

Abstract
Objective:
To examine associations of molecular markers of brain insulin signaling with Alzheimer disease (AD) and cognition among older persons with or without diabetes.
Methods:
This clinical–pathologic study was derived from a community-based cohort study, the Religious Orders Study. We studied 150 individuals (mean age at death =87 years, 48% women): 75 with and 75 without diabetes (matched by sex on age at death and education). Using enzyme-linked immunosorbent assay, immunohistochemistry, and ex vivo stimulation of brain tissue with insulin, we assessed insulin signaling in the postmortem middle frontal gyrus cortex. Postmortem data documented AD neuropathology. Clinical evaluations documented cognitive function proximate to death, based on 17 neuropsychological tests. In adjusted regression analyses, we examined associations of brain insulin signaling with diabetes, AD, and level of cognition.
Results:
Brain insulin receptor substrate-1 (IRS1) phosphorylation (pS307IRS1/total IRS1) and serine/threonine-protein kinase (AKT) phosphorylation (pT308AKT1/total AKT1) were similar in persons with or without diabetes. AKT phosphorylation was associated with the global AD pathology score (p = 0.001). In contrast, IRS1 phosphorylation was not associated with AD (p = 0.536). No other associations of insulin signaling were found with the global AD score, including when using the ex vivo brain insulin stimulation method. In secondary analyses, normalized pT308AKT1 was positively correlated with both the amyloid burden and tau tangle density, and no other associations of brain insulin signaling with neuropathology were observed. Moreover, normalized pT308AKT1 was associated with a lower level of global cognitive function (estimate = −0.212, standard error = 0.097; p = 0.031).
Interpretation:
Brain AKT phosphorylation, a critical node in the signaling of insulin and other growth factors, is associated with AD neuropathology and lower cognitive function.
Metabolic disturbances are common and increasing with contemporary lifestyles. Among the most common metabolic disturbances is insulin resistance, characterized by a blunted response to insulin. Insulin resistance in skeletal muscle, liver, and adipose tissue is a core feature of type 2 diabetes.1 Brain dysfunction, including cognitive impairment and dementia in aging, is a well-recognized complication of diabetes.2 However, beyond cerebrovascular disease, mechanisms underlying the relation of insulin resistance in brain, diabetes, and cognitive impairment remain elusive.3
Insulin has beneficial effects on the brain, including trophic effects on synapses and promotion of dendritic spine formation.4 Insulin signaling also influences the production and turnover of amyloidogenic proteins, including amyloid-β (Aβ)5 and paired-helical filament tau, that disrupt neuronal function. Although the literature on diabetes and Alzheimer disease (AD) neurodegenerative pathology is mixed, some human postmortem data point to a link between brain insulin resistance and increased AD pathology, including increased Aβ deposition. Furthermore, some clinical studies have found that patients with diabetes have altered cerebrospinal fluid Aβ and tau abundance and lower levels of cognition, whereas data from preclinical and small clinical trials suggest, in several but not all studies, that treatment with insulin and agents that promote insulin signaling may reduce neuropathology and improve cognition in diabetes and AD.6–8 Pathophysiologic processes downstream from insulin signaling, such as alterations in serine /threonine-protein kinase (AKT), are also thought to be involved in AD pathology, including Aβ deposition and tau phosphorylation.9
This study examined the associations of key insulin signaling molecules (insulin receptor substrate-1 [IRS1] and AKT) with AD neuropathology and cognitive function in persons with or without diabetes. We used brain specimens from a group of 150 older persons (75 with, and 75 without diabetes; matched 1:1), who were part of a community-based clinical–pathologic study of aging, to investigate insulin signaling measures in postmortem middle frontal gyrus (MFC) cortex, using biochemical, immunohistochemical, and ex vivo brain insulin stimulation approaches. With adjusted linear regression analyses, we tested the associations of brain insulin signaling measures with outcomes of global and specific AD neuropathology measures, and global cognitive function and cognitive domains proximate to death.
Subjects and Methods
Cohort, and Clinical and Pathologic Data
The Religious Orders Study (ROS) is an ongoing, prospective, community-based clinical–pathologic cohort study of aging.10 Subjects signed an informed consent and were asked to sign an anatomical gift act to donate their brain at the time of death. The study was approved by the Rush University Institutional Review Board. ROS began enrolling Catholic nuns, priests, and brothers in 1994, from about 40 convents and monasteries across the USA. Of 1,194 people enrolled at the time of analyses, 669 died and 622 came to autopsy, of whom 613 had neuropathologic data collection complete and available for analysis at the time of this study initiation.
All subjects underwent annual clinical evaluations, which included a medical history, physical examination, and neuropsychological testing.10 A battery of neuropsychological tests assessed a broad range of cognitive abilities, as previously described.11 Seventeen tests were grouped to form composite measures of global cognition and 5 cognitive domains. To create each composite score, individual tests were converted to z scores, using the baseline mean and standard deviation from the entire cohort, and z scores for all tests in a given domain were averaged. Annual clinical data were used to determine the presence of diabetes, based on the medical history and visually inspected medications (antidiabetes medications), as previously published.12 The follow-up rate in ROS is >90% among survivors. Classification of dementia status was made at the time of death, based on clinical data from all prior evaluations, as in our prior publications.10 Data on depressive symptoms, based on the 10 items of the Center for Epidemiological Studies Depression scale, were also available.10 Apolipoprotein E ε4 (APOE ε4) genotype data were collected as previously described.10
Brain autopsies followed a uniform and standardized procedure, with tissues from one hemisphere being frozen (−80°C) and from the other, being fixed (paraformaldehyde).10 Postmortem neuropathologic evaluations were conducted on autopsied brains (ROS autopsy rate > 90%; mean postmortem interval [PMI] = 9.6 hours), blinded to clinical data, as previously described.13 Briefly, a uniform gross and histologic evaluation examined for common age-related neuropathologies. A continuous, overall AD pathology standardized measure summarized counts of neuritic plaques, diffuse plaques, and neuronal neurofibrillary tangles from a 1mm2 area (of greatest density), from sections of hippocampus and entorhinal, MFC (Brodmann area BA46/9), middle temporal gyrus (BA21), and inferior parietal/angular gyrus (BA39) cortices, using a modified silver stain.13 Furthermore, immunohistochemical measures of global Aβ burden and tau tangle density were also available, including as a summary measure across different brain regions, as well as in the MFC specifically.14 For analyses, given that the 3 AD pathology measures were not normally distributed, we transformed these data using the square root.
Selection of Cases with and without Diabetes
Of the 613 persons with neuropathologic data collection complete and available for analysis, we applied a series of exclusion criteria to select cases for this study. Of the 613, we excluded 5 persons younger than 70 years at the time of death, because younger age at death is uncommon in this cohort and the study design involved matching of subjects by age. We then excluded 99 persons because of incomplete clinical data on cognition within 18 months of death (n = 89), final summary clinical diagnosis pending (n = 9), and missing diabetes status (n = 1). From the remaining 509 persons, 2 were excluded because of missing PMI data, and 21 because of pending Aβ or tangle data. Detailed manual review of data by a physician researcher (Z.A.) led to exclusion of 26 cases with either specific and/or less common clinical diseases (eg, Lewy body disease) or other exclusionary pathologies (eg, brain tumor). Thus, a total of 454 persons were available for randomization for this study (352 had a PMI ≤ 12 hours). A total of 75 with and 75 without diabetes were matched on continuous measures of age at death and education, using a propensity score-based algorithm. Matching was then performed within sex groups (ie, men-men and women-women). The algorithm also ensured that at least 39 pairs (39 matched with and 39 without diabetes) with PMI ≤ 12 hours were available for the ex vivo stimulation experiments.
We included in this study 150 autopsied subjects with complete data on essential clinical and pathologic measures of interest, and available MFC tissue samples (all with PMI ≤ 27 hours) for further study. The sample included 75 subjects with diabetes and 75 without diabetes (1:1). The pairs were selected by matching by sex based on a propensity score greedy algorithm, and based on age at death and education (each as a continuous variable). The mean age difference within pairs was 0.12 years, and the mean education difference was 0.03 years (both not significantly different). Several measures were taken to increase study rigor, including testing pairs of tissues from matched subjects simultaneously in the laboratories (eg, at the same time, on the same equipment), blinded to diabetes status. The pairwise difference of the obtained markers was calculated for each marker individually. We then calculated the mean pairwise difference and its 95% confidence intervals. We used a simple linear regression to verify whether the difference in age at death and education within pairs was associated with the pairwise difference in markers. There was no association for any of the markers, supporting the scientific rigor used to match the pairs of persons with and without diabetes.
Brain Insulin Signaling by Enzyme-Linked Immunosorbent Assay
Previously frozen MFC brain samples were thawed and homogenized in lysis buffer containing protease and phosphatase inhibitors, and supernatant aliquots containing 50pg protein were assayed in duplicate using PathScan enzyme-linked immunosorbent assay (ELISA) kits (Cell Signaling Technology, Danvers, MA). The following ELISAs were performed: total IRS1, catalog #7328; pIRS1 (S307), catalog #7287; AKT1, catalog #7170; pAKT1 (Thr308), catalog #7252. The PathScan ELISA kit is a solid-phase sandwich ELISA. After incubation with lysates, the protein was captured by the coated antibody. Following extensive washing, a specific antibody was added to detect the captured protein, antirabbit IgG, horseradish peroxidase (HRP)-linked antibody was added to recognize the bound detection antibody, and HRP substrate, tetramethylbenzidine, was added to develop color, and the absorbance was read at 450nm. The plates were read on an Epoch Microplate Spectrophotometer (BioTek Instruments, Winooski, VT).
Brain Immunohistochemistry
Paraformaldehyde-fixed, paraffin-embedded blocks of MFC were used. Immunohistochemistry for pS616IRS1 in tissue sections was conducted as previously described (Fig 1).15,16 Briefly, 10μm-thick sections were dewaxed in xylenes, rehydrated in descending ethanols, and quenched of endogenous peroxidase activity in 5% H2O2 and treated for epitope retrieval by boiling in 1mM ethylenediaminetetraacetic acid (pH 8.0) for 10 minutes. Tissues were rinsed in buffer, blocked in 10% normal horse serum, and incubated in primary antibody against pS616IRS (44550G, Invitrogen, Carlsbad, CA; rabbit 1:500) overnight at 4°C. Sections were incubated in secondary antibody followed by avidin–biotin–peroxidase complex for 1 hour, and finally reacted with a 0.05% diaminobenzidine (DAB)–0.03% hydrogen peroxide solution for 10 minutes. The immunoreaction signal was enhanced by adding NiSO4 (0.25% final dilution) to the DAB solution. Sections were then rinsed in water, dehydrated in ascending ethanols, cleared in xylenes, and coverslipped. For pS616IRS1 quantitation, we used high-throughput computer-assisted image analysis count determinations, as previously described. Briefly, under uniform lighting conditions, grayscale photomicrographs at ×200 covering the cortical region of interest were acquired on an automated microscopy digital image capture instrument (Glissando Slide Scanner, Objective Imaging, Cambridge, UK). Using Image-Pro Plus software (Media Cybernetics, Rockville, MD), cortical gray matter expanses of interest were manually delineated, and an image filtering algorithm was applied to differentiate contiguous immunostained pixels from background unstained tissue, followed by a size filter to identify immunoreactive neuronal cytoplasmic profiles, which were then counted and expressed as a density (per mm2). Analyses used the semiquantitative variable of the number of pS616IRS1 cell profiles/mm2.
FIGURE 1:
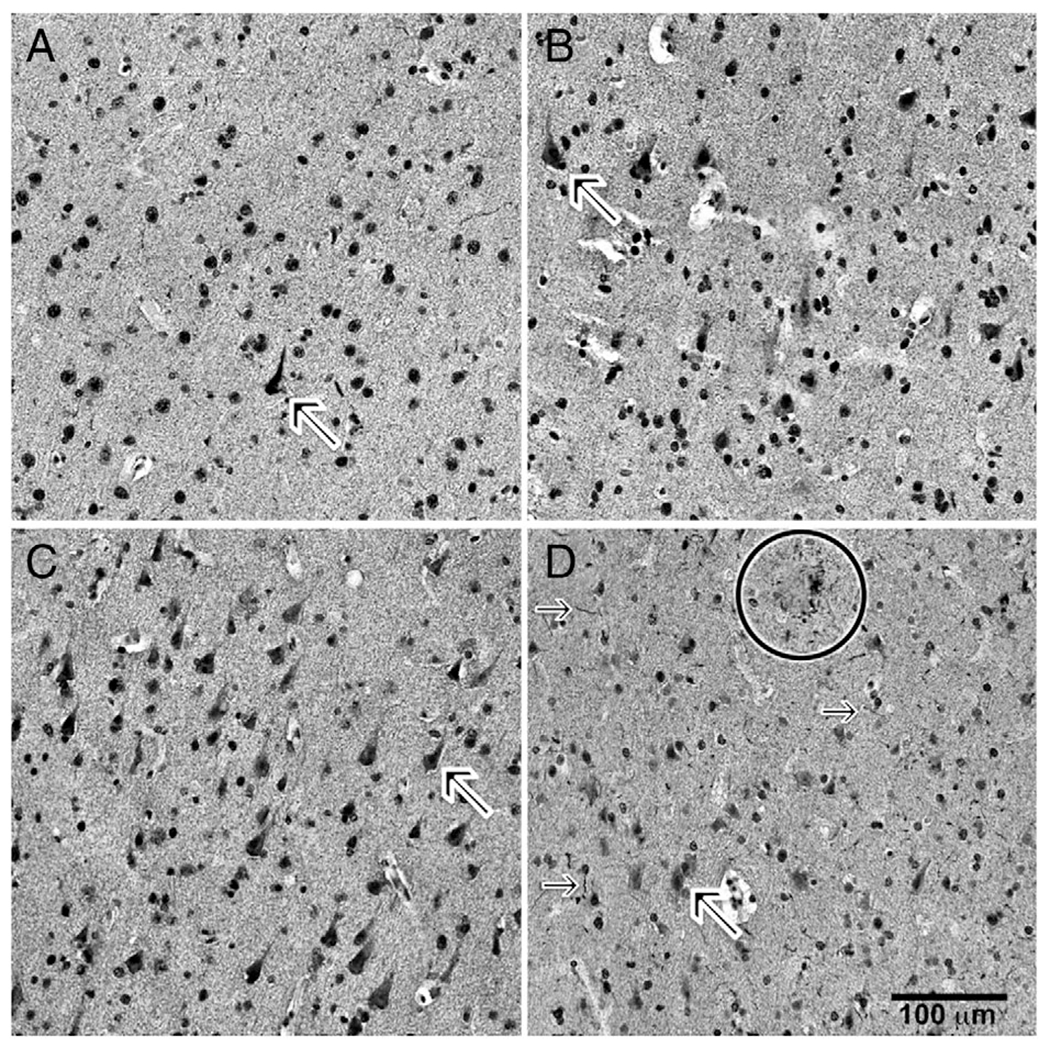
Immunohistochemical labeling of sections for pS616IRS1. Photomicrographs of layer III of middle frontal gyrus cortex immunohistochemically labeled for pS616IRS1 are shown. (A) A 90-year-old woman without cognitive impairment or diabetes. (B) A 84-year-old man without cognitive impairment but with diabetes. (C) A 86-year-old woman with cognitive impairment and without diabetes. (D) A 90-year-old woman with cognitive impairment and diabetes. In all photomicrographs, note frequency of nonnuclear cytoplasmic immunolabeling (diagonal arrows in each of A–D, and especially in D). Also note immunolabeled neuritic threads (horizontal arrows) and neuritic plaque (circle).
Brain Insulin Signaling ex vivo Stimulation Testing
While selecting the total sample of 150 subjects for IRS1 and AKT ELISA studies, we included a randomization algorithm such that we would have at least 78 subjects with or without diabetes (39 pairs matched for age at death, sex, and education) with a PMI ≤ 12 hours. This PMI cutoff was previously determined to be adequate to conduct measurements of ex vivo stimulation of human brain tissue using hormones.15,17 Frozen MFC samples were used. Our control experiments indicate that frozen postmortem human brain tissues were responsive to insulin within a PMI of 12 hours, and we have previously described no difference between frozen and fresh tissues in a simulated postmortem delay study conducted in rats.15,17
Three complementary indicators of insulin-induced insulin receptor activation were measured: (1) IRS1 recruitment to IRβ, (2) phosphorylation of IRβ at tyrosines 1150/1151 (pY1150/1151IRβ), and (3) phosphorylation of IRβ at tyrosine 960 (pY960IRβ). The values for each of these indicators were expressed as the ratio of the measure after incubation with 1nM insulin (IN) to the measure after incubation in Krebs–Ringer solution (KR), all normalized to total IRβ immunoprecipitated by anti-IRβ antibodies, as previously described.17 Briefly, gradually thawed 20mg of 100μm × 100μm × 3mm MFC slices were incubated with 1μM insulin or KR (nonstimulated) incubated for 30 minutes at 37°C. Brain slices were collected and then solubilized in immunoprecipitation buffer containing 0.5% digitonin, 0.2% sodium cholate, and 0.5% NP-40 for 60 minutes at 4°C to prepare lysate. Total IRβ content was immunoprecipitated with 1 βg of immobilized anti-IRβ antibodies (20739, Santa Cruz Biotechnology, Santa Cruz, CA). For (1) IRS1 recruitment to IRβ, the levels of IRS1 associated (coimmunoprecipitated) with IRβ were determined by immunoblotting with anti-IRS1 antibodies (1:750, Santa Cruz Biotechnology 8038). The optical density of IRS1 and total IRβ was quantified using a GS-800 calibrated densitometer (Bio-Rad Laboratories, Hercules, CA) and ratios of IRS1 to total IRβ levels were calculated. One nanomolar insulin elicited a 4.67 ± 0.76-fold increase in the amount of IRS1 recruited to IRβ in control subjects. For (2) IRβ phosphorylation (pY1150/1151IRβ), the levels of pY1150/1151IRβ in the anti-IRβ immunoprecipitate were determined by immunoblotting with anti-pY1150/1151IRβ antibodies (1:1,000, Santa Cruz 81500). One nanomolar insulin induced a 4.67 ± 0.74-fold increase in the pY1150/1151IRβ levels. For (3) IRβ phosphorylation (pY960IRβ), the levels of pY960IRβ in the anti-IRβ immunoprecipitate were determined by immunoblotting with anti-pY960IRβ antibodies (1:1,000, Invitrogen 44800G). One nanomolar insulin induced a 4.55 ± 0.67-fold increase in the pY960IRβ levels.
Similarly, 2 indicators of insulin-induced AKT1 activation were measured: (1) AKT1 phosphorylation at serine 473 (pS473AKT1) and (2) AKT1 phosphorylation at threonine 308 (pT308AKT1). The values for each of these indicators were expressed as the ratio of measure with incubation in 1nM insulin to measures with incubation in KR, all normalized to total AKT1 immunoprecipitated by 1.2βg anti-AKT1 antibodies (Santa Cruz Biotechnology 5298). For (1) pS473AKT1, the levels of pS473AKT1 were determined by immunoblotting with anti-pS473AKT1 antibodies (1:750, Santa Cruz Biotechnology 514032). Stimulation with 1nM insulin elicited a 4.47 ± 0.59-fold increase in the pS473AKT compared to incubation in KR. For (2) pT308AKT1, the levels of pTY308AKT1 in the anti-AKT1 immunoprecipitate were determined by immunoblotting with anti-pT308AKT1 antibodies (1:750, Santa Cruz Biotechnology 271964). One nanomolar insulin induced a 4.67 ± 0.74-fold increase in the pT308AKT1 levels.
Statistical Approach
Measures of brain insulin signaling were collected to examine associations with postmortem AD neuropathology and cognition proximate to death. With such novel and complex data, we always precede any modeling with a detailed examination of data distribution, identification of possible extremes, and the correlation structure of the variables of interest. There were 3 categories of brain insulin signaling variables. The first category was ELISA, with 2 main variables: the ratio of pS307IRS1 over total IRS1 and the ratio of pT308AKT1 over total AKT1. The second was immunohistochemistry, with 1 main variable: the ratio of pS616IRS1 cells per mm2. The third category was ex vivo stimulation data, with 5 variables: IRS1 recruitment to IRβ, pY1150/1151IRβ, pY960IRβ, pS473AKT1, and pT308AKT1. Because some categories include multiple variables, we used a Bonferroni-corrected alpha for each category (eg, for ex vivo stimulation, we used a 0.05/5 = 0.01). We used t tests or the Wilcoxon rank sum test to compare brain insulin signaling variables and basic subject characteristics between those with and without diabetes. We fit separate linear regression models with AD neuropathology and cognition proximate to death as outcomes with terms for each of brain insulin signaling, age at death, sex, and education. Age and education were centered at their means for interpretation purposes. Standard diagnostic methods and graphical examination of residuals were used to demonstrate that the assumptions underlying the statistical models were adequately met. Cumulative ordinal models assuming proportional odds were fit for skewed pathology outcomes (amyloid burden and tau tangle density) as a sensitivity analysis. Logistic regressions were used to examine the relation of measures to dementia status. Although the primary idea of the matching was to obtain a balanced sample of diabetic and nondiabetic subjects not only on the variables they were matched on but also on the testing order in the laboratories, we did additional sensitivity analyses applying 1-sample testing for differences within pairs. We predicted the subjects with diabetes would have higher brain insulin signaling compared to those without, but to be conservative, all tests were 2-sided. All analyses were conducted using SAS/STAT software, version 9.4 of the SAS system for Linux (SAS Institute, Cary, NC).
Results
Sample Characteristics
Demographic, cognitive, and neuropathologic characteristics of the 150 subjects included in the study are shown in Table 1. The mean age at baseline was 86.6 years, and education 18.1 years; half were women. Per the study design of matching subjects with and without diabetes, the demographic characteristics (age at death, sex, education) were not different between the 2 diagnostic groups. Cognitive function and AD pathology levels were similar between the 2 groups (Table 1). There was no association between APOE ε4 and diabetes (χ21 = 1.316, p = 0.251). The pairwise difference (diabetes minus nondiabetes) in last-valid or study-average depressive symptoms was not significant (respectively, t74 = 0.46, p = 0.65 and t74 = 0.37, p = 0.72).
TABLE 1.
Characteristics of Subjects by Diabetes Status
| Characteristic | Total, n = 150 | Diabetes, n = 75 | No Diabetes, n = 75 |
|---|---|---|---|
| Demographic | |||
| Age at death, yr (SD) | 86.6 (6.1) | 86.6 (5.9) | 86.7 (6.3) |
| Women, n (%) | 72 (48%) | 36 (48%) | 36 (48%) |
| Education, yr (SD) | 18.1 (3.3) | 18.2 (3.1) | 18.1 (3.4) |
| Cognitive Scorea | |||
| Global cognitive function score | −0.871 (1.203) | −0.920 (1.183) | −0.822 (1.229) |
| Episodic memory | −0.828 (1.465) | −0.876 (1.459) | −0.781 (1.480) |
| Working memory | −0.607 (1.028) | −0.650 (1.047) | −0.564 (1.015) |
| Semantic memory | −0.792 (1.367) | −0.779 (1.268) | −0.804 (1.468) |
| Perceptual speed | −1.229 (1.202) | −1.341 (1.186) | −1.117 (1.215) |
| Visuospatial ability | −0.668 (0.940) | −0.640 (0.914) | −0.697 (0.970) |
| Neuropathologic | |||
| Alzheimer disease pathology | |||
| Global score | 0.6 (0.6) | 0.7 (0.6) | 0.6 (0.5) |
| Amyloid score, median (IQR) | 1.6 (0.2, 4.5) | 2 (0.3, 5.3) | 1.4 (0.2, 4.1) |
| Tangles score, median (IQR) | 3.2 (1.2, 7.4) | 3.5 (1.1, 7.8) | 2.7 (1.2, 7.0) |
Data are given as mean (SD), unless otherwise specified.
Cognitive data proximate to death.
IQR = interquartile range; SD = standard deviation.
Brain Insulin Signaling and AD Neuropathology
The median values of the 8 measures of interest for insulin signaling and related measures were consistently higher in persons with diabetes compared to persons without diabetes (Table 2, Fig 2). Pairwise differences between persons with and without diabetes were positive for almost all measures of interest. However, only pS307IRS1/total IRS1, pS473AKT1, and pT308AKT1 (respectively, 0.05, 0.24, and 0.26 higher for persons with diabetes) were statistically significant. When we examined these pairwise differences by sex, we detected a pairwise difference in pS307IRS1/total IRS1, IRS1 cells/mm2, and pS473AKT1 among women, but we could not detect a pairwise difference in any of the markers among men. These sex differences need further investigation with larger samples. In additional analyses, there was no association between APOE ε4 and pT308AKT1/total AKT1 in a model adjusting for age, sex, and education (estimate = 0.022, standard error [SE] = 0.057, p = 0.707).
TABLE 2.
Descriptive Data on Insulin Signaling and Related Measures
| Median (interquartile range) | |||||
|---|---|---|---|---|---|
| Variable | n | Total | Diabetes | Nondiabetes | Pairwise Difference Mean (95% CI) |
| ELISA | |||||
| pS307IRS1 | 150 | 0.10 (0.09, 0.10) | 0.10 (0.09, 0.10) | 0.10 (0.09, 0.10) | – |
| Total IRS1 | 150 | 0.28 (0.19, 0.40) | 0.26 (0.17, 0.40) | 0.33 (0.21, 0.44) | – |
| pT308AKT1 | 150 | 0.15 (0.13, 0.18) | 0.15 (0.13, 0.18) | 0.15 (0.13, 0.18) | – |
| Total AKT1 | 150 | 0.40 (0.20, 0.71) | 0.36 (0.20, 0.64) | 0.43 (0.23, 0.85) | – |
| pS307IRS1/total IRS1a | 150 | 0.34 (0.24, 0.49) | 0.39 (0.26, 0.53) | 0.31 (0.23, 0.46) | 0.05 (0.01, 0.09) |
| pT308AKT1/total AKT1a | 150 | 0.41 (0.23, 0.65) | 0.42 (0.24, 0.68) | 0.36 (0.20, 0.60) | 0.05 (−0.04, 0.14) |
| Immunohistochemistry | |||||
| IRS1 cells/mm2 | 139 | 5.72 (4.50, 7.29) | 5.90 (4.64, 7.98) | 5.30 (4.50, 6.91) | −0.8 (−2.63, 1.02) |
| Ex vivo stimulation | |||||
| IRS1 recruitment to IRβ | 78 | 4.66 (4.27, 5.10) | 4.69 (4.33, 5.12) | 4.55 (4.04, 5.10) | 0.19 (−0.13, 0.51) |
| pS473AKT1 | 78 | 4.45 (4.08, 4.81) | 4.67 (4.22, 4.93) | 4.35 (4.00, 4.69) | 0.24 (0.01, 0.46) |
| pT308AKT1 | 78 | 4.5 (4.09, 4.92) | 4.55 (4.16, 4.99) | 4.46 (3.87, 4.80) | 0.26 (0.04, 0.48) |
| pY1150/1151IRβ | 78 | 4.67 (4.21, 5.24) | 4.64 (4.27, 5.26) | 4.68 (4.11, 5.24) | 0.12 (−0.19, 0.44) |
| pY960IRβ | 78 | 4.53 (4.11, 4.97) | 4.64 (4.11, 5.33) | 4.42 (4.10, 4.88) | 0.18 (−0.11, 0.46) |
Among the ELISA measures, the ratios were the primary variables of interest.
CI = confidence interval; ELISA = enzyme-linked immunosorbent assay.
FIGURE 2:
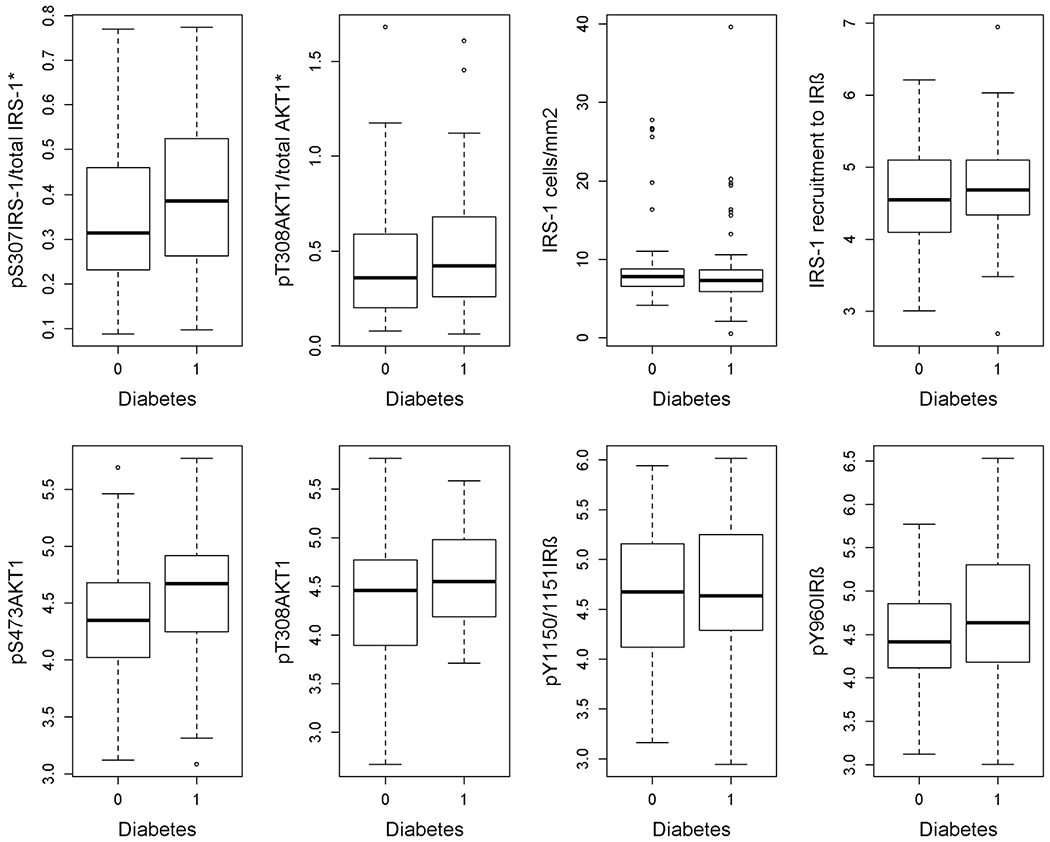
Box-and-whiskers plots of brain insulin signaling for subjects with diabetes (diabetes = 1) and without diabetes (diabetes = 0).
Because diabetes increases dementia risk, we examined the association of insulin signaling levels in brain tissue with AD pathology as the most common pathology in dementia. In Table 3, using the ELISA measures in 150 subjects, a higher level of normalized pT308AKT1 was associated with a higher level of the composite global AD pathology score. Secondary analyses of separate global amyloid burden and tau tangle density found higher pT308AKT1/total AKT1 level was associated with both amyloid and tangles. In additional secondary analyses using AD pathology outcome measures restricted to the MFC region, results were consistent. We used ordinal logistic regressions (AD measures were skewed even after transformation and were therefore categorized into quartiles) and observed weak evidence of an association of pT308AKT1/total AKT1 level with a higher amyloid burden in the MFC (estimate = 1.283, SE = 0.952, p = 0.101), and pT308AKT1/total AKT1 was associated with a higher tangle density in the MFC (estimate = 1.596, SE = 1.156, p = 0.005). There was no association of pS307IRS1/total IRS1 with AD pathology. In the immunohistochemical assessment of pS616IRS1 expression, there was no association of pS616IRS1-positive cell density with AD pathology. In the subset of 79 subjects included in the ex vivo stimulation experiments, there was no significant association observed between any of the 5 measures of insulin signaling with AD pathology (with corrected α = 0.01). Noting the directionality of relations, there was a consistent inverse relationship in all analyses between AD pathology and activation of insulin signaling. Specifically, with more AD pathology, there was less insulin activation. Because of the skewness of the AD pathology outcomes, we repeated the analyses with models using ordinal logistic regression assuming proportional odds and quartiles of pathology as the outcomes. These results were consistent (Table 4).
TABLE 3.
Relation of Diabetes, Insulin Signaling, and Related Measures to AD Neuropathology
| Estimate (SE), p | |||
|---|---|---|---|
| Predictor | Global AD Score | Amyloid Burden | Tau Tangle Density |
| Diabetes | 0.117 (0.091), 0.200 | 0.086 (0.159), 0.591 | 0.129 (0.194), 0.508 |
| ELISA | |||
| pS307IRS1/total IRS1 | 0.020 (0.032), 0.536 | 0.068 (0.084), 0.417 | 0.033 (0.093), 0.724 |
| pT308AKT1/total AKT1 | 0.104 (0.030), 0.001a | 0.192 (0.081), 0.019a | 0.257 (0.088), 0.004a |
| Immunohistochemistry | |||
| IRS1 cells/mm2 | 0.020 (0.035), 0.559 | −0.035 (0.089), 0.699 | 0.026 (0.103), 0.806 |
| Ex vivo stimulation | |||
| IRS1 recruitment to IRβ | −0.034 (0.047), 0.461 | −0.097 (0.113), 0.391 | −0.059 (0.145), 0.687 |
| pS473AKT1 | −0.067 (0.046), 0.147 | −0.152 (0.112), 0.182 | −0.131 (0.143), 0.365 |
| pT308AKT1 | −0.054 (0.045), 0.237 | −0.102 (0.111), 0.359 | −0.152 (0.141), 0.284 |
| pY1150/1151IRβ | −0.047 (0.046), 0.314 | −0.155 (0.112), 0.172 | −0.024 (0.145), 0.871 |
| pY960IRβ | −0.098 (0.045), 0.032 | −0.177 (0.111), 0.116 | −0.216 (0.142), 0.132 |
Separate linear regression models (1 per row) are adjusted for age at death, sex, and education.
Statistically significant.
AD = Alzheimer disease; ELISA = enzyme-linked immunosorbent assay; SE = standard error.
TABLE 4.
Relation of Insulin Signaling and Related Measures to AD Neuropathology Quartiles
| Odds Ratio (95% CI), p | ||
|---|---|---|
| Predictor | Amyloid Burdena | Tau Tangle Densitya |
| ELISA | ||
| pS307IRS1/total IRS1 | 1.106 (0.817, 1.497), 0.516 | 0.847 (0.626, 1.148), 0.285 |
| pT308AKT1/total AKT1 | 1.350 (1.000, 1.823), 0.050 | 1.373 (1.016, 1.855), 0.039 |
| Immunohistochemistry | ||
| IRS1 cells/mm2 | 1.010 (0.731, 1.386), 0.970 | 1.068 (0.775, 1.472), 0.686 |
| Ex vivo stimulation | ||
| IRS1 recruitment to IRβ | 0.827 (0.543, 1.260), 0.377 | 0.844 (0.555, 1.282), 0.425 |
| pS473AKT1 | 0.709 (0.463, 1.086), 0.114 | 0.923 (0.609, 1.398), 0.705 |
| pT308AKT1 | 0.750 (0.495, 1.135), 0.173 | 0.894 (0.594, 1.345), 0.591 |
| pY1150/1151IRβ | 0.703 (0.456, 1.083), 0.110 | 0.877 (0.577, 1.333), 0.539 |
| pY960IRβ | 0.650 (0.423, 0.999), 0.050 | 0.757 (0.498, 1.152), 0.194 |
Separate ordinal logistic regression models (1 per row) are adjusted for age at death, sex, and education.
Quartiles of the outcome.
CI = confidence interval; ELISA = enzyme-linked immunosorbent assay.
Linear regression models were also repeated adding a term for the interaction between each measure and diabetes. Interaction terms were not statistically significant (Table 5), suggesting that the presence of diabetes did not affect the relationship of the measures with pathology.
TABLE 5.
Interaction of Diabetes with Insulin Signaling and Related Measures, in Models with AD Neuropathology as Outcome
| Estimate (SE), p | |||
|---|---|---|---|
| Predictor | Global AD Score | Amyloid Burden | Tau Tangle Density |
| ELISA | |||
| pS307IRS1/total IRS1 | 0.034 (0.090, 0.706) | −0.041 (0.157, 0.792) | 0.022 (0.191, 0.908) |
| pT308AKT1/total AKT1 | 0.139 (0.088, 0.117) | 0.039 (0.158, 0.808) | 0.368 (0.189, 0.053) |
| Immunohistochemistry | |||
| IRS1 cells/mm2 | 0.055 (0.088, 0.529) | 0.172 (0.152, 0.259) | 0.243 (0.185, 0.191) |
| Ex vivo stimulation | |||
| IRS1 recruitment to IRβ | 0.090 (0.144, 0.534) | 0.061 (0.216, 0.777) | 0.139 (0.296, 0.639) |
| pS473AKT1 | 0.014 (0.146, 0.926) | −0.080 (0.221, 0.719) | 0.248 (0.303, 0.417) |
| pT308AKT1 | −0.054 (0.145, 0.713) | −0.362 (0.216, 0.100) | 0.371 (0.298, 0.217) |
| pY1150/1151IRβ | −0.019 (0.140, 0.893) | −0.073 (0.207, 0.725) | −0.156 (0.287, 0.588) |
| pY960IRβ | 0.038 (0.147, 0.795) | −0.112 (0.225, 0.619) | 0.225 (0.306, 0.465) |
Separate linear regression models (1 per row) are adjusted for age at death, sex, and education.
AD = Alzheimer disease; ELISA = enzyme-linked immunosorbent assay; SE = standard error.
Brain Insulin Signaling and Cognitive Function
We next examined the association of insulin signaling with cognition proximate to death, in models adjusting for age at death, sex, and education (Table 6). We found that a higher value on the ELISA measure of pT308AKT1/total AKT1 was associated with a lower level of global cognitive function. Fifty-six persons had a diagnosis of dementia proximate to death. Although the ELISA measure of pT308AKT1/total AKT1 was not significantly associated with dementia, there was a trend for an association and the direction of the association was consistent with results for global cognition (odds ratio = 1.36, 95% confidence interval = 0.95–1.95, p = 0.087). There was no association of any of the other measures, including by ELISA (pS307IRS1/total IRS1), immunohistochemistry (pS616IRS1cells/mm2), or ex vivo stimulation, with global cognitive function.
TABLE 6.
Relation of Insulin Signaling and Related Measures to Level of Cognitive Function Proximate to Death
| Predictor | Estimate (SE) p | |||||
|---|---|---|---|---|---|---|
| Global Cognition | Cognitive Domains | |||||
| Episodic Memory | Working Memory | Semantic Memory | Perceptual Speed | Visuospatial Abilities | ||
| ELISA | ||||||
| pS307IRS1/total IRS1 | 0.086 (0.101) 0.396 | 0.081 (0.123) 0.511 | −0.000 (0.087) 0.999 | 0.170 (0.116) 0.145 | 0.188 (0.098) 0.058 | 0.072 (0.078) 0.362 |
| pT308AKT1/total AKT1 | −0.212 (0.097) 0.031a | −0.285 (0.118) 0.017a | −0.234 (0.082) 0.005a | −0.105 (0.113) 0.356 | −0.166 (0.096) 0.085 | −0.052 (0.076) 0.496 |
| Immunohistochemistry | ||||||
| IRS1 cells/mm2 | −0.003 (0.019) 0.875 | −0.006 (0.023) 0.790 | 0.008 (0.017) 0.611 | −0.007 (0.022) 0.750 | −0.010 (0.019) 0.600 | −0.006 (0.015) 0.688 |
| Ex vivo stimulation | ||||||
| IRS1 recruitment to IRβ | 0.015 (0.148) 0.922 | 0.048 (0.174) 0.782 | 0.007 (0.121) 0.954 | 0.117 (0.175) 0.506 | −0.074 (0.141) 0.600 | 0.095 (0.112) 0.403 |
| pS473AKT1 | 0.094 (0.147) 0.525 | −0.035 (0.173) 0.839 | 0.057 (0.121) 0.639 | 0.226 (0.173) 0.196 | 0.178 (0.139) 0.205 | 0.151 (0.111) 0.180 |
| pT308AKT1 | 0.097 (0.145) 0.507 | 0.048 (0.170) 0.778 | −0.001 (0.119) 0.994 | 0.265 (0.169) 0.122 | 0.176 (0.137) 0.212 | 0.071 (0.111) 0.521 |
| pY1150/1151IRβ | 0.156 (0.147) 0.294 | 0.205 (0.172) 0.239 | 0.170 (0.120) 0.160 | 0.294 (0.173) 0.093 | 0.143 (0.140) 0.311 | 0.127 (0.112) 0.262 |
| pY960IRβ | 0.083 (0.147) 0.575 | −0.026 (0.173) 0.883 | 0.065 (0.120) 0.593 | 0.124 (0.174) 0.481 | 0.005 (0.141) 0.974 | 0.105 (0.112) 0.349 |
Separate linear regression models were adjusted for age at death, sex, and education.
Statistically significant.
ELISA = enzyme-linked immunosorbent assay; SE = standard error.
We conducted secondary analyses, similarly adjusted, of the relation of the measures with 5 separate cognitive domains. We found that a higher value on the ELISA measure of pT308AKT1/total AKT1 was associated with a lower level of function in working memory (estimate = −0.234, SE = 0.082, p = 0.005) and in episodic memory (estimate = −0.285, SE = 0.118, p = 0.017), but not with the other 3 cognitive domains, including semantic memory, perceptual speed, and visuospatial abilities (Table 6). There were no other associations of the other 7 measures with the 5 cognitive domains.
Discussion
Insulin resistance in peripheral tissues (eg, liver, muscle, adipose) is a characteristic feature of type 2 diabetes, but less is known about the brain in persons with diabetes.18 Some data from the literature suggest the presence of insulin resistance in the prefrontal cortex in animal models of diabetes.19 Based on these data, we initially hypothesized that there would be a difference in measures of insulin resistance in brain between persons with and without diabetes, as is seen in peripheral tissues, and that this difference might explain or contribute to lower cognitive function and/or to more neuropathology of cognitive impairment. In this study, we examined the relation of MFC insulin signaling and related molecular measures to postmortem AD neuropathology and cognitive function proximate to death, in 150 community-dwelling older persons with diabetes matched to persons without diabetes. We found that levels of phosphorylated IRS1 (pS307IRS1) and AKT1 (pT308AKT1) measured by ELISA were not statistically different among persons with and without diabetes, although trends suggestive of insulin resistance were observed. Our result of no difference has various possible explanations. It is possible that the diabetes milieu in brain is different from other tissues, that there are competing effects of other stimuli of the pathway we focus on (eg, IGF-1, IGF-2, other AKT simulators) or other brain-intrinsic compensatory mechanisms in the IRS1-AKT signaling pathway, or that impaired signaling in diabetes may be more evident in brain regions other than the MFC. Another possibility is that various agonal and postmortem factors may attenuate the differences present in vivo. Most of the subjects with diabetes in this study were treated with antidiabetes drugs, and it is possible this may also have attenuated signaling impairments and thus our ability to find an association between insulin signaling, brain pathology, and cognition. Other potential selection biases could be that persons with diabetes in our study may have been more “resilient” to disease/death than typically observed. The age at death was older than expected among persons with diabetes. On the other hand, persons in the control group may have had prediabetes or undiagnosed diabetes, despite our annual assessment for diabetes over the years. Finally, studies with yet larger sample sizes may be needed to detect whether the trend we observed toward insulin resistance is meaningful.
Because diabetes increases the risk of dementia,12 and recent literature, particularly from animal models of AD, suggests that brain insulin resistance may precede peripheral insulin resistance,20 we examined the relation of the canonical IRS1-AKT insulin signaling pathway with AD pathology in persons with and without diabetes. We found that increased AKT1 phosphorylation at T308 was associated with a measure of global AD pathology. Secondary analyses identified that increased pT308AKT1 was associated with both increased amyloid burden and tau tangle density, and with tangle density in the MFC in particular. Furthermore, elevated pT308AKT1 was also associated with lower cognitive function proximate to death, including on a measure of global cognitive function, and there was a trend for an association with dementia. Elevated pT308AKT1 was associated with lower function in 2 cognitive domains, working memory and episodic memory, but not other domains. Impairment in these domains is consistent with the hypothesized effect of neural dysfunction in this specific brain region, MFC, and with our prior study in which insulin resistance was associated with memory.15 Our finding of higher pT308AKT, but not S307IRS1, associated with AD pathology raises the possibility that it may not be insulin effects per se, but rather the AKT regulated by other multiple upstream and feedback signaling pathways that plays a more prominent role in AD pathobiology.
In contrast to some prior data, including our own,15,21 we did not find that serine phosphorylation of IRS1 was associated with AD pathology in this sample. There were also no other associations of insulin resistance indicators and related measures, including those derived from the ex vivo stimulation data, with the global AD score. Work by our and other groups indicates that phosphorylation of S307IRS1 is primarily inhibitory to IRS1 function, but its influence on the levels of insulin resistance is complex and tissue-dependent.15 In a cell model study, microRNA was shown to inhibit Aβ-induced pS307IRS1 expression and restore insulin signaling in human neuroblastoma cells.9 Further studies are needed to examine the link of insulin signaling with AD RNA transcript regulators, including using bioinformatics and computational neuroscience approaches. Another explanation for discordant findings between our current study and previously published work15 is that insulin signaling may vary by brain region, with some data suggesting greater insulin activity in the hippocampus rather than the cerebral cortex. Finally, we recognize that there are other IRS isoforms that we did not measure in this study (eg, IRS2) and other downstream insulin signaling molecules that are affected by diabetes and may be related to AD. For instance, insulin affects extracellular signal-regulated kinase independently of IRS. These and other pathways would be interesting to investigate.
Insulin is transported into the brain and binds to insulin receptors expressed in glia and neurons, particularly in dendrites and presynaptic terminals.22 The binding of insulin induces autophosphorylation of the insulin receptor and recruitment and activation of intracellular IRS1 proteins via tyrosine phosphorylation.22 Tyrosinephosphorylated IRS1 activates PI3K, a heteromeric protein consisting of a p110 catalytic subunit and a p85 regulatory subunit. The p110 subunit of PI3K converts phosphatidylinositol (3,4)-bisphosphate into phosphatidylinositol (3,4,5)-trisphosphate (PIP3), leading to activation of the AKT family of serine/threonine kinase AKT1 and AKT3 isoforms in neurons, and AKT2 mostly in astrocytes.23,24 AKT1 interacts with the pleckstrin homology domain of PIP3 that enables phosphoinositide-dependent protein kinase 1 to phosphorylate threonine 308/309/305 residues of AKT1. The full activation of AKT1 requires phosphorylation of serine 473/474/472, respectively, by the mammalian target for rapamycin complex 2.25 The AD brain is associated with reduced PI3K-AKT signaling manifest as decreased total and/or phosphorylation levels.26,27 Some postmortem analyses of AD brain regions have shown increased levels of phosphorylated AKT,15,26 consistent with our ELISA result. Elevated steady state phosphorylated AKT levels, indicating insulin resistance, have been correlated positively with Aβ and tau lesions and negatively with cognition.15,28
Whereas brain insulin signaling is known to be implicated in neuronal function and brain behavior, including cognition,29,30 less is known about insulin resistance in human brain tissue and its relation to the level of cognitive function. Several but not all studies, and mostly in animal models, have found that brain insulin resistance correlates with poor cognition.31 Furthermore, some data, including from our group, suggest the presence of brain insulin resistance as assessed by increased IRS1 phosphorylation at a particular serine site (S616) in human tissue is associated with lower cognition, including in episodic memory, perhaps even in the absence of diabetes.15,32 Using the complementary assessment methods of ELISA, immunohistochemistry, and ex vivo brain stimulation in human tissue, we did not find an association of IRS1 phosphorylation at the S307 or most other related measurements we collected with global cognition or any of the 5 cognitive domains proximate to death. However, in keeping with some prior data,33 we found that increased brain AKT1 phosphorylation at the T308, a critical node in the signaling of insulin and a marker of insulin resistance, is associated with a lower level of global cognition. We further observed that increased AKT phosphorylation correlates with lower levels of working memory and episodic memory. This specificity may reflect the brain region selection (MFC) for our study. However, we believe the MFC region is a particularly useful brain region to investigate based on several factors, including that this region demonstrates (1) a range of AD pathology, (2) importance for cognitive function, and (3) a role in insulin signaling as shown in our prior paper.34 These data extend our understanding of the relation of brain insulin resistance and related molecular pathways in brain dysfunction. More research will further characterize the molecular pathways linking metabolism in peripheral and brain insulin resistance, as well as diabetes, with cognitive impairment and dementia. Of particular interest is to explore the link between peripheral and central insulin signaling and inflammation, and the role of altered cholinergic signaling by age, sex, and disease state (eg, AD and diabetes), including how altered cholinergic signaling may affect inflammation.35 Another avenue of future research is to examine the link of altered nutritional status in AD, and how nutrition may influence insulin signaling. Finally, there are widespread changes in gene expression in AD postmortem brain in relation to both disease pathology and its clinical expression.36 Preclinical models of amyloid, tau, and other pathologies have also been instructive in delineating genes associated with these pathologies. Type 2 diabetes and obesity-related transcription changes have been found in both AD brains and AD mouse models.37 Interestingly, these alterations in the diabetes-related genes in AD brains have been reported to occur independently of peripheral diabetes-related gene abnormalities. In a monkey model, aging-related gene expression in the hippocampus proper was associated with metabolic syndrome variables such as insulin resistance, and Aβ42 has been found to increase gene expression of insulinlike growth factor binding proteins (in particular IGFBP5).38,39 More research will characterize the relation of insulin pathways and gene expression in AD brain.
There are several study weaknesses to consider in interpreting our data. First, diabetes status was not determined using published criteria and we were not able to ascertain diabetes type. However, diabetes status relied on the annual medical history and documentation of the use of antidiabetes medications, and most subjects with diabetes were likely to have had type 2 diabetes associated with insulin resistance. Also, we did not use quantifiable measures of diabetes such as blood glucose or insulin. Other studies will inform on the relation of peripheral insulin and related measures to brain insulin signaling and brain pathology and function. Second, we did not consider the potentially modulating effects of other hormones in the brain, such as adipokines (including leptin) or incretins, which are involved in diabetes. Metabolism is complex, and many factors not considered in this study are likely to play a role in the regulation of the effects of insulin signaling on brain processes.40 Third, this study did not consider other brain pathologies such as cerebrovascular disease, which is likely implicated in diabetes among older persons,41,42 or consider other biologically informative variables or more clinically relevant factors compared to end-of-life cognition.
Nonetheless, there are several important strengths to this study. We used a comprehensive approach to examine insulin and related measures. In addition to collecting quantitative biochemical and semiquantitative immunohistochemical measures, we carefully characterized insulin signaling using a powerful ex vivo brain stimulation paradigm in which we directly stimulated tissue with insulin. Furthermore, the new metabolic data were derived from human MFC tissue, from biospecimens with a short PMI, which we previously validated for these specific experiments. Also, we purposefully applied several strategies to ensure high scientific rigor, including in subject selection (eg, randomization, matching), laboratory measurements (eg, replication studies), and standard statistical analyses (eg, with adjustments). Finally, systematically collected data were available on brain neuropathology (including both amyloid burden and tau tangle density), and among very well clinically characterized subjects with detailed neuropsychological test data (global and 5 domain measures) obtained proximate to death.
Acknowledgment
This work was funded by National Institute on Aging and National Institute of Neurological Disorders and Stroke grants P30 AG10161, R01 AG15819, R01 NS084965, and RF1 AG059621.
We thank the ROS participants from across the USA for their tremendous altruism in taking part in this ongoing study for the past two and a half decades; and all Rush Alzheimer’s Disease Center staff and faculty, in particular the study manager (T. Colvin), data manager (J. Gibbons), laboratory manager (K. Skish), and biostatistical team (in particular, D. Esbjornson and A. Hodges).
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med 2017;23:804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee S, Peters SA, Woodward M, et al. Type 2 diabetes as a risk factor for dementia in women compared with men: a pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care 2016;39:300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oskarsson ME, Paulsson JF, Schultz SW, et al. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. Am J Pathol 2015;185: 834–846. [DOI] [PubMed] [Google Scholar]
- 4.Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology 2011. ;61:867–879. [DOI] [PubMed] [Google Scholar]
- 5.Kwon OH, Cho YY, Kim TW, Chung S. O-GlcNAcylation of amyloid-β protein precursor by insulin signaling reduces amyloid-β production. J Alzheimers Dis 2019;69:1195–1211. [DOI] [PubMed] [Google Scholar]
- 6.Long-Smith CM, Manning S, McClean PL, et al. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromolecular Med 2013;15:102–114. [DOI] [PubMed] [Google Scholar]
- 7.Kang S, Kim CH, Jung H, et al. Agmatine ameliorates type 2 diabetes induced-Alzheimer’s disease-like alterations in high-fat diet-fed mice via reactivation of blunted insulin signalling. Neuropharmacology 2017;113:467–479. [DOI] [PubMed] [Google Scholar]
- 8.Craft S, Claxton A, Baker LD, et al. Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: a pilot clinical trial. J Alzheimers Dis 2017;57:1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li HH, Lin SL, Huang CN, et al. miR-302 attenuates amyloid-β-induced neurotoxicity through activation of Akt signaling. J Alzheimers Dis 2016;50:1083–1098. [DOI] [PubMed] [Google Scholar]
- 10.Bennett DA, Buchman AS, Boyle PA, et al. Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis 2018;64: S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193. [PubMed] [Google Scholar]
- 12.Arvanitakis Z, Wilson RS, Bienias JL, et al. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 2004;61:661–666. [DOI] [PubMed] [Google Scholar]
- 13.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer’s disease and mild cognitive impairment. Ann Neurol 2009;66:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arvanitakis Z, Schneider JA, Wilson RS, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 2006;67:1960–1965. [DOI] [PubMed] [Google Scholar]
- 15.Talbot K, Wang HY, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 2012; 122:1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarchoan M, Toledo JB, Lee EB, et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol 2014;128: 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang HY, Capuano AW, Khan A, et al. Insulin and adipokine signaling and their cross-regulation in postmortem human brain. Neurobiol Aging 2019;84:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 2018;14:168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdul-Rahman O, Sasvari-Szekely M, Ver A, et al. Altered gene expression profiles in the hippocampus and prefrontal cortex of type 2 diabetic rats. BMC Genomics 2012;13:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velazquez R, Tran A, Ishimwe E, et al. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol Aging 2017;58:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frolich L, Blum-Degen D, Riederer P, Hoyer S. A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer’s disease. Ann N Y Acad Sci 1999;893:290–293. [DOI] [PubMed] [Google Scholar]
- 22.Kim B, Feldman EL. Insulin resistance in the nervous system. Trends Endocrinol Metab 2012;23:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. AJP Endocrinol Metab 2009;296: E581–E591. [DOI] [PubMed] [Google Scholar]
- 24.Levenga J, Wong H, Milstead RA, et al. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. Elife 2017;6: e30640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noguchi M, Suizu F. Regulation of AKT by phosphorylation of distinct threonine and serine residues In: Berhardt LV, ed. Advances in medicine and biology. New York, NY: Nova Science Publishers, 2012:139–162. [Google Scholar]
- 26.Steen E, Terry BM, Rivera EJ, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis 2005;7:63–80. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Liu F, Grundke-Iqbal I, et al. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol 2011;225: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Aβ oligomers. J Clin Invest 2012;122:1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao WQ, Alkon DL. Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol 2001;177:125–134. [DOI] [PubMed] [Google Scholar]
- 30.Ghasemi R, Haeri A, Dargahi L, et al. Insulin in the brain: sources, localization and functions. Mol Neurobiol 2013;47:145–171. [DOI] [PubMed] [Google Scholar]
- 31.Kothari V, Luo Y, Tornabene T, et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta Mol Basis Dis 2017;1863:499–508. [DOI] [PubMed] [Google Scholar]
- 32.Moloney AM, Griffin RJ, Timmons S, et al. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010;31: 224–243. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Pan Y, Li X, Wang X. Atorvastatin attenuates cognitive deficits through Akt1/caspase-3 signaling pathway in ischemic stroke. Brain Res 2015;1629:231–239. [DOI] [PubMed] [Google Scholar]
- 34.Williams VJ, Trombetta BA, Jafri RZ, et al. Task-related fMRI BOLD response to hyperinsulinemia in healthy older adults. JCI Insight 2019;5:e129700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang K, Chen Q, Wu N, et al. Berberine ameliorates spatial learning memory impairment and modulates cholinergic anti-inflammatory pathway in diabetic rats. Front Pharmacol 2019;10:1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milind N, Preuss C, Haber A, et al. Transcriptomic stratification of late-onset Alzheimer’s cases reveals novel genetic modifiers of disease pathology. PLoS Genet 2020;16:e1008775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hokama M, Oka S, Leon J, et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex 2014;24:2476–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blalock EM, Grondin R, Chen KC, et al. Aging-related gene expression in hippocampus proper compared with dentate gyrus is selectively associated with metabolic syndrome variables in rhesus monkeys. J Neurosci 2010;30:6058–6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barucker C, Sommer A, Beckmann G, et al. Alzheimer amyloid peptide afi42 regulates gene expression of transcription and growth factors. J Alzheimers Dis 2015;44:613–624. [DOI] [PubMed] [Google Scholar]
- 40.Capuano AW, Wilson RS, Honer WG, et al. Brain IGFBP-5 modifies the relation of depressive symptoms to decline in cognition in older persons. J Affect Disord 2019;250:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abner EL, Nelson PT, Kryscio RJ, et al. Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 2016;12:882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pruzin JJ, Schneider JA, Capuano AW, et al. Diabetes, hemoglobin A1C, and regional Alzheimer disease and infarct pathology. Alzheimer Dis Assoc Disord 2017;31:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]