

Abstract
Background:
Deep brain stimulation (DBS) is an effective therapy for reducing the motor symptoms of Parkinson’s disease, but the mechanisms of action of DBS and neural correlates of symptoms remain unknown.
Objective:
To use the neural response to DBS to reveal connectivity of neural circuits and interactions between groups of neurons as potential mechanisms for DBS.
Methods:
We recorded activity evoked by DBS of the subthalamic nucleus (STN) in humans with Parkinson’s disease. In follow up experiments we also simultaneously recorded activity in the contralateral STN or the ipsilateral globus pallidus from both internal (GPi) and external (GPe) segments.
Results:
DBS local evoked potentials (DLEPs) were stereotyped across subjects, and a biophysical model of reciprocal connections between the STN and the GPe recreated DLEPs. Simultaneous STN and GP recordings during STN DBS demonstrate that DBS evoked potentials were present throughout the basal ganglia and confirmed that DLEPs arose from the reciprocal connections between the STN and GPe. The shape and amplitude of the DLEPs were dependent on the frequency and duration of DBS and were correlated with resting beta band oscillations. In the frequency domain, DLEPs appeared as a 350 Hz high frequency oscillation (HFO) independent of the frequency of DBS.
Conclusions:
DBS evoked potentials suggest that the intrinsic dynamics of the STN and GP are highly interlinked and may provide a promising new biomarker for adaptive DBS.
Introduction
Evoked potentials are generated by neural activity in response to an applied stimulus and can be used to probe neural connectivity and function [1]. For example, visual evoked potentials recorded over the occipital cortex reveal important characteristics of visual neural circuits [2], and somatosensory evoked potentials assess the integrity of spinal cord circuits [3]. Similarly, evoked potentials generated by electrical stimulation of either the auditory nerve or spinal cord - compound action potentials - can be used to assess nerve integrity and guide selection of stimulation parameters [4, 5]. We characterized the evoked potentials generated by deep brain stimulation (DBS) in humans, and subsequently combined computational modeling and additional measurements in humans to determine the neural elements generating the evoked potentials.
DBS is used to treat a variety of neurological disorders and is an effective therapy for the cardinal motor symptoms of PD [6–8], but the underlying mechanisms of action of DBS remain unclear. One of the challenges to deciphering the mechanisms of DBS is that the neural elements mediating symptom relief are not defined [8]. DBS is typically delivered in subcortical nuclei to treat movement disorders, and although DBS evoked potentials have been recorded in the cortex [9, 10], recordings in the stimulated nucleus are rare due to the challenges of large stimulation artifacts saturating amplifiers and obscuring the underlying neural signal [11, 12]. Subcortical DBS evoked potentials appear to be correlated with improvement in UPDRS score [11, 13], but the origin of the signal, while clearly neural [14], remains elusive.
We implemented a novel amplifier configuration [15] and recorded short-latency evoked potentials in the STN of patients with PD undergoing STN DBS, termed DBS local evoked potentials (DLEPs). We used a computational model to determine the underlying neural elements that can reproduce DLEPs. We subsequently tested the predictions of the model with simultaneous recordings from the STN and GP during STN DBS in additional patients with PD. Finally, we determined that DLEPs were correlated with pathological oscillatory neural activity in the basal ganglia and this suggests that they may have utility as feedback signals for closed-loop DBS [16]. Some results were presented previously in abstract form [17].
Materials and Methods
Human Participant Information
The Duke University IRB approved all protocols, and all participants provided written informed consent. Persons with PD undergoing DBS lead implantation in the STN were recruited to participate in the study on a volunteer basis. Nine participants were included in the analysis of STN data, two participants in bilateral STN data, and six participants in simultaneous STN and GP data (Table 1). Participants withheld PD medications for 12 h prior to surgery, and short-acting sedation was administered as part of routine medical care but ceased at least 60 min prior to research activities. Participants were assessed for tremor or bradykinesia during preoperative care. Spontaneous beta band local field potentials from 7 Participants (1, 3, 4, 5, 8, 10, and 11) were analyzed for burst activity and previously reported [18].
Table 1.
Subject Information
| Subject | Cohort | Age - Gender | Target | AMP (V) | PW (μs) | SD (μs) | ED (μs) | Stimulation Contact |
|---|---|---|---|---|---|---|---|---|
| 1 | Unilateral STN | 70 - M | L-STN | 3 | 90 | 20 | 500 | 2 |
| 2 | Unilateral STN | 67 - F | L-STN | 3 | 90 | 20 | 500 | 1 |
| 3 | Unilateral STN | 58 - M | L-STN | 2.5 | 90 | 20 | 500 | 1 |
| 4 | Unilateral STN | 56 - M | L-STN | 2 | 90 | 20 | 500 | 1 |
| 5 | Unilateral STN | 71 - F | R-STN | 2 | 90 | 20 | 400 | 1 |
| 6 | Unilateral STN | 72 - M | L-STN | 2.5 | 90 | 20 | 500 | 2 |
| 7 | Unilateral STN | 62 - M | L-STN | 3 | 90 | 20 | 20 | 1 |
| 8 | Unilateral STN | 71 - M | R-STN | 1.5 | 90 | 20 | 100 | 1 |
| 9 | Unilateral STN | 71 - M | R-STN | 2.5 | 90 | 20 | 100 | 2 |
| 10 | Bilateral STN | 62 - M | L/R-STN | 1.5/1 | 60/60 | 20 | 300/100 | 1/1 |
| 11 | Bilateral STN | 64 - M | L-STN | 2 | 90 | 20 | 100 | 1 |
| 12 | STN + GP | 68 - M | L-STN | 1.25 | 90 | 20 | 300 | 1 |
| 13 | STN + GP | 66 - M | L-STN | 3.5 | 60 | 20 | 300 | 1 |
| 14 | STN + GP | 65 - M | L/R-STN | 2.5/2.5 | 60/60 | 20 | 400/300 | 1/1 |
| 15 | STN + GP | 61 - M | L/R-STN | 2/2 | 60/60 | 20 | 200/300 | 1/1 |
| 16 | STN + GP | 55 - F | L/R-STN | 3/3 | 60/60 | 20 | 400/200 | 2/2 |
| 17 | STN+GP | 65 - M | L/R-STN | 3.5/4.5 | 60/60 | 20 | 200/200 | 1/1 |
Participants where both STN were stimulated are written as Left/Right. M: male; F: female; L: left; R: right; STN: subthalamic nucleus; GP: Globus Pallidus; AMP: stimulation amplitude; SD: blanking start delay; ED: blanking end delay.
Participants were recruited to one of three cohorts depending on the year of surgery. For the unilateral STN cohort, all participants received a Medtronic 3387 (1.27 mm diameter, 1.5 mm contacts, 1.5 mm inter-contact spacing) lead placed in the STN confirmed by microelectrode recordings and postoperative CT, as was standard clinical practice for 2014. In 2018, the simultaneous bilateral STN cohort, all participants received a Medtronic 3389 (1.27 mm diameter, 1.5 mm contacts, 0.5 mm inter-contact spacing) lead placed in the STN of both hemispheres, confirmed by microelectrode array recordings and intraoperative as well as post-operative CT. In 2019–2020, the simultaneous STN + GP cohort, all participants received Medtronic 3389 leads placed in the STN of both hemispheres, confirmed by microelectrode array recordings and post-operative CT. The border between GPi/e was primarily localized by microelectrode recordings. Typical firing activity of GPe cells (so called “pausers” and “bursters”) was identified. While proceeding ventrally (i.e., deeper), there was an approximately 0.5 mm region with limited activity followed by higher amplitude activity indicative of a dense cell population. Approximately 4–5 mm below the border of GPi/e the ventral edge of GPi was located by limited activity, closer to the base of the frontal lobe. We recorded differentially between the most ventral contacts 0 and 1 for putative GPi and most dorsal contacts 2 and 3 for putative GPe recordings. All simultaneous STN + GP participants, except participant 12, had a Medtronic 3387 lead placed in the GP verified by microelectrode recording and post-operative CT. Participant 12 had a Medtronic 3389 lead placed temporarily in the GP using stereotactic technique without microelectrode recordings or post-operative CT, and this temporary GP lead was removed after research was completed.
Experimental Design
The intraoperative research protocol began after completion of microelectrode recordings, DBS lead placement and clinical assessment of therapeutic benefit by the attending neurologist. A sterile connection was made between the DBS lead and the stimulation and recording equipment. A counter-electrode was placed on the chest to simulate the conditions of monopolar stimulation where the implantable pulse generator case acts as the return. DBS pulse trains were generated by the DAQ used for digitization (USB6216, National Instruments, Austin, TX) and isolated using a bp isolator (FHC, Bowdoin, ME). Symmetric biphasic pulses were delivered through one of the two middle contacts on the DBS electrode at an amplitude and pulse width sufficient for therapeutic benefit as determined by the attending neurologist. One of three attending neurologists was present during the participants surgical procedures, and the assessment of clinical responses to stimulation generally started with tremor first, if present, followed by rigidity, and finally bradykinesia. Pulse width was typically held at either 60 or 90 μs, and the stimulation amplitude was varied to determine the therapeutic window. This clinically derived, safe, stimulation amplitude was then using for the subsequent research. During research stimulation, one of the middle contacts was used in monopolar configuration, flanked by a pair of contacts for differential recording, as this configuration greatly reduces the DBS artifact [19]. In the unilateral STN cohort, those considered tremor dominant (five of nine) received four different DBS frequencies (5, 20, 45, and 130 Hz) for 60 s each with interleaved 60 s trials of DBS OFF. The four participants considered bradykinesia dominant received only 45 Hz and 130 Hz DBS for 300 s each with interleaved 300 s trials of DBS OFF, as the slower response kinetics of bradykinesia required longer trial durations. We applied 185 Hz, a frequency commonly used in clinical DBS, in the bilateral STN cohort. For the STN + GP cohort, we applied 60 s trials 130 Hz DBS for therapeutic stimulation and either 10, 20 or 50 Hz for low frequency DBS, with interleaved 60 s trials of DBS OFF (except Participant 13 who received 120 s of DBS ON and OFF while recording from left GPe only). Alternating polarity stimulation (anodic phase first followed by cathodic phase first) was delivered in Participant 5, and to all participants in the bilateral STN cohort and the STN + GP cohort (see Supplemental Methods).
Differential recordings were made between the two contacts flanking the monopolar stimulation contact. The lead implant cannula or burr hole retractor (Participant 5) served as the reference for the recorded signals. The recording instrumentation was developed to record evoked potentials in the presence of DBS [15]. Briefly, the signal was passed through battery-powered amplifiers (SR560, Stanford Research System) with diode clamps between the amplification stages to limit stimulation artifact amplitude (at ±700 mV). The second and third stage amplifiers were blanked 20 μs before, during the pulse, and 20–500 μs after each stimulation pulse to enable greater signal amplification. The sampling rate for the unilateral STN cohort was 100 kHz. For the simultaneous bilateral STN cohort, LFPs were amplified by two biopotential amplifiers with ±190 mV diode clipping between stages and digitized at 50 kHz. The second stage amplifier was blanked as described above. For the simultaneous STN + GP cohort, signals were digitized with the same equipment and parameters as the bilateral STN cohort. During intraoperative data collection a single side was recorded at a time. After data collection on the first side, the STN and GP leads were placed for the second side complete with microelectrode and clinical testing before intraoperative data collection on the second side.
LFP and DLEP signal processing
All data were analyzed in MATLAB 2019a (MathWorks, Natick, MA) using custom scripts. Power spectral density was calculated by Welch’s method using the either the initial or final portions of the recording (as labelled in the figures) using 8 segments and 50 % overlap. Resting beta band power was then calculated by integrating over 12 – 35 Hz of the final 20 s of the resting (DBS OFF) recording preceding the stimulation trial. The last 20 s were selected to isolate LFP with the least residual effect of the previous trial of DBS and best capture the untreated state. High frequency power was calculated by integrating from 200 – 400 Hz after blanking power in a 4 Hz band around each harmonic of the DBS frequency (e.g. 258 – 262 Hz and 388 – 392 Hz for 130 Hz DBS). High frequency power was calculated in 10 s windows at the start of DBS (0 – 10 s) and after 1 minute (50 – 60 s) of continued DBS to align with similar analysis of the DLEPs.
Evoked responses were inverted (polarity of differential contacts changed) for several participants to align to the responses of the entire cohort for that brain region. For visualization purposes, we subtracted the mean value of each evoked response during the period not contaminated by residual stimulation artifact (2 ms – 7.68 ms, in the case of 130 Hz). The mean across responses was then taken to produce an average template for each 1 s bin and each 10 s bin. The mean responses were low pass filtered at 1 kHz (non-causal, Butterworth, 3rd order) and a peak finding algorithm (MATLAB’s findpeaks()) was used to identify the positive and negative peaks between 3.2 ms after the stimulation pulse and the beginning of the following pulse. The peak-to-peak amplitude of each period of the DLEP was calculated by the Pi potential minus the Ni potential (i.e., VP1 – VN1 for the first period). The instantaneous frequency of each period was calculated as 1 divided by twice the time from Pi to Ni. The peak-to-peak values and latency of the templates for 0 – 10 s were then compared to those for 50 – 60 s using Wilcoxon signed rank test. Best fit lines were calculated using the fit function in MATLAB with the poly1 fit type.
Computational Modeling
Modeling was conducted in NEURON (7.2) [20]. The model contained biophysically-based representations of STN and GPe neurons as well as afferent cortical axons representing the hyperdirect pathway to STN. Model subthalamic (Accession: 151460) and pallidal (Accession: 114685) neurons were retrieved from ModelDB (http://senselab.med.yale.edu/modeldb/). These models contain realistic geometry, ion channels, and membrane properties that were derived from experimental studies [21, 22]. Axons from both cell types were modified to be straight and have lengths corresponding to the center-to-center-distance between STN and GPe in humans (~1.2 cm). Axon diameters were also increased to speed conduction velocity to match experimental conduction times (~1–1.2 ms) [23]. Hyperdirect pathway axons were the same diameter (2.5 μm) as STN and GPe axons but modeled as 1.2 cm straight axon segments. Each stimulation unit contained 10 of each cell type to allow divergent and convergent projections and intrapallidal inhibition. Fifty simulation units yielded 500 STN and GPe neurons, and the STN neurons were distributed randomly and uniformly in a 6 × 2 × 8 mm prism representing the relevant physical extent of the human STN.
Action potentials at model axon terminals triggered synaptic currents in postsynaptic model neurons. All synapses were single exponentials triggered by presynaptic action potentials with a delay of 0.5 ms and other parameters selected to match experimental data. Ten model AMPA receptor synapses were randomly distributed on the dendrites of each model STN neuron with a relative probability of 1:2 for placement on proximal and distal dendrites [24]. Proximal dendrites were defined as dendritic segments within five segments of the soma. AMPA receptor-mediated synapses had a reversal potential of 0 mV and a maximum conductance of 1 pS that decayed with a time constant of 2.5 ms [25]. One hundred GABAA-mediated inhibitory synapses from GPe were distributed on model STN neurons with proportions in accordance with experimental measurements: soma (30%), proximal dendrites (40%), and distal dendrites (30%) [26, 27]. The inhibitory synapses had a reversal potential of −84 mV and maximum conductance of 1 nS [27–29]. The exponential decay constant (0.7 ms) was tuned to match the fast component of GPe IPSCs in rat brain slices [28]. One hundred excitatory synapses from STN were randomly distributed on distal dendrites on each model GPe neuron [30]. The properties of these synapse were the same as the cortico-subthalamic synapses, except the decay time constant was 5 ms [31]. Intra-pallidal inhibition on proximal neuronal components is also a key feature in the GPe [32], and one hundred inhibitory synapses were randomly distributed on somatic segments of model GPe neurons.
Stimulation was delivered in the model via the electrode representing contact 1. To match the 3387 lead used in the initial, unilateral STN cohort, the model stimulating electrode was flanked by the recording electrodes with 3 mm spacing, and point electrodes were used to represent the contacts [12]. The ventral recording electrode was placed below the STN, and the stimulating and dorsal electrodes placed within the STN, consistent with the targeted locations of the contacts 0–2 during contemporary lead implantation at Duke University Medical Center. Symmetric, biphasic current pulses of 100 μs/phase at 3 mA were delivered assuming an infinite, homogenous, isotropic medium with a conductivity of 0.3 S/m. Potentials at all the neural elements were calculated using the equation for the potential at a distance r away from a point source:
Where I is the current delivered through the source, σ is the tissue conductivity and r is the distance from the source.
Differential recordings were made from the electrodes representing contacts 0 and 2. Neurons that contained a neural element less than 500 μm from any of the point electrode contacts were discarded to mimic the exclusion arising from the presence of the DBS lead, leaving 311/500 STN neurons contributing to stimulation evoked potentials. Potentials observed at each recording electrode were calculated as:
where ik and rk are the transmembrane current and distance from electrode for each model STN neural element k, and σ is the tissue conductivity (0.3 S/m). Potentials at contact 2 were subtracted from those at contact 0 to simulate the differential recording used experimentally.
Statistical Analyses
With two exceptions, we used ranked statistical tests throughout. Therefore, comparisons are performed by Wilcoxon signed rank and correlations by Spearman correlation. Medians and 95% confidence intervals are reported. The 95% confidence interval was estimated using a bootstrapping method (Matlab’s bootstrap()) to estimate the 2.5th and 97.5th percentile of possible medians. As an exception to ranked analyses, we fitted lines to the P1 and N1 latencies. The resulting p and R2 values of the goodness of fit are analogous to Pearson correlation. The other exception was reported mean and standard deviation (SD) when quantifying noise in our recording equipment (see Supplemental Methods). P values are reported without correction for multiple comparisons.
Results
Characterization of DBS Local Evoked Potentials (DLEPs)
We recorded DLEPs from the STN in 8 of 9 participants in the unilateral STN cohort (Table 1) in response to up to 4 stimulation frequencies in epochs of either 60 or 300 s. The immediate response to stimulation (0 – 8 ms, averaged over 0 – 10 s) was a multiphasic response (Fig. 1A, Supplemental Fig. 1) that was similar across participants with a positive deflection ~4 ms (P1) after the start of the stimulation pulse, followed by a large negative deflection after ~6 ms (N1) and a second positive deflection at a latency of 7 – 8 ms. Some responses evoked by 130 Hz DBS also exhibited a short positive deflection at a latency of 2 – 3 ms (gray arrow, Fig. 1A). DBS at lower stimulation frequencies (5, 20, and 45 Hz, Fig. 1B and Supplemental Fig. 2) revealed longer duration DLEPS, and the response duration (i.e., number of periods) varied between participants but not between stimulation frequencies.
Figure 1.

Deep brain stimulation (DBS) local evoked potentials (DLEPs) recorded in STN in response to STN DBS. A) Average short-latency (8 ms) DLEPs during the first 10 s of stimulation of STN DBS at different frequencies. Arrow indicates the short latency positive going phase that was most pronounced with 130 Hz stimulation. B) Average long-latency (25 ms) DLEPs during the first 10 s of STN DBS. The DLEPs often, but not always, contained multiple cycles of high frequency oscillations (see Supplemental Figure 2). C) Evoked potentials in the STN ipsilateral (blue) and contralateral (orange, 10 times gain) to stimulation from Participant 10. There was no response observed in the STN contralateral to stimulation (see Supplemental Figure 3). Data from panels A and B are from Participant 4.
We confirmed that multiphasic DLEPs were physiological responses and not an electrical artifact by observing consistent responses when the polarity of the symmetric biphasic stimulation pulse was reversed (Supplemental Methods). Further, DLEPs exhibited time- (see below) and DBS frequency-dependent changes that would not occur for a stereotyped electrical artifact, and DLEPs were quite different from evoked potentials recorded in thalamus [19] but similar to reported responses recorded in STN with different equipment [11]. We subsequently recorded bilateral STN DLEPs in a different cohort of subjects (Participants 10 and 11) and observed no DLEP in the hemisphere contralateral to stimulation (Fig. 1C, Supplemental Fig. 3, n = 3 hemispheres) confirming both the biological and local origin of the recorded signals.
Time-Dependent Changes in DLEPs
During the first 100 ms of DBS we observed a rapid increase in the amplitude and reduction in the latency of DLEPs (Fig. 2A), while over a longer timescale the amplitudes of P1 and N1 decreased and the latencies increased (Fig. 2B, Supplemental Figs. 4 & 5). The amplitude of P1 and N1 (Fig. 2C) increased quickly to a maximum at approximately 3 s and then declined to plateau at a minimum value approximately 100 s into stimulation. We quantified the peak-to-peak amplitude of DLEPs averaged over 10 s intervals from 0 – 10 s of DBS and from 50 – 60 s of DBS and observed an amplitude reduction for 130 Hz but not 45 Hz DBS (Fig. 2D, −97.43 [−594.1 – −97.43] μV (median [95% confidence interval]), p = 0.0156 for 130 Hz DBS and 3.35 [−14.65 – 25.17] μV, p = 0.688 for 45 Hz DBS, n = 7 participants, Wilcoxon signed rank). The P1 and N1 latencies decreased at the onset of stimulation and then increased with continued 130 Hz DBS (Fig. 2E). The initial latency of P1 but not N1 was shorter in response to 130 Hz DBS than 45 Hz (0.16 [0.15 – 0.49] ms and 0.19 [−0.09 – 1.59] ms, p = 0.0156 and p = 0.219 for P1 and N1 respectively, n = 7 participants). The latency of both the P1 and N1 features increased by the 50 – 60 s timepoint during 130 Hz DBS (Fig. 2F), but not during 45 Hz DBS (P1 latency: 0.42 [0.40 – 0.70] ms, p = 0.0156 for 130 Hz and 0.02 [0.01 – 0.04] ms, p = 0.313 for 45 Hz, N1 latency: 0.8 [0.46 – 0.87] ms, p = 0.0156 for 130 Hz and 0.08 [0.05 – 0.28] ms, p = 0.297 for 45 Hz, n = 7 participants). N1 latency was significantly longer after 50 – 60 s of 130 Hz stimulation (0.1 [−0.3 – 0.28] ms and 0.32 [−0.09 – 1.45] ms, p = 0.313 and 0.0313 for P1 and N1 respectively, n = 7 participants).
Figure 2.

Temporal dynamics of the amplitude and latency of DLEPs during continuous DBS. A) The amplitude quickly increased, and the latency decreased during the first 100 ms of stimulation (blue to yellow). B) DLEPs averaged over 10 s intervals during the first 110 s of stimulation. C) Voltages of the P1 and N1 features from DLEPs averaged over 1 s intervals during a single application of 130 Hz DBS. The P1 and N1 amplitudes quickly increased, reaching a peak value 2–3 s after the start of stimulation, and then declined, reaching a minimum value at approximately 2 min into stimulation. D) Peak-to-peak amplitudes for all participants derived from DLEPs averaged over 10 s intervals at 5, 55, and 295 s. DLEP amplitude was reduced after 55 s during 130 Hz DBS (left) but not 45 Hz DBS (right). E) The P1 and N1 latency during a single trial of 130 Hz DBS. F). Latency of P1 for all participants calculated from DLEPs averaged over 10 s intervals. 130 Hz DBS increased P1 latency while 45 Hz DBS did not. * p < 0.05. N.S. Not significant. See Results for medians, confidence intervals and p-values. The data in Panels A, B, C, and E are from Participant 3.
We next quantified the second-by-second changes in latency, amplitude, and instantaneous frequency of the DLEPs (Fig 3A). We fit lines to the P1 and N1 latency to examine the linear independence of the two measures. For some participants, these relationships were well explained by a straight line, however for others a linear relationship explained much less of the variation (Fig 3B, Supplemental Figs. 6 and 7, R2 = [0.51 – 0.984] range, p < 10−3, n = 7 participants). Conversely, the relationship between the instantaneous frequency and amplitude of the DLEPs was nonlinear (Fig. 3C, Supplemental Fig. 8), and some DLEP amplitudes were observed for several different values of DLEP frequency. This implies that the DLEP frequency and amplitude were independent or covaried with some other factor(s). We then quantified the DLEP amplitude and frequency for all periods during low frequency DBS (Fig. 3D). We did not observe any systematic effect of low frequency DBS on DLEP amplitude and frequency over time, but the first periods within the response to a DBS pulse (i.e., P1–N1 frequency and amplitude compared to P2–N2 frequency and amplitude) were visually separable from the others (Fig 3D right, Supplemental Fig. 9).
Figure 3.
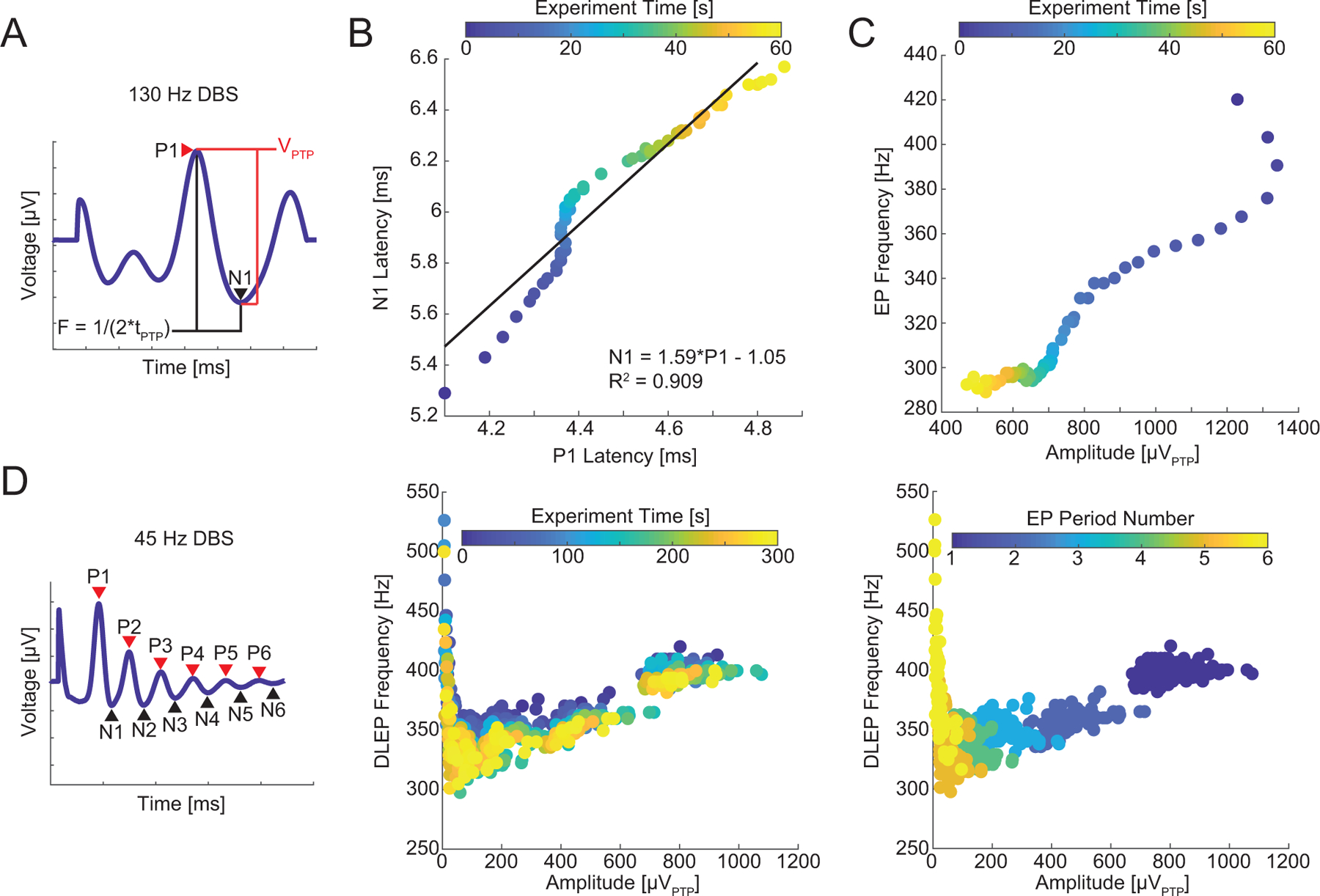
Change in DLEP parameters during continuous DBS. A) An example template demonstrating the calculation of peak-to-peak amplitude and instantaneous frequency using the P1 and N1 points. B) The increases in the latency of P1 and N1 during 130 Hz DBS and best fit line. However, there were deviations from linearity, e.g. note how N1 increases from approximately 5.8 to 6.0 ms while P1 remains at roughly 4.35 ms. Several other participants exhibited a higher degree of nonlinearity (Supplemental Figs. 6 & 7). C) The changes in peak-to-peak amplitude and instantaneous frequency of the DLEP during continuous DBS exhibited a nonlinear relationship. D) Left: We calculated the peak-to-peak amplitude and instantaneous frequency for all periods in the long-latency DLEPs averaged over 1 s intervals during 45 Hz DBS. Center: The amplitude and frequency of each cycle of the response. There does not appear to be separation by experiment time. Right: The same data plotted with color encoding of the period of the DLEP (1–6 periods). The first period (the amplitude and frequency calculated from P1 to N1) was clearly separable from subsequent periods. Data from panels B and C are from participant 1. Data from panel D are from Participant 3. R2: goodness of fit calculated by linear least squares corresponding p < 10−3.
Model-Based Analysis of DLEP Origin
We implemented a biophysical model of STN DBS to investigate the minimum neural elements necessary to generate DLEPs (Fig. 4A–B), and the parsimonious model included the STN, GPe, their synaptic interconnectivity, and afferent cortical-STN axons. Model-generated DLEPs in response to 45 Hz and 130 Hz DBS were very similar to DLEPs recorded from humans with PD (Fig. 4C–D). All phases of the human DLEP were present in the model DLEP. Model STN neurons were relatively quiescent during the IPIs due to strong inhibitory tone from the highly active model GPe neurons (Fig. 4E–F), consistent with reports that STN neurons are inhibited during high frequency stimulation [23]. The increased probability of model STN neuron firing 3 – 5 ms after a DBS pulse matched the timing of the late positive deflection (P1) and consistent with reports of STN neuron activity during IPIs of high frequency DBS [33]. DBS also directly excited the model STN axons, which synaptically excited the model GPe neurons. The highly synchronous STN excitatory input and the recurrent inhibitory connections within the GPe caused GPe neurons to fire repeatedly at intervals of 3 – 4 ms (Fig. 4G–H), and synaptic inhibition of the STN from periodic GPe firing was responsible for the repeated late DLEP phases. Similar quasi-periodic firing of GPe neurons was also observed in Parkinsonian monkeys during STN DBS [34]. The responses in the biophysical model suggest that the reciprocal connections between STN and GPe alone are sufficient to generate the multiphasic DLEPs.
Figure 4.
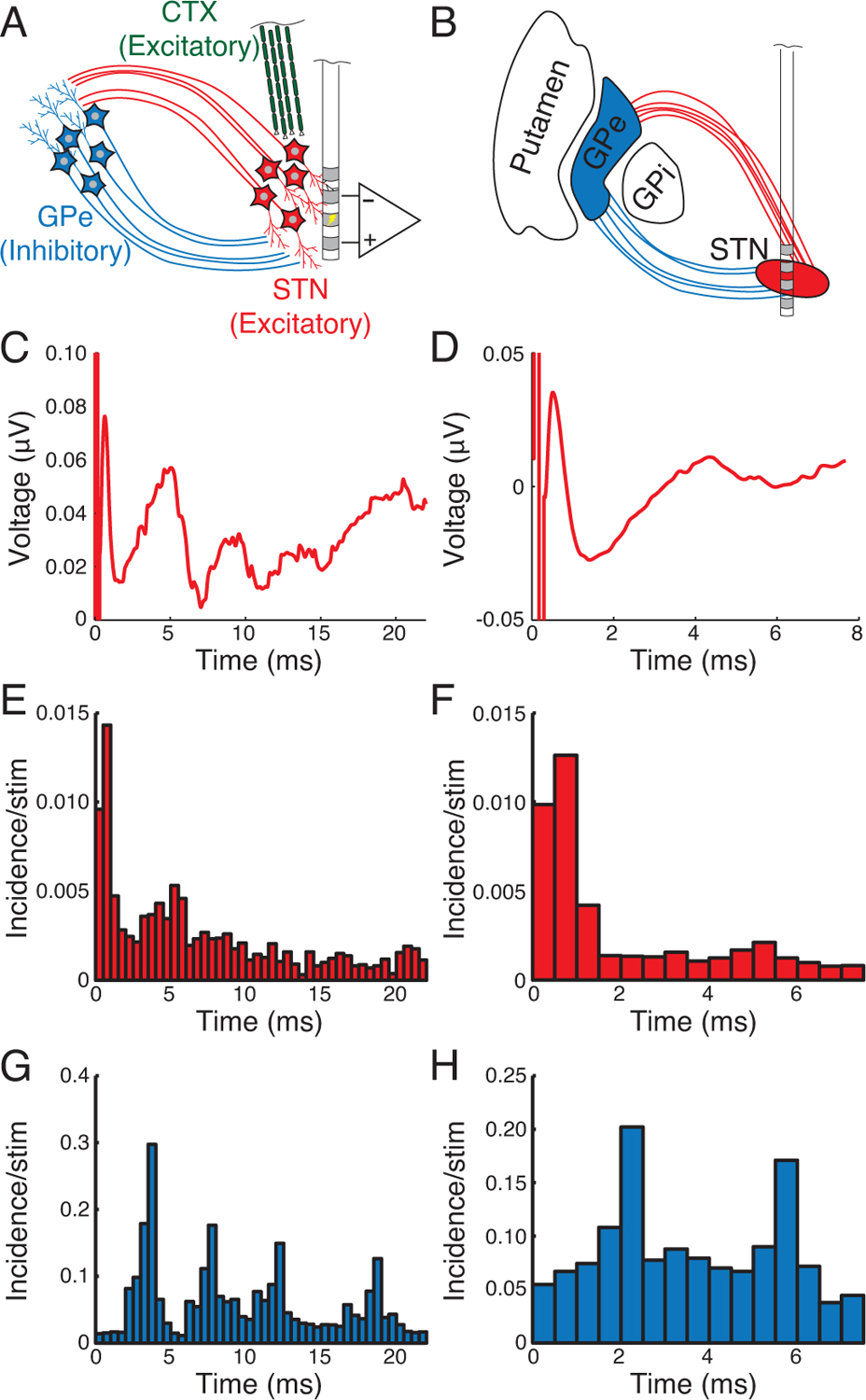
Computational model predicts that reciprocal connections between STN and GPe produce DLEPs. A) The biophysical model included excitatory subthalamic (red) and inhibitory pallidal neurons (blue) in addition to excitatory cortical axons of the hyperdirect pathway (green). B) We simulated the placement of contacts with monopolar stimulation on contact 1 and differential recording on 0 and 2. C & D) DLEPs calculated from the computational model were similar to those observed clinically for 45 Hz (C) and 130 Hz (D) DBS. Post-stimulus time histograms for STN firing during 45 Hz (E) and 130 Hz (F) DBS revealed modest direct excitation from stimulation and relatively strong inhibition in the interpulse interval with slight increases in firing coincident with the positive phases of the DLEPs. G & H) GPe spiking was periodic following the strong excitation via STN afferents with peaks in the PSTH at 3–4 ms intervals.
Simultaneous STN and GP Recordings in Research Participants
To test the predictions of the model on the neural origins of DLEPs, we next recorded simultaneously from STN and GP during ipsilateral STN DBS in 6 participants (9 hemispheres with DLEPs). As with the unilateral STN recordings, we quantified the responses in the GP averaged over the first 10 s of stimulation (Fig. 5, Supplemental Fig. 10). Similar to the STN DLEPs, the responses in the GP to STN DBS exhibited multiple quasi-periodic phases, and the character and latencies of the GPe responses were consistent with those in the biophysical model. The peak-to-peak amplitude of the GP responses decreased in GPi but not in GPe during 130 Hz STN DBS (11.40 [4.46 – 15.31] μV and −0.29 [−3.08 – 0.04] μV, p = 0.023 and 0.945 n = 8 and 8 for GPi and GPe, respectively, Fig. 6A, Supplemental Fig. 11). The P1 latency in both GPi and GPe increased during 130 Hz DBS (Fig. 6B, 0.52 [0.30 – 0.66] ms and 0.72 [0.36 – 1.56] ms, p = 0.016 and 0.008, n = 8 and 9 for GPi and GPe, respectively). The P1 responses in the GPi preceded those of the STN but this comparison only trended towards significance for GPe (0.64 [0.37 – 1.04] ms and 0.43 [0.29 to 0.60] ms, p = 0.002 and 0.070, n = 7 and 8 hemispheres * 2 timepoints for GPi and GPe, respectively). The amount of time by which the GP lead the STN did not change between the initial response to DBS and after a minute of DBS (Fig 6C, −0.24 [−1.26 – 0.06] ms and −0.22 [−0.31 – 0.27] ms, p = 0.219 and 0.547, n = 7 and 8 hemispheres for GPi and GPe, respectively). The P1 feature occurred with shorter latency in GPi than in GPe (Fig. 6D, 0.34 [0.20 – 0.54] ms, p = 0.003, n = 8 hemispheres * 2 timepoints) and did not change over time (Fig. 6D, −0.21 [−0.54 – 0.15] ms, p = 0.383, n = 8 hemispheres). These results agree with the prediction from the model that the DLEP arises from recurrent activity between the STN and GPe mediated by their reciprocal monosynaptic connectivity.
Figure 5.

Simultaneous recordings in the STN and GP during STN DBS. A) DBS electrode and contact locations. B) Evoked potentials in the STN, GPi and GPe normalized by their respective maximum amplitudes for comparison of latencies. C) Left: STN DLEPs. Note the high degree of similarity for repeated trials of STN DBS (blue and red or yellow and green). Right: Evoked responses in GPi or GPe to STN DBS. The EPs were different depending on the recording contacts chosen. For all participants see Supplemental Fig. 9. L/R: Recording and stimulation in the left/right hemisphere. GPi/GPe: Simultaneous recording with GP lead selected to record differentially between contacts 0 and 1 (GPi) or contacts 2 and 3 (GPe). All data depicted are from Participant 15.
Figure 6.
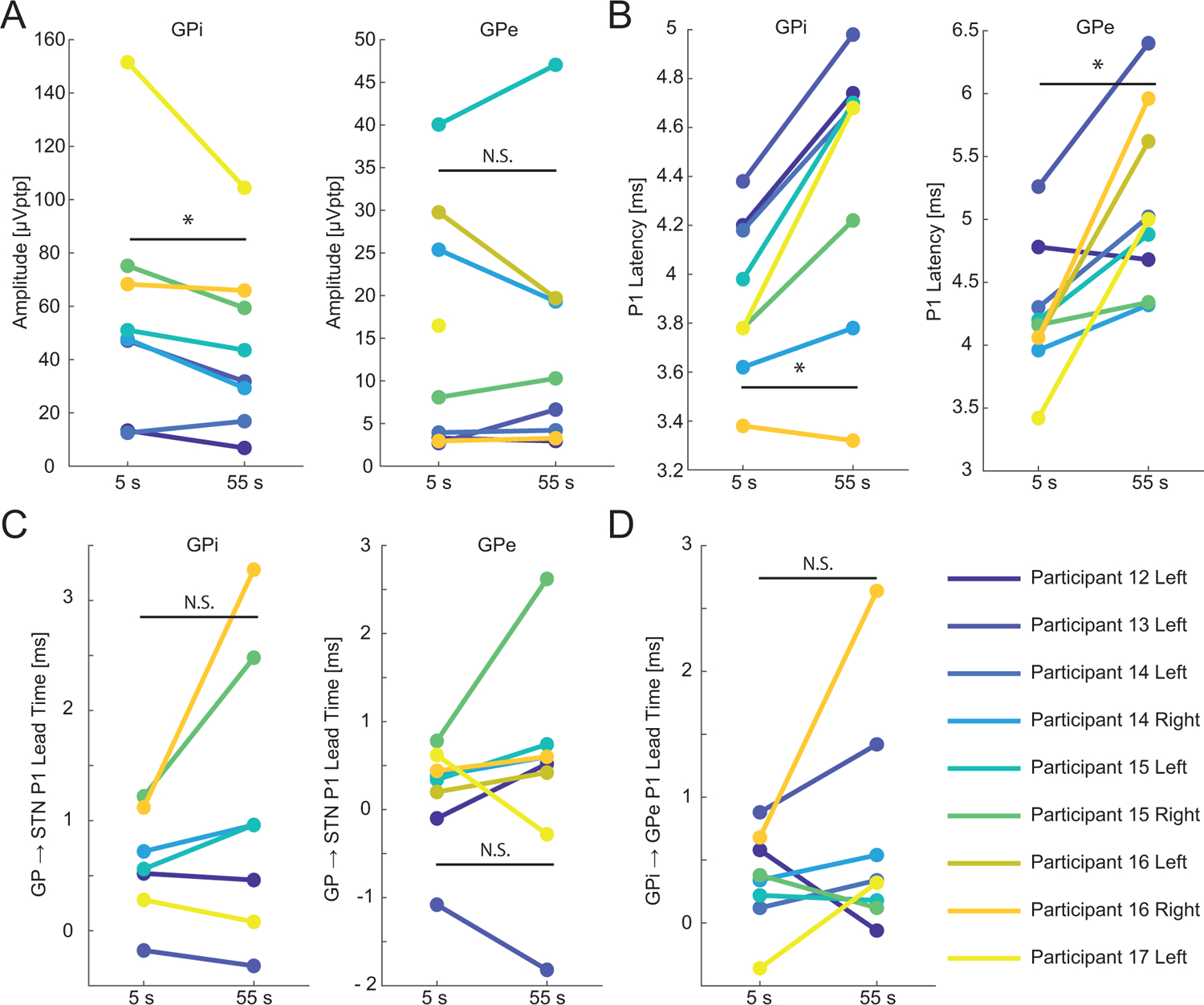
Changes in evoked potentials recorded in GP during 60 s of STN DBS. A) Peak-to-peak amplitude of the evoked responses in the GP from responses averaged over 10 s intervals. As stimulation continued the amplitude decreased in GPi but did not decrease in GPe. B) P1 latency increased in both GPi and GPe with stimulation time. C) The lead time between the P1 feature in GP referenced and the P1 feature in STN. Both regions of GP responded before STN but lead time did not change significantly with continued stimulation. D) GPi responded before GPe. The lead time did not change with continued stimulation. *: p < 0.05. N.S: not significant. See Results for medians, confidence intervals and p-values.
Relation of DLEPs to Oscillatory Activity
We quantified the relationship between DLEPs and beta band oscillatory activity in the local field potential. There was significant correlation between the largest DLEP amplitude (averaged over 0 – 10 s after stimulation onset) to power in the beta band with DBS off (r = 0.668, p = 0.008, n = 15 hemispheres, Spearman’s rho; Fig. 7A–B). We next examined the high end of the frequency spectra during DBS. We hypothesized that the high-amplitude DLEP activity should be visible in the LFP power spectrum despite its non-stationary nature. Indeed, we observed a pronounced 300–350 Hz oscillation in most participants at the onset of both high frequency (Fig. 7C, Supplemental Fig. 12) and low frequency DBS (Supplemental Fig. 13). This contrasts with the lack of HFOs observed in the absence of DBS (Supplemental Fig 14). We integrated the power spectral density between 200 – 400 Hz and observed that the power of the HFO decreased by the 50 – 60 s timepoint of 130 Hz DBS (Fig. 7D, − 1.32 [−3.91 – −1.09] μV2,p = 0.0156, n = 7 participants). By contrast, the power of the HFO did not change during continued low frequency DBS (0.52 [−0.23 – 1.06] μV2, p = 0.313, n = 8 participants).
Figure 7.

Relationship between STN DLEPs and oscillatory activity in the STN local field potential. A) Power spectral density of the LFP during the first and last μs of a DBS OFF trial. B) The peak-to-peak amplitude of the average DLEP during the first 10 seconds of stimulation was rank-correlated with beta power while stimulation was off. C) High frequency power spectral densities immediately after the onset of 130 Hz DBS (blue), one minute after onset of 130 Hz DBS (orange), and after 1 minute with DBS off (gray). A prominent 350 Hz oscillation was evoked by DBS which decreased in power and frequency. D) Comparison of power in the 200 – 400 Hz band. Continued 130 Hz DBS reduced power in this band compared to the power at the onset of DBS while 45 Hz DBS did not. Data in panels A & C are from Participant 4. PSD: power spectral density. *: p < 0.05. N.S.: not significant. See Results for medians, confidence intervals and p-values.
Discussion
Evoked potentials generated by DBS in the human STN by STN DBS exhibited a distinctive, multiphasic signature that arose from the reciprocal connectivity between the STN and GPe. We replicated previous observations of similar responses [11, 17, 35] and used computational modeling and simultaneous STN and GP recordings to determine the neural circuitry underlying the DLEPs. The DLEPs exhibited stimulation frequency- and time-dependent features, and the initial DLEP amplitude was also correlated to resting beta power with DBS off. Previous studies demonstrated that DLEP amplitude and time to reach steady state (minimum) amplitudes were correlated with improvement in symptoms [11, 13]. These observations, and the similarity between DLEP dynamics and temporal changes in bradykinesia, tremor and rigidity at the onset or termination of DBS [36–40], suggest that DLEPs may find utility as feedback control signal for closed-loop DBS.
Origin of DLEPs
The STN receives inhibitory input from the pallidum and excitatory input from the cortex via the hyperdirect pathway. The DLEPs reflect the interaction of these two inputs following local stimulation. The early positive phase (before P1, obscured by the artifact in vivo but visible in the computational model) of the DLEP resulted from direct short latency antidromic excitation of STN neurons, and the early negative phase reflected strong proximal inhibition of STN neurons evoked by activation of pallidal terminals. Excitatory axons were also directly activated by DBS, but their effects on STN firing were observed later in the interpulse interval due to the distal distribution of excitatory synapses on STN neurons [30]. In the model, the DBS-evoked activity of cortical axon terminals provided the initial excitatory drive that activated the reciprocal STN/GPe connections, and the hyperdirect pathway was required in the model to initiate the DLEP.
Oscillations caused by quasi-periodic pallidal inhibition of the STN every 3 – 4 ms were the most distinctive feature of the DLEPs. These oscillations are consistent with single-unit recordings revealing that pallidal neurons fire periodically in 3 – 4 ms intervals in response to STN DBS [23]. Further, STN single unit recordings exhibit action potentials during the interpulse interval with latencies that match the positive deflections of the DLEP signal at a latency of approximately 6 ms [33]. Thus, the late DLEP oscillations reflect excitatory synaptic currents and resulting STN action potentials interrupted by brief periods of inhibition by pallidal afferents. The P1 feature occurred with shorter latency in the GPi compared to the GPe. A simple explanation may be the shorter conduction distance from STN to GPi, but we were not able to test this hypothesis.
Time course of DLEPs
The dynamics of DLEPs during continuous high frequency DBS reflect changes in the subcortical motor circuit that may contribute to DBS efficacy. The increase in P1 magnitude over the first 100 ms reflected temporal summation of excitatory synaptic currents resulting in increased excitation of the STN. The P1 amplitude then decreased substantially during stimulation, which may reflect depletion of excitatory synapses in the STN [41, 42]. The P1 and N1 latencies throughout stimulation were very nearly linearly related, suggesting that both latencies were largely determined by a common underlying mechanism: the delay in pallidal inhibition that was evoked by excitation of the STN efferents. This implies that the latency of orthodromically-driven action potentials in the pallidum increased over time during high frequency STN DBS. Indeed this has been observed [43], and the time course of the increase in latency matched the increase in P1 and N1 latency observed here. Therefore, changes in P1 and N1 latency likely reflect changes in pallidal excitation latency and not local changes within the STN. However, the P1 to N1 instantaneous frequency also changed with stimulation (Fig. 3B–C, Supplemental Figs. 6–8), allowing for additional biological mechanisms.
Oscillations and DLEPs
Levodopa-induced changes in beta amplitude in the STN are correlated with bradykinesia and rigidity [44]. The beta oscillation has therefore been tested as a biomarker in pilot studies of adaptive DBS [45–47]. We observed a correlation between resting beta amplitude in absence of DBS and the peak-to-peak amplitude of the DLEP during the first 10 s of DBS (Fig 7A). Sinclair and colleagues [11] observed higher amplitude evoked potentials in the dorsal part of the STN, localizing the DLEP with hypersynchronized beta oscillations [48, 49]. It is therefore possible that the DLEP is mediated by the same cells as the beta oscillatory activity. However, if true, this does not necessarily mean that DBS causes the beta synchronization to shift to higher frequencies. A recent study by Wiest and colleagues [13] suggests that because DLEPs reach their minima after ~70 s of DBS and beta is reduced within the first 500 ms of DBS, these are separate phenomena. Low frequency DBS does not reduce, and indeed sometimes increases pathological low frequency oscillations in the basal ganglia [50, 51]. Here we show that low frequency DBS also produces DLEPs suggesting that DLEPs and pathological beta oscillations may coexist.
HFOs are observed in both the STN and GP of persons with PD, and we observed pronounced evoked oscillatory activity at 300 – 350 Hz, which was reduced during one minute of 130 Hz DBS, but remained constant during one minute of 45 Hz DBS. 250 Hz oscillations are reported in the absence of treatment with levodopa or DBS [52–56], while with levodopa, and especially when combined with movement, a 350 Hz HFO was observed [52, 53, 56, 57]. However, with one exception, we did not observe such HFOs in the absence of DBS. It is possible that the difference could be in the details of the recording. HFOs are often observed on macroelectrodes during postoperative sessions with the participant seated [52, 53, 56, 57]. Although it remains unclear if spontaneous and DBS evoked HFOs are generated by the same mechanism, we note that after one minute of 130 Hz DBS we observed less power in the HFO band and a decrease in the frequency of the HFOs compared to the first 10 s of 130 Hz DBS. For endogenous HFOs such changes would correspond to increased symptoms [52]. However, our result that the frequency of the evoked HFO decreases with continued stimulation aligns with those of Sinclair and colleagues [35] for pre- and post-DBS spontaneous HFOs.
Implications for DBS therapy
DLEPs reveal the neural elements engaged by STN DBS in humans with PD. First, DLEPs support that STN efferents are excited by DBS, and that activity in STN somata and axons can be functionally decoupled during stimulation [58]. Second, the effects of stimulating the efferent STN axons can be observed via the returning inhibition from GPe even after several minutes of DBS, indicating that STN efferents are not silenced by persistent DBS. Third, the amplitude of P1–N1 declined with a time course similar to the amelioration of some symptoms of PD by DBS suggesting that synaptic depletion, particularly of excitatory synapses, may contribute to the effects of DBS [41].
There is disagreement whether high frequency stimulation is unique in its ability to produce time-locked responses in the basal ganglia. In rat STN slices, only high frequency stimulation produced stimulation-locked responses [59], and in awake freely moving rats stimulation-locked effects were stronger during high frequency stimulation [51]. However, pallidal single-unit recordings during pallidal microstimulation exhibited time-locked responses to both low and high stimulation frequencies [60]. Our results provide further evidence that low frequency stimulation produces complex and long-lasting time-locked responses in the STN. Therefore, the therapeutic mechanism of DBS seems unlikely to rely solely on producing time-locked responses in the STN, but likely also includes masking of intrinsic activity [61]. Further, DLEPs provide evidence against chaotic desynchronization of the GP as a mechanism of DBS [62]. The presence of the P1 peak in the GP and previous single unit recordings in pallidum [23] indicate that pallidal neuron firing is tightly regulated, rather than desynchronized, by DBS.
Previous reports of DBS evoked activity in the STN [11, 13, 14, 35] and motor cortex [63, 64] suggested that the evoked activity is due to resonance. However, resonant activity requires a system with a natural frequency, and higher amplitude output results when the input is applied at the natural frequency. The response to low frequency DBS revealed that the instantaneous frequency of P1 to P2 was different than that of P2 to P3 (Fig. 3D, Supplemental Fig. 9) indicating the lack of a natural frequency. Considering the change in frequency between periods within a DLEP and the lack of attractor dynamics at DBS frequency harmonics during continued stimulation (Fig. 3C, Supplemental Fig. 8), it is unlikely that the evoked potential within the basal ganglia is the product of resonant activity. However Li and colleagues did observe resonance in EEG activity in response to 100 – 120 Hz DBS in rats [64], and further studies are needed to confirm the absence or presence of resonant responses in both the cortex and subcortical nuclei.
Considerations for Adaptive DBS
Features of the DLEP may find utility in adaptive DBS. However other biomarkers are being explored in relation to PD: beta and narrowband gamma oscillations. The beta oscillation is most directly correlated with bradykinesia, however bradykinesia often takes more than a minute to be affected by DBS while beta power is reduced in less than a second [13]. Our data and others [13, 35] demonstrate that DLEPs change amplitude over a longer time course. However, it remains to be determined if this time course better correlates with bradykinetic state. While DLEPs do not exhibit the high variability of beta during DBS ON [13], the potential utility of capturing this variability remains to be determined. Initial examinations of beta bursts contain flawed analyses [18] and most patients manage symptoms with dopaminergic medication which changes neural activity on the time scale of hours. Finally, calculation of the oscillatory power (FFT, wavelet convolution, and especially Hilbert transform with FIR filtering) are computationally inefficient compared to calculating the peak-to-peak voltage between 2 and 7 ms after the DBS pulse. Such computational complexity leads to increased drain on the battery and, especially in the case of primary cells, decrease longevity of IPGs. However, we included analysis of the DLEP as an HFO both for the possible relationship to spontaneous HFOs and because present sensing devices, such as the Medtronic RC+S, can calculate power in specific frequency bands with on-board circuitry. These devices could be used to evaluate the HFO as a biomarker with the embedded electronics, while examination of the P1–N1 amplitude and latency will require new devices.
Sensing a biomarker, be it based on the DLEP or oscillatory features, on the stimulation lead comes with the distinct advantage of requiring less hardware, although providing fewer options for complex clinical stimulation across multiple contacts. Further the cortical evoked potential is generally a few microvolts in amplitude [10, 63], while the DLEP can measure greater than a millivolt providing a much higher signal to noise ratio. However, considering the ubiquity of communication through coherence in the nervous system [65], the additional information from a second location may produce gains that offset the cost and complexity of the additional recording.
Limitations
Although there are limitations in the simplified model used to interpret and reconstruct DLEPs, it reproduced with remarkable fidelity the DLEPs from human recordings. The model-based responses were predictive of subsequent simultaneous recordings in STN and GP suggesting that STN and GPe interactions alone are sufficient to generate DLEPs. The model did not include slower synapses, mediated by metabotropic receptors, and the progressive changes with continued DBS may reflect dynamics of these slower processes. Further, the model did not include plasticity and did not capture the time-dependent changes in DLEPs that occurred over the course of minutes. Finally, we did not include both types of cells observed in the GPe [66], and model neurons were based on the more numerous type I pallidal neurons [22].
The intraoperative experiments also have several limitations. We were limited by time to conduct intraoperative experiments and therefore did not apply all DBS frequencies to all participants. Further, limited time precluded us from testing stimulation on the other contacts in the STN or testing GP stimulation. When examining the pallidal responses, we were not able to separate the later cycles of the GP response to STN stimulation during low frequency DBS and so did not perform analysis of those data. We were unable to quantify motor symptoms in the unilateral STN cohort due to only 3 participants with DLEPs tested for bradykinesia and 5 for tremor. In the STN + GP cohort we did not attempt to quantify symptoms, as there was significant insertion effect following the placement of the second lead into the same hemisphere. Similarly, we did not observe spontaneous HFOs here. Due to the limitations of an intraoperative procedure, we were unable to probe spontaneous HFOs with dopaminergic medication or with the participants in a sitting position or actively moving. Only 3 participants with DLEPs received 300 s of DBS and therefore no comparison was performed on the DLEPs from 250 – 300 s. Finally, Participant 12 received a Medtronic 3389-lead placed in the GP without MER or CT. However, we note that the DLEPs in the GP of Participant 12 were not outliers in any measured dimension suggesting that stereotactic placement based on MRI coordinates alone was sufficient, without microelectrode recordings.
Supplementary Material
DBS evoked low-latency responses within the STN, GPi, and GPe.
The amplitude and latency of DLEPs changed with continued DBS.
A biophysical model suggests that DLEPs arise from reciprocal STN GPe connections.
Acknowledgments
The authors thank Jennifer Peters for assistance collecting intraoperative data and recruiting participants. We thank Gilda Mills and Danielle Degoski for laboratory support and Brandon Thio for his valuable input on the analysis.
Funding Sources
This work was funded by UH3 NS103468, R01 NS040894 and R37 NS040894.
Abbreviations
- DBS
deep brain stimulation
- DLEP
DBS Local Evoked Potential
- GPi/e
globus pallidus (internal/external segment)
- HFO
high frequency oscillation
- LFP
local field potential
- Px/Nx
The xth positive or negative deflection after the DBS pulse
- PD
Parkinson’s disease
- STN
subthalamic nucleus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
WMG is cofounder, Director and CSO of Deep Brain Innovations LLC. WMG is Director and CSO of NDI Healthcare Fund and receives compensation for these positions. These relationships are reported to the Conflict of Interest Committee at Duke University.
Citations
- [1].Sutton S, Braren M, Zubin J, John E. Evoked-potential correlates of stimulus uncertainty. Science 1965;150(3700):1187–8. [DOI] [PubMed] [Google Scholar]
- [2].Thorpe S, Fize D, Marlot C. Speed of processing in the human visual system. Nature 1996;381(6582):520–2. [DOI] [PubMed] [Google Scholar]
- [3].Buchsbaum MS, Davis GC, Bunney WE. Naloxone alters pain perception and somatosensory evoked potentials in normal subjects. Nature 1977;270(5638):620–2. [DOI] [PubMed] [Google Scholar]
- [4].Miller CA, Brown CJ, Abbas PJ, Chi S-L. The clinical application of potentials evoked from the peripheral auditory system. Hearing research 2008;242(1):184–97. [DOI] [PubMed] [Google Scholar]
- [5].Parker JL, Karantonis DM, Single PS, Obradovic M, Laird J, Gorman RB, et al. Electrically evoked compound action potentials recorded from the sheep spinal cord. Neuromodulation: Technology at the Neural Interface 2013;16(4):295–303. [DOI] [PubMed] [Google Scholar]
- [6].Rizzone M, Lanotte M, Bergamasco B, Tavella A, Torre E, Faccani G, et al. Deep brain stimulation of the subthalamic nucleus in Parkinson’s disease: effects of variation in stimulation parameters. Journal of neurology, neurosurgery, and psychiatry 2001;71(2):215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Benabid AL, Chabardes S, Mitrofanis J, Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. The Lancet Neurology 2009;8(1):67–81. [DOI] [PubMed] [Google Scholar]
- [8].Miocinovic S, Somayajula S, Chitnis S, Vitek JL. History, applications, and mechanisms of deep brain stimulation. JAMA neurology 2013;70(2):163–71. [DOI] [PubMed] [Google Scholar]
- [9].Baker KB, EB M Jr., Rezai AR, Burgess R, Lüders HO. Subthalamic nucleus deep brain stimulus evoked potentials: Physiological and therapeutic implications. Movement Disorders 2002;17(5):969–83. [DOI] [PubMed] [Google Scholar]
- [10].Miocinovic S, de Hemptinne C, Chen W, Isbaine F, Willie JT, Ostrem JL, et al. Cortical Potentials Evoked by Subthalamic Stimulation Demonstrate a Short Latency Hyperdirect Pathway in Humans. The Journal of neuroscience : the official journal of the Society for Neuroscience 2018;38(43):9129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sinclair NC, McDermott HJ, Bulluss KJ, Fallon JB, Perera T, Xu SS, et al. Subthalamic nucleus deep brain stimulation evokes resonant neural activity. Ann Neurol 2018;83(5):1027–31. doi: 10.02/ana.25234. Epub 2018 May 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kent AR, Grill WM. Analysis of deep brain stimulation electrode characteristics for neural recording. Journal of neural engineering 2014;11(4):046010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wiest C, Tinkhauser G, Pogosyan A, Bange M, Muthuraman M, Groppa S, et al. Local field potential activity dynamics in response to deep brain stimulation of the subthalamic nucleus in Parkinson’s disease. Neurobiology of disease 2020:105019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sinclair NC, Fallon JB, Bulluss KJ, Thevathasan W, McDermott HJ. On the neural basis of deep brain stimulation evoked resonant activity. Biomed Phys Eng Expr 2019;5(5). [Google Scholar]
- [15].Kent AR, Grill WM. Recording evoked potentials during deep brain stimulation: development and validation of instrumentation to suppress the stimulus artefact. Journal of neural engineering 2012;9(3):036004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hoang KB, Cassar IR, Grill WM, Turner DA. Biomarkers and Stimulation Algorithms for Adaptive Brain Stimulation. Front Neurosci 2017;11:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grill WM, Brocker DT, Swan BD, Kent AR, Turner DA, Hickey P. “Local evoked potentials recorded during subthalamic nucleus deep brain stimulation in humans with Parkinson’s disease”. Neuroscience. Chicago; 2015:397.13/E22. [Google Scholar]
- [18].Schmidt SL, Peters JJ, Turner DA, Grill WM. Continuous deep brain stimulation of the subthalamic nucleus may not modulate beta bursts in patients with Parkinson’s disease. Brain stimulation 2020;13(2):433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kent AR, Swan BD, Brocker DT, Turner DA, Gross RE, Grill WM. Measurement of evoked potentials during thalamic deep brain stimulation. Brain stimulation 2015;8(1):42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hines ML, Carnevale NT. The NEURON simulation environment. Neural Comput 1997;9(6):1179–209. [DOI] [PubMed] [Google Scholar]
- [21].Miocinovic S, Parent M, Butson CR, Hahn PJ, Russo GS, Vitek JL, et al. Computational analysis of subthalamic nucleus and lenticular fasciculus activation during therapeutic deep brain stimulation. Journal of neurophysiology 2006;96(3):1569–80. [DOI] [PubMed] [Google Scholar]
- [22].Johnson M, McIntyre C. Quantifying the neural elements activated and inhibited by globus pallidus deep brain stimulation. Journal of neurophysiology 2008;100(5):2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hashimoto T, Elder CM, Okun MS, Patrick SK, Vitek JL. Stimulation of the subthalamic nucleus changes the firing pattern of pallidal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003;23(5):1916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bevan MD, Francis C, Bolam J. The glutamate-enriched cortical and thalamic input to neurons in the subthalamic nucleus of the rat: convergence with GABA-positive terminals. Journal of Comparative Neurology 1995;361(3):491–511. [DOI] [PubMed] [Google Scholar]
- [25].Farries MA, Kita H, Wilson CJ. Dynamic spike threshold and zero membrane slope conductance shape the response of subthalamic neurons to cortical input. The Journal of Neuroscience 2010;30(39):13180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Smith Y, Bevan M, Shink E, Bolam J. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 1998;86(2):353. [DOI] [PubMed] [Google Scholar]
- [27].Baufreton J, Kirkham E, Atherton JF, Menard A, Magill PJ, Bolam JP, et al. Sparse but selective and potent synaptic transmission from the globus pallidus to the subthalamic nucleus. Journal of neurophysiology 2009;102(1):532–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Atherton JF, Menard A, Urbain N, Bevan MD. Short-term depression of external globus pallidus-subthalamic nucleus synaptic transmission and implications for patterning subthalamic activity. The Journal of Neuroscience 2013;33(17):7130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fan KY, Baufreton J, Surmeier DJ, Chan CS, Bevan MD. Proliferation of external globus pallidus-subthalamic nucleus synapses following degeneration of midbrain dopamine neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2012;32(40):13718–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hanson JE, Smith Y, Jaeger D. Sodium channels and dendritic spike initiation at excitatory synapses in globus pallidus neurons. The Journal of neuroscience 2004;24(2):329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Song S, Miller KD, Abbott LF. Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nature neuroscience 2000;3(9):919–26. [DOI] [PubMed] [Google Scholar]
- [32].Sims RE, Woodhall GL, Wilson CL, Stanford IM. Functional characterization of GABAergic pallidopallidal and striatopallidal synapses in the rat globus pallidus in vitro. European Journal of Neuroscience 2008;28(12):2401–8. [DOI] [PubMed] [Google Scholar]
- [33].Carlson JD, Cleary DR, Cetas JS, Heinricher MM, Burchiel KJ. Deep brain stimulation does not silence neurons in subthalamic nucleus in Parkinson’s patients. Journal of neurophysiology 2010;103(2):962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Meissner W, Leblois A, Hansel D, Bioulac B, Gross CE, Benazzouz A, et al. Subthalamic high frequency stimulation resets subthalamic firing and reduces abnormal oscillations. Brain : a journal of neurology 2005;128(Pt 10):2372–82. [DOI] [PubMed] [Google Scholar]
- [35].Sinclair NC, McDermott HJ, Fallon JB, Perera T, Brown P, Bulluss KJ, et al. Deep brain stimulation for Parkinson’s disease modulates high-frequency evoked and spontaneous neural activity. Neurobiology of disease 2019;130:104522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krack P, Fraix V, Mendes A, Benabid AL, Pollak P. Postoperative management of subthalamic nucleus stimulation for Parkinson’s disease. Movement Disorders 2002;17:S188–S97. [DOI] [PubMed] [Google Scholar]
- [37].Volkmann J, Herzog J, Kopper F, Deuschl G. Introduction to the programming of deep brain stimulators. Movement Disorders 2002;17:S181–S7. [DOI] [PubMed] [Google Scholar]
- [38].Lopiano L, Torre E, Benedetti F, Bergamasco B, Perozzo P, Pollo A, et al. Temporal changes in movement time during the switch of the stimulators in Parkinson’s disease patients treated by subthalamic nucleus stimulation. European neurology 2003;50(2):94–9. [DOI] [PubMed] [Google Scholar]
- [39].Cooper SE, Driesslein KG, Noecker AM, McIntyre CC, Machado AM, Butson CR. Anatomical targets associated with abrupt versus gradual washout of subthalamic deep brain stimulation effects on bradykinesia. PLoS One 2014;9(8):e99663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Temperli P, Ghika J, Villemure JG, Burkhard PR, Bogousslavsky J, Vingerhoets FJ. How do parkinsonian signs return after discontinuation of subthalamic DBS? Neurology 2003;60(1):78–81. [DOI] [PubMed] [Google Scholar]
- [41].Anderson TR, Hu B, Iremonger K, Kiss ZHT. Selective Attenuation of Afferent Synaptic Transmission as a Mechanism of Thalamic Deep Brain Stimulation-Induced Tremor Arrest. J Neurosci 2006;26(3):841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Farokhniaee A, McIntyre CC. Theoretical principles of deep brain stimulation induced synaptic suppression. Brain stimulation 2019;12(6):1402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Moran A, Stein E, Tischler H, Belelovsky K, Bar-Gad I. Dynamic Stereotypic Responses of Basal Ganglia Neurons to Subthalamic Nucleus High-Frequency Stimulation in the Parkinsonian Primate. Frontiers in systems neuroscience 2011;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kuhn AA, Kupsch A, Schneider GH, Brown P. Reduction in subthalamic 8–35 Hz oscillatory activity correlates with clinical improvement in Parkinson’s disease. Eur J Neurosci 2006;23(7):1956–60. [DOI] [PubMed] [Google Scholar]
- [45].Little S, Pogosyan A, Neal S, Zavala B, Zrinzo L, Hariz M, et al. Adaptive deep brain stimulation in advanced Parkinson disease. Ann Neurol 2013;74(3):449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Arlotti M, Marceglia S, Foffani G, Volkmann J, Lozano AM, Moro E, et al. Eight-hours adaptive deep brain stimulation in patients with Parkinson disease. Neurology 2018;90(11):e971–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Velisar A, Syrkin-Nikolau J, Blumenfeld Z, Trager MH, Afzal MF, Prabhakar V, et al. Dual threshold neural closed loop deep brain stimulation in Parkinson disease patients. Brain stimulation 2019. [DOI] [PubMed] [Google Scholar]
- [48].Zaidel A, Spivak A, Grieb B, Bergman H, Israel Z. Subthalamic span of beta oscillations predicts deep brain stimulation efficacy for patients with Parkinson’s disease. Brain : a journal of neurology 2010;133(Pt 7):2007–21. [DOI] [PubMed] [Google Scholar]
- [49].Kuhn AA, Trottenberg T, Kivi A, Kupsch A, Schneider GH, Brown P. The relationship between local field potential and neuronal discharge in the subthalamic nucleus of patients with Parkinson’s disease. Experimental neurology 2005;194(1):212–20. [DOI] [PubMed] [Google Scholar]
- [50].Brown P, Mazzone P, Oliviero A, Altibrandi MG, Pilato F, Tonali PA, et al. Effects of stimulation of the subthalamic area on oscillatory pallidal activity in Parkinson’s disease. Experimental neurology 2004;188(2):480–90. [DOI] [PubMed] [Google Scholar]
- [51].McConnell GC, So RQ, Hilliard JD, Lopomo P, Grill WM. Effective deep brain stimulation suppresses low-frequency network oscillations in the Basal Ganglia by regularizing neural firing patterns. The Journal of neuroscience : the official journal of the Society for Neuroscience 2012;32(45):15657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ozkurt TE, Butz M, Homburger M, Elben S, Vesper J, Wojtecki L, et al. High frequency oscillations in the subthalamic nucleus: a neurophysiological marker of the motor state in Parkinson’s disease. Experimental neurology 2011;229(2):324–31. [DOI] [PubMed] [Google Scholar]
- [53].Lopez-Azcarate J, Tainta M, Rodriguez-Oroz MC, Valencia M, Gonzalez R, Guridi J, et al. Coupling between beta and high-frequency activity in the human subthalamic nucleus may be a pathophysiological mechanism in Parkinson’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010;30(19):6667–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang AI, Vanegas N, Lungu C, Zaghloul KA. Beta-Coupled High-Frequency Activity and Beta-Locked Neuronal Spiking in the Subthalamic Nucleus of Parkinson’s Disease. The Journal of Neuroscience 2014;34(38):12816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Meidahl AC, Moll CKE, van Wijk B, Gulberti A, Tinkhauser G, Westphal M, et al. Synchronised spiking activity underlies phase amplitude coupling in the subthalamic nucleus of Parkinson’s disease patients. Neurobiology of disease 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].van Wijk BC, Beudel M, Jha A, Oswal A, Foltynie T, Hariz MI, et al. Subthalamic nucleus phase-amplitude coupling correlates with motor impairment in Parkinson’s disease. Clin Neurophysiol 2016;127(4):2010–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Foffani G, Priori A, Egidi M, Rampini P, Tamma F, Caputo E, et al. 300-Hz subthalamic oscillations in Parkinson’s disease. Brain : a journal of neurology 2003;126(Pt 10):2153–63. [DOI] [PubMed] [Google Scholar]
- [58].McIntyre CC, Grill WM, Sherman DL, Thakor NV. Cellular Effects of Deep Brain Stimulation: Model-Based Analysis of Activation and Inhibition. Journal of neurophysiology 2004;91(4):1457–69. [DOI] [PubMed] [Google Scholar]
- [59].Garcia L, Audin J, D’Alessandro G, Bioulac B, Hammond C. Dual effect of high-frequency stimulation on subthalamic neuron activity. The Journal of neuroscience 2003;23(25):8743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bar-Gad I, Elias S, Vaadia E, Bergman H. Complex Locking Rather Than Complete Cessation of Neuronal Activity in the Globus Pallidus of a 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Treated Primate in Response to Pallidal Microstimulation. J Neurosci 2004;24(33):7410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Grill WM, Snyder AN, Miocinovic S. Deep brain stimulation creates an informational lesion of the stimulated nucleus. Neuroreport 2004;15(7):1137–40. [DOI] [PubMed] [Google Scholar]
- [62].Wilson CJ, Bryce Beverlin I, Netoff T. Chaotic desynchronization as the therapeutic mechanism of deep brain stimulation. Frontiers in systems neuroscience 2011;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Awad MZ, Irwin ZT, Vaden RJ, Guthrie BL, Walker HC. Short latency cortical evoked potentials elicited by subthalamic nucleus deep brain stimulation: Commentary and results from paired pulse studies. Clin Neurophysiol 2019. [DOI] [PubMed] [Google Scholar]
- [64].Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D. Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. Journal of neurophysiology 2007;98(6):3525–37. [DOI] [PubMed] [Google Scholar]
- [65].Fries P A mechanism for cognitive dynamics: neuronal communication through neuronal coherence. Trends Cogn Sci 2005;9(10):474–80. [DOI] [PubMed] [Google Scholar]
- [66].Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, et al. Dichotomous Organization of the External Globus Pallidus. Neuron 2012;74(6):1075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.