

Abstract
The p53 tumor suppressor is a critical mediator of the cellular response to stress. The N-terminal transactivation domain of p53 makes protein interactions that promote its function as a transcription factor. Among those cofactors is the histone acetyltransferase p300, which both stabilizes p53 and promotes local chromatin unwinding. Here, we report the nuclear magnetic resonance solution structure of the Taz2 domain of p300 bound to the second transactivation subdomain of p53. In the complex, p53 forms an α-helix between residues 47 and 55 that interacts with the α1–α2–α3 face of Taz2. Mutational analysis indicated several residues in both p53 and Taz2 that are critical for stabilizing the interaction. Finally, further characterization of the complex by isothermal titration calorimetry revealed that complex formation is pH-dependent and releases a bound chloride ion. This study highlights differences in the structures of complexes formed by the two transactivation subdomains of p53 that may be broadly observed and play critical roles in p53 transcriptional activity.
Graphical Abstract
The p53 tumor suppressor protein functions as a central regulator in the response of mammalian cells to a variety of stresses. As a transcription factor, p53 orchestrates responses that prevent neoplastic transformation: cell-cycle arrest, DNA repair, senescence, and apoptosis.1 Although it is maintained at low levels in the basal state, the protein becomes stabilized and extensively post-translationally modified in response to a number of stresses, including DNA damage, hypoxia, and heat shock (recently reviewed in ref 2). Both the initial accumulation of activated protein and specific functional outcomes are regulated by the interactions of p53 with effector proteins.2 In particular, the N-terminal transactivation domain (TAD) is a critical region for protein interactions that regulate the stability of p53, its activity as a transcription factor, and its ability to effect transcription-independent programmed cell death.3–7
Central to its activity as a transcription factor is the ability of p53 to recruit the histone acetyltransferase p300 or its paralog CREB-binding protein (CBP) to the promoter regions of its target genes.8–11 In addition to the catalytic domain, CBP and p300 are comprised of multiple globular domains joined by disordered linkers.12 Through these domains, they interact with components of the general transcription machinery and a large number of transcription factors, functioning as highly connected nodes in transcription-regulating networks.12 The p53 TAD has been shown to interact with several domains of CBP/p300, including Taz2, Taz1, NCBD, and KIX (ref 13 and references therein). The binding of CBP/p300 to p53 results in both the stabilization of p53 through acetylation of C-terminal lysine residues, thereby preventing their ubiquitination and subsequent protein degradation, and the recruitment of CBP/p300 to promoter regions of p53 target genes, resulting in the acetylation of proximal histones and chromatin unwinding that favors gene transcription.9–11,14,15
The p53 TAD can be subdivided into two subdomains, termed TAD1 (residues 1–39) and TAD2 (residues 40–61), both of which have been shown to be able to independently activate gene transcription.3 Although the two subdomains do not exhibit recognizable sequence homology, both contain the Φ-X-X-Φ-Φ motif common to many acidic activation domains (where Φ represents a hydrophobic amino acid and X represents any amino acid). Studies of knock-in mice demonstrated that TAD1 alone is generally sufficient for p53-dependent cell-cycle arrest and apoptotic responses to acute DNA damage, whereas each transactivation domain can induce senescence and suppress tumor initiation in response to oncogenic signaling.16 Both p53 TAD1 and TAD2 are intrinsically disordered protein domains that adopt a helical conformation for at least part of their length when bound.13 This inherent flexibility allows the TADs to adapt to and bind a broad range of proteins.
To date, structures have been reported for the complex of a p53 TAD with eight different binding partners: the negative regulators MDM2 and MDMX; the Taz2 and NCBD domains of p300 and CBP, respectively; the PH domains of the yeast transcription factor, tfb1, and its human homologue, p62; the largest subunit of replication protein A (RPA70N); and the high-mobility group B1 protein.17–25 In most cases, the bound p53 TAD forms an amphipathic α-helix, though the recent complex of p53 TAD2 bound to human p62 revealed that p53 bound in an extended conformation.22 The complexes exhibit varying degrees of interfacial burial of the conserved signature motif residues and differences in the proportion of electrostatic to hydrophobic interactions that stabilize the complexes. For example, amino acids from p53 TAD1 interact with specific binding pockets on MDM2 and MDMX, giving rise to high-affinity and high-specificity interactions, consistent with their role in negatively regulating p53 stability. In contrast, p53 binds across broad surfaces, without binding to particular pockets, on Taz2 and NCBD; these interactions are consistent with the multiple interaction partners for both domains of CBP/p300.
In our previous examination of the binding of p53 TAD1 to p300 Taz2, we found that both TAD1 and TAD2 can independently interact with Taz2.26 Although the affinity of p53 TAD2 for Taz2 was tighter than that of p53 TAD1 for Taz2, phosphorylation of specific serine/threonine residues in p53 TAD1 increased its affinity for p300 Taz2 such that binding of the two domains was comparable. In contrast, binding of p53 TAD2 to Taz2 was unaffected by phosphorylation.26 To improve our understanding of the complex of p53 TAD2 with p300 Taz2 and further explore functional differences between p53 TAD1 and TAD2, we report here a structural and biochemical characterization of the p53 TAD2–p300 Taz2 complex. This work completes the first structural characterization of the two p53 TAD subdomains binding independently to the same site of a transcriptional co-activator. It provides a physical basis for ongoing efforts to understand the different roles that TAD1 and TAD2 play in the function of p53.
EXPERIMENTAL PROCEDURES
Expression and Purification of Recombinant Proteins.
The cloning, expression, and purification of Taz2 (A1723-K1812/C1738A, C1746A, C1789A, C1790A) have been previously described;26 this protein contains alanine mutations of four cysteine residues not involved in zinc coordination and maintains secondary structure and p53 binding characteristics similar to those of the wild-type protein. Site-directed mutagenesis of Taz2 was performed using the QuikChange Mutagenesis Kit (Stratagene). Uniformly (>98%) 15N-labeled or 15N- and 13C-labeled Taz2 was prepared in Celtone medium (Cambridge Isotopes) supplemented with [15N]ammonium chloride and [13C6]-d-glucose as the sole nitrogen and carbon sources, respectively.
The coding sequence for p53(35–59) was amplified via polymerase chain reaction from a mammalian vector and subcloned into pGEX4T-1 (GE Healthcare) using the BamHI and NotI restriction sites. GST-p53(35–59) was grown in Escherichia coli in LB medium or minimal medium containing [15N]ammonium chloride and [13C]glucose as the sole nitrogen and carbon sources, respectively. 2H- and 15N-labeled p53 was prepared in minimal medium containing [15N]ammonium chloride and 2H2O. The cultures were grown at 37 °C to an OD600 of 0.6 and induced with 0.5 mM IPTG for 3 h. The cells were spun down and resuspended in 50 mM Tris-HCl (pH 8), 120 mM NaCl, 0.5% NP-40 (EBC buffer), and protease inhibitors. The resuspended cells were lysed with a French pressure cell followed by centrifugation at 13000g for 1 h to clear the lysate. The supernatant was incubated with EBC buffer-equilibrated glutathione sepharose for 16 h at 4 °C, and the bound fusion protein was washed with EBC, re-equilibrated in phosphate-buffered saline, and digested with 100 units of thrombin for 4 h at 25 °C. The cleaved p53(35–59) was further purified to >95% purity using reversed-phase high-pressure liquid chromatography (RP-HPLC) on an Eclipse XDB C-18 column (Agilent) with a 0.05% TFA/water/acetonitrile mixture. The mass of the isotopically labeled p53(35–59) was confirmed by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (Waters).
Peptide Synthesis.
Unlabeled p53(35–59) peptide for NMR and binding experiments, as well as all p53(35–59) mutant peptides, was synthesized by the solid-phase method with 9-fluorenylmethoxycarbonyl chemistry. The peptides were cleaved with a solution of 82.5% trifluoroacetic acid (TFA), 5% phenol, 5% thioanisole, 5% water, and 2.5% ethandithiol and then purified to >95% purity by RP-HPLC on a BioAdvantage Pro300 C-4 column (Thompson) with a 0.05% TFA/water/acetonitrile mixture and masses confirmed by MALDI-TOF mass spectrometry.
NMR Spectroscopy.
All NMR experiments were performed at 35 °C in 50 mM MES (pH 6.3), 200 mM NaCl, 0.1 mM ZnCl, and 1 mM dithiothreitol on a Bruker 500 or 700 MHz spectrometer equipped with pulsed-field gradient units and triple-resonance probes. Chemical shifts (1H, 15N, and 13C) and NOEs of Taz2 and p53(35–59) were determined by performing standard triple-resonance experiments.27 Intermolecular NOEs were obtained from 15N-edited nuclear Overhauser enhancement spectroscopy (NOESY) experiments on 15N- and 2H/1H-labeled p53(35–59) complexed with Taz2.28–30 NMR data were processed with NMRPipe/NMRDraw31 and analyzed with NMRView.32
Structure Calculation.
The NOE-derived restraints were subdivided into four classes as strong (1.8–2.7 Å), medium (1.8–3.3 Å), weak (1.8–5.0 Å), and very weak (1.8–6.0 Å) by comparison with NOEs of protons separated by known distances. An additional 0.5 Å was added to the upper distance limit for methyl protons, and 0.2 Å was added to the upper distance limit for NH protons if the NOEs were in the strong and medium classes. Backbone dihedral angle restraints (ϕ and ψ angles) were obtained from analysis of 1Hα, 1HN, 13Cα, 13Cβ, 13CO, and 15N chemical shifts using TALOS+.33 Two constraints per hydrogen bond (dNH-O ≤ 2.2 Å, and dN-O ≤ 3.2 Å) were added after initial secondary structure analysis.
Models of the protein–peptide complex were developed from the constraints by a modified version of the methods used for construction of the RECOORD Database of Recalculated NMR-Determined Solution Structures.34 This utilized version 1.3 of CNS35,36 to perform successive molecular dynamics (MD) protocols. First, approximately 40000 unique structures were produced from the extended chain conformation by means of MD simulated annealing in vacuo, using the “annealing.sh” script contained in the RECOORDscripts-cns1.3 archive available from http://www.ebi.ac.uk/msd/recoord/. The script was modified to use the R-6 rather than SUM method for NOE averaging in CNS, to provide greater interpeptide distance averages and thus produce tighter complexes. Of the resultant structures, the 500 with the lowest total energy were then subjected to additional refinement in the presence of explicit water molecules. This second MD protocol was conducted with the “re_h2o.sh” script using the same averaging method and sets of distance and dihedral angle constraints. From the resultant group, the final ensemble was taken as the 15 structures that both lacked Ramachandran-disallowed backbone conformations and best satisfied the distance constraints (as determined by the lowest NOE energy). The Ramachandran backbone angle statistics were calculated with PROCHECK.37 Residue contacts among the p53 peptide and Taz2 protein chains in the complexes were determined using MONSTER.38
Isothermal Titration Calorimetry (ITC).
ITC measurements were performed using a VP-ITC calorimeter (MicroCal, GE Healthcare). For experiments with p53(35–59) or Taz2 mutant proteins, titrations were performed in 20 mM Tris-HCl (pH 7.2), 100 mM NaCl, and 2 mM β-mercaptoethanol (ITC buffer) at 35 °C. For analysis of the salt dependence of binding, experiments were performed in ITC buffer with 50 or 200 mM NaCl rather than 100 mM NaCl or in buffer containing 50, 100, or 200 mM NaOAc in place of NaCl. In experiments to determine the effect of pH on binding, titrations were performed in 100 mM Tris, 100 mM Bis-Tris, and 100 mM acetate at the stated pH value. In all experiments, the concentrations of the injected peptides and proteins were determined from the absorbance at 280 nm. The protein and peptide solutions were degassed before each experiment. Heats of dilution were subtracted from the raw data. ITC experiments were fit with a 1:1 binding model using ORIGIN with the ITC package. All experiments were performed at least twice.
RESULTS
Determination of the Structure of the Taz2–p53(35–59) Complex.
Using NMR spectroscopy, we determined the solution structure of the p53 TAD2–Taz2 complex (Table 1). The ensemble of lowest-energy structures is shown in Figure 1A. The structure of the Taz2 protein is well-defined for most of its length. The few regions of greater variation are located near the N-terminus (residues 1723–1734) and within two segments near the C-terminus (residues 1795–1802 and 1810–1812). For the ensemble, the root-mean-square deviations (rmsds) for the backbone and all heavy atoms of Taz2 are 0.61 and 1.33 Å, respectively (residues 1735–1809). This conformation is very similar to that of the p300 Taz2 domain in complex with p53 TAD1 and that of the free CBP Taz2 domain.19,39 For example, the backbone rmsd for the p300 Taz2 components in the two p53 complexes is only 1.86 Å (residues 1735–1809). The tertiary structure of Taz2 comprises four core helices in a characteristic arrangement with a single helical turn in the linker between α2 and α3 (Figure 1B).
Table 1.
Structural Statistics of the Taz2–p53(35–59) Complex
| Restraints Used for Structure Calculations | ||
|---|---|---|
| total no. of NOE distances | 1986 | |
| intraresidue | 531 | |
| sequential (|i – j| = 1) | 544 | |
| short-range (1 < |i – j| < 5) | 595 | |
| long-range (5 ≤ |i – j|) | 276 | |
| interprotein | 40 | |
| no. of hydrogen bond distances | 88 | |
| no. of dihedral angles | 180 | |
| Ensemble Statistics | ||
| mean | standard deviation | |
| restraint violations (rmsd) | ||
| NOE (Å) | 0.22 | 0.01 |
| dihedral (deg) | 4.96 | 0.21 |
| deviations from idealized geometry (rmsd) | ||
| bonds (Å) | 0.03 | 0.00 |
| angles (deg) | 3.39 | 0.05 |
| impropers (deg) | 4.03 | 0.15 |
| average pairwise rmsd (Å)a | ||
| all heavy atoms | 1.39 | |
| backbone atoms | 0.64 | |
| Ramachandran analysis (%) | ||
| core | 77.6 | 2.6 |
| allowed | 19.1 | 1.8 |
| generously allowed | 3.3 | 1.5 |
| disallowed | 0.0 | 0.0 |
The analysis was restricted to residues 1735–1809 of Taz2 and residues 48–55 of p53(35–59); 15 structures were used in the calculation..
Figure 1.
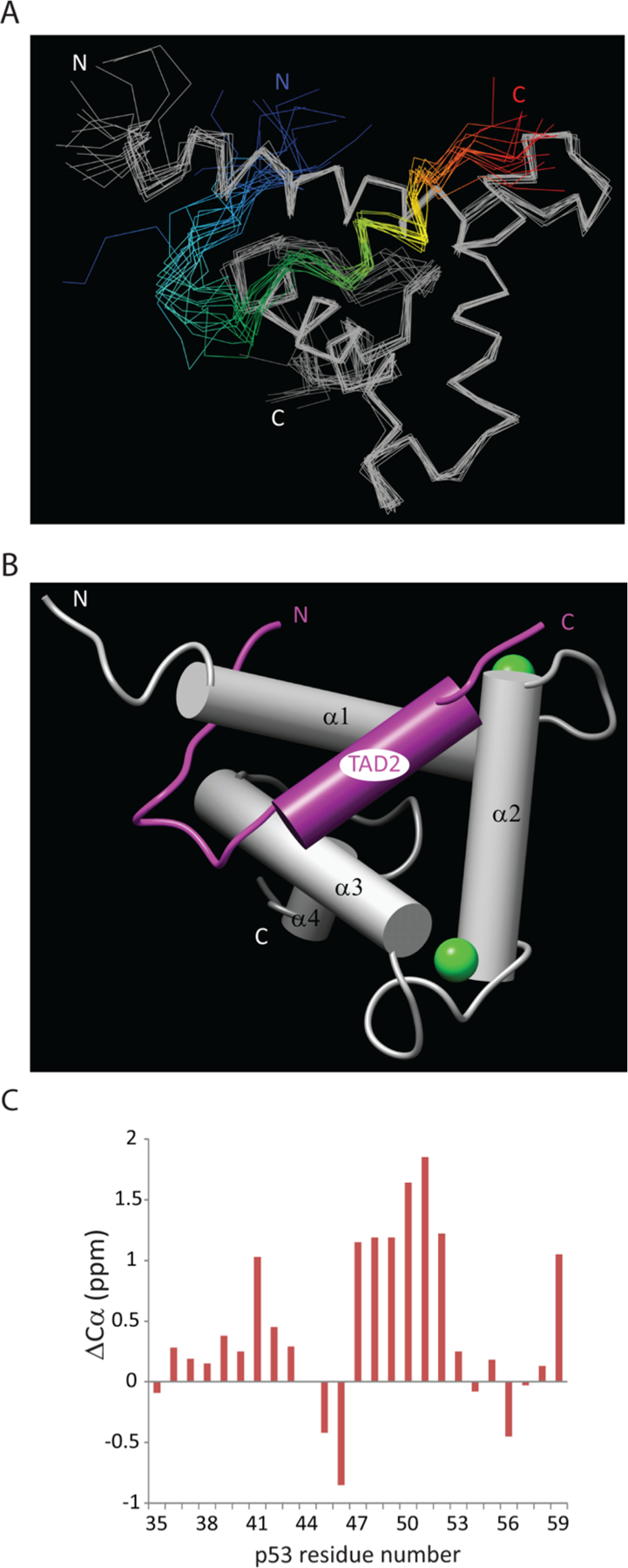
Ensemble of structures and conformation of a representative structure. (A) Overlay of the 15 lowest-energy NMR structures of the complex between p53(35–59) (blue-to-red rainbow) and the Taz2 domain of p300 (gray). The structures are shown as Cα traces. (B) Cylinder model of the conformation of the complex. p53(35–59) is colored purple and Taz2 gray. The zinc ions in the Taz2 fingers are modeled as green spheres. (C) Plot of the changes in the p53 Cα chemical shift when p53 is in complex with Taz2 as compared with random coil values.40
The bound p53(35–59) adopts a helical conformation for part of its length, between Pro47 and Thr55. Comparison of the p53 chemical shifts with random coil values40 indicates that the largest changes in the peptide occur between residues Pro47 and Glu52 (Figure 1C). The p53 helix lies across helices α3, α1, and α2 of Taz2. In the complex, the helical portion of p53(35–59) is more well-defined than the termini; the rmsds for the backbone and all heavy atoms of this helix (residues 48–55) over the ensemble are 0.38 and 1.61 Å, respectively.
Details of the Taz2–p53(35–59) Complex Interface.
The NMR solution structure demonstrates that the Taz2– p53(35–59) complex is stabilized by both hydrophobic and electrostatic interactions. The side chain of Phe54 packs between the nonpolar part of the side chain of Taz2 Arg1737 and the side chain of p53 Thr55 (Figure 2A). An additional stabilizing interaction occurs among partially positively charged aromatic hydrogens of the phenyl ring of p53 Phe54 and the hydroxyl oxygen of Taz2 Ser1741. While the position of Trp53 is also consistent within the ensemble of structures, the side chain is found in two approximately equally populated orientations. In the first orientation, the indole group packs against the side chains of Taz2 Val1764, Ile1781, and the nonpolar portion of Lys1760 (Figure 2A). In some of these structures, the partially positively charged proton of the indole forms a hydrogen bond with the acid group of p53 Asp49. In the alternate conformation, the p53 Trp53 indole ring is flipped but still packs in the same hydrophobic pocket of Taz2 (Figure 2B). In some of these cases, the p53 Asp49 side chain rotates and forms a hydrogen bond with Taz2 Gln1784.
Figure 2.
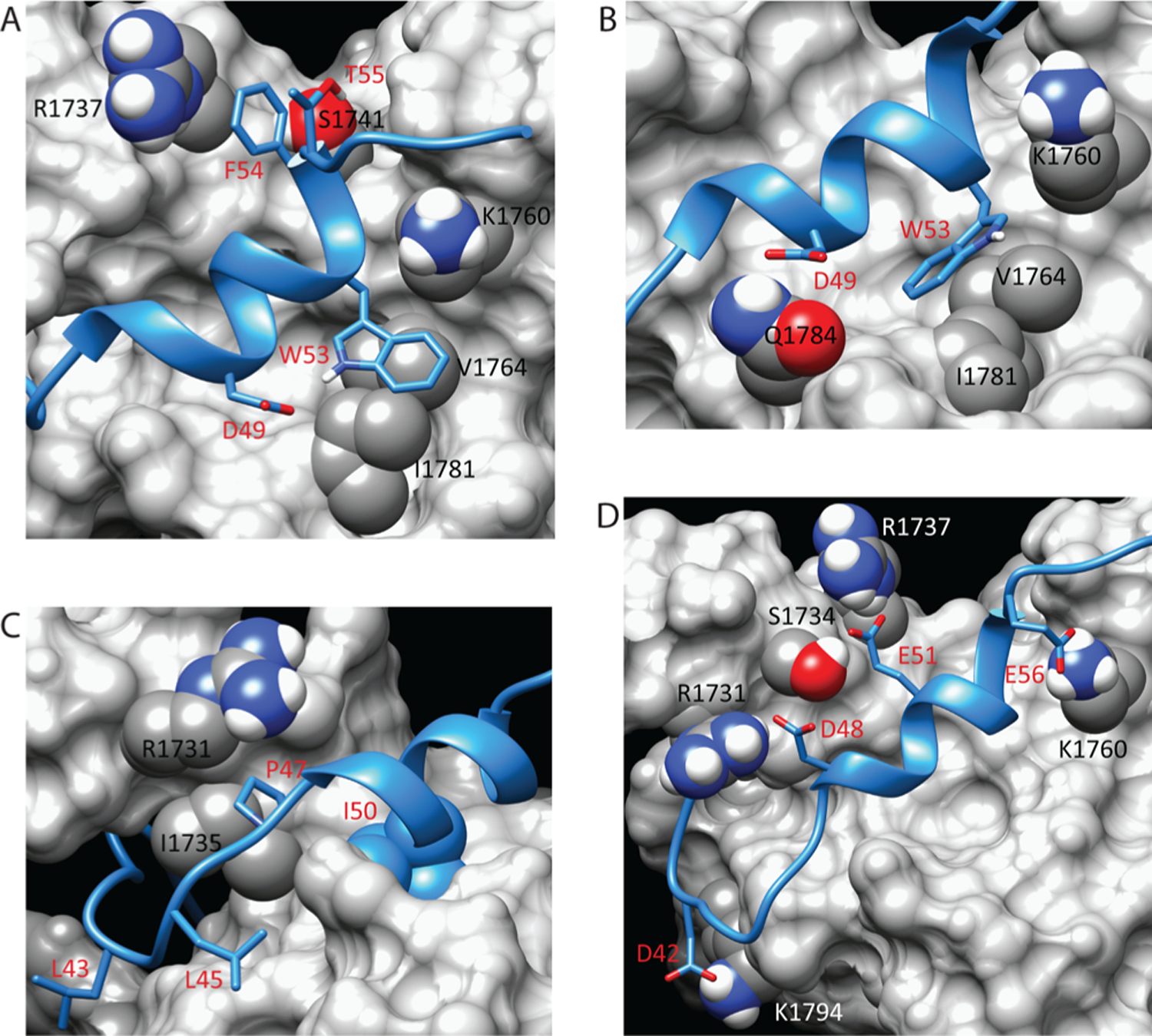
Stabilizing interactions at the interface of the p53(35–59)–Taz2 complex. (A) Model of the Taz2–p53(35–59) interface showing interactions of p53 Trp53 and Phe54. (B) Model of the Taz2–p53(35–59) complex showing the alternate conformation of Trp53 observed in some members of the ensemble. (C) Model of the Taz2–p53(35–59) interface showing positions of other hydrophobic p53 residues. (D) Model of the Taz2–p53(35–59) complex showing primary residues that make salt bridges and hydrogen bonds. In all panels, contacting residues are labeled in black or white for Taz2 (gray surface representation) and red for p53(35–59) (blue ribbon).
Additional hydrophobic interactions that stabilize the complex are represented in Figure 2C. Primary among these is the packing of the p53 Ile50 side chain in a deep hydrophobic pocket in the core of Taz2. Consistent throughout the ensemble, this binding serves to anchor the middle region of the bound p53 helix. The pocket is formed by the side chains and/or backbone portions of Ala1738, Leu1742, Met1761, Val1764, Ile1781, Gln1784, Leu1785, and Leu1788 of Taz2 (not labeled). The packing of the p53 Pro47 ring in a hydrophobic cleft formed by Ile1735 and the nonpolar portion of the Arg1731 side chain of Taz2 anchors the N-terminus of the bound p53 helix in the majority of ensemble structures. Additionally, the side chains of p53 Leu43 and Leu45 bind to nonpolar patches on the surface of Taz2 (Figure 2C). These latter interactions represent one of several binding sites for these residues of p53 within the structural ensemble, reflecting the higher degree of flexibility of the N-terminal segment of p53. Despite this variation, these hydrophobic interactions serve to stabilize the winding of this end of the p53 peptide around the core of Taz2.
Contributing to the electrostatic stabilization of the complex, p53(35–59) contains seven acidic residues that form a series of salt bridges and hydrogen bonds with formal and partial positively charged residues of Taz2. The most consistent of these throughout the ensemble involve p53 Asp48 and Glu51, at the N-terminus and middle of the bound helix, respectively, and p53 Glu56 at the C-terminus (Figure 2D). In a majority of structures, Asp48 interacts with Taz2 Arg1731 and/or Ser1734, Glu51 interacts with Taz2 S1734 and/or R1737, and Glu56 interacts with Taz2 Lys1760. A salt bridge between p53 Asp42 and Taz2 Lys1794 also forms (Figure 2D). Finally, Asp41 can interact with Taz2 Arg1732 and His1795. Of interest in the p53(35–59) peptide are Ser46 and Thr55, which are subject to post-translational phosphorylation and bracket the ends of the bound helix. These residues are mostly unbound in the ensemble, consistent with our previous observation that phosphorylation does not significantly affect binding,26 although in some structures Thr55 hydrogen bonds to Ser1757 of Taz2 and, to a lesser extent, Ser46 interacts with Gln1784 of Taz2 (not shown).
Mutational Analysis of Critical Protein–Protein Contacts.
To improve our understanding of the importance of specific residues in p53(35–59) and Taz2 for complex formation, isothermal titration calorimetry (ITC) binding experiments were performed using selected mutant forms. First, p53 Trp53 was mutated to glutamine, as this mutation maintains the presence of a bulky side chain but alters the hydrophobic, aromatic nature of the side chain. Double mutation of Trp53 to glutamine and Phe54 to serine has previously been shown to have a deleterious effect on p53 transcriptional activation.3 Mutation of p53 Trp53 to glutamine resulted in the loss of detectable binding at 35 °C (Table 2 and Figure S1 of the Supporting Information), consistent with the importance of this highly conserved hydrophobic residue in making critical contacts with Taz2. This result is also consistent with our previous observation that the W53A/F54A double mutation of p53(25–65) abrogated Taz2 binding,26 as well as previous reports of the importance of these residues for TAD2-dependent transactivation.41,42 Interestingly, mutation of p53 Phe54 to serine affected Taz2 binding to a lesser extent. Specifically, this mutation resulted in a 55% reduction in the association constant compared with that of the wild-type p53 peptide (Table 2). The greater importance of Trp53 for complex stability may be due to it being more buried within the Taz2 surface than Phe54.
Table 2.
Binding of p53(35–59) Mutant Peptides to Taz2
| Ka (M−1) | ΔH (kcal/mol) | ΔS (cal mol−1 deg−1) |
|
|---|---|---|---|
| p53 WT | (1.14 ± 0.13) × 106 | −2.70 ± 0.02 | 18.9 ± 0.3 |
| p53 M40A | (1.32 ± 0.05) × 106 | −2.35 ± 0.06 | 20.4 ± 0.3 |
| p53 L45A | (0.91 ± 0.04) × 106 | −3.05 ± 0.28 | 17.4 ± 1.0 |
| p53 D48N | (0.60 ± 0.08) × 106 | −3.13 ± 0.10 | 16.3 ± 0.1 |
| p53 I50A | (0.31 ± 0.01) × 106 | −2.66 ± 0.18 | 16.5 ± 0.6 |
| p53 E51Q | (0.56 ± 0.13) × 106 | −2.19 ± 0.52 | 19.2 ± 1.3 |
| p53 Q52E | (1.29 ± 0.35) × 106 | −2.07 ± 0.03 | 21.2 ± 0.5 |
| p53 W53Q | NDa | NDa | NDa |
| p53 F54S | (0.53 ± 0.09) × 106 | −1.59 ± 0.63 | 21.0 ± 1.7 |
| p53 T55A | (1.16 ± 0.10) × 106 | −2.57 ± 0.03 | 19.4 ± 0.1 |
| p53 pThr55 | (1.30 ± 0.05) × 106 | −1.9 ± 0.46 | 21.8 ± 1.6 |
No binding was detected.
Similar to the effect of the p53 Phe54 mutation, p53(35–59) I50A bound to Taz2 3-fold more weakly than did the wild-type protein. This reduction in binding strength is consistent with burial of Ile50 in a deep hydrophobic pocket of Taz2, beneath the TAD2 helix (Figure 2C). In contrast, the binding affinities for the p53(35–59) M40A and L45A mutant peptides were similar to that of wild-type p53(35–59), consistent with the location of these residues in the flexible, N-terminal region of the peptide. Twofold decreases in binding affinity were also observed following mutation of p53 Asp48 to asparagine and mutation of p53 Glu51 to glutamine (Table 2). As described above and illustrated in Figure 2D, these negatively charged residues form stabilizing, salt bridge bonds with formal and partial positively charged Taz2 residues throughout most of the structural ensemble. The importance of these electrostatic interactions was further demonstrated by mutation of Taz2 Arg1731 to alanine, which resulted in a 3-fold reduction in binding affinity for p53(35–59) (Table 3). Although Taz2 Arg1737 interacts with p53 Glu51 in some members of the structural ensemble, substitution of the Taz2 residue with alanine did not significantly affect the binding affinity. The lack of an effect is likely due to the presence of other proximal residues in Taz1, such as Ser1734 and Arg1731, that can form alternative stabilizing interactions. In contrast, the Taz2 R1732A mutant caused a substantial reduction in binding affinity (Table 3), even though it is only one of several positively charged residues bound by p53 Asp41 and Asp42 in the structure. Finally, we observed that substitution of Taz2 Ser1757 with aspartate reduced the level of binding to p53(35–59) by 2-fold, whereas a similar substitution of Taz2 Ser1741 did not affect binding. The additional negative charge in the Taz2 S1757D mutant may cause repulsion of p53 Glu56 and Asp57 residues, leading to weaker binding, but the Ser1741 mutation may maintain the stabilizing interaction with the partial positively charged ring protons of p53 Phe54.
Table 3.
Binding of p53(35–59) to Mutant Forms of Taz2
| Ka (M−1) | ΔH (kcal/mol) | ΔS (cal mol−1 deg−1) |
|
|---|---|---|---|
| Taz2 WT | (1.14 ± 0.13) × 106 | −2.70 ± 0.02 | 18.9 ± 0.3 |
| Taz2 R1731A | (0.36 ± 0.06) × 106 | −1.05 ± 0.18 | 22.0 ± 0.5 |
| Taz2 R1732A | (0.44 ± 0.08) × 106 | −2.40 ± 0.15 | 18.0 ± 0.2 |
| Taz2 R1737A | (1.10 ± 0.10) × 106 | −1.15 ± 0.15 | 23.9 ± 0.7 |
| Taz2 S1741D | (1.22 ± 0.30) × 106 | −1.59 ± 0.13 | 20.5 ± 1.5 |
| Taz2 S1757D | (0.65 ± 0.08) × 106 | −2.33 ± 0.30 | 20.0 ± 1.3 |
Thermodynamic Analysis of Complex Formation.
We next used ITC to further characterize the p53(35–59)–Taz2 binding interaction. As detailed previously for the p300 Taz2–p53 TAD1 complex,19 a decreased pH of 6.3 was necessary to obtain sufficiently resolved NMR spectra. Because this is significantly more acidic than the nuclear environment, ITC studies were first performed to assess binding over a range of pH values. For these studies, 100 mM Tris, 100 mM Bis-Tris, 100 mM acetate buffer was used to maintain a constant ionic strength under all pH conditions. At 35 °C, all binding reactions were exothermic, consistent with our previous results,26 and were described well by a single-site binding model. As seen in Figure 3A, the binding affinity of p53(35–59) for Taz2 increases substantially with increasing pH between pH 5.9 and 6.8; at higher pH values, the affinity does not change further. At pH 6.8, the Ka was approximately 8-fold higher than at pH 5.9 (12.1 × 106 and 1.4 × 106 M−1 at pH 6.8 and 5.9, respectively). This change is consistent with deprotonation of a residue favoring binding. In contrast, the affinity of p53(1–39) for Taz2 was not dependent upon pH between pH 5.9 and 6.8 (Figure 3A). Thus, there are critical differences in the pH dependence of the binding of p53(1–39) and p53(35–59) to Taz2.
Figure 3.
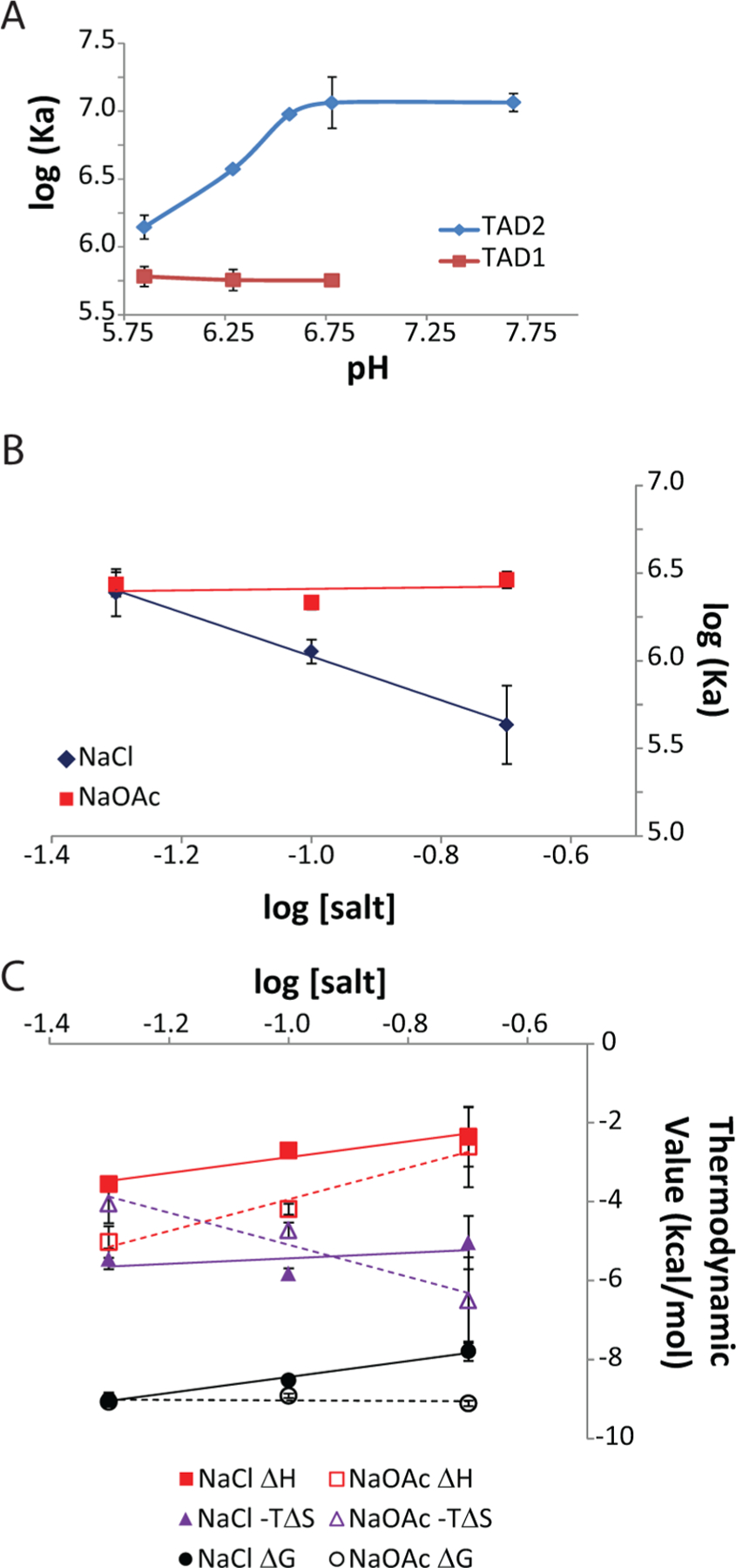
Thermodynamic characterization of the pH and salt dependence of p53–Taz2 interactions. (A) pH dependence of the affinity of the complex formed between Taz2 and p53(1–39) (empty symbols) or p53(35–59) (filled symbols) as measured by ITC at 35°C. Binding experiments were performed as described in Experimental Procedures, with pH values measured at 35 °C. (B) Salt dependence of the binding affinity of the Taz2–p53(35–59) complex as measured by ITC at 35 °C in buffer containing either NaCl (filled symbols) or NaOAc (empty symbols). (C) Salt dependece of the thermodynamic parameters of complexation of p53(25–59) with Taz2 as measured by ITC at 35 °C in buffer containing either NaCl (filled symbols) or NaOAc (empty symbols). Squares, ΔH; triangles, −TΔS; circles, ΔG.
Next, the salt dependence of the Taz2–p53(35–59) interaction was examined. In the first set of experiments, the concentration of NaCl was varied to examine ionic contributions to the binding of p53(35–59) to Taz2. At 50 mM NaCl, the binding of p53(35–59) was 2-fold tighter than that observed at 100 mM NaCl, and a similar corresponding difference was observed when the salt concentration was further increased to 200 mM (Figure 3B). A higher NaCl concentration also resulted in decreased ΔH and ΔS values for complex formation, resulting in a less favorable ΔG at the higher concentrations (Figure 3C). The dependence of the binding affinity on salt concentration can be related to the number of thermodynamically involved ions bound or released in complex formation. For the interaction of p53(35–59) with Taz2, this analysis suggested that complex formation led to the release of approximately one thermodynamically involved ion. Furthermore, extrapolation of the binding constants at 1 M NaCl provides an estimate of the binding affinity without the contribution of the release of thermodynamically involved ions; for this interaction, at 1 M NaCl, the Kd was estimated to be 10 µM.
We observed that the affinity of p53(35–59) for Taz2 was significantly higher when it was measured in buffer containing 100 mM acetate as opposed to 100 mM NaCl at similar pH values (compare panels A and B of Figure 3). To better understand this difference, we next measured the salt dependence of binding in the same buffer described above but replaced the NaCl with NaOAc. At 50 mM NaOAc, the binding of p53(35–59) to Taz2 was similar to that using 50 mM NaCl (Figure 3B). However, in contrast to the effects of NaCl, increasing the NaOAc concentration did not alter the binding affinity. An increased NaOAc concentration led to an increase in ΔH, as observed for binding in NaCl-containing buffer; however, the ΔS increased at higher NaOAc concentrations (Figure 3C). These results suggest that in NaCl-containing buffer, the dependence of binding was due to net release of a chloride ion. As Taz2 is a basic protein, likely this displacement is related to a newly formed electrostatic interaction with p53(35–59), such as those described above (Figure 2D).
DISCUSSION
The p53 TAD is a flexible and intrinsically disordered domain that is critical for many protein–protein interactions that affect p53 activity and stability. Of the several domains of CBP/p300 that interact with the p53 TAD, only structures of the p53 TAD–NCBD and p53 TAD1–Taz2 complexes have been previously described. Here, we expand upon our understanding of p53/CBP–p300 interactions by reporting the NMR solution structure of the p53 TAD2–Taz2 complex. Upon binding, p53(35–59) forms an α-helix between Pro47 and Thr55 that binds across helices α3, α1, α2. The complex is stabilized predominantly by hydrophobic interactions among p53 Ile50, Trp53, and Phe54, with additional contributions from electrostatic bonds between the highly acidic p53(35–59) and the highly basic Taz2. The importance of these interactions was further demonstrated by the decreased binding affinity following mutation of specific residues of p53(35–59) or Taz2.
Although p53 TAD1 and TAD2 bind on the same face of Taz2, the orientation of the two helices is very different. The amphipathic helix of the bound p53 peptide is rotated approximately 90° between the TAD1 and TAD2 complexes (Figure 4A). The rotated orientations allow the conserved phenylalanines of the two p53 domains (Phe19 in TAD1 and Phe54 in TAD2) to bind in similar pockets on the surface of Taz2. The conserved tryptophan residues (Trp23 in TAD1 and Trp53 in TAD2) occupy similar but not identical pockets on Taz2. The summarized interchain interactions in the two complexes indicate that the TAD2–Taz2 complex has a greater proportion of charge-based interactions than the TAD1–Taz2 complex, consistent with the greater number of acidic residues in p53 TAD2 (Figure 4B). There is also a slight shift in the Taz2 residues that interact with the two p53 subdomains, though three of the Taz2 residues that interact with Phe19 in the complex with TAD1 also interact with Phe54 in the complex with TAD2. Likewise, two of the Taz2 residues that interact with Trp23 in the TAD1–Taz2 complex also interact with Trp53 in the TAD2–Taz2 complex. Thus, the change in orientation of the p53 helix in the two complexes reflects the structural changes necessary to maintain these key hydrophobic interactions given the different relative positions of the conserved Phe and Trp residues in the two TADs. Interestingly, a recent study of the binding of the p53 TADs to the anti-apoptotic Bcl-xL/Bcl-2 proteins, using an NMR spectroscopy-based structural model, suggested a similar phenomenon, that two TADs bind to the same surface of Bcl-xL/Bcl-2 but with opposing orientations.43 This observation highlights the importance of the conformational flexibility of the p53 TADs and suggests that the reorientation of the helices may be a more common phenomenon among complexes of the two domains.
Figure 4.
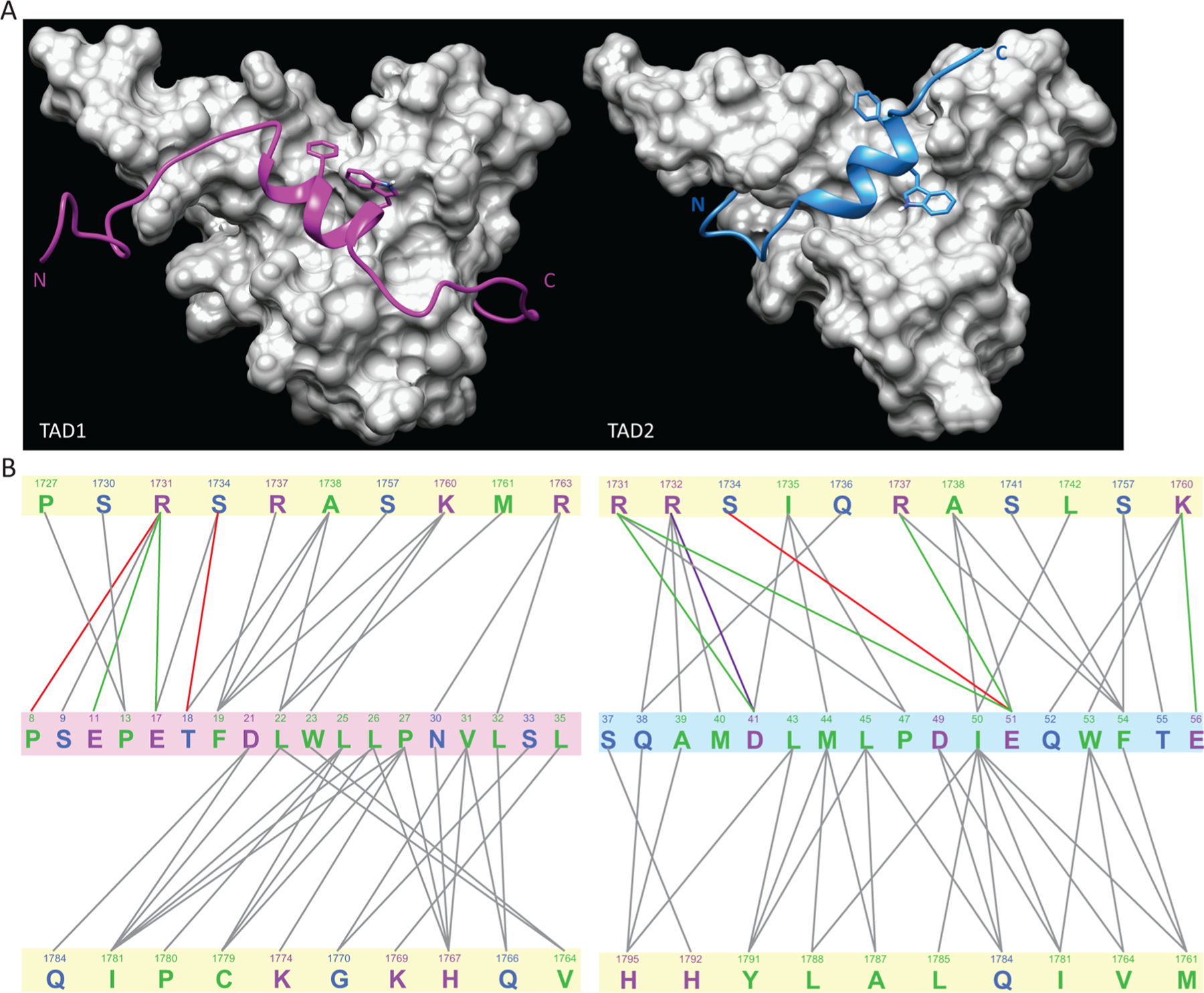
Comparison of the complexes of Taz2 with the p53 TAD1 and TAD2 domains. (A) Comparison of the structures of the Taz2 complexes with p53 TAD1 (left) and TAD2 (right). While both p53 peptides are shown in backbone ribbon format, with TAD1 colored purple and TAD2 cyan, the highly conserved side chains of F19 and W23 of TAD1 and W53 and F54 of TAD2 are also represented. The Taz2 components, shown as a gray surface representation, have the same orientations in both panels. The p53 TAD1–Taz2 structure is from Protein Data Bank entry 2K8F. (B) Schematic comparison of the intermolecular interactions in the p53 TAD1–Taz2 (left) and p53 TAD2–Tax2 (right) complexes. Interactions indicated were detected in >60% of all members of the ensemble. Figures were generated with MONSTER.38 Taz2 residues are highlighted with a pale yellow background in both panels, and the p53 TAD1 residues are highlighted with a pale mauve background; the p53 TAD2 residues are highlighted with a pale blue background. The lines connect interacting residues, with the following color scheme: green, electrostatic; red, hydrogen bonding; purple, salt bridge; gray, hydrophobic. The text colors indicate the type of residue: green, hydrophobic; blue, polar; magenta, charged.
Although both p53 TAD1 and TAD2 are intrinsically disordered when unbound and can form α-helices upon binding, there are notable differences between the two transactivation domains. Unlike the helical conformation of TAD1, which can be observed to form transiently even in the free state, the structured state of TAD2 is more dynamic.44 The greater dynamic behavior of TAD2 can also be discerned in plots of p53 disorder, which show that TAD2 has a greater propensity for disorder than TAD1.45 Whereas both domains can form extended complexes stabilized by hydrophobic interactions, such as observed in the TAD2–Taz2 structure presented here, only TAD2 has also been reported to form electrostatically dominated complexes in which it functions as a single-stranded DNA mimic, such as when bound to RPA70N.17 Moreover, whereas all reported structures of p53 TAD1 complexes exhibit the amphipathic α-helix, with only minor variations in helix length, the structure of bound p53 TAD2 is more variable, including an extended structure without the formation of an α-helix when bound to TFIIH p62.22 These two types of binding complexes suggest a critical, and divergent, role for TAD2 within p53 as compared with TAD1.
Changes in orientation among different complexes have also been observed for several other proteins. Ubiquitin binds to the helical “ubiquitin interacting motif” and “motif interacting with ubiquitin” ubiquitin-binding domains with opposite orientations.46,47 For example, two ubiquitin-binding domains of Rabex-5 have been shown to interact with ubiquitin with superimposable conformations but with opposite orientations.48 Comparison of the interactions of calmodulin with peptides from different interaction partners has suggested that steric and electrostatic features prescribe the orientation of the peptide with respect to the target binding channel formed by the N- and C-terminal domains of calmodulin. Similar to our results presented here, formation of critical electrostatic interactions and steric accessibility are important for determining the orientation of binding, and these features have been used to make predictions about the complexes of interactors without available structures.49 Likewise, the PAH1 and PAH2 domains of the histone deacetylase mSin3a were shown to bind to the same surface on SAP25 but with inverse orientations; the two PAH domains share the same interaction motif, but it is present in the amino acid sequences in reverse order.46
The Taz2 domain interacts with the acidic transactivation domains of numerous transcription factors. In addition to the p53 TAD1 and TAD2 complexes, the interactions of STAT1,50 the E1A oncoprotein,51 B-Myb,52 DNA-bound MEF2,53 and C/EBPε54 with Taz2 have all been structurally characterized. In each case, the acidic transactivation domain forms at least one amphipathic α-helix that interacts with Taz2. STAT1, p53 TAD1, and p53 TAD2 form single helices when they bind to Taz2, whereas E1A forms a well-defined helix with two shorter helical turns; C/EBPε forms two helices in an L conformation. In all complexes, the conformation of Taz2 is similar to that of the free form, with only small local changes observed, and the interactors bind to various surfaces of Taz2. The binding site for the C-terminus of STAT1 overlaps with that of C/EBPε, and the binding site for E1A is similar to that for p53 TAD2 and, to a lesser extent, TAD1. Three MEF2 dimer–DNA complexes are able to bind to Taz2, fully surrounding the protein.53 Thus, Taz2 is a relatively rigid unit that is able to make surface interactions with numerous proteins in different ways.
A recent study of the binding of p53 TAD1 and TAD2 to CBP Taz2 indicated, similar to our findings for p300 Taz2, that both domains bind to the α1, α2, α3 surface of Taz2 with high affinity.55 Moreover, a second, lower-affinity binding site was also proposed proximal to the first. Our interaction site for p53(35–59) on Taz2 corresponds to the “high-affinity” site in this study (Figure S2A of the Supporting Information). Although we do not have evidence of binding to the “low-affinity” site, in all but one structure of the ensemble, the N-terminus of the p53(35–59) peptide wraps around the core of Taz2 and binds proximal to this second site (Figure S2B,C of the Supporting Information). As the N-terminus of the peptide corresponds to the linker between the TAD1 and TAD2 domains, our structure suggests a model in which the two TAD subdomains could simultaneously interact on Taz2 through the “high-affinity” and “low-affinity” sites. Such a complex, in which p53 TAD would create a clamp around the Taz2 domain, would have an overall affinity higher than that of either interaction alone, as has already been demonstrated for binding of p53(1–57) to Taz2,26,56 that would be further increased by potential interactions of the four TADs presented in the p53 tetramer with different domains of CBP/p300. As they are maintained at low levels in the cell, there is significant competition among transcription factors for binding to CBP and p300. The interaction of the full TAD of p53 with the Taz2 domain, which has been shown to have increased affinity,56 would help p53 compete for binding to promote its transcriptional effects. In addition, binding of TAD1 to the secondary site on Taz2, promoted by tighter binding of TAD2 to the primary site, would protect this domain from interaction with the E3 ubiquitin ligase MDM2, further stabilizing p53 and facilitating acetylation of its C-terminal regulatory domain by CBP/p300, similar to the effects observed when cells are treated with nutlin to inhibit binding of MDM2 to TAD1.57
Although mutations in the p53 TAD are rare, both somatic and germ-line mutations in TAD1 and TAD2 have been observed, including mutation of residues important for Taz2 binding, such as Asp21 in TAD1 and Asp48 in TAD2.58 In the case of these mutations, the presence of a second binding site would still allow binding of co-activators. In addition, the two transactivation domains have been shown to respond to different stresses; for example, within the context of the Δ40p53 isoform that lacks TAD1, TAD2 has been shown to be particularly responsible for the response of p53 to ER stress.59 Thus, the two domains provide additional flexibility for p53 to respond to a variety of stress conditions.
Here, we have investigated the structure and thermodynamics of the complex formed between p53 TAD2 and p300 Taz2. We find that TAD2 binds Taz2 on the same interface as TAD1, but with an alternate orientation. Although the two transactivation domains are similar, there are critical differences that give each distinct binding interactions and functions. The results presented further support the importance of the two domains and shed new light on differences in their interactions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ryo Hayashi (Laboratory of Cell Biology, National Cancer Institute) for peptide synthesis.
Funding
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Center Research.
ABBREVIATIONS
- CBP
CREB-binding protein
- CD
circular dichroism
- ITC
isothermal titration calorimetry
- MALDI-TOF
matrix-associated laser desorption ionization time-of-flight
- MD
molecular dynamics
- NOESY
nuclear Overhauser enhancement spectroscopy
- rmsd
root-mean-square deviation
- RP-HPLC
reversed-phase high-pressure liquid chromatography
- TFA
trifluoroacetic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information
Two multipart figures (S1 and S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The final coordinates of the Taz2–p53(35–59) complex have been deposited with the Protein Data Bank as entry 2MZD. The chemical shift assignments and related experimental data have been deposited in the BioMagResBank with accession number 25484.
The authors declare no competing financial interest.
REFERENCES
- (1).Speidel D (2015) The role of DNA damage responses in p53 biology. Arch. Toxicol 89, 501–517. [DOI] [PubMed] [Google Scholar]
- (2).Carvajal LA, and Manfredi JJ (2013) Another fork in the road: Life or death decisions by the tumour suppressor p53. EMBO Rep 14, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Candau R, Scolnick DM, Darpino P, Ying CY, Halazonetis TD, and Berger SL (1997) Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 15, 807–816. [DOI] [PubMed] [Google Scholar]
- (4).Momand J, Zambetti GP, Olson DC, George D, and Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- (5).Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, and Vogelstein B (1993) Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857–860. [DOI] [PubMed] [Google Scholar]
- (6).Wu X, Bayle JH, Olson D, and Levine AJ (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- (7).Xu H, Ye H, Osman NE, Sadler K, Won EY, Chi SW, and Yoon HS (2009) The MDM2-binding region in the transactivation domain of p53 also acts as a Bcl-X(L)-binding motif. Biochemistry 48, 12159–12168. [DOI] [PubMed] [Google Scholar]
- (8).Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, and Kelly K (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89, 1175–1184. [DOI] [PubMed] [Google Scholar]
- (9).Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, and Berger SL (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- (10).Gu W, and Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606. [DOI] [PubMed] [Google Scholar]
- (11).Liu G, Xia T, and Chen X (2003) The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem 278, 17557–17565. [DOI] [PubMed] [Google Scholar]
- (12).Wang F, Marshall CB, and Ikura M (2013) Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell. Mol. Life Sci 70, 3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jenkins LM, Durell SR, Mazur SJ, and Appella E (2012) p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 33, 1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, and Yao TP (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 20, 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, and Appella E (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 12, 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, Karnezis AN, and Attardi LD (2011) Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, Liao JC, Okorokov AL, Milner J, Arrowsmith CH, and Bochkarev A (2005) Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl. Acad. Sci. U.S.A 102, 15412–15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Di Lello P, Jenkins LM, Jones TN, Nguyen BD, Hara T, Yamaguchi H, Dikeakos JD, Appella E, Legault P, and Omichinski JG (2006) Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 22, 731–740. [DOI] [PubMed] [Google Scholar]
- (19).Feng H, Jenkins LM, Durell SR, Hayashi R, Mazur SJ, Cherry S, Tropea JE, Miller M, Wlodawer A, Appella E, and Bai Y (2009) Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure 17, 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, and Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953. [DOI] [PubMed] [Google Scholar]
- (21).Lee CW, Martinez-Yamout MA, Dyson HJ, and Wright PE (2010) Structure of the p53 Transactivation Domain in Complex with the Nuclear Receptor Coactivator Binding Domain of CREB Binding Protein. Biochemistry 49, 9964–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Okuda M, and Nishimura Y (2014) Extended String Binding Mode of the Phosphorylated Transactivation Domain of Tumor Suppressor p53. J. Am. Chem. Soc 136, 14143–14152. [DOI] [PubMed] [Google Scholar]
- (23).Popowicz GM, Czarna A, and Holak TA (2008) Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle 7, 2441–2443. [DOI] [PubMed] [Google Scholar]
- (24).Popowicz GM, Czarna A, Rothweiler U, Szwagierczak A, Krajewski M, Weber L, and Holak TA (2007) Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle 6, 2386–2392. [DOI] [PubMed] [Google Scholar]
- (25).Rowell JP, Simpson KL, Stott K, Watson M, and Thomas JO (2012) HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 20, 2014–2024. [DOI] [PubMed] [Google Scholar]
- (26).Jenkins LM, Yamaguchi H, Hayashi R, Cherry S, Tropea JE, Miller M, Wlodawer A, Appella E, and Mazur SJ (2009) Two distinct motifs within the p53 transactivation domain bind to the Taz2 domain of p300 and are differentially affected by phosphorylation. Biochemistry 48, 1244–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bax AD, and Grzesiek S (1993) Methodological advances in protein NMR. Acc. Chem. Res 26, 131–138. [Google Scholar]
- (28).Iwahara J, Wojciak JM, and Clubb RT (2001) Improved NMR spectra of a protein-DNA complex through rational mutagenesis and the application of a sensitivity optimized isotope-filtered NOESY experiment. J. Biomol. NMR 19, 231–241. [DOI] [PubMed] [Google Scholar]
- (29).Ogura K, Terasawa H, and Inagaki F (1996) An improved double-tuned and isotope-filtered pulse scheme based on a pulsed field gradient and a wide-band inversion shaped pulse. J. Biomol. NMR 8, 492–498. [DOI] [PubMed] [Google Scholar]
- (30).Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, and Kay LE (1997) Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage lambda N-peptide/boxB RNA complex. J. Am. Chem. Soc 119, 6711–6721. [Google Scholar]
- (31).Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (32).Johnson BA, and Blevins RA (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- (33).Cornilescu G, Delaglio F, and Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- (34).Nederveen AJ, Doreleijers JF, Vranken W, Miller Z, Spronk CA, Nabuurs SB, Guntert P, Livny M, Markley JL, Nilges M, Ulrich EL, Kaptein R, and Bonvin AM (2005) RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672. [DOI] [PubMed] [Google Scholar]
- (35).Brunger AT (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- (36).Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, and Warren GL (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54, 905–921. [DOI] [PubMed]
- (37).Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, and Thornton JM (1996) AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486. [DOI] [PubMed] [Google Scholar]
- (38).Salerno WJ, Seaver SM, Armstrong BR, and Radhakrishnan I (2004) MONSTER: Inferring non-covalent interactions in macromolecular structures from atomic coordinate data. Nucleic Acids Res 32, W566–W568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).De Guzman RN, Liu HY, Martinez-Yamout M, Dyson HJ, and Wright PE (2000) Solution structure of the TAZ2 (CH3) domain of the transcriptional adaptor protein CBP. J. Mol. Biol 303, 243–253. [DOI] [PubMed] [Google Scholar]
- (40).Wishart DS, Bigam CG, Holm A, Hodges RS, and Sykes BD (1995) 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR 5, 67–81. [DOI] [PubMed] [Google Scholar]
- (41).Zhu J, Zhang S, Jiang J, and Chen X (2000) Definition of the p53 functional domains necessary for inducing apoptosis. J. Biol. Chem 275, 39927–39934. [DOI] [PubMed] [Google Scholar]
- (42).Zhu J, Zhou W, Jiang J, and Chen X (1998) Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J. Biol. Chem 273, 13030–13036. [DOI] [PubMed] [Google Scholar]
- (43).Ha JH, Shin JS, Yoon MK, Lee MS, He F, Bae KH, Yoon HS, Lee CK, Park SG, Muto Y, and Chi SW (2013) Dual-site interactions of p53 protein transactivation domain with anti-apoptotic Bcl-2 family proteins reveal a highly convergent mechanism of divergent p53 pathways. J. Biol. Chem 288, 7387–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lee H, Mok KH, Muhandiram R, Park KH, Suk JE, Kim DH, Chang J, Sung YC, Choi KY, and Han KH (2000) Local structural elements in the mostly unstructured transcriptional activation domain of human p53. J. Biol. Chem 275, 29426–29432. [DOI] [PubMed] [Google Scholar]
- (45).Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, and Dunker AK (2008) Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 9 (Suppl. 1), S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, and Schneider TR (2006) Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell 124, 1183–1195. [DOI] [PubMed] [Google Scholar]
- (47).Swanson KA, Kang RS, Stamenova SD, Hicke L, and Radhakrishnan I (2003) Solution structure of Vps27 UIM-ubiquitin complex important for endosomal sorting and receptor down-regulation. EMBO J 22, 4597–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sahu SC, Swanson KA, Kang RS, Huang K, Brubaker K, Ratcliff K, and Radhakrishnan I (2008) Conserved themes in target recognition by the PAH1 and PAH2 domains of the Sin3 transcriptional corepressor. J. Mol. Biol 375, 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Osawa M, Tokumitsu H, Swindells MB, Kurihara H, Orita M, Shibanuma T, Furuya T, and Ikura M (1999) A novel target recognition revealed by calmodulin in complex with Ca2+-calmodulin- dependent kinase kinase. Nat. Struct. Biol 6, 819–824. [DOI] [PubMed] [Google Scholar]
- (50).Wojciak JM, Martinez-Yamout MA, Dyson HJ, and Wright PE (2009) Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J 28, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ferreon JC, Martinez-Yamout MA, Dyson HJ, and Wright PE (2009) Structural basis for subversion of cellular control mechanisms by the adenoviral E1A oncoprotein. Proc. Natl. Acad. Sci. U.S.A 106, 13260–13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Oka O, Waters LC, Strong SL, Dosanjh NS, Veverka V, Muskett FW, Renshaw PS, Klempnauer KH, and Carr MD (2012) Interaction of the transactivation domain of B-Myb with the TAZ2 domain of the coactivator p300: Molecular features and properties of the complex. PLoS One 7, e52906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).He J, Ye J, Cai Y, Riquelme C, Liu JO, Liu X, Han A, and Chen L (2011) Structure of p300 bound to MEF2 on DNA reveals a mechanism of enhanceosome assembly. Nucleic Acids Res 39, 4464–4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bhaumik P, Davis J, Tropea JE, Cherry S, Johnson PF, and Miller M (2014) Structural insights into interactions of C/EBP transcriptional activators with the Taz2 domain of p300. Acta Crystallogr D70, 1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Arai M, Ferreon JC, and Wright PE (2012) Quantitative analysis of multisite protein-ligand interactions by NMR: Binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc 134, 3792–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Teufel DP, Freund SM, Bycroft M, and Fersht AR (2007) Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. U.S.A 104, 7009–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, and Vassilev LT (2004) Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol. Chem 279, 53015–53022. [DOI] [PubMed] [Google Scholar]
- (58).Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, and Olivier M (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat 28, 622–629. [DOI] [PubMed] [Google Scholar]
- (59).Bourougaa K, Naski N, Boularan C, Mlynarczyk C, Candeias MM, Marullo S, and Fahraeus R (2010) Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell 38, 78–88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.