthesized to enable cellular uptake of the cell impenetrable Cytc using CTX transporters. A nucleotide sequence containing 1:1 Cytc and CTX was constructed and separated by a hexahistidine-tag and an enterokinase cleavage site. The sequence was cloned into a pBTR1 plasmid, expressed in Escherichia coli, purified via 2-dimensional chromatography. The identity and size of the protein were determined by Western blot and mass spectrometry. Cytc in this soluble hybrid protein has similar structure and stability as human Cytc and the hybrid protein is endocytosed into a glioma cell line, while displaying potent cytotoxicity and a favorable therapeutic index. Its facile, low-cost, and high yield synthesis, biocompatibility, and robustness suggest that the hybrid protein is a promising candidate for antiglioma drug evaluation.
Keywords: cytochrome c-chlorotoxin, antiglioma drug, recombinant hybrid protein, apoptosis, metal-based biomolecule
Graphical Abstract

In this study, a soluble and non-nanoparticle recombinant hybrid cytochrome c-chlorotoxin protein was biosynthesized for antiglioma application. Its synthesis is easy, affordable, and yields high protein content. The protein is effectively taken up by cells and can trigger apoptosis. Being constructured from an endogenous heme-based protein and a therapeutic peptide, the Cytc-CTX hybrid is biocompatible and promising for future human use.
Introduction
Gliomas are a group of tumors that emerge from glial cells including glioblastoma, oligodendroglioma, ependymoma, oligoastrocytoma, amongst others.[1] They account for nearly 70% of the primary brain tumors.[2–3] Malignant gliomas are the third leading cause of cancer mortality for people between between 15 to 34 and result in 2.5% of the global cancer deaths.[4] The conventional method of treatment includes resection (removal of the brain tumor) followed by radio- and chemotherapy.[5–6] Despite advances in treatment methods, clinical equipment, and modern diagnostics, 90% of glioma patients display recurrence around the location of the original tumor after its surgical removal due to the invasive properties and rapid acquired resistance toward radio- and chemotherapy of glioma cells.[7–9] Moreover, the median survival is only between 3 months to 5 years after this treatment regimen.[10–12] Survival is even shorter for glioblastoma patients, roughly 12 to 15 months.[13] A common chemotherapeutic drug used in the treatment of gliomas is temozolomide but it demonstrates many adverse side effects due to a lack of specificity for the tumors[12] and increased tumorigenicity of glioma cells upon long exposure to it due, in part, to rapid acquired resistance.[5, 9] Drugs capable of specifically targeting glioma cells and minimizing resistance are highly warranted.
A molecule that holds tremendous potential to treat gliomas is cytochrome c (Cytc), an endogenous 12 kDa heme protein. It is found in the intermembrane space of mitochondria and plays an important role in cellular respiration as the penultimate protein in the electron transport chain in eukaryotic cells, responsible for transferring electrons from complex III to complex IV.[14] Another major Cytc role is maintaining cellular homeostasis through type II apoptosis.[15] Released into the cytosol, Cytc binds an adaptor protein, Apaf-1, and forms the apoptosome complex which recruits and activates procaspase-9. The latter will activate executioner caspase-3, 6 and 7 which leads to cell death.[14, 16] This property of apoptosis mediation is directly connected with the heme moiety of the protein as the heme-free (apo) form does not exhibit this capacity.[17–18] Exploiting this property has been at the center of different anticancer drug design strategies especially given that several anticancer drugs result in increased expression of the protein as part of their apoptotic mechanism of action.[19–22] A major limitation that needs to be overcome is that Cytc is unable to cross the cytoplasmic membrane.[19–20] Numerous strategies have been taken to address this issue including encapsulating the protein inside nanoparticles,[19–21] nanoparticle formulation of the protein with conjugation to receptor recognition moieties,[23–24] and conjugation of Cytc with the serum proteins transferrin[25] and albumin.[26] While effective in improving cellular uptake of Cytc, many of these strategies involve complex chemical conjugation steps and can result in nonhomogenous products. Furthermore, nanoparticle formulations for anticancer drug delivery have recently been found to possibly contribute to metastasis by a process termed nanomaterials-induced endothelial leakiness (NanEL).[27]
For the purpose of specifically targeting glioma cells, conjugating Cytc to the bioactive peptide chlorotoxin (CTX) in a non-nanoparticle form was sought. CTX is a 36 amino acid peptide (4 kDa) isolated from the venom of the Israeli scorpion Leiurus quinquestriatus. This peptide discovered, purified and characterized in the beginning of the 20th century[28–29] can cross the blood brain barrier (BBB) in mice models.[30] CTX can bind to several receptors and transporters that can facilitate its uptake into cells. It can cross the epithelial and glioma specific chloride channel[9, 31–32] and bind to the glioma specific matrix metalloproteinase-2 (MMP-2) and Annexin A2, which leads to receptor-mediated internalization.[33–35] CTX can selectively bind to glioma cells but not normal brain tissues nor brain tumor cells of non-glial origin.[9] Soroceanu et al. intravenously injected native and recombinant 125I-labeled chlorotoxin into human glioma bearing immuno-deficient mice and found it to specifically accumulate in the tumor. Lyons et al. investigated 79 biopsies of primary human brain tumors using bioactive synthetic CTX containing an N-terminal biotin. Greater than 90% of the biopsies were stained with chlorotoxin while 32 non-brain biopsies showed no or few staining sites.[32] While the peptide can induce paralysis in small insects and invertebrates, it shows no apparent toxicity when it is injected in vertebrates,[36] which bodes well for its potential clinical use in humans. CTX has been explored for various applications in cancer work.[37] A fluorescent form of CTX has been patented (US8778310B2) for cancer diagnosis/imaging. CTX is also studied for cancer therapy. 131I-CTX has reached phase II clinical trials for radiotherapy.[37] Lippard et al. have studied the potential for a CTX-conjugate of a Pt(IV) complex to facilitate targeting of cancer cells and have observed that the conjugates are more potent against cancer cells than the Pt(IV) precursors.[38] To this end, we hypothesize that CTX conjugation to Cytc will facilitate its delivery into glioma tumors. In this work, we present a facile recombinant biology approach to conjugate Cytc with CTX (Figure S1). Our His-tagged 1:1 Cytc-CTX conjugate is robust and displays potent cytotoxicity.
Results and Discussion
In order to express and purify the hybrid recombinant protein Cytc-CTX from E. coli, both nucleotide sequences for Cytc and CTX were included in a single chain, separated by a His6 tag sequence and an enterokinase cleavage sequence (DDDDK) (Figure S1) as a linker and a site to cleave off the CTX. Thus this recombinant protein can serve as an effective route for obtaining CTX alone for future applications. Cytc-CTX was purified using a 2-dimensional chromatographic approach. The His6 tag was first used to purify Cytc-CTX via a Ni(II) affinity column (Figure 1A). This was followed by gel filtration chromatography (Figure 1B). SDS PAGE (Figure 2) of the protein eluted from gel filtration over time reveals high purity protein and a reasonable mass difference of ~6 kDa between Cytc-CTX (~18 kDa) and recombinant human Cytc (~12 kDa). The highest purity batches as indicated by SDS PAGE were pooled. The final yield of purified Cytc-CTX was >12 mg per liter bacteria solution. Incidentally, this corresponds to ~3 mg/L of CTX. Recombinant human Cytc was prepared without a His6 tag and the DDDDK sequence but was purified by gel filtration chromatography. The final yield was ~8 mg/L of culture.
Figure 1.

(A) Elution profile of the cell lysate from a nickel affinity column (His-Trap HP column (5 ml). The bound protein was eluted with an increasing imidazole gradient over ~ 7 column volumes. (B) Elution profile from a Superdex 200 GL column of the combined pooled fractions from the previous His-Trap HP column step. Both profiles were monitored at 410 nm.
Figure 2.

SDS-PAGE gel of recombinant Cytc-CTX obtained from an elution profile from a Superdex 200 GL column (lanes 2–9). Lane 1 is the protein ladder and lane 10 is recombinant human Cytc (lane 10) (lane 1= Protein ladder).
Western blotting and MALDI TOF mass measurement were used to confirm the successful biosynthesis and isolation of the Cytc-CTX protein. In the Western blot experiment, the primary antibody was selected to bind the His6 tag present in the Cytc-CTX protein and this interaction was imaged through immune detection with the secondary antibody (Figure 3). Only the Cytc-CTX protein and not the recombinant human Cytc could be detected. MALDI TOF MS were collected of the intact Cytc-CTX and recombinant human Cytc proteins (Figure 4). The Cytc-CTX protein yielded a size of 17,870 Da versus a size of 12,200 Da for recombinant human Cytc alone. The mass difference of 5,700 Da is consistent with the presence of the CTX peptide (~4 kDa), the His6 tag (~1 kDa) and the enterokinase cleavage sequences (<1 kDa) in the Cytc-CTX protein.
Figure 3.

SDS-PAGE gel (A) and Western blot (B) of samples containing recombinant human Cytc (lanes 1 and 4) and Cytc-CTX (lanes 2 and 3). Only the Cytc-CTX protein is detected due to the presence of the His6 tag sequence.
Figure 4.

MALDI TOF MS of recombinant human Cytc (A) and of Cytc-CTX (B).
The Cytc-CTX protein was structurally and thermally characterized for stability using a variety of techniques and compared with recombinant human Cytc. The UV-Vis absorbance spectrum of Cytc-CTX collected under reducing conditions matches closely with that of recombinant human Cytc. It shows features of reduced mitochondrial c-type cytochrome (Figure 5A). The data indicate that the heme environment of Cytc was not changed with the inclusion of the His6-DDDDK-CTX component.
Figure 5.
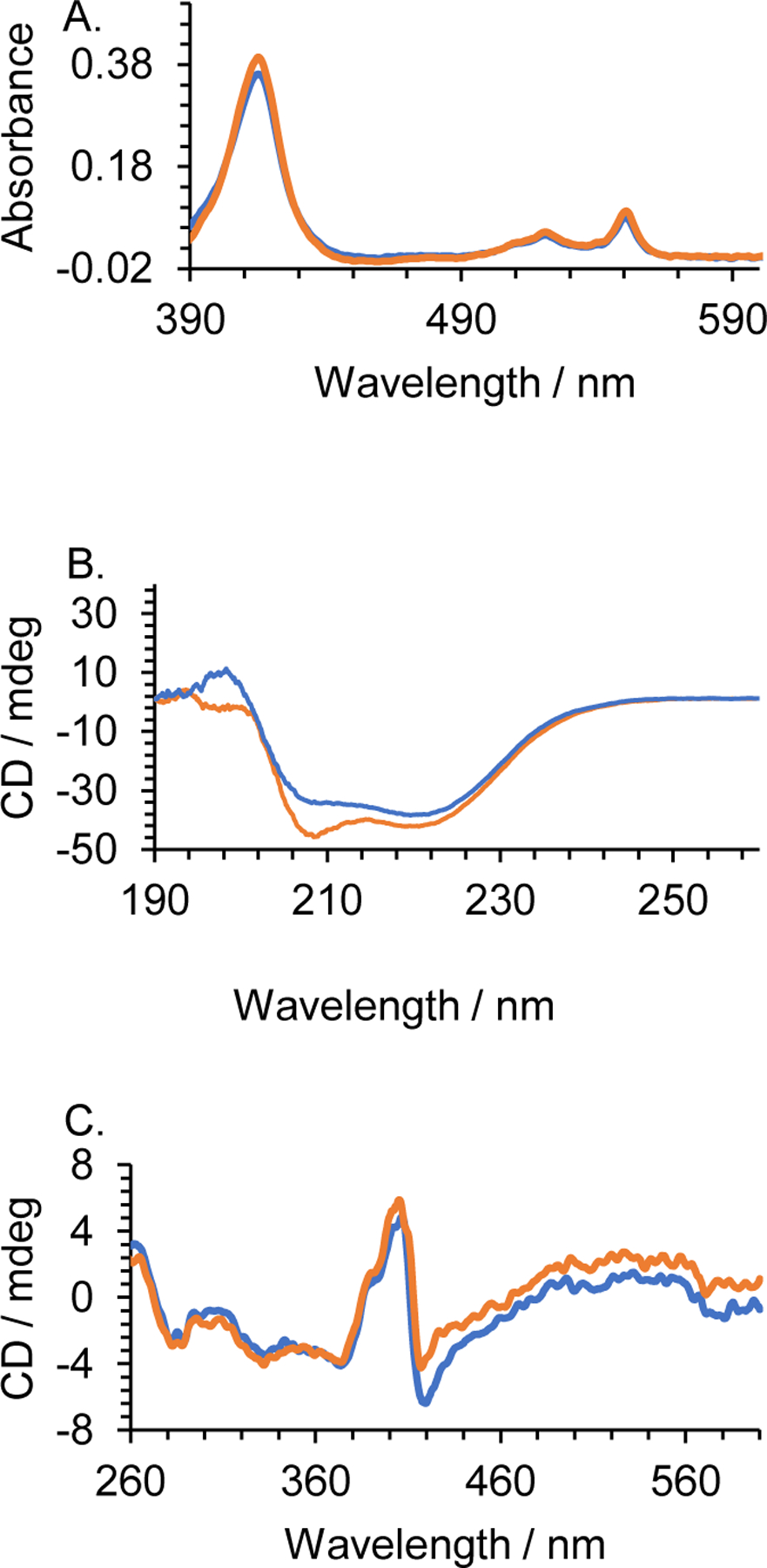
A. UV-Vis spectra of the reduced form of recombinant human Cytc (blue) and the hybrid Cytc-CTX (orange) at 30 μM. B. Far-UV CD spectra. C. Near-UV CD spectra. The CD spectra were collected at 18 μM.
The far-UV CD spectrum of reduced Cytc-CTX exhibits a similar pattern as the one of human Cytc with a slightly more negative minima observed at the 208 nm, which is a well-known alpha helix characteristic peak (Figure 5B).[39] This more pronounced minima might be due to the contribution of the alpha helix of the peptide. The ratio R= [θ]222/[θ]208 was calculated to be 1.1 and 0.92 for the human Cytc and Cytc-CTX proteins, respectively. These values are in agreement with the predominance of alpha helical content in the protein structures.[40] In the near UV CD region (Figure 5C), minima were observed at 282 and 288 nm for both reduced human Cytc and Cytc-CTX,[41] which indicate that the cytochrome c has similar secondary structure in both proteins. The bisignate band for the heme-protein interactions (Figure 5C), indicative of Cytc exhibiting native structure,[42] was observed at 405 and 419 nm in both human Cytc and Cytc-CTX. Similarity of structure does not imply that the two proteins will be identical in activity. Following the method of Pielak et al.,[39, 43] we investigated the stability of reduced human Cytc and Cytc-CTX by monitoring the change in the ellipticity at 222 nm upon heating (Figure 6). The values of the calculated thermodynamic parameters such as thermal denaturation (Tm), the van’t Hoff enthalpy (ΔHm), heat capacity (ΔCp) and the free energy of denaturation (ΔGD) of human Cytc and Cytc-CTX are tabulated in Table 1. A two-state model was adopted to fit a series of 3 data sets per protein (Figure 6). Based on the ΔGD values, Cytc-CTX is only 1.5 kcal less stable than human Cytc. These collective results demonstrate that the conjugation of Cytc to CTX via a recombinant biological approach has virtually insignificant effect on the structure and stability of the human Cytc protein.
Figure 6.

A variable temperature experiment was performed on recombinant human Cytc and Cytc-CTX. The thermal melting curves were measured by focusing on the 222 nm peak. (●) Cytc thermal denaturation data points, (▬) Cytc thermal denaturation fitting curve, (◊) Cytc-CTX thermal denaturation data points, (–) Cytc-CTX thermal denaturation fitting curve.
Table 1.
Thermodynamic parameters obtained from CD thermal melt analysis of recombinant human Cytc and Cytc-CTX. Results are presented as a mean ± SD from 3 independent experiments.
| Proteins | Tm (K) |
ΔHm (kcal mol−1) |
ΔCp (kcal mol−1 K−1) |
ΔGD (kcal mol−1) |
|---|---|---|---|---|
| Cytc-CTX | 341.5±4.3 | 81.7±17.9 | 0.2±0.2 | 9.7±2.5 |
| Cytc | 340.9±3.9 | 93.9±20.4 | 0.3±0.2 | 11.2±1.9 |
We next examined the cellular interaction of the Cytc-CTX protein. Given that gliomas are highly invasive brain tumors, CTX conjugates of different imaging/diagnostic agents have been successfully engineered to cross the blood brain barrier and accumulate in tumors.[30, 44] Furthermore, a CTX conjugate of onconase, a small cytotoxic ribonuclease having a similar molecular weight (12 kDa), charge, and iso-electric point as Cytc (pIonconase = 9.5 and pICytc = 9.6), was previously prepared by chemical crosslinking and shows significant antiproliferative effect toward glioma cells in vitro and sub-cutaneous glioblastoma tumors in nude mice bearing.[45–46] The Cytc-onconase conjugate, however, is not effectively cytotoxic.[45–46] It merely prevents tumor growth. We constructed a Cytc-CTX conjugate in a well-defined 1:1 composition to exploit the role of Cytc in triggering apoptotic cell death and the capacity of CTX to facilitate cellular uptake of Cytc. The glioma cell line 9L/Lacz and noncancerous fibroblast cell line NIH/3T3 were treated with varying concentrations of Cytc and Cytc-CTX, in addition to CTX at the highest concentration. As expected, recombinant human Cytc showed no effect on cell viability owed to its cell impermeability. Also CTX alone showed no effect on viability as was to be expected due to its general lack of toxicity against mammalian cells. The Cytc-CTX protein, however, demonstrated a dose response effect on the viabilities of both cell lines (Figure 7). The Cytc-CTX protein is more potent against the 9L/Lacz cells (IC50 = 17.4±1.2 μM) than the NIH/3 3 cells (IC50 = 21.9 ±1.1 μM). A therapeutic potential (IC50 noncancer/IC50 glioma) was calculated to be 1.3, which is promising although an even higher value is preferred. This behavior is likely owed to the high expression of MMP-2 receptors on fibroblasts cells.[47]
Figure 7.

Comparison of the effect on cell viability of the recombinant human CytC, Cytc--CTX, and CTX on 9/lacZ (A) and NIH/3T3 (B) cells. Results are presented as a mean ± SD from 8 independent experiments.
To examine whether the Cytc-CTX protein induces cytotoxicity, we explored its caspase-9 activation property in 9L/Lacz cells. This activation leads to apoptotic cell death.[14, 16] Figure 8 reveals the potent ability of the protein to activate caspase-9. Both human Cytc and horse Cytc are unable to trigger this activation, which is attributed to their lack of cell permeability and thus inability to interact with the apoptosome. To confirm that the Cytc-CTX protein is taken up by the 9L/Lacz cells, we performed fluorescent microscopy co-localization studies (Figure 9; Figure S2). The Cytc-CTX protein was labeled with the FITC dye to be able to track the protein. The FM-4-64 dye was used to evaluate the endocytotic uptake of the protein and the DAPI dye was used was used to label the nucleus. In fluorescence microscopy-based methods, diffuse cytosolic staining is considered an indication of endosomal release in contrast to exclusive punctate signal, which is indicative of endosomal entrapment.[48] The green fluorescence in the cytosol indicates the internalization of FITC-Cytc-CTX protein. Some of this signal overlaps with the red signal of the labeled endosomes, demonstrating co-localization with endosomes, but a significant amount is spread throughout the cell Figure 9; Figure S2). No background fluorescence is observed in the cells not treated with the dyes (Figure S3). It is important to note that we have previously FITC-labeled Cytc and it was not endocytosed as expected because of the absence of CTX.[25] These observations support that FITC-Cytc-CTX is taken up by the cells via receptor-mediated endocytosis and is released from the endosome into the cytosol. Similar observations have been seen in cellular delivery of Cytc via different formulations by our laboratory.[21, 23–25] However, this was the first time that we have successfully achieved this result using a facile, cost-effective, and reproducible recombinant biology approach, yielding a highly pure and homogenous product that is potently cytotoxic.
Figure 8.

Evaluating caspase-9 activation in 9L/Lacz cells by recombinant Cytc-CTX, recombinant human Cytc, and horse Cytc. Results are presented as a mean ± SD from 8 independent experiments. *** p < 0.001
Figure 9.

Intracellular colocalization of FITC-Cytc-CTX within 9L/Lacz cells after 12 hours of treatment followed by treatment with the dyes DAPI (to label the nuclei) and FM-4–64 (to label endosomes). The fluorescent signals are merged to demonstrate intracellular colocalization.
Conclusions
In this study, it is reported for the first time the biosynthesis, purification and characterization of a soluble and non-nanoparticle recombinant hybrid Cytc-CTX protein. The synthetic approach taken is facile, cost-effective, reproducible, and yields high protein content. The structure and stability of the Cytc-CTX protein is comparable to that of human Cytc alone with slight differences owed to the presence of the CTX peptide. Cytc-CTX demonstrates potent cytotoxicity against a glioma cell line, with a favorable although not ideal therapeutic index based on comparison with a noncancerous cell line. The protein is effectively taken up into cells via CTX facilitated endocytosis and operates by caspase-induced apoptotic activation. The final product is expected to be highly biocompatible because it is constructed from a nontoxic framework consisting of an endogenous heme-based protein and a peptide that is considered safe for human use. Studying this highly promising conjugate against glioma mouse models is warranted and the hope is for the conjugate to be tumor specific and retain its cytotoxic property.
Experimental Section
Materials:
Equine cytochrome c, E. coli strain BL21(DE3), tryptone, yeast extract, glycerol, imidazole, ampicillin, EDTA, lysozyme, DNase I and RNase A, NaCl, DMEM (Dulbecco’s Modified Eagle Medium), sinapinic acid, acetonitrile, Fluorescein isothiocyanate (FITC), Bradford Dye reagent, and trifluoroacetic acid were all purchased from Sigma-Aldrich, (Missouri, USA). Cell counting kit-8 was purchased from MedChemExpress (New Jersey, USA), Caspase-Glo 9 kit from Promega (Wisconsin, USA), and phenylmethylsulfonyl fluoride from Roche (Germany). 9L/Lacz gliosarcoma and NIH/3T3 cell lines were obtained from ATCC (Virginia, USA). Superdex™ 200 and Nickel(II) Histrap HP affinity columns were obtained from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). Mini-PROEAN® TGX™ precast polyacrylamide gels were purchased from BioRad (California, USA). Avestin Emulsiflex C5 homogenizer was purchased from American Lab Trade (Connecticut, USA), NucBlue Fixed Cell (DAPI), FM 4–74 (Pyridinium, 4-[6-[4-(diethylamino)phenyl]-1,3,5-hexatrienyl]-1-[3-(triethylammonio)propyl]-dibromide), iBlot 2 dry transfer system, Bolt LDS sample buffer, and Bolt reducing agent were purchased from Thermo Fisher Invitrogen Life Technologies (Massachusetts, USA). The chlorotoxin peptide was purchased from Biomatik Corporation (Ontario, Canada). High purity buffers, salts, and solvents were purchased from standard suppliers and used as received.
Instruments:
A Thermo Scientific Fiberlite F12–6×500 LEX Rotor (152 mm) and Fiberlite F14–14×50cy Rotor (154 mm) was used in a Sorvall Lynx 4000 centrifuge for pelleting. UV–Vis spectra were collected on a Thermo Scientific Nanodrop 2000/2000c spectrophotometer (0.1 cm pathlength). CD spectra were collected on a Jasco J-1500-150 CD spectrometer equipped with a six-position thermostatted sample changer. Cell plate assays were read using a Tecan Infinite M200 Pro plate reader. Mass spectra were collected using an ABSCIEX 4800 Plus MALDI TOF/OF™ Analyzer. Confocal fluorescent studies were performed using a Nikon Ti-E inverted microscope with A1R confocal microscope. Gel fitration columns were used on a GE AKTA explorer FPLC.
Gene cloning and recombinant Cytc-CTX expression:
The nucleotide sequence of the hybrid construct (made of Cytc and CTX sequences, linked by a His6 tag and enterokinase cleavage sequence (DDDDK) giving rise to Cytc-CTX) was commercially synthesized by Genscript. It was cloned into the vector pBTR1,[39] replacing the human cytochrome c sequence at the cloning sites NcoI/KpnI. The resulting construct was transformed into the E. coli strain BL21(DE3). Similarly, recombinant human Cytc was obtained from the unmodified pBTR1 vector. The protein expression method used here was adapted from a protocol we previously published in our laboratory.[49] Briefly, the bacteria growth media was prepared using 12 g of tryptone, 24 g of yeast extract, 8 mL glycerol, 2.3 g NaH2PO4 and 12.5 g Na2HPO4 and 100 mg of ampicillin for each liter. To inoculate 1 liter of the prepared media, 10 mL of bacteria solution were grown at 37 °C under continuous shaking at 220 rpm. The grown bacteria were harvested by pelleting via centrifugation using a Fiberlite F12–6×500 LEX Rotor (radius 152 mm) for 30 min at 4 °C. The recovered pellet was resuspended in lysis buffer (3 mL of 50 mM Tris-HCl, pH 6.8, 1 mM EDTA per gram of cell paste), followed by the addition of 3 g/L of lysozyme and a pinch of crystals of DNase I and RNase A. The resulting mixture was incubated overnight at 4 °C. To facilitate the cell lysis during the next step, the incubation volume mixture was diluted 3–5 times with lysis buffer followed by addition of phenylmethylsulfonyl fluoride to a final concentration of 1 mM. The latter cell suspension was passed, in two cycles, through the Avestin Emulsiflex C5 homogenizer at a pressure of 117 MPa (17000 psi). During the process, cell suspension and lysate were always maintained in an ice bath and a heat exchanger coupled to the homogenizer tubing was submerged in ice to maintain a constant temperature. The collected lysate was centrifuged at 10000 rpm using a Fiberlite F14–14×50cy Rotor (radius 154 mm) for 30 min at 4 °C to pellet and get rid of cell debris. The red-pink supernatant that contains the recombinant hybrid protein was then dialyzed overnight against 20 mM Na2HPO4, 0.5 M NaCl, 5 mM imidazole, pH 7.4 buffer solution.
Protein purification and characterization:
The dialysate of Cytc-CTX was loaded into a 5 mL Nickel(II) Histrap HP affinity column (flow rate 5 mL/min), previously stripped, then equilibrated with 20 mM Na2HPO4, 0.5 M NaCl, 5 mM imidazole, pH 7.4 buffer solution. The Cytc-CTX protein was eluted with the same buffer solution, using a gradient of 5 mM imidazole to 0.5 M imidazole with the same flow rate as the binding step. 10 kDa molecular weight cut-off filters were used to concentrate and change the buffer to 40 mM phosphate buffer (pH 6.8). The UV-Vis absorbance of the different fractions was monitored. Fractions with absorbance 410 nm/280 nm ratio higher than 2 were used in SDS-gel electrophoresis to verify their purity. The impure fractions were pooled and loaded into a superdex 200 10/300 gel-filtration column to further purify the Cytc-CTX protein. All pure fractions were pooled. A similar approach was taken for the recombinant human Cytc except that the Ni(II) affinity column was not applied because the protein does not contain a His6 sequence. All protein concentrations were determined by the Bradford assay.
UV-Vis spectra analysis.
UV-vis spectra of the reduced form of 30 μM recombinant human Cytc and Cytc-CTX were collected under reducing conditions (0.5 M ascorbate in 0.5 M MES buffer pH 7.3).
SDS-PAGE gel electrophoresis.
SDS-PAGE was achieved by running at 150 V for 1 hour, a 4–15% Mini-PRO EAN® GX™ precast polyacrylamide gel with premixed sample loading buffers (Bio-Rad, USA). 20 μL of sample were mixed with 20 μL of loading buffer and heated for 5 min at 90°C. The gel was stained with 0.02% Coomassie Brilliant Blue G-250 and washed with prepared destaining solution (ethanol, acetic acid, water: 50:40:10 (v/v/v)).
MALDI TOF mass spectrometry.
MALDI TOF mass spectrometry analyses of Cytc-CTX and human Cytc were collected in positive ion reflector mode. The samples were directly applied on a 96 well MALDI plate with a previously prepared matrix made of 10 mg/ml sinapinic acid, in 50% acetonitrile and 0.1% trifluoroacetic acid.
Protein Gels and Western Blotting.
Recombinant Cytc-CTX and recombinant human Cytc were mixed with 10 μL of Bolt LDS sample buffer and 4 μL of Bolt reducing agent. The volumes were adjusted with deionized water to a final volume of 40 μL. The different samples were heated for 10 min at 70 °C, then loaded into a 4–12% Bolt Bis/Tris gel (Invitrogen Life Technologies) and run at 150 V for 22 min. The proteins were transferred onto a nitrocellulose membrane using the iBlot 2 dry transfer system from Invitrogen Life Science at room temperature for 7 min. Primary and secondary antibody binding were performed using the iBind™ Western System (Invitrogen Life Science). 6x-His tag polyclonal antibody was the primary antibody and Anti-rabbit IgG HRP-linked antibody was the secondary antibody. Dilutions of the supplied stock antibody solutions were made according to manufacturer instructions. Immunodetection was performed with the Thermo Scientific SuperSignal West Femto Maximum Sensitivity Substrate.
Circular dichroism spectroscopy (CD).
Far-UV spectra were collected in a 0.1 cm quartz cuvette containing 18 μM protein solution of Cytc-CTX and recombinant human Cytc. Near-UV/Soret spectra were obtained in 1 cm quartz cuvette containing 18 μM of the proteins. The protein solutions were prepared in 0.5 M ascorbate in 0.5 M MES buffer (pH 7.3). A variable temperature experiment was performed on the same samples to determine the thermal melting curves of the proteins from 20 to 90 °C in 2 °C increments, monitored in the far-UV region at 222 nm. The experiment was performed in triplicate. The data were fit using the software CDpal, version 2_18.
Cell-based caspase assays:
The caspase activation property of the hybrid recombinant Cytc-CTX protein was evaluated using the Caspase-Glo 9 kit. The assays were performed as dictated by the manufacturer. Briefly, 9L/lacz cells were grown in High glucose DMEM containing 10% FBS and 1% pen-strep in an incubator at 37°C, under 5% CO2. Cells were passaged twice after 2 to 3 days per passage. On the third passage, 90 μL of DMEM containing 104 cells were seeded into wells of a 96 well plate and equilibrated overnight. The next day, 10 μL of Cytc-CTX, recombinant human Cytc, and horse Cytc solution were added per well to a final concentration of 8.1 μM and 10 μL/well (containing no Cytc or Cytc-CTX) of cell medium was used as blank. Then the cells were incubated for an additional 12 h. An n = 8 was collected per sample condition.
Prior to starting the assay, the caspase reagent was prepared as follows: the content of the bottle of the Caspase-Glo® 9 buffer was transferred into the bottle containing the Caspase-Glo® 9 substrate. The mixture was homogenized by inverting the contents until the substrate is completely dissolved to form the Caspase-Glo® 9 Reagent. The tube containing the MG-132 Inhibitor was added to the reagent and mixed. After the incubation time, the cells and kit reagents were equilibrated for 45 min at room temperature and 100 μL of the reagent were added to each well. The plate was mixed in a shaker for 30 sec at 325 rpm and were incubated for 2 h. The luminescence was then measured using a Tecan plate reader. A Student t-test was performed to determine the p-value when comparing the luminescence intensity produced by Cytc-CTX versus recombinant human Cytc and versus horse Cytc.
Cell viability assays:
Viability studies were performed in a 96 well plate using a cell counting kit-8 (CCK-8) based on the formation of a water-soluble tetrazolium salt, WST-8, that is reduced by dehydrogenase activities in cells to give a yellow-color formazan dye, which is soluble in the tissue culture media and can be monitored at 450 nm. The cell lines used for the assays were 9L/Lacz, a gliosarcoma cell line from the rat, Rattus norvegicus and NIH/3T3, a normal embryo cell line from the mouse, Mus musculus. Briefly, 9L/lacz and NIH/3T3 cell were grown to 80–90% of confluency in Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose, and 1 mM sodium pyruvate 10% fetal bovine serum (FBS), and 1% penicillin in a humidified incubator which is provided with 5% CO2 and 95% air at 37°C. 104 cells/well were seeded in a 96 wells plate in 100 μL of DMEM cell media and left to equilibrate overnight. The next day some cells were treated with varying concentrations (1–50 μM) of recombinant human Cytc and Cytc-CTX, prepared by diluting protein stock solutions (in pH 6.8, 40 mM sodium phosphate buffer) with DMEM cell media. Controls included two lanes of cells without protein samples, of which one lane was to measure background absorbance without addition of CCK-8. An additional control contained all solutions but no cell addition. An n = 8 was collected for all conditions. After equilibrating the cells with samples for 24 h, 10 μL of the CCK-8 reagent were added per well and incubate for another 2 hrs follow by the measurement of the absorbance at 450 nm and the reference absorbance at 800 nm using the Tecan plate reader. Cell viability curves were fitted with the program Origin 8.5 applying the pharmacology dose response curve (Eq. 1). The curve of best fit for the 9l/Lacz cells produced an R-square of 0.9945 and for the NIH/3T3 cells, an R-square of 0.991. The dose response curves suggested cytotoxic behavior given that viable cell levels reach or trend toward 0%.
| (eq.1) |
in which A1 is the maximum viability, A2 is the minimum viability, p is the Hill slope, x is log [protein], and LOGx0 is the IC50 value
FITC-labeling of Cytc-CTX and colocalization studies:
To determine whether Cytc-CTX exerts its toxicity by cell internalization or by binding the cytoplasmic membrane, the protein was labeled with FITC following the manufacturer protocol. Briefly, 16 μM Cytc-CTX was freshly prepared in 0.1 M carbonate buffer (pH 9.0). FITC solution (1 mg/mL in DMSO) was added dropwise into the protein solution to a volume ratio (Vprotein:VFITC) 20:1. The mixture was stirred at 4 °C for 8 hours. Unbound FITC were separated from FITC-Cytc-CTX by repeated centrifugation in a 10 kDa MW cut-off filter tube until the washes showed no fluorescence. The process was done in the dark. 9L/lacz cells were grown as described previously. 4 μM of FITC-Cytc-CTX were incubated with 104 cells for 12 hours. The media was then removed and the cells were washed 3 times with 1X PBS. The cells were then incubated with a solution of 5 μg/mL of the endosomal marker FM-4–64 for 5 min. Subsequently, cells were fixed with 4% formaldehyde and washed with Hanksʼ balanced salt solution (HBSS). Thereafter, the cells nuclei were stained with DAPI. The cells were covered with glycerol and stored in the dark until confocal visualization. The treated coverslips were examined under a Nikon Ti-E inverted microscope with A1R confocal microscope. For the analysis, FITC was excited at 495 nm and its emission was detected at 525 nm. DAPI was excited at 360 nm and its emission was detected at 460 nm. FM-4–64 was excited at 515 nm and its emission was detected at 640 nm.
Supplementary Material
Acknowledgments
We are quite grateful to the different sources of funding that supported this work. L.J.D. was supported by the UPR RP FIPI Grant from the office of the DEGI. A.D.T. was supported by the NIH 5SC1CA190504 grant and the NSF REU 1560278 (as was J.M.). H.P. was supported by the STEP-UP HS program under the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH grant R25DK098067. J.R.-P. and J.C.V.-S. were supported by the RISE program of UPR RP under the NIH 5R25GM061151-16 and 5R25GM061151-18 grants, respectively. We also thank the UPR RP Molecular Sciences Research Center (UPR RP MSRC) and Facundo Bueso facilities for instrumentation access. A special thanks is given to Bismark Madera from the MSRC facility for his help with confocal microscopy experiments. The confocal microscopy work was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under Award Number P20GM103642. We would also like to thank Silvia Planas, José A. González-Feliciano, José A. Rodríguez-Cordero, Luxène Belfeur, and Dr. José Lasalde and the members of his laboratory. An extra thanks to José Alberto Santiago Espinoza and Josué A. Benjamín-Rivera for help with the TOC figure and other contributions.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS, Neuro. Oncol 2018, 20(suppl_4), iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ohgaki H, Kleihues P, Acta Neuropathol 2005, 109(1), 93–108. [DOI] [PubMed] [Google Scholar]
- [3].Lukas RV, Wainwright DA, Ladomersky E, Sachdev S, Sonabend AM, Stupp R, Oncology 2019, 33(3), 91–100. [PMC free article] [PubMed] [Google Scholar]
- [4].Silantyev AS, Falzone L, Libra M, Gurina OI, Kardashova KS, Nikolouzakis TK, Nosyrev AE, Sutton CW, Mitsias PD, Tsatsakis A, Cells 2019, 8(8), 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].William D, Walther M, Schneider B, Linnebacher M, Classen CF, PLoS One 2018, 13(1), e0191511–e0191511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yin LT, Fu YJ, Xu QL, Yang J, Liu ZL, Liang AH, Fan XJ, Xu CG, Biochem. Biophys. Res. Commun 2007, 362(2), 225–229. [DOI] [PubMed] [Google Scholar]
- [7].Demuth T, Berens ME, J. Neurooncol 2004, 70(2), 217–228. [DOI] [PubMed] [Google Scholar]
- [8].Loeffler JS, Alexander E 3rd, Hochberg FH, Wen PY, Morris JH, Schoene WC, Siddon RL, Morse RH, Black PM, Int. J. Radiat. Oncol. Biol. Phys 1990, 19(6), 1455–1462. [DOI] [PubMed] [Google Scholar]
- [9].Soroceanu L, Gillespie Y, Khazaeli MB, Sontheimer H, Cancer Res 1998, 58(21), 4871–4879. [PubMed] [Google Scholar]
- [10].Vredenburgh JJ, Desjardins A, Herndon JE, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J, Wagner M, Bailey L, Bigner DD, Friedman AH, Friedman HS, J. Clin. Oncol 2007, 25(30), 4722–4729. [DOI] [PubMed] [Google Scholar]
- [11].Kirkpatrick JP, Sampson JH, Semin. Radiat. Oncol 2014, 24(4), 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bower M, Newlands ES, Bleehen NM, Brada M, Begent RJ, Calvert H, Colquhoun I, Lewis P, Brampton MH, Cancer Chemother. Pharmacol 1997, 40(6), 484–488. [DOI] [PubMed] [Google Scholar]
- [13].Alcedo-Guardia R, Labat E, Blas-Boria D, Vivas-Mejia PE, Curr. Mol. Med 2016. [PMC free article] [PubMed]
- [14].Lopez J, Tait SW, Br. J. Cancer 2015, 112(6), 957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ooi HK, Ma L, BMC Syst. Biol 2013, 7, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Von Ahsen O, Waterhouse NJ, Kuwana T, Newmeyer DD, Green DR, Cell Death Differ 2000, 7(12), 1192–1199. [DOI] [PubMed] [Google Scholar]
- [17].Garcia Martin A, Fearnhead H, J. Biol. Chem 2003, 277, 50834–50841. [DOI] [PubMed] [Google Scholar]
- [18].Martin AG, Nguyen J, Wells JA, Fearnhead HO, Biochem. Biophys. Res. Commun 2004, 319(3), 944–950. [DOI] [PubMed] [Google Scholar]
- [19].Slowing II, Trewyn BG, Lin VS, J. Am. Chem. Soc 2007, 129(28), 8845–8849. [DOI] [PubMed] [Google Scholar]
- [20].Santra S, Kaittanis C, Perez JM, Mol. Pharm 2010, 7(4), 1209–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Méndez J, Morales Cruz M, Delgado Y, Figueroa CM, Orellano EA, Morales M, Monteagudo A, Griebenow K, Mol. Pharm 2014, 11(1), 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sánchez-Alcázar J, Khodjakov A, Schneider E, Cancer Res 2001, 61, 1038–1044. [PubMed] [Google Scholar]
- [23].Morales-Cruz M, Cruz-Montañez A, Figueroa CM, González-Robles T, Davila J, Inyushin M, Loza-Rosas SA, Molina AM, Muñoz-Perez L, Kucheryavykh LY, Tinoco AD, Griebenow K, Mol. Pharm 2016, 13(8), 2844–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kucheryavykh YV, Davila J, Ortiz-Rivera J, Inyushin M, Almodovar L, Mayol M, Morales-Cruz M, Cruz-Montañez A, Barcelo-Bovea V, Griebenow K, Kucheryavykh LY, Biomolecules 2019, 9(4), 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saxena M, Delgado Y, Sharma RK, Sharma S, Guzmán SLPDL, Tinoco AD, Griebenow K, PLoS One 2018, 13(4), e0195542–e0195542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yeh TH, Wu FL, Shen LJ, J. Drug Target 2014, 22(6), 528–535. [DOI] [PubMed] [Google Scholar]
- [27].Peng F, Setyawati MI, Tee JK, Ding X, Wang J, Nga ME, Ho HK, Leong DT, Nat. Nanotechnol 2019, 14(3), 279–286. [DOI] [PubMed] [Google Scholar]
- [28].DeBin JA, Strichartz GR, Toxicon 1991, 29(11), 1403–1408. [DOI] [PubMed] [Google Scholar]
- [29].DeBin JA, Maggio JE, Strichartz GR, Am. J. Physiol 1993, 264(2 Pt 1), C361–369. [DOI] [PubMed] [Google Scholar]
- [30].Veiseh O, Sun C, Fang C, Bhattarai N, Gunn J, Kievit F, Du K, Pullar B, Lee D, Ellenbogen RG, Olson J, Zhang M, Cancer Res 2009, 69(15), 6200–6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Soroceanu L, Manning TJ Jr., Sontheimer H, J. Neurosci 1999, 19(14), 5942–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lyons S, O’Neal J, Sontheimer H, Glia 2002, 39, 162–173. [DOI] [PubMed] [Google Scholar]
- [33].Deshane J, Garner C, Sontheimer H, J. Biol. Chem 2003, 278, 4135–4144. [DOI] [PubMed] [Google Scholar]
- [34].Stroud MR, Hansen SJ, Olson JM, Curr. Pharm. Des 2011, 17(38), 4362–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kesavan K, Ratliff J, Johnson EW, Dahlberg W, Asara JM, Misra P, Frangioni JV, Jacoby DB, J. Biol. Chem 2010, 285(7), 4366–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dardevet L, Rani D, Aziz TAE, Bazin I, Sabatier J-M, Fadl M, Brambilla E, De Waard M, Toxins 2015, 7(4), 1079–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ojeda PG, Wang CK, Craik DJ, Biopolymers 2016, 106(1), 25–36. [DOI] [PubMed] [Google Scholar]
- [38].Graf N, Mokhtari TE, Papayannopoulos IA, Lippard SJ, J. Inorg. Biochem 2012, 110, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Olteanu A, Patel CN, Dedmon MM, Kennedy S, Linhoff MW, Minder CM, Potts PR, Deshmukh M, Pielak GJ, Biochem. Biophys. Res. Commun 2003, 312(3), 733–740. [DOI] [PubMed] [Google Scholar]
- [40].Toniolo C, Polese A, Formaggio F, Crisma M, Kamphuis J, J. Amer. Chem. Soc 1996, 118(11), 2744–2745. [Google Scholar]
- [41].Davies AM, Guillemette JG, Smith M, Greenwood C, Thurgood AG, Mauk AG, Moore GR, Biochemistry 1993, 32(20), 5431–5435. [DOI] [PubMed] [Google Scholar]
- [42].Wei W, Danielson ND, Biomacromolecules 2011, 12(2), 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cohen DS, Pielak GJ, Protein Sci 1994, 3(8), 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Veiseh M, Gabikian P, Bahrami S-B, Veiseh O, Zhang M, Hackman RC, Ravanpay AC, Stroud MR, Kusuma Y, Hansen SJ, Kwok D, Munoz NM, Sze RW, Grady WM, Greenberg NM, Ellenbogen RG, Olson JM, Cancer Res 2007, 67(14), 6882–6888. [DOI] [PubMed] [Google Scholar]
- [45].Wang X, Guo Z, Cell Biochem. Biophys 2015, 73(2), 389–392. [DOI] [PubMed] [Google Scholar]
- [46].Wang X, Guo Z, Oncol. Lett 2015, 9(3), 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Roomi M, Monterrey J, Kalinovsky T, Rath M, Niedzwiecki A, Oncol. Rep 2009, 21, 821–826. [PubMed] [Google Scholar]
- [48].Yang NJ, Hinner MJ, Methods Mol. Biol 2015, 1266, 29–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Saxena M, Sharma RK, Ramirez-Paz J, Tinoco AD, Griebenow K, BMC Biochem 2015, 16, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.