

Abstract
Tremendous progress has been made in understanding the genetics of pulmonary arterial hypertension (PAH) since its description in the 1950s as a primary disorder of the pulmonary vasculature. Heterozygous germline mutations in the gene coding bone morphogenetic receptor type 2 (BMPR2) are detectable in the majority of cases of heritable PAH, and in approximately 20% of cases of idiopathic pulmonary arterial hypertension (IPAH). However, recent advances in gene discovery methods have facilitated the discovery of additional genes with mutations among those with and without familial PAH. Heritable PAH is an autosomal dominant disease characterized by reduced penetrance, variable expressivity, and female predominance. Biallelic germline mutations in the gene EIF2AK4 are now associated with pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Growing genetic knowledge enhances our capacity to pursue and provide genetic counseling, although the issue remains complex given that the majority of carriers of PAH-related mutations will never be diagnosed with the disease.
Keywords: bone morphogenetic protein receptor type 2, caveolin-1, KCNK3, CBLN2, TBX4, EIF2AK4, pulmonary arterial hypertension, genetic modifiers, hereditary hemorrhagic telangiectasia
Pulmonary arterial hypertension (PAH) was described by Dresdale and colleagues in 1951 as a primary disease of the lungs, termed primary pulmonary hypertension (PPH).1 Shortly thereafter, they published evidence of familial transmission of PPH in a kindred, providing the first known reports of PPH as a heritable condition.2 It is now recognized that PAH comes in many forms, including but not limited to idiopathic PAH, heritable PAH (HPAH), PAH associated with connective tissue disease, and PAH associated with congenital heart disease.3 Subjects with known familial disease, or those with a detectable rare variant (mutation) in a gene known to associate with PAH, have HPAH. However, there is a growing recognition that genetic and genomic variations contribute to multiple forms of PAH, and perhaps pulmonary hypertension more broadly. To date, the major breakthroughs in genetics have occurred for HPAH and IPAH.
In 1984, our group published a study examining the 14 known large families with HPAH in North America, including a family with six deaths due to PAH and a pedigree suggestive of autosomal dominant transmission.4 Careful examination of those families suggested that some patients in the family possessed the risk allele but no evidence of PAH due to a phenomenon known as reduced penetrance—possession of the genetic risk variant of interest does not necessarily result in expression of the relevant disease. This reduced penetrance results in a population of healthy individuals who may transmit the autosomal dominant risk allele to their children, thereby perpetuating the PAH risk through the pedigree. Reduced penetrance also likely explains the discovery that a proportion of cases of PAH which appear to occur as “sporadic” disease actually have a detectable familial basis and/or genetic susceptibility. In fact, the current international classification scheme, which incorporates the label HPAH, does so at least in part to recognize the fact that a large percentage (current estimates remain ~20%) of cases previously thought to be idiopathic (IPAH) harbor deleterious causative mutations and therefore pose a hereditary risk to other family members.5 Due to the significantly higher prevalence of idiopathic PAH compared with PAH due to known familial susceptibility (familial PAH), most PAH patients with a mutation in a PH-specific gene are actually without a known family history. This has significant implications in terms of PAH risk for family members of IPAH cases, as a small but significant percentage of these family members will be at risk of PAH themselves.
BMPR2 Remains the Major Gene Known to be Associated with PAH
In the 1990s, investigators in North America and Europe intensified efforts to determine the gene, or genes, primarily responsible for PAH in families. As noted previously, familial PAH was known to be an autosomal dominant disease, suggesting that it may be caused by a single gene allele mutation shared by family members within a PAH pedigree. Work over the previous 15 to 20 years provided the foundation for family-based studies, including the National Institutes of Health (NIH) prospective registry of the 1980s and work to collect families afflicted by PAH.6 Ultimately, germline mutations in the gene encoding bone morphogenetic protein receptor type-2 (BMPR2), a transforming growth factor-β (TGF-β) superfamily of receptors member, were determined to be responsible for the majority of cases of the autosomal dominant familial disease now known as HPAH.7-10 Today, BMPR2 remains the major gene associated with HPAH, with heterozygous germline mutations accounting for approximately 75% of familial cases.11,12 In addition, BMPR2 mutations are detectable in approximately 20% of patients with IPAH—a finding initially reported by Trembath and colleagues following the discovery of BMPR2 mutations in families.9
Some of the families with HPAH not due to BMPR2 have mutations in genes closely related to the signaling pathways activated by BMPR2 receptor activation. This supports the concept that TGF-β superfamily signaling is relevant to PAH pathogenesis more broadly. For example, a small proportion of familial PAH cases not due to BMPR2 mutations are explained by mutations in the genes associated with hereditary hemorrhagic telangiectasia (HHT)—activin-like kinase type 1 (ALK1) and Endoglin (ENG).13 However, HHT-associated PAH accounts for only a minority of the remaining familial PAH cases not due to BMPR2 mutations.14
Additional Genes Believed to Cause PAH when Pathogenic Mutations Are Present
The discovery of BMPR2, ALK1, and ENG as genes associated with PAH penetrance in families stood alone for many years as the only genes likely to “cause” PAH when mutated. This is somewhat surprising because the TGF-β superfamily of receptors involves multiple genes which, when perturbed potentially, could disrupt signaling in a manner similar to BMPR2 mutations. For example, TGF-β receptors signal in several ways, including via the canonical pathway of SMAD family signaling. Despite a suspicion that multiple SMAD gene mutations contribute to PAH, to date only a few reports of mutations in the SMAD genes have been published in PAH. For example, mutations in SMAD9, which is located on chromosome 13, have been described in PAH by a few reports.15-17 The SMAD9 gene, which encodes a Smad8 protein, is a downstream mediator of canonical BMPR2 activation. SMAD4 mutations have also been described, infrequently, among PAH cases (►Table 1).17
Table 1.
Rare variants (mutations) associated with families and IPAH patients
| Gene name | Specialized information |
|---|---|
| BMPR2 | TGF-β superfamily member ≥80% of familial cases ~20% of IPAH cases |
| ALK1 | TGF-β superfamily member Hereditary hemorrhagic telangiectasia families |
| Endoglin | TGF-β superfamily member Hereditary hemorrhagic telangiectasia families |
| SMAD9 | TGF-β superfamily member |
| CAV1 | Accurate estimate of frequency among PAH patients yet to be determined |
| KCNK3 | Accurate estimate of frequency among PAH patients yet to be determined |
| TBX4 | Accurate estimate of frequency among PAH patients yet to be determined. Prevalence may be higher among pediatric PAH cases. Mutations also associated with small patella syndrome |
| EIF2AK4 | Biallelic mutations in this gene cause PVOD and PCH |
Abbreviations: PAH, pulmonary arterial hypertension; PCH, pulmonary capillary hemangiomatosis; PVOD, pulmonary veno-occlusive disease.
Notes: All PAH patients with detectable germline mutations in these genes have heritable PAH (HPAH) by definition. EIF2AK4 is added to this list—it is associated with PVOD and PCH cases.
Over the past 8 years, with the advent of next-generation sequencing, our understanding of the genes involved in PAH pathogenesis has gradually expanded. Whole exome (and now whole genome) sequencing provides a powerful tool to search rare genetic variants (mutations) of strong effect when studying a well-defined phenotype.18 While these studies in large numbers of idiopathic PAH patients are currently underway at multiple PH centers, the majority of progress to date has been made by studying well characterized families with PAH but no detectable mutation in a PH-specific gene (e.g., BMPR2). The discoveries to date have been highly informative from a pathogenesis perspective, although the number of cases reported has been relatively small.
CAV1 gene mutations.
Mutations in the caveolin-1 (CAV1) gene were the initial discovery in the current next-generation sequencing era via the use of whole exome sequencing. Mutations were found in a large PAH family, and one unrelated IPAH case, without a previously detected mutation in PH-specific genes.19 As with BMPR2, CAV1 mutations are associated with PAH with reduced penetrance, although CAV1 is not a member of the TGF-β superfamily. CAV1 encodes caveolin-1, a membrane protein required to form flask-shaped invaginations of the plasma membrane known as caveolae that function in membrane trafficking, cell signaling, cholesterol homeostasis, and other crucial cellular processes. Interestingly, TGF-β receptors are enriched in caveolae compared with other areas of the plasma membrane.20 Caveolae are abundant in many cells, including fibrocytes, adipocytes, and endothelial cells.21 Prior to the familial PAH CAV1 mutation discovery, investigators had implicated CAV1 in PAH—mice with caveolin-1 defects have airway and pulmonary vascular abnormalities, while rescue expression of caveolin-1 in endothelial cells of caveolin-1 knock out mice reverses several of these defects.22-24 In addition, human studies support the role of CAV1 in PAH, with reduced caveolin-1 expression found to occur in the lungs of PAH patients regardless of mutation, and reduced systemic caveolin-1 levels.25-27 A recent study found a CAV1 mutation in a child also with lipodystrophy, connecting these two metabolic disorders.28 However, the manner by which CAV1 mutations promote PAH pathogenesis in humans remains incompletely elucidated.
KCNK3 gene mutations.
Also using whole exome sequencing, mutations in the gene KCNK3 (Potassium Channel, Subfamily K, Member 3), which encodes the human TASK-1 protein, were initially discovered in three unrelated PAH families and three IPAH patients.29 While studies of KCNK3 continue, it appears that the frequency of KCNK3 mutations among those without detectable mutations in BMPR2, ALK1, ENG, and CAV1 is higher among PAH families than data presented for CAV1 (3.26% [3 out of 92] of PAH families and 1.32% [3 out of 228] of IPAH cases had detectable KCNK3 mutations), but much lower than BMPR2.30-32
The biologic plausibility of the relevance of KCNK3 mutations to PAH is high. KCNK3 encodes the TASK-1 protein, which is a pH-sensitive potassium channel.33 Ion channels play an important role in regulating vascular tone and likely exist in balance with the roles of calcium channels to contribute to vasoconstriction and vascular remodeling in the pulmonary vasculature. Because pharmacological manipulation of currents through TASK-1 channels is possible, the discovery of KCNK3 mutations in PAH patients does suggest that an additional therapeutic approach may one day be possible.
The KCNK3 association has been confirmed by multiple studies in individuals of multiple different ancestry types, although the precise manner by which mutations contribute to PAH pathogenesis remains unknown.11,30-33
TBX4 gene mutations.
While present in children, BMPR2, ALK1, ENG, CAV1, and KCNK3 mutations have all been studied more often in the context of adult PAH. In contrast, mutations in the gene TBX4 have been more closely studied in children than in adults; early work suggests that the prevalence is higher among pediatric than among adult PAH patients.31 As with CAV1, which may cause lipodystrophy, TBX4 mutations highlight the concept that PAH patients may also display manifestations in other organs depending upon the mutation type. TBX4 is a member of the T-box gene family; mutations in this gene may also cause small patella syndrome with or without detectable PAH. Dutch investigators recently found that 30% (6/20) of the pediatric PAH patient demonstrated either a deletion of 17q23.2, the loci containing TBX4, or a point mutation in TBX4. Interestingly, on retrospective evaluation, all patients carrying a TBX4 mutation or deletion demonstrated clinical findings of small patella syndrome; however, a separate cohort of small patella syndrome patients lacked evidence of PAH by transthoracic echocardiogram evaluation.34
Additional gene mutations of interest.
While the genes with mutations associated with PAH described above likely comprise the majority of single-gene mutation associations with PAH, additional findings continue to be reported. These are also likely relevant to PAH pathogenesis, and additional work continues to develop to explore these findings further. For example, de Jesus Perez et al used WES on a cohort of 12 unrelated patients with IPAH to identify mutations inTopBP1, which is involved in DNA repair, as possible contributor to PAH.35 This gene has strong biologic plausibility for PAH related to DNA damage among other contributing factors, although confirmatory studies remain to be reported.
EIF2AK4 Gene Mutations and Pulmonary Veno-occlusive Disease and Pulmonary Capillary Hemangiomatosis
An exciting recent development is the description of a new gene mutation’s association with two diseases which pathologically appear to be two ends of a similar spectrum of pathology: pulmonary veno-occlusive disease (PVOD) and pulmonary capillary hemangiomatosis (PCH). PVOD and PCH are rare causes of pulmonary hypertension characterized by progressive dyspnea, cough, hemoptysis, and fatigue.3 Although PCH is classically described as pulmonary capillary proliferation, while PVOD has small pulmonary venous occlusive lesions, it had been speculated for years that this disease represents different phenotypic expression of the same pathophysiologic processes.36
Similar to PAH, familial cases of both PCH and PVOD have been known for years.37 However, in contrast to BMPR2-associated PAH, the distribution of affected individuals suggested an autosomal recessive mechanism of inheritance.38 Through the use of whole exome sequencing, two groups independently identified the gene EIF2AK4, also known as EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4), as the causal gene for PCH and PVOD in 2013.39,40 Eyries et al focused on 13 families with familial PVOD characterized by an autosomal recessive inheritance pattern. Through the use of WES, the authors demonstrated homozygous mutations in EIF2AK4 of all affected individuals. Interestingly, the investigators also evaluated 20 sporadic cases of PVOD and found that 5 (25%) cases harbored biallelic EIF2AK4 mutations. Similar to BMPR2, different pathogenic mutations in EIF2AK4 were identified, including missense, frameshift, and nonsense mutations.39 Best et al focused on a PCH family with two affected siblings, an unaffected sibling, and unaffected parents. They also analyzed 10 patients with sporadic PCH and 1 patient with familial PCH consistent with an autosomal dominant pattern. WES demonstrated homozygous pathogenic mutations in EIF2AK4 in the affected siblings, as well as the 10 patients with sporadic PCH. The unaffected sibling and parents were all heterozygous for EIF2AK4 mutations. Notably, the patient with autosomal dominant familial PCH did not have a mutation detected, emphasizing the autosomal recessive nature of PVOD–PCH spectrum familial disease.40
As a result, biallelic mutations in the EIF2AK4 gene substantially contribute to heritable PVOD and PCH. A recent study by Montani et al explored the genotype–phenotype correlation between those with a detectable mutation and those without among a cohort of PVOD and PCH patients. Notably, biallelic mutation carriers were younger at presentation than noncarriers, but did not have a more severe hemodynamic profile or functional capacity at diagnosis. Time to death or lung transplantation was also similar, and short, among all patients studied for 1 and 3 years were 63 and 32%, respectively, among mutation carriers and 75 and 34%, respectively, for those without biallelic mutations in EIF2AK4.41
The TGF-β–BMP Signaling Pathway
BMPR2 is ubiquitously expressed and highly conserved throughout nature as a receptor for a family of cytokines known as BMPs. As members of the TGF-β superfamily of receptors, BMPs play a crucial role in the regulation of mammalian development. They are regulators of embryonic lung morphogenesis as well as bone and cartilage development.42 Although genetic studies strongly implicate the TGF-β superfamily in the regulation of pulmonary vascular cell growth and differentiation, the precise molecular mechanisms involved remain unclear.
It is clear that heterodimerization of the serine/threonine transmembrane kinases, BMPR1 and BMPR2, is critical to BMPR2 signaling. Four functional domains comprise BMPR2: ligand binding, kinase, transmembrane, and cytoplasmic domains. Activation of the heterodimeric BMPR2/BMPR1 receptor complex leads to phosphorylation of a series of cytoplasmic mediators, which include the Smad family. Following phosphorylation, Smad proteins 1, 5, and 8 complex with Smad 4 for translocation into the nucleus to regulate target gene transcription in concert with specific nuclear repressors and cofactors. In this manner, the Smad signaling pathway appears to participate in the inhibition of cell growth and the induction of apoptosis. It is postulated that BMPR2 mutations eliminate a critical growth regulatory function in pulmonary vascular cells by disrupting Smad activation, although other substrates related to BMP signaling have also been implicated.42,43
Prevalence of BMPR2 Mutations in Heritable PAH
As stated previously, BMPR2 mutations are responsible for PAH in the vast majority of HPAH, as well as some cases of PAH without family history. Germline BMPR2 mutations appear to cause PAH regardless of ethnic group.5,12 A variety of testing methodologies have been used to detect BMPR2 mutations, including sequencing of genomic DNA, Southern blot analysis, high-performance liquid chromatography, and melting curve analysis. A majority of mutations encode frameshift, nonsense, or splice site donor/acceptance site mutations. Multiplex ligation-dependent probe amplification (MLPA) analysis of genomic DNA with confirmation by real-time PCR has expanded the investigator’s ability to detect BMPR2 mutations.37 It is now generally accepted that approximately 75% or more of families with documented PAH in two or more members have a detectable mutation in BMPR2 in affected individuals.44 This agreement further supports a primary role for BMPR2 in the central pathogenesis of disease in HPAH.11
BMPR2 Mutations in Idiopathic PAH
Given the clinical and pathologic similarity to HPAH, it is not surprising that mutations in BMPR2 cause some cases of IPAH. These mutations may be either inherited low-penetrance alleles or de novo events not present in the parents or siblings.5 While the detection rates vary in part due to the availability of detailed family histories, mutations are typically detected in approximately 20% of IPAH patients (6–40%). Of note, while the precise rate of pathogenic BMPR2 mutations in the general population is unknown, it is extremely low.5
Transmission Patterns and Clinical Expression of HPAH
As noted previously, the penetrance of a BMPR2 mutation is reduced. Reduced penetrance of BMPR2 germline mutations can make establishing familial transmission difficult, as generations of mutation-carrying individuals may not express disease. In families known to harbor BMPR2 mutations, approximately 27% of individuals with a known genetic mutation in BMPR2 will develop detectable PAH.44 In addition to reduced penetrance, expressivity of PAH (variations in the phenotype among those with a given genotype and disease penetrance) is variable. For example, the age of onset can be at any age, from infancy to late adulthood. And, while most patients undergo lung transplant or die within 5 years of diagnosis without therapy, some HPAH patients do survive for many years. Finally, similar to most other forms of PAH, female predominance (~ 2:1 female:male ratio) is another major feature of HPAH.3 The largest family with PAH which we follow up at Vanderbilt is represented in ►Fig. 1.
Fig. 1.

Condensed pedigree of an extended kindred with heritable pulmonary arterial hypertension (HPAH) due to a BMPR2 mutation. This pedigree contains 42 total patients with HPAH, as well as many known BMPR2 mutation carriers without HPAH. Solid symbols represent individuals with disease. Circles represent women, and squares represent men. Line through symbol represents death. Dot inside symbol represents obligate carrier of the BMPR2 mutation. Numbers below symbols represent age at death or current living age. Numbers inside symbols represent numbers of unaffected siblings of each gender. S inside symbol represents stillbirth. Diamond symbol represents sex unknown.
The variable age of onset of HPAH, which can affect individuals at any age from early childhood to late adulthood, reflects the variable expressivity of disease. Recent national and international efforts have demonstrate that BMPR2 mutation carriers with PAH, compared with IPAH cases, are younger at diagnosis and have a more severe hemodynamic condition at the time of diagnosis. The most severe hemodynamic compromise at diagnosis includes higher mean pulmonary artery pressure, lower cardiac index, and higher pulmonary vascular resistance, as well as poor response to acute vasodilator testing.5,45-49 A recent multinational meta-analysis confirmed these findings.12
The BMPR2 mutation location, or type, may also influence the expression of PAH and/or its severity. Recently, Girerd et al noted that single nucleotide mutations in the cytoplasmic tail of BMPR2, as opposed to other domains, may be less severe and result in preserved SMAD signaling. Among 23 cytoplasmic tail mutation patients compared with 148 other BMPR2 mutation carriers with PAH, they found cytoplasmic tail mutation patients to be older at diagnosis and with less severe hemodynamic characteristics. In addition, patients with a cytoplasmic tail domain mutation were more likely to respond to calcium channel blockers for therapy.50 While this study requires replication, it suggests the ability to improve our ability to perform genotype–phenotype determinations by looking at the specific gene variant level and highlights the need to collaboratively investigate PAH across centers because such studies require higher numbers of participants.
Genetic Modifiers of HPAH Due to BMPR2 Mutations
The work highlighted earlier by Girerd et al emphasizes that variable expressivity is a feature of BMPR2-associated PAH. While the precise connection between PAH and BMPR2 signaling is not fully understood, disruption of BMP signaling associates with a loss of cell proliferation and differentiation control in the pulmonary vasculature.51 BMPR2 mutations often cause a condition of protein haploinsufficiency, which presumably is the molecular mechanism by which a BMPR2 mutation predisposes to PAH.52,53 At least half of all reportedly pathogenic mutations are predicted to result in a premature truncation codon, which if it resides in most exonic locations will trigger decay of the mRNA with no protein produced (resulting in a haplosufficient condition).53,54 In addition, many of the reported deletions within exons 2 to 13 of the BMPR2 gene lead to nonfunctional peptides, causing haploinsufficiency of BMPR2 function.55,56 Of course, not all mutations result in haploinsufficiency of the BMPR2 function—some may result in the production of an abnormal protein which may cause a dominant negative effect on BMP signaling.57 For example, functional studies in cells with kinase domain mutations in BMPR2 suggest that the production of malfunctioning BMPR2 protein may be more detrimental than a haploinsufficient state.58 Our group explored the concept that BMPR2 mutation carriers with haploinsufficient mutations had less severe disease than those with dominant negative mutations, and did find phenotypic differences.46 However, this finding was not replicated in a French cohort of BMPR2 mutants with PAH, although the determination of haploinsufficiency was slightly different.45 Given the relatively small numbers of subjects with BMPR2-associated PAH, it may be different to elucidate significant differences according to genotype, although such efforts are ongoing.
However, at a molecular level, BMPR2 production and function may associate with disease penetrance. Hamid et al found that BMPR2 allelic variation in vitro associates with the human phenotype. Specifically, in cells with a haploinsufficient BMPR2 mutation, the level of production of BMPR2 transcript by the wild-type allele (not the mutated allele) modified disease expression. Using immortalized peripheral blood lymphocyte cell lines derived from BMPR2 mutation carriers with four different haploinsufficient mutations, they determined BMPR2 transcript levels from mutated and wild-type alleles. As expected, the mutant alleles contributed very little to the total BMPR2 transcript levels (0–2.5%), so that the wild-type BMPR2 allele was the major determinant of total transcript production. As hypothesized, BMPR2 mutation carriers with PAH had significantly lower wild-type BMPR2 transcript levels compared with unaffected mutation carriers with the same haploinsufficient mutation, as well as reduced BMP signaling. This work suggests that in the setting of a BMPR2 mutation, one modifier of PAH penetrance is the level of wild-type BMPR2 produced.59 As a result, factors which modify BMPR2 expression in general may modify PAH penetrance, and perhaps other factors such as disease severity and progression.
Intriguingly, it has been known for years that IPAH patients, and other types of PAH patients, have reduced protein production of BMPR2 in lung tissues. These subjects, with wild-type BMPR2 genes, may be predisposed to PAH at least in part due to an inherently low BMPR2 production capacity, or posttranslational modifiers of BMPR2 protein expression.60 Thus, whether in the setting of a BMPR2 mutation or not, naturally low expression by the “healthy” wild-type allele may be a risk factor for the pathogenic mechanisms which promote PAH. As a result, factors which modify BMPR2 expression, BMPR2 isoform expression, and BMP signaling more broadly are naturally targets to investigate as modifiers of disease expression, and perhaps as therapeutic targets.61,62
One current approach to correct BMP signaling is the discovery and evaluation of compounds which restore BMPR2 expression and/or function. Spiekerkoetter et al recently employed a molecular discovery approach to find existing FDA-approved compounds which may increase BMPR2 expression. Intriguingly, low-dose FK506 was a top finding in their analyses, and subsequently improved BMPR2 activation and cellular function in vitro.63 As a result, low-dose FK506 (marketed as tacrolimus) has been successfully employed in several patients with severe PAH, with success.64 A phase II randomized clinical trial using FK506 in PAH patients was recently completed, with study results forthcoming (NCT01647945).
Additional mechanisms by which BMPR2 expression and/or function may be modified are an area of interest for many groups, in thinking about PAH among BMPR2 mutation carriers and more broadly. Investigative approaches are varied, including exploring mutations in the BMPR2 promoter region which may modify expression, additional gene mutations (e.g., thrombospondin-1) which may be co-present in a BMPR2 mutation carrier, as well as the presence of multiple mutations in the TGF-β signaling pathway simultaneously.65,66 Overall, there is a growing interest in the concept that no mutation occurs in “isolation”; multiple variations in gene structure, expression, and function, in multiple genes, coalesce to create a cellular phenotype.
Common Variations and PAH
While mutation discovery has been an area of intense focus for the PAH field, it is likely that the PAH condition is also influenced by common variations in relevant genes, as well. As shown in other cardiovascular diseases, confirmation of the contribution of common variants is more challenging, however, given the demand for large numbers of subjects when dealing with variations present at a higher prevalence in the population (i.e., the prevalence of a mutation should be less than 1%, whereas common variants are typically 5% or much higher, in the general population).67 Common genetic variations, like mutations, could influence not only disease development but also severity, response to therapy, and other phenotypic features.
There are few examples of common variations in PAH which have been consistently replicated in multiple studies. However, there are some promising candidates. Germain et al identified a new locus of importance in PAH pathogenesis by conducting the first genome-wide association study of patients (BMPR2 mutation not detected) with familial and idiopathic PAH. They employed two independent case–control studies, totaling 625 PAH cases and 1,525 healthy subjects. Both the discovery and replication stages found a twofold higher odds of PAH among subjects with the common variant rs2217560[G] allele at the 18q22.3 locus. In a subsequent combined population analysis, the rs2217560[G] allele was again associated with a twofold higher odds of PAH (1.59–2.45; p = 7.47 × 10—10).68 While it is difficulty to definitely link a gene with the locus of interest in this study, the cerebellin-2 precursor (CBLN2)68 gene, which lies 52 kb upstream of the SNP of interest, was identified by the investigators.
While this study has prompted great interest in CBLN2 and other loci in that region, the reason for this association remains elusive. Intriguingly, the investigators also found higher CBLN2 mRNA levels in the lungs of humans with PAH compared with controls, a finding particularly striking in endothelial cells from PAH patients (while pulmonary artery smooth muscle cell production of CBLN2 mRNA was reduced). Meanwhile, exposure to increasing concentrations of CBLN2 in vitro inhibited pulmonary artery smooth muscle cell proliferation, suggesting cross-talk between endothelial and smooth muscle cells mediated via CBLN2 levels, although this remains to be determined. While the precise role of CBLN2 variants to PAH remains to be determined, as does its applicability (or not) to other forms of PAH, and validated independently, this discovery identified a novel association that would not have otherwise been uncovered using a smaller whole exome (or whole genome) study.
There are several large-scale studies of PAH patients underway across the globe with dense genomic coverage. If enough patients can be evaluated, both rare and common variations may soon be uncovered, and replicated. Then, the work to understand their association with PAH, their impact on disease onset and progression, the mechanisms to modify these genetic variations, and so on, will need to be pursued.
Genetic Implications for Clinical Evaluation and Therapy
As noted previously, there is a growing body of work to suggest that mutation carriers who carry one of the known PH-specific genes (e.g., BMPR2, ALK1, EIF2AK4) have more severe disease than individuals with PAH who lack a known genetic association. For example, patients with HHT and PAH associated with ALKI or ENG genes have more severe disease, even compared with BMPR2 mutation carriers.69 More recently, Ghigna et al found that BMPR2 mutation carriers with PAH had significantly higher rates of hemoptysis as compared with patients with PAH without BMPR2 mutations.70 In a more global analysis of 1,550 patients from 8 separate cohorts, Evans et al found that patients with PAH and BMPR2 mutations present with more severe disease at a younger age with increased risk of death and transplantation.12 As a possible explanation for the overall worse clinical status seen in BMPR2-associated PAH, van der Bruggen et al demonstrated that mutation carriers presented with decreased RV ejection fraction and cardiac index, which persisted even after treatment was initiated.71
However to date, we have not modified therapeutic approach to PAH patients based on the identification of mutations or common variations in the genetic code. Hopefully, that will change soon, as multiple investigative groups are pursuing this issue. As noted earlier, Fk506 is in clinical trial as a therapeutic agent to treat PAH by improving BMPR2 downstream signaling.63,64 Meanwhile, utilizing mouse models, Reynolds et al, and Harper et al, have demonstrated that BMPR2 gene delivery is a promising therapeutic approach which may soon move to human trials.43,72 Finally, Ataluran, a small molecule that promotes ribosomal read through of premature stop codons, was shown to increase BMP-mediated microRNA and BMPR2 protein levels in lung-derived and blood-derived cells from patients with nonsense mutations in BMPR2 or SMAD9.16
Genetic Testing for Patients and Screening Subjects at Risk
Advances in genetic discovery have amplified the interest of incorporating genetic testing into PAH care as well as screening for at-risk subjects. However, given the lack of mutation-specific therapy, clinic testing of PAH patients remains somewhat controversial. As therapies emerge which modify the care approach, genetic testing may one day become standard of care, such as in the cystic fibrosis care model.73 However, genetic testing as a method to identify familial risk of PAH is growing more rapidly.
With the exception of PVOD and PCH, HPAH is transmitted in autosomal dominant manner. Thus, siblings or children of patients with HPAH, or of known heterozygotes for a BMPR2 mutation, have an overall risk of 50% to inherit the disease-causing mutated gene (allele). The risk of a sibling or child can be estimated without genetic testing, incorporating the recognition that BMPR2-associated PAH is incompletely penetrant. For example, with reduced penetrance (~27% of carriers will develop disease), before genetic testing of an “at-risk” child, one can calculate an estimated risk of 13.5% (50% chance to inherit the gene muta6tion × 27% chance to have PAH penetrance) to express disease for each child of a BMPR2 mutation carrier irrespective of genetic testing for that child.44 Thus, the majority of first-degree (parent, sibling, or child) relatives will be asymptomatic even though 50% will be carriers of a BMPR2 mutation.
However, pursuit of the genetic testing for the asymptomatic but “at-risk” relative modifies this equation. If an “at-risk” subject is tested for a BMPR2 mutation and negative, that individual’s risk of PAH falls from 13.5% to that of the population, which is approximately 1 in 1,000,000 (100,000-fold risk reduction)—this is the ideal situation, in which a person’s risk is dramatically lower due to the absence of a known mutation. But, if an asymptomatic individual at risk is found to carry the known familial BMPR2 mutation, elevation of the risk of PAH in the lifetime doubles from 13.5 to 27% (the percentage of BMPR2 mutation carriers who develop PAH in their lifetime). Of course, it is notable that this risk is actually not equal between the sexes (~40% lifetime risk for females vs. ~15% lifetime risk for males).44
Current clinical care screening recommendations for asymptomatic family members of patients with HPAH include a physical exam and surveillance echocardiogram at 3- to 5-year intervals.5 Uptake of screening is often more common in the pediatric setting, and in our clinic we commonly screen the children and child siblings of known HPAH, and IPAH, cases every 1 to 5 years depending on family preference and clinical factors. However, genetic counseling is routinely offered to subjects at risk, particularly those with a primary relative with HPAH and a known PH-specific mutation (►Fig. 2), including careful consideration of the utility of PAH-specific gene mutation screening to help determine disease risk. However, this remains an area in tremendous need of further study, although new information may soon be available. Investigators in France are conducting a large-scale study of at-risk subjects at the French Referral Centre for Pulmonary Hypertension (NCT01600898). This program offers several research-level approaches to attempt to detect PAH in asymptomatic mutation carriers and will study these subjects over time to identify factors associated with outcome, including disease penetrance.74 Comprehensive approaches such as this may also facilitate preimplantation genetic counseling and diagnosis for asymptomatic but at-risk subjects interested in pursuing a family, as recently demonstrated.75
Fig. 2.
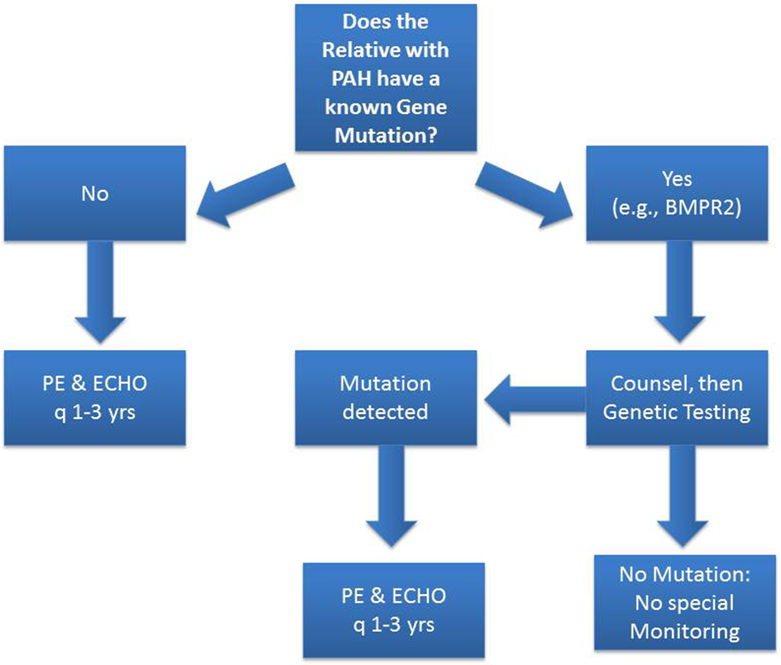
Suggested approach to a relative of a pulmonary arterial hypertension (PAH) patient with known heritable PAH (HPAH). If the asymptomatic but at-risk relative has a detectable PH-specific mutation, that person should undergo clinical evaluation and echocardiographic imaging every 1 to 3 years. However, if the person lacks the family’s PH-specific mutation, further testing can be halted unless new clinical concerns emerge.
Conclusion
Since the discovery of BMPR2 gene mutations nearly 20 years ago, consistent progress has been made to understand the genetics and genomics of PAH. The era of genotype–phenotype correlations, genetic-focused therapeutic approaches, and improved genetic counseling for patients and families is emerging. However, we still do not know the cause(s) of the reduced penetrance and variable expressivity of BMPR2 gene (and other PH-specific gene) mutations. We also do not understand to what degree genetic and genomic factors influence the development of IPAH and other PAH subtypes, as well as the other types of pulmonary hypertension in which genetic studies are more substantially lacking. Hopefully, by improving our understanding of the complex interplay of genetic, genomic, and other molecular factors in PAH, we will diagnose patients earlier, provide improved therapeutic approaches, and focus the overall care system on a more precise way to treat and prevent PAH.
Acknowledgments
The authors thank the many patients and families who graciously contributed to this work, and Ms. Lisa Wheeler, whose service is invaluable as coordinator of the Vanderbilt Familial Pulmonary Arterial Hypertension study.
This work was supported by NIH P01 HL108800.
References
- 1.Dresdale DT, Schultz M, Michtom RJ. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am J Med 1951;11(06):686–705 [DOI] [PubMed] [Google Scholar]
- 2.Dresdale DT, Michtom RJ, Schultz M. Recent studies in primary pulmonary hypertension, including pharmacodynamic observations on pulmonary vascular resistance. Bull N Y Acad Med 1954;30(03):195–207 [PMC free article] [PubMed] [Google Scholar]
- 3.Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(25, Suppl):D34–D41 [DOI] [PubMed] [Google Scholar]
- 4.Loyd JE, Primm RK, Newman JH. Familial primary pulmonary hypertension: clinical patterns. Am Rev Respir Dis 1984;129(01):194–197 [DOI] [PubMed] [Google Scholar]
- 5.Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(25, Suppl):D13–D21 [DOI] [PubMed] [Google Scholar]
- 6.Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987;107(02):216–223 [DOI] [PubMed] [Google Scholar]
- 7.Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31-q32. Circulation 1997;95(12):2603–2606 [DOI] [PubMed] [Google Scholar]
- 8.Morse JH, Jones AC, Barst RJ, Hodge SE, Wilhelmsen KC, Nygaard TG. Familial primary pulmonary hypertension locus mapped to chromosome 2q31-q32. Chest 1998;114(1, Suppl):57S–58S [DOI] [PubMed] [Google Scholar]
- 9.Thomson JR, Machado RD, Pauciulo MW, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 2000;37(10):741–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lane KB, Machado RD, Pauciulo MW, et al. ; International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet 2000;26(01):81–84 [DOI] [PubMed] [Google Scholar]
- 11.Machado RD, Southgate L, Eichstaedt CA, et al. Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat 2015;36(12):1113–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans JD, Girerd B, Montani D, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med 2016;4(02):129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trembath RC, Thomson JR, Machado RD, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001;345(05):325–334 [DOI] [PubMed] [Google Scholar]
- 14.Harrison RE, Flanagan JA, Sankelo M, et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J Med Genet 2003;40(12):865–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drake KM, Comhair SA, Erzurum SC, Tuder RM, Aldred MA. Endothelial chromosome 13 deletion in congenital heart disease-associated pulmonary arterial hypertension dysregulates SMAD9 signaling. Am J Respir Crit Care Med 2015;191(07):850–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drake KM, Dunmore BJ, McNelly LN, Morrell NW, Aldred MA. Correction of nonsense BMPR2 and SMAD9 mutations by Ataluren in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 2013;49(03):403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nasim MT, Ogo T, Ahmed M, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 2011;32(12):1385–1389 [DOI] [PubMed] [Google Scholar]
- 18.Tang H, Desai AA, Yuan JX; Application of Whole-Exome Sequencing to the Study of Pathogenic Mechanisms. Genetic insights into pulmonary arterial hypertension. Am J Respir Crit Care Med 2016;194(04):393–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Austin ED, Ma L, LeDuc C, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 2012;5(03):336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Caveolae and signalling in cancer. Nat Rev Cancer 2015;15(04):225–237 [DOI] [PubMed] [Google Scholar]
- 21.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol 2007;8(03):185–194 [DOI] [PubMed] [Google Scholar]
- 22.Maniatis NA, Shinin V, Schraufnagel DE, et al. Increased pulmonary vascular resistance and defective pulmonary artery filling in caveolin-1-/- mice. Am J Physiol Lung Cell Mol Physiol 2008;294(05):L865–L873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao YY, Malik AB. A novel insight into the mechanism of pulmonary hypertension involving caveolin-1 deficiency and endothelial nitric oxide synthase activation. Trends Cardiovasc Med 2009;19(07):238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao YY, Zhao YD, Mirza MK, et al. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest 2009;119(07):2009–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Achcar RO, Demura Y, Rai PR, et al. Loss of Caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest 2006;129(03):696–705 [DOI] [PubMed] [Google Scholar]
- 26.Zhao YY, Liu Y, Stan RV, et al. Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc Natl Acad Sci U S A 2002;99(17):11375–11380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang KY, Lee MF, Ho HC, et al. Serum caveolin-1 as a novel biomarker in idiopathic pulmonary artery hypertension. BioMed Res Int 2015;2015:173970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han B, Copeland CA, Kawano Y, et al. Characterization of a caveolin-1 mutation associated with both pulmonary arterial hypertension and congenital generalized lipodystrophy. Traffic 2016;17(12):1297–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma L, Roman-Campos D, Austin ED, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 2013;369(04):351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higasa K, Ogawa A, Terao C, et al. A burden of rare variants in BMPR2 and KCNK3 contributes to a risk of familial pulmonary arterial hypertension. BMC Pulm Med 2017;17(01):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navas P, Tenorio J, Quezada CA, et al. Molecular analysis of BMPR2, TBX4, and KCNK3 and genotype-phenotype correlations in Spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev Esp Cardiol (Engl Ed) 2016;69(11):1011–1019 [DOI] [PubMed] [Google Scholar]
- 32.Navas Tejedor P, Tenorio Castaño J, Palomino Doza J, et al. An homozygous mutation in KCNK3 is associated with an aggressive form of hereditary pulmonary arterial hypertension. Clin Genet 2017;91(03):453–457 [DOI] [PubMed] [Google Scholar]
- 33.Antigny F, Hautefort A, Meloche J, et al. Potassium channel subfamily K member 3 (KCNK3) contributes to the development of pulmonary arterial hypertension. Circulation 2016;133(14):1371–1385 [DOI] [PubMed] [Google Scholar]
- 34.Kerstjens-Frederikse WS, Bongers EM, Roofthooft MT, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet 2013;50(08):500–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Jesus Perez VA, Yuan K, Lyuksyutova MA, et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2014;189(10):1260–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montani D, Lau EM, Dorfmüller P, et al. Pulmonary veno-occlusive disease. Eur Respir J 2016;47(05):1518–1534 [DOI] [PubMed] [Google Scholar]
- 37.Montani D, Achouh L, Dorfmüller P, et al. Pulmonary veno-occlusive disease: clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore) 2008;87(04):220–233 [DOI] [PubMed] [Google Scholar]
- 38.Montani D, Price LC, Dorfmuller P, et al. Pulmonary veno-occlusive disease. Eur Respir J 2009;33(01):189–200 [DOI] [PubMed] [Google Scholar]
- 39.Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet 2014;46(01):65–69 [DOI] [PubMed] [Google Scholar]
- 40.Best DH, Sumner KL, Austin ED, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014;145(02):231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montani D, Girerd B, Jaís X, et al. Clinical phenotypes and outcomes of heritable and sporadic pulmonary veno-occlusive disease: a population-based study. Lancet Respir Med 2017;5(02):125–134 [DOI] [PubMed] [Google Scholar]
- 42.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 2012;11(10):790–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng F, Harper RL, Reynolds PN. BMPR2 gene delivery reduces mutation-related PAH and counteracts TGF-β-mediated pulmonary cell signalling. Respirology 2016;21(03):526–532 [DOI] [PubMed] [Google Scholar]
- 44.Larkin EK, Newman JH, Austin ED, et al. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186(09):892–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girerd B, Montani D, Eyries M, et al. Absence of influence of gender and BMPR2 mutation type on clinical phenotypes of pulmonary arterial hypertension. Respir Res 2010;11:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Austin ED, Phillips JA, Cogan JD, et al. Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir Res 2009;10:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenzweig EB, Morse JH, Knowles JA, et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant 2008;27(06):668–674 [DOI] [PubMed] [Google Scholar]
- 48.Elliott CG, Glissmeyer EW, Havlena GT, et al. Relationship of BMPR2 mutations to vasoreactivity in pulmonary arterial hypertension. Circulation 2006;113(21):2509–2515 [DOI] [PubMed] [Google Scholar]
- 49.Sztrymf B, Coulet F, Girerd B, et al. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med 2008;177(12):1377–1383 [DOI] [PubMed] [Google Scholar]
- 50.Girerd B, Coulet F, Jaïs X, et al. Characteristics of pulmonary arterial hypertension in affected carriers of a mutation located in the cytoplasmic tail of bone morphogenetic protein receptor type 2. Chest 2015;147(05):1385–1394 [DOI] [PubMed] [Google Scholar]
- 51.Orriols M, Gomez-Puerto MC, Ten Dijke P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol Life Sci 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomson J, Machado R, Pauciulo M, et al. Familial and sporadic primary pulmonary hypertension is caused by BMPR2 gene mutations resulting in haploinsufficiency of the bone morphogenetic protein type II receptor. J Heart Lung Transplant 2001;20(02):149. [DOI] [PubMed] [Google Scholar]
- 53.Machado RD, Pauciulo MW, Thomson JR, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet 2001;68(01):92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maquat LE. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 2004;5(02):89–99 [DOI] [PubMed] [Google Scholar]
- 55.Cogan JD, Pauciulo MW, Batchman AP, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174(05):590–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cogan JD, Vnencak-Jones CL, Phillips JA III, et al. Gross BMPR2 gene rearrangements constitute a new cause for primary pulmonary hypertension. Genet Med 2005;7(03):169–174 [DOI] [PubMed] [Google Scholar]
- 57.Cogan J, Austin E, Hedges L, et al. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation 2012;126(15):1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rudarakanchana N, Flanagan JA, Chen H, et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet 2002;11(13):1517–1525 [DOI] [PubMed] [Google Scholar]
- 59.Hamid R, Cogan JD, Hedges LK, et al. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum Mutat 2009;30(04):649–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Atkinson C, Stewart S, Upton PD, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002;105(14):1672–1678 [DOI] [PubMed] [Google Scholar]
- 61.Cogan J, Austin E, Hedges L, et al. Role of BMPR2 alternative splicing in heritable pulmonary arterial hypertension penetrance. Circulation 2012;126(15):1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.West J, Austin E, Fessel JP, Loyd J, Hamid R. Rescuing the BMPR2 signaling axis in pulmonary arterial hypertension. Drug Discov Today 2014;19(08):1241–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spiekerkoetter E, Tian X, Cai J, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 2013;123(08):3600–3613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spiekerkoetter E, Sung YK, Sudheendra D, et al. Low-dose FK506 (Tacrolimus) in end-stage pulmonary arterial hypertension. Am J Respir Crit Care Med 2015;192(02):254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Viales RR, Eichstaedt CA, Ehlken N, et al. Mutation in BMPR2 promoter: a ‘second hit’ for manifestation of pulmonary arterial hypertension? PLoS One 2015;10(07):e0133042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maloney JP, Stearman RS, Bull TM, et al. Loss-of-function thrombospondin-1 mutations in familial pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2012;302(06):L541–L554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pierpont ME, Basson CT, Benson DW Jr, et al. ; American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation 2007;115(23):3015–3038 [DOI] [PubMed] [Google Scholar]
- 68.Germain M, Eyries M, Montani D, et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet 2013;45(05):518–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Girerd B, Montani D, Coulet F, et al. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med 2010;181(08):851–861 [DOI] [PubMed] [Google Scholar]
- 70.Ghigna MR, Guignabert C, Montani D, et al. BMPR2 mutation status influences bronchial vascular changes in pulmonary arterial hypertension. Eur Respir J 2016;48(06):1668–1681 [DOI] [PubMed] [Google Scholar]
- 71.van der Bruggen CE, Happé CM, Dorfmüller P, et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: a view on the right ventricle. Circulation 2016;133(18):1747–1760 [DOI] [PubMed] [Google Scholar]
- 72.Harper RL, Reynolds AM, Bonder CS, Reynolds PN. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology 2016;21(04):727–733 [DOI] [PubMed] [Google Scholar]
- 73.Brewington J, Clancy JP. Diagnostic testing in cystic fibrosis. Clin Chest Med 2016;37(01):31–46 [DOI] [PubMed] [Google Scholar]
- 74.Girerd B, Montani D, Jaïs X, et al. Genetic counselling in a national referral centre for pulmonary hypertension. Eur Respir J 2016;47(02):541–552 [DOI] [PubMed] [Google Scholar]
- 75.Frydman N, Steffann J, Girerd B, et al. Pre-implantation genetic diagnosis in pulmonary arterial hypertension due to BMPR2 mutation. Eur Respir J 2012;39(06):1534–1535 [DOI] [PubMed] [Google Scholar]