

Abstract
Herein we report photoinitiated polymerization-induced self-assembly (photo-PISA) of spherical micelles consisting of proapoptotic peptide-polymer amphiphiles. The one-pot synthetic approach yielded micellar nanoparticles at high concentrations and at scale (150 mg/mL) with tunable peptide loadings up to 48 wt. %. The size of the micellar nanoparticles was tuned by varying the lengths of hydrophobic and hydrophilic building blocks. Critically, the peptide-functionalized nanoparticles imbued the proapoptotic “KLA” peptides (amino acid sequence: KLAKLAKKLAKLAK) with two key properties otherwise not inherent to the sequence: 1) proteolytic resistance compared to the oligopeptide alone; 2) significantly enhanced cell uptake permeability by multivalent display of KLA peptide brushes. The result was demonstrated improved apoptosis efficiency in HeLa cells. These results highlight the potential of photo-PISA in the large-scale synthesis of functional, proteolytically resistant peptide-polymer conjugates for intracellular delivery.
Keywords: Peptide-Polymer Conjugates, Scaled Synthesis, Polymerization-Induced Self-Assembly
Graphical Abstract
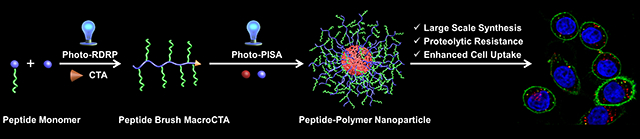
Peptide-Polymer Amphiphiles: Polymerization-induced self-assembly enables the at scale synthesis of nanoparticles with a high-density display of peptide, tunable particle size, tunable peptide loadings. The resulting peptide brush polymer nanoparticles exhibit enhanced stability, cell uptake efficiency and efficacy in comparison with their peptide analogues, highlighting the potential of these peptide-polymer amphiphiles as peptide delivery systems.
Introduction
Synthetic peptides are powerful therapeutics and chemical biology tools because of their biocompatibility, straightforward synthesis, predictable metabolism, and high degree of modularity in molecular design.[1] However, these advantages are typically compromised by natural processes prevalent in cells and tissues that have evolved to degrade them.[2] Traditional approaches for protecting active peptides from enzymatic digestion capitalize on chemical modification of the peptide.[3] These approaches include cyclization,[3a–b] lipidation,[3c] conjugation of PEG,[3d–e] introduction of unnatural amino acids,[3f] peptide backbone modification (e.g., N-methylation),[3g] and capping of N- or C-terminus,[3h] among others.[3i] Consequently, modified peptides are rendered inaccessible to, or unrecognizable by the active site of protease. Nevertheless, the bioactivity or function of modified peptides can be reduced as a result of the alteration of chemical identity and connectivity in amino acids. Moreover, penetration of peptides into cells is typically inefficient unless some special care is taken to engage cell surfaces selectively or through the use of cell penetrating sequences.[4]
Beyond modification of the sequence itself, the three-dimensional spatial arrangement of peptides not only can improve the stability of peptides, but also enhance their biological activities such as cell binding and penetration via multivalent effects.[5] Examples include peptide-coated inorganic nanoparticles,[6] peptide-shell polymer nanoparticles,[7] and peptide brush polymers,[7c, 8] all of which display multiple strands of peptides on a scaffold. However, the synthesis of these materials is not scalable, hindering continued development of these systems to larger animal models and clinical translations.[7d] Standard solvent-switch strategies to access self-assembled nanostructures of peptide-polymer amphiphiles are typically conducted in dilute solution (< 1 wt.%).[7c]
Polymerization-induced self-assembly (PISA) has emerged as a scalable synthetic route to soft nanomaterials at high solids content (up to 50 wt.%).[9] Particularly, photoinitiated PISA (photo-PISA) is promising for the incorporation of biological molecules into nanoparticles, because the reaction can be performed under mild conditions characterized by ambient temperatures, aqueous environments, and metal-free protocols.[10] We reasoned that photo-PISA could be used as a powerful tool for the large-scale synthesis of polymer nanoparticles that exhibit multiple peptides in the hydrophilic shell (Figure 1). Herein, we present a one-pot photo-PISA approach for the preparation of nanoparticles that carry apoptotic peptides with tunable size (36–105 nm in diameter) and loading of peptides ranging from 20 to 48 wt.%. High concentrations of peptide brush polymer nanoparticles of up to 150 mg/mL were achieved because of the nature of PISA. Importantly, both proteolytical resistance and bioactivity, including cell penetration and apoptotic efficiency, were significantly higher for the peptide brush polymer nanoparticles compared to their linear peptide analogue.
Figure 1.
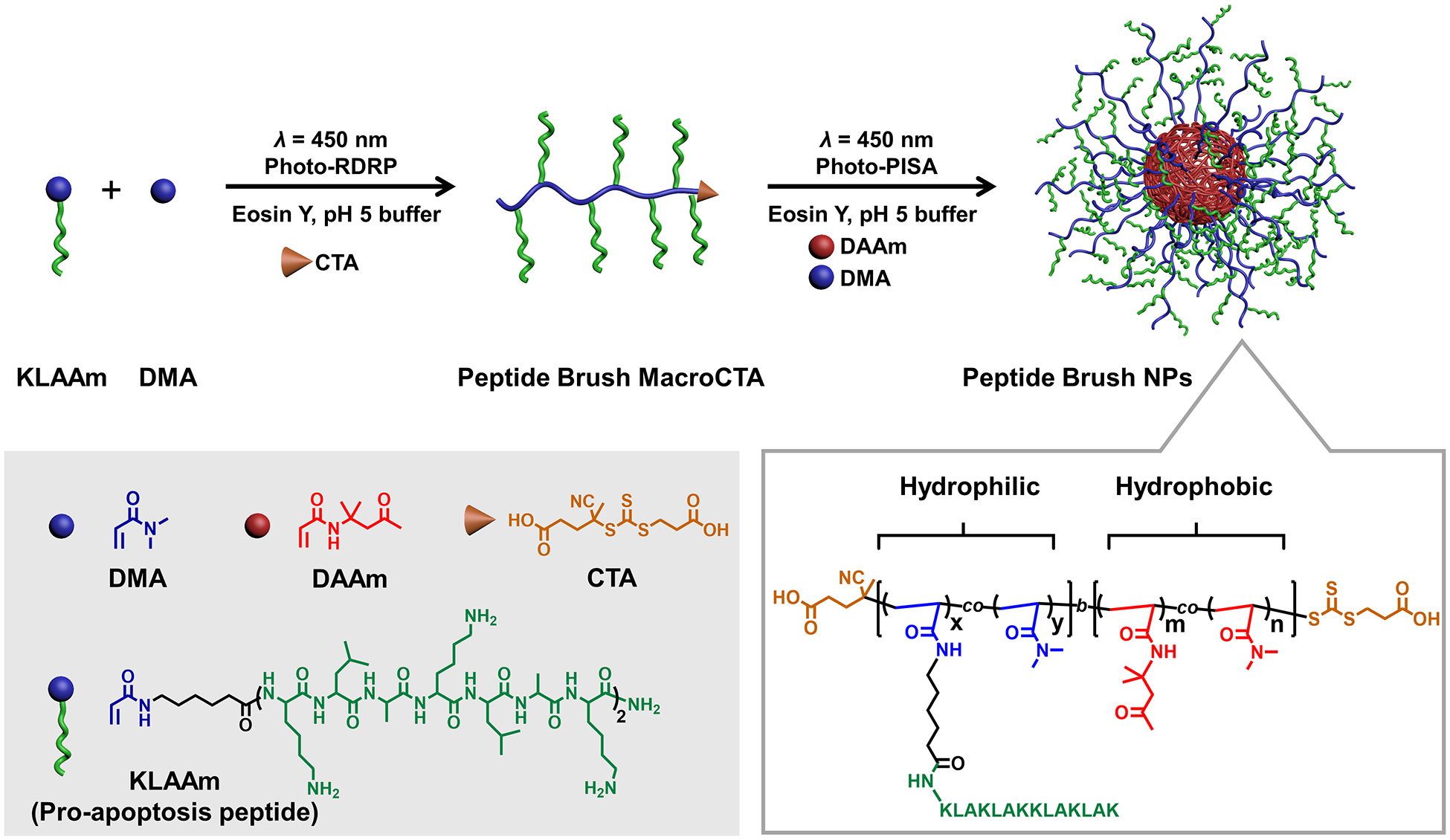
Schematic illustration of the one-pot photo-PISA approach to proapoptotic peptide brush polymer nanoparticles.
Results and Discussion
With the goal of preparing peptide brush polymer nanoparticles, we began our exploration by designing a peptide acrylamide monomer that features the amino acid sequence of KLAKLAKKLAKLAK (i.e., KLA peptide acrylamide). This peptide sequence is well known for inducing rapid apoptosis of cancer cells via disruption of the mitochondrial membrane.[11] The monomer was prepared via the Fmoc solid-phase peptide synthesis procedure.[7c] High performance liquid chromatography (HPLC), NMR spectroscopy, and electrospray ionization mass spectrometry (ESI-MS) verified the identity and purity of the monomer (Figures S1–S3).
Photoinduced reversible-deactivation radical polymerization (Photo-RDRP) [10a,12] was used to prepare the macromolecular chain transfer agents (macroCTAs) (Figure 1). To suppress the nucleophilicity of primary amines that could cause aminolysis of the CTA, an acidic buffer (pH 5.0) was used as the solvent for polymerization. Notably, copolymerization of KLA peptide acrylamide monomer (KLAAm) and a comonomer N, N-dimethylacrylamide (DMA) was conducted in the presence of a biocompatible organic photocatalyst (i.e., eosin Y) at room temperature under visible light irradiation (λ = 450 nm). The feed ratio of KLAAm and DMA was varied to tune the loading and graft density of peptides along the hydrophilic polymer chain. The monomers were fully consumed after photo-polymerization for 12 h (Figures S4 and S5). Since the photo-polymerization was conducted at room temperature, we postulated that side reactions such as hydrolysis of terminal trithiocarbonate would be minimized, thus leading to high end-group fidelity.[13] Indeed, gel permeation chromatography (GPC) analysis with integrated UV detection confirmed our hypothesis by revealing that the GPC trace of the macroCTA exhibited the characteristic trithiocarbonate absorption at 315 nm (Figure S6).
Next, we aimed to perform photo-PISA by chain extension of the macroCTA with a combination of diacetone acrylamide (DAAm) and DMA, which have been shown as readily tunable core-forming monomers in PISA processes (Figure 2). [9f,14] The macroCTA contains primary amines which could potentially form imines with the ketone-containing DAAm. In view of this, we conducted an experiment that involved incubation of KLAAm and DAAm in acidic buffer (pH 5.0). As indicated by HPLC and ESI-MS (Figure S7), no imine products were detected even after 24 h. Therefore, we considered any undesired imine formation during the photo-PISA process would be negligible.
Figure 2.
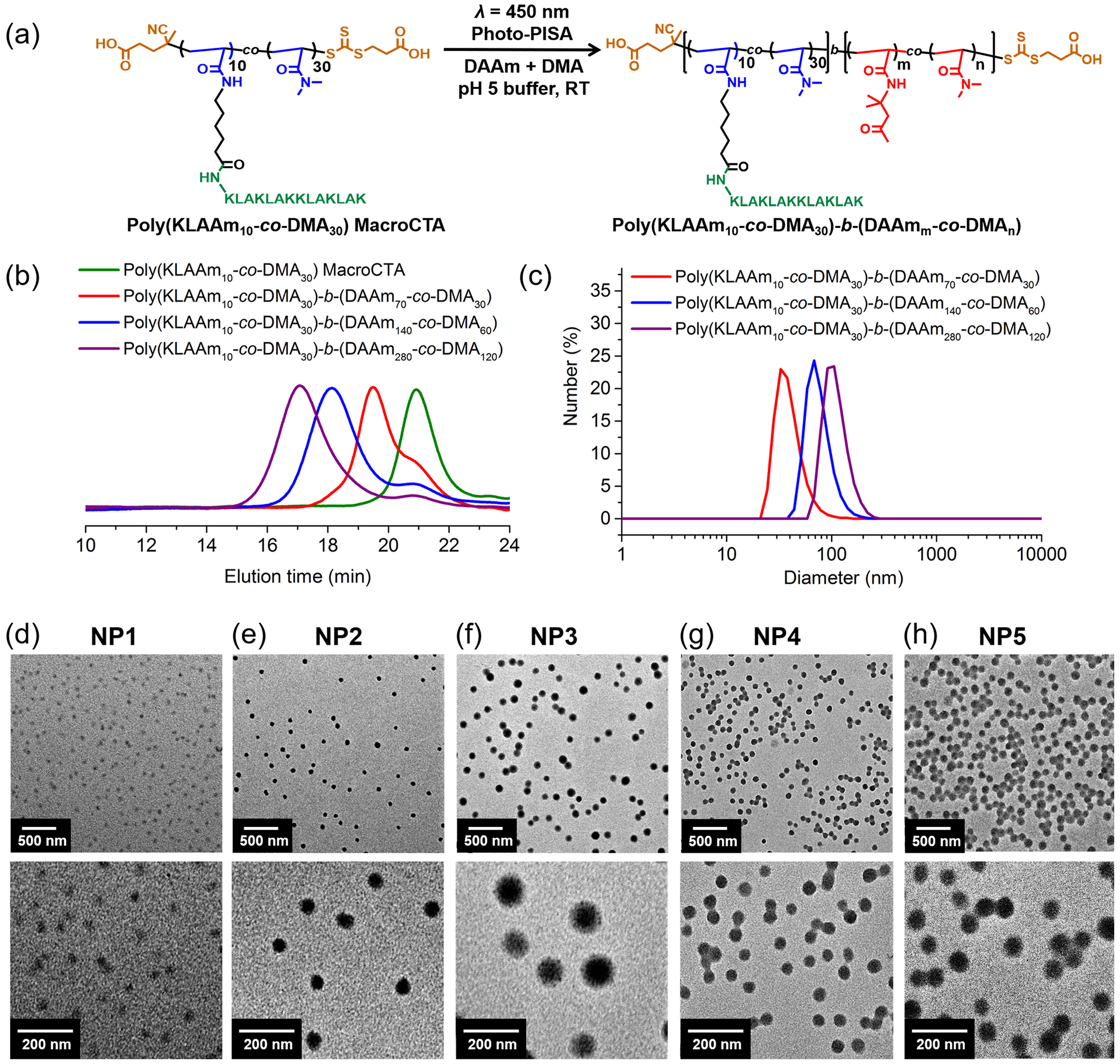
Synthesis and characterization of KLA peptide brush polymer nanoparticles. (a) Synthesis of peptide brush polymer nanoparticles by photo-PISA; (b) GPC analysis of peptide brush polymer macroCTA and resulting amphiphilic block copolymers (NP1-NP3); (c) DLS traces of peptide brush polymer nanoparticles (NP1-NP3); (d-h) TEM images of peptide brush polymer nanoparticles (NP1-NP5) with low and high magnifications.
Photo-PISA was performed under the identical condition used in the macroCTA synthesis (i.e., eosin Y and acidic buffer). In light of this, the one-pot synthesis of nanoparticles was achieved without isolating the macroCTA from the buffer solution in which it was synthesized. The efficiency of photo-PISA was revealed via NMR spectroscopy analysis that indicated quantitative conversion of monomers after 12 h (Figure S8). Moreover, GPC traces of block copolymers have clearly shifted to higher molecular weight regions with narrow MW distribution, indicative of successful chain extensions (Table 1, Table S1, Figure 2a–b, and Figures S9 and S10). We note that the GPC signal of residual macroCTA remained in all block copolymers (Figure 2b). However, the extent of residual macroCTA was significantly decreased as the targeted degree of polymerization (DP) increased. The modularity of peptide brush polymer nanoparticles was examined by tuning variables including compositions in hydrophilic macroCTA and hydrophobic polymer core. Hence, five peptide brush polymer nanoparticles with different sizes and loadings of peptide were achieved (Table 1).
Table 1.
Peptide brush polymer nanoparticles via photo-PISA at solids content of 15 wt.%.
| Entry | Shell-forming | Core-forming | Peptide Loadinga (wt.%) | Mn,theob (g/mol) | Mn,MALSc (g/mol) | Ðc | Dhd (nm) | ||
|---|---|---|---|---|---|---|---|---|---|
| KLAAm | DMA | DAAm | DMA | ||||||
| NP1 | 10 | 30 | 70 | 30 | 48 | 34 980 | 41 700 | 1.29 | 36 |
| NP2 | 10 | 30 | 140 | 60 | 33 | 51 160 | 57 300 | 1.24 | 62 |
| NP3 | 10 | 30 | 280 | 120 | 21 | 80 640 | 98 600 | 1.17 | 105 |
| NP4 | 10 | 10 | 140 | 60 | 38 | 49 180 | 54 900 | 1.03 | 64 |
| NP5 | 10 | 10 | 280 | 120 | 24 | 78 660 | 92 150 | 1.15 | 92 |
Peptide loading was calculated by feed ratios.
Mn,theo = ∑DP of monomers ⨯ MW of monomers + MW of CTA.
Number-average molecular weights of polymers were determined by GPC-MALS.
Hydrodynamic diameters of NPs were determined by DLS.
The hydrodynamic diameters of peptide brush polymer nanoparticles were determined by dynamic light scattering (DLS), which suggested a trend of increasing size as the length of the hydrophobic chain increased (Figure 2c and Figure S11). Transmission electron microscopy (TEM) further revealed the shape and dry-state size of peptide brush polymer nanoparticles (Figure 2d–h). According to the TEM micrographs, all the peptide brush polymer nanoparticles exhibit spherical morphologies and uniform size distributions. In addition, the nanoparticle size increased as the degree of polymerization of the hydrophobic core increased. This is in a good agreement with DLS and cryogenic transmission electron microscopy analysis (Figure S12). Notably, only spherical morphology of nanoparticles was observed even at high DPs of core-forming monomers. This can be attributed to the high surface curvature which stems from the positively charged peptide brush shell.[15] The net charge of the nanoparticles was further assessed (Figure S13). Zeta potentials of those nanoparticles were positive, ranging from 31 mV to 65 mV because KLA peptide brush polymer nanoparticles have an abundant number of free amines. Furthermore, the secondary structures of KLA peptide and nanoparticles were evaluated by circular dichroism spectroscopy, which exhibited consistent patterns with a mixture of α‐helix and random coil conformations (Figure S14).
Despite the promise of proapoptotic KLA peptide as anti-cancer therapeutics, the anti-cancer efficacy of free KLA peptide is significantly impeded by its low proteolytic stability as well as poor cell uptake efficiency.[16] Since peptide brush polymer nanoparticles possess a high-density display of KLA peptides on the nanoparticle surface, we reasoned that the stability of the peptides would potentially be enhanced due to steric hindrance limiting access of the peptides to the active sites of proteases.[7c,17] In view of this, we examined the proteolytic resistance of KLA-containing materials of three kinds; 1) peptide, 2) peptide brush polymer, and 3) peptide brush polymer nanoparticles. For this test, trypsin was used as a potent proteinase typically found in the digestive system and freely capable of cleaving the KLA peptide (Figure 3).[18] The concentration of trypsin was set to 0.1 μM, notably much higher than the level of trypsin in serum.[19] According to HPLC analysis, the KLA peptide underwent fast degradation, reaching 100% cleavage within 1 h (Figure S15). Similarly, rapid degradation was observed for poly(KLAAm10-co-DMA10), for which more than 90% of the side-chain peptides were cleaved within 1 h. On the other hand, in the case of KLA brush polymer nanoparticles, more than 70% of KLA peptide survived during the first hour of cleavage (Figure 3, and Figures S16 and S17). This result confirmed that a high-density array of peptides on nanoparticle surface can endow the dangling peptides with enhanced proteolytic stability.
Figure 3.
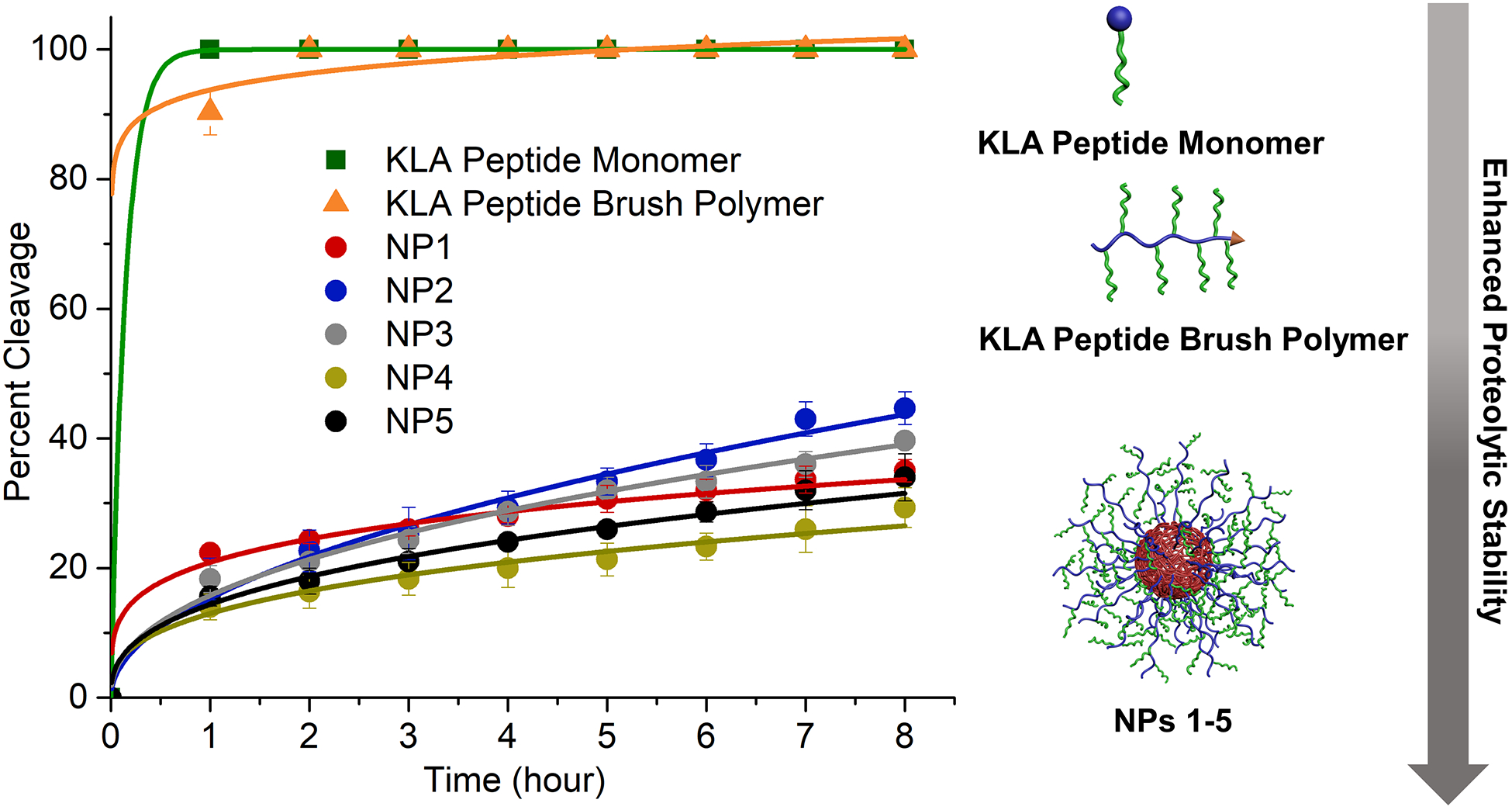
Proteolytic cleavage of KLA peptide monomer, KLA brush polymer (poly(KLAAm10-co-DMA10)), and KLA peptide brush polymer nanoparticles (NPs 1–5) in the presence of trypsin (0.1μM) at 37 °C. All the peptide containing materials had a concentration of 200 μM with respect to peptide in PBS buffer (pH = 7.4). Data displayed as mean ± standard deviation of three independent experiments.
The cytotoxicity of KLA peptide brush nanoparticles and KLA peptide was examined in vitro with human cervical cancer (HeLa) cells (Figure 4). Two nanoparticles including NP3 and NP5 were chosen to compare in the cell studies because of their similar sizes but different grafting density of peptides on the hydrophilic chain. According to the cell viability assay, NP3 and NP5 demonstrated dose-dependent cytotoxicity in HeLa cells, whereas no toxicity was observed for free KLA peptide even at a high concentration of 200 μM. To unequivocally credit the toxicity of KLA brush nanoparticles to the proapoptotic peptides on the nanoparticles, the cytotoxicity of a spherical polymer nanoparticle without carrying the peptides (i.e., polyDMA40-b-poly(DAAm70-co-DMA30)) was further evaluated (Figures S18–S20). Cell viability assay revealed a high viability (> 90%) of HeLa cells in the presence of peptide-free polymer micelles under the investigated concentrations, confirming the cytocompatibility of the polymer nanocarrier.
Figure 4.
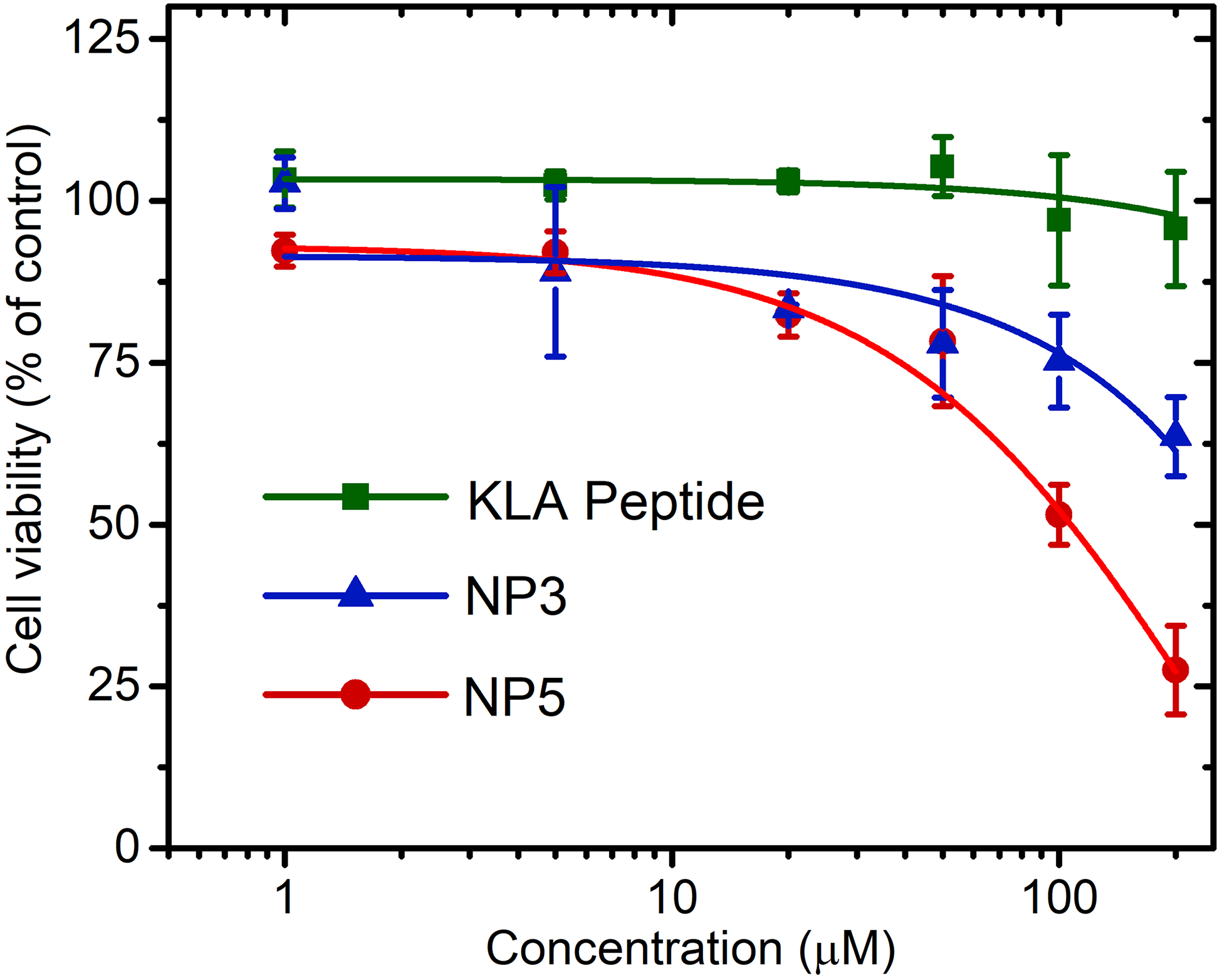
Cytotoxicity of free KLA peptide, poly[(KLAAm10-co-DMA30)-b-(DAAm280-co-DMA120)] (NP3), and poly[(KLAAm10-co-DMA10)-b-(DAAm280-co-DMA120)] (NP5) using a CellTiter-Blue cell viability assay. Concentrations were calculated with respect to the total KLA peptide content. HeLa cells were treated with peptide-containing materials and incubated for 72 h at 37 °C. Data displayed as mean ± standard deviation of three independent experiments.
Notably, the toxicity of NP5 was significantly higher than NP3, potentially the result of the higher graft density of the KLA peptide in the hydrophilic shell on NP5 leading to enhanced multivalent interactions. In addition, the cell uptake efficiency of rhodamine B-labeled KLA peptide and NPs was investigated (Figures S21–S23). Flow cytometry and confocal laser scanning microscopy clearly demonstrated the significantly enhanced cell uptake of the NPs over free KLA peptide.
Finally, to discern the mechanism of cell death, we studied the mitochondrial membrane potential using the turn-on JC-1 probe. Mitochondria are central regulators of cellular energy and metabolism, and have the essential function of ATP synthesis by maintaining a membrane potential gradient.[20] The JC-1 probe is green-fluorescent carbocyanine that forms red-shifted J-aggregates upon accumulation in mitochondria and has very narrow red fluorescence.[5b,7b] Therefore, confocal laser scanning microscopy was utilized to compare the green and red fluorescence at the same excitation wavelength at 488 nm (Figure 5). HeLa cells incubated with free KLA peptide showed strong red fluorescence, similar to cells treated only with media, indicative of healthy mitochondria. As a comparison, almost no red emission from JC-1 J-aggregates was observed in cells treated with KLA brush polymer nanoparticles (NP5) even after 30 min incubation, confirming efficient depolarization of the mitochondria. The behavior of NP5 was similar to the commercial mitochondrial membrane potential disruptor, carbonyl cyanide 3-chlorophenylhydrazone (CCCP).
Figure 5.
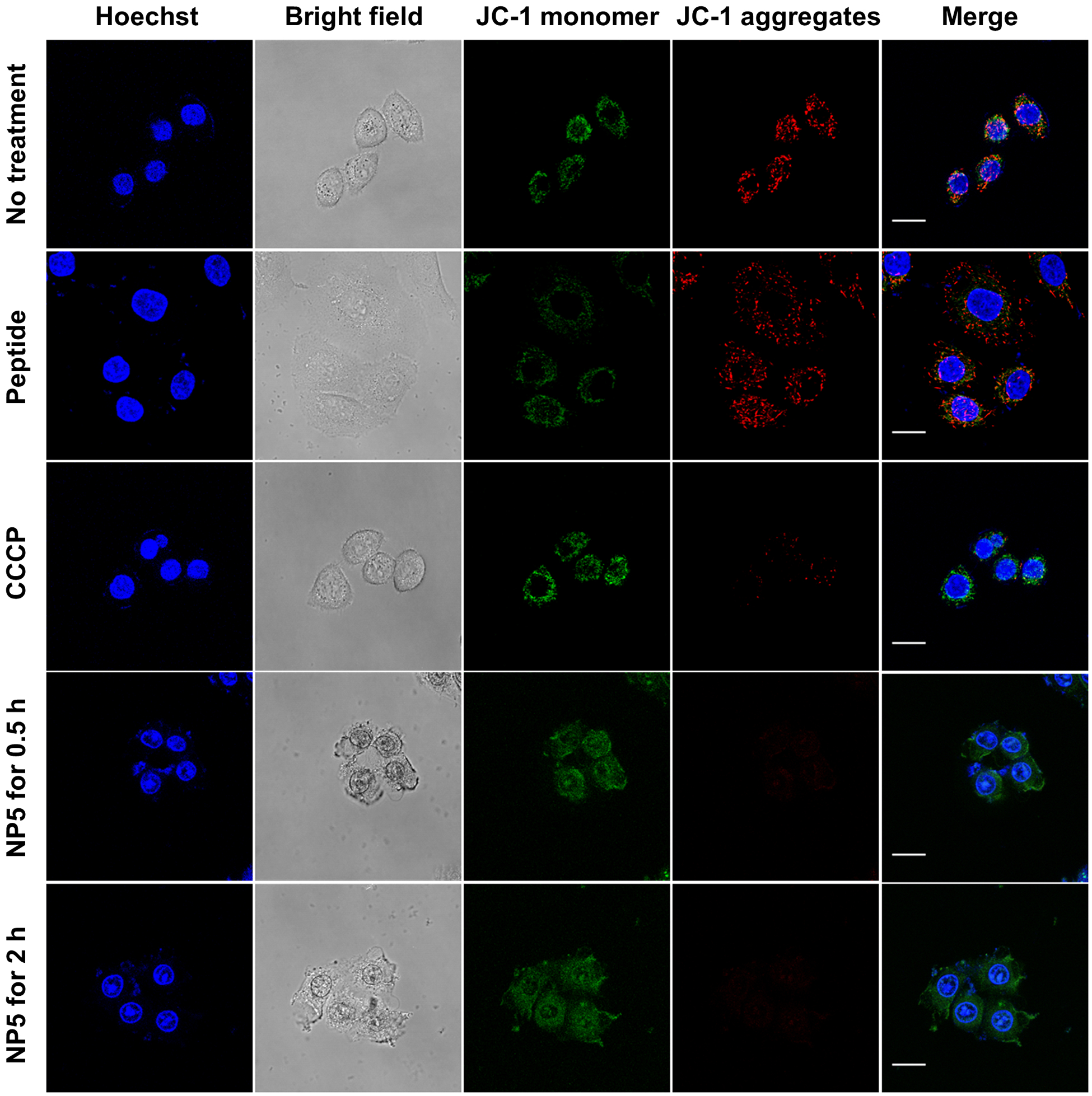
Assessment of mitochondrial dysfunction induced by the peptide-containing materials using JC-1 probe. Live-cell confocal microscopy images of HeLa cells incubated with KLA peptide, CCCP, NP5 for desired periods of time. Prior to imaging, cells were stained with 2 μM of JC-probe (green, monomer, λex/em = 488 nm/510–550 nm; red, J-aggregates, λex/em = 488 nm/585–649 nm) and then Hoechst 33342 (blue, λex/em = 405 nm/ 420–480 nm). Scale bars, 20 μm.
Conclusion
In summary, we developed a scalable and highly modular photo-PISA approach to functional peptides displayed as hydrophilic brushes on polymeric amphiphiles packed to form micellar nanoparticles. This is a robust approach to access nanoparticles with a high-density display of peptides, tunable particle size, tunable peptide loading, and at scale (150 mg/mL). This method for packaging peptides was demonstrated with a proof-of-concept proapoptotic peptide. These results clearly demonstrate the promise of exploiting NPs with high peptide grafting densities to achieve enhanced proteolytic stability, cellular internalization, and cytotoxicity in comparison with free apoptotic peptides. We envision that many other functional peptides such as cell-penetrating and therapeutic peptides would be compatible with the photo-PISA approach to polymer brush amphiphile self-assemblies.
Supplementary Material
Acknowledgements
The authors are grateful for the support of the NIH through the NHLBI (R01HL139001), National Science Foundation fund grants (DMR‐1710105 and DMR-1904631), and an AFOSR MURI (FA9550‐16‐1‐0150). Additional support was provided by the Center for Regenerative Nanomedicine in the Simpson Querrey Institute at Northwestern University. TDC acknowledges the support from an American Australian Association Dow Chemical Company Fellowship.
References
- [1].Yu B, Hwang D, Jeon H, Kim H, Lee Y, Keum H, Kim J, Lee DY, Kim Y, Chung J, Jon S, Angew. Chem. Int. Ed 2019, 58, 2005–2010. [DOI] [PubMed] [Google Scholar]
- [2].Drucker DJ, Nat. Rev. Drug Discov 2020, 19, 277–289. [DOI] [PubMed] [Google Scholar]
- [3].a) Navaratna T, Atangcho L, Mahajan M, Subramanian V, Case M, Min A, Tresnak D, Thurber GM, J. Am. Chem. Soc 2020, 142, 1882–1894; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kasper MA, Glanz M, Oder A, Schmieder P, von Kries JP, Hackenberger CPR, Chem Sci 2019, 10, 6322–6329; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Menacho-Melgar R, Decker JS, Hennigan JN, Lynch MD, J. Control Release 2019, 295, 1–12; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Roberts MJ, Bentley MD, Harris JM, Adv. Drug Deliv. Rev 2012, 64, 116–127; [DOI] [PubMed] [Google Scholar]; e) Webber MJ, Appel EA, Vinciguerra B, Cortinas AB, Thapa LS, Jhunjhunwala S, Isaacs L, Langer R, Anderson DG, Proc. Natl. Acad. Sci. USA 2016, 113, 14189–14194; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Gentilucci L, De Marco R, Cerisoli L, Curr. Pharm. Design 2010, 16, 3185–3203; [DOI] [PubMed] [Google Scholar]; g) Chatterjee J, Gilon C, Hoffman A, Kessler H, Accounts Chem. Res 2008, 41, 1331–1342; [DOI] [PubMed] [Google Scholar]; h) Itoh H, Miura K, Kamiya K, Yamashita T, Inoue M, Angew. Chem. Int. Ed 2020, 59, 4564–4571; [DOI] [PubMed] [Google Scholar]; i) Wu YT, Villa F, Maman J, Lau YH, Dobnikar L, Simon AC, Labib K, Spring DR, Pellegrini L, Angew. Chem. Int. Ed 2017, 56, 12866–12872. [DOI] [PubMed] [Google Scholar]
- [4].Guidotti G, Brambilla L, Rossi D, Trends Pharmacol. Sci 2017, 38, 406–424. [DOI] [PubMed] [Google Scholar]
- [5].a) Pokorski J, Church DC, Angew. Chem. Int. Ed 2020; [Google Scholar]; b) Blum AP, Kammeyer JK, Gianneschi NC, Chem. Sci 2016, 7, 989–994; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Blum AP, Kammeyer JK, Yin J, Crystal DT, Rush AM, Gilson MK, Gianneschi NC, J. Am. Chem. Soc 2014, 136, 15422–15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Suma T, Cui JW, Mullner M, Fu SW, Tran J, Noi KF, Ju Y, Caruso F, J. Am. Chem. Soc 2017, 139, 4009–4018; [DOI] [PubMed] [Google Scholar]; b) Spicer CD, Jumeaux C, Gupta B, Stevens MM, Chem. Soc. Rev 2018, 47, 3574–3620; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chee HL, Gan CRR, Ng M, Low L, Fernig DG, Bhakoo KK, Paramelle D, ACS Nano 2018, 12, 6480–649. [DOI] [PubMed] [Google Scholar]
- [7].a) Adamiak L, Touve MA, LeGuyader CLM, Gianneschi NC, ACS Nano 2017, 11, 9877–9888; [DOI] [PubMed] [Google Scholar]; b) Cheng DB, Zhang XH, Gao YJ, Ji L, Hou DY, Wang ZQ, Xu WH, Qiao ZY, Wang H, J. Am. Chem. Soc 2019, 141, 7235–7239; [DOI] [PubMed] [Google Scholar]; c) Callmann CE, Thompson MP, Gianneschi NC, Accounts Chem. Res 2020, 53, 400–413; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hua S, de Matos MBC, Metselaar JM, Storm G, Front Pharmacol. 2018, 9, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sun H, Choi W, Zang N, Battistella C, Thompson MP, Cao W, Zhou X, Forman C, Gianneschi NC, Angew. Chem. Int. Ed 2019, 58, 17359–17364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Grazon C, Salas-Ambrosio P, Ibarboure E, Buol A, Garanger E, Grinstaff MW, Lecommandoux S, Bonduelle C, Angew. Chem. Int. Ed 2020, 59, 622–626; [DOI] [PubMed] [Google Scholar]; b) Foster JC, Varlas S, Blackman LD, Arkinstall LA, O’Reilly RK, Angew. Chem. Int. Ed 2018, 57, 10672–10676; [DOI] [PubMed] [Google Scholar]; c) Penfold NJW, Yeow J, Boyer C, Armes SP, ACS Macro Lett 2019, 8, 1029–1054; [DOI] [PubMed] [Google Scholar]; d) Canning SL, Smith GN, Armes SP, Macromolecules 2016, 49, 1985–2001; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Oliver AM, Gwyther J, Boott CE, Davis S, Pearce S, Manners I, J. Am. Chem. Soc 2018, 140, 18104–18114; [DOI] [PubMed] [Google Scholar]; f) Figg CA, Carmean RN, Bentz KC, Mukherjee S, Savin DA, Sumerlin BS, Macromolecules 2017, 50, 935–943; [Google Scholar]; g) Wright DB, Touve MA, Thompson MP, Gianneschi NC, ACS Macro Lett 2018, 7, 401–405; [DOI] [PubMed] [Google Scholar]; h) Touve MA, Figg CA, Wright DB, Park C, Cantlon J, Sumerlin BS, Gianneschi NC, ACS Central Sci. 2018, 4, 543–547; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Figg CA, Simula A, Gebre KA, Tucker BS, Haddleton DM, Sumerlin BS, Chem. Sci 2015, 6, 1230–1236; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Le D, Dilger M, Pertici V, Diabate S, Gigmes D, Weiss C, Delaittre G, Angew. Chem. In. Ed 2019, 58, 4725–4731. [DOI] [PubMed] [Google Scholar]
- [10].a) Yeow J, Boyer C, Adv. Sci 2017, 4, 1700137; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blackman LD, Varlas S, Arno MC, Houston ZH, Fletcher NL, Thurecht KJ, Hasan M, Gibson MI, O’Reilly RK, ACS Central Sci. 2018, 4, 718–723; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tan JB, Sun H, Yu MG, Sumerlin BS, Zhang L, ACS Macro Lett 2015, 4, 1249–1253; [DOI] [PubMed] [Google Scholar]; d) Corrigan N, Yeow J, Judzewitsch P, Xu JT, Boyer C, Angew. Chem. Int. Ed 2019, 58, 5170–5189. [DOI] [PubMed] [Google Scholar]
- [11].Cieslewicz M, Tang JJ, Yu JL, Cao H, Zavaljevski M, Motoyama K, Lieber A, Raines EW, Pun SH, Proc. Natl. Acad. Sci. USA 2013, 110, 15919–15924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Xu JT, Jung K, Atme A, Shanmugam S, Boyer C, J. Am. Chem. Soc 2014, 136, 5508–5519.; [DOI] [PubMed] [Google Scholar]; b) Chen M, Zhong MJ, Johnson JA, Chem. Rev 2016, 116, 10167–10211. [DOI] [PubMed] [Google Scholar]
- [13].a) Thomas DB, Sumerlin BS, Lowe AB, McCormick CL, Macromolecules 2003, 36, 1436–1439; [Google Scholar]; b) Thomas DB, Convertine AJ, Hester RD, Lowe AB, McCormick CL, Macromolecules 2004, 37, 1735–1741. [Google Scholar]
- [14].a) Zhou W, Qu QW, Xu YY, An ZS, ACS Macro Lett 2015, 4, 495–499; [DOI] [PubMed] [Google Scholar]; b) Luckerath T, Koynov K, Loescher S, Whitfield CJ, Nuhn L, Walther A, Barner-Kowollik C, Ng DYW, Weil T, Angew. Chem. Int. Ed 2020, doi: 10.1002/anie.201916177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chien MP, Rush AM, Thompson MP, Gianneschi NC, Angew. Chem. Int. Ed 2010, 49, 5076–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Lee J, Oh ET, Lee H, Kim J, Kim HG, Park HJ, Kim C, Bioconjug. Chem 2020, 31, 43–50; [DOI] [PubMed] [Google Scholar]; b) Toft DJ, Moyer TJ, Standley SM, Ruff Y, Ugolkov A, Stupp SI, Cryns VL, ACS Nano 2012, 6, 7956–7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Jia F, Lu XG, Tan XY, Wang DL, Cao XY, Zhang K, Angew. Chem. Int. Ed 2017, 56, 1239–1243; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jia F, Lu XG, Wang DL, Cao XY, Tan XY, Lu H, Zhang K, J. Am. Chem. Soc 2017, 139, 10605–10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ji BA, Logsdon CD, Gastroenterology 2011, 141, 1972–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Artigas JMG, Faure MRA, Garcia ME, Gimeno AMB, Postgrad. Med. J 1981, 57, 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Giacomello M, Pyakurel A, Glytsou C, Scorrano L, Nat. Rev. Mol. Cell Bio 2020, 21, 202–224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.