

Abstract
Background
Carbapenemase-producing Klebsiella pneumoniae (CP-Kp) poses distinct clinical challenges due to extensively drug resistant (XDR) phenotype, and sequence type (ST) 11 is the most dominant blaKPC-2-bearing CP-Kp clone in China. The purpose of this current retrospective study was to explore the genetic factors associated with the success of XDR CP-Kp ST11 strains circulated in the intensive care unit (ICU) of a Chinese tertiary hospital.
Methods
Six ST11 XDR CP-Kp strains were identified between May and December 2014 and validated by minimum inhibitory concentration examination, polymerase chain reaction, and pyrosequencing. The six ST11 XDR CP-Kp, as well as three multi-drug resistant (MDR) and four susceptible strains, were sequenced using single-molecule real-time method. Comprehensively structural and functional analysis based on comparative genomics was performed to identify genomic characteristics of the XDR ST11 CP-Kp strains.
Results
We found that ST11 XDR blaKPC-2-bearing CP-Kp strains isolated from inpatients spread in the ICU of the hospital. Functionally, genes associated with information storage and processing of the ST11 XDR CP-Kp strains were more abundant than those of MDR and susceptible strains, especially genes correlative with mobile genetic elements (MGEs) such as transposons and prophages. Structurally, eleven large-scale genetic regions taken for the unique genome in these ST11 XDR CP-Kp strains were identified as MGEs including transposons, integrons, prophages, genomic islands, and integrative and conjugative elements. Three of them were located on plasmids and eight on chromosomes; five of them were with antimicrobial resistance genes and eight with adaptation associated genes. Notably, a new blaKPC-2-bearing ΔΔTn1721-blaKPC-2 transposon, probably transposed and truncated from ΔTn1721-blaKPC-2 by IS903D and ISKpn8, was identified in all six ST11 XDR CP-Kp strains.
Conclusion
Our findings suggested that together with clonal spread, MGEs identified uniquely in the ST11 XDR CP-Kp strains might contribute to their formidable adaptability, which facilitated their widespread dissemination in hospital.
Keywords: Whole genome sequencing, Carbapenemase-producing Klebsiella pneumoniae, Mobile genetic elements, Antimicrobial resistance genes, Adaptation associated genes
Introduction
The human pathogenic carbapenemase-producing Klebsiella pneumoniae (CP-Kp) bacterium poses distinct clinical challenges due to its extensively drug resistant (XDR) phenotype.[1] CP-Kp causes hospital-acquired and long-term care-related infections that feature high morbidity and mortality.[2–4] It is now known that enzymes like Klebsiella pneumoniae carbapenemase (KPC) are important products of CP-Kp.[5] KPC-2 (with the identical protein sequence to KPC-1) was the first known variant of the KPC enzymes, and it was identified in a K. pneumoniae strain in North Carolina, USA in 1996.[6] In China, the first report of a KPC-producing K. pneumoniae strain was from Zhejiang province in 2007.[7] Since then, this pathogen has been identified in several Chinese provinces; and it has been demonstrated that sequence type (ST) 11 is the dominant clone amongst the CP-Kp strains in China.[8–17] Many researchers have explored the potential molecular factors that govern the success of the ST258 CP-Kp clone,[5,18] which is threatening European and American countries; however, relatively little progress has been made in the ST11 strains; so it is urgent to identify which characteristics have granted ST11 its particularly strong ability to spread rapidly.
Mobile genetic elements (MGEs), including plasmids, insertion sequences (ISs), transposons, integrons, and prophages, as well as integrative and conjugative elements (ICEs) and genomic islands,[19] are important carriers of antimicrobial resistance genes (ARGs) and adaptation associated genes.[20] Previous pan-genome comparative analyses has shown that horizontal gene transfer of MGEs across much of the bacterial world can explain many of the key differences between core genomes (the genes present in all strains within a group).[21] XDR CP-Kp strains of ST11 are common vehicle of MGEs, and thus, we speculate that the successful widespread of XDR CP-Kp strains of ST11 is related to certain MGEs, which are always closely related to the acquisition and spread of resistance and/or adaptation associated genes as well as the broader evolution of bacteria.
Single-molecule real-time (SMRT) based whole genome sequencing (WGS) has emerged as a powerful tool for deep dissection of bacterial genomes and their functions, and its ability to generate long reads (with an average size of 10 Kb) that can cross complex repeat regions has substantially facilitated functional genomics research about many species.[22] In the current study, we report a dissemination of ST11 XDR CP-Kp strains in intensive care unit (ICU) of a Chinese hospital between May 1st and December 31st, 2014 retrospectively. Then we assembled the complete genomes and plasmids of 13 clinical K. pneumoniae isolates based on SMRT sequencing data, including six ST11 CP-Kp XDR group strains, three non-CP-Kp multi-drug resistant (MDR) group strains and four susceptible (S) group strains, and performed comparative genomics analysis to explore the genetic factors unique to the ST11 XDR CP-Kp strains. Our analyses revealed extensive MGEs carrying ARGs and adaptation associated genes that can help to explain the widespread dissemination of XDR CP-Kp strains of ST11, and might facilitate the development of novel targets to these unique genetic factors.
Methods
Ethical approval
The retrospective study was approved by the Ethics Committee of Peking University People's Hospital (2015PHB037-01) and the requirement of written informed consent was waived.
Klebsiella pneumoniae strains and bacterial experiments
We analyzed six patients with CK-Kp infection who were admitted to a five-ward ICU of a Fujian tertiary hospital with 2400 beds in China between May 1st and December 31st, 2014. Six K. pneumoniae strains were isolated from urine, sputum, bile, ascites, end of catheter, and blood. Multi-locus sequence typing, ARGs including blaKPC-2, and later gene confirmation was determined by polymerase chain reaction (PCR) and pyrosequencing.[23] Minimum inhibitory concentrations were determined using a VITEK2 automated system (Biomerieux Vitek, Inc., France), and antibiotic susceptibilities were assessed according to the Clinical and Laboratory Standards Institute 2017 and the European Committee on Antimicrobial Susceptibility Testing 2017. According to an international expert proposal for the interim standard definitions for acquired resistance,[24] the six isolates were defined as XDR ST11 CP-Kp strains. Besides, another seven non-CP-Kp strains were isolated in the same hospital during the same period to compare with XDR CP-Kp strains, and three of these non-CP-Kp strains were defined as MDR and four were defined as susceptible (S) strains.[24]
Genomic DNA extraction, genome sequencing, and assembly
Bacterial cells of a total 13 K. pneumoniae strains were collected at the stationary phase by centrifugation (12,000 ×g) for 10 min at 4°C. Total DNA was extracted using a Qiagen DNA Mini Kit (Qiagen, Valencia, CA, USA) according to its protocol. The integrity of extracted DNA was assessed by gel electrophoresis using 0.7% agarose gel, and the quantification was performed using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Libraries were constructed according to the large SMRT Bell gDNA protocol (Pacific Biosciences, Menlo Park, CA, USA), and the whole genomes of isolates were sequenced via SMRT sequencing with the PacBio RS II platform. Genomes were assembled by using the Short Oligonucleotide Analysis Package (SOAPdenovo 2.3.0). The final contigs were checked for circularization, and the overlapping ends were trimmed.
Pan-genome analysis
The protein coding sequences (CDSs) were predicted using GeneMarkS software (http://topaz.gatech.edu/genemark/genemarks.cgi), and annotated via BLAST analysis against the NCBI non-redundant (NR) protein sequence database. The core genome for XDR, MDR, or S group strains was determined as homologous proteins (as defined by 70% coverage and 50% sequence identity using cd-hit[25]) presenting in all of the isolates inside the group. Each of the three intra-group sets of core gene sequences were created by extracting one sequence for every core gene in each group. Three intra-group sets of core genes were then compared and clustered by cd-hit[25] (using the same coverage and sequence identity above) and the “supercore” genes (core genes that were present in all three groups) were determined. The dispensable genome of XDR, MDR, or S group, designated as DI, DII, or DIII, was created by subtracting the “supercore” genes from each group's core genome. A Venn diagram was generated to depict the number of core genes in each of the three groups as well as the “supercore” genes and the dispensable genes.
Single-nucleotide polymorphisms (SNPs) and phylogenic analysis
The 13 K. pneumoniae genomes we sequenced were used for phylogenic analysis along with 12 K. pneumoniae and two Klebsiella variicola genomes from GenBank. The core SNPs inside each group strains and across the 27 examined genomes were called from the de novo-assembled sequences using the kSNP with k-mers size of 21.[26] The core SNPs across the 27 examined genomes were used to construct a phylogenetic tree using the maximum-likelihood based approach in FastTreeMP with 1000 bootstraps.[27]K. variicola X39[28] and K. variicola DSM 15968[29] were set as the out-group sequences. Local support values for each node were calculated. The tree was visualized in iTOL (https://itol.embl.de/).
Structural and functional analyses of bacterial genomes
All proteins were aligned against specialized databases such as the Clusters of Orthologous Groups (COG),[30] and the results were screened according to the highest hit coverage value. The replicon type of plasmids was identified using tools available at the Institut Pasteur website (https://bigsdb.pasteur.fr/). ISs and transposases within the 13 K. pneumoniae strains were detected using the online platform ISfinder, with an E-value cutoff set to 1e−5 (https://www-is.biotoul.fr/index.php). Prophages were predicted via the web server PHASTER (http://phaster.ca/). Genomic islands were predicted using the IslandPath-DIMOB tool.[31] ARGs were identified using the BLASTP-based Resistance Gene Identifier (RGI) (E-value cutoff set to 1e −30) available at the Comprehensive Antibiotic Resistance Database (CARD).[32] Any annotations to resistance conferred by base mutations were removed to prevent misidentification. To identify candidate adaptation associated genes within the 13 K. pneumoniae strains, proteins inside all the unique MGEs regions of the XDR strains were annotated against the NCBI NR database. A BLASTN-based whole genome comparison was performed and visualized using the BLAST Ring Image Generator (BRIG)[33] to exhibit the architecture and gene repertoire of the K. pneumoniae genomes. The strain F1, including the plasmid sequence pF1_1 and its chromosome sequence, was selected as the reference because F1 was the earliest strain isolated in the study period. We picked the IncFII plasmids carrying blaKPC-2 in the XDR group and the most similar plasmids from the MDR and S groups, and then compared them to pF1_1. Comparison was also performed between the F1 chromosome sequence and other chromosome sequences. Pairwise comparisons of the transposons of blaKPC-2 within K. pneumoniae genomes were visualized using EasyFig v2.3.[34]
Statistical analysis
The patients’ ages, genome sizes, and numbers of predicted CDSs and MGEs, as well as positive genes percentages of the PCR confirmation test, were compared using the Kruskal-Wallis and Mann-Whitney test. Categorical variables of predicted functional genes were compared by the Chi-square test. Using Prism GraphPad software version 6.01 (GraphPad software Inc.; La, Jolla, CA, USA), P ≤ 0.05 was considered significant.
Sequence data accession numbers
The whole genomes we sequenced in this study are available in the NCBI repository under the accession numbers of CP026130-CP026131 (F1), CP026132-CP026135 (F5), CP026136-CP026139 (F77), CP026140-CP026142 (F127), CP026145-CP026148 (F132), CP026149-CP026152 (F138), CP026153-CP026154 (F10(AN)), CP026155-CP026156 (B12(AN)), CP026157-CP026158 (F93-2), CP026162-CP026163 (F13), CP026164-CP026166 (F81), CP026159 (F89-1), and CP026160-CP026161 (F93-1).
Results
Dissemination of six ST11 XDR CP-Kp strains
The six CP-Kp strains, which were resistant to imipenem and meropenem, were isolated from different wards of the ICU in the hospital within 8 months (between May 1st and December 31st, 2014) [Table 1]. They were all identified as the ST11 type, and the capsular type K47. BlaKPC-2 gene was detected in all of the six ST11-type CP-Kp strains, with other β-lactamase genes mainly blaCTX-M-65, blaSHV-12, blaSHV-11, and blaTEM-1 [Supplementary Table 1].
Table 1.
Characteristics of the thirteen Klebsiella pneumoniae strains and their source patients.
However, WGS data confirmed that the six ST11 XDR CP-Kp strains were not identical. First, the sizes of their chromosomes varied from 5,511,418 to 5,539,281 bp. Second, there were 68 core SNPs among them. Third, the antimicrobial resistance phenotypes of the XDR strains were not the same, as shown in Supplementary Table 2. Fourth, analysis of the assembled complete genomes using RGI tool in CARD identified different distributions of ARGs in the six XDR CP-Kp strains [Supplementary Table 3]. Fifth, there were differences of the MGEs, for example, ISs, prophages, and genomic islands, found among the six XDR CP-Kp strains [Supplementary Table 1, Supplementary Table 4, and Supplementary Figure 1]. Lastly, they did not have the same Inc plasmid types. Each of the CP-Kp strains in the XDR group has an IncFII plasmid bearing a blaKPC-2 gene. Besides the IncFII plasmid, additional plasmids of different Inc types were found in the XDR group. For example, a plasmid of IncFI was found in isolate F5, and a plasmid of Incl1 was found in isolate F127. These showed no redundancy of the six ST11 blaKPC-2-bearing K. pneumoniae strains.
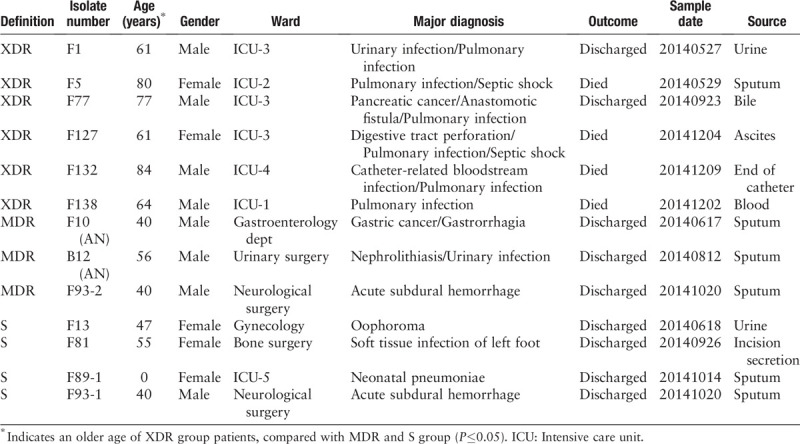
WGS data were then used to analyze the dissemination of the six ST11 XDR CP-Kp strains. Phylogenetic analysis was employed to assess the relationships between the 13 SMRT sequenced strains along with 12 K. pneumoniae and two K. variicola strains from GenBank [Supplementary Table 4]. The six ST11 XDR CP-Kp strains (F1, F5, F77, F127, F132, and F138) were significantly supported as a single clade that was clearly separate from other groups [Figure 1A]. There were fewer core SNPs (68) among the six ST11 XDR CP-Kp strains, while there were much more core SNPs among the three MDR strains (26,131), or the four susceptible strains (32,724). The numbers of core SNPs between each pair of the six ST11 CP-Kp XDR strains ranged from 4 to 55 [Supplementary Figure 2], thus suggesting the phylogenetically high similarity of these strains. Furthermore, there were seven core SNPs between F1 and F127 and 4 to 14 core SNPs among F5, F77, F132, and F138, while there were 50 to 55 core SNPs differentiating the strains of F1 and F127 from the strains of F5, F77, F132, and F138. According to a recently published study, the distance of no more than 23 SNPs between each pair of CP-Kp isolates suggests a local transmission.[35] Therefore we consider that the six ST11 CP-Kp strains we sequenced contain two extremely close clones. They had evolved with chromosomes or plasmids mutation and/or additional acquisition of other plasmids-bearing resistance genes from a common ancestor or clone, and disseminated in the ICU of the hospital at the period we collected.
Figure 1.
(A) The phylogeny analysis of K. pneumoniae strains. SNPs across the 27 examined genomes were called. The core SNPs were used to construct phylogenetic trees using the ML method based approach FastTreeMP with 1000 bootstraps. K. variicola X39 and DSM 15968 was included as the out-group sequences. The branch bearing double hatch marks indicates that it has been truncated and is not proportional to the rest. (B) Venn diagram of the tally of homologous proteins shared by or unique among K. pneumoniae strains of the XDR, MDR, and S groups. The core genome for XDR, MDR, or S group was determined as homologous proteins presenting in all of the isolates inside the group. The “supercore” genome was determined as the core genes that were present in each of the groups. The dispensable genome of each group were created by subtracting the “supercore” from each group's core genome. (C) Distribution of COG functional categories among DI, DII, and DIII. All proteins in DI, DII, and DIII were aligned against the COG database and comparisons among DI, DII, and DIII were conducted. COG: Clusters of Orthologous Groups; K. pneumoniae: Klebsiella pneumoniae; MDR: Multidrug-resistant; ML: Maximum-likelihood; SNPs: Single-nucleotide polymorphisms; XDR: Extensively drug resistant.
Different essential features between XDR strains and MDR and S strains
The essential features between the six XDR group strains and the three MDR group and four S group strains were different [Table 1]. Epidemiologically, consistent with previously reported epidemiological trends,[36–38] we noted that the average age of XDR group patients was older than that of MDR (P = 0.0238) and S (P = 0.0095) group significantly. However, the average age of MDR group patients was similar to that of S group (P = 0.8857). All XDR group patients suffered from pulmonary infection, whereas none of the MDR group and S group patients did. All the XDR group strains were isolated from patients in the ICU, whereas only one strain amongst the MDR group and S group was isolated from a patient in the ICU. Again emphasizing the known increased threat from XDR strains,[1] four XDR group patients died in the hospital, while none of the MDR or S group patients died.
The sizes of six XDR genomes ranged from 5.68 to 5.92 Mb, larger than that of the three MDR genomes (5.20–5.64 Mb) and four S genomes (5.27–5.49 Mb) significantly (P = 0.0238 and 0.0095, respectively), whereas no difference between four S genomes and three MDR genomes (P = 0.6286) [Supplementary Table 1]. The six XDR genomes harbored predicted CDSs between 5545 and 5802, more than three MDR genomes (between 4925 and 5496) and four S genomes (between 5016 and 5256) significantly (P = 0.0238 and 0.0095, respectively), while the four S genomes were similar to three MDR genomes (P = 0.6286). Functional annotation predictions based on COG classification showed that there were between 4214 and 4582 genes assigned to COG categories. On the whole, the distribution of functional categories among the thirteen genomes were similar [Supplementary Figure 3].
Distinct MGE distributions between XDR strains and MDR and S strains
The MGEs carried by the three groups of the 13 examined K. pneumoniae strains [Supplementary Table 1] were dissected. Although the average numbers of ISs in the three groups were discrepant (P = 0.0241), there were not significant differences between XDR (37) and MDR (22) groups (P = 0.5119), neither between S (10) and MDR groups (P = 0.2857). However some ISs in the XDR group, like ISKpn26, IS26, or ISKpn14, which are always present in the genomes of the Klebsiella species,[39,40] was apparently more abundant than that in the MDR and S groups [Supplementary Figure 1]. For instance, there were an average of 15.83 ISKpn26 in the XDR group, compared with that of only 0.33 and 0 in the MDR group and susceptible group, respectively. The number of prophages identified in the strains of XDR (13.33) group was much larger than that of MDR (4.00) and S (4.25) groups (P = 0.0238 and 0.0048, respectively), whereas there was no significant difference between MDR and S groups (P = 0.8857). Similarly, the average numbers of genomic islands in XDR group (21.17) were larger than that in MDR group (15.67) and S group (5.75) significantly (P = 0.0238 and 0.0048, respectively), yet not in the S group compared with that of the three MDR genomes (P = 0.0857). The distributions of prophages and genomic islands of the XDR group strains were similar, but there were significant differences between groups. The detailed information of prophages and genomic islands was listed in Supplementary Table 4.
Different predicted functional genes between XDR strains and MDR and S strains
There were 4264 core genes in the MDR strains and 4155 genes in the S strains, markedly lower than the number (5036) of core genes in the XDR group. Pan-genome analysis based on the three groups showed that there were a total of 5158 genes comprising their pan-genome. In the pan-genome, 4009 genes were identified to be shared by all three groups; these were defined as the supercore genome. The non-supercore genes within the XDR, MDR, and S groups were assigned to dispensable genomes and defined as, respectively, DI (1027), DII (255), and DIII (146) [Figure 1B].
Functional classification analysis of DI, DII, DIII, and the supercore genome was carried out by comparison against the COG database [Figure 1C, Supplementary Table 6]. We classified the COG functional categories into four classes, namely, metabolism, information storage and processing, cellular processes and signaling, and “poorly characterized.” An apparently high proportion of genes in DI (32.47%) were assigned to COG categories involved in information storage and processing, in comparison with that in supercore genome (18.61%), DII (17.60%), and DIII (16.33%) (P = 0.0329, 0.0206 and 0.0124, respectively) [Supplementary Table 6]. In particular, the proportion of COG categories [X] (mobilome: prophages, transposons; 13.89%) within DI is apparently higher than that in the supercore genome, DII, or DIII (P = 0.0002, 0.0054, and 0.0054, respectively), suggesting the presence of a large amount of MGEs and implying that much of the genetic and phenotypic diversity amongst the clinical isolates may be a result of horizontal gene transfer. As expected, a relatively low proportion of genes dispersed in the DI were predicted to be involved in metabolic profiles (32.64%) compared with that in supercore genome (48.90%) (P = 0.0295), reflecting a relatively low proportion of essential genes supporting basic bacterial activities and; therefore, a relative abundance of unessential genes such as MGEs. For genes involved in cellular processes and signaling and in the “poorly characterized” class, there was no significant discrepancy between supercore genome, DI, DII, and DIII.
Linking unique large genomic regions in XDR genomes to MGEs
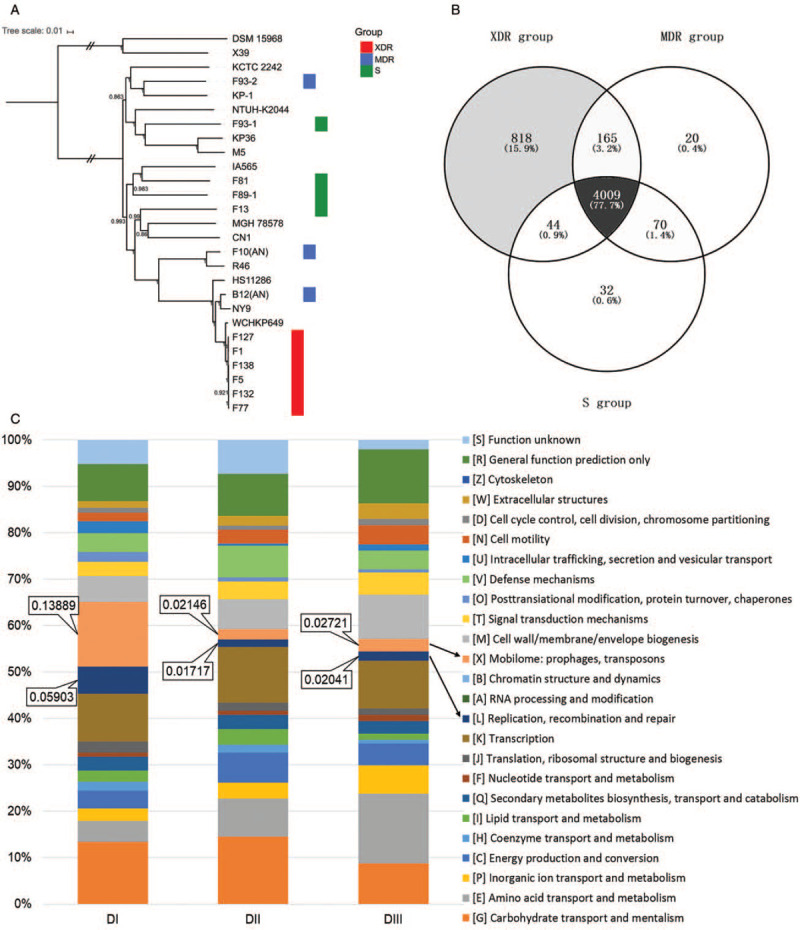
We conducted a further inspection of a large number of genes unique to the XDR ST11 CP-Kp strains with the idea that these unique genomic regions may be related to MGEs that may have contributed to the wide dissemination of these strains. Gene names and genomic positions for the 818 genes unique to the core genome of XDR ST11 CP-Kp strains were extracted and manually inspected: this identified 11 large (>9 Kb) genomic regions that ranged in size from 9.21 to 143.20 Kb. Three regions were in the IncFII plasmids, and eight were in the chromosomes. These 11 unique large genomic regions in the XDR group strains basically match to five MGEs carrying ARGs and eight MGEs carrying adaptation associated genes.
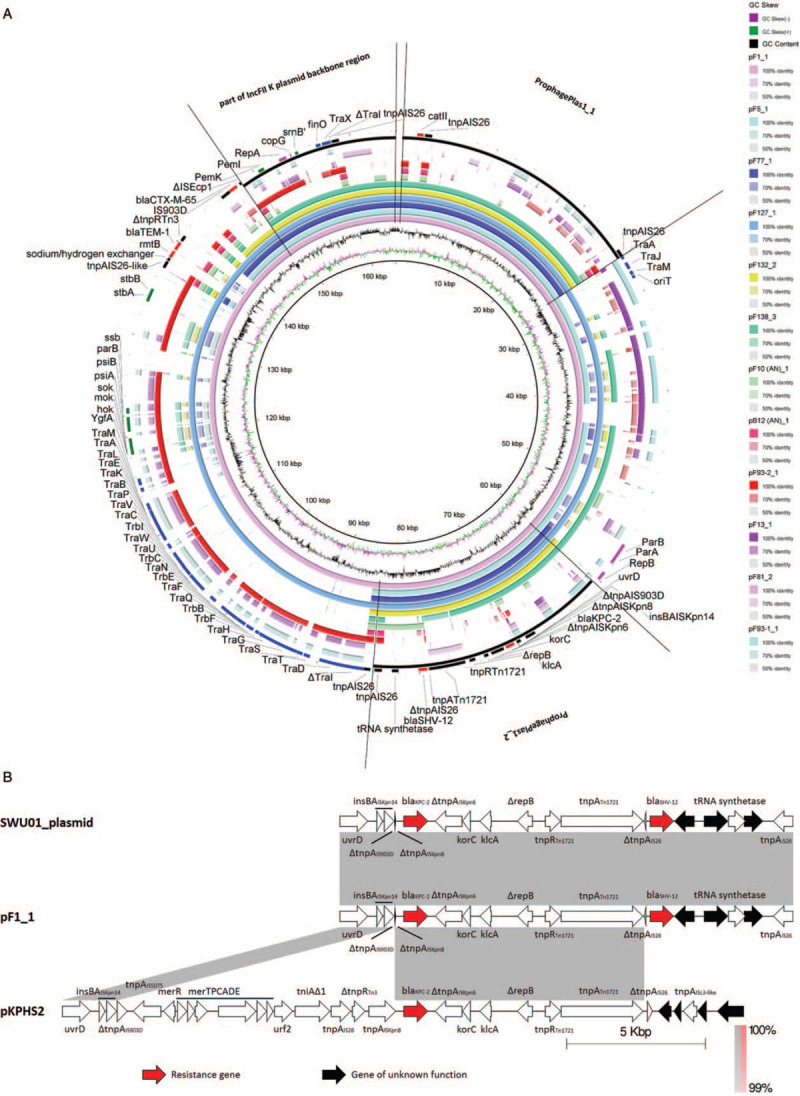
Of the five MGEs carrying ARGs, three were located on IncFII plasmids and two on chromosomes. For plasmids, XDR group-specific MGEs included a 16.05 Kb IncFII plasmid partial backbone and two prophages (ProphagePlas1_1 and ProphagePlas1_2) [Figure 2A, Supplementary Table 7, and Supplementary Table 8]. The IncFII plasmid itself is a large MGE, and many IncFII plasmid carry blaKPC-2 as well as other ARGs.[41] Homologous sequences of this region in GenBank are from K. pneumoniae, Proteus mirabilis, Escherichia coli, and Escherichia albertii which are all opportunistic pathogens of the Enterobacteriaceae. BlaKPC-2 is carried by ProphagePlas1_2 and is located within a transposon that is a variant of the ΔTn1721-blaKPC-2.[19] We named it as ΔΔTn1721-blaKPC-2 [Figure 2B]. The unique 25.45 Kb prophage region (ProphagePlas1_1) carries a chloramphenicol acetyltransferase catII, which can inactivate chloramphenicol.[42] Homologous sequences of this region in GenBank are all from opportunistic pathogens of the Enterobacteriaceae such as K. pneumoniae, E. coli, Citrobacter freundii, or Enterobacter cloacae. The composite transposon of catII (composed of catII and a transferase of IS26, tnpAIS26), the ProphagePlas1_1, and the IncFII plasmid might collectively contribute the horizontal genetic exchange of catII. Besides, the two XDR group-specific chromosome-bearing MGEs carrying ARGs included a class 1 integron In127 (carrying sul1 and aadA) that was carried by ProphageChr1_6, and another class 1 integron In610 (carrying cmlA, ANT(2”)-Ia, APH(3”)-I, APH(6)-I, and a gene for a puromycin acetyltransferase) that was carried by ICE_F1 [Figure 3, Supplementary Table 7, and Supplementary Table 8].
Figure 2.
(A) Alignment of the plasmid sequences of the K. pneumoniae strains. BLASTN-based whole genome comparison was performed and visualized using BRIG to exhibit the architecture and gene repertoire of a total of twelve K. pneumoniae plasmids, using the plasmid sequence of pF1_1 as a reference. Part of the IncFII plasmid backbone region, ProphagePlas1_1, ProphagePlas1_2, and the significant genes are indicated by rectangles. (B) Transposon carrying blaKPC-2 located in our isolates (ΔΔTn1721-blaKPC-2) was compared with that in pKPHS2 (ΔTn1721- blaKPC-2) and that in the unnamed plasmid of SWU01 (ΔΔTn1721-blaKPC-2). Regions of synteny between adjacent schematics are indicated by the shaded areas; the matching percentage nucleotide sequence identity for each such region is indicated. These schematics are drawn to scale. K. pneumoniae: Klebsiella pneumoniae.
Figure 3.
Alignment of the chromosome sequences of the K. pneumoniae strains. BLASTN-based whole genome comparison was performed and visualized using BRIG to exhibit the architecture and gene repertoire of a total of thirteen K. pneumoniae genomes, using the chromosome sequence of F1 as a reference. Regions of prophages, genomic islands, ICEs, and integrons are indicated by rectangles. ICEs: Integrative and conjugative elements; K. pneumoniae: Klebsiella pneumoniae.
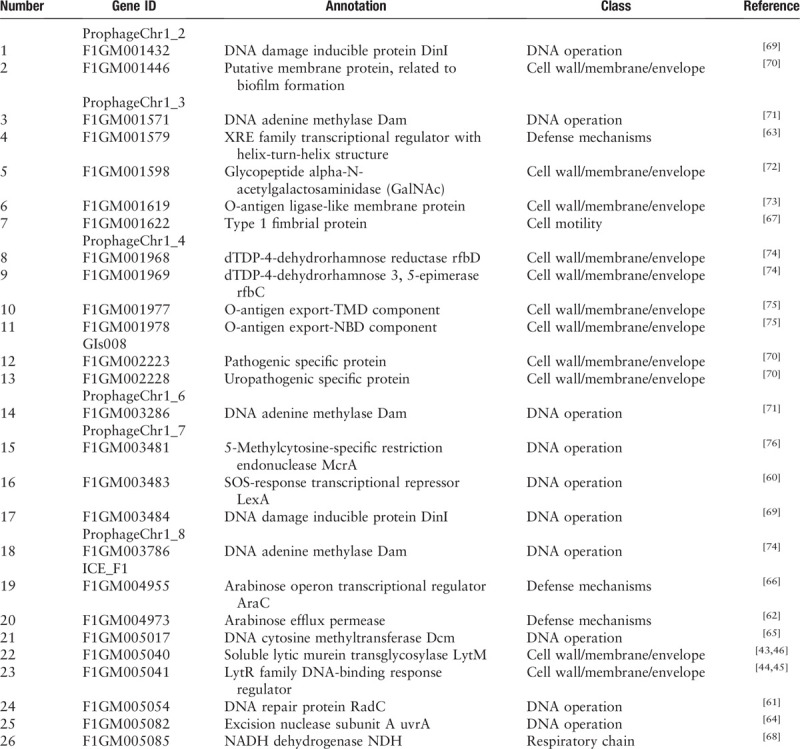
We also detected a variety of adaptation associated genes that were carried by 8 of the eleven XDR group-specific MGEs from chromosomes, including six prophages, one genomic island, and one ICE. The functions of these adaptation associated genes could be classified into five classes: cell wall/membrane/envelope, DNA operation, defense mechanisms, cell motility, and respiratory chain [Table 2]. Of particular note, we found that cell wall/membrane/envelope associated genes—responsible for the biosynthesis of lipopolysaccharide and biofilms—were present in ProphageChr1_2 of the XDR strains. Moreover, we found on ICE_F1 both LytM and the regulator LytR, which are known to participate in the synthesis of bacterial cell wall and the formation of biofilms and which likely enhance virulence, and the ability of anti-innate immune killing[43–45]; these may also participate in maintaining the integrity of bacterial plasma membrane and promoting β-lactam resistance.[46]
Table 2.
Adaptation associated genes on MGEs unique to the chromosomes of XDR Klebsiella pneumoniae strains.
Furthermore, to confirm preliminarily that these MGEs were XDR CP-Kp group specific, we detected some genes of these XDR-specific MGEs by PCR using a total of 98 K. pneumoniae strains we collected subsequently as templates [Supplementary Table 9]. The percentage of positive genes detected in XDR strains was larger than that in MDR (P = 0.0045) and S (P < 0.0001) strains significantly, whereas there was no significant difference between MDR and S strains (P = 0.7081) [Supplementary Table 10].
Discussion
ST11 is a high-risk clone often presented as the XDR phenotype, which is associated with KPC-2 dissemination, and is commonly found in Asian countries especially in China,[16,47–49] and in Latin American countries.[50] The ST11 genomes were found to be highly heterogeneous based on the patterns of SNPs.[51] In the present study, we found and WGS-confirmed a dissemination of ST11 XDR CP-Kp strains in a Fujian tertiary teaching hospital within eight months in 2014, and it has a warning role on the infection prevention and control in this hospital. Two extremely close clones were identified in these ST11 blaKPC-2-bearing K. pneumoniae strains, and they were considered to spread in the ICU of the hospital with open genomes and with evolution of chromosomes or plasmids mutations and/or genetic recombination from a common ancestor or clone.
Then, we found that the unique genes in the XDR group strains are related to MGEs either functionally or structurally. Horizontal gene transfer and subsequent recombination was considered another considerable factor for the widespread dissemination of ST11 XDR CP-Kp strains apart from clonal spread.[52] We will discuss this MGEs from two aspects: MGEs carrying ARGs and MGEs carrying adaptation associated genes.
MGEs specific to the XDR group strains on the IncFII plasmid and on chromosomes (including plasmids themselves, transposons, integrons, prophages, ICEs, and genomic islands) are well-known as prominent carriers of ARGs. All of the blaKPC-2 genes in the XDR group are carried by IncFII plasmids. Note that IncFII type plasmids are of the salient incompatibility group blaKPC-2-harboring plasmids, and commonly have low copy number, harbor multiple replicons, and exist widely in different species of the Enterobacteriaceae,[53,54] especially amongst opportunistic pathogens. We found that blaKPC-2 genes in the ST11 CP-Kp strains are located on transposons nested into prophages and plasmids.
Notably, a ΔΔTn1721-blaKPC-2 transposon carried by ProphagePlas1_2 in all of the six ST11 CP-Kp strains sequenced has never been reported before: it might have been transposed and then truncated by an IS903D and an ISKpn8 that is flanked on the left and right sides of a region encompassing IS5075-ΔTn21 (harboring the anti-mercury operon) -ΔIn2-IS26-ΔTn3 located on pKPHS2 (ΔTn1721- blaKPC-2).[19] A study found that the deletion of tnpRTn3 increased the transposition frequency by 16-fold.[55] Likewise, we speculate that the bacteria proactively lost the aforementioned IS5075-ΔTn21-ΔIn2-IS26-ΔTn3 genetic region to order to increase the transposition frequency, thereby promoting horizontal gene transfer. Additionally, the bacteria might passively lose the genetic region IS5075-ΔTn21-ΔIn2-IS26-ΔTn3 harboring the anti-mercury operon due to the lack of mercury pressure in the environment. Furthermore, considering that the blaKPC-2 bearing transposon found in the most similar plasmid from SWU01[56] resembles ΔΔTn1721-blaKPC-2 in our sequences, we suspect that ΔΔTn1721-blaKPC-2 in our sequences was transposed, with or without ProphagePlas1_2 or IncFII plasmid, to the unnamed plasmid of SWU01. Besides blaKPC-2, there are also other ARGs carried by MGEs. The selective advantage of XDR phenotypes due to these ARGs may have played an important role in promoting ST11 CP-Kp strains to flourish in the face of heavy antibiotic pressure in the healthcare environment and may have favored their dissemination.
Studies found that prophages, genomic islands, and ICEs of CP-Kp strains always contain some proteins which have putative functions associated with bacterial virulence,[57–59] that are speculated to make a contribution to the strong fitness of these strains. XDR group-specific MGEs (on chromosome) including six prophages, one genomic island, and one ICE, carry adaptation associated genes. These adaptation associated genes participate, for example, in SOS responses and help repair damaged DNA,[60,61] potentially protecting ST11 CP-Kp strains from environmental attacks and enhancing of the resistance of these strains to antibiotics,[46,62,63] UV radiation,[64] and other chemical stresses.[65] These adaptation associated genes also likely increase the strain's abilities for host cell invasion and virulence,[66,67] as they have functional annotations relating to the progress of bacterial recognition, adhesion, and pathogenesis.
Due to the lack of available effective antibiotics, infections caused by CP-Kp strains have significantly higher morbidity and mortality than non-CP-Kp strains.[2–4] The higher risk of CP-Kp infection is associated with a number of patient factors, including being admitted to the ICU, being older, or presenting with pulmonary infection.[36–38] It bears emphasis that the XDR group strains isolated and analyzed in our study were from the ICU and that four of the patients from which these strains were isolated eventually died. Thus, the adaptation associated genes which we identified in these strains are attractive potential drug targets in the future.[68] Moreover, it will be interesting to track possible horizontal gene transfer of MGEs from these strains, as such transfer could promote the strong adaptive traits in other bacteria and thereby favoring their survival and spread.
There were some limitations in our research. This retrospective study could not provide immediate assistance to effectively control CP-Kp, and real-time WGS are urgently required to improve the surveillance and management of nosocomial infection. The sample size was limited, and although PCR confirmation was performed with some genes unique to XDR group in another 98 strains, further verifications based on larger-scale bioinformatics analysis and even biological experiments are waiting to be performed to confirm our findings. However, to some extent, our results could illustrate the cause of the success of XDR ST11 CP-Kp strains, and could provide an idea of identifying important genetic factors of this tricky infection caused by XDR ST11 CP-Kp strains clinically.
In summary, to the best of our knowledge, this is a novel study to investigate the widespread mechanism of the dominant ST11 XDR CP-Kp strains based on WGS and comparative genomics in China. Our study demonstrated a dissemination of ST11 XDR CP-Kp strains in a Chinese hospital, indicating that prevention and control strategies for CP-Kp nosocomial infection needed to be investigated. Using SMRT sequencing, which enables analysis of long and repetitive sequence, we identified MGEs carrying ARGs and adaptation associated genes potentially contribute to the strong fitness of the ST11 XDR CP-Kp strains, and helped probe the genetic basis of their widespread dissemination. Meanwhile, a new blaKPC-2-bearing ΔΔTn1721-blaKPC-2 transposon identified in all of the ST11 XDR CP-Kp we sequenced was reported. Both clonal spread and horizontal gene transfer were related to the ST11 XDR CP-Kp strains dissemination. Our study assists to define potential new targets to develop more effective strategies for the control and prevention of CP-Kp.
Data availability statement
The datasets generated for this study can be found in the GenBank (https://www.ncbi.nlm.nih.gov/genome/genomes/815).
Acknowledgements
The authors thank Dr. Xing Shi (Department of Respiratory and Critical Care Medicine, Peking University People's Hospital, Beijing, China) for the technical help in figure-drawing assistance. The authors thank the Company of Novogene, Beijing, China for the technical help in WGS data analysis.
Funding
This work was supported by the Chinese Ministry of Science and Technology (No. 2016YFC0903800), the National Natural Science Foundation of China (No. 81870010), and the Natural Science Foundation of Beijing Municipality (No. 7192217).
Conflicts of interest
None.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
How to cite this article: Li DX, Zhai Y, Zhang Z, Guo YT, Wang ZW, He ZL, Hu SN, Chen YS, Kang Y, Gao ZC. Genetic factors related to the widespread dissemination of ST11 extensively drug-resistant carbapenemase-producing Klebsiella pneumoniae strains within hospital. Chin Med J 2020;133:2573–2585. doi: 10.1097/CM9.0000000000001101
Supplemental digital content is available for this article.
References
- 1.Navon-Venezia S, Kondratyeva K, Carattoli A. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev 2017; 41:252–275. doi: 10.1093/femsre/fux013. [DOI] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR Morb Mortal Wkly Rep 2013; 62:165–170. [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Z, Qin RR, Huang L, Sun LY. Risk factors for carbapenem-resistant Klebsiella pneumoniae infection and mortality of Klebsiella pneumoniae infection. Chin Med J 2018; 131:56–62. doi: 10.4103/0366-6999.221267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munoz-Price LS, Poirel L, Bonomo RA, Schwaber MJ, Daikos GL, Cormican M, et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect Dis 2013; 13:785–796. doi: 10.1016/s1473-3099(13)70190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Mathema B, Chavda KD, DeLeo FR, Bonomo RA, Kreiswirth BN. Carbapenemase-producing Klebsiella pneumoniae: molecular and genetic decoding. Trends Microbiol 2014; 22:686–696. doi: 10.1016/j.tim.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yigit H, Queenan AM, Anderson GJ, Domenech-Sanchez A, Biddle JW, Steward CD, et al. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother 2001; 45:1151–1161. doi: 10.1128/AAC.45.4.1151-1161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei ZQ, Du XX, Yu YS, Shen P, Chen YG, Li LJ. Plasmid-mediated KPC-2 in a Klebsiella pneumoniae isolate from China. Antimicrob Agents Chemother 2007; 51:763–765. doi: 10.1128/AAC.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qi Y, Wei Z, Ji S, Du X, Shen P, Yu Y. ST11, the dominant clone of KPC-producing Klebsiella pneumoniae in China. J Antimicrob Chemother 2011; 66:307–312. doi: 10.1093/jac/dkq431. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Du SX, Zhang JN, Liu SH, Zhou YY, Wang XR. Spreading of extended-spectrum beta-lactamase-producing Escherichia coli ST131 and Klebsiella pneumoniae ST11 in patients with pneumonia: a molecular epidemiological study. Chin Med J 2019; 132:1894–1902. doi: 10.1097/CM9.0000000000000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gu B, Bi R, Cao X, Qian H, Hu R, Ma P. Clonal dissemination of KPC-2-producing Klebsiella pneumoniae ST11 and ST48 clone among multiple departments in a tertiary teaching hospital in Jiangsu Province, China. Ann Transl Med 2019; 7:716.doi: 10.21037/atm.2019.12.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D, Li H, Zhao Y, Qiu Y, Xiao L, He H, et al. Characterization of carbapenem-resistant Klebsiella pneumoniae in a tertiary hospital in Fuzhou, China. J Appl Microbiol 2020; doi: 10.1111/jam.14700 [Published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou K, Xiao T, David S, Wang Q, Zhou Y, Guo L, et al. Novel subclone of carbapenem-resistant Klebsiella pneumoniae sequence type 11 with enhanced virulence and transmissibility, China. Emerg Infect Dis 2020; 26:289–297. doi: 10.3201/eid2602.190594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu X, Zhang W, Zhao Z, Ye C, Zhou S, Wu S, et al. Molecular characterization of carbapenem-resistant Klebsiella pneumoniae isolates with focus on antimicrobial resistance. BMC Genomics 2019; 20:822.doi: 10.1186/s12864-019-6225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chi X, Hu G, Xu H, Li X, Xiao T, Zhou Y, et al. Genomic analysis of a KPC-2-producing Klebsiella pneumoniae ST11 outbreak from a teaching hospital in Shandong Province, China. Infect Drug Resist 2019; 12:2961–2969. doi: 10.2147/IDR.S221788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Zhang Y, Yu SL, Zhou Y, Yang SY, Jin JL, et al. Tracking carbapenem-producing Klebsiella pneumoniae outbreak in an intensive care unit by whole genome sequencing. Front Cell Infect Microbiol 2019; 9:281.doi: 10.3389/fcimb.2019.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Feng Y, Tang G, Lin J, Huang W, Qiao F, et al. Carbapenem-resistant isolates of the Klebsiella pneumoniae complex in Western China: the common ST11 and the surprising hospital-specific types. Clin Infect Dis 2018; 67:S263–S265. doi: 10.1093/cid/ciy662. [DOI] [PubMed] [Google Scholar]
- 17.Sui W, Zhou H, Du P, Wang L, Qin T, Wang M, et al. Whole genome sequence revealed the fine transmission map of carbapenem-resistant Klebsiella pneumonia isolates within a nosocomial outbreak. Antimicrob Resist Infect Control 2018; 7:70.doi: 10.1186/s13756-018-0363-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chmelnitsky I, Shklyar M, Hermesh O, Navon-Venezia S, Edgar R, Carmeli Y. Unique genes identified in the epidemic extremely drug-resistant KPC-producing Klebsiella pneumoniae sequence type 258. J Antimicrob Chemother 2013; 68:74–83. doi: 10.1093/jac/dks370. [DOI] [PubMed] [Google Scholar]
- 19.Bi D, Jiang X, Sheng ZK, Ngmenterebo D, Tai C, Wang M, et al. Mapping the resistance-associated mobilome of a carbapenem-resistant Klebsiella pneumoniae strain reveals insights into factors shaping these regions and facilitates generation of a ’resistance-disarmed’ model organism. J Antimicrob Chemother 2015; 70:2770–2774. doi: 10.1093/jac/dkv204. [DOI] [PubMed] [Google Scholar]
- 20.Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol 2005; 3:722–732. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 21.Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev 2005; 15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Loman NJ, Pallen MJ. Twenty years of bacterial genome sequencing. Nat Rev Microbiol 2015; 13:787–794. doi: 10.1038/nrmicro3565. [DOI] [PubMed] [Google Scholar]
- 23.Diancourt L, Passet V, Verhoef J, Grimont PA, Brisse S. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol 2005; 43:4178–4182. doi: 10.1128/JCM.43.8.4178-4182.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 2012; 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 25.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 26.Gardner SN, Slezak T, Hall BG. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 2015; 31:2877–2878. doi: 10.1093/bioinformatics/btv271. [DOI] [PubMed] [Google Scholar]
- 27.Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5:e9490.doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo Y, Zhai Y, Zhang Z, Li D, Wang Z, Li J, et al. Complete genomic analysis of a kingdom-crossing Klebsiella variicola isolate. Front Microbiol 2018; 9:2428.doi: 10.3389/fmicb.2018.02428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, Li Y, Li S, Tang L, Zheng J, An Q. Genomic identification of nitrogen-fixing Klebsiella variicola, K. pneumoniae and K. quasipneumoniae. J Basic Microbiol 2016; 56:78–84. doi: 10.1002/jobm.201500415. [DOI] [PubMed] [Google Scholar]
- 30.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 2013; 41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langille MG, Hsiao WW, Brinkman FS. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 2008; 9:329.doi: 10.1186/1471-2105-9-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 2017; 45:D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 2011; 12:402.doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics 2011; 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherry NL, Lane CR, Kwong JC, Schultz M, Sait M, Stevens K, et al. Genomics for molecular epidemiology and detecting transmission of carbapenemase-producing Enterobacterales in Victoria, Australia, 2012 to 2016. J Clin Microbiol 2019; 57:e00573-19.doi: 10.1128/JCM.00573-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng B, Dai Y, Liu Y, Shi W, Dai E, Han Y, et al. Molecular epidemiology and risk factors of carbapenem-resistant Klebsiella pneumoniae infections in Eastern China. Front Microbiol 2017; 8:1061.doi: 10.3389/fmicb.2017.01061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiao Y, Qin Y, Liu J, Li Q, Dong Y, Shang Y, et al. Risk factors for carbapenem-resistant Klebsiella pneumoniae infection/colonization and predictors of mortality: a retrospective study. Pathog Glob Health 2015; 109:68–74. doi: 10.1179/2047773215Y.0000000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du J, Cao J, Shen L, Bi W, Zhang X, Liu H, et al. Molecular epidemiology of extensively drug-resistant Klebsiella pneumoniae outbreak in Wenzhou, Southern China. J Med Microbiol 2016; 65:1111–1118. doi: 10.1099/jmm.0.000338. [DOI] [PubMed] [Google Scholar]
- 39.Liu P, Li P, Jiang X, Bi D, Xie Y, Tai C, et al. Complete genome sequence of Klebsiella pneumoniae subsp. pneumoniae HS11286, a multidrug-resistant strain isolated from human sputum. J Bacteriol 2012; 194:1841–1842. doi: 10.1128/JB.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jayol A, Poirel L, Villegas MV, Nordmann P. Modulation of mgrB gene expression as a source of colistin resistance in Klebsiella oxytoca. Int J Antimicrob Agents 2015; 46:108–110. doi: 10.1016/j.ijantimicag.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 41.Fu P, Tang Y, Li G, Yu L, Wang Y, Jiang X. Pandemic spread of blaKPC-2 among Klebsiella pneumoniae ST11 in China is associated with horizontal transfer mediated by IncFII-like plasmids. Int J Antimicrob Agents 2019; 54:117–124. doi: 10.1016/j.ijantimicag.2019.03.014. [DOI] [PubMed] [Google Scholar]
- 42.Murray IA, Martinez-Suarez JV, Close TJ, Shaw WV. Nucleotide sequences of genes encoding the type II chloramphenicol acetyltransferases of Escherichia coli and Haemophilus influenzae, which are sensitive to inhibition by thiol-reactive reagents. Biochem J 1990; 272:505–510. doi: 10.1042/bj2720505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zielinska A, Billini M, Moll A, Kremer K, Briegel A, Izquierdo Martinez A, et al. LytM factors affect the recruitment of autolysins to the cell division site in Caulobacter crescentus. Mol Microbiol 2017; 106:419–438. doi: 10.1111/mmi.13775. [DOI] [PubMed] [Google Scholar]
- 44.Patras KA, Derieux J, Al-Bassam MM, Adiletta N, Vrbanac A, Lapek JD, et al. Group B Streptococcus biofilm regulatory protein A contributes to bacterial physiology and innate immune resistance. J Infect Dis 2018; 218:1641–1652. doi: 10.1093/infdis/jiy341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malm S, Maass S, Schaible UE, Ehlers S, Niemann S. In vivo virulence of Mycobacterium tuberculosis depends on a single homologue of the LytR-CpsA-Psr proteins. Sci Rep 2018; 8:3936.doi: 10.1038/s41598-018-22012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamers RP, Nguyen UT, Nguyen Y, Buensuceso RN, Burrows LL. Loss of membrane-bound lytic transglycosylases increases outer membrane permeability and beta-lactam sensitivity in Pseudomonas aeruginosa. Microbiologyopen 2015; 4:879–895. doi: 10.1002/mbo3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bi W, Liu H, Dunstan RA, Li B, Torres VVL, Cao J, et al. Extensively drug-resistant Klebsiella pneumoniae Causing nosocomial bloodstream infections in China: molecular investigation of antibiotic resistance determinants, informing therapy, and clinical outcomes. Front Microbiol 2017; 8:1230.doi: 10.3389/fmicb.2017.01230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin X, Wu S, Hao M, Zhu J, Ding B, Yang Y, et al. The colonization of carbapenem-resistant Klebsiella pneumoniae: epidemiology, resistance mechanisms, and risk factors in patients admitted to intensive care units in China. J Infect Dis 2020; 221:S206–S214. doi: 10.1093/infdis/jiz622. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Zou MX, Wang HC, Dou QY, Hu YM, Yan Q, et al. An outbreak of infections caused by a Klebsiella pneumoniae ST11 clone coproducing Klebsiella pneumoniae carbapenemase-2 and RmtB in a Chinese Teaching Hospital. Chin Med J 2016; 129:2033–2039. doi: 10.4103/0366-6999.189049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Campos TA, Goncalves LF, Magalhaes KG, de Paulo Martins V, Pappas Junior GJ, Peirano G, et al. A fatal bacteremia caused by hypermucousviscous KPC-2 producing extensively drug-resistant K64-ST11 Klebsiella pneumoniae in Brazil. Front Med (Lausanne) 2018; 5:265.doi: 10.3389/fmed.2018.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong N, Zhang R, Liu L, Li R, Lin D, Chan EW, et al. Genome analysis of clinical multilocus sequence Type 11 Klebsiella pneumoniae from China. Microb Genom 2018; 4:149.doi: 10.1099/mgen.0.000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou T, Zhang Y, Li M, Yu X, Sun Y, Xu J. An outbreak of infections caused by extensively drug-resistant Klebsiella pneumoniae strains during a short period of time in a Chinese teaching hospital: epidemiology study and molecular characteristics. Diagn Microbiol Infect Dis 2015; 82:240–244. doi: 10.1016/j.diagmicrobio.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 53.Villa L, Garcia-Fernandez A, Fortini D, Carattoli A. Replicon sequence typing of IncF plasmids carrying virulence and resistance determinants. J Antimicrob Chemother 2010; 65:2518–2529. doi: 10.1093/jac/dkq347. [DOI] [PubMed] [Google Scholar]
- 54.Comandatore F, Sassera D, Bayliss SC, Scaltriti E, Gaiarsa S, Cao X, et al. Gene composition as a potential barrier to large recombinations in the bacterial pathogen Klebsiella pneumoniae. Genome Biol Evol 2019; 11:3240–3251. doi: 10.1093/gbe/evz236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang Y, Li G, Liang W, Shen P, Zhang Y, Jiang X. Translocation of carbapenemase gene blaKPC-2 both internal and external to transposons occurs via novel structures of Tn1721 and exhibits distinct movement patterns. Antimicrob Agents Chemother 2017; 61:e01151–01117. doi: 10.1128/AAC.01151-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, Li Y, Shen W, Wang SM, Wang G, Zhou Y. Whole-genome sequence of a carbapenem-resistant hypermucoviscous Klebsiella pneumoniae isolate SWU01 with capsular serotype K47 belonging to ST11 from a patient in China. J Glob Antimicrob Resist 2017; 11:87–89. doi: 10.1016/j.jgar.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 57.Bleriot I, Trastoy R, Blasco L, Fernandez-Cuenca F, Ambroa A, Fernandez-Garcia L, et al. Genomic analysis of 40 prophages located in the genomes of 16 carbapenemase-producing clinical strains of Klebsiella pneumoniae. Microb Genom 2020; 6:e000369.doi: 10.1099/mgen.0.000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pina-Iturbe A, Ulloa-Allendes D, Pardo-Roa C, Coronado-Arrazola I, Salazar-Echegarai FJ, Sclavi B, et al. Comparative and phylogenetic analysis of a novel family of Enterobacteriaceae-associated genomic islands that share a conserved excision/integration module. Sci Rep 2018; 8:10292.doi: 10.1038/s41598-018-28537-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen Z, Gao Q, Qin J, Liu Y, Li M. Emergence of an NDM-5-producing hypervirulent Klebsiella pneumoniae sequence type 35 strain with chromosomal integration of an integrative and conjugative element, ICEKp1. Antimicrob Agents Chemother 2019; 64:e01675-19.doi: 10.1128/AAC.01675-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Couve-Deacon E, Jove T, Afouda P, Barraud O, Tilloy V, Scaon E, et al. Class 1 integrons in Acinetobacter baumannii: a weak expression of gene cassettes to counterbalance the lack of LexA-driven integrase repression. Int J Antimicrob Agents 2019; 53:491–499. doi: 10.1016/j.ijantimicag.2018.11.012. [DOI] [PubMed] [Google Scholar]
- 61.Liang PJ, Han WY, Huang QH, Li YZ, Ni JF, She QX, et al. Knockouts of RecA-like proteins RadC1 and RadC2 have distinct responses to DNA damage agents in Sulfolobus islandicus. J Genet Genomics 2013; 40:533–542. doi: 10.1016/j.jgg.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 62.Lee JY, Na IY, Park YK, Ko KS. Genomic variations between colistin-susceptible and -resistant Pseudomonas aeruginosa clinical isolates and their effects on colistin resistance. J Antimicrob Chemother 2014; 69:1248–1256. doi: 10.1093/jac/dkt531. [DOI] [PubMed] [Google Scholar]
- 63.Yan J, Xia Y, Yang M, Zou J, Chen Y, Zhang D, et al. Quantitative proteomics analysis of membrane proteins in Enterococcus faecalis with low-level linezolid-resistance. Front Microbiol 2018; 9:1698.doi: 10.3389/fmicb.2018.01698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kraithong T, Channgam K, Itsathitphaisarn O, Tiensuwan M, Jeruzalmi D, Pakotiprapha D. Movement of the beta-hairpin in the third zinc-binding module of UvrA is required for DNA damage recognition. DNA Repair (Amst) 2017; 51:60–69. doi: 10.1016/j.dnarep.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 65.Militello KT, Mandarano AH, Varechtchouk O, Simon RD. Cytosine DNA methylation influences drug resistance in Escherichia coli through increased sugE expression. FEMS Microbiol Lett 2014; 350:100–106. doi: 10.1111/1574-6968.12299. [DOI] [PubMed] [Google Scholar]
- 66.De Majumdar S, Yu J, Fookes M, McAteer SP, Llobet E, Finn S, et al. Elucidation of the RamA regulon in Klebsiella pneumoniae reveals a role in LPS regulation. PLoS Pathog 2015; 11:e1004627.doi: 10.1371/journal.ppat.1004627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy CN, Mortensen MS, Krogfelt KA, Clegg S. Role of Klebsiella pneumoniae type 1 and type 3 fimbriae in colonizing silicone tubes implanted into the bladders of mice as a model of catheter-associated urinary tract infections. Infect Immun 2013; 81:3009–3017. doi: 10.1128/IAI.00348-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lencina AM, Franza T, Sullivan MJ, Ulett GC, Ipe DS, Gaudu P, et al. Type 2 NADH dehydrogenase is the only point of entry for electrons into the Streptococcus agalactiae respiratory chain and is a potential drug target. MBio 2018; 9:e01034-18.doi: 10.1128/mBio.01034-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yasuda T, Morimatsu K, Kato R, Usukura J, Takahashi M, Ohmori H. Physical interactions between DinI and RecA nucleoprotein filament for the regulation of SOS mutagenesis. EMBO J 2001; 20:1192–1202. doi: 10.1093/emboj/20.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shishodia SK, Tiwari S, Shankar J. Resistance mechanism and proteins in Aspergillus species against antifungal agents. Mycology 2019; 10:151–165. doi: 10.1080/21501203.2019.1574927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fang CT, Yi WC, Shun CT, Tsai SF. DNA adenine methylation modulates pathogenicity of Klebsiella pneumoniae genotype K1. J Microbiol Immunol Infect 2017; 50:471–477. doi: 10.1016/j.jmii.2015.08.022. [DOI] [PubMed] [Google Scholar]
- 72.Aquilini E, Azevedo J, Merino S, Jimenez N, Tomas JM, Regue M. Three enzymatic steps required for the galactosamine incorporation into core lipopolysaccharide. J Biol Chem 2010; 285:39739–39749. doi: 10.1074/jbc.M110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yeh KM, Lin JC, Yin FY, Fung CP, Hung HC, Siu LK, et al. Revisiting the importance of virulence determinant magA and its surrounding genes in Klebsiella pneumoniae causing pyogenic liver abscesses: exact role in serotype K1 capsule formation. J Infect Dis 2010; 201:1259–1267. doi: 10.1086/606010. [DOI] [PubMed] [Google Scholar]
- 74.Artier J, da Silva Zandonadi F, de Souza Carvalho FM, Pauletti BA, Leme AFP, Carnielli CM, et al. Comparative proteomic analysis of Xanthomonas citri ssp. citri periplasmic proteins reveals changes in cellular envelope metabolism during in vitro pathogenicity induction. Mol Plant Pathol 2018; 19:143–157. doi: 10.1111/mpp.12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kalynych S, Morona R, Cygler M. Progress in understanding the assembly process of bacterial O-antigen. FEMS Microbiol Rev 2014; 38:1048–1065. doi: 10.1111/1574-6976.12070. [DOI] [PubMed] [Google Scholar]
- 76.Mulligan EA, Hatchwell E, McCorkle SR, Dunn JJ. Differential binding of Escherichia coli McrA protein to DNA sequences that contain the dinucleotide m5CpG. Nucleic Acids Res 2010; 38:1997–2005. doi: 10.1093/nar/gkp1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for this study can be found in the GenBank (https://www.ncbi.nlm.nih.gov/genome/genomes/815).