

Abstract
With the emergence of multidrug resistant Salmonella strains, the development of anti-Salmonella vaccines is an important task. Currently there are no approved vaccines against Salmonella Paratyphi A, the leading cause of paratyphoid fever. To fill this gap, oligosaccharides corresponding to the O-polysaccharide repeating units from the surface of Salmonella Paratyphi A have been synthesized through convergent stereoselective glycosylations. The synthetic glycan antigen was conjugated with a powerful immunogenic carrier system, the bacteriophage Qβ. The resulting construct was able to elicit strong and long-lasting anti-glycan IgG antibody responses, which were highly selective toward Salmonella Paratyphi A associated glycans. The availability of well-defined glycan antigen enabled the determination that one repeating unit of the polysaccharide is sufficient to induce protective antibodies, and the paratose residue and/or the O-acetyl modifications on the backbone are important for recognition by antibodies elicited by a Qβ-tetrasaccharide conjugate. Immune sera provided excellent protection to mice from lethal challenge with Salmonella Paratyphi A, highlighting the potential of the synthetic glycan-based vaccine.
Keywords: glycosylation, salmonella paratyphi A, synthesis, vaccine
Graphical Abstract
Salmonella Paratyphi A associated cell surface glycans have been synthesized for the first time through convergent stereoselective glycosylation reactions. The synthetic glycan was conjugated with a powerful immunogenic carrier, i.e., bacteriophage Qβ. The resulting conjugate induced high levels of IgG antibodies in mice and rabbits against the glycan. Sera from immunized rabbits effectively protected mice from bacterium induced death.
Introduction
Salmonella enterica subspecies enterica serovar Paratyphi A (S. Paratyphi A) is the most prevalent cause of paratyphoid fever,[1] resulting in an estimated 6 million cases worldwide annually.[2–3] While antibiotic treatment against paratyphoid fever reduces the risk of death,[4] the emergence of fluoroquinolone-resistant or multidrug-resistant (resistant to ampicillin, chloramphenicol, and trimethoprim-sulfamethoxazole) strains is a serious public health concern.[5–6]
Vaccines can provide powerful protection against salmonellosis.[7] However, no vaccines are available against S. Paratyphi A.[8] The cell surface O-polysaccharide (OPS) of S. Paratyphi A is a potential protective antigen and a target for vaccine development.[9–11] S. Paratyphi A OPS consists of a trisaccharide backbone repeat of D-mannose (Man), L-rhamnose (Rha), and D-galactose (Gal), with a paratose (Par) branch at C-3 of Man (Figure 1).[12] The hydroxyl groups at C-2 and C-3 positions of Rha can be acetylated[13] with the O-acetyl groups shown to be important for the antigenicity of the polysaccharides.[14] An additional structural feature of the OPS is that the 6-OH of Gal can be α-glucosylated. However, the extent of glucosylation is variable leading to greater structural heterogeneity of native OPS.
Figure 1:
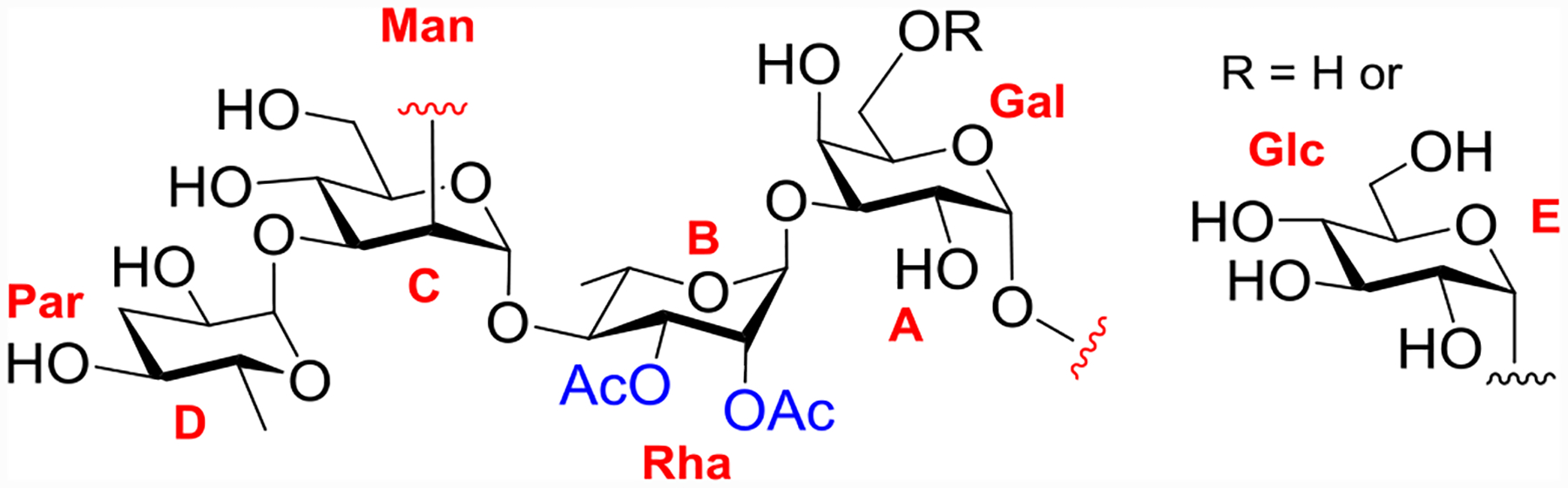
Representative structure of the repeating unit of S. Paratyphi A OPS.
S. Paratyphi A OPS has been investigated as a vaccine antigen.[9–11] However, isolation of polysaccharide from bacteria generally requires large-scale fermentation of pathogenic S. Paratyphi A organisms,[11] followed by thorough detoxification to remove endotoxin. Furthermore, the heterogeneous composition of the isolated polysaccharides renders it challenging to pinpoint the structural features needed for immune protection. With their well-defined structures, synthetic oligosaccharide antigens are attractive alternatives for vaccine development. While the oligosaccharide antigens from several other Salmonella strains have been prepared chemically before,[15–18] S. Paratyphi A glycans have not been synthesized. Herein, we report the first synthesis of S. Paratyphi A associated oligosaccharides 1-3 (Figure 2). As the tetrasaccharide 1 represents a typical repeating unit for S. Paratyphi A, it was evaluated in vaccine studies by conjugating it with a powerful virus like particle-based carrier, i.e., the bacteriophage Qβ, which elicited strong anti-glycan IgG antibody responses. The synthetic glycans helped shine light on the key structural epitope features for antibody recognition. Furthermore, antisera from rabbits immunized with Qβ−1 conjugate provided significant protection to mice against lethal challenge by S. Paratyphi A.
Figure 2.
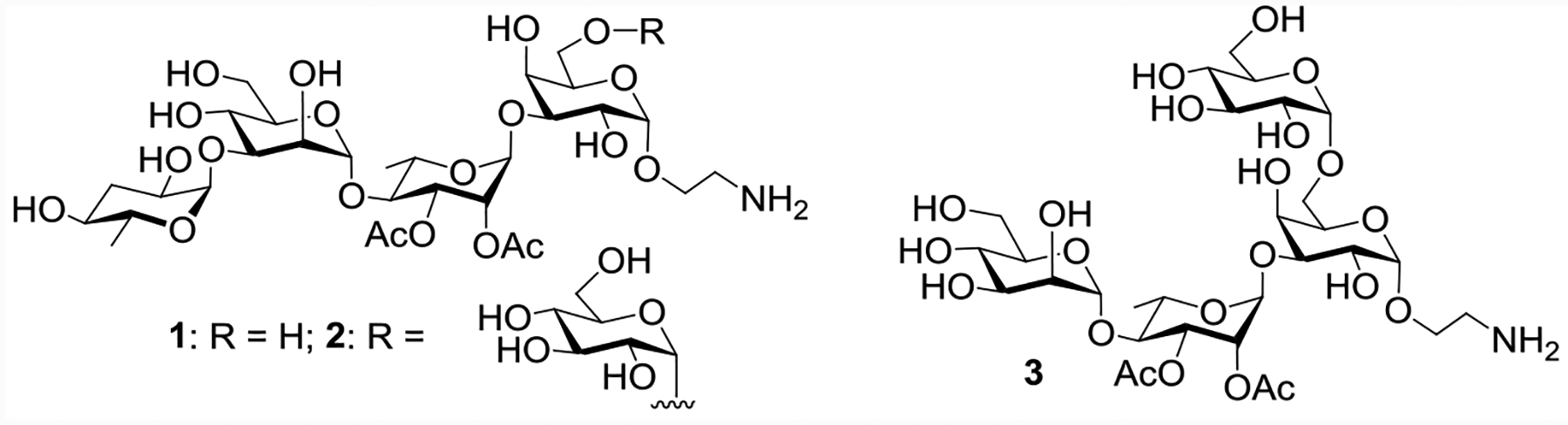
Structures of the S. Paratyphi A associated synthetic oligosaccharides 1-3.
Results and Discussion
Convergent stereoselective synthesis of S. Paratyphi A associated glycans 1–3
A key challenge in the chemical syntheses of S. Paratyphi A associated oligosaccharides is the stereoselective formation of the three 1,2-cis glycosyl linkages of α-paratoside, α-glucoside and α-galactoside. It was envisaged that the target oligosaccharides 1-3 can be obtained by stereoselective glycosylation of suitably functionalized monosaccharide intermediates 4, 5,[19] 6,[20] 7,[21] 8,[22] 9 and 10[23] (Figure 3).
Figure 3:
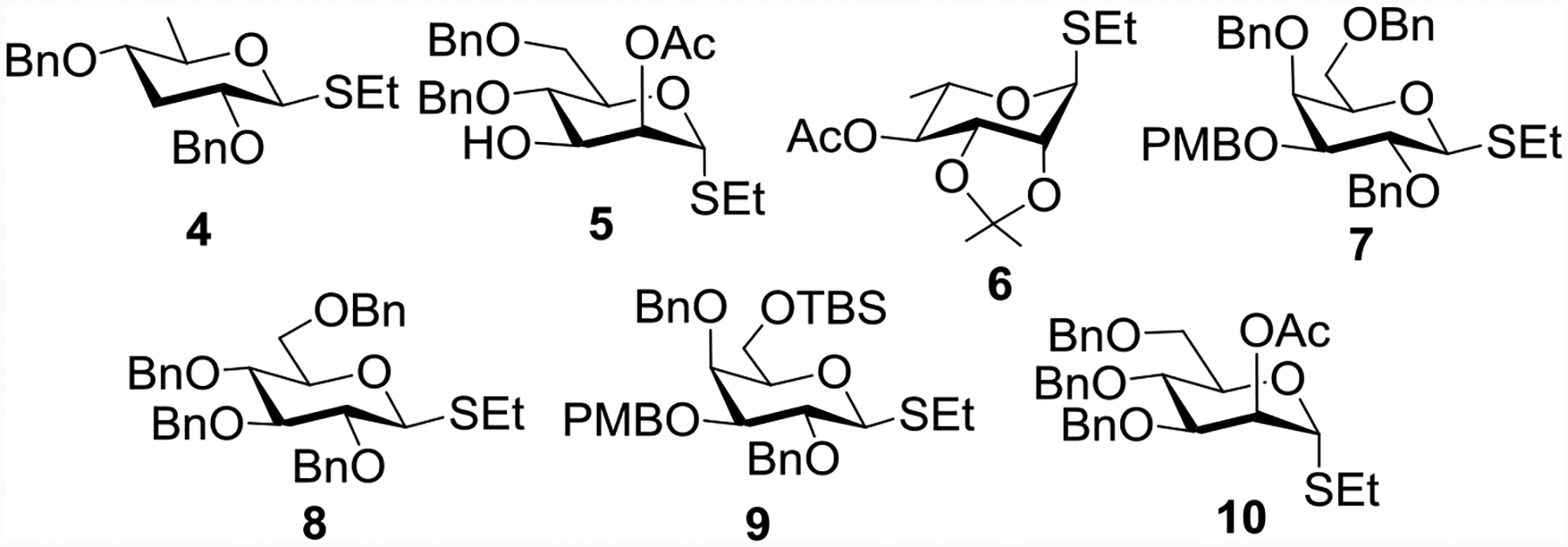
Structures of building blocks needed for S. Paratyphi A associated glycan synthesis.
The rare paratose building block 4 was prepared from ethyl 1-thio-β-D-glucopyranoside 11[24] (Scheme 1). Regioselective silylation[25] of the C3- and C6-OH groups of 11 gave compound 12 in 66% yield. Benzylation[26] of 12 followed by removal of the TBS groups using a catalytic amount of p-toluenesulfonic acid (p-TSA)[27] afforded compound 14. Dideoxygenation of compound 14 via the Barton-McCombie deoxygenation[28–29] method was carried out in one pot furnishing the dideoxy paratose donor 4 in 45% overall yield (Scheme 1).
Scheme 1:
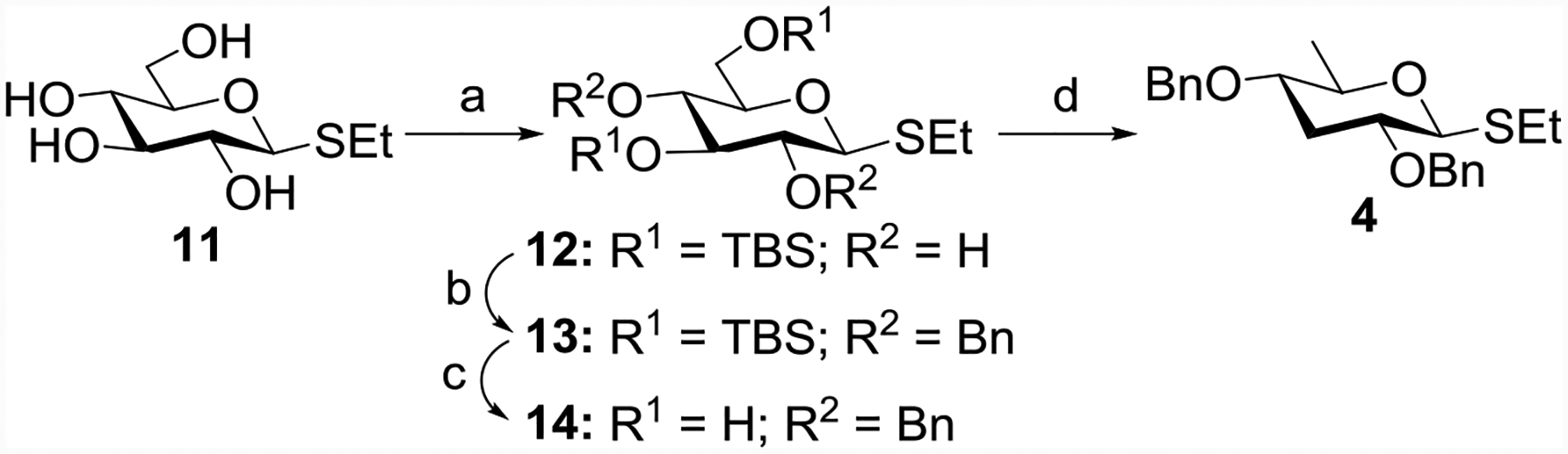
Reagents and conditions: (a) TBSCl, imidazole, DMF, r t, 12 h, 66%; (b) BnBr, NaH, DMF, 2 h, 0 °C, 88%; (c) p-TSA, CH3OH, r t, 2 h, 89%; (d) (i) NaH, imidazole, CS2, CH3I, THF, r t; (ii) tri-n-butyltin hydride (TBTH), azobis-isobutyronitrile (AIBN), reflux, 45% yield in one pot.
To prepare the thiogalactosyl donor 9, regioselective dibutyltin oxide mediated p-methoxybenzylation[30] at the C-3 hydroxyl group of ethyl 1-thio-β-D-galactopyranoside 15[31] followed by protection of C6-OH using t-butyldimethylsilyl chloride (TBSCl) in the presence of imidazole[32] gave compound 16 in 66% overall yield (Scheme 2). Compound 16 was benzylated to give the galactosyl thioglycoside donor 9 in 95% yield.
Scheme 2:
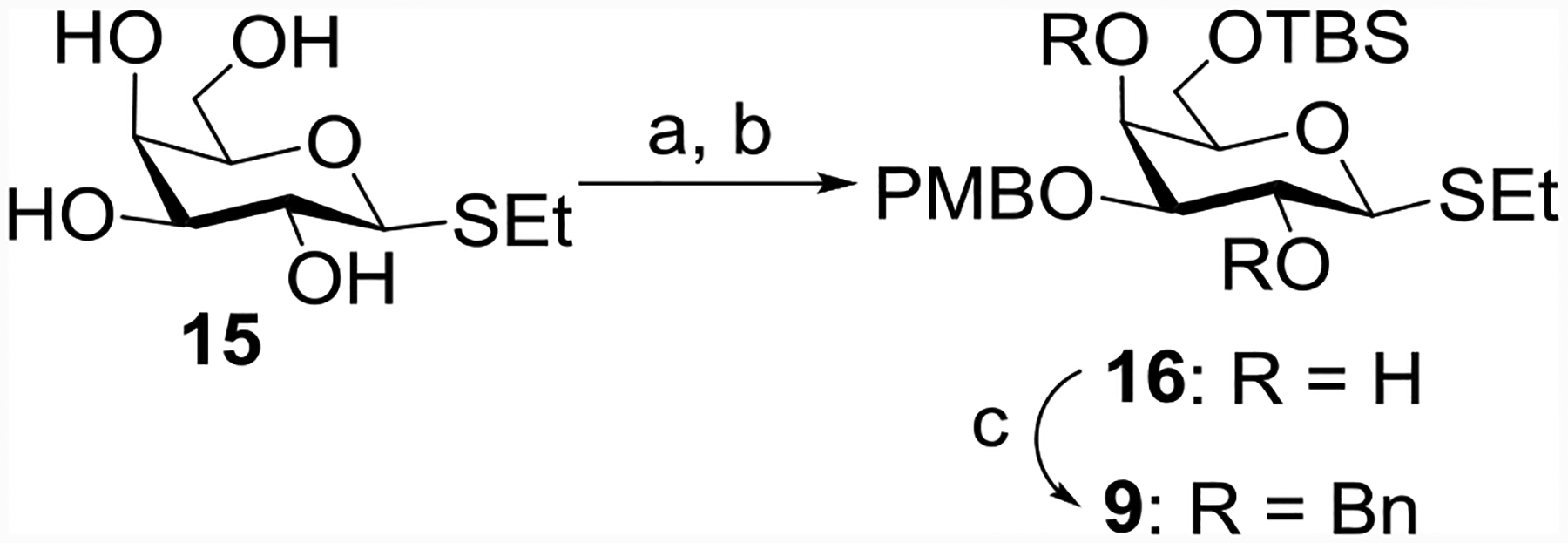
Reagents and conditions: (a) Bu2SnO, CH3OH, 70 °C, 3 h, then PMBCl, toluene, 60 °C, 14 h; (b) TBSCl, imidazole, DMF, r t, 16 h, 66 % in two steps; (c) BnBr, NaH, DMF, 0 °C, 2 h, 95%.
We next investigated the key α-paratosylation reaction using donor 4. Following the “armed-disarmed” glycosylation concept,[33–34] chemoselective activation of di-deoxy paratose donor 4 was achieved by a combination of N-iodosuccinimide (NIS) and trifluoromethane sulfonic acid (TfOH),[35–36] which glycosylated acceptor 5 leading to paratose containing disaccharides. Unexpectedly, the major anomer formed was the β-paratosylated disaccharide 17 (60%) together with the desired α-paratosylated disaccharide 18 (15%) as a minor product (Scheme 3). NMR signals for 17 were observed at δ 5.26 (d, J = 1.5 Hz, H-1C), 4.44 (d, J = 7.5 Hz, H-1D) in 1H NMR and at δ 101.6 (C-1D), 82.1 (C-1C) in its 13C NMR spectrum. The assignment of signals to rings C and D was made based on the typical chemical shift values of anomeric carbons of similar disaccharide thioglycosides.[37–38] From the anomeric carbon, the H-C correlations in 2D HMQC spectrum helped to assign the anomeric 1Hs. The relatively large coupling constant of H-1D suggested that stereochemical linkage was 1,2-trans between rings C and D. In comparison, NMR signals at δ 5.33 (d, J = 1.0 Hz, H-1C), 4.89 (d, J = 3.5 Hz, H-1D) in 1H NMR and at δ 98.3 (C-1D), 81.9 (C-1C) in 13C NMR were observed for compound 18, consistent with the assigned 1,2-cis linkage between rings C and D. Changing the parameters such as temperature, promoter and reaction solvents did not improve the yield of α-paratosylated product 18.
Scheme 3:
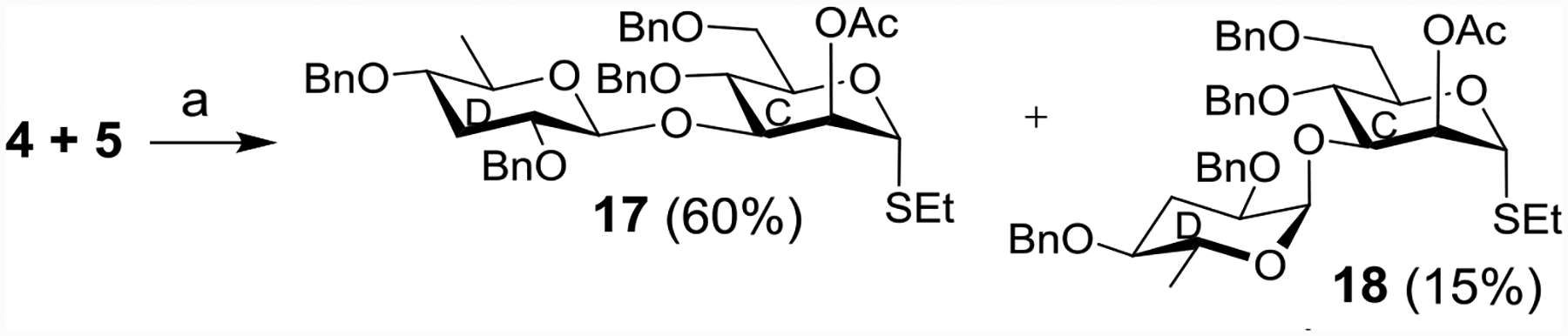
Reagents and conditions: NIS, TfOH, MS 4Å, CH2Cl2-Et2O (1:3), −20 °C, 20 min.
To overcome the challenge of α-paratosylation, an indirect approach was explored. Interestingly, glycosylation between the thioglucoside donor 13 and the acceptor 5 in the presence of NIS and TfOH promoter[35–36] gave a high selectivity of the 1,2-cis product 19 (72% yield) together with a minor quantity (~5%) of the β-anomer (Scheme 4).
Scheme 4:
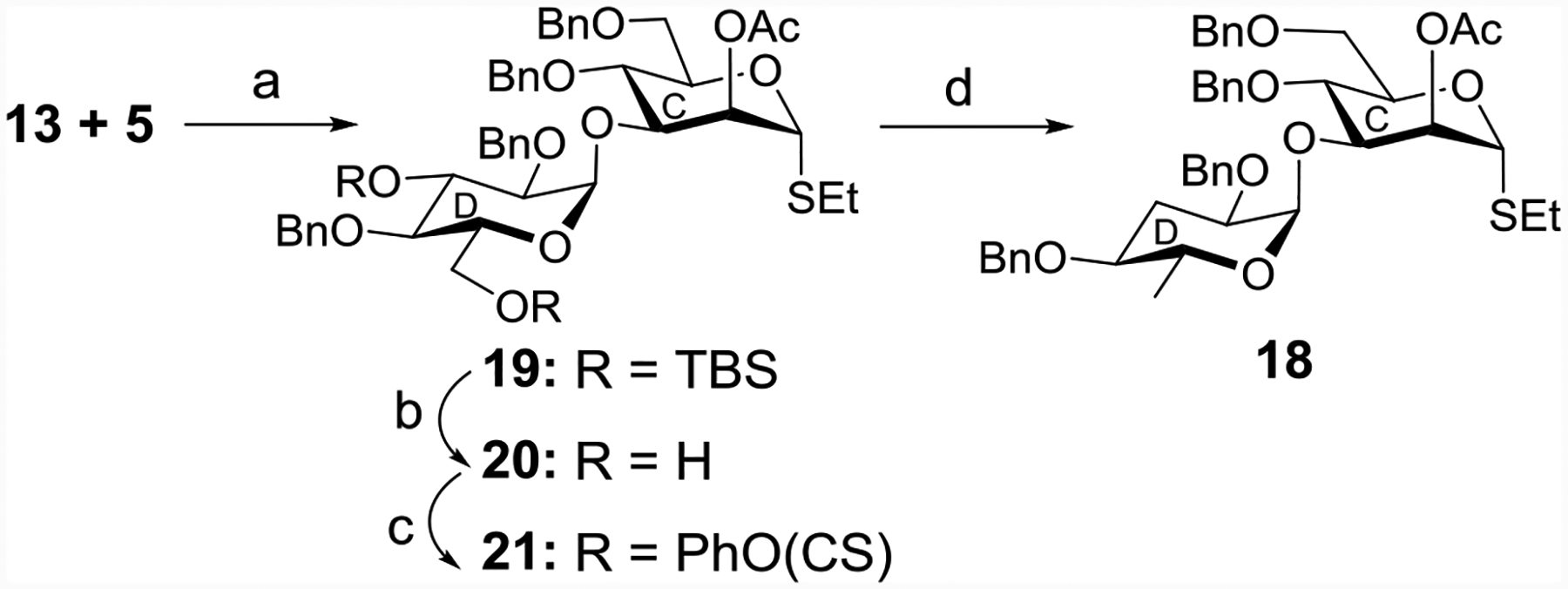
Reagents and conditions: (a) NIS, TfOH, MS 4Å, CH2Cl2, −40 °C, 15 min, 72%; (b) p-TSA, CH3OH, r t, 2 h, 79%; (c) PhOCSCl, pyridine, r t, 4 h, 91%; (d) TBTH, AIBN, toluene, reflux, 4 h, 55%.
Multiple factors can impact stereochemical control in glycosylation, especially in the absence of neighbouring group participation. Recently, a 2-deoxyglucosyl donor was shown to be more α-selective as compared to the corresponding thioglucoside donor,[39] which is different from our results where the paratose deoxy donor gave a lower α-selectivity than the glucoside. Crich and Vinogradova reported that while the 4,6-O-benzylidene protected glucosyl and mannosyl donors gave high 1,2-cis selectivities in glycosylation, the corresponding 2- or 3-deoxy donors led to erosion of stereoselectivities, which was explained in terms of the interactions between the C2-O2 and C3-O3 bonds on the formation of the glycosyl oxacarbenium ion intermediates.[40] The differential stereoselectivity observed between paratose donor 4 and glucoside 13 is possibly due to changes in stereochemical preferences on the nucleophile approaching the purported glycosyl oxacarbenium ions.
Removal of the TBS group from disaccharide 19 with a catalytic amount of p-TSA[27] followed by treatment with phenyl chlorothionoformate in pyridine[41] furnished the thionoformate derivative 21. The reduction of 21 with TBTH in the presence of AIBN[28, 42] provided the paratose disaccharide 18 in 55% yield. The configuration of the paratose disaccharide 18 was confirmed by the 1HNMR [4.89 (d, J = 3.5 Hz, 1 H, H-1D)] (Scheme 4).
Parallel to the preparation of disaccharide 18, the glycosylation reaction of the thiogalactoside donor 7[21] with 2-azidoethanol with NIS/TfOH promoter[35–36] followed by in situ removal of the PMB-group[43] by warming up the reaction provided compound 22 in 62% yield (Scheme 5). The configuration of the α-isomer 22 was confirmed by the 1H-NMR coupling constant (JH1-H2 = 3.5 Hz). The condensation reaction of 22 with 6 enabled the access to disaccharide 23 in 76% yield. De-O-acetylation of compound 23 using sodium methoxide[44] gave the disaccharide acceptor 24 in 92% yield, which was glycosylated stereoselectively by the paratose disaccharide donor 18 producing tetrasaccharide 25 in 71% yield as a single isomer. Global deprotection of tetrasaccharide derivative 25 was accomplished using a five step procedure involving (a) deacetylation with sodium methoxide;[44] (b) benzylation of the free hydroxyl groups using benzyl bromide and sodium hydride;[26] (c) hydrolysis of the acetonide group with 80% AcOH;[45] (d) acetylation of the C2-OH and C3-OH; and (e) hydrogenolysis over Pd(OH)2-C[46] to furnish tetrasaccharide 1. The anomeric configuration of the tetrasaccharide 1 was confirmed by NMR analysis[47] (δ 5.14 (d, J = 1.5 Hz, 1 H, H-1B), 5.05 (d, J = 3.5 Hz, H-1D, H-1A), 5.02 (d, J = 1.5 Hz, 1 H, H-1C)).
Scheme 5:
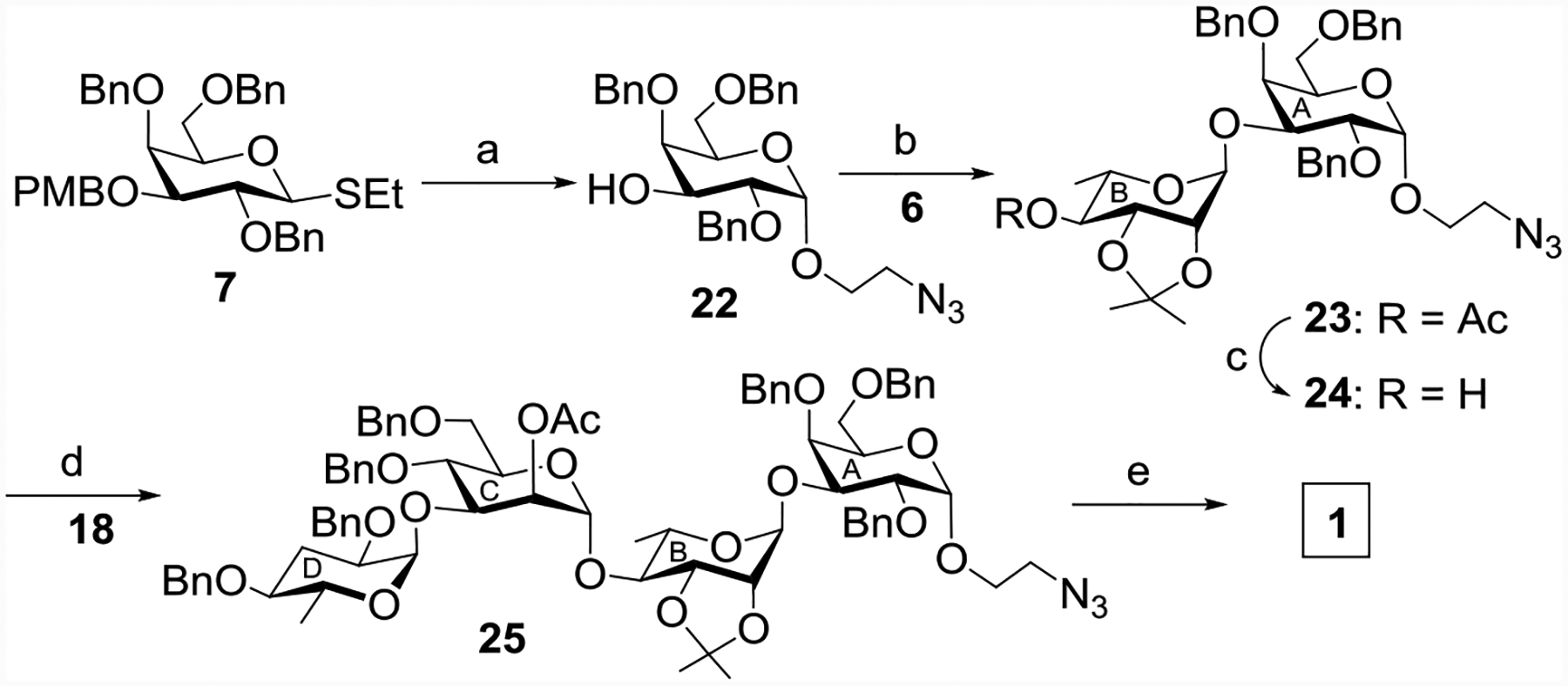
Reagents and conditions: (a) 2-azidoethanol, NIS, TfOH, MS 4Å, CH2Cl2-ether (1:4), −20 °C, 30 mins, then 0 °C, 1 h, 62%; (b) NIS, TMSOTf, MS 4Å, CH2Cl2, −20 °C, 30 min, 76%; (c) CH3ONa, CH3OH, r t, 2 h, 92%; (d) NIS, AgOTf, 4Å MS, CH2Cl2, −20 °C, 45 min, 71%; (e) (i) CH3ONa, CH3OH, r t, 2 h; (ii) BnBr, NaH, DMF, r t, 2 h; (iii) 80% AcOH, 80 °C, 2 h; (iv) Ac2O, pyridine, r t, 3 h; (v) 20% Pd(OH)2-C, H2, CH3OH, rt, 24 h, overall yield 47%.
In order to synthesize pentasaccharide 2, the coupling reaction of the 6-TBS protected thiogalactosyl donor 9 with 2-azidoethanol provided compound 26 in 70% yield (Scheme 6). The removal of the TBS protecting group from 26 using tetrabutylammonium fluoride (TBAF) in THF[32] resulted in compound 27 in 90% yield. Stereoselective 1,2-cis glycosylation of compound 27 with compound 8 gave the disaccharide derivative 28 in 72% yield together with minor quantity (~5%) of its anomer, which was separated by column chromatography. Stereoselective glycosylation of 28 by the rhamnoside donor 6 gave the protected trisaccharide derivative 29 in 76% yield. Saponification of the compound 29 using sodium methoxide[44] provided the trisaccharide acceptor 30 in 92% yield. Convergent synthesis of the pentasaccharide derivative 31 was achieved in 74% yield as a single isomer by stereoselective [2+3] glycosylation of the disaccharide donor 18 and the trisaccharide acceptor 30 promoted by a combination of NIS and AgOTf (Scheme 6).[48] Separately, the trisaccharide acceptor 30 was glycosylated by the ethyl 2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranoside 10[23] furnishing tetrasaccharide 32 in 77% yield (Scheme 7). Global deprotections of pentasaccharide 31 and tetrasaccharide 32 were achieved similarly to that of 26 providing the pentasaccharide 2 and tetrasaccharide 3 in 53% and 47% overall yields respectively (Schemes 6 and 7).
Scheme 6:
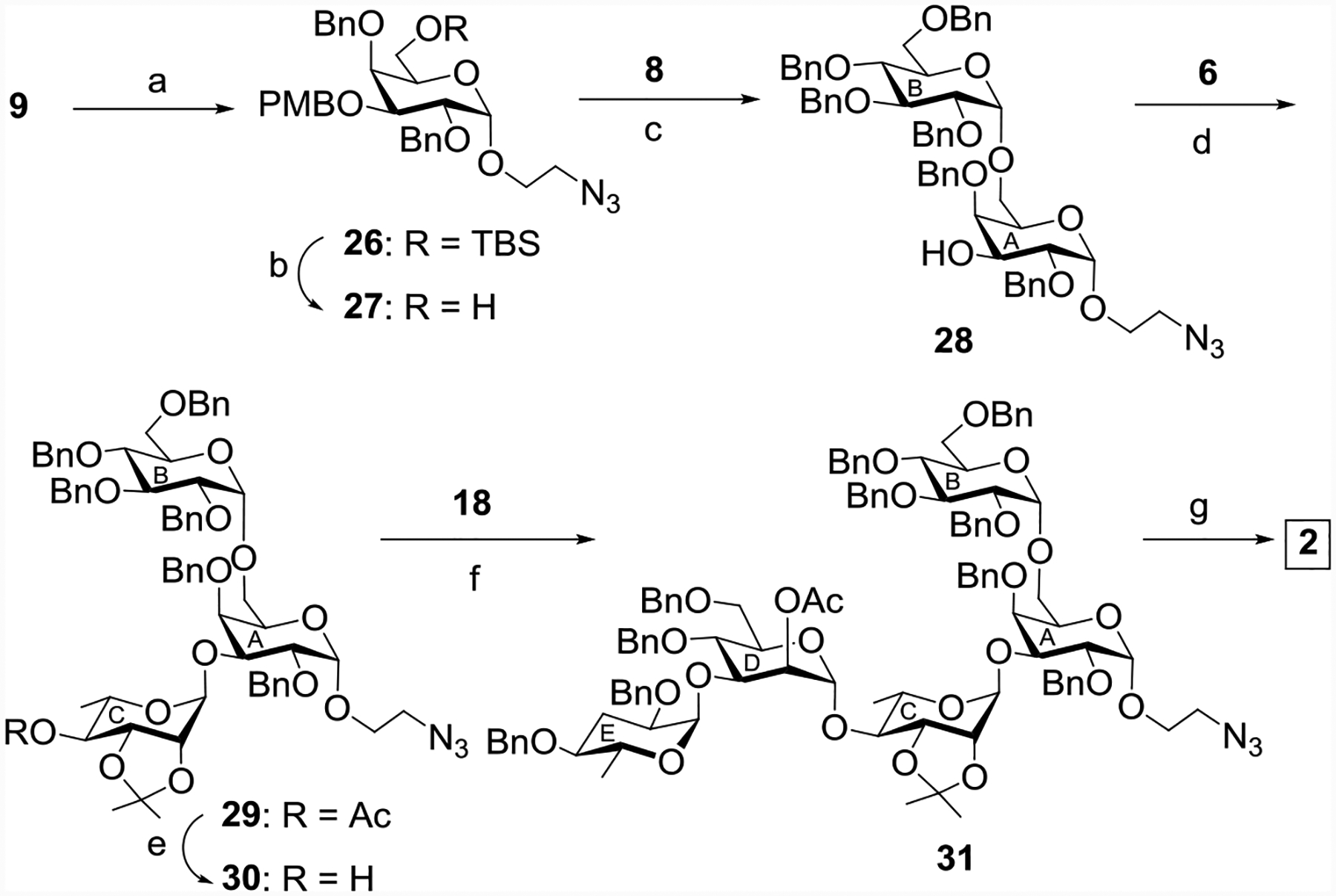
Reagents and conditions: (a) 2-azidoethanol, NIS, TMSOTf, MS 4Å, CH2Cl2-ether (1:4), −20 °C 1.5 h, 83%; (b) TBAF, THF, r t, 4 h, 90%; (c) NIS, TfOH, MS 4Å, CH2Cl2-ether (1:4), −20 °C, 30 min, then 0 °C, 1 h, 72%; (d) NIS, TMSOTf, MS 4Å, CH2Cl2, −20 °C, 40 min, 76%; (e) CH3ONa, CH3OH, r t, 4 h, 92%; (f) NIS, AgOTf, MS 4Å, CH2Cl2, −20 °C, 45 min, 74%; (g) (i) CH3ONa, CH3OH, r t, 2 h; (ii) BnBr, NaH, DMF, r t, 2 h; (iii) 80% AcOH, 80 °C, 2 h; (iv) Ac2O, pyridine, 3 h; (v) 20% Pd(OH)2-C, H2, CH3OH, r t, 24 h, overall yield 53%.
Scheme 7:
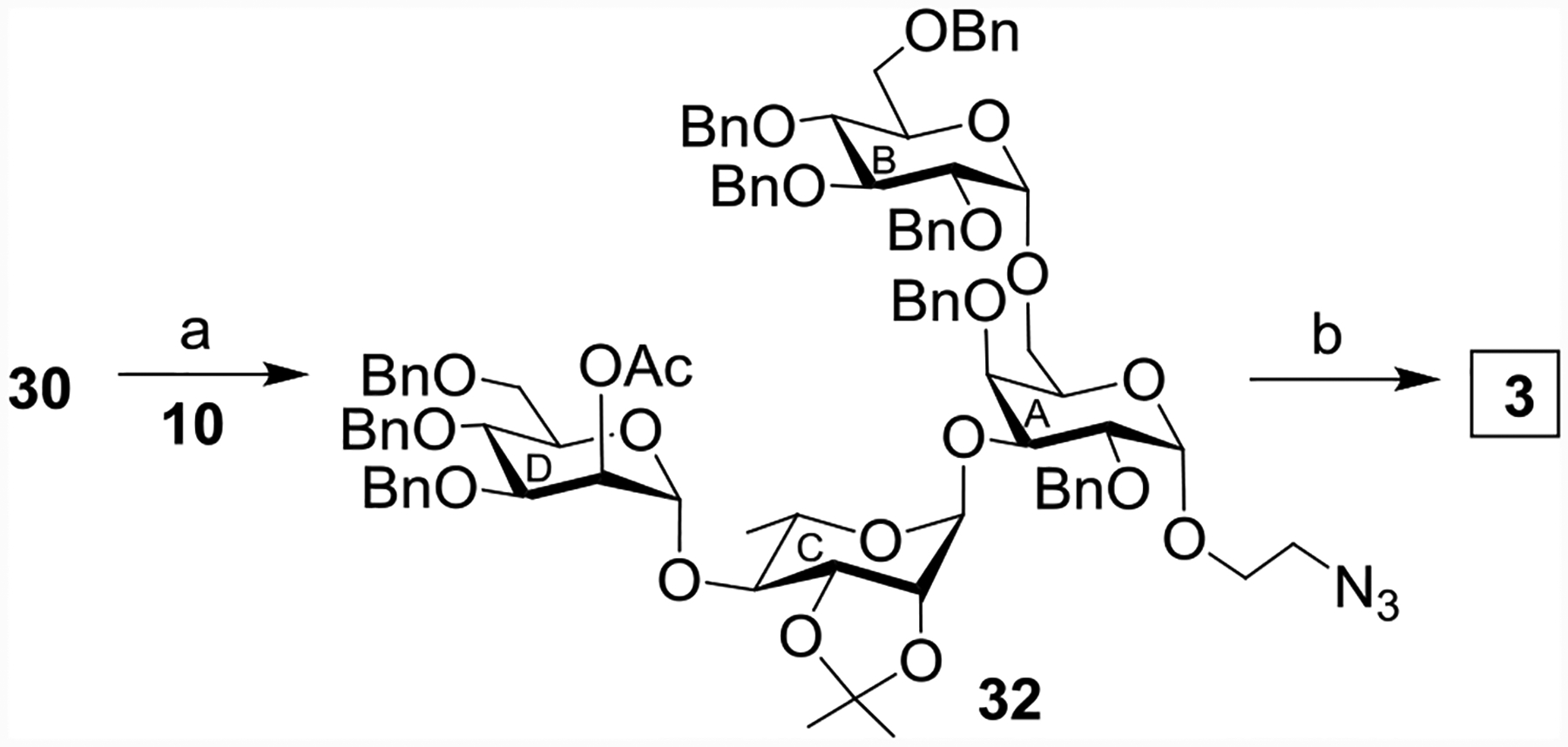
Reagents and conditions: (a) NIS, AgOTf, MS 4Å, CH2Cl2, −30 °C, 30 min, 77%; (b) (i) CH3ONa, CH3OH, r t, 2 h; (ii) BnBr, NaH, DMF, r t, 2 h; (iii) 80% AcOH, 80 °C, 2 h; (iv) Ac2O, pyridine, 3 h; (v) 10% Pd-C, H2, CH3OH, r t, 24 h, overall yield 47%.
Bioconjugation of S. Paratyphi A glycans to enable vaccine studies
Bioconjugation of tetrasaccharide 1 with the bacteriophage Qβ virus-like particle was explored next for vaccine studies. As carbohydrate antigens are typically unable to activate helper T cells, which are critical for production of high affinity and long-lasting anti-glycan IgG antibodies, antigen conjugation to T-cell-dependent carriers is critical in overcoming this limitation.[49–50] Bacteriophage Qβ is a powerful carrier for glycoconjugate-based vaccine development due to its ability to display antigens on its capsid in a highly organized manner for potent antibody generation,[51–53] which has translated into higher antibody responses than those observed with the popular benchmark carrier keyhole limpet hemocyanin (KLH).[54–55] In this work, tetrasaccharide 1 was derivatized with a bifunctional linker 33, and subsequently conjugated with bacteriophage Qβ (Scheme 8a). Mass spectrometry analysis of the resulting Qβ-glycan 1 conjugate showed that there were on average 290 copies of glycan per capsid (Figure S1).
Scheme 8:
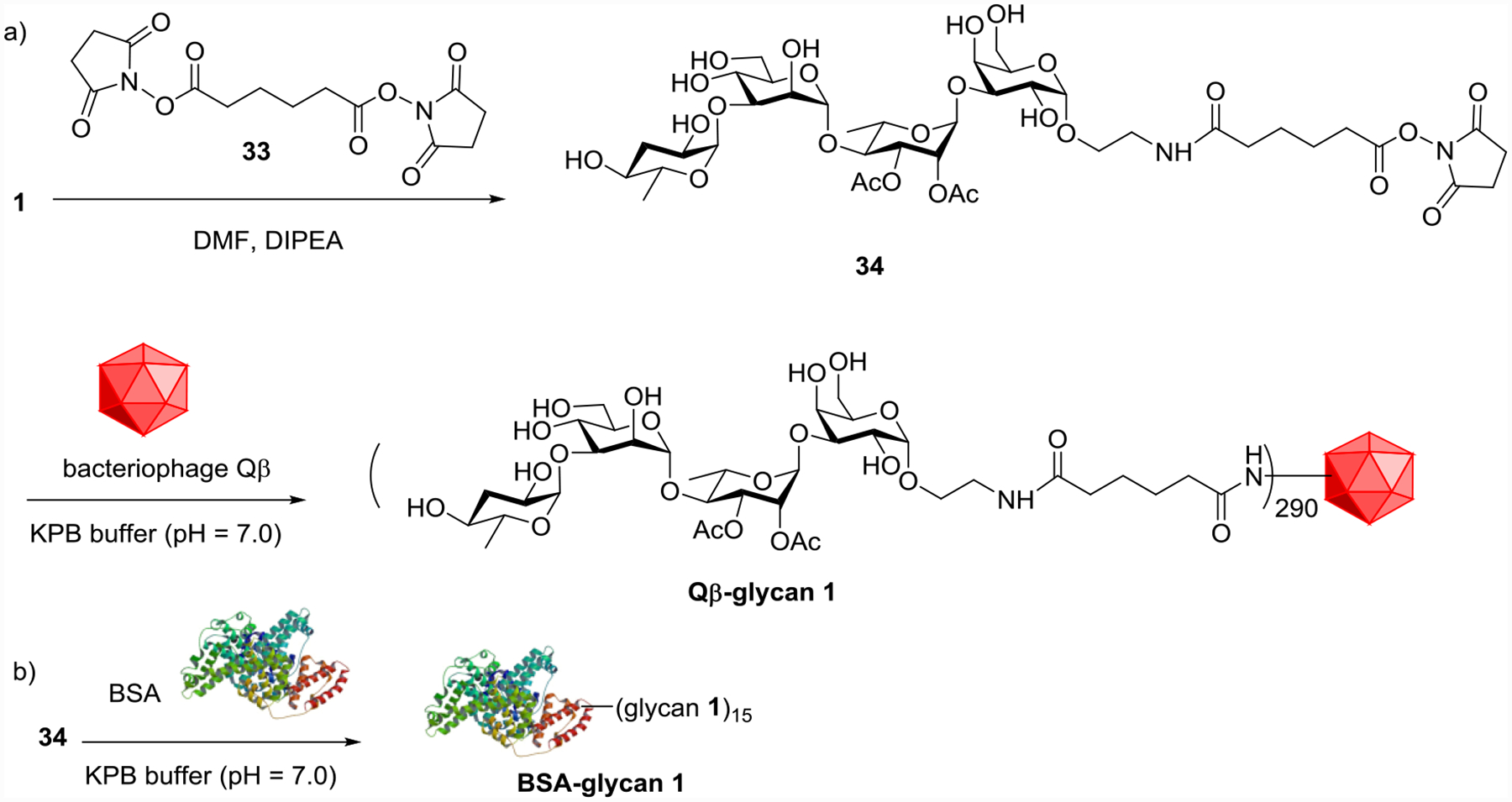
Conjugation of S. Paratyphi A glycan 1 with a) Qβ carrier, and b) BSA.
To aid in the immunological analysis, S. Paratyphi A glycan 1 was conjugated to bovine serum albumin (BSA) for use as a coating antigen in enzyme linked immunosorbent assays (ELISA) to specifically quantify the anti-glycan antibody responses while excluding antibodies recognizing the Qβ carrier (Scheme 8b). In a similar manner, BSA conjugates with pentasaccharide 2 and tetrasaccharide 3 were synthesized. In addition, tetrasaccharide 35 from Salmonella Enteritidis (S. Enteritidis), which contains a tyvelose moiety on the same trisaccharide backbone, was conjugated with BSA.[18]

Qβ-glycan 1 conjugate can elicit potent and selective IgG antibody responses against the glycan
With Qβ-glycan 1 construct in hand, its immunogenicity was first evaluated in mice. Groups of 5 mice were immunized three times with Qβ-glycan 1 (1 μg or 4 μg of carbohydrate per injection) on days 0, 14 and 28. As controls, mice received either Qβ or glycan 1 alone following the same immunization protocol. High levels of anti-glycan 1 IgG antibodies were observed 35 days after the initial immunization, with an average IgG titer of 2.2 × 106 and 3.1 × 106 ELISA units for Qβ-glycan 1 immunized groups receiving 1 μg and 4 μg of glycan, respectively (Figure 4). In contrast, mice immunized with Qβ or glycan 1 alone only mounted anti-glycan IgG titers around 2,000 ELISA units, which were similar to pre-vaccination titers. Evaluation of IgG subtypes raised against 4 μg of Qβ-glycan 1 showed that all major IgG subclasses (IgG1, IgG2b, IgG2c and IgG3) were induced against glycan 1, while IgM antibody levels were insignificant in comparison (Figure S3).
Figure 4:
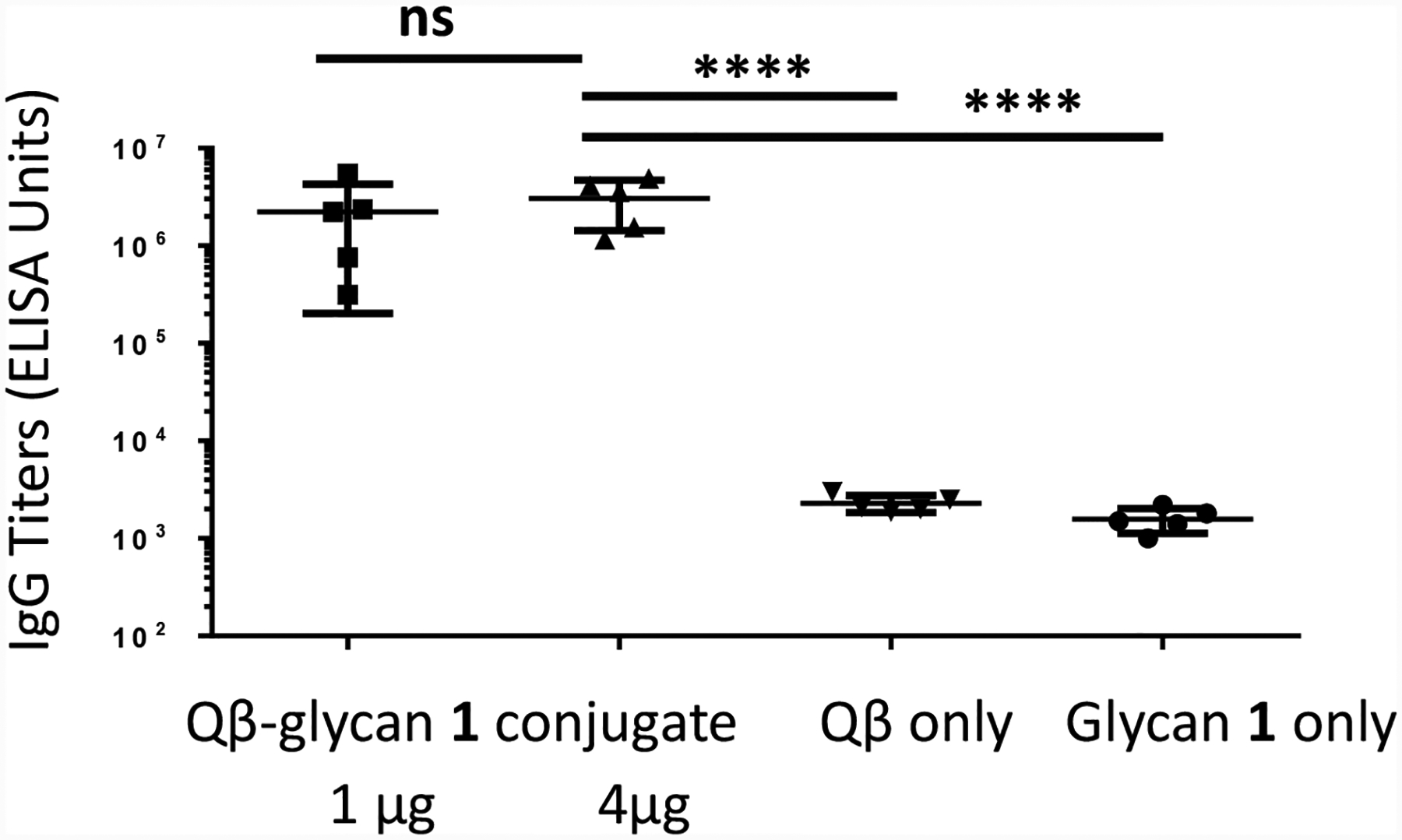
Robust anti-glycan 1 antibody responses were induced by Qβ-glycan 1 in mice. Anti-glycan 1 IgG titers on day 35 after the first immunization from Qβ-glycan 1 (1 and 4 μg of glycan) immunized and control groups. Each symbol represents one mouse. Geometric mean titers are indicated by the longer horizontal line ± standard deviation. Statistical analysis was performed using a 2-tailed Student’s t-test. ns: p > 0.05; ****: p < 0.0001.
To assess the structural selectivity and specificity of antibodies induced by Qβ-glycan 1, the binding of the post-immunization sera with BSA conjugates of pentasaccharide 2, tetrasaccharide 3 as well as S. Enteritidis OPS tetrasaccharide 35[18] were analysed (Figure 5). Antiserum binding to the S. Enteritidis tetrasaccharide 35 was much weaker, suggesting the paratose and/or the backbone O-acetyl moieties are likely part of the immunodominant epitopes. This was further confirmed by the weak antiserum recognition of tetrasaccharide 3, which lacks the dideoxy sugar residue. On the other hand, comparison of immune sera binding to tetrasaccharide 1 vs pentasaccharide 2 indicate that the reducing end α-glucoside moiety is not as critical for antibody recognition.
Figure 5:
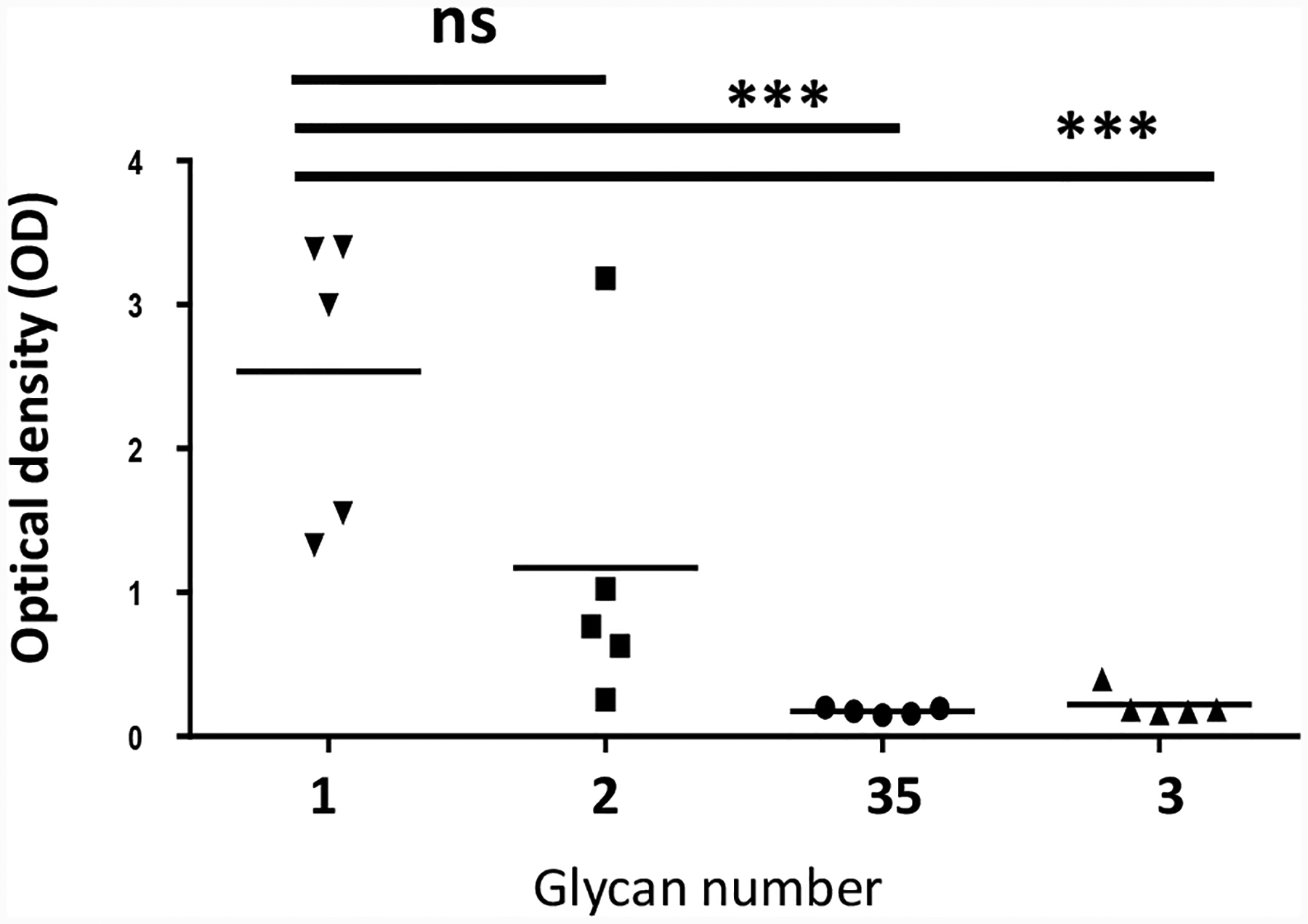
IgG antibodies in sera (serum dilution at 102,400-fold) from mice immunized with Qβ-glycan 1 were selective towards glycan 1 and glycan 2, while recognition of glycans 3 and 35 were much weaker, when assessed against various synthetic Salmonella glycans by ELISA. Each symbol represents an individual mouse, and solid bars indicate the group’s geometric mean value. Statistical analysis was performed using 2-tailed Student’s t-test. ns: p > 0.05; ***: p < 0.001.
An important consideration for an effective vaccine is whether the immune responses are long lasting. To evaluate this, the levels of anti-glycan IgG antibodies induced by Qβ-glycan 1 were monitored over time. As shown in Figure 6, the average IgG titers from Qβ-glycan 1 immunized mice were the highest on day 56, reaching 5.5 × 106 ELISA units. The strong IgG antibody responses were persistent with an average IgG titer of 1 million ELISA units remaining on day 477. Control mice immunized with Qβ only showed low anti-glycan titers (~ 2,000 ELISA units) throughout.
Figure 6.
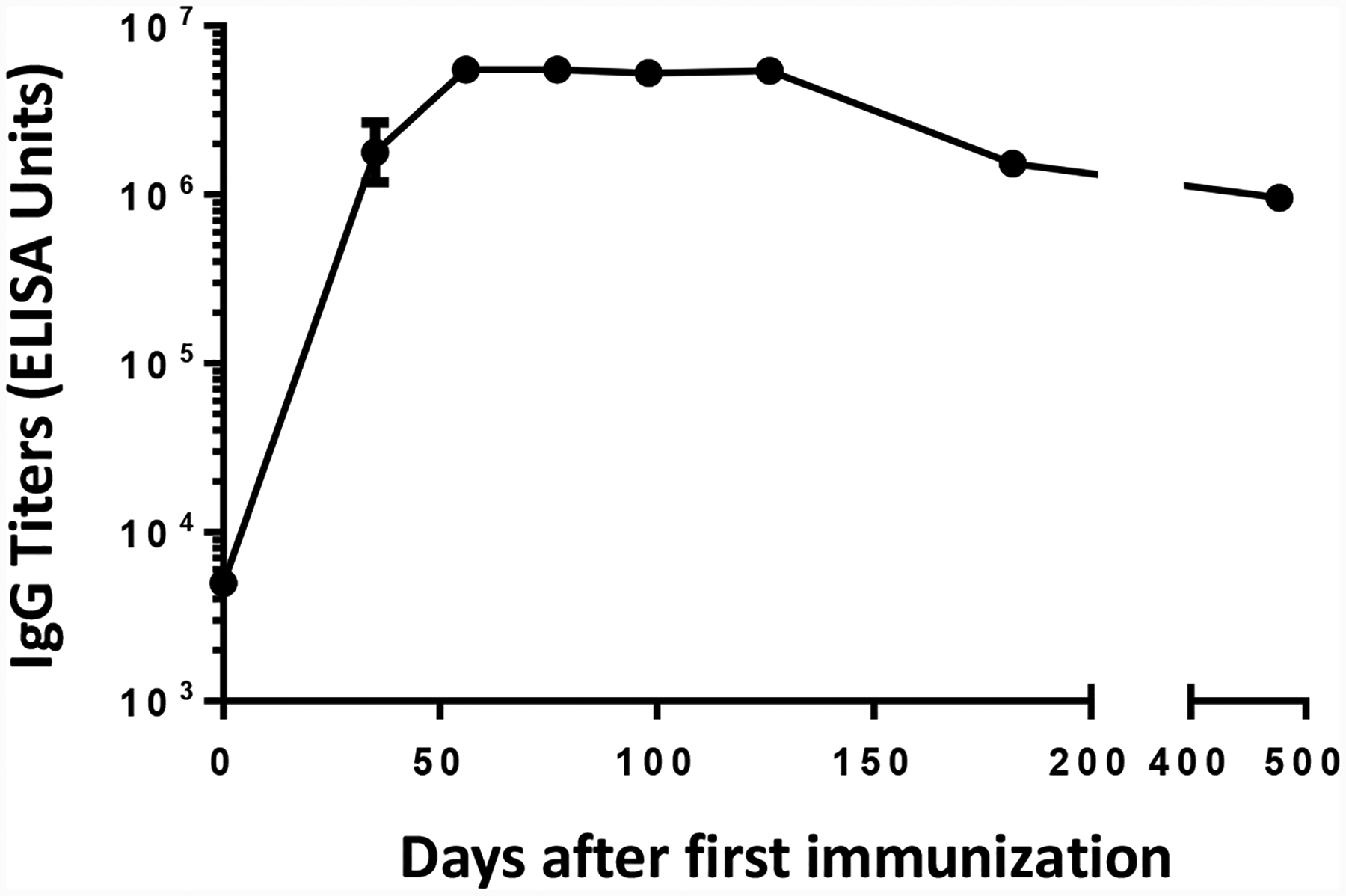
Immunization with Qβ-glycan 1 conjugate (4 μg) induced long lasting anti-glycan IgG antibody responses. There were five mice at each time-point with the geometric mean titer ± standard deviation shown.
Immunization with Qβ-glycan 1 elicited high titers of anti-glycan IgG antibodies capable of protecting mice from lethal challenge with S. Paratyphi A
To demonstrate immunogenicity in multiple species, we immunized rabbits subcutaneously with Qβ-glycan 1 following similar protocols as the mouse immunization. ELISA analysis showed that while the pre-immunization sera had little anti-glycan IgG antibodies (titers below 2,000), the average IgG titers in post-immunization sera reached over 10 million ELISA units, suggesting strong antibody responses were produced by vaccination in rabbits (Table S1a). Furthermore, the post-immunization sera also strongly recognized the native OPS isolated from S. Paratyphi A strain ATCC 9150 (Table S1b). Compared to rabbit sera, mouse serum recognition of S. Paratyphi A OPS was much weaker (data not shown), which was consistent with our prior results with a synthetic S. Enteritidis Qβ-glycan vaccine.[18] This is possibly due to differences in the paratope repertoire between mice and rabbits, or structural differences in the binding pockets of rabbit compared to mouse antibodies. Thus, rabbit sera were utilized for further analyses.
The ability of post-immunization rabbit sera to recognize native S. Paratyphi A bacteria was analysed by flow cytometry. As shown in Figure 7a, negligible binding was found for pre-immunization control sera even at a 1:40 dilution as compared to samples without sera. In contrast, the IgG antibodies elicited by Qβ-glycan 1 in rabbits potently bound to S. Paratyphi A (Figure 7a). The binding of sera from Qβ-glycan 1 immunized rabbits to S. Paratyphi A was dose dependent (Figures 7b,c).
Figure 7:
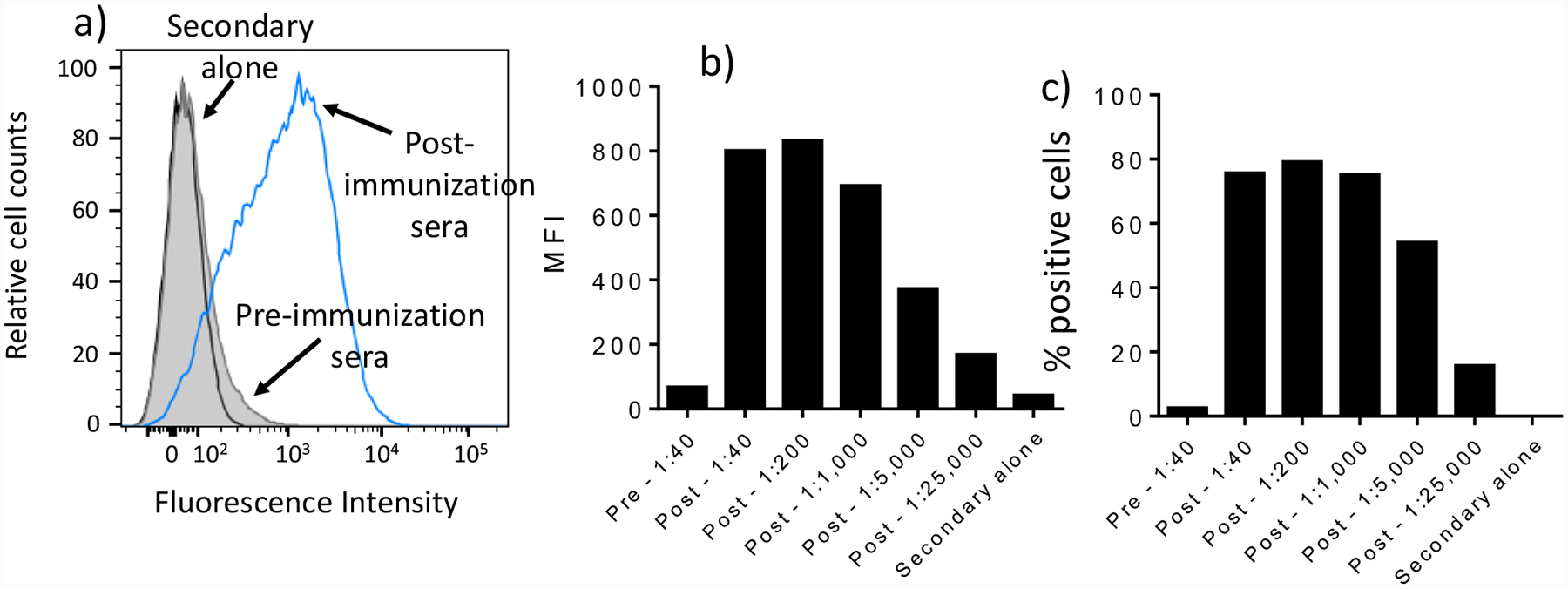
Qβ-glycan 1 induced rabbit antibodies bind S. Paratyphi A bacteria. a) Flow cytometry graphs of post-immunization sera (1:40 dilution) binding with S. Paratyphi A strain ATCC 9150, as compared to anti-rabbit IgG secondary antibody alone or pre-immunization rabbit sera (1:40 dilution). b) Mean fluorescence intensities (MFI) of S. Paratyphi A cells, and c) percentages of positive S. Paratyphi A cells stained by various dilutions of sera as quantified by flow cytometry. Positive cells are defined as fluorescence intensities of cells higher than 405 upon antibody binding. Pre-, pre-immunization sera; post-, post-immunization sera. Secondary alone refers to cells treated with secondary antibody only without any sera.
Having established that immune sera strongly recognized intact S. Paratyphi A, we next assessed the ability of vaccine-induced antibodies to opsonize S. Paratyphi A and mediate phagocytosis by macrophages.[56] Opsonophagocytic antibody activity is an important functional mechanism for antibody-mediated clearance of S. Paratyphi A.[57] Antisera from Qβ-glycan 1 vaccinated rabbits markedly enhanced uptake of S. Paratyphi A bacteria by murine J774 macrophage, as compared to pre-immunization sera from these rabbits as well as sera generated after immunization with Qβ particles alone (Figure 8).
Figure 8:
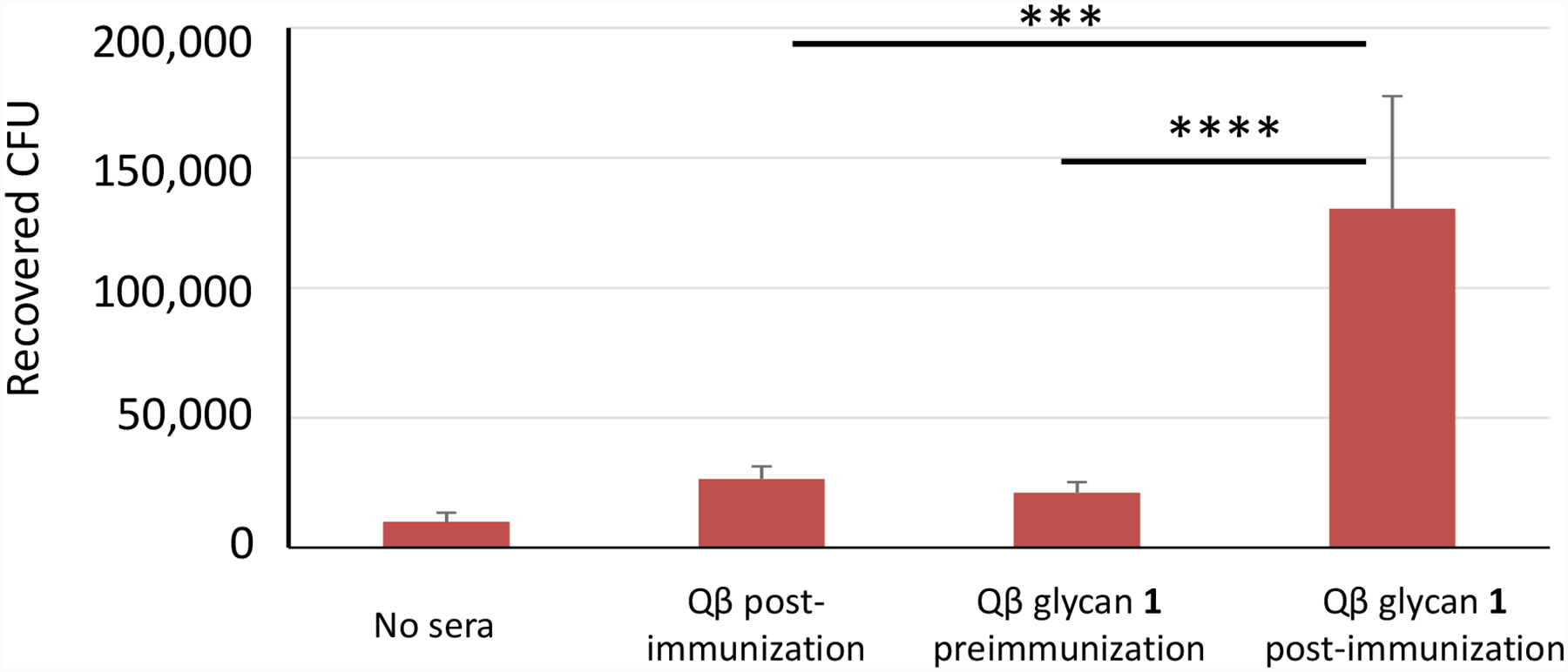
Rabbit anti-Qβ-glycan 1 sera enhance opsonophagocytosis of S. Paratyphi A. Log-phase cultures of S. Paratyphi A strain ATCC 9150 were first opsonized with heat-inactivated rabbit sera and then added to partially confluent monolayers of J774 macrophages. Gentamicin was then applied to kill non-phagocytosed bacteria, and internalized bacteria (represented as colony forming units [CFUs]) were enumerated by serial dilution and plating. Bars indicate the mean + standard deviation and are derived from four independent experiments. Statistical analysis was performed using a 2-tailed Student’s t-test. ***: p < 0.001; ****: p < 0.0001.
To demonstrate the protective efficacy of anti-Qβ-glycan 1 rabbit antibodies, we used a S. Paratyphi A murine challenge model.[58] BALB/c mice were passively administered pooled pre- or post-immunization sera or phosphate buffered saline (PBS) as a negative control via the intraperitoneal (i.p.) route and then challenged by the same route with S. Paratyphi A strain ATCC 9150 in 10% hog gastric mucin (HGM) (Figure 9). A group of 5 mice was administered PBS with 10% HGM as a control. By day 3 post infection, 16.7% (3/18) mice receiving pre-immunization sera had survived, which was similar to the survival rate for the PBS/HGM control group (30% [3/10]). In comparison, 94.4% (17/18) of the mice that received post-immunization sera were protected from bacterial challenge with a vaccine efficacy of 93.3% (p < 0.0001) (Figure 9).
Figure 9:
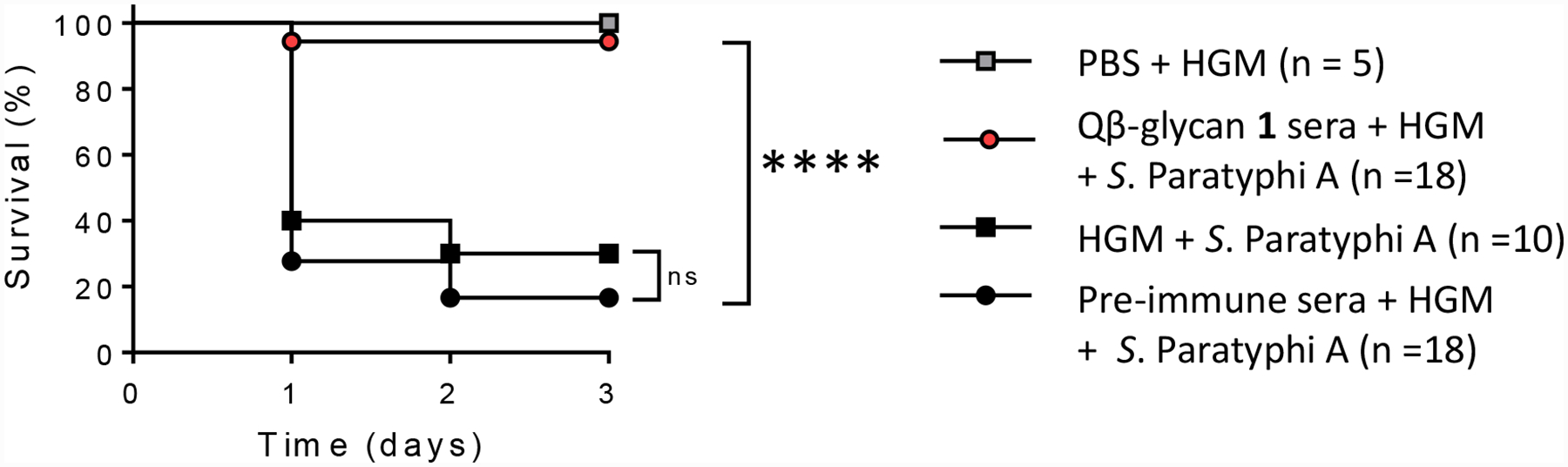
Passive transfer of Qβ-glycan 1 immune rabbit sera protects mice against lethal S. Paratyphi A challenge. Pools of rabbit pre- and post-immunization sera were heat-inactivated and diluted 1:10 in PBS. These sera were then administered via the i.p. route to BALB/c mice (n=18/group). PBS was used as a negative control (n=10). Mice were challenged i.p. 6 hours later with 7.3 × 103 CFU of S. Paratyphi A strain ATCC 9150 in 10% HGM. A separate group of mice (n=5) was pre-treated with PBS followed by 10% HGM without S. Paratyphi A. Statistical significance was determined by a 2-tailed Fisher’s exact test, and comparisons between groups are indicated with brackets. P-values of <0.05 were considered statistically significant. **** p < 0.0001; ns: non-significant.
Conclusion
Oligosaccharides associated with S. Paratyphi A OPS have been synthesized using concise synthetic strategies, which involve convergent stereoselective [2+2] and [2+3] block glycosylation reactions. The tetrasaccharide antigen 1 was conjugated to bacteriophage Qβ, which was found to induce potent IgG antibody responses in both mice and rabbits, with high selectivities toward S. Paratyphi A associated glycans. Antibodies produced in immunized rabbits were able to recognize native S. Paratyphi A bacteria and promote their clearance by macrophages. Excitingly, passive transfer of antisera from rabbits immunized with Qβ-glycan 1 provided close to complete protection against lethal S. Paratyphi A challenge in mice.
The availability of structurally well-defined glycan antigens enabled us to establish that the paratose residue and/or the O-acetyl modifications on the backbone are important for recognition by antibodies produced through the Qβ−1 conjugate. One unit from the repeating polysaccharide sequence was sufficient to elicit protective antibodies, which suggests a promising direction to develop synthetic antigen based anti-Salmonella vaccines.
Experimental Section
General methods
All chemicals were reagent grade and were used as received from the manufacturer, unless otherwise noted. All reactions were monitored by thin layer chromatography over silica gel coated TLC plates. The spots on TLC were visualized by warming ceric sulphate (2% Ce(SO4)2 in 2N H2SO4) sprayed plates in hot plate. Silica gel 230–400 mesh was used for column chromatography. NMR spectra were recorded on Bruker Avance 500 MHz using CDCl3 as solvent and TMS as internal reference unless stated otherwise. Chemical shift value is expressed in δ ppm. The complete assignment of proton and carbon spectra was carried out by using a standard set of NMR experiments, e.g. 1H NMR, 13C NMR, 13C DEPT 135, 2D COSY and 2D HSQC. Mass spectra were obtained by ESI mass spectra (Water Xevo G2-S Q-TOF LC-MS instrument). Optical rotations were recorded in a Jasco P-2000 polarimeter. For purification of Qβ, centrifugal filter units of 10, 000 and 100, 000 molecular weight cut-off (MWCO) were purchased from EMD Millipore. ESI-TOF LC-MS analysis was performed on each purified Qβ sample (1 μL, 1 mg/mL) after denaturing (Waters Xevo G2-XS UPLC/MS/MS). MALDI-TOF MS analysis was performed on each purified BSA sample (10 μL, 1 mg/mL) after denaturing and desalting the sample using Cleanup C18 Pipette Tips (Agilent Technologies). The mixture (0.6 μL) and matrix solution (0.6 μL, 10 mg/mL sinapic acid in 50/50/0.1 CH3CN/H2O/TFA) was spotted on a MALDI plate, air-dried, and analysed by MALDI-TOF mass spectrometry (Applied Biosystems Voyager DE STR). Protein concentration was measured using the Coomassie Plus Protein Reagent (Bradford Assay, Pierce) with bovine serum albumin (BSA) as the standard.
Characterization of anomeric stereochemistry.
The stereochemistries of the newly formed glycosidic linkages were determined by 3JH1,H2 through 1H-NMR and/or 1JC1,H1 through gHMQC 2-D NMR (without 1H decoupling). For the galactoside linkages, smaller coupling constants of 3JH1,H2 (around 3 Hz) indicate α linkages and larger coupling constants 3JH1,H2 (7.2 Hz or larger) indicate β linkages. For all glycosyl linkages including mannoside, rhamnoside and paratoside, 1JC1,H1 around 170 Hz suggests α-linkages and 160 Hz suggests β-linkages.[47]
Ethyl 3,6-di-O-(tert-butyldimethylsilyl)-1-thio-β-D-glucopyranoside (12):
To a solution of compound 11 (11.4 g, 51 mmol) in DMF (100 ml) were added imidazole (8.6 g, 127 mmol) and TBSCl (19 g, 127 mmol) at 0 °C. The mixture was stirred at room temperature for 12 h and then quenched with CH3OH (5 ml). The mixture was diluted with EtOAc (200 ml) and washed with 1 M HCl, satd. NaHCO3, and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. The crude product was purified by column chromatography over SiO2 using hexane-EtOAc (5:1) as the eluant to give the pure compound 12 (15 g, 66%). Colorless oil; [α]D25 −12 (c 1.0, CHCl3); IR (neat): 3339, 2925, 2850, 1649, 1609, 1554, 1363, 1264, 1073, 1026, 755, 697 cm−1; 1H NMR (500 MHz, CDCl3): δ 4.43 (d, J = 9.5 Hz, 1 H, H-1), 4.00–3.93 (m, 2 H, H-6ab), 3.63–3.57 (m, 2 H, H-3, H-4), 3.47–3.40 (m, 2 H, H-2, H-5), 2.90 (br s, 1 H, OH), 2.86–2.73 (m, 2 H, SCH2CH3), 2.42 (br s, 1 H, OH), 1.42 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.02, 1.01 (2 s, 18 H, Si(CH3)2C(CH3)3), 0.26, 0.20 (2 s, 12 H, Si(CH3)2C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 85.7 (C-1), 78.7 (C-3), 76.9 (C-4), 72.7 (C-2), 72.5 (C-5), 64.5 (C-6), 25.9, 25.8 (6 C, Si(CH3)2C(CH3)3), 24.2 (SCH2CH3), 15.3, (2 C, Si(CH3)2C(CH3)3), 15.1 (SCH2CH3), − 4.3, −4.4, −4.5, −5.3 (4 C, Si(CH3)2C(CH3)3); HRMS calcd. for C20H44NaO5SSi2: [M+Na]+ 475.2340; found: 475.2349.
Ethyl 2,4-di-O-benzyl-3,6-di-O-(tert-butyldimethylsilyl)-1-thio-β-D-glucopyranoside (13):
To a solution of compound 12 (14 g, 30.8 mmol) in DMF (50 ml) were added BnBr (9.4 ml, 77.4 mmol) and NaH (60% in mineral oil, 3.4 g, 149 mmol) at 0 °C. The mixture was stirred for 2 h at 0 °C and quenched with CH3OH (2 ml). The mixture was diluted with Et2O (200 ml) and washed with water and brine. The organic layer was dried over MgSO4, filtered, and evaporated to dryness. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (9:1) as eluant to give pure compound 13 (17.2 g, 88%). Colorless oil; [α]D25 +12 (c 1.0, CHCl3); IR (neat): 3032, 2925, 2857, 1495, 1359, 1251, 1149, 1086, 788, 698 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.53–7.33 (m, 10 H, Ar-H), 5.04 (d, J = 10.5 Hz, 1 H, PhCH2), 4.94 (d, J = 11.5 Hz, 1 H, PhCH2), 4.82 (d, J = 10.5 Hz, 1 H, PhCH2), 4.80 (d, J = 11.5 Hz, 1 H, PhCH2), 4.55 (d, J = 9.5 Hz, 1 H, H-1), 3.94 (dd, J = 1.5, 11.5 Hz, 1 H, H-6a), 3.88 (dd, J = 4.0, 11.5 Hz, 1 H, H-6b), 3.82 (t, J = 8.5 Hz, 1 H, H-3), 3.54 (t, J = 9.0 Hz, 1 H, H-4), 3.36–3.30 (m, 2 H, H-2, H-5), 2.92–2.77 (m, 2 H, SCH2CH3), 1.44 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.07, 1.01 (2 s, 18 H, Si(CH3)2C(CH3)3), 0.14, 0.12, 0.11, 0.10 (4 s, 12 H, Si(CH3)2C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 128.3–127.4 (Ar-C), 84.4 (C-1), 82.1 (C-2), 79.4 (C-5), 78.6 (C-3), 78.4 (C-4), 75.1 (PhCH2), 74.6 (PhCH2), 62.4 (C-6), 26.1, 25.9 (6 C, Si(CH2)2C(CH3)3), 24.4 (SCH2CH3), 17.4 (2 C, Si(CH2)2C(CH3)3), 15.1 (SCH2CH3), −3.9, −4.9, −5.3 (4 C, Si(CH2)2C(CH3)3); HRMS calcd. for C34H56NaO5SSi2: [M+Na]+ 655.3279; found: 655.3303.
Ethyl 2,4-di-O-benzyl-1-thio-β-D-glucopyranoside (14):
To a solution of compound 13 (16 g, 25.2 mmol) in CH3OH (50 ml) was added p-TSA (420 mg, 2.4 mmol) and the mixture was stirred at room temperature for 2 h and quenched with Et3N (1 ml). The mixture was evaporated, and the crude product was purified over SiO2 column chromatography using hexane-EtOAc (7:1) as eluant to give pure compound 15 (9 g, 89%). White solid; m.p. 93–95 °C [EtOH]; [α]D25 +52 (c 1.0, CHCl3); IR (KBr): 3432, 2958, 2875, 1498, 1492, 1363, 1265, 1207, 1090, 696 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.50–7.37 (m, 10 H, Ar-H), 5.08 (d, J = 11.0 Hz, 1 H, PhCH2), 4.96 (d, J = 11.0 Hz, 1 H, PhCH2), 4.81 (d, J = 11.0 Hz, 1 H, PhCH2), 4.78 (d, J = 10.5 Hz, 1 H, PhCH2), 4.58 (d, J = 4.5 Hz, 1 H, H-1), 3.99–3.96 (m, 1 H, H-6b), 3.86–3.79 (m, 2 H, H-3, H-6a), 3.59 (t, J = 9.5 Hz, 1 H, H-4), 3.46–3.43 (m, 1 H, H-5), 3.36 (t, J = 9.5 Hz, 1 H, H-2), 2.90–2.82 (m, 2 H, SCH2CH3), 2.49 (d, J = 2.0 Hz, 1 H, 3-OH), 2.01 (t, J = 6.5, 7.0 Hz, 3 H, 6-OH), 1.46 (t, J = 7.5 Hz, 3 H, SCH2CH3); 13C NMR (125 MHz, CDCl3): δ 128.6–127.9 (Ar-C), 84.8 (C-1), 81.4 (C-2), 78.9 (C-5), 78.4 (C-3), 77.1 (C-4), 75.2 (PhCH2), 74.6 (PhCH2), 62.1 (C-6), 25.2 (SCH2CH3), 15.1 (SCH2CH3); HRMS calcd. for C22H28NaO5S: [M+Na]+ 427.1549; found: 427.1540.
Ethyl 2,4-di-O-benzyl-3,6-dideoxy-1-thio-β-D-ribo-hexopyranoside (4):
To a solution of compound 14 (9 g, 22.2 mmol) and imidazole (1.5 g, 22.2 mmol) in anhydrous THF (100 ml) was added NaH (60% in mineral oil, 2.67 g, 111 mmol) by small portions. After stirring at room temperature for 1.5 h, CS2 (5.6 ml, 89.1 mmol) was added to it and the mixture was stirred at room temperature for 3.5 h. To the reaction mixture was added CH3I (5.6 mmol, 89.1 mmol) and stirring was continued for another 1.5 h. The reaction mixture was cooled in ice and quenched with CH3OH (5 ml). The solvents were removed under reduced pressure and the residue was dissolved in EtOAc (500 ml). The organic layer was successively washed with 1N HCl (200 ml), satd. NaHCO3 (200 ml) and brine (200 ml), dried over anhydrous Na2SO4 and evaporated. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (6:1) as eluant to give xanthate derivative. To the solution of the xanthate derivative in anhydrous toluene (100 ml) were added AIBN (200 mg, 1.0 mmol) and tributyltin hydride (19.4 ml, 66.8 mmol) and the reaction mixture was heated to reflux for 3 h. The solvent was removed under reduced pressure and the crude product was purified over SiO2 column chromatography using hexane-EtOAc (10:1) as eluant to give pure compound 4 (4.4 g, 45%). Colourless oil; [α]D25 +45 (c 1.0, CHCl3); IR (neat): 2980, 2932, 2875, 1495, 1451, 1371, 1262, 1204, 1098, 860 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.40–7.27 (m, 10 H, Ar-H), 4.72 (d, J = 12.0 Hz, 1 H, PhCH2), 4.64 (d, J = 11.5 Hz, 1 H, PhCH2), 4.63 (d, J = 11.5 Hz, 1 H, PhCH2), 4.47 (d, J = 11.5 Hz, 1 H, PhCH2), 4.43 (d, J = 9.5 Hz, 1 H, H-1), 3.40–3.36 (m, 1 H, H-5), 3.32–3.27 (m, 1 H, H-2), 3.12–3.06 (m, 1 H, H-4), 2.78–2.67 (m, 2 H, SCH2CH3), 2.63–2.59 (m, 1 H, H-3a), 1.53–1.46 (m, 1 H, H-3b), 1.32 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.27 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 128.4–127.6 (Ar-C), 86.2 (C-1), 77.4 (2 C, C-4, C-5), 75.5 (C-2), 72.1 (PhCH2), 71.1 (PhCH2), 36.3 (C-3), 24.6 (SCH2CH3), 18.1 (CCH3), 15.0 (SCH2CH3); HRMS calcd. for C22H28NaO3S: [M+Na]+ 395.1651; found: 395.1638.
Ethyl 3-O-p-methoxybenzyl-6-O-(tert-butyldimethylsilyl)-1-thio-β-D-galactopyranoside (16):
A solution of compound 15 (5.7 g, 25.5 mmol) in dry CH3OH (60 ml) was added Bu2SnO (7.5 g, 30.6 mmol) and the reaction mixture was stirred at 70 °C for 3 h. The solvent was removed under reduced pressure to give the crude stannylidene acetal derivative. To a solution of the stannylidene acetal derivative in dry DMF (50 ml) were added p-methoxybenzyl chloride (3.78 ml, 28.0 mmol) and the reaction mixture was allowed to stir at 60 °C for 14 h. The reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The residue was dissolved in CH2Cl2 (200 ml) and washed with 1N HCl, satd. NaHCO3 and water in succession, dried (Na2SO4) and concentrated to give the crude product. To a solution of the crude product in DMF (100 ml) were added imidazole (2 g, 30.6 mmol) and TBSCl (4.5 g, 30.6 mol) at 0 °C and the mixture was stirred at room temperature for 16 h and quenched with CH3OH (2 ml). The mixture was diluted with EtOAc (200 ml) and washed with 1M HCl, satd. NaHCO3, and brine. The organic layer was dried with MgSO4, filtered, and evaporated to dryness. The crude product was purified over SiO2 column chromatography using hexane-CH2Cl2 (9:1) as eluant to give pure compound 16 (7.7 g, 66%). White solid; m.p. 158–160 °C; [α]D25 − 4 (c 1.0, CHCl3); IR (KBr): 3366, 2933, 2869, 1615, 1518, 1255, 1094, 1052, 984, 825 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.41 (d, J = 8.5 Hz, 2 H, Ar-H), 6.98 (d, J = 8.5 Hz, 2 H, Ar-H), 4.81–4.80 (m, 2 H, PhCH2), 4.36 (d, J = 10.0 Hz, 1 H, H-1), 4.11 (d, J = 2.5 Hz, 1 H, H-4), 3.97–3.88 (m, 6 H, H-5, H-6ab, OCH3), 3.54 (t, J = 8.5 Hz, 1 H, H-2), 3.48 (dd, J = 9.0, 3.0 Hz, 1 H, H-3), 2.85–2.80 (m, 2 H, SCH2CH3), 1.43 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.00 (s, 9 H, Si(CH3)2C(CH3)3), 0.18 (s, 6 H, Si(CH3)2C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 129.5, 113.9 (Ar-C), 86.0 (C-1), 81.2 (C-3), 78.5 (C-2), 71.7 (PhCH2), 69.3 (C-4), 66.5 (C-5), 62.2 (C-6), 55.1 (OCH3), 25.9 (3 C, Si(CH3)2C(CH3)3), 23.6 (SCH2CH3), 15.3 (2 C, Si(CH3)2C(CH3)3, SCH2CH3), −5.3, −5.4 (2 C, Si(CH3)2C(CH3)3); HRMS calcd. for C22H38NaO6SSi: [M+Na]+ 481.2050; found: 481.2045.
Ethyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-6-O-(tert-butyldimethylsilyl)-1-thio-β-D-galactopyranoside (9):
To a solution of compound 16 (6 g, 13.1 mmol) in DMF (40 ml) were added BnBr (3.8 ml, 31.4 mmol) and NaH (753 mg, 31.4 mmol) at 0 °C and the mixture was stirred at 0 °C for 2 h and quenched with CH3OH (1 ml). The mixture was diluted with Et2O (150 ml) and extracted with water and brine. The organic layer was dried with MgSO4, filtered and evaporated to dryness. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (9:1) as eluant to give pure compound 9 (7.9 g, 95%). Yellow oil; [α]D25 +15 (c 1.0, CHCl3); IR (neat): 2929, 2857, 1611, 1454, 1361, 1247, 1202, 1090, 795 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.51–7.35 (m, 12 H, Ar-H), 6.95 (d, J = 8.5 Hz, 2 H, Ar-H), 5.09 (d, J = 11.5 Hz, 1 H, PhCH2), 5.00 (d, J = 10.5 Hz, 1 H, PhCH2), 4.91 (d, J = 10.5 Hz, 1 H, PhCH2), 4.81–4.72 (m, 3 H, PhCH2), 4.50 (d, J = 9.5 Hz, 1 H, H-1), 3.97 (d, J = 2.0 Hz, 1 H, H-4), 3.91–3.88 (m, 4 H, H-5, OCH3), 3.81–3.75 (m, 2 H, H-6ab), 3.64 (dd, J = 9.5, 3.0 Hz, 1 H, H-3), 3.50 (t, J = 8.5 Hz, 1 H, H-2), 2.90–2.79 (m, 2 H, SCH2CH3), 1.43 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.00 (s, 9 H, Si(CH3)2C(CH3)3), 0.16 (s, 6 H, Si(CH3)2C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 159.2–113.7 (Ar-C), 85.1 (C-1), 83.4 (C-3), 78.8 (C-2), 78.4 (C-4), 75.6 (PhCH2), 74.4 (PhCH2), 73.6 (C-5), 72.4 (PhCH2), 61.6 (C-6), 55.1 (OCH3), 25.9 (3 C, Si(CH3)2C(CH3)3), 24.4 (SCH2CH3), 18.2 (Si(CH3)2C(CH3)3, 15.1 (SCH2CH3), −5.3 (2 C, Si(CH3)2C(CH3)3); HRMS calcd. for C36H50NaO6SSi: [M+H]+ 639.3175; found: 639.3178.
Ethyl (2,4-di-O-benzyl-3,6-dideoxy-β-D-ribo-hexopyranosyl)-(1→3)-2-O-acetyl-4,6-di-O-benzyl-1-thio-α-D-mannopyranoside (17):
To a solution of compound 4 (1 g, 2.69 mmol) and compound 5 (1 g, 2.22 mmol) in anhydrous CH2Cl2-Et2O (40 ml; 1:3) was added MS 4Å (1 g) and the reaction mixture was stirred at room temperature for 45 min and cooled to −20 °C under argon. To the cooled reaction mixture was added NIS (635 mg, 2.82 mmol) followed by TfOH (20 μl) and then allowed to stir at −20 °C for 20 min. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 ml). The organic layer was washed with 5% aq. Na2S2O3 (100 mL), satd. NaHCO3 (100 mL) and water (100 mL) in succession, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (8:1) as eluant to give pure compound 17 (1.03 g) and α-glycosylated product (18) (285 mg) (total yield 76%). Colorless oil; [α]D25 +81 (c 1.0, CHCl3); IR (neat): 3028, 2865, 1738, 1603, 1372, 1263, 1094, 1075, 749, 696 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.32–7.20 (m, 20 H, Ar-H), 5.27 (br s, 1 H, H-2C), 5.26 (d, J = 1.5 Hz, 1 H, H-1C), 4.93 (d, J = 10.5 Hz, 1 H, PhCH), 4.74 (d, J = 12.5 Hz, 1 H, PhCH), 4.67 (d, J = 12.0 Hz, 1 H, PhCH), 4.58 (d, J = 12.0 Hz, 2 H, 2 PhCH), 4.47 (d, J = 12.5 Hz, 1 H, PhCH), 4.44 (d, J = 7.5 Hz, 1 H, H-1D), 4.42 (d, J = 10.5 Hz, 1 H, PhCH), 4.40 (d, J = 12.0 Hz, 1 H, PhCH), 4.27 (dd, J = 3.0, 9.5 Hz, 1 H, H-3C), 4.10 (dd, J = 2.0, 9.0 Hz, 1 H, H-5C), 3.93 (t, J = 9.5 Hz, 1 H, H-4C), 3.81 (dd, J = 11.0, 4.0 Hz, 1 H, H-6aC), 3.61–3.58 (m, 1 H, H-6bC), 3.41–3.38 (m, 1 H, H-5D), 3.19–3.17 (m, 1 H, H-2D), 3.01–2.98 (m, 1 H, H-4D), 2.66–2.55 (m, 2 H, SCH2CH3), 2.43–2.40 (m, 1 H, H-3aD), 1.90 (s, 3 H, COCH3), 1.46–1.43 (m, 1 H, H-3bD), 1.29 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.25 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 169.9 (CH3CO), 138.6–127.2 (Ar-C), 101.6 (C-1D), 82.1 (C-1C), 77.4 (C-4D), 75.5 (C-3C), 75.4 (C-2D), 74.6 (PhCH2), 74.5 (C-5D), 73.8 (C-4C), 73.3 (PhCH2), 71.7 (PhCH2), 71.6 (C-5C), 70.9 (PhCH2), 70.9 (C-2C), 68.7 (C-6C), 34.5 (C-3D), 29.8 (SCH2CH3), 20.9 (CH3CO), 18.1 (CCH3), 15.0 (SCH2CH3); HRMS calcd. for C44H52NaO9S: [M+Na]+ 779.3224; found: 779.3252.
Ethyl (2,4-di-O-benzyl-3,6-di-O-(tert-butyldimethylsilyl)-α-D-glucopyranosyl)-(1→3)-2-O-acetyl-4,6-di-O-benzyl-1-thio-α-D-mannopyranoside (19):
To a solution of compound 13 (5.1 g, 8.07 mmol) and compound 5 (3 g, 6.72 mmol) in anhydrous CH2Cl2 (100 ml) was added MS 4Å (4 g) and the reaction mixture was stirred at room temperature for 1 h and then cooled to −40 °C under argon. To the cooled reaction mixture was added NIS (1.9 g, 84.7 mmol) followed by TfOH (70 μl, 0.12 mmol) and then allowed to stir at −40 °C for 15 min. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 ml). The organic layer was washed with 5% aq. Na2S2O3, satd. NaHCO3 and water in succession, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (10:1) as eluant to give pure compound 19 (4.92 g, 72%). White solid; m.p. 84–86 °C [EtOH]; [α]D25 +75 (c 1.0, CHCl3); IR (KBr): 3032, 2925, 2857, 1742, 1454, 1363, 1232, 1156, 1071, 778 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.29–7.14 (m, 20 H, Ar-H), 5.28 (s, 1 H, H-1C), 5.20 (d, J = 11.5 Hz, 1 H, PhCH), 5.09 (br s, 1 H, H-2C), 4.82 (d, J = 11.5 Hz, 1 H, PhCH), 4.68 (d, J = 11.5 Hz, 1 H, PhCH), 4.64 (d, J = 13.0 Hz, 1 H, PhCH), 4.61 (d, J = 5.0 Hz, H-1D), 4.60 (d, J = 13.0 Hz, 1 H, PhCH), 4.50–4.43 (m, 3 H, PhCH), 4.12 (dd, J = 2.5, 9.5 Hz, 1 H, H-5C), 4.01 (t, J = 9.0, 9.5 Hz, 1 H, H-3D), 3.94 (t, J = 9.5 Hz, 1 H, H-4C), 3.81–3.77 (m, 3 H, H-3C, H-6aC, H-6bD), 3.61–3.48 (m, 3 H, H-5D, H-6bC, H-6aD), 3.45 (t, J = 10.0 Hz, H-4D), 3.17 (dd, J = 9.5, 3.5 Hz, 1 H, H-2D), 2.64–2. 61 (m, 2 H, SCH2CH3), 2.09 (s, 3 H, COCH3), 1.32 (t, J = 7.0 Hz, 3 H, SCH2CH3), 0.92, 0.89 (2 s, 18 H, Si(CH3)2C(CH3)3), 0.04, 0.02 (2 s, 12 H, Si(CH3)2C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 169.7 (COCH3), 139.0–127.0 (Ar-C), 99.7 (C-1D), 81.7 (C-1C), 80.9 (C-3C), 80.6 (C-2D), 78.2 (C-4D), 74.9 (PhCH2), 74.4 (PhCH2), 74.0 (2 C, C-2C, C-4C), 73.7 (PhCH2), 73.4 (C-3D), 73.3 (PhCH2), 72.6 (C-5D), 72.0 (C-5C), 68.9 (C-6C), 61.2 (C-6D), 29.7 (SCH2CH3), 26.1, 25.8 (6 C, Si(CH3)2C(CH3)3), 21.1 (CH3CO), 18.4, 18.2 (2 C, Si(CH3)2C(CH3)3), 15.0 (SCH2CH3), −3.8, −4.0, −4.8, −5.3 (4 C, Si(CH3)2C(CH3)3); HRMS calcd. for C56H80NaO11SSi2: [M+NH4]+ 1034.5298; found: 1034.5344.
Ethyl (2,4-di-O-benzyl-α-D-glucopyranosyl)-(1→3)-2-O-acetyl-4,6-di-O-benzyl-1-thio-α-D-mannopyranoside (20):
To a solution of compound 19 (4.5 g, 4.42 mmol) in CH3OH (50 ml) was added p-TSA (100 mg) and the mixture was stirred for 2 h and quenched by adding Et3N (1 ml). The solvents were removed under reduced pressure and the crude product was purified over SiO2 column chromatography with hexane-EtOAc as eluant to give the pure compound 20 (2.7 g, 79%). Colorless oil; [α]D25 +43 (c 1.0, CHCl3); IR (neat): 3476, 3032, 2925, 2872, 1740, 1494, 1372, 1234, 1069, 834 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.32–7.13 (m, 20 H, Ar-H), 5.27 (d, J = 1.5 Hz, 1 H, H-1C), 5.12 (dd, J = 1.5, 2.5 Hz, 1 H, H-2C), 5.08 (d, J = 11.5 Hz, 1 H, PhCH), 4.86 (d, J = 11.5 Hz, 1 H, PhCH), 4.83 (d, J = 3.5 Hz, 1 H, H-1D), 4.69 (d, J = 11.5 Hz, 1 H, PhCH), 4.61 (d, J = 11.5 Hz, 1 H, PhCH2), 4.58–4.41 (m, 4 H, 4 PhCH), 4.12–4.05 (m, 2 H, H-3D, H-5D), 3.95 (t, J = 9.5 Hz, 1 H, H-4C), 3.89 (dd, J = 9.5, 3.0 Hz, 1 H, H-3C), 3.78 (dd, J = 10.5, 4.0 Hz, 1 H, H-6aD), 3.67–3.59 (m, 4 H, H-5C, H-6abC, H-6bD), 3.42 (t, J = 9.0 Hz, 1 H, H-4D), 3.25 (dd, J = 10.0, 3.5 Hz, 1 H, H-2D), 2.64–2.59 (m, 2 H, SCH2CH3), 2.36 (br s, 1 H, 3-OH), 2.10 (s, 3 H, COCH3), 1.55 (br s, 1 H, 6-OH), 1.28 (t, J = 7.0 Hz, 3 H, SCH2CH3); 13C NMR (125 MHz, CDCl3): δ 170.5 (CH3CO), 138.7–127.2 (Ar-C), 99.0 (C-1D), 81.8 (C-1C), 80.5 (C-3C), 79.3 (C-2D), 77.2 (C-4D), 74.9 (PhCH2), 74.5 (PhCH2), 73.9 (2 C, C-4C, C-3D), 73.3 (PhCH2), 73.1 (C-2C), 73.0 (PhCH2), 72.1 (C-5D), 71.6 (C-5C), 68.8 (C-6D), 61.7 (C-6C), 25.8 (SCH2CH3), 21.2 (COCH3), 15.0 (SCH2CH3); HRMS calcd. for C44H52NaO11S: [M+Na]+ 811.3122; found: 811.3159.
Ethyl (2,4-di-O-benzyl-3,6-di-phenoxythiocarbonyl-α-D-glucopyranosyl)-(1→3)-2-O-acetyl-4,6-di-O-benzyl-1-thio-α-D-mannopyranoside (21):
To a solution of compound 20 (2.5 g, 3.2 mmol) in anhydrous CH2Cl2 (15 ml) and pyridine (15 ml) was added dropwise phenyl chlorothionocarbonate (1.1 ml, 7.7 mmol) at room temperature. After stirring for 4 h, the mixture was concentrated and filtered through a short SiO2 column using acetone as eluant. The residue was then purified over SiO2 column chromatography using hexane-EtOAc (3:1) as eluant to give pure compound 21 (3 g, 91%). Yellow oil; [α]D25 +89 (c 1.0, CHCl3); IR (neat): 3062, 3028, 2925, 1742, 1588, 1488, 1452, 1370, 1285, 1370, 1285, 1202, 1075, 1020 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.40–7.07 (m, 28 H, Ar-H), 6.91 (d, J = 8.0 Hz, 2 H, Ar-H), 6.20 (t, J = 9.0 Hz, 1 H, H-3D), 5.26 (br s, 1 H, H-1C), 5.25 (d, J = 2.0 Hz, 1 H, H-2C), 5.12 (d, J = 11.5 Hz, 1 H, PhCH), 5.02 (d, J = 3.5 Hz, 1 H, H-1D), 4.80 (d, J = 11.5 Hz, 1 H, PhCH), 4.70 (dd, J = 12.0, 3.0 Hz, 1 H, H-6aD), 4.62–4.41 (m, 7 H, H-6bD, PhCH2), 4.13–4.07 (m, 2 H, H-5C, H-5D), 4.00–3.93 (m, 2 H, H-3C, H-4C), 3.84 (t, J = 9.5 Hz, 1 H, H-4D), 3.76 (dd, J = 4.5, 11.0 Hz, 1 H, H-6aC), 3.64–3.62 (m, 2 H, H-2D, H-6bC), 2.64–2.59 (m, 2 H, SCH2CH3), 2.16 (s, 3 H, COCH3), 1.29 (t, J = 7.5 Hz, 3 H, SCH2CH3); 13C NMR (125 MHz, CDCl3): δ 194.6, 194.2 (2 C, 2 PhOCSO), 170.0 (COCH3), 153.5–122.0 (Ar-C), 99.3 (C-1D), 83.7 (C-3D), 81.8 (C-1C), 80.7 (C-3C), 77.6 (C-2D), 75.1 (C-4D), 74.8 (PhCH2), 74.5 (PhCH2), 73.9 (C-4C), 73.6 (C-2C), 73.3 (PhCH2), 73.1 (PhCH2), 72.1 (C-5C), 71.3 (C-6D), 69.4 (C-5D), 68.7 (C-6C), 25.7 (SCH2CH3), 21.1 (COCH3), 14.9 (SCH2CH3); HRMS calcd. for C58H60NaO13S3: [M+Na]+ 1083.3088; found: 1083.3138.
Ethyl (2,4-di-O-benzyl-3,6-dideoxy-α-D-ribo-hexopyranosyl)-(1→3)-2-O-acetyl-4,6-di-O-benzyl-1-thio-α-D-mannopyranoside (18):
To the solution of compound 21 (2.9 g, 2.7 mmol) in anhydrous toluene (100 ml) was added AIBN (20 mg) and Bu3SnH (2.3 ml, 8.2 mmol) and the reaction mixture was heated to reflux for 4 h. The reaction mixture was concentrated under reduced pressure and the crude product was purified over SiO2 using hexane-EtOAc (8:1) as eluant to give compound 18 (1.1 g, 55%). Colorless oil; [α]D25 +53 (c 1.0, CHCl3); IR (neat): 3028, 2922, 2857, 1740, 1494, 1452, 1369, 1319, 1232, 1071 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.32–7.11 (m, 20 H, Ar-H), 5.33 (d, J = 1.0 Hz, 1 H, H-1C), 5.20 (dd, J = 3.0, 1.5 Hz, 1 H, H-2C), 5.13 (d, J = 11.5 Hz, 1 H, PhCH), 4.89 (d, J = 3.5 Hz, 1 H, H-1D), 4.63–4.59 (m, 2 H, 2 PhCH), 4.47–4.43 (m, 5 H, 5 PhCH), 4.15 (dd, J = 9.5, 2.5, Hz, 1 H, H-5C), 4.04–3.95 (m, 2 H, H-3C, H-4C), 3.80 (dd, J = 10.5, 4.0 Hz, 1 H, H-6aC), 3.68–3.62 (m, 1 H, H-5D, H-6bC), 3.38–3.34 (m, 1 H, H-2D), 3.01–2.97 (m, 1 H, H-4D), 2.64–2.57 (m, 2 H, SCH2CH3), 2.28–2.21 (m, 1 H, H-3aD), 2.15 (s, 3 H, COCH3), 1.86 (q, J = 11.5 Hz, 1 H, H-3bD), 1.30 (t, J = 7.5 Hz, 3 H, SCH2CH3), 1.14 (d, J = 6.0 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.0 (COCH3), 138.3–127.1 (Ar-C), 98.3 (C-1D), 81.9 (C-1C), 79.7 (C-3C), 77.7 (C-4D), 77.1 (C-2D), 74.9 (PhCH2), 74.2 (C-4C), 74.1 (C-2C), 73.3 (PhCH2), 71.9 (C-5C), 70.8 (PhCH2), 70.6 (PhCH2), 68.9 (C-6C), 68.2 (C-5D), 29.9 (C-3D), 25.7 (SCH2CH3), 21.2 (CH3CO), 17.7 (CCH3), 14.9 (SCH2CH3); HRMS calcd. for C44H52NaO9S: [M+Na]+ 779.3224; found: 779.3262.
2-Azidoethyl 2,4,6-tri-O-benzyl-α-D-galactopyranoside (22):
To a solution of compound 7 (4 g, 6.51 mmol) and 2-azidoethanol (1.5 ml) in anhydrous CH2Cl2-Et2O (100 ml; 1:4) was added MS 4Å (4 g) and the reaction mixture was cooled to −20 °C under argon. To the cooled reaction mixture was added NIS (1.6 g, 7.17 mmol) followed by TfOH (80 μl) and it was allowed to stir at −20 °C for 30 min. The reaction mixture was warmed up to 0 °C and stirred at 0 °C for 1 h. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% aq. Na2S2O3, satd. NaHCO3 and water in succession, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 22 (2.1 g, 62%). White solid; m.p. 53–54 °C [EtOH]; [α]D25 +48 (c 1.0, CHCl3); IR (KBr): 3445, 3028, 2918, 2110, 1603, 1378, 1283, 1253, 1158, 1113, 1041, 972, 741 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.31–7.19 (m, 15 H, Ar-H), 4.81 (d, J = 12.0 Hz, 1 H, PhCH), 4.72 (d, J = 3.5 Hz, 1 H, H-1), 4.65 (d, J = 12.0 Hz, 1 H, PhCH), 4.58 (d, J = 12.0 Hz, 1 H, PhCH), 4.53 (d, J = 12.0 Hz, 1 H, PhCH), 4.44 (d, J = 12.0 Hz, 1 H, PhCH), 4.36 (d, J = 11.5 Hz, 1 H, PhCH), 4.01 (d, J = 10.0 Hz, 1 H, H-3), 3.91 (t, J = 6.5 Hz, 1 H, H-5), 3.63 (d, J = 2.5 Hz, 1 H, H-4), 3.76 (dd, J = 10.0, 3.5 Hz, 1 H, H-2), 3.72–3.68 (m, 1 H, H-6a), 3.45 (d, J = 6.5 Hz, 2 H, OCH2), 3.40–3.34 (m, 2 H, H-6b, NCH2), 3.31–3.29 (m, 1 H, NCH2), 2.35 (br s, 1 H, OH); 13C NMR (125 MHz, CDCl3): δ 138.6–127.5 (Ar-C), 97.3 (C-1), 77.4 (C-2), 76.3 (C-4), 75.0 (PhCH2), 73.3 (PhCH2), 72.8 (PhCH2), 70.0 (C-3), 69.5 (C-5), 69.0 (OCH2), 66.8 (C-6), 50.6 (NCH2); HRMS calcd. for C29H33N3NaO6: [M+Na]+ 542.2261; found: 542.2265.
2-Azidoethyl (4-O-acetyl-2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-2,4,6-tri-O-benzyl-α-D-galactopyranoside (23):
To a solution of compound 22 (1.5 g, 2.89 mmol) and compound 6 (1 g, 3.46 mmol) in anhydrous CH2Cl2 (20 ml) was added MS 4Å (2 g) and the reaction mixture was cooled to −20 °C under argon. To the cooled reaction mixture was added NIS (850 mg, 3.79 mmol) followed by TMSOTf (60 μl) and then allowed to stir at −20 °C for 30 min. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% aq. Na2S2O3 (50 ml), satd NaHCO3 (50 ml) and water (50 ml) in succession, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 23 (1.6 g, 76%). Yellow oil; [α]D25 +27 (c 1.0, CHCl3); IR (neat): 2942, 2933, 2106, 1742, 1647, 1495, 1452, 1374, 1230, 1092, 1043 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.33–7.23 (m, 15 H, Ar-H), 5.35 (br s, 1 H, H-1B), 4.80–4.76 (m, 2 H, H-3B, PhCH), 4.79 (d, J = 3.5 Hz, 1 H, H-1A), 4.67 (d, J = 12.0 Hz, 1 H, PhCH), 4.61 (d, J = 11.5 Hz, 1 H, PhCH), 4.55 (d, J = 11.5 Hz, 1 H, PhCH), 4.50 (d, J = 12.0 Hz, 1 H, PhCH), 4.44 (d, J = 12.0 Hz, 1 H, PhCH), 4.15 (dd, J = 3.0, 10.5 Hz, 1 H, H-3A), 4.09 (d, J = 5.5 Hz, 1 H, H-2B), 3.99–3.94 (m, 3 H, H-2A, H-5A, H-4B), 3.79–3.75 (m, 2 H, H-4A, H-6aA), 3.70–3.67 (m, 1 H, H-5B), 3.52–3.42 (m, 4 H, H-6bA, OCH2, NCH2), 3.38–3.31 (m, 1 H, NCH2), 2.10 (s, 3 H, COCH3), 1.51, 1.28 (2 s, 6 H, C(CH3)2), 1.13 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 169.2 (COCH3), 138.5–127.5 (Ar-C), 109.5 (C(CH3)2), 98.6 (C-1B), 97.5 (C-1A), 77.4 (C-4A), 76.9 (C-2A), 75.9 (C-2B), 75.5 (C-3A), 75.2 (C-4B), 74.9 (PhCH2), 74.3 (C-3B), 73.3 (PhCH2), 72.7 (PhCH2), 69.6 (C-5A), 68.6 (OCH2), 67.0 (C-6A), 64.7 (C-5B), 50.0 (NCH2), 27.7, 26.5 (2 C, C(CH3)2), 20.9 (COCH3), 16.9 (CCH3); HRMS calcd. for C40H49N3NaO11: [M+Na]+ 770.3259; found: 770.3293.
2-Azidoethyl (2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-2,4,6-tri-O-benzyl-α-D-galactopyranoside (24):
A solution of compound 23 (1.5 g, 2.00 mmol) in 0.1 M CH3ONa in CH3OH (20 ml) was allowed to stir at room temperature for 2 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered and concentrated. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (2:1) as eluant to give pure compound 24 (1.3 g, 92%). Yellowish oil; [α]D25 +13 (c 1.0, CHCl3); IR (neat): 3461, 2979, 2929, 2106, 1495, 1452, 1380, 1243, 1219, 1132, 1043 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.33–7.22 (m, 15 H, Ar-H), 5.30 (br s, 1 H, H-1B), 4.80 (d, J = 10.5 Hz, 1 H, PhCH), 4.79 (d, J = 3.5 Hz, 1 H, H-1A), 4.68 (d, J = 12.0 Hz, 1 H, PhCH), 4.61 (d, J = 12.0 Hz, 1 H, PhCH), 4.54 (d, J = 12.0 Hz, 1 H, PhCH), 4.49 (d, J = 12.0 Hz, 1 H, PhCH), 4.42 (d, J = 12.0 Hz, 1 H, PhCH), 4.15 (dd, J = 3.0, 10.0 Hz, 1 H, H-3A), 4.10–4.08 (m, 1 H, H-2B), 3.98–3.94 (m, 2 H, H-2A, H-3B), 3.87–3.85 (m, 1 H, H-5A), 3.79–3.75 (m, 2 H, H-4A, H-6aA), 3.69–3.61 (m, 1 H, H-5B), 3.51–3.42 (m, 4 H, H-6bA, OCH2, NCH2), 3.25–3.21 (m, 2 H, H-4B, NCH2), 1.48, 1.29 (2 s, 6 H, C(CH3)2), 1.26 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 138.5–127.4 (Ar-C), 109.2 (C(CH3)2), 98.7 (C-1B), 97.5 (C-1A), 78.4 (C-4A), 77.4 (C-2A), 76.8 (C-3B), 75.8 (C-3A), 75.1 (C-2B), 74.9 (PhCH2), 74.5 (C-4B), 73.3 (PhCH2), 72.7 (PhCH2), 69.7 (C-5A), 68.7 (OCH2), 67.0 (C-6A), 66.3 (C-5B), 50.5 (NCH2), 28.1, 26.2 (2 C, C(CH3)2), 17.3 (CCH3); HRMS calcd. for C38H47N3NaO10: [M+Na]+ 728.3153; found: 728.3187.
2-Azidoethyl (2,4-di-O-benzyl-3,6-dideoxy-α-D-ribo-hexopyranosyl)-(1→3)-(2-O-acetyl-4,6-di-O-benzyl-α-D-mannopyranosyl)-(1→4)-(2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-2,4,6-tri-O-benzyl-α-D-galactopyranoside (25):
A mixture of compound 24 (500 mg, 0.70 mmol), compound 18 (605 mg, 0.78 mmol) and MS 4Å (0.75 g) in dry CH2Cl2 (15 ml) was stirred at room temperature for 1 h and cooled to −20 °C. To the reaction mixture were added NIS (195 mg, 0.85 mmol) and AgOTf (40 mg, 0.16 mmol) sequentially and the reaction was stirred under an argon atmosphere at −20 °C for 45 min. The reaction mixture was filtered and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% Na2S2O3 (50 mL), satd. NaHCO3 (50 mL) and water (50 mL) in succession, dried (Na2SO4) and concentrated under vacuum. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (4:1) as eluant to give pure compound 25 (705 mg, 71%). Colorless oil; [α]D25 +38 (c 1.0, CHCl3); IR (neat): 3028, 2929, 2872, 2102, 1742, 1495, 1454, 1369, 1236, 1143, 1143, 1092, 1044 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.33–7.10 (m, 35 H, Ar-H), 5.31 (s, 1 H, H-1C), 5.16 (d, J = 11.0 Hz, 1 H, PhCH), 5.10 (br s, 1 H, H-2C), 4.97 (s, 1 H, H-1B), 4.90 (d, J = 3.5 Hz, 1 H, H-1D), 4.80 (d, J = 2.5 Hz, 1 H, H-1A), 4.79 (d, J = 11.0 Hz, 1 H, PhCH), 4.67–4.57 (m, 5 H, 5 PhCH), 4.51–4.41 (m, 7 H, 7 PhCH), 4.12–4.03 (m, 5 H, H-2B, H-3A, H-3C, H-4C, H-5C), 3.98–3.95 (m, 2 H, H-2A, H-5A), 3.93 (t, J = 7.0 Hz, 1 H, H-3B), 3.86 (dd, J = 2.5, 10.5 Hz, 1 H, H-6aC), 3.79–3.72 (m, 2 H, H-4A, H-6bA), 3.69–3.55 (m, 3 H, H-5B, H-5D, H-6bC), 3.52–3.40 (m, 4 H, H-6aA, OCH2, NCH2), 3.36–3.32 (m, 3 H, H-2D, H-4B, NCH2), 3.02–2.96 (m, 1 H, H-4D), 2.30–2.23 (m, 1 H, H-3aD), 2.15 (s, 3 H, COCH3), 1.86 (q, J = 11.5 Hz, 1 H, H-3bD), 1.43 (s, 3H, C(CH3)2), 1.27 (d, J = 6.5 Hz, 3 H, CCH3), 1.24 (s, 3 H, C(CH3)2), 1.13 (d, J = 6.0 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.3 (COCH3), 139.1–127.0 (Ar-C), 108.8 (C(CH3)2), 98.6 (C-1D), 98.4 (C-1C), 97.6 (C-1B), 97.5 (C-1A), 80.5 (C-4B), 79.6 (C-2B), 77.7 (C-4D), 77.0 (C-4A), 76.9 (C-2A), 76.6 (C-3B), 76.0 (C-3A), 75.0 (C-3C), 75.0 (PhCH2), 74.7 (PhCH2), 74.1 (C-2D), 73.5 (C-2C), 73.5 (PhCH2), 73.3 (PhCH2), 72.7 (PhCH2), 72.4 (C-4C), 71.5 (C-5C), 70.7 (PhCH2), 70.4 (PhCH2), 69.8 (C-5A), 68.7 (OCH2), 68.4 (C-6C), 68.1 (C-5D), 67.0 (C-6A), 65.7 (C-5B), 50.5 (NCH2), 29.9 (C-3D), 28.2, 26.5 (2 C, C(CH3)2), 21.1 (COCH3), 17.8 (CCH3), 17.3 (CCH3); HRMS calcd. for C80H93N3NaO19: [M+Na]+ 1422.6295; found: 1422.6322.
2-Azidoethyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-6-O-(tert-butyldimethylsilyl)-α-D-galactopyranoside (26):
To a solution of compound 9 (6 g, 9.86 mmol) and 2-azidoethanol (2 ml) in anhydrous CH2Cl2-Et2O (150 ml; 1:4) was added MS 4Å (4.0 g) and the reaction mixture was cooled to −20 °C under argon. To the cooled reaction mixture was added NIS (2.4 g, 10.8 mmol) followed by TMSOTf (0.25 ml) and it was allowed to stir at −20 °C for 1.5 h. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% aq. Na2S2O3 (50 ml), satd. NaHCO3 (50 ml) and water (50 ml) in succession, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (4:1) as eluant to give pure compound 26 (5.1 g, 83%). Colorless oil; [α]D25 +26 (c 1.0, CHCl3); IR (neat): 2926, 2866, 2103, 1642, 1513, 1454, 1340, 1245, 1071 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.47–7.35 (m, 12 H, Ar-H), 6.96 (d, J = 8.5 Hz, 2 H, Ar-H), 5.05 (d, J = 11.5 Hz, 1 H, PhCH), 4.96 (d, J = 12.0 Hz, 1 H, PhCH), 4.89 (d, J = 11.0 Hz, 1 H, PhCH), 4.87 (d, J = 3.5 Hz, 1 H, H-1), 4.78 (d, J = 12.0 Hz, 1 H, PhCH), 4.76 (d, J = 11.5 Hz, 1 H, PhCH), 4.68 (d, J = 11.0 Hz, 1 H, PhCH), 4.12 (dd, J = 10.0, 3.5 Hz, 1 H, H-2), 4.04 (dd, J = 10.0, 3.5 Hz, 1 H, H-3), 3.95 (br s, 1 H, H-4), 3.92–3.89 (m, 4 H, OCH3, OCH2), 3.87 (t, J = 6.5 Hz, 1 H, H-5), 3.72–3.61 (m, 4 H, H-6ab, NCH2, OCH2), 3.49–3.42 (m, 1 H, NCH2), 1.01 (s, 9 H, Si(CH3)2C(CH3)3), 0.08 (s, 6 H, Si(CH3)2C(CH3)3); 13C NMR (125 MHz, CDCl3): δ 129.2–113.7 (Ar-C), 98.1 (C-1), 78.5 (C-3), 76.4 (C-2), 75.1 (C-4), 74.7 (PhCH2), 73.4 (PhCH2), 73.0 (PhCH2), 71.5 (C-5), 66.7 (OCH2), 62.0 (C-6), 55.0 (OCH3), 50.6 (NCH2), 25.9 (3 C, Si(CH3)2C(CH3)3), 10.2 (Si(CH3)2C(CH3)3, −5.3 (2 C, Si(CH3)2C(CH3)3); HRMS calcd. for C36H49N3NaO7Si: [M+Na]+ 686.3232; found: 686.3257.
2-Azidoethyl 2,4-di-O-benzyl-3-O-p-methoxybenzyl-α-D-galactopyranoside (27):
To a solution of compound 26 (5 g, 7.89 mmol) in THF (50 ml) was added a 1M solution of TBAF in THF (11.4 ml, 11.8 mmol), and the reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure and the residue was purified over SiO2 column chromatography using hexane-EtOAc (2:1) to give pure compound 27 (3.6 g, 90%). Colorless oil; [α]D25 +41 (c 1.0, CHCl3); IR (neat): 3510, 2928, 2868, 2106, 1640, 1514, 1454, 1336, 1245, 1071, 967 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.49–7.37 (m, 12 H, Ar-H), 6.99 (d, J = 8.5 Hz, 2 H, Ar-H), 5.07 (d, J = 11.5 Hz, 1 H, PhCH), 4.97 (d, J = 12.5 Hz, 1 H, PhCH), 4.92 (d, J = 3.5 Hz, 1 H, H-1), 4.91 (d, J = 11.5 Hz, 1 H, PhCH), 4.80–4.71 (m, 3 H, PhCH), 4.15 (dd, J = 10.0, 3.5 Hz, 1 H, H-2), 4.07 (dd, J = 10.5, 3.0 Hz, 1 H, H-3), 3.95–3.87 (m, 6 H, H-4, H-5, OCH3, OCH), 3.80 (dd, J = 11.5, 6.0 Hz, 1 H, H-6a), 3.69–3.59 (m, 3 H, H-6b, OCH, NCH), 3.47–3.42 (m, 1 H, NCH); 13C NMR (125 MHz, CDCl3): δ 129.2–113.7 (Ar-C), 98.2 (C-1), 78.4 (C-3), 76.4 (C-2), 75.1 (C-4), 74.4 (PhCH2), 73.4 (PhCH2), 73.3 (PhCH2), 70.7 (C-5), 66.9 (OCH2), 62.4 (C-6), 55.1 (OCH3), 50.6 (NCH2); HRMS calcd. for C30H35N3NaO7: [M+Na]+ 572.2367; found: 572.2374.
2-Azidoethyl (2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1→6)-2,4-di-O-benzyl-α-D-galactopyranoside (28):
To a solution of compound 27 (3 g, 5.78 mmol) and 8 (4 g, 6.93 mmol) in anhydrous CH2Cl2-Et2O (150 ml; 1:4) was added MS 4Å (3 g) and the reaction mixture was cooled to −20 °C under argon. To the cooled reaction mixture was added NIS (1.71 g, 7.63 mmol) followed by TfOH (120 μl, 1.36 mmol) and it was allowed to stir at −20 °C for 30 min. The reaction mixture was warmed up to 0 °C and stirred at 0 °C for 1 h. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% aq. Na2S2O3 (50 mL), satd. NaHCO3 (50 mL) and water (50 mL) in succession, dried (Na2SO4) and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (2:1) as eluant to give pure compound 28 (3.96 g, 72%). White solid; m.p. 80–82 °C [EtOH]; [α]D25 +97 (c 1.0, CHCl3); IR (KBr): 3411, 3032, 2914, 2098, 1603, 1495, 1365, 1272, 1211, 1098, 887, 739, 696 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.47–7.22 (m, 30 H, Ar-H), 5.07 (d, J = 11.0 Hz, 1 H, PhCH), 4.97 (d, J = 12.0 Hz, 1 H, PhCH), 4.94 (d, J = 3.5 Hz, 1 H, H-1A), 4.93–4.91 (m, 2 H, PhCH), 4.87 (d, J = 3.5 Hz, 1 H, H-1B), 4.86–4.70 (m, 6 H, 6 PhCH), 4.58–4.55 (m, 2 H, 2 PhCH), 4.20 (dd, J = 10.0, 3.0 Hz, 1 H, H-3A), 4.12 (t, J = 8.5 Hz, 1 H, H-4B), 4.06–4.04 (m, 2 H, H-3B, H-4A), 3.93–3.87 (m, 2 H, H-2A, H-5A), 3.85–3.74 (m, 5 H, H-5B, H-6abB, OCH2), 3.67 (dd, J = 9.5, 3.5 Hz, 1 H, H-2B), 3.61–3.59 (m, 1 H, H-6aA), 3.54–3.52 (m, 1 H, H-6bA), 3.42–3.40 (m, 2 H, NCH2); 13C NMR (125 MHz, CDCl3): δ 128.5–127.6 (Ar-C), 97.1 (2 C, C-1A, C-1B), 82.0 (C-3B), 79.7 (C-2B), 77.5 (C-2A), 77.1 (C-5B), 76.5 (C-4A), 75.5 (PhCH2), 75.0 (2 C, PhCH2), 73.4 (PhCH2), 73.3 (PhCH2), 72.7 (PhCH2), 70.4 (C-3A), 70.0 (C-5A), 69.5 (C-4B), 68.3 (OCH2), 66.8 (C-6A, C-6B), 50.5 (NCH2); HRMS calcd. for C56H61N3NaO11: [M+Na]+ 974.4198; found: 974.4244.
2-Azidoethyl (4-O-acetyl-2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1→6)]-2,4-di-O-benzyl-α-D-galactopyranoside (29):
To a solution of compound 28 (3 g, 3.15 mmol) and compound 6 (1 g, 3.78 mmol) in anhydrous CH2Cl2 (30 ml) was added MS 4Å (2 g) and the reaction mixture was stirred at room temperature for 45 min under argon atmosphere and cooled to −20 °C. To the cooled reaction mixture was added NIS (853 mg, 3.79 mmol) followed by TMSOTf (60 μl) and then allowed to stir at −20 °C for 40 min. The reaction mixture was filtered through a Celite® bed and washed with CH2Cl2 (100 ml). The organic layer was washed with 5% Na2S2O3 (50 ml), satd. NaHCO3 (50 ml) and water (50 ml) in succession, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 using hexane-EtOAc (3:1) as eluant to give pure compound 29 (2.8 g, 76%). Colorless oil; [α]D25 +19 (c 1.0, CHCl3); IR (neat): 3030, 2928, 2105, 1639, 1497, 1449, 1371, 1243, 1217, 1133, 1067, 975 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.34–7.06 (m, 30 H, Ar-H), 5.40 (s, 1 H, H-1C), 4.94 (d, J = 11.0 Hz, 1 H, PhCH), 4.85 (d, J = 3.0 Hz, 1 H, H-1A), 4.82–4.78 (m, 5 H, 5 PhCH), 4.77 (d, J = 3.0 Hz, 1 H, H-1B), 4.65–4.60 (m, 4 H, 4 PhCH), 4.47–4.44 (m, 2 H, 2 PhCH), 4.18 (dd, J = 9.0, 2.5 Hz, 1 H, H-3A), 4.17–4.14 (m, 2 H, H-4B, H-4C), 4.04–3.94 (m, 4 H, H-2A, H-2C, H-3B, H-3C), 3.83–3.60 (m, 8 H, H-4A, H-5A, H-5B, H-5C, H-6abB, OCH2), 3.56 (dd, J = 10.0, 3.5 Hz, 1 H, H-2B), 3.49–3.46 (m, 2 H, H-6abA), 3.33–3.28 (m, 2 H, NCH2), 2.10 (s, 3 H, COCH3), 1.56, 1.32 (2 s, 6 H, C(CH3)2), 1.14 (d, J = 6.0 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 169.7 (COCH3), 138.6–127.6 (Ar-C), 109.6 (C(CH3)2), 98.7 (C-1C), 97.3 (C-1A), 97.1 (C-1B), 82.0 (C-3B), 79.8 (C-2B), 77.6 (C-4A), 77.5 (C-5B), 76.6 (C-4B), 75.9 (C-3A), 75.6 (C-2A), 75.5 (C-4C), 74.9 (PhCH2), 74.8 (PhCH2), 74.3 (PhCH2), 73.4 (PhCH2), 73.4 (C-2C), 73.2 (PhCH2), 72.6 (PhCH2), 70.4 (C-5A), 69.7 (C-3C), 68.3 (OCH2), 66.9 (C-6B), 66.6 (C-6A), 64.7 (C-5C), 50.4 (NCH2), 27.7, 26.4 (2 C, C(CH3)2), 21.0 (COCH3), 16.8 (CCH3); HRMS calcd. for C67H81N4O16: [M+NH4]+ 1197.5642; found: 1197.5699.
2-Azidoethyl (2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1→6)]-2,4-di-O-benzyl-α-D-galactopyranoside (30):
A solution of compound 29 (2.5 g, 2.12 mmol) in 0.1 M CH3ONa in CH3OH (25 ml) was allowed to stir at room temperature for 4 h. The reaction mixture was neutralized with Dowex 50W-X8 (H+) resin, filtered and concentrated. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (2:1) as eluant to give pure compound 30 (2.2 g, 92%). Colorless oil; [α]D25 +19 (c 1.0, CHCl3); IR (neat): 3472, 3028, 2925, 2106, 1639, 1495, 1452, 1369, 1243, 1217, 1135, 1069 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.34–7.07 (m, 30 H, Ar-H), 5.36 (br s, 1 H, H-1C), 4.95 (d, J = 11.0 Hz, 1 H, PhCH), 4.84–4.81 (m, 3 H, 3 PhCH), 4.81 (d, J = 3.0 Hz, 1 H, H-1A), 4.76 (d, J = 3.0 Hz, 1 H, H-1B), 4.75 (d, J = 12.0 Hz, 1 H, PhCH), 4.66–4.60 (m, 5 H, 5 PhCH), 4.47–4.44 (m, 2 H, 2 PhCH), 4.18 (dd, J = 2.5, 9.5 Hz, 1 H, H-3A), 4.15 (m, 1 H, H-4B), 4.04 (t, J = 8.5 Hz, 1 H, H-3C), 4.00–3.90 (m, 3 H, H-2A, H-2C, H-3B), 3.84–3.63 (m, 8 H, H-4A, H-5A, H-5B, H-5C, H-6abB, OCH2), 3.55 (dd, J = 9.5, 3.0 Hz, 1 H, H-2B), 3.51–3.42 (m, 2 H, H-6abA), 3.38 (t, J = 9.5 Hz, 1 H, H-4C), 3.33–3.24 (m, 2 H, NCH2), 1.54, 1.34 (2 s, 6 H, C(CH3)2), 1.29 (d, J = 6.0 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 138.8–127.5 (Ar-C), 109.8 (C(CH3)2), 98.8 (C-1C), 97.4 (C-1A), 97.0 (C-1B), 82.0 (C-3B), 79.7 (C-2B), 78.4 (C-2C), 77.6 (C-5B), 77.5 (C-4A), 76.5 (C-2A), 75.9 (C-3A), 75.5 (PhCH2), 75.4 (C-4B), 74.9 (PhCH2), 74.8 (PhCH2), 74.6 (C-4C), 73.4 (PhCH2), 73.2 (PhCH2), 72.6 (PhCH2), 70.4 (C-5A), 69.8 (C-3C), 68.3 (OCH2), 66.9 (C-6B), 66.7 (C-6A), 66.3 (C-5C), 50.4 (NCH2), 28.1, 26.2 (2 C, C(CH3)2), 17.2 (CCH3); HRMS calcd. for C65H75N3NaO15: [M+Na]+ 1160.5090; found: 1160.5144.
2-Azidoethyl (2,4-di-O-benzyl-3,6-dideoxy-α-D-ribo-hexopyranosyl)-(1→3)-(2-O-acetyl-4,6-di-O-benzyl-α-D-mannopyranosyl)-(1→4)-(2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1→6)]-2,4-di-O-benzyl-α-D-galactopyranoside (31):
A mixture of compound 30 (600 mg, 0.53 mmol), compound 18 (440 mg, 0.58 mmol) and MS 4Å (1 g) in CH2Cl2 (15 ml) was stirred at room temperature for 1 h and cooled to −20 °C. To the reaction mixture were sequentially added NIS (145 mg, 0.64 mmol) and AgOTf (30 mg, 0.11 mmol) and the reaction mixture was stirred at −20 °C for 45 min under argon. The reaction mixture was filtered and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% Na2S2O3 (50 ml), satd. NaHCO3 (50 ml) successively, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (4:1) as eluant to give pure compound 31 (715 mg, 74%). Colorless oil; [α]D25 +23 (c 1.0, CHCl3); IR (neat): 3062, 3028, 2967, 2925, 2106, 1744, 1603, 1495, 1450, 1370, 1234, 1098, 1029, 997 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.31–7.07 (m, 50 H, Ar-H), 5.30 (br s, 1 H, H-1D), 5.16 (d, J = 11.0 Hz, 1 H, PhCH), 5.12 (br s, 1 H, H-2D), 4.99 (s, 1 H, H-1C), 4.92–4.87 (m, 2 H, 2 PhCH), 4.80 (d, J = 12.0 Hz, 1 H, PhCH), 4.78 (d, J = 3.5 Hz, 1 H, H-1A), 4.76 (d, J = 3.5 Hz, H-1B), 4.74 (d, J = 12.0 Hz, 1 H, PhCH), 4.68 (d, J = 3.5 Hz, 1 H, H-1E), 4.65–4.56 (m, 8 H, 8 PhCH), 4.49–4.39 (m, 7 H, 7 PhCH), 4.15–4.05 (m, 5 H, H-3A, H-3D, H-4B, H-4D, H-5D), 4.01–3.85 (m, 4 H, H-2A, H-2C, H-3B, H-3C), 3.79–3.70 (m, 4 H, H-4A, H-5A, H-5B, H-6bB), 3.68–3.57 (m, 6 H, H-5C, H-6aB, H-6abD, OCH2), 3.50 (dd, J = 9.0, 3.5 Hz, 1 H, H-2B), 3.47–3.41 (m, 2 H, H-5E, H-6aA), 3.37–3.27 (m, 3 H, H-2E, H-4C, H-6bA), 3.24–3.21 (m, 2 H, NCH2), 2.99–2.92 (m, 1 H, H-4E), 2.28–2.22 (m, 1 H, H-3aE), 2.13 (s, 3 H, COCH3), 1.85 (q, J = 11.0 Hz, 1 H, H-3bE), 1.43 (s, 3 H, C(CH3)2), 1.28–1.05 (m, 9 H, C(CH3)2, 2 CCH3); 13C NMR (125 MHz, CDCl3): δ 170.4 (COCH3), 139.1–127.0 (Ar-C), 108.9 (C(CH3)2), 98.6 (C-1D), 98.5 (C-1A), 97.6 (C-1C), 97.2 (C-1B), 97.0 (C-1E), 81.9 (C-3B), 80.4 (C-2B), 79.8 (C-4C), 79.6 (C-3D), 77.7 (C-5C), 77.5 (C-4E), 77.3 (C-4A), 77.1 (C-2A), 77.6 (C-3A), 76.0 (C-2C), 75.5 (C-2D), 75.4 (PhCH2), 75.0 (PhCH2), 74.8 (PhCH2), 74.6 (PhCH2), 74.1 (C-4D), 73.5 (C-4B), 73.5 (PhCH2), 73.4 (PhCH2), 73.2 (PhCH2), 72.5 (PhCH2), 72.4 (C-2E), 71.5 (C-5D), 70.7 (PhCH2), 70.5 (PhCH2), 70.3 (C-3C), 70.0 (C-5A), 68.4 (2 C, C-6D, OCH2), 68.1 (C-5B), 67.1 (C-6B), 66.9 (C-6A), 65.8 (C-5E), 50.4 (NCH2), 29.9 (C-3E), 28.1, 26.4 (2 C(CH3)2), 21.2 (COCH3), 17.7, 17.2 (CCH3); HRMS calcd. for C107H125N4O24: [M+NH4]+ 1849.8678; found: 1849.8770.
2-Azidoethyl (2-O-acetyl-3,4,6-tri-O-benzyl-1-thio-α-D-mannopyranosyl)-(1→4)-(2,3-O-isopropylidene-α-L-rhamnopyranosyl)-(1→3)-[(2,3,4,6-tetra-O-benzyl-α-D-glucopyranosyl)-(1→6)]-2,4-di-O-benzyl-α-D-galactopyranoside (32):
A mixture of compound 30 (450 mg, 0.39 mmol), 10 (320 mg, 0.59 mmol) and MS 4Å (1 g) in CH2Cl2 (15 ml) was stirred at room temperature for 1 h and cooled to −30 °C. To the reaction mixture were sequentially added NIS (145 mg, 0.65 mmol) and AgOTf (30 mg, 0.12 mmol) and the reaction mixture was stirred at −30 °C for 30 min under argon. The reaction mixture was filtered and washed with CH2Cl2 (50 ml). The organic layer was washed with 5% Na2S2O3, satd. NaHCO3 successively, dried (Na2SO4), and concentrated. The crude product was purified over SiO2 column chromatography using hexane-EtOAc (4:1) as eluant to give pure compound 32 (490 mg, 77%). Colorless oil; [α]D25 +43 (c 1.0, CHCl3); IR (neat): 3059, 3025, 2937, 2106, 1744, 1603, 1498, 1450, 1371, 1234, 1102, 1027, 996 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.34–7.09 (m, 45 H, Ar-H), 5.31 (s, 1 H, H-1D), 5.29 (dd, J = 3.0, 1.5 Hz, 1 H, H-2D), 4.95 (br s, 1 H, H-1C), 4.93 (d, J = 11.0 Hz, 1 H, PhCH), 4.87 (d, J = 12.0 Hz, 1 H, PhCH), 4.81–4.76 (m, 4 H, 4 PhCH), 4.76 (d, J = 3.0 Hz, 1 H, H-1A), 4.71 (d, J = 3.0 Hz, 1 H, H-1B), 4.62–4.53 (m, 6 H, 6 PhCH), 4.51–4.42 (m, 6 H, 6 PhCH), 4.13 (dd, J = 9.0, 3.0 Hz, 1 H, H-3A), 4.08–4.01 (m, 4 H, H-3C, H-3D, H-4B, H-4D), 3.96–3.89 (m, 5 H, H-2A, H-2C, H-3B, H-5B, H-5D), 3.78–3.72 (m, 4 H, H-4A, H-5A, OCH2), 3.70–3.58 (m, 5 H, H-5C, H-6abB, H-6abD), 3.51 (dd, J = 9.0, 3.0 Hz, 1 H, H-2B), 3.44–3.36 (m, 2 H, NCH2), 2.14 (s, 3 H, COCH3), 1.45, 1.33 (2 s, 6 H, C(CH3)2), 1.30 (d, J = 7.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, CDCl3): δ 170.2 (COCH3), 138.7–127.4 (Ar-C), 108.9 (C(CH3)2), 98.7 (C-1D), 98.4 (C-1C), 97.3 (C-1A), 97.1 (C-1B), 82.0 (C-3B), 80.6 (C-4C), 79.8 (C-2B), 78.0 (C-2A), 77.6 (C-4A), 77.5 (C-5C), 76.6 (2 C, C-2C, C-3D), 76.0 (C-5D), 75.8 (C-3A), 75.4 (PhCH2), 75.3 (PhCH2), 75.2 (PhCH2), 75.0 (PhCH2), 74.5 (C-4B), 73.5 (PhCH2), 73.4 (PhCH2), 73.3 (PhCH2), 72.6 (PhCH2), 71.7 (PhCH2), 71.5 (C-4D), 70.4 (C-5A), 69.9 (C-3C), 68.8 (C-2D), 68.4 (OCH2), 68.3 (C-6D), 66.9 (2 C, C-6A, C-6B), 65.8 (C-5B), 50.4 (NCH2), 28.1, 26.4 (C(CH3)2), 21.0 (COCH3), 17.4 (CCH3); HRMS calcd. for C94H105N3NaO21: [M+Na]+ 1634.7139.; found: 1634.7167.
2-Aminoethyl (3,6-dideoxy-α-D-ribo-hexopyranosyl)-(1→3)-(α-D-mannopyranosyl)-(1→4)-(2,3-di-O-acetyl-α-L-rhamnopyranosyl)-(1→3)-α-D-galactopyranoside (1):
A solution of compound 25 (290 mg, 0.21 mmol) in 0.1 M CH3ONa in CH3OH (10 ml) was stirred at room temperature for 2 h, neutralized with Dowex 50W X8 (H+), filtered, and concentrated. To a solution of the de-O-acetylated product in DMF (2 ml) was added BnBr (30 μL, 0.25 mmol) and NaH (15 mg, 0.31 mmol, 50% oil coated) and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with CH3OH and the solvents were evaporated to dryness. A solution of the dry mass in 80% AcOH (5 ml) was stirred at 80 °C for 2 h and concentrated under reduced pressure. A solution of the residue in Ac2O (1 ml) and pyridine (1 ml) was stirred at room temperature for 3 h, concentrated and passed through a short pad of SiO2. To a solution of the resulting mass in methanol (10 ml) was added 20% Pd(OH)2-C (100 mg) and the mixture was stirred under a positive pressure of hydrogen for 24 h. The reaction mixture was filtered through a Celite® bed and the filtering bed was washed with a mixture of CH3OH and H2O. The combined filtrate was concentrated and the product was purified on a Sephadex G-25 gel column using water as the eluant. Lyophilisation of the product fractions afforded pure compound 1 (72 mg, 47%). White powder; [α]D25 +12.3 (c 1.0, H2O); 1H NMR (500 MHz, D2O): δ 5.41 (dd, J = 3.5, 1.5 Hz, H-2B), 5.32 (dd, J = 10.0, 3.5 Hz, 1 H, H-3B), 5.14 (d, J = 1.5 Hz, 1 H, H-1B), 5.05 (2 d, J = 3.5 Hz each, H-1A, H-1D), 5.02 (d, J = 1.5 Hz, 1 H, H-1C), 4.13–4.02 (m, 6 H, H-2A, H-2C, H-3A, H-3C, H-5B, OCH), 4.00–3.91 (m, 3 H, H-4A, H-4C, H-5C), 3.87–3.82 (m, 3 H, H-2D, H-4B, H-6aA), 3.81–3.73 (m, 6 H, H-5A, H-5D, H-6bA, H-6abC, OCH), 3.45–3.39 (m, 1 H, H-4D), 3.33–3.31 (m, 2 H, NCH2), 2.20 (s, 3 H, COCH3), 2.15–2.13 (m, 4 H, H-3aD, COCH3), 1.82 (q, J = 11.5 Hz, 1 H, H-3bD), 1.41 (d, J = 6.0 Hz, 3 H, CCH3), 1.25 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, D2O): δ 173.3, 172.9 (COCH3), 101.0 (C-1C), 99.5 (C-1D), 98.9 (C-1B), 98.4 (C-1A), 78.1 (C-4B), 77.9 (C-4A), 77.1 (C-2A), 73.3 (C-5A), 71.2 (C-4C), 70.8 (C-3B), 70.2 (C-3A), 69.8 (C-4D), 69.7 (C-3C), 69.1 (C-2B), 68.8 (C-2C), 68.3 (C-2D), 67.3 (C-5D), 67.0 (C-5B), 65.2 (C-5C), 63.8 (OCH2), 61.0 (C-6C), 60.2 (C-6A), 39.2 (NCH2), 34.4 (C-3D), 20.6, 20.1 (2 C, COCH3), 17.0, 16.4 (2 C, CCH3); HRMS calcd. for C30H52NO20: [M+H]+ 746.3077; found: 746.3111.
2-Aminoethyl (3,6-dideoxy-α-D-ribo-hexopyranosyl)-(1→3)-(α-D-mannopyranosyl)-(1→4)-(2,3-di-O-acetyl-α-L-rhamnopyranosyl)-(1→3)-[(α-D-glucopyranosyl)-(1→6)]-α-D-galactopyranoside (2):
A solution of compound 31 (350 mg, 0.191 mmol) in 0.1 M CH3ONa in CH3OH (10 ml) was stirred at room temperature for 2 h, neutralized with Dowex 50W X8 (H+), filtered, and concentrated. To a solution of the de-O-acetylated product in DMF (3 ml) was added BnBr (30 μl, 0.23 mmol) and NaH (16 mg, 0.34 mmol, 50% oil coated) and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with CH3OH and the solvents were evaporated to dryness. A solution of the dry mass in 80% AcOH (5 ml) was stirred at 80 °C for 2 h. A solution of the residue in Ac2O (1 ml) and pyridine (1 ml) was stirred at room temperature for 3 h, concentrated and passed through a short pad of SiO2. To a solution of the resulting mass in methanol (10 ml) was added 20% Pd(OH)2-C (100 mg) and the mixture was stirred under a positive pressure of hydrogen for 24 h. The reaction mixture was filtered through a Celite® bed and the filtering bed was washed with a mixture of CH3OH and H2O. The combined filtrate was concentrated and the product was purified on a Sephadex G-25 gel column using water as the eluant. Lyophilisation of the product fractions afforded pure compound 2 (91 mg, 53%). White powder; [α]D25 +21 (c 1.0, H2O); 1H NMR (500 MHz, D2O): δ 5.41 (dd, J = 3.0, 2.0 Hz, 1 H, H-2C), 5.32 (dd, J = 9.5, 3.5 Hz, 1 H, H-3C), 5.14 (br s, 1 H, H-1C), 5.05 (2 d, J = 3.5 Hz each, 2 H, H-1A, H-1E), 5.02 (d, J = 1.5 Hz, 1 H, H-1D), 4.97 (d, J = 3.5 Hz, 1 H, H-1B), 4.17–4.12 (m, 2 H, H-3A, H-5C), 4.10–4.03 (m, 4 H, H-2A, H-2D, H-4C, H-6aD), 3.96–3.84 (m, 7 H, H-3D, H-4A, H-4D, H-5D, H-6abB, OCH), 3.79–3.67 (m, 9 H, H-2E, H-3B, H-5A, H-5B, H-5E, H-6abA, H-6bD, OCH), 3.61 (d, J = 9.5, 3.5 Hz, 1 H, H-2B), 3.46–3.40 (m, 2 H, H-4B, H-4E), 3.33–3.31 (m, 2 H, NCH2), 2.20 (s, 3 H, COCH3), 2.17–2.14 (m, 1 H, H-3aE), 2.13 (s, 3 H, COCH3), 1.82 (q, J = 11.5 Hz, 1 H, H-3bE), 1.42 (d, J = 6.5 Hz, 3 H, CCH3), 1.25 (d, J = 6.0 Hz, 3 H, CCH3); 13C NMR (125 MHz, D2O): δ 172.2, 171.8 (2 C, COCH3), 101.0 (C-1D), 99.5 (C-1E), 98.9 (C-1C), 98.4 (C-1A), 98.0 (C-1B), 78.2 (C-4D), 77.5 (C-2A), 77.1 (C-4A), 73.3 (C-2E), 73.0 (C-3B), 71.8 (C-5E), 71.1 (C-2B), 70.8 (C-3C), 70.2 (C-3A), 69.8 (C-4C), 69.7 (C-2C), 69.4 (C-4B), 69.1 (C-4E), 69.0 (C-3D), 68.9 (C-5C), 68.3 (C-2D), 67.2 (C-5A), 67.0 (C-5B), 66.4 (OCH2), 65.2 (C-5D), 63.8 (C-6D), 60.5 (C-6B), 60.2 (C-6A), 39.2 (NCH2), 34.4 (C-3E), 20.6, 20.1 (2 C, COCH3), 17.0, 16.4 (2 C, CCH3); HRMS calcd. for C36H61NNaO25: [M+Na]+ 930.3425; found: 930.3462.
2-Aminoethyl (α-D-mannopyranosyl)-(1→4)-(2,3-di-O-acetyl-α-L-rhamnopyranosyl)-(1→3)-[(α-D-glucopyranosyl)-(1→6)]-α-D-galactopyranoside (3):
A solution of compound 32 (380 mg, 0.23 mmol) in 0.1 M CH3ONa in CH3OH (10 ml) was stirred at room temperature for 2 h, neutralized with Dowex 50W X8 (H+), filtered, and concentrated. To a solution of the de-O-acetylated product in DMF (3 ml) was added BnBr (35 μl, 0.28 mmol) and NaH (22 mg, 0.46 mmol, 50% oil coated) and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with CH3OH and the solvents were evaporated to dryness. A solution of the dry mass in 80% AcOH (5 ml) was stirred at 80 °C for 2 h. A solution of the residue in Ac2O (2 ml) and pyridine (2 ml) was stirred at room temperature for 3 h, concentrated and passed through a short pad of SiO2. To a solution of the resulting mass in CH3OH (10 ml) was added 20% Pd(OH)2-C (100 mg) and the mixture was stirred under a positive pressure of hydrogen for 24 h. The reaction mixture was filtered through a Celite® bed and the filtering bed was washed with a mixture of CH3OH and H2O. The combined filtrate was concentrated and the product was purified on a Sephadex G-25 gel column using water as the eluant. Lyophilisation of the product fractions afforded pure compound 3 (95 mg, 52%). White powder; [α]D25 +27 (c 1.0, H2O); 1H NMR (500 MHz, D2O): δ 5.41 (br s, 1 H, H-2C), 5.29 (dd, J = 9.5, 3.0 Hz, 1 H, H-3C), 5.14 (br s, 1 H, H-1C), 5.05 (d, J = 3.5 Hz, 1 H, H-1A), 5.04 (d, J = 1.5 Hz, 1 H, H-1D), 4.97 (d, J = 3.5 Hz, 1 H, H-1B), 4.17–4.10 (m, 2 H, H-4A, H-5C), 4.08–4.00 (m, 5 H, H-2A, H-2D, H-4D, H-5A, H-6aD), 3.96–3.81 (m, 5 H, H-4C, H-6aA, H-6abB, OCH), 3.79–3.67 (m, 8 H, H-3A, H-3B, H-3D, H-5B, H-5D, H-6bA, H-6bD, OCH), 3.61 (dd, J = 9.5, 3.5 Hz, 1 H, H-2B), 3.46 (t, J = 9.5 Hz, 1 H, H-4B), 3.32–3.30 (m, 2 H, NCH2), 2.19, 2.12 (2 s, 6 H, COCH3), 1.41 (d, J = 6.5 Hz, 3 H, CCH3); 13C NMR (125 MHz, D2O): δ 173.3, 172.9 (2 C, 2 COCH3), 100.9 (C-1D), 98.9 (C-1C), 98.4 (C-1A), 98.0 (C-1B), 77.4 (C-2A), 76.6 (C-4C), 73.1 (C-3B), 73.0 (C-3A), 71.8 (C-2B), 71.1 (C-3D), 70.7 (C-3C), 70.1 (C-2D), 70.0 (C-4D), 69.8 (C-4A), 69.4 (C-4B), 69.0 (C-2C), 68.9 (C-5B), 68.5 (C-5C), 67.2 (C-5A), 66.3 (OCH2), 66.0 (C-5D), 63.8 (C-6D), 60.5 (C-6B), 60.4 (C-6A), 39.2 (NCH2), 20.6, 20.0 (2 C, COCH3), 17.0 (CCH3); HRMS calcd. for C30H52NO22: [M+H]+ 778.2976; found: 778.3013.
Synthesis and characterization of Qβ-glycan conjugate 1.
A solution of adipate bisNHS ester 33 (13 mg in 0.5 ml DMF, 0.037 mmol) was added into a solution of tetrasaccharide 1 (5 mg in 0.5 ml DMF, 0.007 mmol), which was followed by addition of di-isopropylethylamine (DIPEA, 1 μl, 0.007 mmol). The reaction mixture was stirred for 4 h at room temperature, concentrated under reduced pressure and purified by washing with DCM 3 times to give pure compound 34 (5 mg, 75%). White solid; 1H NMR (500 MHz, CD3OD): δ 8.18 (brs, 1H, NH), 5.39 (s, 1H), 5.30 (dd, J = 9.5, 3 Hz, 1H), 5.03 (s, 1H), 4.93 (d, J = 3.0 Hz, 1H), 4.91 (s, 1H), 4.82 (d, J = 3.5 Hz, 1H), 4.12–4.05 (m, 1H), 4.00–3.92 (m, 5H), 3.83–3.61 (m, 11H), 3.62–3.46 (m, 3H), 3.17 (td, J = 11.5, 3.5 Hz, 1H), 2.83 (s, 4H, CH2 in NHS), 2.67 (t, J = 6.0 Hz, 2H, COCH2), 2.26 (t, J = 6.5 Hz, 2H, COCH2), 2.10 (s, 3H, COCH3), 2.06–2.01 (m, 4H, H-3aD, COCH3), 1.85–1.77 (m, 1H, H-3bD), 1.77–1.71 (m, 4H), 1.34 (d, J = 6.0 Hz, 3H, CCH3), 1.17 (d, J = 6.5 Hz, 3H, CCH3). To a solution of bacteriophage Qβ (5 mg, expressed and purified as previously described[59–60]) in potassium phosphate buffer (0.1 M, pH=7, 2.5 ml) was added a solution of 34 (20 mg/ml in DMSO, 0.25 mL, 0.006 mmol). The reaction mixture was stirred overnight at room temperature on a tube rotator. The conjugate was purified by repeated filtration using an Amicon Ultra 100 kDa MW cut-off device against PBS buffer. Total protein content was quantified by Bradford assay against BSA standards. An average loading of 290 tetrasaccharide 1 per Qβ particle was determined by ESI-TOF LC-MS analysis (Figure S1).
Synthesis and characterization of BSA-glycan conjugate 1.
A solution of 34 (20 mg/ml in DMSO, 0.1 ml, 0.002 mmol) was added to a solution of BSA (2 mg) in potassium phosphate buffer (0.1 M, pH=7, 1 ml). The reaction mixture was stirred overnight at room temperature on a tube rotator. The conjugate was purified by repeated filtration using an Amicon Ultra 10 kDa MW cut-off device against PBS buffer. Total protein content was quantified by Bradford assay against BSA standards. A loading of 15 tetrasaccharides per BSA was determined using MALDI-TOF MS analysis (Figure S2).
Procedure for mouse immunization.
Pathogen-free C57BL6 female mice aged 6–10 weeks were purchased from Charles River and maintained in the University Laboratory Animal Resources facility of Michigan State University. These animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of Michigan State University. C57BL6 mice were injected subcutaneously under the scruff on day 0 with 0.1 mL Qβ-glycan 1 construct (at 1 μg and 4 μg glycan respectively) as emulsions in Complete Freund’s Adjuvant according to manufacturer’s instructions. Boosters were given subcutaneously under the scruff on days 14 and 28 mixed with Incomplete Freund’s Adjuvant. Serum samples were collected on days 0 (before immunization), 7 and 35.
Procedure for rabbit immunization.
Rabbit immunization studies were performed by ProSci Inc (Poway, CA). Two New Zealand rabbits were injected subcutaneously on day 0 with 0.1 mL Qβ-glycan 1 constructs (at 8 μg glycan) as emulsions in Complete Freund’s Adjuvant according to manufacturer’s instructions. Boosters were given subcutaneously on days 14, 28 and 42 mixed with Incomplete Freund’s Adjuvant. Serum samples were collected on days 0 (before immunization), 35, 49 and 56.
Evaluation of antibody titers by ELISA.
A 96-well Nunc microtiter plate was first coated with a solution of BSA-glycan conjugate in NaHCO3/Na2CO3 buffer (pH = 9.5) and incubated at 4 °C overnight. The plate was washed with PBST (4 × 200 μL), blocked with 1% BSA/PBS (200 μL/well) for 1 h at room temperature, washed with PBST (4 × 200 μL), and incubated with serial dilutions of anti-sera from immunized mice or rabbits in 0.1% BSA/PBS (100 μL/well, 4 wells for each dilution). The plate was incubated for 2 h at 37 °C and then washed with PBST (4 × 200 μL). A 1:2000 dilution of HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Jackson ImmunoResearch Laboratory) in 0.1% BSA/PBS (100 μL) was added to the wells respectively to determine the levels of antibodies generated. The plate was incubated for 1 h at 37 °C. A solution of enzymatic substrate was prepared by dissolving TMB (5 mg) in a mixture of DMSO (2 mL) and citric acid buffer (18 mL) in a 50 mL centrifuge tube covered with aluminium foil. H2O2 (20 μL) was added and the mixture was homogenized by vortexing. The plate was washed with PBST (4 × 200 μL) and a solution of enzymatic substrate was added (200 μL). Color was allowed to develop for 15 min and 0.5 M H2SO4 (50 μL) was added to quench the reaction. The absorbance was measured at 450 nm using a microplate reader. The titer was determined by regression analysis with log10 dilution plotted with optical density. ELISA titers were calculated as the highest fold of serum dilution that gave optical density of 0.1 higher than the background in ELISA assays.
Bacterial strains, medium, and growth conditions.
S. Paratyphi A ATCC 9150, a wild-type enteric fever isolate, was used to show binding of antibodies by flow cytometry, in opsonophagocytosis assays and protection studies in mice. S. Paratyphi A CVD 1902 is a live attenuated vaccine strain with mutations in guaBA and clpX. This strain was used as the source of OPS. All strains were maintained in Hy-Soy (HS) media (5 g/L sodium chloride, 10 g/L soytone [Teknova, CA, USA], 5 g/L Hy-yest [Kerry, WI, USA]) at 37 °C. Growth medium for all guaBA mutants was supplemented with 0.001% w/v guanine (Sigma-Aldrich, MO).
S. Paratyphi A OPS used as an ELISA antigen were purified from fermentation cultures of CVD 1902 as described previously[61] by extraction with acetic acid and purification by sequential tangential flow filtration, anion exchange chromatography, and ammonium sulfate precipitation.
Procedure for flow cytometry.
The flow cytometry assay was performed using procedures similar to those described previously.[18] Briefly, a single colony of S. Paratyphi A strain ATCC 9150 was used to inoculate an overnight HS broth culture (37 °C, 220 rpm). The next day, the bacterial suspension was diluted to an OD600 of 0.8 and cooled on ice. Pooled pre-immunization and day 56 post-vaccination rabbit sera were heat-inactivated at 56 °C for 30 min. These serum pools were then diluted to a final concentration of 1:40 (pre-immunization) or 1:40, 1:250, 1:1000, 1:5000, and 1:25000 (post-immunization) in 250 μL of flow buffer (1% heat-inactivated FBS [Gemini Bio, CA, USA], 0.02% sodium azide [Sigma-Aldrich] in PBS). Then, a 250 μL aliquot of the bacterial suspension was stained for 1 h at 4 °C with the diluted rabbit sera. Bound antibodies were then detected with 1 μg/mL of AlexaFluor 488-conjugated donkey anti-rabbit IgG (Thermo Fisher, IL), which was incubated for 1 h at 4 °C. Bacteria were then fixed with 2% formaldehyde and read using an LSR-II flow cytometer (BD Biosciences, NJ, USA) with 5 × 104 events collected.
Procedure for opsonophagocytosis assay.
J774 mouse macrophage cells (American Type Culture Collection, VA, USA) were maintained in Dulbecco’s modified Eagle medium (DMEM) containing 4.5 g/L D-glucose and L-glutamine (Gibco, MD, USA) supplemented with 1 mM sodium pyruvate, 25 mM HEPES (Sigma-Aldrich), and 10% v/v heat-inactivated FBS (Gemini Bio) hereafter referred to as D10. For subculture, 1% v/v antibiotic/antimycotic (Gemini Bio) was added to D10 to prevent contamination. 24-well plates were seeded with 1 × 105 J774 cells/well and incubated for 48 h at 37 °C with 5% CO2. Partially confluent monolayers of J774 cells were then washed three times with sterile PBS, and the media was replaced with 500 μL of fresh D10. Bacteria were prepared in the following manner: a single colony of S. Paratyphi A strain ATCC 9150 was used to inoculate an overnight HS broth culture (37 °C, 220 rpm). The next day, log-phase cultures were prepared by diluting the overnight culture 1:50 in fresh HS broth (37 °C, 220 rpm). At 3 h, the culture was adjusted to an OD600 of 0.7. To microcentrifuge tubes, 22.5 μL of the diluted bacteria (~9 × 106 CFU), 2 μL of pooled rabbit sera, and 75.5 μL of PBS were added. Bacteria were opsonized for 20 min at room temperature with periodic flicking. The sera for this experiment were collected from Qβ-glycan 1-immunized rabbits (pre-immunization and day 56 post-immunization) and Qβ-immunized rabbits (day 56 post-immunization). Afterward, 10 μL of the opsonized bacteria (~9 × 105 CFU) were added to duplicate wells of the 24-well plate, which were then swirled to ensure adequate mixing. Following a 45 min incubation at 37 °C/5% CO2, plates were washed once with sterile PBS, which was replaced with 500 μL of D10 supplemented with 100 μg/mL of gentamicin (Gemini Bio). Plates were returned to the 37 °C/5% CO2 incubator for 1 h. Afterward, gentamicin-treated plates were washed three times with sterile PBS, and cells were lysed with 500 μL of sterile water (10 min, 250 rpm). Next, wells were scraped with a pipette tip to ensure complete lysis. Lysates were then serially diluted in PBS and plated on HS to determine viable counts (100 μL spread on duplicate plates).
Protection of mice from lethal challenge with S. Paratyphi A.
All animal studies were performed in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and in compliance with policies established by the US Department of Agriculture Animal Welfare Act and Public Health Service. These animal experiments were conducted with approval from the Institutional Animal Care and Use Committee at the University of Maryland, School of Medicine. Pre-immunization and day 56 post-immunization rabbit sera were heat-inactivated at 56 °C for 30 min and diluted 1:10 in PBS. Naïve 6–8 week old female BALB/c mice (Charles River Laboratories, MA, USA) were injected i.p. with 100 μL of the diluted rabbit sera or PBS. After 5–6 hours, recipient mice were challenged i.p. with 7.3 × 103 CFU of S. Paratyphi A strain ATCC 9150 (prepared from an overnight HS plate) diluted in 500 μL of sterile 10% hog-gastric mucin (Sigma-Aldrich). Infected mice were monitored twice a day for three days, and animals that were moribund and/or had sustained 20% weight loss were euthanized and recorded as having succumbed to challenge. Vaccine efficacy for passively immunized mice was determined based on the proportionate attack rate (AR) in vaccinated and PBS control mice: ((ARcontrol – ARvaccinated)/ARcontrol) × 100.
Supplementary Material
Acknowledgments:
We are grateful for financial support from the National Institute of Allergy and Infectious Diseases, NIH (R01AI146210), Michigan State University, and the Council of Scientific and Industrial Research (CSIR), India (AKM) [Project No. 02(0237)/15/ EMR-II].
References
- [1].Crump JA and Mintz ED, Clin. Infect. Dis 2010, 50, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Whitaker JA, Franco-Paredes C, del Rio C and Edupuganti S, J. Travel Med 2009, 16, 46–52. [DOI] [PubMed] [Google Scholar]
- [3].Gupta SK, Medalla F, Omondi MW, Whichard JM, Fields PI, Gerner-Smidt P, Patel NJ, Cooper KLF, Chiller TM and Mintz ED, Clin. Infect. Dis 2008, 46, 1656–1663. [DOI] [PubMed] [Google Scholar]
- [4].Kanungo S, Dutta S and Sur D, J. Infect. Dev. Countr 2008, 2, 454–460. [DOI] [PubMed] [Google Scholar]
- [5].Britto CD, John J, Verghese VP and Pollard AJ, Indian J. Med. Res 2019, 149, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Verma S, Thakur S, Kanga A, Singh G and Gupta P, Indian J. Med. Microbiol 2010, 28, 51–53. [DOI] [PubMed] [Google Scholar]
- [7].MacLennan CA, Martin LB and Micoli F, Hum. Vaccin. Immunother 2014, 10, 1478–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sahastrabuddhe S, Carbis R, Wierzba TF and Ochiai RL, Expert Rev. Vaccines 2013, 12, 1021–1031. [DOI] [PubMed] [Google Scholar]
- [9].Kaur J and Jain SK, Microbiol. Res 2012, 167, 199–210. [DOI] [PubMed] [Google Scholar]
- [10].Konadu EY, Lin F-YC, Hó VO, Thuy NTT, van Bay P, Thanh TC, Khiem HB, Trach DD, Karpas AB, Li J, Bryla DA, Robbins JB and Szu S, Infect. Immun 2000, 68, 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Micoli F, Rondini S, Gavini M, Lanzilao L, Medaglini D, Saul A and Martin LB, PLoS One 2012, 7, e47093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ravenscroft N, Cescutti P, Gavini M, Stefanetti G, MacLennan CA, Martin LB and F. M, Carbohydr. Res 2015, 404, 108–116. [DOI] [PubMed] [Google Scholar]
- [13].Hellerqvist C-G, Hoffman J, Lindberg AA, Lindberg B and Svensson S, Acta Chem. Scand 1972, 26, 3282–3286. [DOI] [PubMed] [Google Scholar]
- [14].Konadu E, Shiloach J, Bryla DA, Robbins JB and Szu SC, Infect. Immun 1996, 64, 2709–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Olsson JDM, Landström J, Rönnols J, Oscarson S and Widmalm G, Org. Biomol. Chem 2009, 7, 1612–1618 [DOI] [PubMed] [Google Scholar]
- [16].Zhang G-L, Wei M-M, Song C, Ma Y-F, Zheng X-J, Xiaong D-C and Ye X-S, Org. Chem. Front 2018, 5, 2179–2188. [Google Scholar]
- [17].Torgov VI, Panossian CA and Shibaev VN, Carbohydr. Res 1987, 161, 97–112. [DOI] [PubMed] [Google Scholar]
- [18].Huo C-X, Dhara D, Baliban SM, Tahmasebi Nick S, Tan Z, Simon R, Misra AK and Huang X, Chem. Commun 2019, 55, 4519–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sarbajna S, Misra AK and Roy N, Synth. Commun 1998, 28, 2559–2570. [Google Scholar]
- [20].Lowary TL, Eichler E and Bundle DR, J. Org. Chem 1995, 60, 7316–7327. [Google Scholar]
- [21].Dhara D, Mandal PK and Misra AK, Tetrahedron: Asymm. 2014, 25, 263–267. [Google Scholar]
- [22].Andersson F, Fugedi P, J. GP and Nashed M, Tetrahedron Lett. 1986, 27, 3919–3922. [Google Scholar]
- [23].Dhara D and Misra AK, Tetrahedron: Asymm. 2013, 24, 1488–1494. [Google Scholar]
- [24].Lemieux RU, Can. J. Chem 1951, 29, 1079–1091. [DOI] [PubMed] [Google Scholar]
- [25].Heuckendorff M, Pedersen CM and Bols M, J. Org. Chem 2013, 78, 7234–7248. [DOI] [PubMed] [Google Scholar]
- [26].Brimacombe JS, Methods Carbohydr. Chem 1972, 6, 376–378. [Google Scholar]
- [27].Thomas EJ and Williams AC, J. Chem. Soc., Chem. Commun 1987, 992–993. [Google Scholar]
- [28].Barton DHR and McCombie SW, J. Chem. Soc., Perkin Trans 1 1975, 1574–1585. [Google Scholar]
- [29].Robins MJ, Wilson JS and Hansske F, J. Am. Chem. Soc 1983, 105, 4059–4065. [Google Scholar]
- [30].Kiyoi T, Nakai Y, Kondo H, Ishida H, Kiso M and Hasegawa A, Bioorg. Med. Chem 1996, 4, 1167–1176. [DOI] [PubMed] [Google Scholar]
- [31].Chatterjee S, Moon S, Hentschel F, Gilmore K and Seeberger PH, J. Am. Chem. Soc 2018, 140, 11942–11953. [DOI] [PubMed] [Google Scholar]
- [32].Corey EJ and Venkateswarlu A, J. Am. Chem. Soc 1972, 6190–6191. [Google Scholar]
- [33].Fraser-Reid B and Lopez JC, Top. Curr. Chem 2011, 301, 1–29. [DOI] [PubMed] [Google Scholar]
- [34].Koeller KM and Wong C-H, Chem. Rev 2000, 100, 4465–4494 and references cited therein. [DOI] [PubMed] [Google Scholar]
- [35].Konradsson P, Udodong UE and Fraser-Reid B, Tetrahedron Lett. 1990, 31, 4313–4316. [Google Scholar]
- [36].Veeneman GH, van Leeuwen SH and van Boom JH, Tetrahedron Lett. 1990, 31, 1331–1334. [Google Scholar]
- [37].Escopy S, Singh Y and Demchenko AV, Org. Biomol. Chem 2019, 17, 8379–8383. [DOI] [PubMed] [Google Scholar]
- [38].Gemma E, Lahmann M and Oscarson S, Carbohydr. Res 2005, 340, 2558–2562. [DOI] [PubMed] [Google Scholar]
- [39].Chang CW, Wu CH, Lin MH, Liao P-H, Chang C-C, Chuang H-H, Lin S-C, Lam S, Verma VP, Hsu C-P and Wang C-C, Angew. Chem. Int. Ed 2019, 58, 16775–16779. [DOI] [PubMed] [Google Scholar]
- [40].Crich D and Vinogradova O, J. Org. Chem 2006, 71, 8473–8480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Castro EA, Cubillos M and Santos JG, J. Org. Chem 2004, 69, 4802–4807. [DOI] [PubMed] [Google Scholar]
- [42].Clive DLJ, Chittattu G and Wong CK, J. Chem. Soc., Chem. Commun 1978, 41–42. [Google Scholar]
- [43].Bhattacharyya S, Magnusson BG, Wellmer U and Nilsson UJ, J. Chem. Soc. Perkin Trans 1 2001, 886–890. [Google Scholar]
- [44].Zemplén G, Ber. Dtsch. Chem. Ges 1926, 59, 1254–1266. [Google Scholar]
- [45].Gent PA and Gigg R, J. Chem. Soc., Perkin Trans I 1974. 1446–1455. [Google Scholar]
- [46].Pearlman WM, Tetrahedron Lett. 1967, 17, 1663–1664. [Google Scholar]
- [47].Bock K and Pedersen C, J. Chem. Soc. Perkin Trans 2 1974, 293–297. [Google Scholar]
- [48].Saksena R, Zhang J and Kovac P, J. Carbohydr. Chem 2002, 21, 453–470. [Google Scholar]
- [49].Guo ZW and Wang QL, Curr. Opin. Chem. Biol 2009, 13, 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yin Z and Huang X, J. Carbohydr. Chem 2012, 31, 143–186 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bachmann MF and Jennings GT, Nat. Rev. Immunol 2010, 10, 787–796. [DOI] [PubMed] [Google Scholar]
- [52].Bachmann MF and Zinkernagel RM, Annu. Rev. Immunol 1997, 15, 235–270. [DOI] [PubMed] [Google Scholar]
- [53].Yin Z, Comellas-Aragones M, Chowdhury S, Bentley P, Kaczanowska K, BenMohamed L, Gildersleeve JC, Finn MG and Huang X, ACS Chem. Biol 2013, 8, 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wu X, Yin Z, McKay C, Pett C, Yu J, Schorlemer M, Gohl T, Sungsuwan S, Ramadan S, Baniel C, Allmon A, Das R, Westerlind U, Finn MG and Huang X, J. Am. Chem. Soc 2018, 140, 16596–16609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang P, Huo C-X, Lang S, Caution K, Nick ST, Dubey P, Deora R and Huang X, Angew. Chem. Int. Ed 2020, 59, 6451–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Simon R and Levine MM, Hum. Vaccine Immunother 2012, 8, 494–498 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ramachandran G, Boyd MA, MacSwords J, Higginson EE, Simon R, Galen JE, Pasetti MF, Levine MM and Tennant SM, Clin. Vaccine Immunol 2016, 23, 520–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gat O, Galen JE, Tennant S, Simon R, Blackwelder WC, Silverman DJ, Pasetti MF and Levine MM, PLoS Negl. Trop. Dis 2011, 5, e1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wu X, McKay C, Pett C, Yu J, Schorlemer M, Ramadan S, Lang S, Behren S, Westerlind U, Finn MG and Huang X, ACS Chem. Biol 2019, 14, 2176–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yin Z, Wu X, Kaczanowska K, Sungsuwan S, Comellas-Aragones M, Pett C, Yu J, Baniel C, Westerlind U, Finn MG and Huang X, ACS Chem. Biol 2018, 13, 1668–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Baliban SM, Yang M, Ramachandran G, Curtis B, Shridhar S, Laufer RS, Wang JY, Van Druff J, Higginson EE, Hegerle N, Varney KM, Galen JE, Tennant SM, Lees A, MacKerell AD, Levine MM and Simon R, PLoS Negl. Trop. Dis 2018, 11, e0005493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.