

Transcriptomic analyses of several wheat species with different ploidies reveal alternative splicing changes in response to wheat domestication or polyploidization.
Abstract
Alternative splicing (AS) occurs extensively in eukaryotes as an important mechanism for regulating transcriptome complexity and proteome diversity, but variation in the AS landscape in response to domestication and polyploidization in crops is unclear. Hexaploid wheat (AABBDD, Triticum aestivum) has undergone two separate allopolyploidization events, providing an ideal model for studying AS changes during domestication and polyploidization events. In this study, we performed high-throughput transcriptome sequencing of roots and leaves from wheat species with varied ploidies, including wild diploids (AbAb, Triticum boeoticum) and tetraploids (AABB, Triticum dicoccoides), domesticated diploids (AmAm, Triticum monococcum) and tetraploids (AABB, Triticum dicoccum), hexaploid wheat (AABBDD, T. aestivum), as well as newly synthesized hexaploids together with their parents. Approximately 22.1% of genes exhibited AS, with the major AS type being intron retention. The number of AS events decreased after domestication in both diploids and tetraploids. Moreover, the frequency of AS occurrence tended to decrease after polyploidization, consistent with the functional sharing model that proposes AS and duplicated genes are complementary in regulating transcriptome plasticity in polyploid crops. In addition, the subgenomes exhibited biased AS responses to polyploidization, and ∼87.1% of homeologs showed AS partitioning in hexaploid wheat. Interestingly, substitution of the D-subgenome modified 42.8% of AS patterns of the A- and B-subgenomes, indicating subgenome interplay reprograms AS profiles at a genome-wide level, although the causal–consequence relationship requires further study. Conclusively, our study shows that AS variation occurs extensively after polyploidization and domestication in wheat species.
Polyploids experience whole-genome duplication and often exhibit exceptional hybrid vigor and enhanced stress tolerance compared to their progenitors (Chen, 2010; Yang et al., 2014; Han et al., 2016). This mechanism has been recognized as a major force driving and shaping the evolution and speciation of vascular plants (Otto, 2007; Madlung, 2013; Van de Peer et al., 2017), because it supplies a vast reservoir of raw genetic materials for mutation, selection, and evolution. In addition to gene duplication (GD), alternative splicing (AS) is also widely accepted as a major mechanism for transcriptome plasticity and proteomic complexity during plant polyploidization (Filichkin et al., 2010). AS refers to a mechanism of post-transcriptional RNA processing whereby multiple distinct transcripts can derive from a single gene, mainly including intron retention (IR), exon skipping (ES), alternative 5′ splicing site (A5), and A3 (Syed et al., 2012). AS can result in alterations of protein structures, functions, or subcellular locations (Gracheva et al., 2011; Kriechbaumer et al., 2012), and thus contributes to diverse regulation of biological process. Up to ∼92% to 94% of human (Homo sapiens) genes and ∼21.2% to 63% of plant genes undergo AS (Wang et al., 2008; Zhang et al., 2016). ES accounts for the largest proportion of AS in humans, whereas IR is the primary form in plants (Reddy et al., 2013).
AS and GD are two main processes responsible for protein functional diversity expansion. What is the evolutionary relationship between them, and do they evolve independently, or can they be regarded as interchangeable repositories of protein diversity? Genome-wide analyses report a negative correlation between the number of AS events and the size of protein families in human, mouse (Mus musculus), Drosophila melanogaster, and Caenorhabditis elegans (Kopelman et al., 2005; Su et al., 2006; Talavera et al., 2007; Iñiguez and Hernández, 2017), and that larger gene families tend to have fewer genes subjected to AS. In addition, duplicates may undergo fewer AS events than single copy genes (singletons). Furthermore, AS is likely to gain an acceleration in divergence after polyploidization in that AS events are lost in recent gene duplicates, plus novel AS events are gained in ancient homologs (Zhang et al., 2010; Xu et al., 2012). These findings are generalized as the functional sharing model for AS after GD, which illustrates the asymmetric AS evolution and subfunctionalization of homologous genes, where one homolog would retain a certain number of ancestral AS events and the other ones adopt the rest of the isoforms. However, this AS-duplication equivalence interpretation is debated by the accelerated AS model, which argues that an age-dependent progressive acquisition of new AS occurs after GD probably due to the relaxation of selection (Jin et al., 2008; Chen et al., 2011; Roux and Robinson-Rechavi, 2011). To reconcile the observation of negative correlation between gene family size and AS event number, the model proposes that genes with fewer AS events prefer to duplicate more frequently during evolutionary history, which accounts for why ancient homologs persist with increased AS events compared to recent duplicated ones (Roux and Robinson-Rechavi, 2011).
It is particularly interesting to investigate the interplay of AS and GD in plants, because most plant species have undergone polyploidization during their evolutionary history. However, most of the above conclusions were drawn from vertebrates, and the relationship between AS and GD is still ambiguous in plants. Lin et al. (2008) reported that duplicated genes tend to have more AS forms than those of singletons in both rice (Oryza sativa) and Arabidopsis (Arabidopsis thaliana) by analyzing families of paralogous genes. However, another study demonstrates that ∼63% of multiexonic genes are subjected to AS in paleopolyploid soybean and shows a decrease of AS events in polyploidization-derived duplicates compared with singletons, causing a negative correlation between the amount of AS events and the size of the gene family (Shen et al., 2014). Furthermore, extensive changes of AS patterns occur to duplicated genes in both Arabidopsis and Brassica napus (Zhang et al., 2010; Zhou et al., 2011), and both loss of ancestral splicing forms and gain of new splicing forms are observed in polyploidization duplicates and tandem duplicates (Tack et al., 2014). In addition, AS patterns of homeologs in resynthesized polyploid B. napus are greatly distinct from that of natural lines, presenting more AS pattern changes than does natural B. napus compared with the progenitors (Zhou et al., 2011).
Hexaploid wheat (AABBDD, Triticum aestivum) is a typical allopolyploid with three distinct subgenomes, involving two separate polyploidization events. The progenitors and polyploids are extant, and the phylogenetic relationships between each other have been well established. In addition, synthetic polyploid wheat can be readily obtained, which enables us to mimic the polyploidization process and investigate the transcriptomic and genomic variation at the onset of polyploidy. Furthermore, there are plenty of Triticum boeoticum and Triticum dicoccoides as well as Triticum monococcum and Triticum dicoccum in natural environments that can be used to interpret wheat domestication events. Thus, the Aegilops–Triticum system provides an ideal model for studying the genetic and epigenetic events that occur early during the evolutionary history of polyploids. Our previous study demonstrates that AS occurs extensively in wheat, and homeologs exhibit partitioned AS patterns in response to abiotic stresses (Liu et al., 2018b). Here, we studied AS divergence during wheat domestication and polyploidization events and investigated the potential effects of subgenome interplay on AS profile using T. boeoticum, T. monococcum, T. dicoccoides, T. dicoccum, T. aestivum, and synthetic polyploid wheat. Our results also provide evidence of an interesting relationship between AS and GD in response to polyploidization.
RESULTS
RNA Sequencing, Transcriptome Assembly, and AS Identification
Our interest here was to identify potential AS events in wheat and to investigate their changes in response to domestication and polyploidization events at a genome-wide level. To this end, we performed high-throughput RNA sequencing (RNA-seq) for 40 libraries using the Illumina platform, including leaf and root tissues at the seedling stage for wild diploid and tetraploid wheat, domesticated diploid and tetraploid wheat, hexaploid wheat, and resynthesized hexaploid wheat (see “Materials and Methods”). After adaptor sequence trimming and data processing, 2.4 billion high-quality clean reads were obtained, with an average of 28.3 million reads per subgenome (Supplemental Dataset S1). By comparison of unique mapping rate, we selected WEWv1 as the reference genome for T. boeoticum, T. monococcum, and T. dicoccum (Supplemental Fig. S1), whereas the AETv4 genome sequence was used for reference-guided transcript assembly for Aegilops tauschii and IWGSCv1 for T. aestivum ssp. yunnanense ‘King’ (TKM), T. aestivum, extracted tetraploid wheat (ETW), and synthesized allohexaploid wheat (Dobin et al., 2013; Avni et al., 2017; Appels et al., 2018; Ling et al., 2018).
After read mapping and transcript assembly, we excluded low-abundance transcripts (less than five normalized supporting junction reads per splicing site per sample) and obtained 46,837, 20,902, and 81,942 unique assembled transcripts involving 35,836, 16,654, and 60,079 annotated genes based on WEWv1, AETv4, and IWGSCv1 genomes, respectively (Fig. 1A). Subsequently, we used custom scripts to perform AS analysis of those filtered transcripts and only those genes exhibiting AS in both biological replicates were considered (Supplemental Fig. S2). In total, we identified 7,782 AS events in WEWv1 (A-subgenome: 4,880; B-subgenome: 2,902), 3,064 AS events in AETv4 (D-genome), and 18,358 AS events in IWGSCv1 (A-subgenome: 6,553; B-subgenome: 7,622; D-subgenome: 4,183; Fig. 1B; Supplemental Dataset S2) with 2,691 to 10,831 AS events identified for different wheat species (Supplemental Fig. S3; Supplemental Dataset S3). Seven major types of AS events were identified, including IR, ES, A5, A3, alternative first exon (FE), alternative last exon (LE), and mutually exclusive exons (ME). Of these, IR was the most abundant type, representing 64% of the total, followed by A3, A5, ES, FE, LE, and ME (Fig. 1B; Supplemental Fig. S3), which was consistent with the observation in Arabidopsis and rice (Syed et al., 2012).Cumulative distribution analysis showed that 55.3% of annotated genes possessed only one major transcript although with multiple AS isoforms (percent spliced index [PSI] > 0.75 or PSI < 0.25; the PSI measures the efficiency of splicing a sequence into the transcript population of a gene, see “Materials and Methods”), whereas 44.7% of the genes simultaneously exhibited two or more predominant transcripts (0.25 ≤ PSI ≤ 0.75; Fig. 2A). Pfam enrichment analysis showed that these genes were significantly enriched in an RNA recognition motif gene family, consistent with the fact that splicing requires a large number of RNA-binding proteins that have one or more RNA-recognition motifs, indicating AS genes (ASGs) are involved in the regulation of AS occurrence in wheat, such as Ser/Arg-rich genes in Arabidopsis (Fig. 2B; Supplemental Dataset S4). Further investigation indicated GC content might be correlated with occurrence of ES and IR, that is, skipped exons exhibited a significantly lower GC content than that of other exons (46.9% versus 50.5%, Student’s t test, P < 0.001), whereas retained introns had a significantly higher GC content (42.1% versus 39.1%, Student’s t test, P < 0.001; Fig. 2C). In addition, the length of skipped exons or retained introns was shorter than that of the counterpart (195.3 versus 402.8 nucleotides for ES and 377.3 versus 511.5 nucleotides for IR, Student’s t test, P < 0.001 respectively; Fig. 2D).To obtain an overview of the landscape of AS events in the wheat genome, we investigated their distributions along the chromosomes and observed a significant enrichment of ASGs in the arm regions that was positively associated with gene distribution (P = 8.825e−09 and 2.2e−16 from Pearson correlation test, respectively; Fig. 3), which was consistent with a previous report in soybean (Shen et al., 2014). Interestingly, we observed a significant difference in the number of ASGs among subgenomes in hexaploid wheat (Kruskal–Wallis test, P = 0.01639), but not in tetraploid (Wilcoxon-signed–rank test P = 0.875). In addition, a large proportion of homeologous genes exhibited AS partitioning in polyploid wheat (see details below); only 303 (3.6%) and 286 (4%) groups of homeologs exhibited similar AS patterns in tetraploid (IW136 and DM4) and hexaploid wheat (TAA10, TKM, and CS).
Figure 1.

AS event identification in wheat species with different ploidies. A, Column diagram shows the number of assembled genes (yellow), transcripts (light gray), alternative spliced genes (orange), and AS transcripts (dark gray) on each subgenome. Pie charts represent the proportion of total assembled genes with single-transcript or multi-transcripts. Green, blue, yellow, brown, and purple indicate genes with one, two, three, four, and greater than five transcripts. B, Structural sketches of different AS events and their corresponding number and proportion on each subgenome.
Figure 2.

Characterization of ASGs in wheat species with different ploidies. A, Density distribution of PSI of AS events. A large portion of ASGs with low PSI values indicate that these genes express one major transcript. B, The top-10 significantly enriched Pfam domains in ASGs ranked by hypergeometric test. Red broken line represents calculated –log10 (P value). Dark gray and light gray bars represent the observed and the expected ASG number, respectively. C, Density distribution of GC content between skipped exons (1,395) and other exons (328,525), as well as between retained introns (9,226) and other introns (249,641). The vertical dashed lines represent the average value of the data corresponding to each color. D, The comparison of length of skipped exons (1,395) and other exons (328,525) as well as retained introns (9,226) and other introns (249,641). The horizontal solid line represents the median length of the exon or intron in the AS event, and the horizontal dashed line represents the median length of counterparts. Asterisks indicate significant difference by Student’s t test (***P < 0.001).
Figure 3.

Chromosome distribution of ASGs in wheat species with different ploidies. A and B, Chromosome distribution of ASGs in hexaploid wheat (A) as well as in diploid and tetraploid wheat species (B). Lane a, chromosome visualization. Lane b, chromosome distribution of high-confidence annotated genes per megabit from reference genome. Lane c, density histogram of assembled genes per 500 kb from our transcriptome sequencing data. Lane d, distribution of ASGs per 500 Kb along chromosome. Lane e, conservation of AS between homeologous genes. Orange line in A indicates that AS patterns are conserved among all three homoeologs of hexaploid. Gray line in A indicates that AS patterns are conserved in only two homeologs of hexaploid. Orange line in B represents that AS patterns are conserved in tetraploid wheat.
AS Alteration in Response to Wheat Domestication
To explore AS changes responsive to domestication, we compared the AS patterns of 19,590 and 35,836 pairs of genes between AA2 (T. boeoticum, AbAb) and AA11 (T. monococcum, AmAm) as well as between IW136 (T. dicoccoides, AABB) and DM4 (T. dicoccum, AABB), respectively. We identified 1,531 and 1,350 ASGs with 2,691 and 2,318 AS events in wild diploid wheat AA2 and cultivated diploid wheat AA11, respectively, and thus found that the proportion of AS events decreased by 13.9% after domestication in diploid wheat. In addition, a total of 2,996 (5,137 events) and 1,663 (2,546 events) ASGs were revealed from wild emmer IW136 and domesticated tetraploid wheat DM4, respectively. Of these, 1,503 and 862 ASGs resided in subgenome A, whereas 1,493 and 801 were located in subgenome B, and the number of AS events was reduced by 50.4% in response to domestication in tetraploid wheat (Fig. 4A). Specifically, 1,481 and 3,481 AS events were lost, whereas 1,108 and 890 were newly generated in diploid and tetraploid wheat, respectively, during the domestication process (Fig. 4B).
Figure 4.

AS changes in diploid and tetraploid wheat. A, Histogram showing the number of AS events in T. boeoticum (AA2), T. monococcum (AA11), T. dicoccoides (IW136), and T. dicoccum (DM4). The Venn diagram at the top shows the comparison of ASGs between diploid or tetraploid wheat. B, The number of AS events in response to domestication in diploid and tetraploid wheat, including lost AS events (red), new AS events (blue), QDASE (green), and ATI (tan) after domestication. C, Display and validation of AS changes in response to domestication in diploid wheat. AA2, AA4, AA15, AA21, and AA1 represent wild diploid accessions, whereas AA11, AA19, AA28, AA30, and AA31 represent domesticated diploid wheat accessions. D, Display and validation of AS changes in response to domestication in tetraploid wheat. IW136, IW92, IW95, IW47, and IW104 represent wild tetraploid accessions, whereas DM4, Z39, CX03, CX04, and CX05 represent domesticated tetraploid wheat. The gray (isoform1) and black (isoform2) squares in the upper representation represent the structure and transcriptional direction of different isoforms of ASGs. The lower representation shows the results of RNA-seq data mapping at gene splicing sites and the number of normalized junction supporting reads of different isoforms. The red arrows represent the location and direction of primers (F, forward; R, reverse) designed for RT-PCR, and agarose gel electrophoresis on the right verifies the AS.
Due to insufficient read coverage, genome variation, and species–specific AS occurrence, a proportion of the eliminated AS events mentioned above were probably not due to domestication. To minimize artifacts, we next focused on the quantitative differential AS events (QDASEs) and altering transcript isoform (ATI) before and after domestication. QDASE refers to one splicing isoform exhibiting differential transcript abundance, which was defined by these criteria: junction-supporting reads based on a false discovery rate < 0.05 and > 20% variation of PSI (ΔPSI; see “Materials and Methods”) after domestication. ATI indicates an ASG showing changes in splicing isoform in response to domestication. In total, 293 AS events from 218 genes were exploited as domestication-responsive ASGs in diploid wheat, including 218 QDASEs and 75 ATIs. We also investigated the divergence of AS events in tetraploid wheat in response to domestication. By pairwise comparison, 419 responsive AS events from 322 genes were identified in tetraploid wheat, including 325 QDASEs and 94 ATIs (Fig. 4B; Supplemental Dataset S5). To confirm the altered AS patterns in response to domestication identified by RNA-seq data, we performed RT-PCR using conserved primer pairs amplifying different splicing variants in a single reaction across five wild diploid species, five domesticated diploid species, five wild tetraploid species, and five domesticated tetraploid species (Fig. 4, C and D). As expected, most of the examined alternative ASGs showed consistent splicing patterns with the prediction. For example, two transcript isoforms of WEWv1_A_gene.77369.0 (armadillo-type-fold-domain–containing gene) were expressed in all examined domesticated diploid wheat, whereas only one isoform was detected in 60% (3 of 5) wild diploid wheat (Fig. 4C). Similarly, WEWv1_A_gene.80316.0, encoding a phosphoinositide phosphatase gene, transcribed one more isoform in response to domestication in most domesticated tetraploid wheat. Whereas WEWv1_B_gene.13387.3, encoding an ATPase, had two major transcript isoforms before domestication, but only one major was detected after domestication (Fig. 4D). To explore the underlying molecular mechanism of AS event reduction in response to domestication, we analyzed the expression abundance of genes involved in AS regulation in wild and domesticated wheat species, and 17 and 21 AS-regulation related genes were downregulated after domestication in diploid and tetraploid wheat, respectively, whereas only seven and 11 genes were upregulated (Supplemental Dataset S6). RT-qPCR results further confirmed our observation that the three examined genes exhibited decreased expression abundance in domesticated varieties compared with wild ones (Supplemental Fig. S4). Therefore, we propose that the reduced expression level of splicing regulators might contribute to decreased splicing efficiency in domesticated species compared with wild species, which may lead to a decrease of AS occurrence in response to domestication in wheat.
To analyze conservation of AS changes in response to domestication in both diploid and tetraploid wheat, we compared 293 domestication-responsive AS events (both QDASEs and ATI) from diploid wheat (A-genome) and 237 from the A-subgenome of tetraploid wheat. A total of 27 AS events (including 26 QDASEs and one ATI) from 18 ASGs commonly occurred in both wheat species in response to domestication that were significantly enriched in gene ontology (GO) terms of “nucleic acid binding” (GO:0003676; Supplemental Fig. S5; Supplemental Dataset S4). Of these ASGs, WEWv1_A_gene.112770.0 and WEWv1_A_gene.1336.0 encoded Ser/Arg-rich splicing factors highly similar to RSZ22 (AT4G31580) and RS2Z32 (AT3G53500) in Arabidopsis, respectively, which are required for the regulation of gene AS (Golovkin and Reddy, 1998; Lopato et al., 2002; Lorkovic et al., 2004). To confirm the observation, we verified the AS patterns in multiple diploid and tetraploid wheat lines. The two transcript isoforms of the RSZ22 homolog (WEWv1_A_gene.112770.0) showed similar expression level in wild diploid and tetraploid lines (except IW47 and IW104), whereas isoform 1 transcript exhibited higher expression abundance than that of isoform 2 in all examined domesticated wheat. In contrast, isoform 2 of the RS2Z32 homolog (WEWv1_A_gene.1336.0) showed a higher expression level than that of isoform 1 after domestication (Supplemental Fig. S6). Thus, we propose that the splicing variation of AS regulators might also contribute to AS occurrence efficiency after domestication in wheat, which needs further investigation.
To further investigate the biological significance of the domestication-responsive ASGs, we performed translation analysis and Pfam assignment of different transcript isoforms of intron-retained genes. AS resulted in the change of coding sequence, thus subsequently leading to 590 and 850 pairs of isoforms showing differences in Pfam domain in diploid and tetraploid wheat, respectively (Supplemental Dataset S7). Dormancy is a desired trait for wild species to prevent seed germination during unsuitable external conditions, whereas domesticated crops have undergone artificial selection against seed dormancy (Liu et al., 2018a). Of these genes, we identified two ABA-responsive transcription factors (WEWv1_A_gene.9008.0 and WEWv1_A_gene.75640.0, similar to DI19-3 and BZIP63 in Arabidopsis, respectively) that are involved in regulating seed germination under stress conditions (Qin et al., 2014; Veerabagu et al., 2014) and one dormancy/auxin-associated protein (WEWv1_A_gene.66541.0 homolog to DRMH2 in Arabidopsis) subjected to AS in response to domestication in both diploid and tetraploid wheat (Supplemental Dataset S5). In addition, rachis brittleness and seed shattering are also typical characteristics altered during the wheat domestication process, whereas the Q gene is an important domestication gene involved in pleiotropical influences on phenotypic variation, including rachis fragility and seed threshability (Simons et al., 2006). Previous genetic and molecular evidence indicates that the phenotypic effect of the Q gene on domestication-related traits is associated with the presence of mutation at the miRNA binding site (which affects transcript abundance) and the 161-bp transposon insertion (which results in a deletion of conserved domains; Simons et al., 2006; Debernardi et al., 2017; Jiang et al., 2019b). Interestingly, the homolog of the Q gene (WEWv1_A_gene.90708.0) in diploid and tetraploid wheat was also subjected to differential AS, and the IR retention isoform abundance decreased in domesticated species compared to wild species in both diploid and tetraploid wheat (Supplemental Fig. S7).
AS Changes in Response to Wheat Polyploidization
To determine whether AS patterns changed after wheat polyploidization, we examined the AS landscape in ETW, TQ18, and XX329. Allohexaploid XX329 was resynthesized by the maternal parent ETW and the paternal parent TQ18 (A. tauschii). ETW was ploidy-reversed from hexaploid cultivar wheat TAA10 with high karyotype stability, produced by recurrent backcrossing (Kerber, 1964; Zhang et al., 2014). In total, we identified 8,713 AS events from 4,374 genes in ETW (A-subgenome: 2,090; B-subgenome: 2,284), 2,536 AS events from 1,583 genes in TQ18, and 9,922 AS events from 5,450 genes in XX329 (A-subgenome: 1,863; B-subgenome: 2,053; D-subgenome: 1,534; Fig. 5A). Compared with ETW (AABB) and TQ18 (DD), 2,661 and 587 AS events were lost, whereas 1,394 and 527 AS events were newly formed after polyploidization in XX329. Accordingly, the proportion of AS events in resynthesized allohexaploid wheat reduced by 11.8% compared to the counterpart in the parental lines, with 15.7%, 13.5%, and 2.4% decreases for subgenomes A, B, and D, respectively (Fig. 5A). We also examined QDASEs and ATIs in response to polyploidization according to the mentioned criteria above and identified 478 polyploidization-responsive QDASEs and 39 polyploidization-responsive ATIs in wheat (Fig. 5B; Supplemental Dataset S5). The splicing patterns were also confirmed by RT-PCR in three sets of synthesized wheat accessions. For example, we only detected the band of isoform 1 of Regulator of Chromosome Condensation1 (IWGSCv1_B_gene.25853.0) in tetraploid wheat, while both isoform-1 and -2 bands amplified in hexaploid wheat after polyploidization (Supplemental Fig. S8A). Isoform 1 of the glycosyltransferase related gene (IWGSCv1_B_gene.137552.0) only amplified in synthetic hexaploid wheat XX329 and SXS (Supplemental Fig. S8B).
Figure 5.

AS changes in response to wheat polyploidization. A, The number of AS events in TAA10, ETW, TQ18, and XX329. Allohexaploid XX329 was resynthesized by the maternal parent ETW and the paternal parent TQ18. ETW was ploidy-reversed from hexaploid cultivar wheat TAA10 with high karyotype stability, produced by recurrent backcrossing. B, The number of polyploidization-responsive AS events in wheat, including lost AS events, new AS events, QDASE, and ATI. C, Comparison of density distribution of PSI before and after wheat polyploidization. P value was calculated using the Kolmogorov–Smirnov test. Blue, before polyploidization; red, after polyploidization. D, GO enrichment analysis of ASGs responsive to polyploidization. The color scale from white (low) to dark red (high) represents differential log10(P value) of GO enrichment, and only significantly enriched terms (P < 0.05) are indicated.
In addition, density curve analysis of PSI of AS events showed that the distribution of PSI was biased to one in subgenomes A and B after polyploidization, which was obviously different from the corresponding distribution in subgenome D (Kolmogorov–Smirnov test, P = 1.4e−12) but similar to the trend in A- and B-subgenomes (Kolmogorov–Smirnov test, P = 0.098) during domestication (Fig. 5C). GO enrichment analysis also confirmed our observation that AS occurrence distributed unevenly among three subgenomes, because polyploidization-responsive ASGs in subgenome D were significantly enriched in two transfer RNA-related terms (GO:0004822 and GO:0006428), whereas the responsive ASGs in subgenomes A and B were enriched in 10 other terms, including transmembrane transport (GO:0055085; Fig. 5D). These results further suggested that the subgenomes exhibited different AS responses between each other, with two or more predominant transcripts from one ASG in subgenome D after polyploidization but less in subgenomes A and B. To better understand why AS occurrence decreased after polyploidization, we examined the expression profile of key AS regulators and found that 13 splicing related factors were differentially expressed after polyploidization. For example, the homologs of U2 small nuclear ribonucleoprotein auxiliary factor 65 (U2AF65A, IWGSCv1_A_gene.121408.0) were significantly downregulated in synthesized hexaploid wheat (XX329/DXY/SXS) compared with the tetraploid (ETW/DM4/SCAUP) parents, which interact with U2AF35 to promote U2snRNP for the recognition of the premRNA 3′ splice site during early spliceosome assembly in Arabidopsis (Chusainow et al., 2005; Heiner et al., 2010). In addition, the homolog of RS2Z32 (AT3G53500) in the Ser/Arg-rich protein family, which controls splice site selection and spliceosome assembly in Arabidopsis (Kalyna et al., 2006; Barta et al., 2010), was also downregulated after polyploidization in wheat (Supplemental Fig. S9). Therefore, we speculate that the differential expression of splicing-related genes after polyploidization, especially the downregulation of important splicing factors, is one of the causes for the decrease of the number of AS events. This observation is consistent with the functional sharing model that reduced AS events and duplicated genes are complementary in regulating transcriptome plasticity in polyploid crops.
Previous studies revealed a complex interplay of subgenomes in the regulation of gene expression in polyploid wheat (Pfeifer et al., 2014). To determine if subgenome interplay influences AS occurrence, we surveyed expression abundance of AS isoforms between XX329 and TAA10, which were predicted to contain >99.8% identical A- and B-subgenomic components and only differ from each other in the origin of the D-subgenome (Wang et al., 2016). We revealed 16.2% (1,583 of 9,781) XX329-specifc and 23.9% (2,335 of 9,781) TAA10-specific AS events on A- and B-subgenomes, respectively (Supplemental Fig. S10A). Among the coexisting AS events, 100 and 144 AS events were identified as QDASEs, and 14 and 15 were characterized as ATIs on subgenomes A and B (Fig. 5B). We revealed that the homologs of splicing regulators U2AF65A and U2AF35B (IWGSCv1_A_gene.121408.0 and IWGSCv1_A_gene.14304.0) were downregulated in XX329 compared with TAA10 (Supplemental Fig. S10B), which might be involved in the regulation of AS variation between each other. In addition, differential AS types of AS-regulating genes were examined between XX329 and TAA10. For example, the transcripts of IWGSCv1_A_gene.121408.0 deleted the fifth exon in TAA10 but retained it in XX329, whereas IWGSCv1_B_gene.67777.0, encoding a DEAD-box ATP-dependent RNA helicase involved in the mRNA splicing process, expressed two transcripts in TAA10 but only one in XX329 (Supplemental Fig. S11A). These lines of evidence suggested that the AS profile of subgenomes A and B was significantly affected due to substitution of the subgenome D in polyploid wheat, possibly due to differential expression patterns and altered AS profiles of AS-regulating genes.
AS Partitioning among Homeologs in Polyploidy Wheat
To investigate whether AS partitioning occurs among homeologs in hexaploid wheat, we comprehensively assayed AS patterns in hexaploid wheat of TAA10, TKM, and CS and identified 10,831 (5,588 genes), 10,797 (6,019 genes), and 8,949 (5,150 genes) AS events, respectively (Fig. 6A). Of these AS events, 28.3% (4,679 to 16,515) were commonly present among the three wheat lines, whereas 43.2% (7,132 of 16,515) showed accession-specific AS patterns (Supplemental Fig. S11B).
Figure 6.

AS partitioning among homeologs in hexaploid wheat. A, The number of AS events identified in TAA10 (green), TKM (blue), and CS (red). A, B, and D, Subgenomes in wheat. B, Comparison of AS patterns of homeologs among TAA10, TKM, and CS. Venn diagram shows conservation of AS triplets with same splicing type in three genotypes of hexaploid wheat. C, Ternary plots show AS partitioning among homeologous genes with same AS type in hexaploid wheat (TAA10 = 243, TKM = 220, CS = 181). Each dot represents a triad’s coordinate based on the PSI value.
Next, we focused on the AS variation among homeologous triplets, which have exactly one representative gene on subgenomes A, B, and D at a similar genomic region. In total, 1,501, 1,518, and 1,386 AS triplets were identified in TAA10, TKM, and CS, respectively (Supplemental Dataset S8), with at least one of the three homeologs showing AS. We observed that AS patterns were partitioned extensively, with only 14.4% (TAA10:243, TKM:220, and CS:181) of triplets showing simultaneous AS in three homeologs. Of these triplets, on average, 89.4% (TAA10:213, TKM:197, and CS:166) exhibited the same AS pattern and 10.6% (TAA10:30, TKM:23, and CS:15) showed different AS types, respectively (Fig. 6B). For example, a triplet cluster (IWGSCv1_A_gene.150363.0, IWGSCv1_B_gene.169279.0, and IWGSCv1_D_gene.65226.0) associated with the phosphoglucosamine mutase family has an AS event with the second exon skipped in all A-, B-, and D-subgenomes (Supplemental Fig. S12A). Another gene with a WW/Rsp5/WWP domain contained triplets showing IR in subgenomes A and B but A5 in subgenome D (Supplemental Fig. S12B). Among the 89.4% of homeologs displaying conservative AS types, a ternary plot (using a triad’s position based on the proportion of AS events abundance) classified them into three categories: (1) balanced category with similar proportion of AS transcripts for all three homeologs; (2) homeologous-dominant category with a significantly higher PSI value of an AS event in one homeolog than the other two; and (3) homeologous-suppressed category with a significantly lower PSI value of an AS event in one homeolog than the other two (Ramírez-González et al., 2018). The resulting plot showed that 17.4% (37 of 213), 24.9% (49 of 197), and 21.1% (35 of 166) of homeologous ASGs in TAA10, TKM, and CS, respectively, showed partitioned AS patterns among subgenomes (Fig. 6C), although most homeologous ASGs were assigned to the AS-balanced category. Thus, in total, 87.1% (85.6% + 14.4% × 10.6%) of homeologous triplets showed biased or different splicing patterns, suggesting AS changes occur extensively among homeologous genes in hexaploid wheat. In addition, we identified 225 homologous AS triplets from WEWv1 and AETv4 reference genomes, of which 198 (88%) showed the same AS type. Ternary plots by averaging the PSI value of the corresponding sample showed most of these conservative AS triplets (165 of 198 = 83.3%) in WEWv1 and AETv4 are assigned to the balanced category with a similar proportion of AS transcripts for A-, B-, and D-homeologs (Supplemental Fig. S13).
DISCUSSION
AS refers to a mechanism by which multiple mRNA isoforms are generated from one primary type of precursor molecule. Large-scale genomics studies have revealed that AS occurs extensively in plants, accounting for 42% to 61% of genes (Marquez et al., 2012; Reddy et al., 2013; Klepikova et al., 2016). AS expands the repertoire of functional complexity in plant genomes by increasing the diversity of the transcriptome, and growing evidence indicates that AS participates in a large fraction of biological pathways in plants (Staiger and Brown, 2013; Wang et al., 2015). Despite increasing interest in how AS is regulated during plant development, limited information is available in wheat. The question of how AS is influenced by subgenome interplay and what trends occur during evolution and domestication history have not been answered. Our results provided interesting suggestions for biologists to better understand how AS responds in plants, that is, AS evolutionary diverged during wheat domestication and the hexaploidization process; AS and GD might be functionally complementary in hexaploid wheat; and subgenome interplay significantly affects AS occurrence in hexaploid wheat and AS patterns partitioned extensively among homeologs.
AS Patterns Dramatically Changed in Response to Wheat Domestication
Domestication has been proven to strongly reduce genomic diversity through population bottlenecks in numerous cultivated crop species, resulting in a series of human-preferred syndromes, including loss of rachis brittleness, changes in seed dormancy, increases in seed size, etc. Reduced variation in gene expression patterns was observed in response to domestication by comparative transcriptomes between maize (Zea mays) and teosinte (Zea mays ssp. parviglumis) as well as between domesticated accessions of common bean (Phaseolus vulgaris) and its wild counterpart (Bellucci et al., 2014; Huang et al., 2015). Besides, AS profiles significantly changed during the domestication episode from the wild relatives to the cultivated species, although with different trends: Increased AS complexity occurred in domesticated maize, but decreased AS diversity occurred in cultivated sorghum (Sorghum bicolor; Huang et al., 2015; Ranwez et al., 2017; Smith et al., 2018). AS participates in the crop domestication process, e.g. a splice-site mutation in the fourth intron of the sh1 gene leads to the removal of the adjacent exon in sorghum varieties of SC35 and SC265, and subsequently accelerates its agronomic improvement with the non-shattering trait (Lin et al., 2012), and a C-to-A splice-site mutation of OsSSH1 (SNB) alters the splicing of its mRNA, causing reduced shattering of the ssh1 mutant by altering the development of the abscission layer and vascular bundle in rice (Jiang et al., 2019a).
In this study, we compared the AS profiles between wild (AbAb) and domesticated diploid wheat species (AmAm) as well as tetraploid wheat species (AABB). Not surprisingly, AS patterns dramatically changed during wheat domestication history, and a significant decrease of AS event abundance occurred in cultivated wheat species compared with wild species, consistent with the findings in sorghum (Ranwez et al., 2017). Expression levels of several AS regulators were significantly downregulated in domesticated wheat species compared with wild ones, which might result in decreased splicing efficiency and reduced AS occurrence in response to domestication in wheat under this circumstance. In addition, AS patterns of RS2Z32 and RSZ22, which are required for the regulation of splicing, were obviously different before and after domestication, indicating another potential contributor to the decreased amount of ASGs after domestication. Interestingly, although previous studies report that rachis brittleness and seed shattering variation are attributed to the presence of mutation at the miRNA binding site and a 161-bp transposon insertion of the Q gene (Simons et al., 2006; Debernardi et al., 2017; Jiang et al., 2019b), we revealed that the homolog of the Q gene was subjected to differential AS in diploid wheat and tetraploid wheat (Supplemental Fig. S7). Furthermore, we found a similar trend for AS variation during domestication events in diploid and tetraploid wheat species, and 16.6% (660 of 3,983) of AS events exhibited conserved domestication-responsive splicing patterns, including AS elimination and new AS formation, which is similar to a previous report (Ranwez et al., 2017). AS loss, in this case, results in a decrease of protein-coding genes in domesticated wheat species (∼1.4% and 3.2% for diploids and tetraploids, respectively), and consequently reduces the genomic diversity, which may be negatively corresponded to adaptive ability. However, the “less-is-more” hypothesis proposes that gene loss is recognized as a pervasive source of genetic variation that can cause adaptive phenotypic diversity (Albalat and Cañestro, 2016), suggesting that AS may not be just a noisy consequence of domestication. However, the question remains open whether AS actively influences the plant domestication event or if it is just a consequence of this evolution process. Because we do not have convincing biological evidence to support this hypothesis based on this study, the hypothesis merits further investigation in the future.
The Contribution of AS to Specialization during Wheat Polyploidization
GD and AS are considered two main contributors involved in the increase of protein diversity during the polyploidization process. The evolutionary relationship between the two processes is far from being understood, although several models have been proposed based on the studies in mammals. Of these, the functional sharing model suggests an asymmetric subfunctionalization of homologous genes such that one homolog would adopt part of the ancestral AS events whereas the other homeolog(s) adopt the rest; thus, on average, the number of AS events per gene decreases after GD (Kopelman et al., 2005; Su et al., 2006; Iñiguez and Hernández, 2017). The accelerated AS model indicates an increased number of AS events per gene probably due to relaxed selection pressure and an acquisition of AS for duplicated genes over time (Iñiguez and Hernández, 2017). In contrast, the independent model is established to explain a lack of relationship between AS and GD events (Su and Gu, 2012; Iñiguez and Hernández, 2017).
Hexaploid wheat (AABBDD, T. aestivum) is a widespread, typical allopolyploid with three distinct subgenomes and has undergone two separate allopolyploidization events. Functional diversification of duplicated genes has widely occurred among homeologous genes in polyploid wheat, probably caused by sequence variation or gene expression pattern change (Bottley et al., 2006; Zhang et al., 2011; Sakuma et al., 2019). In this study, we proposed that AS might also participate in the regulation of subfunctionalization of homeologs, because we revealed that AS patterns were dramatically changed in response to polyploidization at a genome-wide level and the variations in AS patterns diverged significantly among homeologous genes. Furthermore, consistent with the functional sharing model, we observed that the proportion of ASGs dramatically decreased in resynthesized allohexaploid wheat compared with their counterparts in the parental lines. The above results were in accordance with a previous study in resynthesized allotetraploid B. napus, which reported that more than one-quarter of the duplicated genes showed diverged AS patterns compared with the parents based on RT-PCR validation, with most cases of AS loss from one homeolog after polyploidy (Zhou et al., 2011). However, an increased level of AS was reported in tetraploid watermelon compared with the diploid in all vegetative tissues but fruit (Saminathan et al., 2015), but the authors did not discriminate homeologs in polyploids, which might not be able to differentiate AS changes for each homeolog. In conclusion, we therefore propose that AS and GD may not evolve independently in wheat. Another major outcome from this study is the finding that subgenome interplay reprograms AS occurrence in polyploid wheat. We took advantage of allopolyploid wheat (wherein chromosome sets are derived from different species), allowing us to distinguish subgenome origin of transcripts using single nucleotide polymorphism information among subgenome sequence. The results show that AS patterns in the subgenomes A and B are substantially dysregulated due to the substitution of subgenome D. However, the molecular mechanisms underlying this observation are still unknown, which is a topic that merits further investigation.
MATERIALS AND METHODS
Plant Materials
Wild diploid wheat AA1, AA2, AA4, AA15, and AA21 (AbAb, Triticum boeoticum) domesticated diploid wheat AA11, AA19, AA28, AA30, and AA31 (AmAm, Triticum monococcum), diploid goatgrass lines TQ18, SQ523, and Y199 (DD, Aegilops tauschii), tetraploid wheat species (AABB, Triticum dicoccoides [IW92, IW95, IW47, IW104, and IW136], Triticum dicoccum [DM4, Z39, CX03, CX04, CX05, and SCAUP]; ETW), and four hexaploid wheat species (AABBDD, Triticum yunnanense ‘King’ [TKM]; Triticum aestivum [CS]; T. aestivum [TAA10]; and a resynthesized allohexaploid wheat [XX329]) were grown in a growth chamber with 22°C/18°C (day/night), 16-h/8-h (light/dark), and 50% humidity. TKM is a hexaploid wheat subspecies with wrapped glumes and fragile spikes endemic to Yunnan Province, China. ETW is a ploidy-reversed form of tetraploid wheat from hexaploid wheat TAA10, produced by recurrent backcrossing. The hexaploid wheat XX329 was resynthesized by maternal parent ETW and paternal parent TQ18 (A. tauschii; Kerber, 1964); The hexaploid SXS was resynthesized by maternal parent SCAUP (Triticum turgidum) and paternal parent SQ523 (A. tauschii); The hexaploid wheat DXY was resynthesized by maternal parent DM4 (T. dicoccum) and paternal parent Y199 (A. tauschii).
RNA Extraction and Sequencing
Total RNA of leaf and root tissues from 10-d-old seedings (20 samples × 2 replicates) was extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA concentration was measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). RNA integrity was assessed on a model no. 2100 Bioanalyzer (Agilent Technologies). Then 125-bp (ETW, TQ18, XX329, and TAA10) or 150-bp (AA2, AA11, IW136, DM4, TKM, and CS) paired-end sequencing libraries with an average insert size of 280 bp were prepared with a TruSeq RNA Sample Preparation Kit v2 (Illumina) and sequenced on a HiSeq2500 (125 bp; Illumina) or a Novaseq6000 (150 bp; Illumina) according to the manufacturer’s standard protocols. Raw data obtained from Illumina sequencing were processed and filtered using the Illumina pipeline (https://www.Illumina.com) to generate Fastq files. Finally, ∼304.4G high-quality clean reads were generated from 40 libraries after quality detection (FastQC v0.11.5; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and filtering (Trimmomatic, v0.36; Bolger et al., 2014). Next, the high-quality reads were aligned to the respective reference genomes using the program STAR (v2.5.3a; https://github.com/alexdobin/STAR), and then the program Scallop (Shao and Kingsford, 2017; De Clerck et al., 2018) was employed to perform transcriptome assembly from merged mapping results by the program SAMtools (Li et al., 2009). Illumina reads of all samples were deposited with the Bioproject at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/bioproject) under the accession number PRJNA553311.
Reads Mapping, Transcript Structure Assembly, and Gene Expression Analysis
An overview of the bioinformatics pipeline used for data analysis is shown in Supplemental Figure S2. High-quality paired-end RNA-seq reads from each library were mapped to the appropriate reference genome with transcriptome general transfer format annotation files using the program STAR (v2.5.3a) with stringent parameters ′–alignEndsType EndToEnd–outFilterMultimapNmax 1–alignIntronMax 20000–alignIntronMin 20–outSJfilterCountUniqueMin 4 2 2 2–outSJfilterCountTotalMin 4 2 2 2–alignSJoverhangMin 8–alignSJDBoverhangMin 2′ (Dobin et al., 2013). Due to lack of a uniform reference genome for AA2, AA11, and DM4, we first mapped all their sequencing reads to IWGSCv1, WEWv1, and Tuv2 genomes using the program STAR and chose the best according to the unique mapping rate (Supplemental Fig. S1). Finally, the WEWv1_A genome was used as a reference for AA2 and AA11, the WEWv1_A&B genome was used for IW136 and DM4, the IWGSCv1_A&B genome was used for ETW, AETv4 was used for TQ18, IWGSCv1_A&B&AETv4 was used for XX329, and IWGSCv1 was used for TAA10, TKM, and CS.
The mapped reads were merged according to the mapping location of different reference subgenomes by the program SAMtools (v0.1.19; Li et al., 2009). Merged mapping results were assembled into putative transcripts with the program Scallop (v0.10.2) with the parameters ′–min_transcript_coverage 5–min_single_exon_coverage 50–min_flank_length 6′ (Shao and Kingsford, 2017). Next, transcripts with exons or introns <20 bp were filtered out, and only those transcripts with more than five normalized junction supporting reads for each junction in at least two biological replicates of one sample were retained for the following analysis using custom Perl scripts. The number of genes or ASGs per 1 Mb on the genome were counted and the program R was used (https://www.r-project.org/) to perform statistical tests. The number of mapping reads and Fragments per Kilobase Million on each gene were calculated by the tool featureCount (v1.5.2; http://bioinf.wehi.edu.au/featureCounts/) and an in-house script, respectively. The differentially expressed genes were characterized by the R package DESeq2 (v1.24.0; Love et al., 2014) with an absolute value of log2-fold change ≥ 1 and a false discovery rate < 0.05 as cutoffs. Moreover, only the reads of the AABB or DD subgenome in XX329 were counted to compare with ETW and TQ18, respectively, to analyze the differentially expressed genes before and after polyploidization.
AS Event, QDASE, and ATI Identification
To identify AS events, exon–exon and exon–intron junction information was extracted from the general transfer format file generated by the program Scallop and filtered via custom Perl scripts. AS events in all reference genomes, consisting of IR, A3, A5, ES, FE, LE, and ME, were identified based on the junction information by custom Perl scripts using the methods described by Brooks et al. (2011). Then, custom Perl scripts were used to count the number of supporting reads for each detected junction based on BAM files generated by the program STAR, and this number was multiplied by the normalized coefficient (average sequencing base/the sample sequencing base) based on the sequencing bases of the sample. To minimize artifacts, only unique mapped reads with ≤5 mismatches and ≥5 overhang bases were considered. The number of normalized junction supporting reads in these AS events was used to measure the percentage of a gene’s mRNA transcript that includes a specific exon or splice site, namely, PSI (Supplemental Fig. S2; Katz et al., 2010; Liu et al., 2018b; Park et al., 2018):
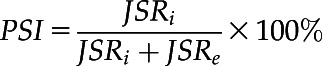 |
JSRi represents the number of junction reads that support inclusion of alternative exons (ES and IR) or longer exons (A3, A5, FE, LE, and ME) in an AS event, where the JSRi in IR type AS events was normalized according to the length of reads and introns:
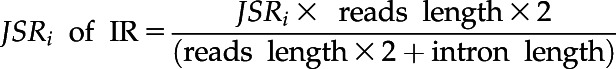 |
JSRe represents the number of junction reads that support exclusion of alternative exons (ES and IR) or shorter exons (A3, A5, FE, LE, and ME) in an AS event. Only those with the value of PSI between 0.05 and 0.95, and more than five normalized junction-supporting reads for each junction in both biological replicates, were considered AS events in this study. If the PSI value was close to 0 or 1, the ASG possessed one major transcript; if the PSI value was close to 0.5, the ASG tended to express both splicing transcripts.
To identify domestication-responsive and polyploidization-responsive AS events, Fisher’s exact tests in R (https://www.r-project.org/) were performed on the comparison of junction read counts supporting different splice isoforms. Next, ∆PSI (∆PSI = |PSIsample1-PSIsample2| × 100%) representing the degree of deviation in the AS events between different samples was calculated. Finally, AS events with ∆PSI > 20%, false discovery rate-adjusted P values (Benjamini and Hochberg’s method) < 0.05 in two biological replicates were identified as QDASEs or ATIs (Dong et al., 2018; Liu et al., 2018b).
Functional Annotation and Enrichment
The longest transcripts of assembled genes were aligned against IWGSCv1 reference genes with fairly good functional annotations by a local BLASTN program (Camacho et al., 2009). The functional description of the IWGSCv1 gene was derived from the functional description of the homologous genes of rice (Oryza sativa ssp. Japonica; IRGSP1.0, https://rapdb.dna.affrc.go.jp/) and Arabidopsis (Arabidopsis thaliana; The Arabidopsis Information Resource, https://www.arabidopsis.org/) by a local BLASTP program. The best hit with e value < 1e−5 and percentage of alignment amino acid length > 50% (BLASTP) was retained for functional annotation. GO annotations of IWGSCv1 genes were obtained from Ensembl Plant Biomart (http://plants.ensembl.org/). Significantly overrepresented GO terms were implemented by the software goseq (v1.30.0; http://bioconductor.org/packages/release/bioc/html/goseq.html), and all ASGs or assembled genes were used as background genes for domestication- and polyploidization-responsive ASGs or differentially expressed genes, respectively. Translation prediction of transcripts was realized by the Transcoder program (v5.3.0; https://github.com/TransDecoder/TransDecoder), and supplemented by the search of the Pfam 32.0 database (https://pfam.xfam.org/). Pfam annotations were built and annotated by HMMER from Pfam 32.0 (Johnson et al., 2010). Pfam enrichment analysis of ASGs was carried out with a custom Python script (https://www.python.org/).
Identification of Homeologous Triplets and AS Triplets in Wheat
Wheat homeologous genes were first compared against each other using BLASTN (v2.6.0+; ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.6.0/) with e-value cutoff of 1e−5, and only candidate alignments with a minimum of 200-bp sequence coverage and >90% sequence identity were considered. Next, all the aligned sequences were clustered, and only clusters that had exactly one representative member from each subgenome, and were located on similar position on three chromosomes (triplets), were considered. Then, the AS types of triplet members were analyzed, and if all three homeologous members possessed the same AS type at similar sequence position, the triplet was classified as having the conservative AS type. Otherwise, AS events occurring in all three triplet genes but showing an inconsistent type of splicing event or a similar AS type, but different splicing site locations, were classified as different types of AS triplet. Visualization of homeologous triplets was achieved by using the software circos v0.69 (http://circos.ca/) and R package ggtern (https://cran.r-project.org/web/packages/ggtern/index.html).
RT-PCR, Semi-RT-qPCR, and RT-qPCR Validation of AS Events or Genes
Total RNA treated with DNase I was reverse-transcribed with oligo(dT) primers using Reverse Transcriptase M-MLV (TaKaRa) following the manufacturer’s instructions. RT-PCR was performed in a 10-μL reaction volume and a primer pair for each gene was designed to amplify both splice variants (isoforms 1 and 2) in a single reaction. Then 2% (w/v) agarose gel electrophoresis was used to demonstrate different isoforms. For semi-qRT and PCR, gene-specific primers across junctions were used to amplify WEWv1_A_gene.90708.0 (Q), WEWv1_A_gene.112770.0 (AT4G31580/RSZ22), and WEWv1_A_gene.1336.0 (AT3G53500/RS2Z32). Wheat β-ACTIN gene (WEWv1_A_gene.7189.0 /TraesCS1A02G179000) was chosen as an internal control for normalization. The PCR conditions for the amplification were as follows: 5 min at 94°C, followed by 32 cycles of 20 s at 94°C, 30 s at 58°C, and 20 s at 72°C. RT-qPCR was performed using a CFX96 real-time PCR detection system and SYBR Green Supermix solution (Bio-Rad; http://www.bio-rad.com/). Each sample was quantified at least in triplicate and normalized using β-ACTIN as an internal control. The gene-specific primers used in this research are listed in Supplemental Dataset S9. The visualization of gene structure and RNA-seq reads alignment results was accomplished using the software IGV (v.2.4.10; Robinson et al., 2011).
Accession Numbers
Genes referenced in this article can be found in the Ensembl Plants database (https://plants.ensembl.org/index.html) under the accession numbers given in Supplemental Dataset S10.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Determination of reference genome for different wheat species based on unique read mapping rate.
Supplemental Figure S2. Schematic diagram representing the procedure of AS identification.
Supplemental Figure S3. Structural sketches of different AS events and their corresponding number and proportion in different wheat species.
Supplemental Figure S4. Expression analysis of AS-regulating genes after domestication in diploid and tetraploid wheat.
Supplemental Figure S5. GO enrichment analysis of ASGs responsive to domestication in diploid and tetraploid wheat.
Supplemental Figure S6. Verification of AS differences of splicing regulating genes before and after domestication in diploid and tetraploid wheat.
Supplemental Figure S7. Display and validation of AS changes of the Q gene (WEWv1_A_gene.90708.0) in response to domestication in diploid and tetraploid wheat species.
Supplemental Figure S8. Display and validation of AS changes in response to wheat polyploidization.
Supplemental Figure S9. Expression variation of AS-regulating genes in response to polyploidization in wheat.
Supplemental Figure S10. Comparison of AS events between TAA10 and XX329.
Supplemental Figure S11. Differential expression level and AS patterns of ASGs in wheat caused by the substitution of the D-subgenome in hexaploid wheat.
Supplemental Figure S12. AS conservation and partitioning among homeologs in hexaploid wheat.
Supplemental Figure S13. Ternary plot showing AS partitioning among homeologous genes with same AS type in triplets using WEWv1 and AETv4 as reference genomes.
Supplemental Dataset S1. Overview of RNA-seq data and reads mapping.
Supplemental Dataset S2. Identified AS events from merged RNA-seq mapping results.
Supplemental Dataset S3. AS events in each sample.
Supplemental Dataset S4. PFAM and GO enrichment of ASGs.
Supplemental Dataset S5. Domestication or polyploidization response AS events and genes in wheat.
Supplemental Dataset S6. Differentially expressed genes in this study.
Supplemental Dataset S7. Pfam changes of domestication or polyploidization-responsive ASGs by IR.
Supplemental Dataset S8. Homologous genes with only one copy on each subgenome (triplet cluster) and their AS type in hexaploid wheat.
Supplemental Dataset S9. Primers used in this research.
Supplemental Dataset S10. Accession numbers of major genes mentioned in this article.
Acknowledgments
We thank Bao Liu (Northeast Normal University) for providing the seeds and for information about ETW, TQ18, and XX329; Chaojie Xie (China Agricultural University) for providing the seeds and for information about T. boeoticum, T. monococcum, and T. dicoccoides; and Pengfei Qi (Sichuan Agricultural University) and Wei Chen (Huazhong Agricultural University) for providing the seeds and for information about T. dicoccum.
Footnotes
This work was supported by the Major Program of the National Natural Science Foundation of China (grant nos. 31991210 and 91935304).
Articles can be viewed without a subscription.
References
- Albalat R, Cañestro C(2016) Evolution by gene loss. Nat Rev Genet 17: 379–391 [DOI] [PubMed] [Google Scholar]
- Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak CJ, Stein N, Choulet F, Distelfeld A, et al. (2018) Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361: 661. [DOI] [PubMed] [Google Scholar]
- Avni R, Nave M, Barad O, Baruch K, Twardziok SO, Gundlach H, Hale I, Mascher M, Spannagl M, Wiebe K, et al. (2017) Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357: 93–97 [DOI] [PubMed] [Google Scholar]
- Barta A, Kalyna M, Reddy ASN(2010) Implementing a rational and consistent nomenclature for serine/arginine-rich protein splicing factors (SR proteins) in plants. Plant Cell 22: 2926–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci E, Bitocchi E, Ferrarini A, Benazzo A, Biagetti E, Klie S, Minio A, Rau D, Rodriguez M, Panziera A, et al. (2014) Decreased nucleotide and expression diversity and modified coexpression patterns characterize domestication in the common bean. Plant Cell 26: 1901–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B(2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottley A, Xia GM, Koebner RM(2006) Homoeologous gene silencing in hexaploid wheat. Plant J 47: 897–906 [DOI] [PubMed] [Google Scholar]
- Brooks AN, Yang L, Duff MO, Hansen KD, Park JW, Dudoit S, Brenner SE, Graveley BR(2011) Conservation of an RNA regulatory map between Drosophila and mammals. Genome Res 21: 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL(2009) BLAST+: Architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wu TH, Ng WV, Lin WC(2011) Interrogation of alternative splicing events in duplicated genes during evolution. BMC Genomics 12(Suppl 3): S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ.(2010) Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci 15: 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chusainow J, Ajuh PM, Trinkle-Mulcahy L, Sleeman JE, Ellenberg J, Lamond AI(2005) FRET analyses of the U2AF complex localize the U2AF35/U2AF65 interaction in vivo and reveal a novel self-interaction of U2AF35. RNA 11: 1201–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debernardi JM, Lin H, Chuck G, Faris JD, Dubcovsky J(2017) microRNA172 plays a crucial role in wheat spike morphogenesis and grain threshability. Development 144: 1966–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clerck O, Kao SM, Bogaert KA, Blomme J, Foflonker F, Kwantes M, Vancaester E, Vanderstraeten L, Aydogdu E, Boesger J, et al. (2018) Insights into the evolution of multicellularity from the sea lettuce genome. Curr Biol 28: 2921–2933 e2925 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR(2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, He F, Berkowitz O, Liu J, Cao P, Tang M, Shi H, Wang W, Li Q, Shen Z, et al. (2018) Alternative splicing plays a critical role in maintaining mineral nutrient homeostasis in rice (Oryza sativa). Plant Cell 30: 2267–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, Wong WK, Mockler TC(2010) Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res 20: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovkin M, Reddy AS(1998) The plant U1 small nuclear ribonucleoprotein particle 70K protein interacts with two novel serine/arginine-rich proteins. Plant Cell 10: 1637–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracheva EO, Cordero-Morales JF, González-Carcacía JA, Ingolia NT, Manno C, Aranguren CI, Weissman JS, Julius D(2011) Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature 476: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Zhang P, Ryan PR, Rathjen TM, Yan Z, Delhaize E(2016) Introgression of genes from bread wheat enhances the aluminium tolerance of durum wheat. Theor Appl Genet 129: 729–739 [DOI] [PubMed] [Google Scholar]
- Heiner M, Hui J, Schreiner S, Hung LH, Bindereif A(2010) HnRNP L-mediated regulation of mammalian alternative splicing by interference with splice site recognition. RNA Biol 7: 56–64 [DOI] [PubMed] [Google Scholar]
- Huang J, Gao Y, Jia H, Liu L, Zhang D, Zhang Z(2015) Comparative transcriptomics uncovers alternative splicing changes and signatures of selection from maize improvement. BMC Genomics 16: 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñiguez LP, Hernández G(2017) The evolutionary relationship between alternative splicing and gene duplication. Front Genet 8: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Ma X, Zhao S, Tang Y, Liu F, Gu P, Fu Y, Zhu Z, Cai H, Sun C, et al. (2019a) The APETALA2-like transcription factor SUPERNUMERARY BRACT controls rice seed shattering and seed size. Plant Cell 31: 17–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YF, Chen Q, Wang Y, Guo ZR, Xu BJ, Zhu J, Zhang YZ, Gong X, Luo CH, Wu W, et al. (2019b) Re-acquisition of the brittle rachis trait via a transposon insertion in domestication gene Q during wheat de-domestication. New Phytol 224: 961–973 [DOI] [PubMed] [Google Scholar]
- Jin L, Kryukov K, Clemente JC, Komiyama T, Suzuki Y, Imanishi T, Ikeo K, Gojobori T(2008) The evolutionary relationship between gene duplication and alternative splicing. Gene 427: 19–31 [DOI] [PubMed] [Google Scholar]
- Johnson LS, Eddy SR, Portugaly E(2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11: 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyna M, Lopato S, Voronin V, Barta A(2006) Evolutionary conservation and regulation of particular alternative splicing events in plant SR proteins. Nucleic Acids Res 34: 4395–4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB(2010) Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber ER.(1964) Wheat—reconstitution of tetraploid component (AABB) of hexaploids. Science 143: 253–255 [DOI] [PubMed] [Google Scholar]
- Klepikova AV, Kasianov AS, Gerasimov ES, Logacheva MD, Penin AA(2016) A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J 88: 1058–1070 [DOI] [PubMed] [Google Scholar]
- Kopelman NM, Lancet D, Yanai I(2005) Alternative splicing and gene duplication are inversely correlated evolutionary mechanisms. Nat Genet 37: 588–589 [DOI] [PubMed] [Google Scholar]
- Kriechbaumer V, Wang P, Hawes C, Abell BM(2012) Alternative splicing of the auxin biosynthesis gene YUCCA4 determines its subcellular compartmentation. Plant J 70: 292–302 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R(2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Ouyang S, Egan A, Nobuta K, Haas BJ, Zhu W, Gu X, Silva JC, Meyers BC, Buell CR(2008) Characterization of paralogous protein families in rice. BMC Plant Biol 8: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Li X, Shannon LM, Yeh CT, Wang ML, Bai G, Peng Z, Li J, Trick HN, Clemente TE, et al. (2012) Parallel domestication of the Shattering1 genes in cereals. Nat Genet 44: 720–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling HQ, Ma B, Shi X, Liu H, Dong L, Sun H, Cao Y, Gao Q, Zheng S, Li Y, et al. (2018) Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li Q, Xing Y(2018a) Genes contributing to domestication of rice seed traits and its global expansion. Genes (Basel) 9: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Qin J, Tian X, Xu S, Wang Y, Li H, Wang X, Peng H, Yao Y, Hu Z, et al. (2018b) Global profiling of alternative splicing landscape responsive to drought, heat and their combination in wheat (Triticum aestivum L.). Plant Biotechnol J 16: 714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopato S, Forstner C, Kalyna M, Hilscher J, Langhammer U, Indrapichate K, Lorković ZJ, Barta A(2002) Network of interactions of a novel plant-specific Arg/Ser-rich protein, atRSZ33, with atSC35-like splicing factors. J Biol Chem 277: 39989–39998 [DOI] [PubMed] [Google Scholar]
- Lorkovic ZJ, Lopato S, Pexa M, Lehner R, Barta A(2004) Interactions of Arabidopsis RS domain containing cyclophilins with SR proteins and U1 and U11 small nuclear ribonucleoprotein-specific proteins suggest their involvement in pre-mRNA Splicing. J Biol Chem 279: 33890–33898 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S(2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlung A.(2013) Polyploidy and its effect on evolutionary success: old questions revisited with new tools. Heredity 110: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez Y, Brown JW, Simpson C, Barta A, Kalyna M(2012) Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res 22: 1184–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP.(2007) The evolutionary consequences of polyploidy. Cell 131: 452–462 [DOI] [PubMed] [Google Scholar]
- Park E, Pan Z, Zhang Z, Lin L, Xing Y(2018) The expanding landscape of alternative splicing variation in human populations. Am J Hum Genet 102: 11–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer M, Kugler KG, Sandve SR, Zhan B, Rudi H, Hvidsten TR, Mayer KFX, Olsen OA; International Wheat Genome Sequencing Consortium (2014) Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 345: 1250091. [DOI] [PubMed] [Google Scholar]
- Qin LX, Li Y, Li DD, Xu WL, Zheng Y, Li XB(2014) Arabidopsis drought-induced protein Di19-3 participates in plant response to drought and high salinity stresses. Plant Mol Biol 86: 609–625 [DOI] [PubMed] [Google Scholar]
- Ramírez-González RH, Borrill P, Lang D, Harrington SA, Brinton J, Venturini L, Davey M, Jacobs J, van Ex F, Pasha A, et al. (2018) The transcriptional landscape of polyploid wheat. Science 361: 662. [DOI] [PubMed] [Google Scholar]
- Ranwez V, Serra A, Pot D, Chantret N(2017) Domestication reduces alternative splicing expression variations in sorghum. PLoS One 12: e0183454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AS, Marquez Y, Kalyna M, Barta A(2013) Complexity of the alternative splicing landscape in plants. Plant Cell 25: 3657–3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP(2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux J, Robinson-Rechavi M(2011) Age-dependent gain of alternative splice forms and biased duplication explain the relation between splicing and duplication. Genome Res 21: 357–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma S, Golan G, Guo Z, Ogawa T, Tagiri A, Sugimoto K, Bernhardt N, Brassac J, Mascher M, Hensel G, et al. (2019) Unleashing floret fertility in wheat through the mutation of a homeobox gene. Proc Natl Acad Sci USA 116: 5182–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saminathan T, Nimmakayala P, Manohar S, Malkaram S, Almeida A, Cantrell R, Tomason Y, Abburi L, Rahman MA, Vajja VG, et al. (2015) Differential gene expression and alternative splicing between diploid and tetraploid watermelon. J Exp Bot 66: 1369–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M, Kingsford C(2017) Accurate assembly of transcripts through phase-preserving graph decomposition. Nat Biotechnol 35: 1167–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Zhou Z, Wang Z, Li W, Fang C, Wu M, Ma Y, Liu T, Kong LA, Peng DL, et al. (2014) Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26: 996–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons KJ, Fellers JP, Trick HN, Zhang Z, Tai YS, Gill BS, Faris JD(2006) Molecular characterization of the major wheat domestication gene Q. Genetics 172: 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CCR, Tittes S, Mendieta JP, Collier-Zans E, Rowe HC, Rieseberg LH, Kane NC(2018) Genetics of alternative splicing evolution during sunflower domestication. Proc Natl Acad Sci USA 115: 6768–6773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiger D, Brown JWS(2013) Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25: 3640–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Gu X(2012) Revisit on the evolutionary relationship between alternative splicing and gene duplication. Gene 504: 102–106 [DOI] [PubMed] [Google Scholar]
- Su Z, Wang J, Yu J, Huang X, Gu X(2006) Evolution of alternative splicing after gene duplication. Genome Res 16: 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed NH, Kalyna M, Marquez Y, Barta A, Brown JW(2012) Alternative splicing in plants—coming of age. Trends Plant Sci 17: 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack DC, Pitchers WR, Adams KL(2014) Transcriptome analysis indicates considerable divergence in alternative splicing between duplicated genes in Arabidopsis thaliana. Genetics 198: 1473–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera D, Vogel C, Orozco M, Teichmann SA, de la Cruz X(2007) The (in)dependence of alternative splicing and gene duplication. PLOS Comput Biol 3: e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Mizrachi E, Marchal K(2017) The evolutionary significance of polyploidy. Nat Rev Genet 18: 411–424 [DOI] [PubMed] [Google Scholar]
- Veerabagu M, Kirchler T, Elgass K, Stadelhofer B, Stahl M, Harter K, Mira-Rodado V, Chaban C(2014) The interaction of the Arabidopsis response regulator ARR18 with bZIP63 mediates the regulation of PROLINE DEHYDROGENASE expression. Mol Plant 7: 1560–1577 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB(2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang H, Li Y, Zhang Z, Li L, Liu B(2016) Transcriptome asymmetry in synthetic and natural allotetraploid wheats, revealed by RNA-sequencing. New Phytol 209: 1264–1277 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu J, Huang BO, Xu Y-M, Li J, Huang L-F, Lin J, Zhang J, Min Q-H, Yang W-M, et al. (2015) Mechanism of alternative splicing and its regulation. Biomed Rep 3: 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Guo C, Shan H, Kong H(2012) Divergence of duplicate genes in exon-intron structure. Proc Natl Acad Sci USA 109: 1187–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Zhao L, Zhang H, Yang Z, Wang H, Wen S, Zhang C, Rustgi S, von Wettstein D, Liu B(2014) Evolution of physiological responses to salt stress in hexaploid wheat. Proc Natl Acad Sci USA 111: 11882–11887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Yang H, Yang H(2016) Evolutionary character of alternative splicing in plants. Bioinform Biol Insights 9(Suppl 1): 47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhu B, Qi B, Gou X, Dong Y, Xu C, Zhang B, Huang W, Liu C, Wang X, et al. (2014) Evolution of the BBAA component of bread wheat during its history at the allohexaploid level. Plant Cell 26: 2761–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PG, Huang SZ, Pin AL, Adams KL(2010) Extensive divergence in alternative splicing patterns after gene and genome duplication during the evolutionary history of Arabidopsis. Mol Biol Evol 27: 1686–1697 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Belcram H, Gornicki P, Charles M, Just J, Huneau C, Magdelenat G, Couloux A, Samain S, Gill BS, et al. (2011) Duplication and partitioning in evolution and function of homoeologous Q loci governing domestication characters in polyploid wheat. Proc Natl Acad Sci USA 108: 18737–18742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Moshgabadi N, Adams KL(2011) Extensive changes to alternative splicing patterns following allopolyploidy in natural and resynthesized polyploids. Proc Natl Acad Sci USA 108: 16122–16127 [DOI] [PMC free article] [PubMed] [Google Scholar]