

Abstract
We identified a family with a UMOD gene mutation (C106F) resulting in glomerular inflammation and complement deposition. To determine if the observed phenotype is due to immune system activation by mutant uromodulin, a mouse strain with a homologous cysteine to phenylalanine mutation (C105F) in the UMOD gene was generated using CRISPR-Cas9 gene editing and the effect of this mutation on mononuclear phagocytic cells was examined. Mutant mice developed high levels of intracellular and secreted aggregated uromodulin, resulting in anti-uromodulin antibodies and circulating uromodulin containing immune complexes with glomerular deposition and kidney fibrosis with aging. F4/80+ and CD11c+ kidney cells phagocytize uromodulin. Differential gene expression analysis by RNA sequencing of F4/80+ phagocytic cells revealed activation of the activating transcription factor 5 (ATF5)-mediated stress response pathway in mutant mice. Phagocytosis of mutant uromodulin by cultured dendritic cells resulted in activation of the endoplasmic reticulum stress response pathway and markers of cell inactivation, an effect not seen with wild-type protein. Mutant mice demonstrate a twofold increase in T-regulatory cells, consistent with induction of immune tolerance, resulting in decreased inflammatory response and improved tissue repair following ischemia-reperfusion injury. The C105F mutation results in autoantibodies against aggregated misfolded protein with immune complex formation and kidney fibrosis. Aggregated uromodulin may induce dendritic cell tolerance following phagocytosis through an unfolded protein/endoplasmic reticulum stress response pathway, resulting in decreased inflammation following tissue injury.
Uromodulin (encoded by the UMOD gene), alias Tamm-Horsfall glycoprotein, is the most abundant protein in normal urine. Uromodulin is produced by epithelial cells of the thick ascending limb, where it is targeted to the apical membrane and secreted into the urine. Clear roles of urine uromodulin as a defense against urinary tract infections and in regulation of apical membrane sodium and potassium transport and blood pressure control are supported by studies of UMOD knockout mice.1,2
Immunohistochemical studies have shown that uromodulin is also targeted to the basolateral membrane and secreted into the interstitium and blood.3,4 Both human and mouse studies have detected ng/mL levels of circulating protein.5,6 Interstitial uromodulin has been hypothesized to regulate innate immune responses. In vitro studies demonstrate that uromodulin activates macrophage inflammasomes through toll-like receptor 3 signaling.7 UMOD knockout mice studies support both an anti-inflammatory and proreparative effect of uromodulin following ischemia-reperfusion injury (IRI)8 and an inflammatory effect associated with decreased kidney injury molecule-1 (Kim-1) expression and apoptotic cell scavenging following chronic injury with unilateral ureteral obstruction.9 These disparate effects have been hypothesized to be due to differences in epithelial and immune cell responses to uromodulin released from the basolateral membrane.
Patients expressing mutant uromodulin protein as a result of autosomal dominant missense mutations in cysteine residues in the UMOD gene develop chronic tubulointerstitial disease that typically progresses to end-stage kidney disease between the fourth and seventh decade of life.10 Several mouse models transgenic for cysteine missense mutations using various genetic methods have been used to investigate the mechanisms by which these mutations cause kidney disease.11, 12, 13, 14 In all models, intracellular uromodulin accumulation due to protein misfolding resulted in decreased urine excretion and endoplasmic reticulum (ER) stress, leading to an unfolded protein response and, in most models, apoptosis in the thick ascending limb and subsequent interstitial fibrosis.
We identified a family with multiple members from several generations affected by a cysteine 106 to phenylalanine (C106F) UMOD mutation. The clinical course and kidney biopsy findings of affected members, including hematuria, proteinuria, and glomerular and interstitial inflammation, were atypical of previously described mutations. To determine how this mutation results in kidney disease, a mouse model with a homologous cysteine to phenylalanine mutation was generated and the effects of this mutation on the innate and adaptive immune system were studied with normal aging and following acute injury.
Materials and Methods
Study Approval
All animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee of Central Arkansas Veterans Healthcare System. All patients consented to the use of their DNA for genotyping using a Central Arkansas Veterans Healthcare System Institutional Review Board approved protocol (941985-1).
Protein Structure Simulation
The full-length structure of human UMOD (isoform 4) was predicted using fold recognition and Ab Initio software version 3.1.5 (Ab Initio Software Corporation, Lexington, MA). After validating the structure, the predicted structure was energy minimized using a GROMACS simulation package. All structures, including native (control), were simulated using Desmond simulation package from Schrodinger for 200 ns. The resulting trajectories were analyzed for differences in structure, including root mean square deviation, amino acid fluctuations (root mean square fluctuation), and secondary structure.
Generation of Mutant Mice
A construct expressing Cas9 and single-guide RNA in the pX330 vector, along with a single-stranded DNA homology donor, was injected into the pronuclei of fertilized C57BL/6J mouse eggs at a final concentration of 5 and 10 ng/μL, respectively. Microinjected eggs were then implanted in the oviducts of pseudopregnant ICR mice. Founders were screened for the desired mutation using a TaqMan genotyping assay with the following forward primers: allele 1: VIC/MGB-NFQ: 5′-GTCTGTG[C]AGCCGA-3′; and allele 2: FAM/MGB-NFQ: 5′-GTCTGTG[A]AGCCGA-3′. Sanger sequencing of 300 bp genomic PCR products confirmed the identity of two founders with the desired mutation. A single founder mouse with the correct mutation was crossed with C57BL/6 mice, and the backcross continued for a total of seven generations for the mice used in this study. The presence of the correct mutation in the F1 offspring was confirmed using the genotyping assay described below. For all experiments, 10- to 12-week–old male C57BL/6J (stock number: 000664; Jackson Laboratory, Bar Harbor, ME) mice were used, with the exception of 9-month–old mice for the effect of aging.
TaqMan Genotyping Assay
DNA was isolated from tail tissue using a QIAamp fast tissue kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. Concentrations (ng/μL) and OD ratios (260/280 nm) of DNA were determined using the Nanodrop UV/VIS spectrophotometer (Thermo Fisher Scientific, Waltham, MA). All DNA specimens had OD ratios between 1.70 and 1.95. Relative quantitative PCR was performed using the Applied Biosystems (Foster City, CA) 7900 fast real-time PCR system withSequence Detection System software version 2.4.1 (Applied Biosystems) followed by an allelic discrimination post-read run. All quantitative PCRs were performed in a final volume of 10 μL containing 1× of TaqMan Genotyping Master mix (Applied Biosystems), 1× of Custom TaqMan SNP Genotyping Assay (FAM-MGB dyes), and 10 ng cDNA in sterile molecular-grade water. The standard cycling conditions were 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Quantitative PCR was performed in triplicate to ensure quantitative accuracy, and 0.037 was the CT threshold.
Site-Directed Mutagenesis and LLC-PK1 Cell Transfection
A full-length human uromodulin transcript variant 1 cDNA clone in a pCMV-entry vector was purchased from Origene (Rockville, MD). Site-directed mutagenesis was performed with a Q5 site-directed mutagenesis kit (New England Biolabs, Ipswich, MA). Primers were selected for amplification of mutant plasmid using the NEBaseChanger program (https://nebasechanger.neb.com, last accessed October 1, 2018): forward: 5′-GGTCTCGGCTtCACAGACGTGGATG-3′ (t = base change for cysteine to phenylalanine) and reverse: 5′-GGGCGACAGGCGGAAGCC-3′. Plasmid sequences were verified using Sanger sequencing. Plasmids were transfected into LLC-PK1 cells (CL-101; ATCC, Manassas, VA) using TransIT-X2 reagent (Mirus Bio, Madison, WI). Cells were grown in medium 199 with 10% fetal bovine serum with addition of G418 (800 μg/mL) for selection of stable transfectants. Individual colonies were selected by limiting dilution and grown to confluence before transferring to Transwell clusters for apical and basolateral medium assays. Cell confluence was tested by transepithelial electrical resistance using an EVOM2 meter. Cells were grown on Permanox chamber slides (ThermoFisher) for immunofluorescence.
Immunoprecipitation
Rabbit monoclonal uromodulin (Abcam, Cambridge, MA; Ab207170) was conjugated to Protein G Dynabeads (ThermoFisher) and cross-linked with BS3 reagent (ThermoFisher), according to the manufacturer's directions. Mouse plasma or cell lysates were precleared with agarose A/G (Santa Cruz Biotechnology, Dallas, TX) and incubated with anti-uromodulin antibody conjugated magnetic beads for 1 hour at room temperature. Following washes with phosphate-buffered saline (PBS)/Tween, antibody was eluted and used for Western blot analysis. Protein was detected using anti-rabbit VeriBlot for IP secondary antibodies (Abcam; ab131366). Positive control uromodulin 64.22-kDa peptide was purchased from LSBio (Seattle, WA; LS-G23183).
Plasma Creatinine Assay
Creatinine was measured in heparinized plasma samples (n = 10 per group) by liquid chromatography–tandem mass spectrometry at the University of Alabama at Birmingham O'Brien Center for Acute Kidney Injury Research Bioanalytical Core C.
MP Isolation
PBS perfused kidneys were minced and digested with Liberase TL (ThermoFisher; 0.25 mg/mL) and RNAse free DNAse 1 (100 μ/mL) in Dulbecco’s modified Eagle’s medium with rotation at 225 rpm at 37°C for 30 minutes. Tissue digests were filtered through a 40-μm nylon mesh (BD Bioscience, San Jose, CA) and washed with PBS. Dead cells were removed with an immunomagnetic Dead Cell Removal Kit (Miltenyi, Auburn, CA). A total of 1 × 107 live cells were incubated with anti-mouse F4/80 magnetic beads (Miltenyi) for positive selection of mononuclear phagocyte (MP) cells. Pelleted cells were lysed in RLT buffer followed by RNA isolation using an RNeasy micro kit with genomic DNA (gDNA) eliminator columns (Qiagen). RNA was quantified by a Qubit assay. RNA samples with Agilent bioanalyzer RNA integrity number scores ≥8.0 were used for construction of cDNA libraries.
DC Isolation
Wild-type (WT) mouse spleens were digested with 2 mg/mL collagenase IV (ThermoFisher) for 30 minutes at 37°C and passed through 100 μm followed by 40-μm nylon filters. Dendritic cells (DCs) were isolated by negative selection using a pan DC immunomagnetic kit (Miltenyi; 130-100-875), according to the manufacturer's directions. A total of 500,000 cells/mL were cultured in RPMI 1640 medium with 5% fetal bovine serum and 20 ng/mL granulocyte-macrophage colony-stimulating factor for 24 hours before addition of uromodulin. Assays were performed 24 hours after addition of uromodulin.
Flow Cytometry
Following dead cell removal, live cells were incubated with FC block (Miltenyi) followed by anti-CD45 microbeads (Miltenyi) for positive and negative hematopoietic cell selection. Cells were incubated with primary antibodies in flow cytometry buffer (PBS with 5% fetal bovine serum, 2 mmol/L EDTA, and 0.02% sodium azide) for 30 minutes at 4°C and analyzed using a BD Fortessa flow cytometer (BD Biosciences). Primary antibodies were purchased from ThermoFisher/eBioscience and include the following: anti: F4/80 (clone BM8), major histocompatibility complex (MHC) II (clone M5/114.15.2), CD86 (clone GL1), and CD11c, and a mouse regulatory T-cell staining kit 1 (CD4, CD25, and Foxp3). Cells were analyzed with a BD Fortessa flow cytometer.
Ischemia-Reperfusion Injury
The 10- to 12-week–old male mice were anesthetized with isoflurane. Mice were placed on a heating pad at 37°C. The left renal hilum was identified, and the renal vessels were dissected and clamped for 28 minutes. The vessels of the right kidney were ligated with sutures, and the kidney was subsequently removed. Kidney ischemia and reperfusion were verified by expected changes in tissue color. The midline incision was closed with 3-0 silk suture. A total of 10 mice for each genotype were used for each time point.
Western Blots
Protein samples were separated by electrophoresis on 5% to 20% Tris-glycine gels (Novex, Invitrogen/ThermoFisher, Waltham, MA). Proteins were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA) using an Xcell miniblot system (Invitrogen, Carlsbad, CA). Membranes were blocked with Tris-buffered saline/Tween 20 with 5% non-fat dry milk for 1 hour and incubated overnight at 4°C with primary antibodies. Proteins were detected using horseradish peroxidase–conjugated anti-rabbit secondary antibodies at 1:2000 dilution (Cell Signaling, Danvers, MA) and SignalFire ECL reagent (Cell Signaling). For quantification of protein levels, autoradiographs were scanned, and densitometry was performed with ImageJ software version 1.51 (NIH, Bethesda, MD; https://imagej.nih.gov/ij). Results were corrected for variations in the amount of protein loaded on each lane using corresponding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels. Rabbit primary antibodies used include the following: anti-uromodulin (Abcam; ab207170), GAPDH (Cell Signaling Technologies; 14c10), NOD-, LRR- and pyrin domain–containing protein (NLRP3 (Cell Signaling Technologies; D4D8T), phosphorylated p65 (Cell Signaling Technologies; 93H1), and caspase 11 (Abcam; ab180673).
ELISAs
Mouse interferon γ (Abcam; 100690), mouse uromodulin (Abcam; ab245726), mouse Kim-1 (R&D Systems, Minneapolis, MN; MKM100), and mouse IL-β (Invitrogen; 88-7013-22) enzyme-linked immunosorbent assays (ELISAs) were performed using commercial kits, according to the manufacturer's instructions. For anti-uromodulin antibody ELISA, Nunc Maxisorb 96-well plates were coated with recombinant mouse uromodulin amino acid 25-588 (LS Bio) at 2 μg/mL in coating buffer overnight at 4°C. Following washes with PBS, wells were blocked with 0.25% casein in PBS, pH 7.4, for 1 hour at room temperature. Following washes with PBS/Tween 0.05%, plasma diluted 1:200 in blocking buffer and a mouse anti-uromodulin antibody standard (Santa Cruz Biotechnology; B-2; sc-271022; diluted 1:20,000 to 1:1,280,000) were incubated overnight at 4°C. Following washes in PBS/Tween 0.05%, wells were incubated with anti-mouse IgG-biotin (Invitrogen; 1:30,000) followed by streptavidin–horseradish peroxidase (Abcam; 1:15,000) and developed with TMB substrate (Abcam; high sensitivity). Results were expressed as means of triplicate samples (n = 8 per genotype).
Immunofluorescence and Immunohistochemistry
Immunofluorescence and immunohistochemistry were performed as previously described.15 Antibodies include the following: rabbit anti–α-smooth muscle actin (Abcam; ab124964), anti–4-hydroxynonenal (4HNE) (Abcam; ab46544), anti-FoxP3 (Novus Biologicals, Littleton, CO; NB100-39002), anti-calnexin (Novus Biologicals; NBP1-37774), rat anti-mouse F4/80 (BM8; eBioscience), anti-Mo Ly-6G/Gr-1 (eBioscience; R86-BC5), and anti-Tim1 (3809; ProSci, Poway, CA). Fluorescence slides were imaged using a Zeiss LSM880 microscope with Airyscan (Zeiss, White Plains, NY). Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed on paraffin-embedded kidney sections using an HRP In Situ Apoptosis Detection Kit (Abcam; ab206386), according to the manufacturer's instructions. Masson-Trichrome staining was performed with a Dako/Agilent Technologies kit (Santa Clara, CA), and stained areas were measured using ImageJ software.
RNA Sequencing
The library was prepared using a NEBNext Ultra II Directional RNA Library kit (New England Biolabs). A total of 75 ng of total RNA was mRNA enriched using oligodT beads. The mRNA-stranded libraries were pre-amplified with PCR incorporating dual index primers and purified with magnetic beads. The library's size distribution was validated and quality inspected on a Tapestation Bioanalyzer (Agilent Technologies). Next-generation sequencing was performed by Macrogen using a Novaseq sequencer (Illumina, San Diego, CA). A total of 30 million reads (50-bp paired-end reads) were generated and mapped to the Mus musculus reference genome GRCm38, with annotation reference Ensembl_70. HISAT2 was used to align sequence reads to the reference genome. After read mapping, Stringtie was used for transcript assembly. Expression profile was calculated for each sample and transcript/gene as read count and fragment per kilobase of transcript per million mapped reads. Differentially expressed gene analysis was performed on comparison pairs [MUT (TG)_vs_WT] using DESeq2. The results showed 220 genes that satisfied |fc|≥2 & nbinomWaldTest raw P < 0.05 conditions in comparison pair.
Biological process gene ontology enrichment analysis was also performed using open source software from the Gene Ontology Consortium (https://geneontology.github.io/docs/go-enrichment-analysis, last accessed October 30, 2019).16 Detailed RNA-sequencing information, including access to raw data, meeting Minimum Information About a Microarray Experiment requirements, has been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo; accession number GSE139585).
Kidney Tubule Epithelial Cell Cultures
Kidney medullas and outer cortexes were dissected from mouse kidneys, minced, and digested with Liberase TL (0.25 mg/mL) and RNAse free DNAse 1 (100 units/mL) in Dulbecco’s modified Eagle’s medium with rotation at 225 rpm at 37°C for 30 minutes. Tissue digests were filtered through a 100-μm nylon mesh (BD Bioscience) and washed with PBS. Tissue retained on the filters was centrifuged and plated on type I collagen-coated 6-well plates (Corning 354400; ThermoFisher). Cells were grown in Dulbecco’s modified Eagle’s medium/F12 supplemented with a Renal Epithelial Basal Cell Growth Kit (ATCC; PCS-400-040) until confluent. RNA was prepared from passage 1 of all cultures within 3 to 4 days of cell isolation using an RNEasy plus mini kit with gDNA eliminator columns (Qiagen).
Uromodulin Purification
Uromodulin was prepared from 1-mL samples of pooled WT mouse urine using diatomaceous earth, as previously described.17 Following precipitation using 0.5 mol/L NaCl, pelleted protein was resuspended in PBS, pH 7.2. Uromodulin from homozygous mutant mice was prepared from the radioimmunoprecipitation assay buffer insoluble pellet following NaCl precipitation. All samples were run on a silver-stained polyacrylamide gel (Pierce Silver Stain Kit; Thermo Scientific) to verify purification, and protein concentrations were determined using a bicinchoninic acid assay (Thermo Scientific).
Gene Expression Validation by Real-Time Quantitative PCR
Total RNA was isolated from macrophages, tubules, and DCs using the RNeasy Plus Micro kit (Qiagen), according to the manufacturer's instructions. gDNA was removed with gDNA eliminator columns. Concentrations (ng/μL) and OD ratios (260/280 nm) of total RNA were determined using the Nanodrop UV/VIS spectrophotometer (Thermo Fisher). RNA integrity number scores, which are a ratio of ribosomal RNAs 18S and 28S in total RNA samples, were obtained using the Agilent 2100 Bioanalyzer with the Agilent RNA 6000 Nano Kit (Santa Clara, CA). All total RNA specimens had OD ratios of 1.85 to 2.10 and RNA integrity number scores of >7.4. The quantitative conversion of 250 ng of total RNA to single-stranded cDNA in a single 30-μL reaction was performed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using QuantStudio 12K Flex real-time PCR system software version 1.3 (Applied Biosystems). All quantitative PCRs were performed in a final volume of 10 μL containing 1× of TaqMan Gene Expression Master mix with UNG (Applied Biosystems), 1× of each TaqMan Gene Expression Assay (FAM-MGB dyes), and 10 ng cDNA in sterile molecular-grade water (Table 1). The standard cycling conditions were 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Quantitative PCR was performed in duplicate to ensure quantitative accuracy. The results were analyzed using Expression Suite Software version 1.0 (Applied Biosystems). The comparative threshold cycle values (ΔΔCT) were normalized for the GAPDH reference gene.
Table 1.
Significant GO Processes and Corresponding Genes and Significance Values
| GO term | Gene symbol | Fold change, mut/+/wt | Adjusted P value |
|---|---|---|---|
| Response to unfolded protein | Trib3 | 54 | 2.27 × 10−52 |
| ATF5 | 7.8 | 6.24 × 10−29 | |
| Asns | 7.3 | 3.38 × 10−20 | |
| Chac1 | 9.5 | 6.55 × 10−12 | |
| Mthfd2 | 3.9 | 4.5 × 10−11 | |
| Ddit3 | 3.3 | 9.54 × 10−8 | |
| ATF4 | 2.0 | 0.0003 | |
| Hspb8 | 1.8 | 0.03 | |
| Regulation of developmental process | Wnt10a | 8.6 | 1.16 × 10−15 |
| Stc2 | 2.9 | 0.0004 | |
| Cxcl10 | 4.0 | 0.001 | |
| Ifrd1 | 2.7 | 0.001 | |
| Tgif1 | 1.8 | 0.004 | |
| Hmox1 | 1.7 | 0.01 | |
| Egf | −6.8 | 1.38 × 10−9 | |
| Wnt11 | −2.2 | 0.0002 |
P < 0.01 was considered to be significant.
GO, gene ontology; wt, wild type.
TaqMan Gene Expression Assay identifiers and gene symbols were as follows: Mm04179654_m1 (Atf5), Mm99999915_g1 (Gapdh), Mm01324470_m1 (Lcn2), Mm00454879_m1 (Trib3), Mm00437325_m1 (Wnt10a), Mm00486868_m1 (CD83), Mm03048248_m1 (CD274), Mm00439683_m1 (Inhibin A), and Mm00545913_m1 (Socs3).
Statistical Analysis
All data are presented as the means ± SD. Significance of difference between groups was evaluated by two-way analysis of variance with Bonferroni post-hoc analysis or t-test, where appropriate (SigmaPlot version 14.0; SYSTAT version 13.2; Systat Software, Inc., San Jose, CA). P < 0.05 was considered statistically significant.
Results
Kidney Biopsy Pathology in Three Family Members Shows Glomerular Inflammation
Kidney biopsy reports of three family members were available for review (Supplemental Table S1). In all cases, mesangial proliferation and matrix deposition were detected, and two of the three biopsies showed glomerular inflammatory cell infiltration and complement deposition.
The C106F Mutation Results in Simulated Protein Misfolding in the Zona Pellucida Domain
Whole exome sequencing of DNA isolated from the proband, two additional affected family members, and the proband's unaffected mother was performed, and mutations were identified using Ingenuity Variant Analysis. A guanine to thymidine (TGC to TTC) mutation, resulting in a cysteine to phenylalanine substitution (C106F) in the UMOD gene, was the only pathologic mutation identified in all affected family members. This mutation was originally reported in the National Center for Biotechnology Information ClinVar database as dbSNP ID 398123697 in an uncharacterized individual and more recently in two families following genetic testing for hereditary focal segmental glomerulosclerosis.18 The mutated cysteine is located in a region between epithelial growth factor domains 2 (amino acids 65 to 103) and 3 (amino acids 108 to 149; https://www.ncbi.nlm.nih.gov/protein; reference sequence NP_003352.2). Computer stimulation of protein folding demonstrated marked fluctuations (root mean square fluctuation) at amino acids 300 to 600 with predicted misfolding at the C-terminal zona pellucida domain. The backbone root mean square deviation between the final simulated structures of WT and C106F uromodulin was 18.30 angstroms, indicating a large structural change due to this single amino acid mutation (Supplemental Figure S1). Misfolding in this area would be expected to result in altered protein polymerization.19
Expression of Mutant Uromodulin in Kidney Epithelial Cell Cultures Results in Increased Basolateral Secretion of Aggregated Protein
To assess if the C106F mutation affects uromodulin polymerization, membrane targeting, and secretion, WT and C106F mutant constructs produced by in vitro mutagenesis of a full-length human uromodulin clone were stably expressed alone and in combination in LLC-PK1 cells, a porcine kidney epithelial cell line that has previously been shown to process transfected uromodulin for apical targeting and formation of extracellular filaments.3 Transfected cells expressed WT and mutant uromodulin protein at equal concentrations (Figure 1A). Western blot analysis of apical and basolateral medium prepared from confluent cells grown on Transwell filters (Figure 1B) demonstrated apical secretion of full-length protein and lower levels of basolateral secreted uromodulin in mutant construct transfected cells. Cells expressing both WT and mutant protein secreted higher levels of a 55-kDa N′ terminal truncated protein (verified by mass spectroscopy) in both apical and basolateral medium. This truncated protein also appeared at lower levels in WT basolateral medium. Deglycosylation treatment of apical medium from WT and mutant transfected cells with PNGase H resulted in detection of 65- and 85-kDa proteins (Figure 1C), consistent with the size of the precursor protein. Uromodulin membrane localization was examined by confocal microscopy (Figure 1D). WT and combined WT and mutant transfected cells displayed apical uromodulin protein with fibril formation. Mutant and combined WT and mutant transfected cells also displayed basolateral uromodulin localization. To determine the effect of the mutation on protein polymerization, apical and basolateral medium from cell cultures were examined by nondenaturing Western blot and electron microscopy (Figure 1E). Fibril formation was detected only in apical medium from WT cells by electron microscopy and correlated with an inability to enter the gel by electrophoresis. Mutant and WT and mutant medium showed only protein aggregates under these conditions, and protein aggregates at several higher molecular weights in medium from mutant transfected cells were noted on a nondenaturing Western blot.
Figure 1.

Expression of mutant uromodulin (Umod) in kidney epithelial cell cultures demonstrates increased basolateral secretion of aggregated protein. A: Western blot analysis of Umod using LLC-PK1 cell lysates with stable transfection of wild-type (WT) and/or mutant (mut) uromodulin expression plasmids as indicated. Control (cntrl) cells were transfected with empty plasmid. B: Western blot analysis of uromodulin using 10× concentrated cell culture medium removed from apical (A) and basolateral (BL) sides of stable transfected LLC-PK1 cells as indicated. C: Western blot analysis of uromodulin using 10× concentrated cell culture medium removed from apical chamber and untreated (−) or treated (+) with PNGase. Untreated medium from cells transfected with WT and mutant plasmids was added for size comparison. D: Confocal images of uromodulin immunofluorescence using LLC-PK1 cells transfected as indicated. Left column: Apical membrane plane. Right column: Lateral plane. E: Nondenaturing Western blot analysis of uromodulin using A and BL medium (arrow indicates high-molecular-weight protein; and arrowheads indicate uromodulin multimers) and corresponding electron microscopy images. Scale bars: 5 μm (D); 100 nm (E). sup, supernatant.
A Mouse Model of the C106F Uromodulin Mutation Demonstrates Secretion of Intracellular Aggregated Protein into the Urine and Blood
Alignment of human and mouse uromodulin protein sequences demonstrated a conserved cysteine at amino acid position 105 in the mouse (Supplemental Figure S2). To generate a guanine to thymine point mutation at this location in the mouse gene, resulting in cysteine to phenylalanine transition, a plasmid encoding a CRISPR RNA guide and the Cas9 protein, along with a 153-bp single-stranded donor DNA fragment containing the mutation, was microinjected into C57BL/6J mouse zygotes. Resulting offspring carrying the C105F mutation were identified using a TaqMan single-nucleotide polymorphism genotyping assay (Supplemental Figure S3) that demonstrated allelic discrimination. The mutation site was verified by Sanger sequencing of gDNA (data not shown). Sequencing results identified 20% of founders with the correctly targeted mutations. Positive founders were bred with WT mice to produce F1 animals. The heterozygous mice of the F1 generation (mutant/+) were found to have germ-line transmission of 50%. Mutant/+ mice were backcrossed with C57BL/6J mice for an additional seven generations to eliminate possible off-target mutations. A cohort of mutant/+, homozygous (mutant/mutant), and WT littermate controls were evaluated at 12 weeks to examine the effects of the mutation on uromodulin localization and aggregation.
WT mice displayed both intracellular and luminal surface uromodulin expression (Figure 2A), and mutant/+ mice (Figure 2B) showed protein in intracellular compartments that was not colocalized with calnexin (Figure 2, C and D), an ER membrane marker expressed in the distal tubule. Mutant/+ mice exhibited increased urine uromodulin excretion measured by ELISA compared with WT littermates (Figure 2E), and the mutant protein migrated at the expected glycosylated size by denaturing Western blot analysis. To determine whether mutant protein was secreted in the urine, urine from homozygous mutant/mutant mice was examined. Mutant uromodulin was detected at low levels in unspun urine but was present at high levels in the pelleted fraction at the expected glycosylated size following solubilization in 8 mol/L urea buffer (Figure 2E), indicating urine protein aggregation in these mice.
Figure 2.

A mouse model of the C106F uromodulin mutation demonstrates secretion of intracellular aggregated protein into the urine and blood. A–D: Confocal immunofluorescence images of wild-type (WT) and mutant (mut)/+ kidney sections stained for uromodulin (A and B) and uromodulin (green) and calnexin (red; C and D). E: Western blot (W) analysis for uromodulin in unprocessed urine and urine sediment solubilized in 8 mol/L urea buffer from three mouse genotypes, as indicated, and corresponding uromodulin enzyme-linked immunosorbent assay (ELISA). F: Western blot analysis for uromodulin in kidney lysates solubilized in buffers with increasing stringency, as indicated from three mouse genotypes. G: Western blot analysis of uromodulin using immunoprecipitates (IPs) of kidney lysates and plasma from indicated genotypes using uromodulin-conjugated dynabeads (IgG indicates control nonspecific rabbit IgG; rUMOD IP, 100 ng recombinant uromodulin peptide IP; and rUmod, 10 ng non-IP). H: Western blot analysis of uromodulin using PNGase treated (+) and untreated (−) IP of kidney plasma from a WT mouse. I: Western blot analysis of uromodulin using IP of plasma from WT and mut/mut genotypes, as indicated. J: Western blot analysis of uromodulin using IP of plasma used in I with additional treatment with 0.1 mol/L dithiothreitol and boiling. K: Western blot analysis of uromodulin using IP of plasma filtered with 200-kDa cutoff polysulphone concentrator with corresponding Ponceau S staining of membrane of Western blot of flow-through fraction showing albumin (arrow) and IgG heavy and light chains. L: Uromodulin ELISA of plasma samples (P value for mut/mut versus WT). n = 6 samples per genotype (E); n = 6 to 7 (L). ∗P < 0.05, ∗∗P < 0.01 versus wt. Scale bars = 10 μm (A–D). RIPA, radioimmunoprecipitation assay.
Western blot analysis of kidney protein lysates in increasing stringent denaturing buffers was used to determine if relatively insoluble intracellular protein aggregates formed in mutant mice. Insoluble pellets from the radioimmunoprecipitation assay buffer lysates of mutant/+ and mutant/mutant mice yielded large amounts of uromodulin multimers and protein corresponding with the size of the 85-kDa precursor monomer when dissolved in 8 mol/L urea buffer (Figure 2F). To determine if mutant protein also undergoes basolateral secretion into blood, protein was immunoprecipitated from plasma of WT, mutant/+, and mutant/mutant mice. A 105-kDa uromodulin protein was precipitated from both WT and mutant/+ plasma, corresponding in size with kidney lysate protein (Figure 2G). Protein bands were verified to be uromodulin by mass spectroscopy (data not shown). Glycosylation of the circulating protein was evaluated by PNGase treatment (Figure 2H), resulting in a shift to the deglycosylated weight. Immunoprecipitation of protein from mutant/mutant mice demonstrated a 105-kDa protein in denaturing gels, but also showed a larger amount of protein at approximately 267 kDa (Figure 2I). Additional denaturation steps resulted in shifting of this aggregate to the monomer weight (Figure 2J). The presence of circulating high-molecular-weight uromodulin multimers in mutant mice was confirmed by immunoprecipitation of protein from plasma filtered through a 200-kDa molecular weight cutoff polysulfone filter. Uromodulin was detected by Western blot in filtered plasma from mutant/+ and mutant/mutant mice but not WT mice (Figure 2K). Protein staining of the flow-through fraction (Figure 2K) showed equal amounts of albumin and IgG in all three samples. Plasma levels of uromodulin, measured by ELISA, were similar between WT and mutant/+ mice but were significantly lower in mutant/mutant mice (Figure 2L). This difference may be due to failure of the ELISA to detect aggregated protein, as has previously been suggested.20
C105F Mutant Mice Produce Autoantibodies against Uromodulin with Immune Complex Formation
The detection of intracellular and circulating mutant aggregated uromodulin suggested the possibility that C105F mice may form autoantibodies against mutant protein.17,21 Western blot analysis of lysates from cultured WT and mutant/+ outer medullary tubular epithelial cells using plasma from WT and mutant/mutant mice demonstrated the presence of anti-uromodulin antibodies in mutant plasma (Figure 3A). Plasma anti-uromodulin antibody levels were quantified by ELISA using plates coated with recombinant WT uromodulin (Figure 3E). Anti-uromodulin antibodies were detected at ng/mL levels that were not significantly different between mutant/+ and mutant/mutant mice with no detectable antibody in WT mice. WT and mutant/+ plasma were also incubated with cultured WT and mutant/+ medullary tubular epithelial cells expressing uromodulin and analyzed by flow cytometry, demonstrating a threefold increase in binding of mutant/+ plasma antibodies to mutant/+ cells (Figure 3D). To determine if these antibodies resulted in circulating immune complex formation, C1q was immunoprecipitated from plasma of WT, mutant/+, and mutant/mutant mice, demonstrating uromodulin co-immunoprecipitation in mutant mice by Western blot analysis (Figure 3B). These results were verified by an ELISA for circulating immune complexes showing a sixfold increase in levels in mutant/mutant mice (Figure 3C). Deposition of immune complexes in mutant/+ mice was evaluated by electron microscopy and immunofluorescence of glomeruli (Figure 4). Subepithelial (Figure 4B) and mesangial electron-dense immune complex deposits were detected in mutant/+ kidneys along with glomerular basement membrane deposits of C3 (Figure 4D) and IgG (Figure 4F) using these techniques in comparison with WT glomeruli (Figure 4, A, C, and E). Tubular basement membrane IgG was also diffusely positive, although the specificity of this was uncertain because of low-level background staining in WT mice (Figure 4E).
Figure 3.

C105F mutant mice produce autoantibodies against uromodulin and immune complex formation A: Western blot (WB) analysis of lysates of wild-type (WT) and mutant (mut)/+ cultured medullary tubular epithelial cells using plasma from WT and mut/mut mice diluted 1:1000. B: Immunoprecipitation of WT and mut/+ and mut/mut plasma using anti-C1q antibodies followed by Western blot analysis with anti-uromodulin antibody. Control immunoprecipitated (IP) with rabbit IgG. C: Enzyme-linked immunosorbent assay (ELISA) for circulating immune complexes using WT, mut/+, and mut/mut plasma. D: Flow cytometry analysis of cultured WT and mut/+ medullary tubular epithelial incubated with plasma from WT and mut/+ mice diluted 1:1000. Control cells were incubated with purified mouse IgG. E: ELISA for anti-uromodulin antibodies using plates coated with recombinant mouse uromodulin amino acid 25 to 588 at 2 μg/mL and plasma from WT, mut/+, and mut/mut mice diluted 1:100. WT plasma was identical to background controls, and levels are not shown. Data are expressed as means ± SD (C and E). n = 7 per genotype (C); n = 8 per genotype (E). ∗P < 0.05, ∗∗∗∗∗P < 0.00001 versus wt.
Figure 4.
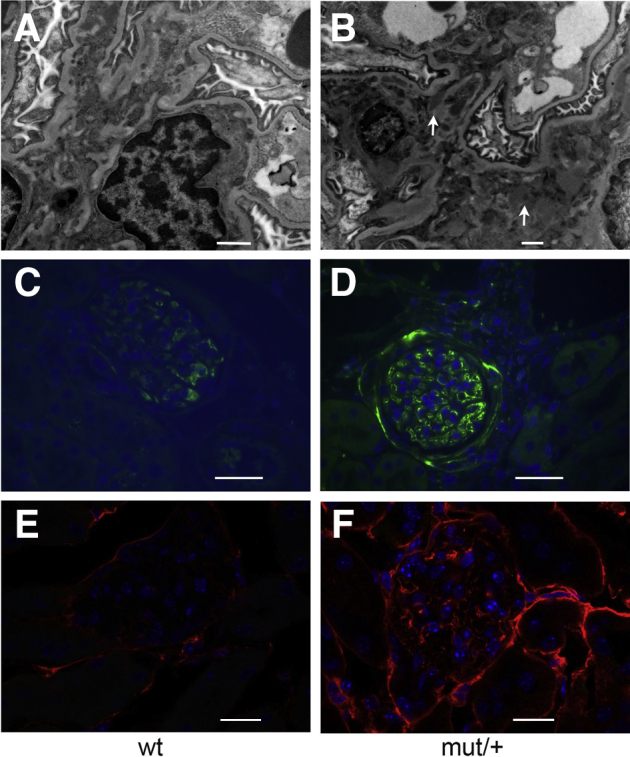
Immune complex deposition in mutant mouse kidneys. Electron micrographs of wild-type (WT; A) and mutant (mut)/+ (B) kidneys. Arrows indicate electron-dense subepithelial deposits. Immunofluorescence of WT and mut/+ kidneys for C3 (C and D) and IgG (E and F). Scale bars: 1 μm (A); 2 μm (B); 100 μm (C and D); 50 μm (E and F).
Transcriptional Analysis of F4/80+ Cells Isolated from Mutant/+ Kidneys Reveals an Integrated Stress Response with ATF4 Regulated Gene Expression
To determine if mutant uromodulin interaction with the innate immune system contributes to the pathogenesis of disease, mRNA sequencing of F4/80+ mononuclear phagocytic cells isolated from 8- to 12-week–old mutant and WT littermate control mice was performed using anti-F4/80 conjugated magnetic microbeads (https://www.ncbi.nlm.nih.gov/geo; accession number GSE139585). A total of 70% to 90% of cells collected with this method expressed the F4/80 and CD11b markers expressed by kidney macrophages and DCs22 (Supplemental Figure S4). Hierarchical clustering and principal component analysis showed differentiation of WT and mutant gene expression (Supplemental Figure S5). A total of 121 significantly up-regulated or down-regulated genes (log2 fold-difference > 1 with Padjusted < 0.05) were detected and displayed as a heat map in Supplemental Figure S6. There was a marked activation of downstream effectors of the integrated stress response, including activating transcription factor 4 (ATF4) regulated genes [Trib3, ATF4 and ATF5, Ddit3 (CHOP), Chac 1, and Asns], genes associated with both a proreparative or developmental macrophage phenotype (Arg2, Lcn2, Hmox1, EGF, Ptch1, Wnt10a, Wnt11, Cxcl10, and Stc2), and down-regulation of interferon-regulated genes (Ifit1bl1 and Ifit1bl2). Gene ontology analysis of the 121 significantly differentially expressed genes confirmed significant up-regulation of the response to stress and toxic substance and nephron and tubule developmental processes (Table 1). To verify that the observed changes in gene expression were not due to admixture of tubule epithelial cells, RNA from primary cultures of outer medullary tubule epithelial cells from WT and mutant/+ mouse kidneys was used for TaqMan relative quantification PCR assays of four of the most differentially expressed genes (Trib3, ATF5, Lcn2, and WNT10a) and compared with RNA from F4/80 magnetic bead purified cells. No differences in gene expression were detected between WT and mutant/+ tubule epithelial cells, whereas increased gene expression was detected in F4/80+ purified cells (Supplemental Figure S7A). Expression of uromodulin in these tubule epithelial cells was confirmed by Western blot analysis (Supplemental Figure S7B).
DCs Express ER Stress and Immune Tolerance Markers in Response to Phagocytosis of Mutant Uromodulin
DCs survey and respond to cellular damage and link the innate and adaptive immune responses. Uromodulin has been hypothesized to act as a danger-associated molecular pattern,7,23 suggesting that DC response to this protein may regulate the immune system following kidney injury. To test this hypothesis, DCs were isolated from WT mouse spleens using a pan-DC immunomagnetic bead isolation kit and incubated with 1 μg/mL uromodulin purified from WT or mutant/mutant mouse urine using diatomaceous earth or as the insoluble aggregated pellet, respectively. Uromodulin uptake by CD11C+ cells was confirmed by immunofluorescence (Figure 5, A–F), demonstrating large mutant uromodulin aggregates that colocalized with the ER chaperone and ER stress/unfolded protein response activator binding immunoglobin protein (BiP)/glucose regulatory protein 78 (GRP78) (Figure 5, J–L). No BiP colocalization was seen with uptake of WT uromodulin (Figure 5, G–I), and increased BiP expression was also demonstrated by Western blot analysis (Figure 5M). TaqMan assays were used to quantify relative gene expression of ER stress markers TRIB3 and ATF 4 and 5 and demonstrated significant relative increased expression in cells incubated with mutant uromodulin (Figure 5N). The ATF4/ER stress response has previously been shown to inhibit dendritic cell maturation.24 To determine if phagocytosis of uromodulin affects the antigen presentation and stimulatory capability of these cells, DC maturation markers were examined by TaqMan gene expression assays and flow cytometry. Uptake of mutant uromodulin by CD11c+ cells resulted in increased expression of inactivation or tolerance markers CD274/PD-125 and SOCS326 and decreased expression of inhibin A27 compared with control and DCs incubated with WT uromodulin (Figure 5O), consistent with decreased maturity of mutant protein treated cells. CD83, a marker of DC maturation, was unchanged. Flow cytometry showed decreased expression levels of MHCII and CD86 (Figure 5, P–S) in mutant uromodulin treated cells, also consistent with decreased maturation.
Figure 5.

Dendritic cells (DCs) express endoplasmic reticulum (ER) stress and immune tolerance markers in response to phagocytosis of mutant uromodulin. A–L: Immunofluorescence of cultured DCs 24 hours after addition of wild-type (WT; A–C and G–I) or mutant (mut; D–F and J–L) uromodulin at 1 μg/mL. Control cells incubated with vehicle only. A–F: Uromodulin, purple; CD11c, green; and DAPI, blue. G–L: Uromodulin, purple; BiP, green; and DAPI, blue. M: Western blot of BiP/GRP78 levels in lysates of DCs incubated with WT or mutant uromodulin. N and O: TaqMan gene expression assays for ER stress markers (N) and DC tolerogenic/maturation markers (O) displayed as relative quantification (RQ) values for cells incubated with WT or mutant uromodulin from three independent experiments. P–R: Flow cytometry analysis of expression of DC maturation markers major histocompatibility complex (MHC) II and CD86 in DCs incubated with control (con; P), WT (Q), and mutant (R) uromodulin. S: CD11C+ cells were gated for analysis (results representative of three separate cell purifications) and percentage MHCIIhi/CD86hi for indicated condition (P value for mutant versus WT uromodulin). Data are expressed as means ± SD (N, O, and S). ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001, and ∗∗∗∗∗P < 0.00001 versus wt. Scale bars = 10 μm (A–L). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 6.

Increased T-regulatory (Treg) cells and decreased interferon-γ (IFN-γ) levels in mutant kidneys. A: Flow cytometry analysis of CD45+ microbead isolated hematopoietic cells from wild-type (WT) and mutant (mut)/+ kidneys gated for CD25 expression and further evaluated for CD4/FoxP3 expression. Results are representative of three separate experiments. B: Rare cells were detected in WT kidneys. C: Anti-FoxP3 immunohistochemistry showing staining for Treg cells (arrows) in mut/+ kidneys. D: Interferon-γ enzyme-linked immunosorbent assay of kidney lysates from WT and mutant mice, as indicated. n = 5 per group (D). ∗P < 0.05, ∗∗P < 0.01 versus wt. Scale bars = 80 μm (B and C).
Increased numbers of immature or tolerogenic DCs would be expected to result in increased numbers of T-regulatory cells in mutant mice. To examine this possibility, CD45+ immunomagnetic beads were used to isolate hematopoietic cells from 12-week–old WT and mutant/+ kidneys. Lymphocyte subpopulation analysis by flow cytometry revealed a twofold increased percentage of CD4/CD25/FoxP3 T-regulatory cells (Figure 6A). Increased T-regulatory numbers in mutant mice were confirmed by immunohistochemical staining of WT (Figure 6B) and mutant kidneys (Figure 6C) for FoxP3. RNA sequencing results showed decreased expression of interferon-regulated genes and increased numbers of T-regulatory cells that reduce interferon-γ production,14 suggesting that mutant kidneys may have reduced levels of this cytokine, a type 1 helper T-cell product and activating factor of inflammation. This was confirmed by an interferon-γ ELISA of kidney lysates (Figure 6D) demonstrating a 1.5-fold and fourfold decrease in mutant/+ and mutant/mutant, respectively compared with WT mice.
The C105F Mutation Leads to Improved Tubular Repair 7 Days after IRI
DC tolerance improves recovery following kidney injury.28,29 To determine if the tolerogenic effect of mutant uromodulin on DCs affects the outcome of acute kidney injury, WT and mutant/+ mice were examined following IRI. Immunofluorescence of normal mutant/+ kidney showed intracellular uromodulin in MHCII+ and CD11c mononuclear phagocytes (Figure 7, A and B). Although uromodulin was not detected in normal WT kidney mononuclear phagocytes (data not shown), uptake was seen in these cells in WT mice at 7 days following IRI (Figure 7, C and D).
Figure 7.
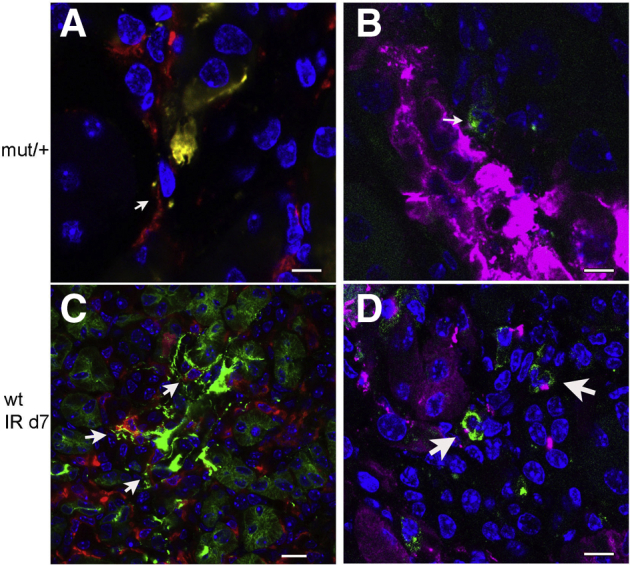
Kidney mononuclear phagocytic cell uptake of uromodulin. Immunofluorescence of uromodulin and mononuclear phagocyte (MP) marker colocalization in kidney sections from a 12-week–old mutant (mut)/+ mouse (A and B) and 12-week–old wild-type (WT) mouse 7 days following ischemia-reperfusion (IR) injury (C and D). Uromodulin indicates green signal (A and C) and purple signal (B and D); major histocompatibility complex II indicates red signal (A and C); and CD11c indicates green signal (B and D). Arrows indicate MPs with uptake of uromodulin. Scale bars: 10 μm (A, B, and D); 40 μm (C).
At 24 hours after injury, Western blot analysis showed mutant/+ mice had a threefold increase in kidney lysate uromodulin (Figure 8A). WT mice showed no significant change in uromodulin levels, with several mice demonstrating a marked reduction in levels by Western blot analysis and immunofluorescence (Figure 8, A and B). WT and mutant/+ sham operated mice had similar plasma uromodulin levels. At 24 hours after injury, plasma levels were unchanged in mutant/+ mice but increased up to fivefold in WT mice (Figure 8C), correlating with increased release from the thick ascending limb.
Figure 8.

The C105F mutation leads to improved tubular repair 7 days following ischemia-reperfusion injury (IRI). Mice with indicated genotype underwent unilateral ischemia (28 minutes) and contralateral nephrectomy followed by reperfusion for 24 hours or 7 days. A: Western blot analysis of uromodulin (Umod) expression and corresponding densitometry in whole kidney lysates from indicated genotypes. B: Immunofluorescence of uromodulin expression in wild-type (WT) and mutant (mut) kidneys, as indicated. C: Enzyme-linked immunosorbent assay of plasma uromodulin levels at 24 hours following IRI in sham operated (control) and experimental mice. n = 10 per group (C). ∗P < 0.05, ∗∗∗P < 0.001 versus wt con. Scale bars = 50 μm (B). con, control; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Following IRI, the repair phase begins 3 days following injury, leading to resolution of inflammation.30 At 7 days following IRI, WT mice demonstrated increased uromodulin expression by Western blot analysis (Figure 9A) and persistently increased plasma levels compared with a reduction to baseline levels in mutant/+ mice (Figure 9B). Histologic examination showed areas of continued tubular injury in WT kidneys that displayed TUNEL-positive tubular epithelial cells (Figure 9, C, D, and F). In contrast, mutant/+ kidneys had complete tubular repair and no residual TUNEL-positive cells (Figure 9, E and F). Tubular repair in WT compared with mutant kidneys was also evaluated by Kim-1 immunohistochemistry and urine levels by ELISA, demonstrating elevation at 7 days following injury in WT (Figure 9, G and I) versus mutant/+ mice (Figure 9, H and I). Mutant/+ mice displayed increased numbers of apoptotic or necrotic tubular cells by TUNEL staining and increased peritubular neutrophil infiltration at 24 hours after IRI (Supplemental Figure S8), indicating that improved tubular repair in these mice was not due to a lower level of initial injury. Although NLRP3 levels were not significantly different, downstream inflammatory markers, phosphorylated NF-κB p65 and cleaved caspase 11, were persistently elevated in WT kidneys (Figure 9J) along with increased WT kidney lysate Il-1β (Figure 9K), a key cytokine produced by inflammasome activation. In contrast, mutant/+ mice showed a return of these inflammatory markers to baseline. These results correlated with plasma creatinine measurements demonstrating no differences in increased levels between genotypes at 24 hours but with significantly improved levels in mutant/+ mice compared with continued elevation in WT mice at 7 days following injury (Supplemental Figure S9).
Figure 9.

Mice with indicated genotype were examined 7 days following ischemia-reperfusion injury (IRI). A: Western blot analysis of uromodulin (Umod) expression and corresponding densitometry in whole kidney lysates from indicated genotypes. B: Enzyme-linked immunosorbent assay (ELISA) of plasma uromodulin levels at 7 days following IRI. C–F: Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining of wild-type (WT; C and D) and mutant (mut)/+ kidney (E) (D) with corresponding positive cell counts (F). Boxed area in C is shown at higher magnification in D. G–I: Kim 1 immunohistochemistry of kidney sections of WT (arrows indicate Kim-1 expressing tubules; G) and mut/+ mice (H) and corresponding urine Kim-1 ELISA (I) at day 7 following IRI (P value compared with sham operated WT mice). J: Western blot of NLRP3, phosphorylated p65 (p-p65), and caspase 11 expression in whole kidney lysates from indicated genotypes. K: IL-1β ELISA assay of whole kidney lysates at indicated time points following IRI (P value for WT IR 24 hours versus WT control (con), mut/+ IR 24 hours versus mut/+ con, and WT IR day 7 versus WT con). n = 10 per group (K). ∗P < 0.05 versus wt con; †P < 0.05, ††P < 0.01 versus wt IR D7; ‡‡‡P < 0.001 versus tg ir d7; §§P < 0.01 versus control. Scale bars: 100 μm (C and E); 50 μm (D); 500 μm (G and H). Cr, creatinine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LPF, low-power field.
The C105F Mutation Causes Myofibroblast Proliferation, Matrix Deposition, and Chronic Renal Failure with Aging
Mutant/+ and WT littermates were examined at 40 weeks of age to determine the effects of the mutation on the development of kidney disease with aging. Kidney function in mutant/+ deteriorated with aging with a doubling of plasma creatinine (Figure 10A). Mutant/+ mice developed interstitial fibrosis characterized by a 10-fold increase in outer medullary collagen deposition (Figure 10C) and peritubular myofibroblasts detected by α-smooth muscle actin immunohistochemistry (Figure 10, E and F) in comparison with WT mice (Figure 10, B and D). Histologic examination showed no evidence of tubular atrophy (Figure 10C) or apoptosis by TUNEL staining (data not shown). Additional markers of tubular injury and oxidative stress were also absent, including neutrophil gelatinase associated lipocalin (NGAL) and 4-HNE (Supplemental Figure S10). Despite the presence of persistent C3 and immune complex deposition, no histologic evidence of glomerulosclerosis, including mesangial matrix deposition or a significant increase in albuminuria, occurred in mutant mice at this time point.
Figure 10.

The C105F mutation causes myofibroblast proliferation, matrix deposition, and chronic renal failure with aging. A: Plasma creatinine levels measured by liquid chromatography–mass spectrometry. B, C, and F: Masson trichrome staining (B and C) and corresponding percentage area staining (F). D and E: α-Smooth muscle actin immunohistochemistry. n = 10 mice per group (A). ∗P < 0.05, ∗∗P < 0.01 versus wt 9 mo. Scale bars = 100 μm (B, C, D, and E). Mut, mutant; WT, wild type.
Discussion
The C105F mouse homolog of the human C106F mutation demonstrates that misfolded and aggregated uromodulin engages the kidney's innate and adaptive immune system, resulting in autoantibody and immune complex formation. Previous mouse models have shown that mutant uromodulin is retained in the endoplasmic reticulum, reducing its trafficking to the apical membrane.11, 12, 13,31, 32, 33 As a result, both uromodulin associated kidney disease patients and mutant mouse models have reduced amounts of uromodulin in urine.12,31 These uromodulin trafficking defects cause renal tubular epithelial cell ER stress, inflammation, and apoptosis, resulting in tubulointerstitial fibrosis and chronic kidney disease.11, 12, 13,32,33 In contrast, the C105F mutation results in markedly increased intracellular levels of aggregated uromodulin precursor protein that are secreted in the urine and cross the basolateral membrane and interact with kidney immune cells. Release of misfolded, aggregated protein has previously been shown in ER stressed neurons, and may be a mechanism to reduce cell stress.34 The C105F mouse model provides the first demonstration of both a similar mechanism in the kidney and autoimmune antibody production to a misfolded kidney-specific protein. Although family members with the C106F mutation showed biopsy evidence of immune complex and complement deposition, it is currently unknown if this is a result of anti-uromodulin antibodies, as observed in the mouse model. Despite the presence of glomerular immune complex and complement deposition in 10- to 12-week–old mice, no histologic evidence of glomerulosclerosis or development of proteinuria occurred at 9 months of age. Additional studies using older mice are therefore necessary to determine if glomerular injury eventually occurs, corresponding with the pathologic findings in some of the C106F family members.
A focus of this study was to determine if the chronic kidney disease and fibrosis with aging was due to direct tubular injury by misfolded protein and/or the release of mutant uromodulin into the interstitium with engagement of innate immune cells. A previous study using Crispr Cas9 generated C147T mutant mice demonstrated that misfolded uromodulin causes ER stress that activates unfolded protein response mechanisms, leading to apoptotic cell death in renal tubule epithelial cells by 24 weeks of age.12 In comparison, C105F mice showed no evidence of tubular injury at any age up to 9 months using various methods, including histology, TUNEL staining for apoptosis, NGAL staining, or immunohistochemical markers for oxidative injury. Mediators of the unfolded protein response, including Trib3, CHOP, and ATF4, were not detected in isolated tubular epithelial cells by relative quantification PCR, correlating with the absence of thick ascending limb apoptosis in these mice. Additional studies, including the effect of B-cell depletion in mutant mice, are needed to determine if anti-uromodulin antibodies and immune complexes are responsible for kidney fibrosis with aging.
Aggregated mutant protein was detected in plasma and within MHCII- and CD11c-positive peritubular MPs. Flow cytometric assays of surface markers, immunofluorescence, and gene expression profiling of kidney MPs were used to characterize their phenotype in mutant mice and the effects of uromodulin uptake in cultured dendritic cells were examined. Together, these studies demonstrated that uptake of misfolded uromodulin results in markedly increased expression of genes associated with the ATF4-mediated integrated stress response, including a 50-fold increase in Tribbles 3 expression.
The TRIB (Tribbles 1 to 3) family of genes encodes a group of highly conserved pseudokinase proteins that act as adaptors and regulate key inflammatory signaling pathways by inhibition of multiple kinases and interacting with ubiquitin ligases. Tribbles 1 and 2 decrease expression of inflammatory cytokines35,36 in monocytes and macrophages, promoting a proreparative or M2 phenotype. Although ER stress induces TRIB3 expression, its function in regulating MP inflammatory response is unknown. In adipocytes, Tribbles 3 inhibits AKT and terminal differentiation and has an anti-inflammatory effect. Additional studies are needed to determine how uromodulin uptake causes ER stress and how increased Tribbles 3 in MP interferes with maturation and promotes an anti-inflammatory phenotype and if this promotes kidney repair following injury.19,30 Because the kidney F4/80+ MP population used in the RNA sequencing was heterogeneous, it will be important to examine the effect of uromodulin uptake in different MP subtypes, such as tissue-resident macrophages and dendritic cells.
Gene expression profiling also showed changes associated with inflammatory responses, including increased lipocalin 2 and Wnt10a and decreased epithelial growth factor and interferon-stimulated gene expression. Interferon-γ promotes phagocytotic cell toll-like receptor ligand stimulation, resulting in increased production of proinflammatory cytokines, and enhances antigen processing and presentation in macrophages by up-regulating the MHC class I and II antigen presentation pathways. The reduced kidney tissue interferon-γ levels suggest an underlying mechanism for the myofibroblast proliferation,37,38 matrix deposition,39,40 and decreased renal function observed with aging in mutant mice.
An interesting question is how can anti-uromodulin autoantibody production continue despite the impaired activation of dendritic cells by uptake of mutant protein and induction of markers of immune tolerance? In vitro assays demonstrate that uromodulin is a toll-like receptor ligand.7 Immune complexes containing toll-like receptor ligands lead to systemic inflammation through the activation of innate immune cells by this mechanism and binding to Fc receptors.37 Inflammation and continued antigen exposure may impair tolerance mechanisms and promote B-cell clonal expansion,41 resulting in continued anti-uromodulin antibody production. The effect of mutant uromodulin on the ability of renal dendritic cells to mature, however, may result in decreased inflammation following acute tubular injury and improved tubular repair.
In conclusion, the C106F (human) and homologous C105F (mouse) uromodulin mutation results in a novel mechanism of chronic kidney disease that involves both production of anti-uromodulin antibodies and immune complexes with resulting inflammatory kidney disease and an MP stress response to uptake of aggregated uromodulin with expression of markers of immune tolerance. The improved kidney repair following injury supports a role for uromodulin as a signaling molecule that engages resident kidney immune cells following release from injured cells.
Acknowledgments
We thank Dr. Meenakshisundaram Balasubramaniam [University of Arkansas for Medical Sciences (UAMS)] for performing the protein stability and folding simulations; Andrea Harris (UAMS) for expert assistance with flow cytometry; Todd Fite [Central Arkansas Veterans Healthcare System (CAVHS)] for expert assistance with ischemia-reperfusion injury procedures; Dr. Jason Stumhofer (UAMS) for assistance with magnetic bead cell separation; and Dr. Terry Harville (UAMS) for helpful discussions.
Footnotes
Supported by a Dialysis Clinics Incorporated (DCI) Paul Teschan grant FP53001 (M.P.); the Veterans Administration; NIH grant P20GM125503 (C.A.O.); the John L. McClellan VA Hospital; and the Pharmacogenomics Analysis Laboratory (Research Service, Central Arkansas Veterans Healthcare System, Little Rock, AR), a core research laboratory funded by the Cooperative Studies Program, Research and Development, Department of Veterans Affairs.
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2020.08.015.
Author Contributions
M.P. designed and performed the experiments, prepared the figures, and wrote the manuscript; A.S. performed the experiments and prepared the figures; C.O. designed the experiments; and J.G., J.W., S.S., and W.K. performed the experiments.
Supplemental Data
Supplemental Figure S1.

Protein folding simulations of wild type and C106F uromodulin (UMOD). A: Root mean square fluctuations (RMSFs), demonstrating marked differences in structure at the zona pellucida/polymerization region of mutant protein. B: Simulated ribbon structure, demonstrating folding differences in the polymerization region (arrow).
Supplemental Figure S2.

Amino acid alignment of human (bottom) and mouse N′ terminal sequence, demonstrating homologous cysteine residues (green) and mutated C105 residue (red; arrow). EGF, epithelial growth factor; Sbjct, subject; UMOD, uromodulin.
Supplemental Figure S3.

TaqMan single-nucleotide polymorphism genotyping assay of tail genomic DNA, demonstrating allele discrimination of wild-type (WT; blue), heterozygous [mutant (MT)/+; green], and homozygous (mutant/mutant; red) mice. NTC, no template control; UMOD, uromodulin.
Supplemental Figure S4.
Flow cytometry analysis of F4/80 and CD11b expression in F4/80 microbead isolated kidney cells.
Supplemental Figure S5.
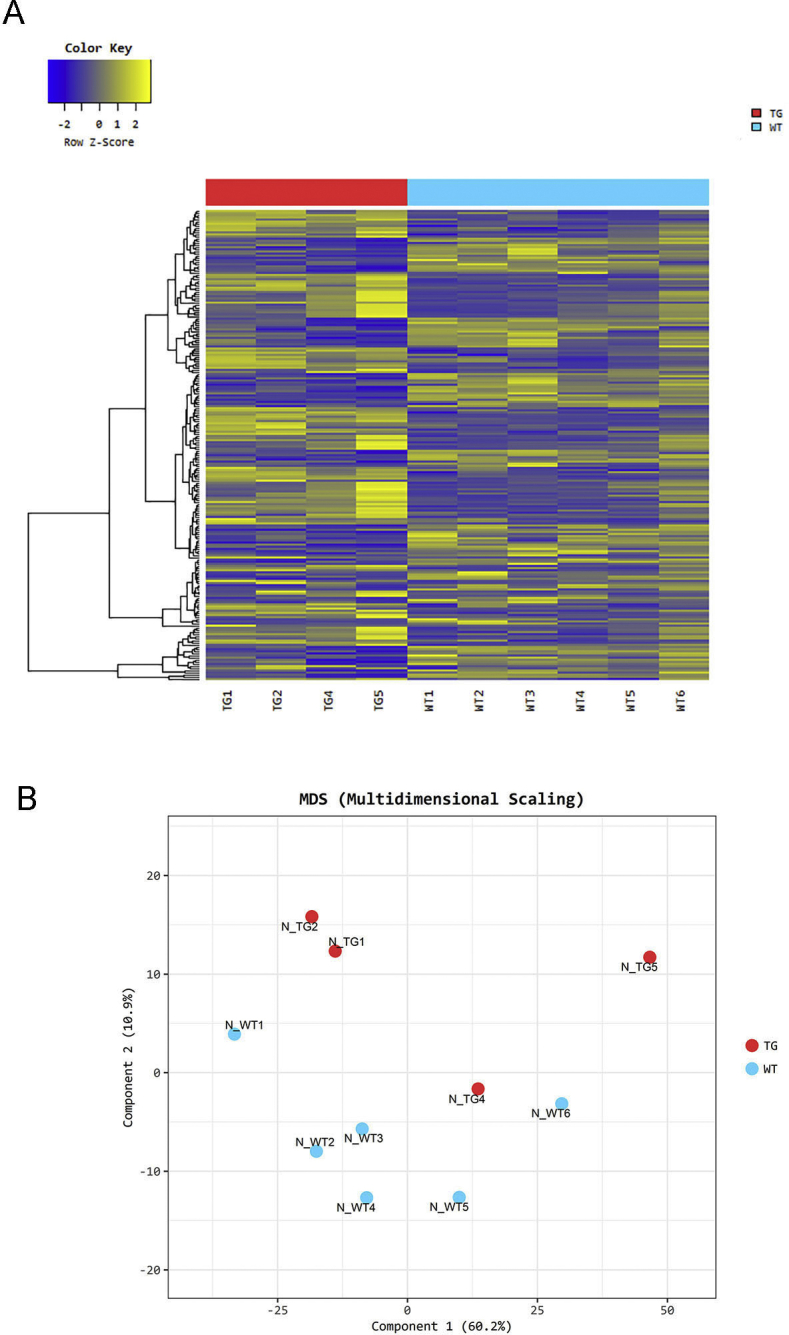
A: Heat map of one-way hierarchical clustering analysis (euclidean method, complete linkage), which clusters the similarity of genes and samples by expression level (normalized value) of significantly differentially expressed gene using Z-score for normalized value (log2 based). B: Multidimensional scaling analysis: using each sample's normalized value, the similarity between samples is displayed in a two-dimensional plot to show the variability of the total data. TG, transgenic or mutant genotype; WT, wild type.
Supplemental Figure S6.
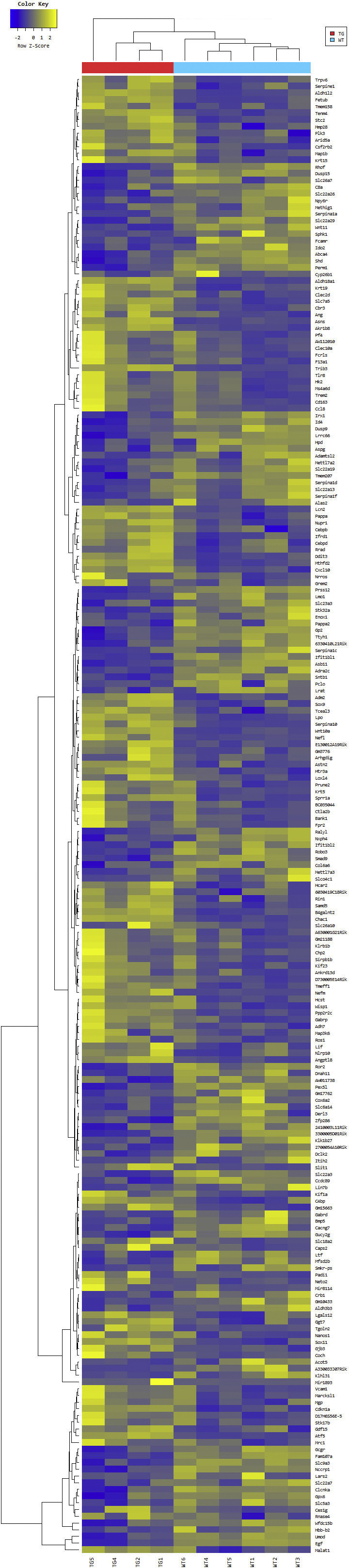
Transcriptional analysis of F4/80+ cells isolated from C105F/+ kidneys reveals an integrated stress response with ATF4 regulated gene expression. Heat map of the one-way hierarchical clustering using Z-score for normalized value between mutant (MT)/+/wild type of significantly up-regulated (yellow) or down-regulated (yellow) genes (log2 FD > 1 with Padjusted < 0.05) using Benjamini-Hochberg correction for multiple comparisons. There were 220 genes satisfying with fc2 and raw.p. TG, transgenic or mutant genotype.
Supplemental Figure S7.

A: TaqMan RT-PCR with RNA isolated from primary cultures of wild-type (WT) and mutant (mut)/+ medullary tubule epithelial cells and F4/80 mononuclear phagocytes isolated from WT and mut/+ kidneys. Results displayed as relative quantification (RQ) versus WT samples. B: Western blot analysis of uromodulin expression in tubule epithelial cell cultures at time of RNA isolation of PCR. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. TG, transgenic or mutant genotype.
Supplemental Figure S8.

Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining for apoptotic cells in kidney inner cortex of wild-type (WT; A) and mutant (mut)/+ (B) kidneys 1 day (D1) following ischemia-reperfusion (IR) injury and corresponding cell counts (H). Anti-Ly6G immunofluorescence staining for neutrophils in WT and mut/+ kidneys at low magnification (B and C) and at higher magnification (D and E) with double labeling for uromodulin (green) and Ly6G (red; F) and corresponding cell counts (G). Arrows indicate LY6G-positive neutrophils. ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001. Scale bars: 30 μm (A–D); 25 μm (E and F). LPF, low-power field; pos, positive.
Supplemental Figure S9.

Plasma creatinine (Cr) levels (horizontal bars indicate means) from sham operated age-matched mice [control (con)] and sacrificed at 24 hours [ischemia-reperfusion (IR) D1] and 7 days (IR D7) following IR injury. n = 8 to 10 per group. ∗∗P < 0.01, ∗∗∗∗P < 0.0001, and ∗∗∗∗∗P < 0.00001. Mut, mutant; WT, wild type.
Supplemental Figure S10.

Ngal immunofluorescence in kidney section from wild type (WT) 24 hours following ischemia-reperfusion injury (IRI; A) and mutant (mut)/+ control mice (green, uromodulin; red, Ngal; B) and 4-HNE and uromodulin double labeling in kidney sections from WT day 7 following IRI [arrows, double-positive (yellow) tubules; C] and mut/+ control (D) mice. Scale bars: 40 μm (A and B); 80 μm (C and D).
References
- 1.Bachmann S., Mutig K., Bates J., Welker P., Geist B., Gross V., Luft F., Alenina N., Bader M., Thiele B., Prasadan K., Raffi H., Kumar S. Renal effects of Tamm-Horsfall protein (uromodulin) deficiency in mice. Am J Physiol Renal Physiol. 2005;288:F559–F567. doi: 10.1152/ajprenal.00143.2004. [DOI] [PubMed] [Google Scholar]
- 2.Raffi H.S., Bates J.M., Laszik Z., Kumar S. Tamm-Horsfall protein acts as a general host-defense factor against bacterial cystitis. Am J Nephrol. 2005;25:570–578. doi: 10.1159/000088990. [DOI] [PubMed] [Google Scholar]
- 3.Jennings P., Aydin S., Kotanko P., Lechner J., Lhotta K., Williams S., Thakker R., Pfaller W. Membrane targeting and secretion of mutant uromodulin in familial juvenile hyperuricemic nephropathy. J Am Soc Nephrol. 2007;18:264–273. doi: 10.1681/ASN.2006020158. [DOI] [PubMed] [Google Scholar]
- 4.Bachmann S., Koeppen-Hagemann I., Kriz W. Ultrastructural localization of Tamm-Horsfall glycoprotein (THP) in rat kidney as revealed by protein A-gold immunocytochemistry. Histochemistry. 1985;83:531–538. doi: 10.1007/BF00492456. [DOI] [PubMed] [Google Scholar]
- 5.Steubl D., Buzkova P., Garimella P.S., Ix J.H., Devarajan P., Bennett M.R., Chaves P., Shlipak M., Bansal N., Sarnak M. Association of serum uromodulin with mortality and cardiovascular disease in the elderly-the Cardiovascular Health Study. Nephrol Dial Transplant. 2020;35:1399–1405. doi: 10.1093/ndt/gfz008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Micanovic R., Khan S., Janosevic D., Lee M.E., Hato T., Srour E.F., Winfree S., Ghosh J., Tong Y., Rice S., Dagher P., Wu X., El-Achkar T. Tamm-Horsfall protein regulates mononuclear phagocytes in the kidney. J Am Soc Nephrol. 2018;29:841–856. doi: 10.1681/ASN.2017040409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darisipudi M.N., Thomasova D., Mulay S.R., Brech D., Noessner E., Liapis H., Anders H. Uromodulin triggers IL-1β-dependent innate immunity via the NLRP3 inflammasome. J Am Soc Nephrol. 2012;23:1783–1789. doi: 10.1681/ASN.2012040338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Achkar T.M., Wu X.R., Rauchman M., McCracken R., Kiefer S., Dagher P.C. Tamm-Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am J Physiol Renal Physiol. 2008;295:F534–F544. doi: 10.1152/ajprenal.00083.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maydan O., McDade P.G., Liu Y., Wu X.R., Matsell D.G., Eddy A.A. Uromodulin deficiency alters tubular injury and interstitial inflammation but not fibrosis in experimental obstructive nephropathy. Physiol Rep. 2018;6:e13654. doi: 10.14814/phy2.13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bleyer A.J., Kidd K., Živná M., Kmoch S. Autosomal dominant tubulointerstitial kidney disease. Adv Chronic Kidney Dis. 2017;24:86–93. doi: 10.1053/j.ackd.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernascone I., Janas S., Ikehata M., Trudu M., Corbelli A., Schaeffer C., Rastaldi M., Devuyst O., Rampoldi L. A transgenic mouse model for uromodulin-associated kidney diseases shows specific tubulo-interstitial damage, urinary concentrating defect and renal failure. Hum Mol Genet. 2010;19:2998–3010. doi: 10.1093/hmg/ddq205. [DOI] [PubMed] [Google Scholar]
- 12.Johnson B.G., Dang L.T., Marsh G., Roach A.M., Levine Z.G., Monti A., Reyon D., Feigenbaum L., Duffield J., Uromodulin p. Cys147Trp mutation drives kidney disease by activating ER stress and apoptosis. J Clin Invest. 2017;127:3954–3969. doi: 10.1172/JCI93817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piret S.E., Olinger E., Reed A.A., Nesbit M.A., Hough T.A., Bentley L., Devuyst O., Cox R., Thakker R. Mouse model for inherited renal fibrosis associated with endoplasmic reticulum stress. Dis Model Mech. 2017;10:773–786. doi: 10.1242/dmm.029488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kemter E., Rathkolb B., Rozman J., Hans W., Schrewe A., Landbrecht C., Klaften M., Ivandic B., Fuchs H., Gailus-Durner V., Klingenspor M., de Angelis M., Wolf E., Wanke R., Aigner B. Novel missense mutation of uromodulin in mice causes renal dysfunction with alterations in urea handling, energy, and bone metabolism. Am J Physiol Renal Physiol. 2009;297:F1391–F1398. doi: 10.1152/ajprenal.00261.2009. [DOI] [PubMed] [Google Scholar]
- 15.Lee D., Shenoy S., Nigatu Y., Plotkin M. Id proteins regulate capillary repair and perivascular cell proliferation following ischemia-reperfusion injury. PLoS One. 2014;9:e88417. doi: 10.1371/journal.pone.0088417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gold M., Pul R., Bach J.P., Stangel M., Dodel R. Pathogenic and physiological autoantibodies in the central nervous system. Immunol Rev. 2012;248:68–86. doi: 10.1111/j.1600-065X.2012.01128.x. [DOI] [PubMed] [Google Scholar]
- 18.Chun J., Wang M., Wilkins M.S., Knob A.U., Benjamin A., Bu L., Pollak M. Autosomal dominant tubulointerstitial kidney disease-uromodulin misclassified as focal segmental glomerulosclerosis or hereditary glomerular disease. Kidney Int Rep. 2020;5:519–529. doi: 10.1016/j.ekir.2019.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bokhove M., Nishimura K., Brunati M., Han L., de Sanctis D., Rampoldi L., Jovine L. A structured interdomain linker directs self-polymerization of human uromodulin. Proc Natl Acad Sci U S A. 2016;113:1552–1557. doi: 10.1073/pnas.1519803113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youhanna S., Weber J., Beaujean V., Glaudemans B., Sobek J., Devuyst O. Determination of uromodulin in human urine: influence of storage and processing. Nephrol Dial Transplant. 2014;29:136–145. doi: 10.1093/ndt/gft345. [DOI] [PubMed] [Google Scholar]
- 21.Arase N., Arase H. Cellular misfolded proteins rescued from degradation by MHC class II molecules are possible targets for autoimmune diseases. J Biochem. 2015;158:367–372. doi: 10.1093/jb/mvv093. [DOI] [PubMed] [Google Scholar]
- 22.Krüger T., Benke D., Eitner F., Lang A., Wirtz M., Hamilton-Williams E.E., Engel D., Giese B., Müller-Newen G., Floege J., Kurts C. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J Am Soc Nephrol. 2004;15:613–621. doi: 10.1097/01.asn.0000114553.36258.91. [DOI] [PubMed] [Google Scholar]
- 23.Anders H.J., Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. 2014;25:1387–1400. doi: 10.1681/ASN.2014010117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luís A., Martins J.D., Silva A., Ferreira I., Cruz M.T., Neves B.M. Oxidative stress-dependent activation of the eIF2α–ATF4 unfolded protein response branch by skin sensitizer 1-fluoro-2,4-dinitrobenzene modulates dendritic-like cell maturation and inflammatory status in a biphasic manner [corrected] Free Radic Biol Med. 2014;77:217–229. doi: 10.1016/j.freeradbiomed.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Wölfle S.J., Strebovsky J., Bartz H., Sähr A., Arnold C., Kaiser C., Dalpke A., Heeg K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur J Immunol. 2011;41:413–424. doi: 10.1002/eji.201040979. [DOI] [PubMed] [Google Scholar]
- 26.Li Y., Chu N., Rostami A., Zhang G.X. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol. 2006;177:1679–1688. doi: 10.4049/jimmunol.177.3.1679. [DOI] [PubMed] [Google Scholar]
- 27.Olguín-Alor R., de la Fuente-Granada M., Bonifaz L.C., Antonio-Herrera L., García-Zepeda E.A., Soldevila G. A key role for inhibins in dendritic cell maturation and function. PLoS One. 2016;11:e0167813. doi: 10.1371/journal.pone.0167813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim M.G., Boo C.S., Ko Y.S., Lee H.Y., Cho W.Y., Kim H.K., Jo S. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2010;25:2908–2921. doi: 10.1093/ndt/gfq183. [DOI] [PubMed] [Google Scholar]
- 29.Zhang T., Song N., Fang Y., Teng J., Xu X., Hu J., Zhang P., Chen R., Lu Z., Yu X., Ding X. Delayed ischemic preconditioning attenuated renal ischemia-reperfusion injury by inhibiting dendritic cell maturation. Cell Physiol Biochem. 2018;46:1807–1820. doi: 10.1159/000489366. [DOI] [PubMed] [Google Scholar]
- 30.Bonventre J.V., Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams S.E., Reed A.A., Galvanovskis J., Antignac C., Goodship T., Karet F.E., Kotanko P., Lhotta K., Morinière V., Williams P., Wong W., Rorsman P., Thakker R. Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum. Hum Mol Genet. 2009;18:2963–2974. doi: 10.1093/hmg/ddp235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaeffer C., Merella S., Pasqualetto E., Lazarevic D., Rampoldi L. Mutant uromodulin expression leads to altered homeostasis of the endoplasmic reticulum and activates the unfolded protein response. PLoS One. 2017;12:e0175970. doi: 10.1371/journal.pone.0175970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trudu M., Schaeffer C., Riba M., Ikehata M., Brambilla P., Messa P., Martinelli-Boneschi F., Rastaldi M., Rampoldi L. Early involvement of cellular stress and inflammatory signals in the pathogenesis of tubulointerstitial kidney disease due to UMOD mutations. Sci Rep. 2017;7:7383. doi: 10.1038/s41598-017-07804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melentijevic I., Toth M.L., Arnold M.L., Guasp R.J., Harinath G., Nguyen K.C., Parker J.A., Neri C., Gabel C.V., Hall D.H., Driscoll M.C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature. 2017;542:367–371. doi: 10.1038/nature21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arndt L., Dokas J., Gericke M., Kutzner C.E., Müller S., Jeromin F., Thiery J., Burkhardt R. Tribbles homolog 1 deficiency modulates function and polarization of murine bone marrow-derived macrophages. J Biol Chem. 2018;293:11527–11536. doi: 10.1074/jbc.RA117.000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satoh T., Kidoya H., Naito H., Yamamoto M., Takemura N., Nakagawa K., Yoshioka Y., Morii E., Takakura N., Takeuchi O., Akira S. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature. 2013;495:524–528. doi: 10.1038/nature11930. [DOI] [PubMed] [Google Scholar]
- 37.Poosti F., Bansal R., Yazdani S., Prakash J., Post E., Klok P., van den Born J., de Borst M., van Goor H., Poelstra K. Selective delivery of IFN-γ to renal interstitial myofibroblasts: a novel strategy for the treatment of renal fibrosis. FASEB J. 2015;29:1029–1042. doi: 10.1096/fj.14-258459. [DOI] [PubMed] [Google Scholar]
- 38.Oldroyd S.D., Thomas G.L., Gabbiani G., El Nahas A.M. Interferon-gamma inhibits experimental renal fibrosis. Kidney Int. 1999;56:2116–2127. doi: 10.1046/j.1523-1755.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- 39.Jimenez S.A., Freundlich B., Rosenbloom J. Selective inhibition of human diploid fibroblast collagen synthesis by interferons. J Clin Invest. 1984;74:1112–1116. doi: 10.1172/JCI111480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Granstein R.D., Flotte T.J., Amento E.P. Interferons and collagen production. J Invest Dermatol. 1990;95 Suppl:75S–80S. doi: 10.1111/1523-1747.ep12874789. [DOI] [PubMed] [Google Scholar]
- 41.Suurmond J., Diamond B. Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest. 2015;125:2194–2202. doi: 10.1172/JCI78084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.