

Abstract
While many life critical reactions would be infeasibly slow without metal cofactors, a detailed understanding of how protein structure can influence catalytic activity remains elusive. Using de novo designed 3-stranded coiled coils (TRI and Grand peptides formed using a heptad repeat approach), we examine how the insertion of a three residue discontinuity, known as a stammer insert, directly adjacent to a (His)3 metal binding site alters catalytic activity. The stammer, which locally alters the twist of the helix, significantly increases copper catalyzed nitrite reductase activity (CuNiR). In contrast, the well-established zinc catalyzed carbonic anhydrase activity (p-nitrophenyl acetate, pNPA) is effectively ablated. This study illustrates for the first time how perturbation of the protein sequence using non-coordinating and non-acid base residues in the helical core can perturb metalloenzyme activity through the simple expedient of modifying the helical pitch adjacent to the catalytic center.
Keywords: Protein Design, Stammer, Coiled Coils, Cu Nitrite Reductase, Carbonic Anhydrase
Graphical Abstract
Metalloenzymes are workhorses in cells, catalyzing essential redox and non-redox transformations.[1] For this reason, significant attention has been focused on elucidating how the type of metal, its first coordination sphere structure, and the surrounding protein environment control catalytic activity.[2] Most of these studies have focused on characterizing specific metal ligands or identifying nearby residues that may serve as acid/base catalysts. Remarkably few studies directly perturb the protein structure in a designed fashion while leaving these important first and second coordination sphere residues unaltered. This is, in part, a consequence of the fact that native metalloenzyme centers often reside in highly evolved scaffolds where it is difficult to tease out the impact of specific changes due to the evolutionary baggage that accompanies native proteins. Thus, one can best probe fundamental questions pertaining to the influence of protein structure on metal-based catalysis in simplified protein constructs developed through protein design.[3]
α-Helical systems are ubiquitous throughout biology and have served as one of the favorite templates to build de novo designed systems.[4] Metal sites occur naturally in helical bundles and can be designed into simplified versions such as designed coiled coils.[5] A key feature of de novo protein design is the development of protein models that share a similar metal environment in a very dissimilar scaffold than is seen in the native enzyme. The ability to recapitulate the mechanistic requirements of enzymatic activity, therefore, is a thorough test of our understanding of these enzymes.[6] Our lab has a long-standing history of investigating the metal binding properties of three-fold symmetric metal-binding sites within three stranded coiled coils (3SCC).[7] These units are formed when sequences of 30 (TRI) or 37 (Grand, GR) amino acid long peptides containing heptad repeats assemble into parallel 3SCCs (Table 1). We first described a de novo designed carbonic anhydrase (CA) model, in which we replaced a core leucine side chain with histidine at the 23rd position of our TRI scaffold – TRIW-H.[8] Not only did this scaffold allow the recapitulation of the Zn(His)3(H2O) first coordination sphere of the native enzyme (which has a markedly different fold around the metal) but it was also able to catalyze the hydration of CO2 and the hydrolysis of esters such as p-nitrophenylacetate (pNPA). Even though CA is one of the fastest known enzymes, the simple Zn(TRIW-H)3 reached efficiencies for both hydration and esterase activities that were within a few hundred-fold of the fastest Human CAs. Not only did Zn(TRIW-H)3 show remarkably similar metal binding site geometry and rates to CAs, it significantly outperformed small molecule models in aqueous environments.
Table 1.
Sequences and apparent dissociation constants of peptides discussed in this work.
| Peptide[a] | Sequence | Zn(II) | Kd,app (M) Cu(I) | Cu(II) |
|---|---|---|---|---|
| abcdefg abcdefg abcdefg abcdefg | ||||
| TRIW | G WKALEEK LKALEEK LKALEEK LKALEEK G | N/A[b] | N/A | N/A |
| TRIW-H | G WKALEEK LKALEEK LKALEEK HKALEEK G | (6.0 ± 0.1) × 10−10[c] | (3.1 ± 0.7) × 10−9[d] | (4.0 ± 0.8) × 10−8[d] |
| TRIW-HL19A | G WKALEEK LKALEEK LKAAEEK HKALEEK G | N.D.[e] | (2.3 ± 0.7) × 10−12[f] | (1.3 ± 0.2) × 10 5[f] |
| abcdefg abcdefg abcdefg abcdefg abcdefg | ||||
| GRW | G WKALEEK LKALEEK LKALEEK LKALEEK LKALEEK G | N/A | N/A | N/A |
| GRW-H | G WKALEEK LKALEEK LKALEEK LKALEEK HKALEEK G | N.D. | N.D. | N.D. |
| abcdefg abcdefg abcdefg abcdb′c′d′efg abcdefg | ||||
| GRW27stamAELL33H | G WKALEEK LKALEEK LKALEEK LKALA E L EEK HKALEEK G | (4.2 ± 0.1) × 10−9[g] | (15 ± 3) × 10−8[g] | (7.5 ± 0.4) × 10−7[g] |
Given the high hydrolysis rates afforded by Zn(TRIW-H)3, and the idea that enzyme active sites are optimized for specific reactions, it came as a surprise that substituting the Zn with Cu allowed us to report the best non-electrode catalyzed nitrite reductase activity in aqueous solution.[9] Copper nitrite reductase (CuNiR) catalyzes the reduction of nitrite to nitric oxide. This enzyme possesses two copper sites: a Type 1 Cu electron transfer site and a Type 2 catalytic copper center (T2Cu). The T2Cu site is structurally similar to that of the Zn site from CA, with the Cu coordinated to a (His)3(OH2) environment.[10] It is remarkable that this de novo designed protein can catalyze both the hydrolytic carbonic anhydrase activity using zinc as well as the redox conversion of nitrite to nitric oxide using copper, with no other modification than metal substitution. We have thus been exploring structural and functional models of the T2Cu center from CuNiR in our TRI scaffolds.[7, 9]
It is clear from these studies that the first coordination sphere of the metal is critical for efficient catalysis, but one might conclude that incorporation of the metal into a coiled coil rather than a markedly different secondary structure was less important. For this reason, we investigated the role of amino acid substitutions both to the exterior[11] and interior[12] of the metal binding site in CuNiR activity in TRIW-H system. These studies illustrated that a two order of magnitude range in activity could be obtained through outer sphere modifications. Additionally, modifications to the interior residues demonstrated that changes to the hydrophobic packing above or below the T2Cu site modulates the NiR activity. These previous studies, as well as numerous structurally focused works on non-catalytic metal sites in 3SCC scaffolds[13] have demonstrated that modification of the packing around the metal binding site can alter the metal binding geometries as well as solvent (and substrate) accessibility.[14] Therefore, we sought new strategies to include in our de novo designs to change the packing around our catalytically active sites.
Discontinuities in the heptad repeats of α-helical coiled coils modify the layer packing or helical pitch in the core.[15] These discontinuities can be either insertions of three (or deletions of four) or insertions of four residues (or deletions of three), named stutters and stammers, respectively.[16] Stutters, stammers and other discontinuities are well established in coiled coils associated with filamentous and cytoskeletal proteins and other systems where they serve to influence the aggregation state of protein assemblies.[16c] We have commented how such inserts may be important structural determinants at putative metal binding regions within 3SCC proteins such as the ORF1p of the LINE-1 Human Retrotransposon.[13b, 17] A traditional α-helix requires 7 residues over approximately 2 turns – at 100 degrees each per residue, the helix is 20 degrees short of completing the full two turns, so the 3SCC adopts a supercoil. A stammer insert places an additional three residues in the heptad forming an additional hydrophobic contact layer in the 3SCC interior abcdb’c’d’efg pattern. This insert forces the α-helices to overwind an extra turn (10 residues over 3 turns) into a local 310-helix and thus the 3SCC bundle must supercoil even tighter. The insertion of three residues therefore changes the overall topology of the 3SCC bundle, the spacing between the Leu layers above and below the stammer insertion, and the residue packing both on the interior and exterior of the three-helix bundle. Because we have shown previously that these structural features play a significant role in metal binding and catalytic properties of our scaffolds[13b, 18], we hypothesized that metal binding sites in TRI scaffolds that contain nearby stammer insertions could enhance catalysis.
Our hypothesis was that the geometry changes upon stammer inclusion would alter substrate and solvent access to the active site, thereby changing the overall rates of our zinc catalyzed ester hydrolysis and copper catalyzed nitrite reductase activities. We designed a TRI scaffold incorporating a stammer insert immediately adjacent to the (His)3 metal binding site, GRW27stamAELL33H. This design places the stammer discontinuity as close to the metal binding site as possible – and thus we expected to observe maximal impact on steric packing and disruption to the metal binding properties and catalysis. To ensure stability, we added the stammer insert to our larger 3SCC GR scaffold, which includes an additional heptad compared to TRI (Table 1). Using the X-ray crystal structures of the 3SCC structure of GrandCoilSer (GRCS)L30H[13b], a crystallographic analogue of GRW-H, and that of the stammer region of the 3SCC domain of ORF1p[17], we created a model of our GRW27stamAELL33H peptide. The GAFF[20] with Antechamber[21] geometry minimized structure is shown in Figure 1. The model illustrates how the stammer causes a distortion of the helices in proximity to the stammer; overlays of the parent sequence with the model are shown in the SI (Figure S1). The glutamate of the stammer (AEL) points away from the helical core, salt bridging with an interstrand Lys residue and is not able to interact with the copper site or copper binding ligands directly. CD spectra and guanidinium unfolding experiments of the purified GRW27stamAELL33H showed the double well spectra characteristic of well folded and stable α-helical structure (Figure S2).
Figure 1.
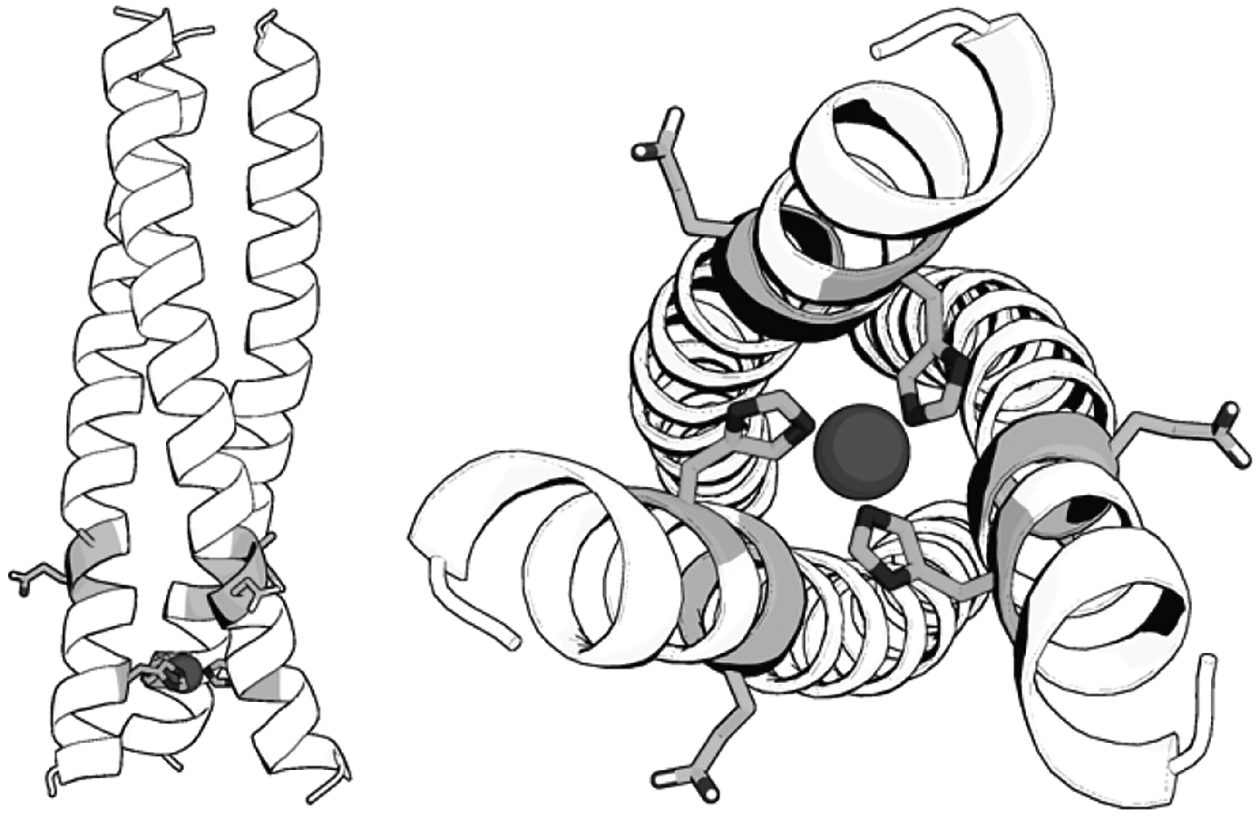
Model of GRW27stamAELL33H. The position of the stammer relative to the metal site (Left) and looking from the N-termini of the 3SCC (Right). The model was created by inserting the stammer (residues 91–93) of ORF1p CCD (6FIA[17]) into the 5KB0[13b] structure of GRCSL16C30H to create a chimeric structural model (complete details including colored representations are provided in the SI). The sidechains of Ala27 and Glu28 of the stammer and the zinc-coordinating His33 are shown as shaded sticks.
The metal binding affinities of GRW27stamAELL33H for Cu(I), Cu(II) and Zn(II) were determined using competitive titration with bathocuproine sulfonate (BCS), tryptophan quenching, and competition with zincon, respectively (Table 1 and Figure S3). Each of these metals bind to GRW27stamAELL33H with sub-micromolar apparent Kd values at the pH’s of the catalytic assays, indicating that the peptides are fully metalated under the conditions of the catalytic assays and other experiments.
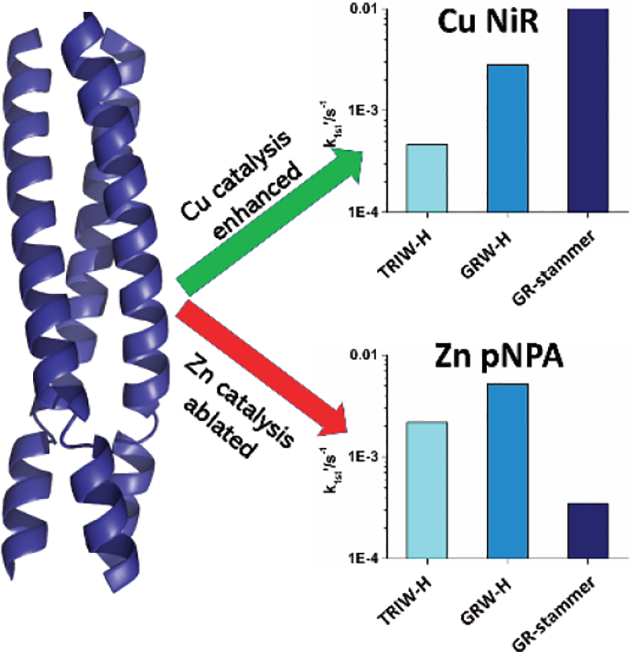
The CuNiR activity (as pseudo-first order rate constants) of GRW27stamAELL33H and GRW-H were measured according to established procedures at pH 5.8 and these rates are shown in Figure 2 and Table 2 in comparison with NiR rates we have previously obtained using our TRIW scaffold. The addition of the extra heptad to GRW-H from TRIW-H showed approximately a six-fold increased NiR rate (4.7 × 10−4 s−1 for TRIW-H vs. 2.8 × 10−3 s−1). The addition of the stammer insert showed an even higher enhancement to the NiR rate with approximately 4-fold increase over GRW-H (1.1 × 10−2 s−1) which is also within 3-fold of our highest rate using only natural amino acids in TRIW-HL19A (Figure 1). This peptide had a second sphere mutation within the interior of the 3SCC, allowing for increased solvent and substrate access adjacent to the His3 site with no change to the helical pitch.
Figure 2.
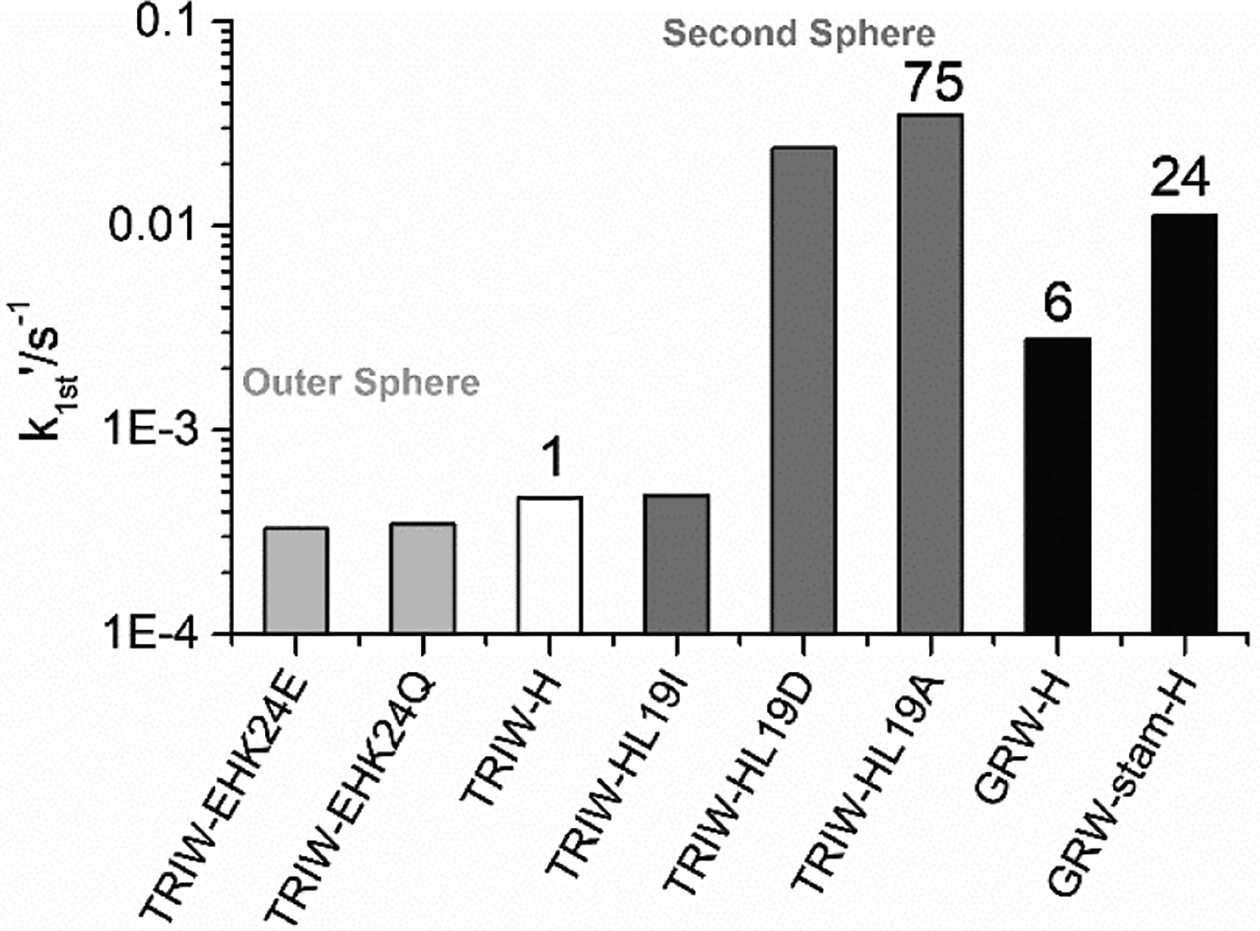
Pseudo-first order rate constants for Cu catalyzed NiR activity at pH 5.8 for the original construct TRIW-H (white)[9], previously described modifications to the exterior (light grey)[11] and interior (dark grey)[12] compared to those determined for GRW-H and GRW27stamAELL33H (black).
Table 2.
Pseudo-first order rate constants for zinc catalyzed pNPA hydrolysis at pH 9.5 and Cu catalyzed NiR activity at pH 5.8.
| Construct | CuNiR (×10−4 s−1) | pNPA rate (×10−4 s−1) |
|---|---|---|
| TRIW-H | 4.7 | 22 |
| GRW-H | 28 | 52 |
| GRW27stamAELL33H | 112 | 3.5 |
As we observed enhancement of the CuNiR activity, we hypothesized that we might observe a similar enhancement to zinc catalyzed pNPA hydrolysis and determined the esterase activity of both the longer scaffold GRW-H and the stammer GRW27stamAELL33H at pH 9.5. As with the CuNiR activity, we observed a significant increase (~2.3x) in the rate of pNPA hydrolysis with the elongated scaffold over the parent TRIW-H. However, we measured a significantly reduced rate (nearly 15-fold) for the 3SCC with the stammer insert compared to GRW-H (Table 2); this activity is nearly completely ablated, with esterase activity close to background levels. That we saw an increase in the rate with the elongated scaffold, but a decrease with the addition of the stammer indicates that this decrease in rate is entirely due to the inclusion of the stammer insert and not changes to the overall helical stability by addition of the extra heptad. Thus, the addition of the stammer insert inhibits pNPA activity while enhancing CuNiR activity.
To characterize the structural differences between these metal binding sites further, we collected X-ray absorption spectroscopic (XAS) data for GRW27stamAELL33H to compare to those we have previously described for our TRI constructs (Table 3 and S1 and Figure S4). These data were processed and fit to models to determine the best fitting coordination mode for each of the metals at the pH of the respective activity assay. It is important to note that numerous models fit the EXAFS data satisfactorily as demonstrated in the SI. In all cases, the metals are coordinated with 2–3 of the histidine residues at typical M-N distances. As with our earlier analysis on TRIW-H mutants, we observe an increase in the best fit coordination number upon Cu oxidation in the more open helical interface of GRW27stamAELL33H. That is, by including the stammer insert above the metal binding site, we have reduced the steric packing above the copper, permitting Cu(I) to adopt a more linear geometry. This observation is significant as it suggests that this more open complex could allow for better substrate access. The Cu(I) pre-edge feature, whose intensity is dependent on coordination number, of GRW27stamAELL33H has similar intensity to TRIW-HL19A which we proposed to have reduced steric packing (L19A mutation is less sterically demanding, opening space for solvent or substrate above the copper) and has been assigned as two-coordinate (Figure 3). The GRW27stamAELL33H similarly altered steric packing. We propose this effect to be the result of changes to the pitch and packing of residues around the copper binding site due to the stammer insert distorting that local geometry (Figure 1 and S1). We have previously shown that higher NiR rates also show a slight increase in the peak energy of this feature, and again this trend is preserved, with the GRW27stamAELL33H showing both a higher intensity (more indicative of a two coordinate species) as well as a shift to higher energy compared to TRIW-H and in agreement with TRIW-H L19A.
Table 3.
GRW27stamAELL33H EXAFS fitting parameters.
| Metal | pH | Model | M-O R (Å) | M-O σ2 × 10−3 (Å2) | M-N R (Å) | M-N σ2 × 10−3 (Å2) |
|---|---|---|---|---|---|---|
| Cu(I) | 5.8 | CuHis2 | 1.89 | 7.3 | ||
| Cu(II) | 5.8 | CuHis2O | 1.91 | 1.5 | 1.95 | 7.4 |
| Zn(II) | 9.5 | ZnHis3O | 1.99 | 3.2 | 1.98 | 10.6 |
Figure 3.
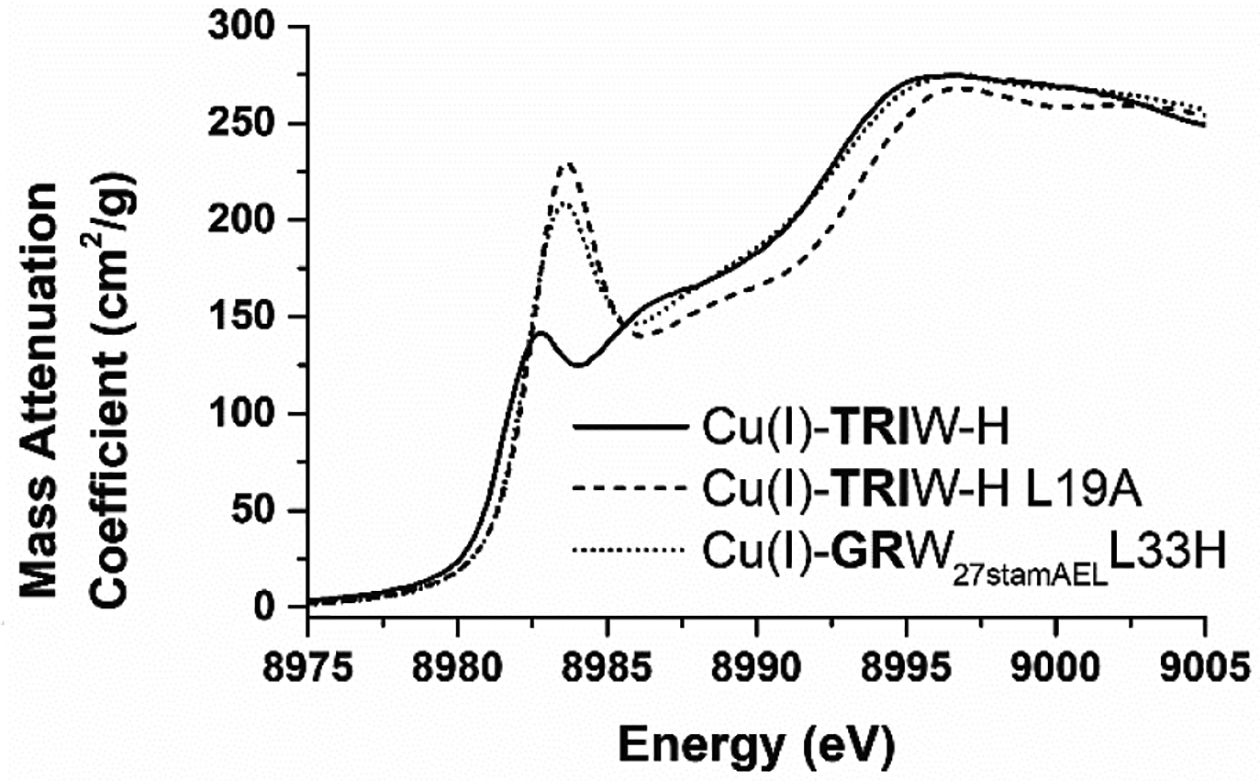
XANES of Cu(I) bound GRW27stamAELL33H compared to the parent TRIW-H and our His3 construct with the highest CuNiR activity using only naturally occurring amino acids, TRIW HL19A.
The results from XAS suggest that the insertion of the stammer in the 3SCC above the (His)3 metal binding site has altered the structure of the packing around the (His)3 site. This is consistent with previous studies showing that α-helix discontinuities such as stammer inserts alter the packing of α-helices.[16c] Thus, we observed a lower coordination number with the GRW27stamAELL33H permitting the Cu(I) to adopt the preferred linear geometry. The Cu(II) XAS data demonstrate that coordination geometry from the protein is likely preserved with the increase in coordination number due to binding of water to the metal upon oxidation. EPR collected for Cu(II) bound GRW27stamAELL33H provides additional evidence to support the Cu geometry of GRW27stamAELL33H, which is similar to that of other 3SCC CuNiR catalysts we have reported with g-tensor and parallel hyperfine coupling constant values matching those of Cu(II)TRIW-H (Table S2 and Figure S5). Finally, the Zn(II) XAS show stereotypical four-coordinate His3O with the imidazole nitrogens and oxygen bound at ~2 Å. Taken together, these XAS data show that the metals are bound in typical coordination geometries in GRW27stamAELL33H and suggest that the enhancement of the CuNiR rate is due to secondary coordination sphere effects, while zinc is bound with the correct geometry to perform pNPA esterase activity, but no trend in this data suggests a reason for the decreased activity. At this point, it is worth considering why we see a marked increase in activity for the Cu system while a significant decrease in esterase behavior is observed for Zn. For the redox active copper, it appears that the Cu(II) structure is retained; however, in the reduced form, the increased twisting afforded by the stammer appears to shift the structural preference more toward a 2-coordinate system that has been associated previously with higher catalytic activity. In this case, the over-winding appears to alter the reduced state geometry sufficiently to enhance reactivity and, since nitrite is a relatively small substrate, the tighter rotation does not block substrate access. In contrast, the Zn(II) coordination pseudo tetrahedral geometry appears to be retained. This suggests that the over-rotation of the helices either discriminates against an intermediate of higher coordination number, blocks access of the sterically bulkier pNPA molecule to the Zn(II), or a combination of these two factors. We can conclude that the simple insertion of three amino acids to our generic sequence can influence either first or second coordination sphere properties that may enhance or ablate catalytic activities. Future work will investigate the parameters and effects of stammer and stutter inserts and deletions on reactivity for development towards improved de novo designed 3SCC catalysts.
Supplementary Material
Acknowledgements
We thank Winston Pitts for the Cu(II) visible spectral data and helpful discussion. T.B.J.P. acknowledges the Natural Sciences and Engineering Research Council of Canada for support in the form of a Postdoctoral Fellowship. V.L.P thanks the National Institutes of Health for financial support of this research (ES012236). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Andreini C, Bertini I, Rosato A, Acc. Chem. Res 2009, 42, 1471–1479; [DOI] [PubMed] [Google Scholar]; b) Buccella D, Lim MH, Morrow JR, Metals in Biology: From Metallomics to Trafficking, ACS Publications, 2019; [DOI] [PubMed] [Google Scholar]; c) Maret W, Int. J. Mol. Sci 2016, 17, 66; [Google Scholar]; d) Crichton RR, Biological inorganic chemistry: a new introduction to molecular structure and function, Elsevier, 2012; [Google Scholar]; e) Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y, Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Dudev T, Lim C, Annu. Rev. Biophys 2008, 37, 97–116; [DOI] [PubMed] [Google Scholar]; b) Hosseinzadeh P, Lu Y, Biochim. Biophys. Acta 2016, 1857, 557–581; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Riordan J, Vallee B, in Protein- Metal Interactions, Springer, 1974, pp. 33–57; [Google Scholar]; d) Waldron KJ, Rutherford JC, Ford D, Robinson NJ, Nature 2009, 460, 823; [DOI] [PubMed] [Google Scholar]; e) Waldron KJ, Robinson NJ, Nat. Rev. Microbiol 2009, 7, 25–35; [DOI] [PubMed] [Google Scholar]; f) Lu Y, Berry SM, Pfister TD, Chem. Rev 2001, 101, 3047–3080. [DOI] [PubMed] [Google Scholar]
- [3].a) Lu Y, Yeung N, Sieracki N, Marshall NM, Nature 2009, 460, 855; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Korendovych IV, DeGrado WF, Q. Rev. Biophys 2020, 53; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Dawson WM, Rhys GG, Woolfson DN, Curr. Opin. Chem. Biol 2019, 52, 102–111. [DOI] [PubMed] [Google Scholar]
- [4].a) Hodges RS, Biochem. Cell Biol 1996, 74, 133–154; [DOI] [PubMed] [Google Scholar]; b) Cohen C, Parry DA, Proteins: Struct. Funct. Bioinform 1990, 7, 1–15; [DOI] [PubMed] [Google Scholar]; c) Woolfson DN, in Fibrous Proteins: Structures and Mechanisms, Springer, 2017, pp. 35–61. [Google Scholar]
- [5].a) Peacock AF, Curr. Opin. Chem. Biol 2013, 17, 934–939; [DOI] [PubMed] [Google Scholar]; b) Masuda T, Goto F, Yoshihara T, Mikami B, J. Biol. Chem 2010, 285, 4049–4059; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Moutevelis E, Woolfson DN, J. Mol. Biol 2009, 385, 726–732; [DOI] [PubMed] [Google Scholar]; d) Eshaghi S, Niegowski D, Kohl A, Molina DM, Lesley SA, Nordlund P, Science 2006, 313, 354–357. [DOI] [PubMed] [Google Scholar]
- [6].Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL, Chem. Rev 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pinter TBJ, Koebke KJ, Pecoraro VL, Angew. Chem. Int. Ed 2020, 59, 7678–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL, Nat. Chem 2012, 4, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL, Proc. Natl. Acad. Sci. U. S. A 2012, 109, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Godden J, Turley S, Teller DC, Adman ET, Liu M, Payne W, LeGall J, Science 1991, 253, 438–442. [DOI] [PubMed] [Google Scholar]
- [11].Yu F, Penner-Hahn JE, Pecoraro VL, J. Am. Chem. Soc 2013, 135, 18096–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Koebke KJ, Yu F, Salerno E, Van Stappen C, Tebo AG, Penner-Hahn JE, Pecoraro VL, Angew. Chem. Int. Ed 2018, 57, 3954–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Pecoraro VL, Ruckthong L, Stuckey JA, Chem. Eur. J 2019; [Google Scholar]; b) Ruckthong L, Zastrow ML, Stuckey JA, Pecoraro VL, J. Am. Chem. Soc 2016, 138, 11979–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Lombardi A, Pirro F, Maglio O, Chino M, DeGrado WF, Acc. Chem. Res 2019, 52, 1148–1159; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nastri F, D’Alonzo D, Leone L, Zambrano G, Pavone V, Lombardi A, Trends Biochem. Sci [DOI] [PubMed] [Google Scholar]
- [15].Brown JH, Cohen C, Parry DA, Proteins: Struct. Funct. Bioinform 1996, 26, 134–145. [DOI] [PubMed] [Google Scholar]
- [16].a) Lupas A, Trends Biochem. Sci 1996, 21, 375–382; [PubMed] [Google Scholar]; b) Gruber M, Lupas AN, Trends Biochem. Sci 2003, 28, 679–685; [DOI] [PubMed] [Google Scholar]; c) Lupas AN, Bassler J, Trends Biochem. Sci 2017, 42, 130–140. [DOI] [PubMed] [Google Scholar]
- [17].Khazina E, Truffault V, Büttner R, Schmidt S, Coles M, Weichenrieder O, Nat. Struct. Mol. Biol 2011, 18, 1006–1014. [DOI] [PubMed] [Google Scholar]
- [18].a) Lee K-H, Matzapetakis M, Mitra S, Marsh ENG, Pecoraro VL, J. Am. Chem. Soc 2004, 126, 9178–9179; [DOI] [PubMed] [Google Scholar]; b) Lee KH, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL, Angew. Chem. Int. Ed 2006, 45, 2864–2868; [DOI] [PubMed] [Google Scholar]; c) Ruckthong L, Peacock AFA, Pascoe CE, Hemmingsen L, Stuckey JA, Pecoraro VL, Chem. Eur. J 2017, 23, 8232–8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yu F, Doctoral thesis, University of Michigan; (Ann Arbor, Michigan, USA: ), 2014. [Google Scholar]
- [20].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- [21].Wang J, Wang W, Kollman PA, Case DA, Mol J. Graphics Modell. 2006, 25, 247–260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.