

Abstract
Asparagine-linked glycosylation 13 homolog (ALG13) encodes a nonredundant, highly conserved, X-linked uridine diphosphate (UDP)-N-acetylglucosaminyltransferase required for the synthesis of lipid linked oligosaccharide precursor and proper N-linked glycosylation. De novo variants in ALG13 underlie a form of early infantile epileptic encephalopathy known as EIEE36, but given its essential role in glycosylation, it is also considered a congenital disorder of glycosylation (CDG), ALG13-CDG. Twenty-four previously reported ALG13-CDG cases had de novo variants, but surprisingly, unlike most forms of CDG, ALG13-CDG did not show the anticipated glycosylation defects, typically detected by altered transferrin glycosylation. Structural homology modeling of two recurrent de novo variants, p.A81T and p.N107S, suggests both are likely to impact the function of ALG13. Using a corresponding ALG13-deficient yeast strain, we show that expressing yeast ALG13 with either of the highly conserved hotspot variants rescues the observed growth defect, but not its glycosylation abnormality. We present molecular and clinical data on 29 previously unreported individuals with de novo variants in ALG13. This more than doubles the number of known cases. A key finding is that a vast majority of the individuals presents with West syndrome, a feature shared with other CDG types. Among these, the initial epileptic spasms best responded to adrenocorticotropic hormone or prednisolone, while clobazam and felbamate showed promise for continued epilepsy treatment. A ketogenic diet seems to play an important role in the treatment of these individuals.
Keywords: congenital disorders of glycosylation, epilepsy, N-linked glycosylation, whole exome sequencing
1 |. INTRODUCTION
Congenital disorders of glycosylation (CDG) are a group of nearly 140 rare metabolic disorders which can present with a broad, nonspecific spectrum of clinical symptoms. The vast majority of CDG are autosomal recessive disorders, but several are caused by de novo variants. One such type is ALG13-CDG, which frequently presents as an early infantile epileptic encephalopathy.1–5
Asparagine-linked glycosylation 13 homolog (ALG13) encodes a highly conserved X-linked uridine diphosphate (UDP)-N-acetylglucosaminyltransferase required for the transfer of N-acetylglucosamine (GlcNAc) onto the extending lipid-linked oligosaccharide (LLO) structure, dolichol-P-P GlcNAc6–8 (Figure 1A). Studies in yeast show that this early step in the LLO synthesis pathway is essential for proper N-linked glycosylation.6,7 Given its critical role in N-linked glycosylation, it is not surprising that ALG13 and its UDP-GlcNAc transferase activity are conserved across all eukaryotic species.
FIGURE 1.
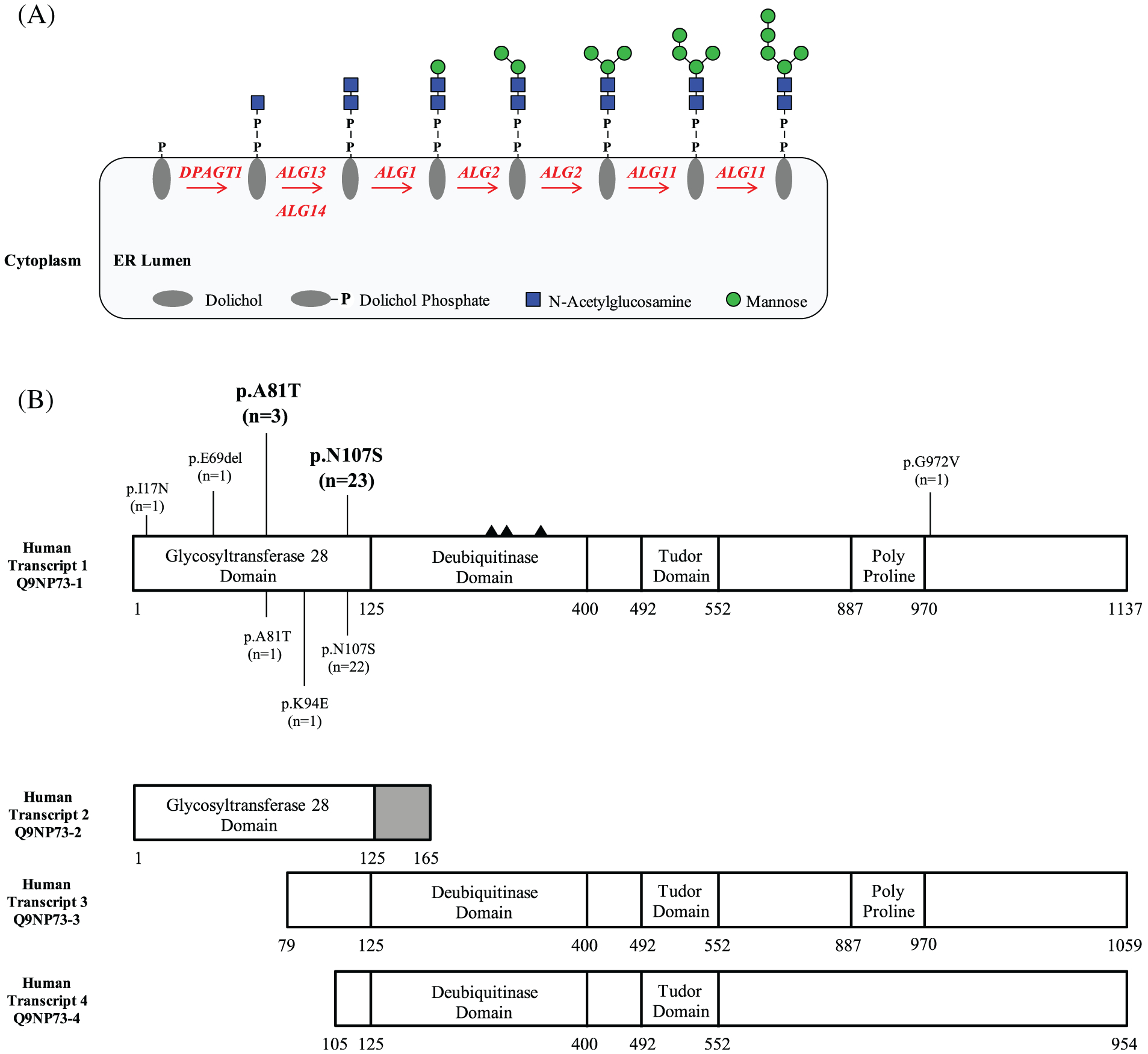
Lipid linked oligosaccharide pathway highlighting the role of ALG13 and a schematic showing the location of de novo variants identified in ALG13. A, Schematic showing the role of ALG13 in LLO synthesis. B, Schematic showing the four primary ALG13 transcripts with the positions of each de novo variant within the ALG13 protein. Variants identified in this study have been placed on the top portion, while previously reported variants are on the lower portion. The number of individuals identified is also listed as (n=). The solid triangles denote the catalytic triad required for the deubiquitinase domain active site D239, C242, H345
Nearly all our knowledge about the function of ALG13 is based on biochemical and genetic analyses in Saccharomyces cerevisiae. The majority of yALG13 protein localizes to the cytoplasm with only a portion in the endoplasmic reticulum (ER) through dimerization with yALG14, an ER transmembrane protein.6,7 This ER-localized yALG13-yALG14 heterodimer is essential for both cell viability and proper N-glycosylation, since abolishing this interaction results in profound defects in both.6,7,9
Much less is known about the function of ALG13 in higher eukaryotes, although its strong evolutionary conservation would presumably indicate its role in the proper synthesis of LLO.
Within the LLO pathway, at least 35 genes have been identified to cause a glycosylation related disorder.8 To date, 24 individuals have been identified with pathogenic de novo variants in ALG13 resulting in a neurodevelopmental disorder primarily characterized by early infantile epileptic encephalopathy.1–5,10–20 Most of these cases were identified in sequencing studies where clinical and variant information is summarized in the supplemental material. Nearly all reported affected persons are female and harbor an apparently recurrent de novo variant (c.320G>A; p.N107S). Surprisingly, glycosylated serum transferrin, which is a commonly used biomarker for CDG, showed a normal glycosylation pattern in the few ALG13-CDG individuals who have been tested.8
Here we present molecular data on 29 individuals with de novo variants in ALG13, including three novel variants not yet reported in the literature. Clinical information was available for 26 of these individuals, with detailed neurological findings for 24. We address the use of serum transferrin as a biomarker and show that two recurrent variants, p.A81T and p.N107S, that affect highly conserved residues, impact the function of ALG13 in a yeast complementation assay. Molecular modeling shows their potential interactions with the substrate, UDP-GlcNAc.
2 |. METHODS AND MATERIALS
2.1 |. Clinical data
Inclusion criteria for this study required the presence of de novo variants in ALG13 (Genbank NM_001099922.2, UniProt Q9NP73). Ultimately, we identified 29 individuals for which retrospective clinical data were obtained. Written consent was provided for all families in accordance with each individual’s primary physician, neurologist or when required, Sanford Burnham Prebys Medical Discovery Institute approval IRB-2014-038-17.
2.2 |. Carbohydrate-deficient transferrin Analysis
As previously described.21
2.3 |. Next-generation sequencing
Next-generation sequencing (NGS) consisted of either epileptic spasms (ES), genome sequencing (GS), or targeted gene panels. NGS and analysis was performed via each institutions or clinical lab services own standardized method. These methods are available upon request.
2.4 |. ALG13 structural modeling
A structural model of human ALG13 (hALG13) was generated using the Phyre2 server with the yALG13 structure (PDB code: 2JZ). MurG is the closest biological ortholog of ALG13/14 for which a high-resolution structure is currently available while bound to its substrate UDP-GlcNAc. Therefore, to get an indication of how UDP-GlcNAc might be positioned in the hALG13 active site, we aligned the structural model of hALG13 with the structure of MurG (PDB code: 3S2U) using PyMOL.
2.5 |. Western blot analysis of ALG13 protein
Fibroblasts from control and affected ALG13 individuals were grown as previously described in 1 g/L glucose Delbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Sigma). Western blot analysis was also performed as previously described using a polyclonal antibody to ALG13 (Proteintech 20 810–1-AP) and a monoclonal alpha-tubulin (12G10-DSHB hybridoma bank).22
2.6 |. Yeast complementation assay
Isolation and characterization of a yALG13 mutant strain was previously described.7 Survival of this strain is dependent on the expression of wild type yAlg13 protein under the control of a GAL1 promoter which is repressed by glucose and induced by galactose. Growth assays and carboxypeptidase Y (CPY) glycosylation analysis were both previously described.7,23,24 The expression plasmid pRS305 containing yALG13 with a C-terminal 3× FLAG tag driven off a glucose responsive promoter was used as a template to introduce either the p.A118T (p.A81T) or p.N144S (p.N107S) yeast-specific mutants. Insertion of the p.A118T (p.A81T) or p.N144S (p.N107S) mutants was carried out using a NEB Q5 site-directed mutagenesis kit.
3 |. RESULTS
3.1 |. Molecular analysis
Due to the lack of a reliable biomarker for screening and identifying ALG13-CDG, all previously reported cases (N = 24) were identified via NGS. In our cohort, this trend held true with all 29 individuals being identified by NGS (21 ES, 2 GS, 6 gene panel) (Table 1). We identified two recurrent de novo variants that accounted for the majority of identified individuals. The c.241G>A (p. A81T) variant was observed in 3/29 (10%) individuals, while the c.320A>G (p.N107S) variant was observed in the vast majority 23/29 (79%) (Figure 1B, Table 1). The c.320A>G (p.N107S) variant was also the most frequent (22/24 (92%) in previously reported cases (Figure 1B). We also identified several novel de novo variants including c.50T>A (p.I17N), c.207_209delAGA (p.E69del) and c.2915G>T (p.G972V) (Figure 1B, Table 1). None of the mentioned variants included in this cohort are present in gnomAD v2.1.1 or v3 (accessed May 20, 2020). In silico modeling each specific variant was performed using the combined annotation-dependent depletion (http://cadd.gs.washington.edu/) scoring method and showed each variant to have a score above 20 (p.I17N [25.7], p.A81T [25.3], p.N107S [20.7], p.G972V [30]) placing all five in the top 1% of deleterious variants in the human genome.
TABLE 1.
Genotypes identified in 29 individuals found to have de novo variants in ALG13
| CDG-ID | Status | Sex | Inheritance | cDNA position | Protein position | Method of detection | |
|---|---|---|---|---|---|---|---|
| 1 | CDG-0075 | Affected | F | De novo | c.320A>G | p.N107S | Panel |
| 2 | CDG-0077a | Affected | F | De novo | c.320A>G | p.N107S | Panel |
| 3 | CDG-0078a | Unaffected | F | De novo | c.320A>G | p.N107S | Panel |
| 4 | CDG-0079 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 5 | CDG-0080 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 6 | CDG-0081 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 7 | CDG-0082 | Affected | F | De novo | c.320A>G | p.N107S | GS |
| 8 | CDG-0083 | Affected | M | De novo | c.320A>G | p.N107S | ES |
| 9 | CDG-0085 | Affected | F | De novo | c.241G>A | p.A81T | Panel |
| 10 | CDG-0086 | Affected | F | De novo | c.320A>G | p.N107S | Panel |
| 11 | CDG-0088 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 12 | CDG-0089 | Affected | F | De novo | c.241G>A | p.A81T | ES |
| 13 | CDG-0092 | Affected | F | De novo | c.320A>G | p.N107S | Panel |
| 14 | CDG-0101b | Affected | M | De novo | c.2915G>T | p.G972V | ES |
| 15 | CDG-0125 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 16 | CDG-0133 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 17 | CDG-0134 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 18 | CDG-0135 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 19 | CDG-0136 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 20 | CDG-0139 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 21 | CDG-0140b | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 22 | CDG-0141 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 23 | CDG-0417 | Affected | F | De novo | c.241G>A | p.A81T | ES |
| 24 | CDG-0431 | Affected | F | De novo | c.320A>G | p.N107S | GS |
| 25 | CDG-0453 | Affected | F | De novo | c.50T>A | p.I17N | ES |
| 26 | CDG-0456 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 27 | CDG-0457 | Affected | F | De novo | c.207_209del AGA | p.E69del | ES |
| 28 | CDG-0458 | Affected | F | De novo | c.320A>G | p.N107S | ES |
| 29 | CDG-1017 | Affected | F | De novo | c.320A>G | p.N107S | ES |
Note: Genotypes from 29 individuals along with their sex, inheritance status, ALG13 variant and the method of detection are listed. Nucleotide numbering for cDNA uses +1 as the A of the ATG translation initiation codon in the reference sequence (Genbank: NM_001099922.2, UniProt: Q9NP73), with the initiation codon as codon 1.
Abbreviations: CDG, congenital disorder of glycosylation; ES, epileptic spasms; GS, genome sequencing.
CDG-0077 and CDG-0078 are identical twins.
CDG-0101 and CDG-0140 were found to be mosaic.
3.2 |. Clinical phenotype
ALG13 deficiency presents primarily as a neurodevelopmental disorder of varying severity. In our cohort, we were able to review clinical records for 26 individuals and found the most frequently seen symptoms were developmental delays in 26/26 (100%), seizures/epilepsy in 24/26 (92%), intellectual disability in 22/24 (92%) who could be assessed for this feature, and hypotonia in 22/26 (85%) (Figure 2).
FIGURE 2.
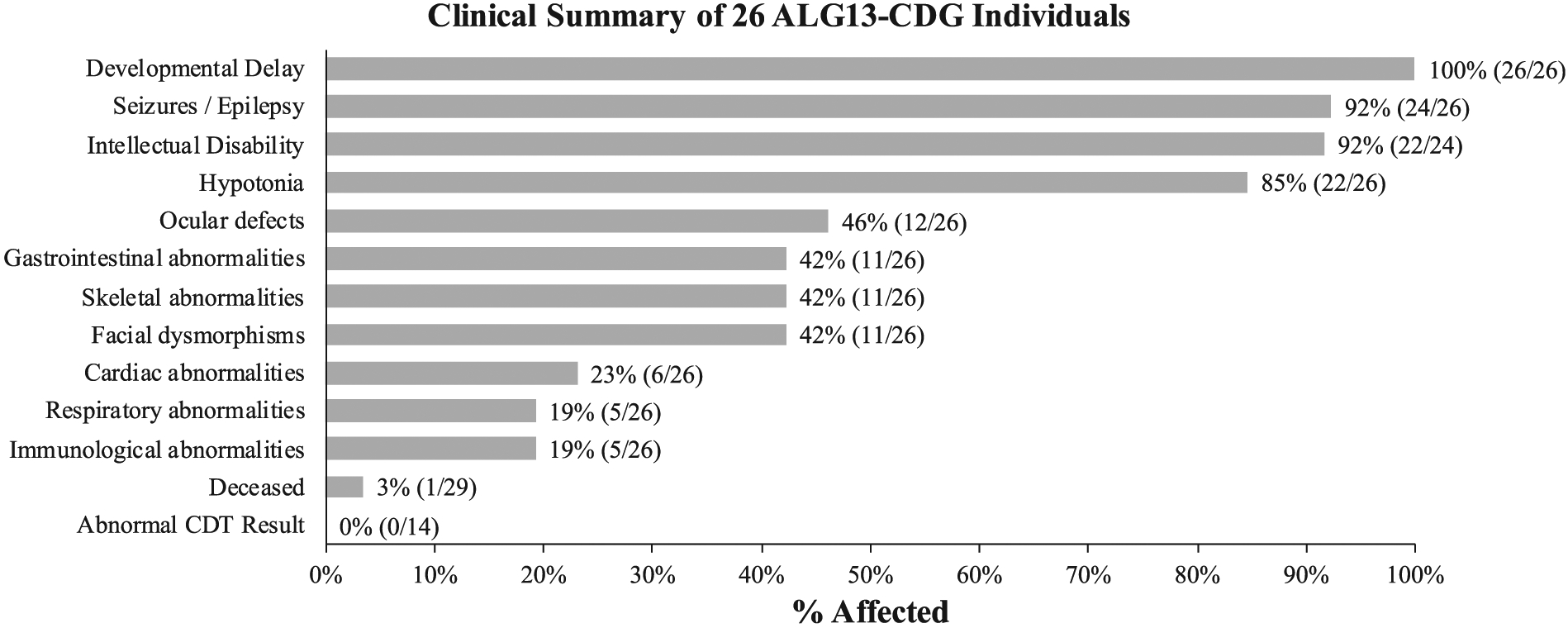
Clinical summary for 26 individuals with de novo variants in ALG13. General clinical summary for 26 individuals found to have de novo variants in ALG13
Due to the severe epileptic encephalopathy previously reported in this disease, a more in-depth analysis of the epileptic manifestations was performed in those individuals (n = 24) for whom detailed information was available. In the 24 individuals who presented with seizures, the mean age of onset was 6.5 (confidence interval [CI] 4.3–8.7) months. The semiology was very consistent, with epileptic spasms (ES) in 20/23 (87%) individuals as the presenting semiology, and hypsarrhythmia as the initial electroencephalogram (EEG) finding in 20/23 (87%) individuals with recorded initial EEG changes (Table 2). The spasms were treated either with adrenocorticotropic hormone (ACTH), prednisolone (Pred) or vigabatrin (VGB). ACTH and/or Pred was described as an effective treatment for ES in 11 individuals, and ineffective in five individuals, whereas VGB was effective in five individuals and was ineffective, or sometimes even aggravated seizure activity, in four individuals. One subject showed VGB-induced changes on brain magnetic resonance imaging (MRI) scan symmetrical diffusion restriction in the thalamus and globus pallidus), which reversed once VGB was discontinued. A plethora of antiseizure medications was used, illuminating the pronounced pharmaco-resistance of this disorder. Benzodiazepines (clobazam [CLB]), clonazepam, nitrazepam seemed effective in a rather high proportion of individuals (5/8–63%) where the most commonly used was CLB (used in 7%/8%−88%) (Table 2). Felbamate (FBM) was only used in two individuals but was reported to be effective in both. Topiramate was used more frequently (7 individuals), however, none of the affected individuals had a favorable outcome with this treatment. Levetiracetam, valproate, and lamotrigine showed a minimal effectiveness, with a reported positive effect in 2/8, 1/7, and 1/6 individuals, respectively. Cannabidiol was used in five subjects of whom two had a positive response. Most individuals continued to show signs of an epileptic encephalopathy after the initial spasms were treated. In six individuals, a diagnosis of Lennox-Gastaut syndrome (LGS) was made following the initial diagnosis of infantile spasms (IS). Five individuals remain seizure-free on current treatment. In addition to pharmaceuticals, a ketogenic diet (KD) was widely used in our cohort (12%/23%−52%); eight had a sustained positive effect of the diet and one had an initial response (Table 2). Two individuals had placement of a vagus nerve stimulator (VNS), only one of which had a favorable response. All individuals had an MRI scan, but no consistent findings were observed. Most were either normal or had nonspecific and vaguely described findings such as cerebral atrophy and benign enlargement of the subarachnoid spaces (Table 2). No structural abnormalities or dysplasias have been reported as potential epileptogenic foci.
TABLE 2.
Neurological summary for 42 individuals with de novo variants in ALG13
| Case | Sex | Mutation | Age at sz start | Initial semiology | EEG at diagnosis | MRI findings | Current and previous AED drugs | Other interv entions | Epilepsy outcome | DD/ID |
|---|---|---|---|---|---|---|---|---|---|---|
| CDG-0075 | F | p.N107S | 6 mo | GTCS | Mu/G | Normal | VPA | No | N/A | Yes |
| CDG-0077 | F | p.N107S | 10 mo | IS | H | BESS | ACTH, VGB, LEV, TPM | No | N/A | Yes |
| CDG-0079 | F | p.N107S | 6 mo | IS | H | Normal | ACTH, LEV, ESX, TPM, GBP, LTG | Keto | Myoclonic sz, moderate control | Yes |
| CDG-0080 | F | p.N107S | 3 mo | IS | H | Normal | ACTH, Pred, VGB, B6 | No | Poor | Yes |
| CDG-0082 | F | p.N107S | 6 mo | IS | H | Lack of WM | Pred, VGB, ACTH, LEV, VPA, ZON, LCM, CBD, NZM | Keto | N/A | Yes |
| CDG-0083 | M | p.N107S | 7 mo | IS | H | Cerebral atrophy | ACTH, VGB, VPA, ESX, CLB, LTG | Keto | Frontal lobe sz/Nocturnal sz. | Yes |
| CDG-0085 | F | p.A81T | 9 mo | IS | H | Normal | ACTH | No | sz free w/o AED | Yes |
| CDG-0086 | F | p.N107S | 2 mo | IS? | H? | BESS/normal | ZON, LTG, RUF, LEV, CLB, CBD | No | Myoclonic sz, head drops, LGS; mild | Yes |
| CDG-0088 | F | p.N107S | 6 mo | IS | H | VGB changes | VGB, TPM, ACTH, FBM, Pred | No | N/A | Yes |
| CDG-0089 | F | p.A81T | 1 mo | IS | H | BESS, thinning CC | N/A | Keto | Good control on keto | Yes |
| CDG-0092 | F | p.N107S | 1 mo | IS | H | Normal | ACTH, Pred, B6, VGB, PHB, TPM | Keto | IS and GTCS, controlled on AED/keto | Yes |
| CDG-0125 | F | p.N107S | 4–5 m | IS | H | Mild cerebral atrophy | CBD, CLB, CZP, LEV, Pred, LCM | No | N/A | Yes |
| CDG-0133 | F | p.N107S | No sz | no | normal | Normal | N/A | N/A | N/A | Yes |
| CDG-0134 | F | p.N107S | 4 mo | IS | H | Normal | Pred, VGB | No | sz free w/o AED | Yes |
| CDG-0136 | F | p.N107S | 3 mo | IS | H | PVL | VPA, TPM, CZP, CLB, LEV, NZM, LTG, ESX, PHB | VNS, Keto | LGS | Yes |
| CDG-0139 | F | p.N107S | 5 mo | IS | H | Normal | Pred, CZP, B6, VGB | Keto | sz free on Keto | Yes |
| CDG-0140 | F | p.N107S | 18 mo | ES | H | Normal | ZON, LEV | no | GC | Yes |
| CDG-0141 | F | p.N107S | 6 mo | N/A | N/A | Cortical atrophy | LTG, VPA, PER | no | Myoclonic sz, LGS, refractory | Yes |
| CDG-0417 | F | pA81T | 1 mo | IS | H | Progressive atrophy | Pred, VGB, LEV, CLB, TPM, VPA, OXC, LTG | Keto, inital effect | Focal tonic sz | Yes |
| CDG-0431 | F | p.N107S | 5 mo | IS | H | Mild cerebral atrophy | ACTH, CLB, LEV, TPM, LTG, OXC | Keto | N/A | Yes |
| CDG-0453 | F | p.I17N | 10 mo | Eye deviation | Mu | Normal | LEV, CLB, CBD | VNS | LGS, sz free on treatment | Yes |
| CDG-0456 | F | p.N107S | 6 mo | absence w eye flutter | Fo | BESS | PHB, TPM, ACTH | Keto | M/GT, sz free on keto | Yes |
| CDG-0457 | F | p.E69del | 24 mo | IS | evolving H | Normal | VGB, VPA, FBM | no | LGS | Yes |
| CDG-0458 | F | p.N107S | 6 mo | IS | H | Cerebral atrophy | Pred, VGB, ACTH, ZON, CBD | Keto | LGS, refractory | Yes |
| CDG-1017 | F | p.N107S | 7 mo | IS | H | Normal | ACTH, LEV, LTG | Keto | Complex partial sz | Yes |
| Madaan et al18 | F | p.N107S | 5 mo | IS | H | Normal | ACTH | No | infrequent sz | Yes |
| Fung et al14 | F | p.N107S | 4 mo | IS | N/A | N/A | 3 (nonspecified) | N/A | 50% reduction on treatment | Severe |
| Ortega-Moreno et al19 | F | p.N107S | 5 mo | IS | N/A | N/A | N/A | N/A | LGS | N/A |
| Bastaki et al11 | F | p.N107S | N/A | IS | H | N/A | N/A | N/A | N/A | Yes |
| Galama et al15 | M | p.N107S | 4.5 mo | IS | H | Hypoplasia of CC/DM | VGB, Pred, VPA, NZM, LTG, LEV | no | Pred stopped ES; LEV stable GTCS | Yes |
| Deciphering Developmental Disorders1 | F | p.N107S | N/A | N/A | N/A | N/A | N/A | N/A | Sz | Yes |
| Deciphering Developmental Disorders1 | F | p.N107S | N/A | N/A | N/A | N/A | N/A | N/A | Sz | Yes |
| Hamici et al16 | F | p.N107S | N/A | IS | H | N/A | N/A | N/A | N/A | Yes |
| Epi et al3 | F | p.N107S | 1–2 mo | Tonic/IS | H | VGB changes | VGB, Pred, TPM, LEV, CZP, ZON | no | Ongoing sz myoclonic | Profound |
| Kobayashi et al17 | F | p.N107S | 6 mo | IS | H | Cerebral atrophy | ACTH | N/A | Reduced sz on ACTH | Yes |
| Dimassi et al13 | F | p.N107S | 2 mo | IS | H | Mild global atrophy | N/A | N/A | Spasms continue | Yes |
| Smith-Packard et al20 | F | p.N107S | 8 mo | IS | H | N/A | ACTH | Keto | Complex partial sz at 5 years | Severe |
| Michaud et al5 | F | p.N107S | 4 mo | IS | H | Cerebral atrophy | N/A | N/A | Focal sz, multifocal EEG | Severe |
| Epi KC et al3 | F | p.N107S | 1 mo | IS | H | BESS | ACTH | N/A | Spasms returned on taper | Yes |
| Epi et al3 | F | p.N107S | 4 mo | IS | H | Normal | N/A | N/A | LGS | Severe |
| de Ligt et al2 | F | p.N107S | N/A | N/A | N/A | Atrophy, delayed myelin | N/A | N/A | Seizure unspecified | Profound |
| Timal et al10 | M | p.K94E | N/A | N/A | N/A | N/A | N/A | N/A | Refractory sz | N/A |
Note: Detailed neurological summary for 25 individuals in our cohort found to have de novo variants in ALG13. Additionally, available information for 17 previously reported individuals is also provided. Bolded drugs indicate a favorable response effect, non-bolded ones indicate uncertain response and gray indicates no response or unacceptable side effects.
Abbreviations: ACTH, adrenocorticotropic hormone; B6, pyridoxine; BESS, benign enlargement of the subarachnoidal spaces; CBD, cannabidiol; CC, corpus callosum; CLB, clobazam; CZP, clonazepam; DM, delayed myelinization; ESX, ethosuximide; FBM, felbamate; Fo, focal; GTCS, generalized tonic-clonic seizure; GBP, gabapentin; H, hypsarrhythmia; IS, infantile spasms; LCM, lacosamide; LGS, Lennox-Gastaut syndrome; LEV, levetiracetam; LTG, lamotrigine; Keto, ketogenic diet; Mu, multifocal; N/A, not available; NZM, nitrazepam OXC, oxcarbazepine; PHB, phenobarbital; Pred, prednisolone; PVL, Periventricular leukomalacia; RUF, rufinamide; SZ, seizure; TPM, topiramate; VGB, vigabatrin, VNS, vagus nerve stimulator; VPA, valproate; WM, white matter; ZON, zonisamide.
In addition to characterizing the prominent neurological phenotype, we queried other clinical symptoms occurring in these subjects.
In our cohort, ocular deficits were noted in 12/26 (46%) with cortical visual impairment present in eight of those individuals (Figure 2). Gastrointestinal symptoms characterized by either vomiting, gastroesophageal reflux disease (GERD), reflux and the need for G-tube placement were seen in 11/26 (42%). Skeletal defects, primarily scoliosis (6/11) or osteopenia (2/11), were seen in a total of 11/26 (42%) individuals. Dysmorphic features, mainly facial were seen in 11/26 (42%) and included coarse features, high arched palate or prominent forehead to name a few (Figure 2). Cardiac (6/26), respiratory (5/26) and immunologic abnormalities (5/26) were all seen in <25% of cases (Figure 2).
Out of the group of individuals carrying p.N107S variant, three individuals (CDG-0078, 0081 and 0135) were not included in the final clinical summary. CDG-0078 and CDG-0077 are identical twins and both carry the de novo p.N107S. However, in contrast to her affected sister, CDG-0078 does not have any clinical history of developmental and intellectual delays or seizure activity and is considered to be “unaffected.” One explanation for the dramatic clinical discrepancy between these two identical sisters is skewed X-inactivation of mutant ALG13 in the “healthy” sister. However, no additional testing could be performed. For CDG-0081, we were not able to obtain a complete clinical history, although it is known that she had intractable seizures and, at 13 years of age, is the only individual in our cohort who has died (Figure 2). Finally, for CDG-0135, we were only provided variant information for reporting.
We did not see a consistent ALG13-CDG-specific phenotype that could be used to help differentiate this disorder from other CDG types or even other epilepsy-related disorders. This highlights the importance of finding an ALG13-CDG-specific biomarker.
3.3 |. Carbohydrate-deficient transferrin analysis
ALG13 is a critical component of the N-linked glycosylation pathway and it is logical to use carbohydrate-deficient transferrin (CDT) analysis as a biomarker since the great majority of N-linked defects have an abnormal CDT. However, it was previously noted that several affected females who carried the recurrent p.N107S variant were tested and found to have a normal CDT result, indicating normal N-linked glycosylation, at least in hepatocytes.20 In our cohort of 29 individuals, 14 had CDT testing with 14/14 (100%) found to be normal (Figure 2). Two individuals (CDG-0417, CDG-1017) were found to have a CDT profile suggestive of a type I CDG. CDG-0417 had two mildly abnormal CDT results detected by capillary zone electrophoresis (initial CDT at age 1 year 11 months). However, we should note that over the course of a month, this amount of this abnormal peak improved for no clear reason. A follow-up analysis 4 years later using the more sensitive liquid chromatography mass spectrometry method showed that the abnormal peak was still detectable, but it was within the normal reference range. CDG-1017 also had a detectable abnormal peak (initial CDT at age 8 years 10 months) suggesting a type I pattern and, like CDG-0417, the amount was within the normal reference range. While only 14/29 individuals had CDT analysis performed, it is encouraging that CDG was considered as a possible cause in nearly half the subjects. These data suggest that CDT is unlikely to be a reliable biomarker for ALG13-CDG caused by de novo variants, and further work will be needed to identify one.
3.4 |. ALG13 structural modeling
The protein structure for S. cerevisiae yALG13 and the Pseudomonas aeruginosa ALG13 ortholog, MurG, are both known.25,26 MurG is a single polypeptide whose N-terminal domain has a high degree of homology to ALG14 and a C-terminal domain to ALG13.24 Furthermore, MurG is required to carry out a similar enzymatic reaction as the human ALG13:ALG14 heterodimer. Due to the structural conservation of yALG13 and MurG to human ALG13, we aimed to model and then determine the potential impact the p.A81T and p.N107S variants may have on human ALG13 protein.
From this alignment, human p.A81 and p.N107 are both situated close to the predicted position of the GlcNAc moiety of UDP-GlcNAc, the donor substrate (Figure 3A,B). The p.N107 in human ALG13 is predicted to be positioned close to the homologous p.Q288 in P. aeruginosa MurG27 (Figure 3B), which hydrogen bonds with the C3 hydroxyl group, and is part of the DDHQ motif that is homologous to the NNHQ motif in human ALG13. The p.A81 in human ALG13 is homologous to p. A260 in P. aeruginosa MurG, which is part of a hydrogen bond network with p.N291 and p.A125, that could be important in the correct positioning of p.N124, which binds to the C4 and C6 hydroxyls of the GlcNAc moiety. Further enzymatic and biophysical analyses are needed to determine the roles that p.N107 and p.A81 play in human ALG13 function, as well as how the disease-causing mutations affect this.
FIGURE 3.
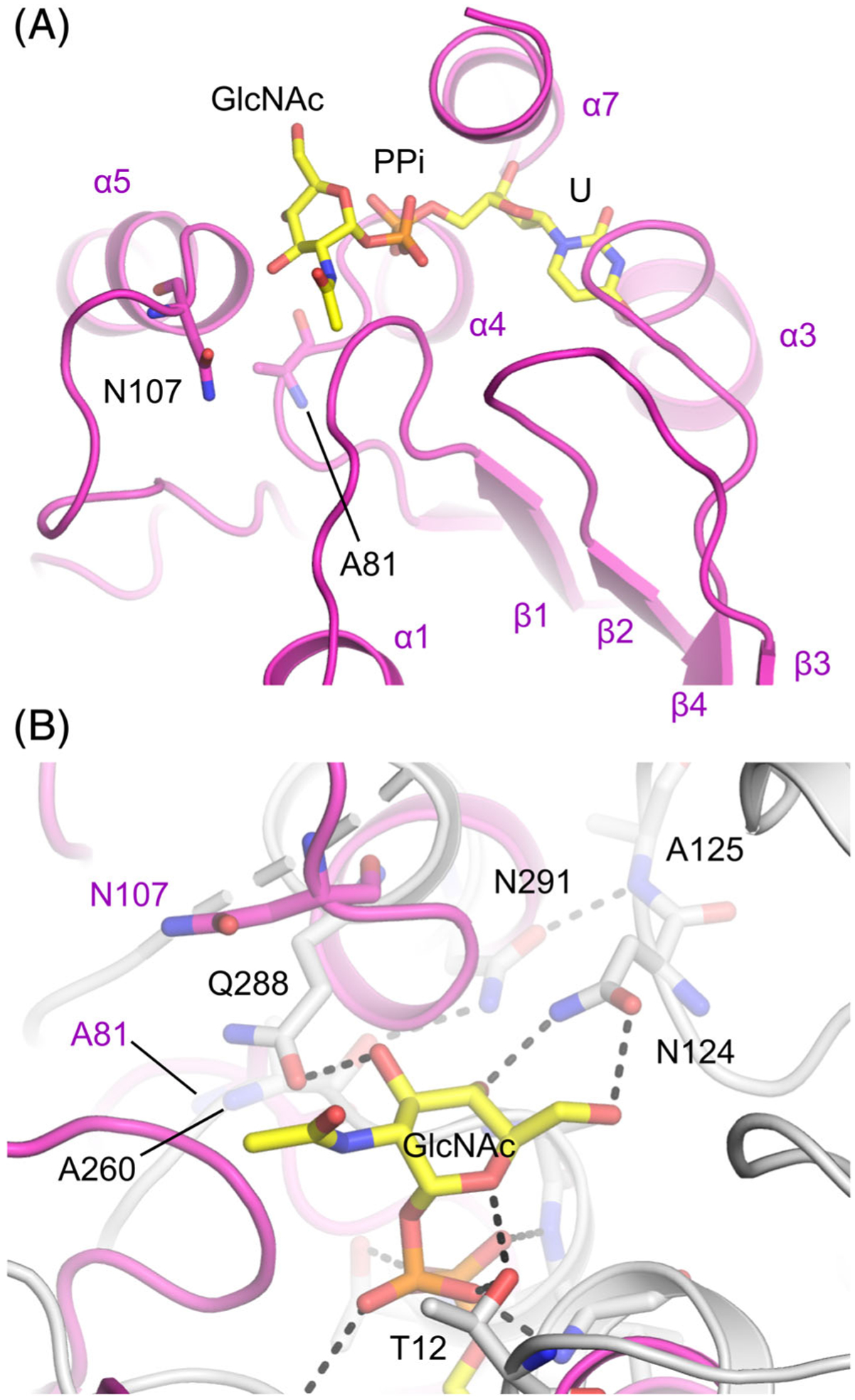
Structural homology model of ALG13, showing the predicted positions of recurrent de novo mutations relative to UDP-GlcNAc. A, N107, A81, and UDP-GlcNAc shown as sticks, colored according to the element, with carbon represented in yellow in UDP-GlcNAc and mauve in ALG13. Structural elements of ALG13 labeled according to Reference 26. B, An overlay of ALG13 in mauve, and MurG (PDB: 3S2U, PMID: 22973843) in gray, with H-bonds observed in the structure represented by dashed black lines
Hence, significant changes to the side chain chemistry of either of these residues is likely to affect the structure of the ALG13 active site. We speculate these variants could affect their affinity and/or specificity for the activated monosaccharide carried by the UDP. What is clear is that neither the p.A81T nor the p.N107S variants affect the stability of mutant ALG13 protein (Figure 4).
FIGURE 4.
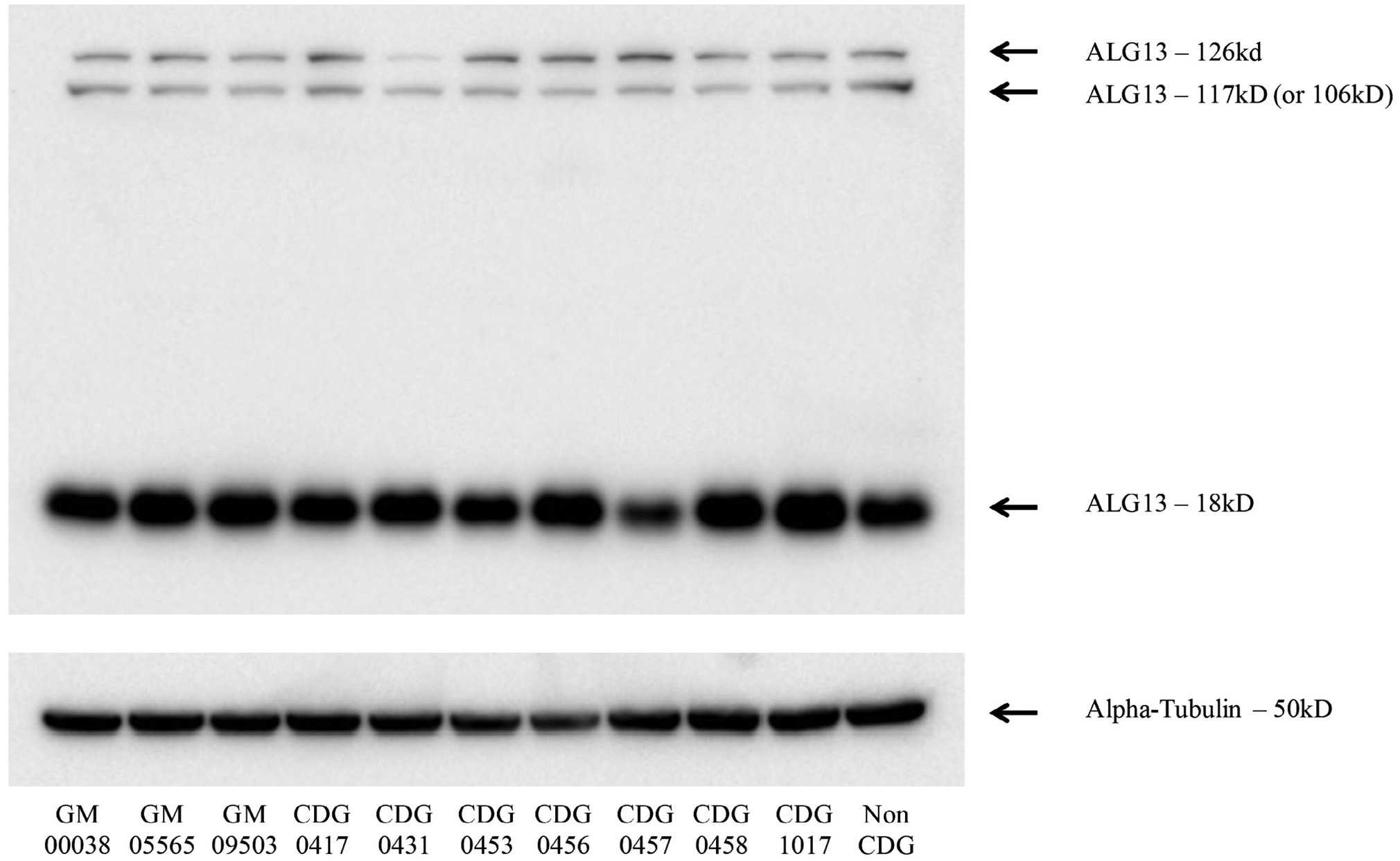
Western blot analysis of ALG13 from fibroblasts. Available fibroblasts were used for whole cell extracts to detect endogenous ALG13 protein levels. Alpha tubulin was used as a loading control to assure equal protein levels
3.5 |. Yeast complementation assay
Deletion of yALG13 causes a severe growth defect and ultimately lethality (https://www.yeastgenome.org/locus/S000003015). A conditional null mutant yALG13 strain is available, but its survival depends on the presence of wild type yALG13 driven off a GAL1-responsive promoter (ie, when galactose is provided in the absence of glucose, the strain will grow). We took advantage of a previously described method using this mutant strain and the ability to express yALG13 under the control of a glucose responsive promoter.7 When we expressed highly conserved equivalent yALG13 mutants and shifted to selection under glucose (ie, the rescued yALG13 under galactose is repressed), the p.A118T and p.N144S mutants both rescued the growth defect in a similar fashion to wild type (Figure 5). However, unlike wild type yALG13, neither the p.A118T nor p.N144S mutants were capable of restoring glycosylation of a commonly used biomarker (CPY) for yeast glycosylation mutants (Figure 5).28 Importantly, Western blot analysis determined neither expressed mutation affected the stability of yALG13 when compared to wild type (Figure 5). These data suggest in yeast the p.A118T and p.N144S variants likely affect the function of ALG13.
FIGURE 5.
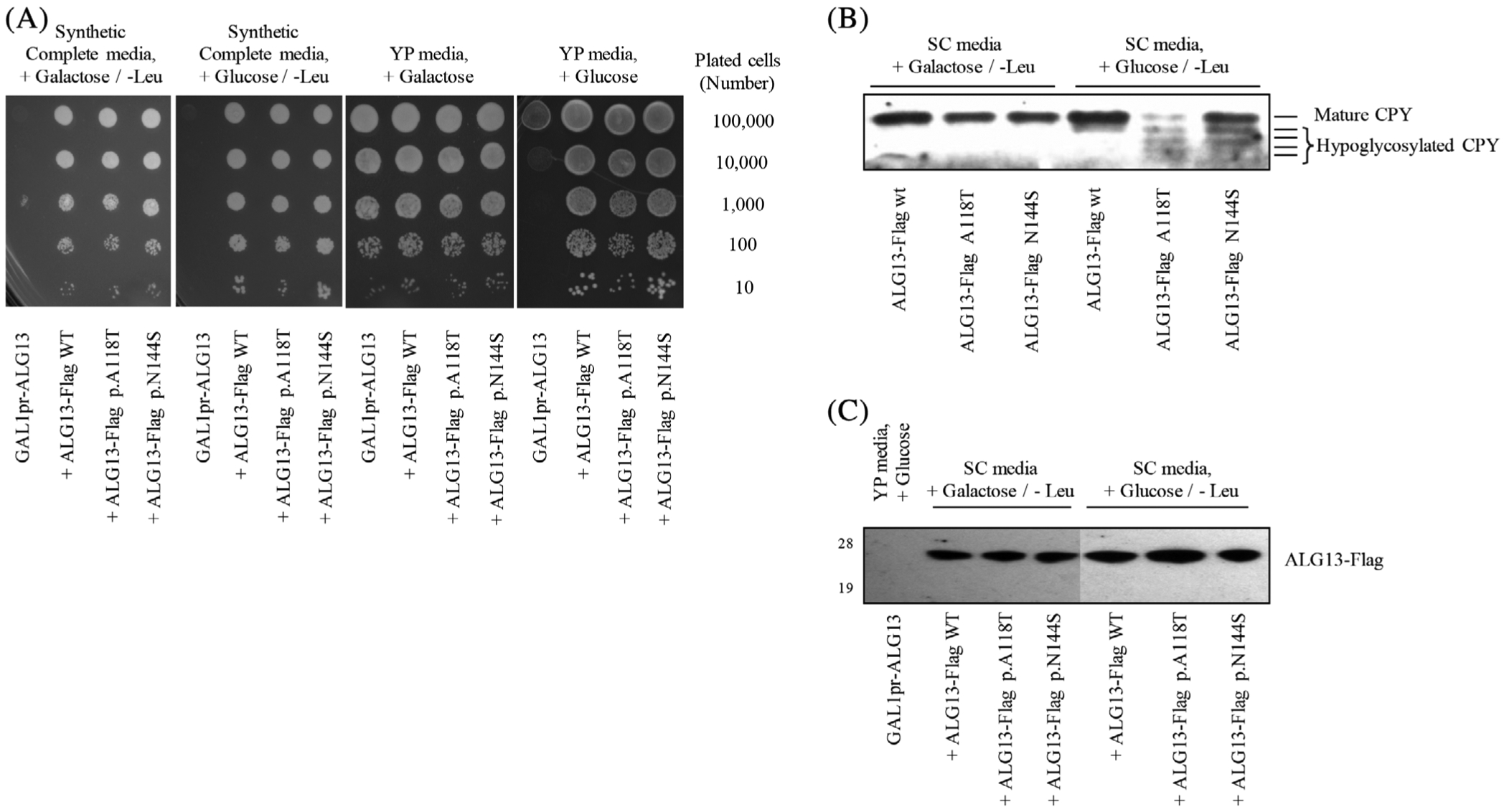
Yeast complementation assay using an ALG13 deficient yeast strain. A, An ALG13 deficient yeast strain was grown under selection conditions allowing for expression of wild type yALG13 when grown in the presence of galactose. Transfection and expression of various yALG13 mutants were performed as previously described.7 B, Western blot analysis of glycosylated carboxypeptidase Y under various complementation and growth conditions was performed in triplicates with representative data shown. C, Western blot analysis showing the transfected levels of yALG13-Flag tagged protein
4 |. DISCUSSION
Here we present data on 29 newly reported individuals who were found to have de novo variants in ALG13, which more than doubles the number of known cases. Previously de novo variants in ALG13 were shown to cause a neurodevelopmental disorder characterized by varying degrees of developmental and intellectual disabilities and epilepsy. Together with our novel cases, the total number of individuals affected with ALG13-CDG who have been identified is now 53.
The epileptic phenotype in the ALG13-CDG subjects described here is strikingly homogenous and is consistent with previous publications.14,18,19 It emerges in the usual time for ES, with a mean age of 6.5 months at the debut of the seizures, where the peak age at the debut of spasms in the whole group is 6–8 months.29 Most individuals show the electrophysiological pattern of hypsarrhythmia in their first EEG, and all show developmental arrest, thus fulfilling the criteria for West syndrome. This age-dependent epileptic encephalopathy syndrome often develops later on into another age-dependent syndrome, LGS, and many of the individuals in our cohort displayed a fully developed LGS or partial symptoms thereof.30 Only five individuals remained seizure-free on treatment. Traditionally, three main treatment approaches to ES exist, corticosteroids (usually Pred or ACTH), and the gamma aminobutyric acid (GABA) aminotransferase inhibitor VGB (γ-vinyl-GABA). In a recent review of all clinical studies involving these agents, ACTH seems to be the most effective single treatment, whereas a combination of VGB and ACTH also shows promise.31 In our cohort ACTH and Pred seemed superior to VGB in both effectiveness and side effects, which is also supported by previous case descriptions.3,17,18 We therefore suggest that ACTH or Pred to be used as the first-line treatment of ALG13-related ES, possibly with an extended period of weaning as several individuals had seizure recurrence during tapering. In the continued care for these individuals, a large number of different AEDs have been used in our cohort, highlighting widespread pharmaco-resistance.
Among the different drugs, two stood out as potentially favorable alternatives, benzodiazepines (most commonly CLB and FBM). FBM is a drug initially approved for LGS that was previously restricted due to unusual cases of fatal aplastic anemia and hepatic failure,32 but now is seeing a revival as a rescue agent in unresponsive IS33; its use in ALG13-related epileptic encephalopathy should certainly be further studied. A KD is a powerful treatment option in some epileptic encephalopathies such as Dravet syndrome,34 and other metabolic conditions such as glucose transporter 1 (GLUT1) deficiency.35 In ALG13-CDG with epileptic encephalopathy, we only found one report of successful treatment using a KD,20 and it is unclear from the other reports whether it has been tried on this cohort of individuals. In our cohort, however, as many as 12 individuals were being treated with, or were previously treated with a KD; 8 showed a sustainable response, whereas 1 showed an initial response to the diet. This is very encouraging and suggest a KD may be an important potential alternative/complement to pharmaceuticals in this disease.
One important issue we were not able to fully address was the role X-chromosome inactivation (XCI) plays in ALG13 deficiency. In our cohort, only three individuals were reported to have XCI analysis and all were found to have random XCI from whole blood samples. Because of the strong neurological phenotype, XCI in whole blood may not fully represent what is happening in the brain.
Despite the clear role of ALG13 in glycosylation, the common CDG biomarker transferrin was not as reliable for ALG13-CDG as it is for other CDG types. This is reminiscent of SLC35A2-CDG, which is also an X-linked disorder caused by de novo variants and like ALG13-CDG can give unreliable or unexpected CDT results. However, unlike SLC35A2-CDG, which is due to loss of function variants, we hypothesize that the recurrent p.N107S and other de novo variants are likely gain of function variants. While it is unclear what that gain of function is, this could potentially explain why CDT is not abnormal.
As is the case with all LLO synthesis proteins, ALG13 is an essential component of the glycosylation machinery. In both yeast and humans, it is the only known enzyme capable of carrying out the transfer of a GlcNAc onto Dol-P-P-GlcNAc to generate Dol-P-P-GlcNAc-GlcNAc, which serves as a substrate for ALG1, the next enzyme in LLO biosynthesis (Figure 1A). Human deficiencies in ALG13 should cause a profound glycosylation defect like that seen in the preceding (DPAGT1-CDG) and subsequent (ALG1-CDG) LLO steps.36,37 However, despite the proven role of ALG13 in glycosylation, individuals with de novo variants do not demonstrate a clear glycosylation abnormality, at least not in serum glycoproteins or skin fibroblasts (data not shown). Interestingly, this is also true for the ALG13-specific binding partner, ALG14, where biallelic variants cause both congenital myasthenic syndrome-15 (ALG14-CMS) and also a disorder characterized as an early lethal neurodegeneration with myasthenic and myopathic features.38,39
While ALG13 and ALG14 are both ubiquitously expressed proteins, it is possible that deficiencies in either could cause a tissue-specific disorder. This has for example been seen with a few CDG types that primarily affect the liver (eg, MPI-CDG, TMEM199-CDG, CDCC115-CDG, ATP6AP1-CDG).40
While ALG13’s role in N-glycosylation has been well documented in yeast, its role in humans is less clear and more complicated because, at some point within its evolution from S. cerevisiae to Homo sapiens, there was a dramatic change in the ALG13 protein. S. cerevisiae ALG13 (Uniprot P53178) is a small 202 amino acid (aa) protein consisting only of a glycosyltransferase 28 (GT28) domain. However, in humans, multiple transcripts of ALG13 (Uniprot Q9NP73) occur with the canonical transcripts encoding a 1137aa protein, two intermediate forms of 1059aa and 954aa, and the smallest form of 165aa. This smaller 165aa form most closely resembles the single, essential yeast protein. Interestingly, the canonical human isoform contains not only the glycosyltransferase 28 domain, but also several other domains including an ovarian tumor (OTU) deubiquitinase domain (OTUD), a TUDOR domain and a Proline rich domain (Figure 1B).
Transcript 3 lacks 78 amino acid of the N-terminal GT28 domain just before the p.A81T hotspot, while transcript 4 lacks 105 amino acid of the N-terminal GT28 domain prior to the p.N107S hotspot. (Figure 1B). Both of these proteins likely lack GT28 activity due to the loss of many critical amino acids required for substrate binding and catalytic activity. We are able to detect three of four isoforms (Figure 4); however, it is very possible isoform 4 is not detectable due to the loss of the antibody epitope.
It is unclear what roles the different transcripts play in the pathology of ALG13 deficiency. We hypothesis that only the long and short forms could potentially harbor a functional GT28 domain, but to date no functional studies have proven the long form has catalytic activity. Thus, it is unclear if the long and short forms compensate for one another. Studies have shown it is possible to completely delete the long isoform form, but not the short form, suggesting the short form is the essential glycosyltransferase required for glycosylation.41
The functional significance of these additional domains within ALG13 is unknown. These other, non-glycosyltransferase domains are found together as a separate gene in zebrafish (https://zfin.org). In zebrafish, zgc:92907 is most similar to the small ALG13 isoform seen across all organisms, while the gene annotate as ALG13 does not contain the GT28 domain required for GT28 activity but does contain the OTUD family domain. It is unclear when during evolution these quite different genes fused into the single gene seen in humans and other vertebrates.
It is tantalizing to speculate what functions these additional domains within the long isoform of ALG13 are performing. For example, the ovarian tumor deubiquitinase (OTUD) family domain of ALG13 contains the conserved catalytic triad of amino acids (Asp239, Cys242, His345) required for deubiquitinase (DUB) activity.42 However, when expressed in bacteria, the purified ALG13 OTUD domain lacked DUB activity toward a Ub-propargylamide substrate but did have activity toward an artificial haloalkyl substrate.42 Is it possible that ALG13-dependent DUB activity is restricted to a very small select set of protein targets, like those in the LLO pathway? Interestingly, DPAGT1, which catalyzes the proceeding step to ALG13, has been found to be ubiquitinated at Lys48 within a critical cytoplasmic loop required for UDP-GlcNAc binding.43 Could the long isoform of ALG13 DUB activity regulate DPAGT1 activity via deubiquitination? These, and many other questions remain unanswered about the function of ALG13.
In conclusion, we present data on 29 individuals found to specifically harbor de novo variants in ALG13, allowing us to expand both the clinical phenotype and molecular understanding of this disorder. Clinical and pharmacological data suggest certain antiseizure medications could potentially be prioritized as a first line therapies, while others could be avoided. Furthermore, nonpharmaceutical alternatives such as a KD could have beneficial effects on suppression of seizures and should be considered. We identified several novel de novo variants, additional cases of the recurrent p.N107S and p. A81T. Structural modeling predicts both the p.A81T and p.N107S variants may affect the ALG13 active site and UDP-GlcNAc interface. Finally, we show expression of either recurrent variant in an ALG13 mutant yeast model restores the observed growth defect but does not correct the N-glycosylation abnormality, suggesting that both residues are important for normal glycosylation in yeast.
SYNOPSIS.
Novel and recurrent de novo variants in ALG13 cause a neurodevelopmental disorder.
ACKNOWLEDGMENTS
We would like to thank all the families for their continued support and for providing valuable biological specimens. We thank Dr Neta Dean for the ALG13 deficient yeast strain and expression plasmids. We also thank Mrs Krista Williams for her supporting the ALG13 family Facebook support group. This work was supported in part by the Intramural Research Program of the National Human Genome Research Institute. We thank Jamie Smolin for technical help. This work was also supported by the Rocket Fund, National Institutes of Health (NIH) grants R01DK099551 (to H. H. F) and partial funding from U54 NS115198. Regional funding, Region Skåne, Sweden (to E. A. E). JPB Foundation (to W. K. C), SFARI (to W. K. C.). Research reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG007708 and U01HG010218. The University of Washington Center for Mendelian Genomics through NHGRI and NHLBI grants UM1 HG006493 and U24 HG008956. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. I. E. S. has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, Biomarin and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Anavex Life Sciences and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics and UCB. She receives/has received research support from the National Health and Medical Research Council of Australia, Health Research Council of New Zealand, CURE, Australian Epilepsy Research Fund, March of Dimes and NIH/NINDS.
Footnotes
CONFLICT OF INTEREST
Bobby G. Ng, Erik A. Eklund, Sergey A. Shiryaev, Yin Y. Dong, Mary Alice Abbott, Carla Asteggiano, Michael J. Bamshad, Eileen Barr, Jonathan A. Bernstein, Shabeed Chelakkadan, John Christodoulou, Wendy K. Chung, Michael A. Ciliberto, Janice Cousin, Fiona Gardiner, Suman Ghosh, William D. Graf, Stephanie Grunewald, Katherine Hammond, Natalie S. Hauser, George E. Hoganson, Kimberly M. Houck, Jennefer N. Kohler, Eva Morava, Austin A. Larson, Sujana Madathil, Colleen McCormack, Naomi J.L. Meeks, Rebecca Miller, Deborah A. Nickerson, Gabriela Magali Papazoglu, Beth A. Pletcher, Ingrid E. Scheffer, Andrea Beatriz Schenone, Leah J. Rowe, Alvaro H. Serrano Russi, Rossana Sanchez Russo, Farouq Thabet, Allysa Tuite, María Mercedes Villanueva, Raymond Y. Wang, Richard I. Webster, Dorcas Wilson, Alice Zalan, Lynne A. Wolfe, and Hudson H. Freeze declare that they have no conflict of interest. Kristin G. Monaghan, Timothy Blake Palculict, Rhonda E. Schnur, Yue Si and Lindsay Rhodes are employees of GeneDx, Inc. Pengfei Liu and Jill A. Rosenfeld are employed by the Department of Molecular and Human Genetics at Baylor College of Medicine who receives revenue from clinical genetic testing conducted at Baylor Genetics.
ETHICS STATEMENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000.6 Informed consent was obtained from all individuals being included in the study. Sanford Burnham Prebys Medical Discovery Institute (IRB-2014-038-17).
ANIMAL RIGHTS
This article does not contain any studies with animal subjects.
REFERENCES
- 1.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–438. 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–1929. 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 3.Epi KC, Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013; 501:217–221. 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heyne HO, Artomov M, Battke F, et al. Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genet Med. 2019;21:2496–2503. 10.1038/s41436-019-0531-0. [DOI] [PubMed] [Google Scholar]
- 5.Michaud JL, Lachance M, Hamdan FF, et al. The genetic landscape of infantile spasms. Hum Mol Genet. 2014;23:4846–4858. 10.1093/hmg/ddu199. [DOI] [PubMed] [Google Scholar]
- 6.Bickel T, Lehle L, Schwarz M, Aebi M, Jakob CA. Biosynthesis of lipid-linked oligosaccharides in Saccharomyces cerevisiae: Alg13p and Alg14p form a complex required for the formation of GlcNAc(2)-PP-dolichol. J Biol Chem. 2005;280:34500–34506. 10.1074/jbc.M506358200. [DOI] [PubMed] [Google Scholar]
- 7.Gao XD, Tachikawa H, Sato T, Jigami Y, Dean N. Alg14 recruits Alg13 to the cytoplasmic face of the endoplasmic reticulum to form a novel bipartite UDP-N-acetylglucosamine transferase required for the second step of N-linked glycosylation. J Biol Chem. 2005;280:36254–36262. 10.1074/jbc.M507569200. [DOI] [PubMed] [Google Scholar]
- 8.Ng BG, Freeze HH. Perspectives on glycosylation and its congenital disorders. Trends Genet. 2018;34:466–476. 10.1016/j.tig.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Averbeck N, Keppler-Ross S, Dean N. Membrane topology of the Alg14 endoplasmic reticulum UDP-GlcNAc transferase subunit. J Biol Chem. 2007;282:29081–29088. 10.1074/jbc.M704410200. [DOI] [PubMed] [Google Scholar]
- 10.Timal S, Hoischen A, Lehle L, et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet. 2012;21:4151–4161. 10.1093/hmg/dds123. [DOI] [PubMed] [Google Scholar]
- 11.Bastaki F, Bizzari S, Hamici S, et al. Single-center experience of N-linked congenital disorders of glycosylation with a summary of molecularly characterized cases in Arabs. Ann Hum Genet. 2018;82:35–47. 10.1111/ahg.12220. [DOI] [PubMed] [Google Scholar]
- 12.Demos M, Guella I, DeGuzman C, et al. Diagnostic yield and treatment impact of targeted exome sequencing in early-onset epilepsy. Front Neurol. 2019;10:434 10.3389/fneur.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimassi S, Labalme A, Ville D, et al. Whole-exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clin Genet. 2016;89:198–204. 10.1111/cge.12636. [DOI] [PubMed] [Google Scholar]
- 14.Fung CW, Kwong AK, Wong VC. Gene panel analysis for non-syndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open. 2017;2:236–243. 10.1002/epi4.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galama WH, Verhaagen-van den Akker SLJ, Lefeber DJ, Feenstra I, Verrips A. ALG13-CDG with infantile spasms in a male patient due to a de novo ALG13 gene mutation. JIMD Rep. 2018;40:11–16. 10.1007/8904_2017_53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamici S, Bastaki F, Khalifa M. Exome sequence identified a c.320A>G ALG13 variant in a female with infantile epileptic encephalopathy with normal glycosylation and random X inactivation: review of the literature. Eur J Med Genet. 2017;60:541–547. 10.1016/j.ejmg.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi Y, Tohyama J, Kato M, et al. High prevalence of genetic alterations in early-onset epileptic encephalopathies associated with infantile movement disorders. Brain Dev. 2016; 38:285–292. 10.1016/j.braindev.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 18.Madaan P, Negi S, Sharma R, Kaur A, Sahu JK. X-linked ALG13 gene variant as a cause of epileptic encephalopathy in girls. Indian J Pediatr. 2019;86:1072–1073. 10.1007/s12098-019-03059-3. [DOI] [PubMed] [Google Scholar]
- 19.Ortega-Moreno L, Giraldez BG, Soto-Insuga V, et al. Molecular diagnosis of patients with epilepsy and developmental delay using a customized panel of epilepsy genes. PLoS One. 2017;12: e0188978 10.1371/journal.pone.0188978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith-Packard B, Myers SM, Williams MS. Girls with seizures due to the c.320A>G variant in ALG13 do not show abnormal glycosylation pattern on standard testing. JIMD Rep. 2015;22: 95–98. 10.1007/8904_2015_416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacey JM, Bergen HR, Magera MJ, Naylor S, O’Brien JF. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem. 2001;47:513–518. [PubMed] [Google Scholar]
- 22.Ferreira CR, Xia ZJ, Clement A, et al. A recurrent de novo heterozygous COG4 substitution leads to Saul-Wilson syndrome, disrupted vesicular trafficking, and altered proteoglycan glycosylation. Am J Hum Genet. 2018;103:553–567. 10.1016/j.ajhg.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Averbeck N, Gao XD, Nishimura S, Dean N. Alg13p, the catalytic subunit of the endoplasmic reticulum UDP-GlcNAc glycosyltransferase, is a target for proteasomal degradation. Mol Biol Cell. 2008;19:2169–2178. 10.1091/mbc.E07-10-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao XD, Moriyama S, Miura N, Dean N, Nishimura S. Interaction between the C termini of Alg13 and Alg14 mediates formation of the active UDP-N-acetylglucosamine transferase complex. J Biol Chem. 2008;283:32534–32541. 10.1074/jbc.M804060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu Y, Chen L, Ha S, et al. Crystal structure of the MurG:UDP-GlcNAc complex reveals common structural principles of a superfamily of glycosyltransferases. Proc Natl Acad Sci USA. 2003;100:845–849. 10.1073/pnas.0235749100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Weldeghiorghis T, Zhang G, Imperiali B, Prestegard JH. Solution structure of Alg13: the sugar donor subunit of a yeast N-acetylglucosamine transferase. Structure. 2008;16:965–975. 10.1016/j.str.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown K, Vial SC, Dedi N, et al. Crystal structure of the Pseudomonas aeruginosa MurG: UDP-GlcNAc substrate complex. Protein Pept Lett. 2013;20:1002–1008. 10.2174/0929866511320090006. [DOI] [PubMed] [Google Scholar]
- 28.Avaro S, Belgareh-Touze N, Sibella-Arguelles C, Volland C, Haguenauer-Tsapis R. Mutants defective in secretory/vacuolar pathways in the EUROFAN collection of yeast disruptants. Yeast. 2002;19:351–371. 10.1002/yea.838. [DOI] [PubMed] [Google Scholar]
- 29.Riikonen R Epidemiological data of West syndrome in Finland. Brain Dev. 2001;23:539–541. 10.1016/s0387-7604(01)00263-7. [DOI] [PubMed] [Google Scholar]
- 30.Lombroso CT. A prospective study of infantile spasms: clinical and therapeutic correlations. Epilepsia. 1983;24:135–158. 10.1111/j.1528-1157.1983.tb04874.x. [DOI] [PubMed] [Google Scholar]
- 31.Riikonen R Infantile spasms: outcome in clinical studies. Pediatr Neurol DOI. 2020;108:54–64. 10.1016/j.pediatrneurol.2020.01.015. [DOI] [PubMed] [Google Scholar]
- 32.Shah YD, Singh K, Friedman D, Devinsky O, Kothare SV. Evaluating the safety and efficacy of felbamate in the context of a black box warning: a single center experience. Epilepsy Behav. 2016;56:50–53. 10.1016/j.yebeh.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Dozieres-Puyravel B, Nasser H, Bellavoine V, Ilea A, Delanoe C, Auvin S. Felbamate for infantile spasms syndrome resistant to first-line treatments. Dev Med Child Neurol. 2020; 62:581–586. 10.1111/dmcn.14427. [DOI] [PubMed] [Google Scholar]
- 34.Dressler A, Trimmel-Schwahofer P, Reithofer E, et al. Efficacy and tolerability of the ketogenic diet in Dravet syndrome-comparison with various standard antiepileptic drug regimen. Epilepsy Res. 2015;109:81–89. 10.1016/j.eplepsyres.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Kass HR, Winesett SP, Bessone SK, Turner Z, Kossoff EH. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure. 2016;35:83–87. 10.1016/j.seizure.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Ng BG, Shiryaev SA, Rymen D, et al. ALG1-CDG: clinical and molecular characterization of 39 unreported patients. Hum Mutat. 2016;37:653–660. 10.1002/humu.22983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng BG, Underhill HR, Palm L, et al. DPAGT1 deficiency with encephalopathy (DPAGT1-CDG): clinical and genetic description of 11 new patients. JIMD Rep. 2019;44:85–92. 10.1007/8904_2018_128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cossins J, Belaya K, Hicks D, et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain. 2013; 136:944–956. 10.1093/brain/awt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schorling DC, Rost S, Lefeber DJ, et al. Early and lethal neurodegeneration with myasthenic and myopathic features: a new ALG14-CDG. Neurology. 2017;89:657–664. 10.1212/WNL.0000000000004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marques-da-Silva D, Dos Reis FV, Monticelli M, et al. Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J Inherit Metab Dis. 2017;40: 195–207. 10.1007/s10545-016-0012-4. [DOI] [PubMed] [Google Scholar]
- 41.Gao P, Wang F, Huo J, et al. ALG13 deficiency associated with increased seizure susceptibility and severity. Neuroscience. 2019; 409:204–221. 10.1016/j.neuroscience.2019.03.009. [DOI] [PubMed] [Google Scholar]
- 42.Mevissen TE, Hospenthal MK, Geurink PP, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–184. 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Udeshi ND, Svinkina T, Mertins P, et al. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12: 825–831. 10.1074/mcp.O112.027094. [DOI] [PMC free article] [PubMed] [Google Scholar]