

Abstract
Intermedin (IMD) is a calcitonin/calcitonin-related peptide that elicits cardioprotective effects in a variety of heart diseases, such as cardiac hypertrophy and heart failure. However, the molecular mechanism of IMD remains unclear. The present study investigated the effects of IMD on neonatal rat ventricular myocytes treated with thapsigargin. The results of the present study demonstrated that thapsigargin induced apoptosis in cardiomyocytes in a dose- and time-dependent manner. Thapsigargin induced endoplasmic reticulum stress, as determined by increased expression levels of 78-kDa glucose-regulated protein, C/EBP-homologous protein and caspase-12, which were dose-dependently attenuated by pretreatment with IMD. In addition, IMD treatment counteracted the thapsigargin-induced suppression of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity and protein expression levels, and cytoplasmic Ca2+ overload. IMD treatment also augmented the phosphorylation of phospholamban, which is a crucial regulator of SERCA. Additionally, treatment with the protein kinase A antagonist H-89 inhibited the IMD-mediated cardioprotective effects, including SERCA activity restoration, anti-Ca2+ overload, endoplasmic reticulum stress inhibition and antiapoptosis effects. In conclusion, the results of the present study suggested that IMD may protect cardiomyocytes against thapsigargin-induced endoplasmic reticulum stress and the associated apoptosis at least partly by activating the protein kinase A/SERCA pathway.
Keywords: intermedin, thapsigargin, endoplasmic reticulum stress, sarcoplasmic reticulum Ca2+-ATPase, phospholamban, protein kinase A
Introduction
Intermedin (IMD), also termed adrenomedullin-2, is a member of the calcitonin/calcitonin gene-related peptide family and is widely expressed in the heart, blood vessels, brain, hypothalamus, kidney, lung, spleen, thymus, ovary and adipose tissue (1). The plasma levels of IMD are relatively low, between 100 and 200 pg/ml (2). Following proteolytic cleavage, pre-proIMD generates IMD1-53, IMD1-40 and IMD1-47, the major active fragments that act through the calcitonin-like receptor/receptor activity-modifying protein complexes to induce multiple biological effects, such as increased prolactin release, antidiuretic and natriuretic effects, and reduced food intake (3). Accumulating evidence has indicated extensive functions of IMD in maintaining cardiovascular homeostasis, such as accelerating angiogenesis, anti-apoptotic and fibroblast activity, and increasing cardiac contractility and perfusion (4–6). The plasma levels of IMD and the endogenous IMD in cardiomyocytes have been reported to be significantly increased in response to heart failure and acute cardiac infarction stimuli, and IMD supplementation inhibits cardiomyocyte injury induced by heart failure and cardiac infarction (4,7,8). These results also suggested that IMD may be a potential endogenous protector of the heart. Although these studies have confirmed the cardioprotective effects of IMD, the underlying protective mechanisms are still unclear. Teng et al (3) and Zhang et al (9) have reported that IMD attenuates tunicamycin and dithiothreitol-induced myocardial injury in rats by inhibiting endoplasmic reticulum (ER) stress. Tunicamycin and dithiothreitol induce ER stress via inhibition of protein glycosylation and disulfide bond formation, respectively. Similar to tunicamycin or dithiothreitol, thapsigargin is also an inducer of ER stress. Thapsigargin, originally isolated from the plant Thapsia garganica, is a specific and potent inhibitor for sarco/endoplasmic reticulum calcium ATPase (SERCA) (10). By inhibiting SERCAs, thapsigargin interferes with the ER lumen Ca2+ flux that subsequently leads to ER stress (10,11). SERCA has been reported to exert important effects in the heart, and a number of heart diseases are associated with SERCA dysfunction, such as cardiac hypertrophy and heart failure (12,13). Thapsigargin induces ER stress-related apoptosis by inhibiting SERCA activity to partially simulate the pathophysiological processes of various cardiovascular diseases (14).
The SERCA that is present in all organisms is a 110 kDa transmembrane protein encoded by three homologous genes (SERCA1, SERCA2 and SERCA3), with a dominant expression of SERCA2a in cardiomyocytes (15). SERCA2a is crucial for regulating Ca2+ homeostasis by transporting cytosolic Ca2+ into the sarcoplasmic reticulum (16,17). Phospholamban (PLB) located in the cardiac sarcoplasmic reticulum and, as an endogenous SERCA inhibitor, compromises SERCA affinity for Ca2+ (18). PLB phosphorylation is the primary regulator of SERCA activity in cardiomyocytes (19,20).
To date, there have been no published reports on the effects of IMD on cardiomyocyte injury induced by thapsigargin to the best of our knowledge. Therefore, the preset study focused on the role of IMD in thapsigargin-induced cardiomyocyte apoptosis and examined SERCA activity during thapsigargin treatment to explore whether IMD restored ER stress in cardiomyocytes via a SERCA-dependent mechanism. Protein kinase A (PKA) has also been reported to increase SERCA activity (21); to further explore the underlying mechanisms through which IMD regulates SERCA activity and ER stress, the present study examined the involvement of the PKA pathway in the IMD-mediated protective effects in cardiomyocytes.
Materials and methods
Cell culture and treatment
The experimental protocols were approved by the Ethical Committee of Shanxi Medical University (Taiyuan, China) and complied with internationally accepted principles of laboratory animal care and use (22). Animals were housed at 23°C with 50% humidity, 12-h light/dark cycles, and free access to food and water. Briefly, 16 neonatal SD rats (male and female; age, 1–3 days; Laboratory Animal Center of Shanxi Medical University) were anesthetized by pentobarbital sodium (100 mg/kg) and decapitated for cardiac tissue harvesting. Left ventricular tissues were digested with collagenase II (Sigma-Aldrich; Merck KGaA) in Hank's balanced salt solution (Ca2+- and Mg2+-free) for 1 h at 37°C, as described previously (23). After centrifugation at 500 × g for 10 min at 4°C, the supernatants were discarded, and the cells were resuspended; ~90 min later, non-myocytes were attached to the dishes. The viable non-attached cells were collected and plated on 60-mm culture dishes at a density of 4×106 cells per dish and cultured in DMEM supplemented with 1% penicillin-streptomycin and 20% FBS (all purchased from Sigma-Aldrich; Merck KGaA) supplemented with 0.1 mM bromodeoxyuridine (Sigma-Aldrich; Merck KGaA). Using this strategy, a myocyte population was obtained, and all experiments were performed within 3 days after incubation. To evaluate the effects of IMD on thapsigargin-induced ER stress, cardiomyocytes were divided into 6 groups: i) Control; ii) IMD, cells incubated with 100 nM IMD for 24 h at 37°C (100 nM; Phoenix Pharmaceuticals, Inc.) under normoxic conditions (5% CO2; iii) thapsigargin, cells incubated with 3 µM thapsigargin (Sigma-Aldrich; Merck KGaA), a classic ER stress inducer, for 24 h; iv-vi) thapsigargin +1, 10 and 100 nM IMD, cells incubated with the indicated dose of IMD (thapsigargin was administered 30 min after the addition of IMD). To determine the role of PKA in IMD-mediated cardioprotection, cardiomyocytes were pretreated with 10 µM protein kinase A antagonist H-89 (Sigma-Aldrich; Merck KGaA) for 24 h at 37°C.
Western blot analysis
Cardiomyocytes were lysed in the presence of 10 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich; Merck KGaA), and the protein concentration of the lysates was determined using a BCA Protein Assay kit (Beyotime Institute of Biotechnology). Proteins from each sample (~30 µg) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene fluoride membranes. Following incubation with 5% bovine serum albumin for 2 h at room temperature, the membranes were incubated overnight at 4°C with primary antibodies against 78 kDa glucose-regulated protein (GRP78; 1:200; cat. no. sc-13539), C/EBP-homologous protein (CHOP; 1:200; cat. no. sc-7351), caspase-12 (1:200; cat. no. sc-21747), GAPDH (1:500; cat. no. sc-32233; Santa Cruz Biotechnology, Inc.), SERCA2 (1:500; cat. no. S1439), phosphorylated (p-)PLB (1:200; cat. no. SAB1305590), total (t-)PLB (1:200; cat. no. HPA026900; Sigma-Aldrich; Merck KGaA). Subsequently, the blots were incubated with the appropriate secondary antibodies (cat. no. sc-2004; Santa Cruz Biotechnology, Inc.) for 3 h at 37°C. The membranes were developed with an enhanced chemiluminescence detection system (Amersham; Cytiva). Signal intensities were analyzed using a Gel Imaging System (Bio-Rad Laboratories, Inc.). All assays were performed in triplicate. The relative protein expression levels were determined by normalization to GAPDH.
Measurement of intracellular Ca2+ concentration
Intracellular Ca2+ concentration ([Ca2+]i) was measured using the Ca2+-specific fluorescent probe fluo-3/AM (cat. no. 50013; Biotium, Inc.). Cardiomyocytes were loaded with 5 µM fluo-3/AM for 30 min at 37°C. The mean fluorescence intensity of the cells was monitored using an Olympus FV1000 laser scanning confocal microscope (Olympus Corporation). Fluo-3/AM was excited at 488 nm, and the emission intensity was measured at 528 nm. All experiments were repeated at least four times.
Measurement of SERCA activity
The sarcoplasmic reticulum of cardiomyocytes was obtained using an ER isolation kit (cat. no. ER0100; Sigma-Aldrich; Merck KGaA) according to the manufacturer's instructions. SERCA activity was measured using a Ca2+-ATPase Assay kit (Nanjing Jiancheng Bioengineering Institute) according to the manufacturer's instructions, normalized to protein concentration and expressed as nanomoles of phosphorus ions per milligram of protein per minute (nmol Pi/mg prot/min).
Flow cytometry analysis
Apoptotic cells were detected using an Annexin-V-FITC kit (BD Biosciences) with propidium iodide (PI) staining and flow cytometric analysis according to the manufacturer's instructions. The double-negative (Annexin V-negative/PI-negative) cells were defined as viable, the single-positive populations were considered to be early apoptotic (Annexin V-positive/PI-negative) or necrotic (Annexin V-negative/PI-positive) cells, and double-positive (Annexin V-positive/PI-positive) cells were considered to be in a late stage of apoptosis. Apoptotic cells were analyzed using a FACScan flow cytometer (BD Biosciences) and CellQuest Pro software (version 5.1; BD Biosciences). Apoptosis was calculated as the total of early and late apoptotic cells.
Statistical analysis
Data are expressed as the mean ± standard deviation. Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, Inc.). All data on dose response and time series were analyzed with a one-way ANOVA followed by a Tukey's post hoc test. Additional data were analyzed with student's t-test for two group comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
Thapsigargin induces cardiomyocyte apoptosis in a dose- and time-dependent manner
Neonatal rat cardiomyocytes were incubated with 1.5, 3 or 6 µM thapsigargin for 24 h to determine the working concentration of thapsigargin. Cell viability was determined using an MTT assay. As demonstrated in Fig. 1A, treatment with increasing concentrations of thapsigargin led to a dose-dependent loss of cardiomyocyte viability compared with the control group. Following exposure to 3 µM thapsigargin, 69.33±4.63% of the cardiomyocytes were viable relative to the control group (P<0.01). However, with 6 µM thapsigargin, the viability of cells was significantly reduced to 44.52±6.88% (Fig. 1A). Apoptosis was analyzed by flow cytometry using the Annexin V/PI double staining assay. The percentages of apoptotic cells were 9.8±7.55, 26.47.0±10.31 and 50.87±13.51% in the 1.5, 3 and 6 µM thapsigargin groups, respectively (Fig. 1B and C).
Figure 1.

Viability and apoptosis of cells treated with TG. Cultured neonatal rat cardiomyocytes were left untreated or treated with 1.5, 3 or 6 µM TG for 24 h. Following this, (A) cell viability was determined using an MTT assay, (B) apoptosis was determined using flow cytometry and (C) the rates of apoptotic cells are presented in bar graphs. Cultured neonatal rat cardiomyocytes were left untreated or treated with 3 µM TG for 12, 24 or 48 h. Following this, (D) cell viability was determined using an MTT assay. (E) Apoptosis was assessed using flow cytometry analysis and (F) the rates of apoptotic cells are presented in bar graphs. In the flow cytometry plots, the horizontal axis represents the Annexin V intensity, and the vertical axis represents PI staining. Lower left quadrant, living cells; upper left quadrant, necrotic cells; right quadrants, apoptotic cells. All data on dose response and time series were analyzed with a one-way ANOVA followed by a Tukey's test. Data are presented as the mean ± SD. n=3. *P<0.05 and **P<0.01. TG, thapsigargin; PI, propidium iodide.
In order to further study the effects of thapsigargin, cardiomyocytes were treated with 3 µM thapsigargin for 12, 24 or 48 h. After exposure to 3 µM thapsigargin for the indicated times, cell viability was significantly decreased in the 24 and 48 h groups compared with that of the control group (Fig. 1D). The apoptotic rates in the 24 and 48 h groups, but not the 12 h group, were significantly higher compared with those in the control group (Fig. 1E and F). Thapsigargin induced apoptosis in cardiomyocytes in a dose- and time-dependent manner; thus, 3 µM thapsigargin treatment for 24 h was used in subsequent experiments.
IMD inhibits thapsigargin-induced ER stress in cardiomyocytes
Cardiomyocytes were incubated with 3 µM thapsigargin in the presence or absence of IMD for 24 h. CHOP, GRP78 and caspase-12 are molecular markers specific for ER stress (24). Compared with the control group, the thapsigargin group exhibited significantly upregulated protein expression levels of GRP78, CHOP and caspase-12, indicating that thapsigargin treatment induced ER stress. IMD (10 and 100 nM) reduced the thapsigargin-mediated upregulation of ER stress in a dose-dependent manner. Pretreatment with 100 nM IMD resulted in a strong protective effect against thapsigargin-induced injury (Fig. 2A-D). Collectively, these results demonstrated that IMD pretreatment inhibited the thapsigargin-induced ER stress in a dose-dependent manner.
Figure 2.

IMD suppresses TG-induced ER stress in cardiomyocytes. Neonatal rat cardiomyocytes were treated with 3 µM TG and the indicated dose of IMD for 24 h. The expression levels of ER stress markers (GRP78, CHOP and caspase-12) were detected by western blotting. (A) Representative western blots for GRP78, CHOP and caspase-12. Semi-quantification of the protein levels of (B) GRP78, (C) CHOP and (D) caspase-12. Data on dose response were analyzed with a one-way ANOVA followed by the Tukey test. Data are presented as the mean ± SD. n=3. **P<0.01; #P<0.05, ##P<0.01. IMD, intermedin; ER, endoplasmic reticulum; TG, thapsigargin; GRP78, 78 kDa glucose-regulated protein; CHOP, C/EBP-homologous protein.
IMD attenuates SERCA suppression and [Ca2+]i overload induced by thapsigargin in cardiomyocytes
Thapsigargin is a highly selective inhibitor of SERCA. Therefore, the present study examined SERCA activity in cardiomyocytes treated with thapsigargin in the presence or absence of IMD (100 nM) to further investigate the possible relationship between IMD and SERCA. As demonstrated in Fig. 3A and B, thapsigargin treatment significantly reduced the activity and protein expression levels of SERCA2, which were reversed by IMD treatment.
Figure 3.

IMD inhibits TG-induced endoplasmic reticulum stress by regulating SERCA activity and [Ca2+]i homeostasis. Cardiomyocytes were treated with 3 µM TG in the presence or absence of 100 nM IMD for 24 h. (A) SERCA activity. (B) Representative western blots and semi-quantification of SERCA2 protein levels. (C and D) [Ca2+]i in cardiomyocytes. The intensity of [Ca2+]i fluorescence was measured using fluo-3AM with a laser scanning confocal microscope. Data were analyzed with student's t-test for two group comparison. Data are presented as the mean ± SD. n=3. *P<0.05 and **P<0.01. IMD, intermedin; SERCA, sarco/endoplasmic reticulum calcium ATPase; [Ca2+]i, intracellular Ca2+ concentration; TG, thapsigargin; nmol Pi/mg prot/min, nanomoles of phosphorus ions per milligram of protein per minute.
Since SERCA serves a crucial role in Ca2+ homeostasis, subsequent experiments were performed to determine whether IMD decreased cytosolic Ca2+ overload in cardiomyocytes using the fluorescent indicator fluo-3AM. As presented in Fig. 3C and D, the intensity of [Ca2+]i fluorescence was significantly elevated in the thapsigargin group compared with that in the vehicle group. This increase in [Ca2+]i fluorescence was significantly reduced by IMD pretreatment (Fig. 3C and D).
IMD improves SERCA function by regulating PLB phosphorylation
PLB is a key regulatory protein of SERCA (25). Unphosphorylated PLB binds SERCA and inhibits its activity, which is abolished upon PLB phosphorylation (25). Therefore, elucidating the changes in the SERCA regulatory protein PLB may help understand the protective mechanisms of IMD. Thus, t-PLB and p-PLB protein levels were analyzed in the present study by western blotting. As demonstrated in Fig. 4, the levels of p-PLB, which is the active form of the SERCA regulatory protein, were reduced in the thapsigargin group compared with those in the vehicle group. This reduction was reversed by IMD pretreatment (Fig. 4).
Figure 4.
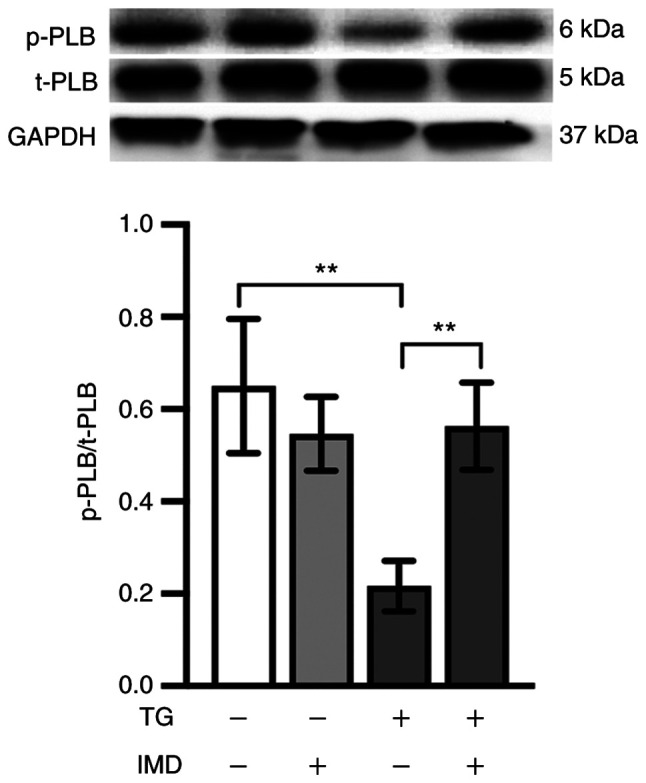
IMD improves SERCA activity by regulating PLB phosphorylation. Cardiomyocytes were treated with 3 µM TG in the presence or absence of 100 nM IMD for 24 h. p-PLB and t-PLB protein expression levels were assayed by western blot analysis. Representative western blots and semi-quantification of p-PLB and t-PLB. Data were analyzed with a student's t-test for two group comparison. Data are presented as the mean ± SD. n=3. **P<0.01. IMD, intermedin; SERCA, sarco/endoplasmic reticulum calcium ATPase; PLB, phospholamban; p-, phosphorylated; t-, total; TG, thapsigargin.
PKA contributes to SERCA function and ER stress regulated by IMD
As PKA regulates PLB phosphorylation and SERCA activity (21,26), the present study further examined whether the PKA signaling pathway was involved in IMD-mediated cardioprotection, PLB phosphorylation and SERCA function. As presented in Fig. 5A-D, co-treatment with the PKA inhibitor H-89 (10 µM) inhibited the effects of IMD on PLB phosphorylation and SERCA activity in cardiomyocytes treated with thapsigargin. These results suggested that IMD-induced PLB phosphorylation and restoration of SERCA activity were mediated in part by the PKA pathway.
Figure 5.

PKA signaling pathway is involved in the effects of IMD on SERCA activity and endoplasmic reticulum stress. Cardiomyocytes were treated with TG in the presence or absence of 100 nM IMD or 10 µM PKA inhibitor H-89 for 24 h. (A) SERCA activity. (B) Representative western blot images of p-PLB, t-PLB, SERCA2, GRP78, CHOP and caspase-12. Semi-quantification of (C) p-PLB/t-PLB, (D) SERCA2, (E) GRP78, (F) CHOP and (G) caspase-12 protein levels. Data were analyzed with a student's t-test for two group comparison. Data are presented as the mean ± SD. n=3. *P<0.05 and **P<0.01. IMD, intermedin; SERCA, sarco/endoplasmic reticulum calcium ATPase; PLB, phospholamban; p-, phosphorylated; t-, total; GRP78, 78 kDa glucose-regulated protein; CHOP, C/EBP-homologous protein; PKA, protein kinase A; TG, thapsigargin; nmol Pi/mg prot/min, nanomoles of phosphorus ions per milligram of protein per minute.
PKA inhibitor H-89 treatment also abolished the protective effects of IMD on ER stress and ER stress-related cardiomyocyte apoptosis induced by thapsigargin, as demonstrated by western blotting and Annexin V/PI double staining assay (Figs. 5B, 5E-G, 6A and 6B). These results suggested that IMD may ameliorate ER stress and ER stress-related cardiomyocyte apoptosis induced by thapsigargin at least in part via the PKA/SERCA pathway.
Figure 6.

IMD reduces endoplasmic reticulum stress-mediated apoptosis in cardiomyocytes. Neonatal rat cardiomyocytes were treated with TG in the presence or absence of IMD or H-89 for 24 h. Apoptotic rates were assessed using flow cytometry analysis. (A) Representative flow cytometric dot plots for each treatment group. Horizontal and vertical axis represent Annexin V and PI staining. Lower left quadrant, living cells; upper left quadrant, necrotic cells; and right quadrants, apoptotic cells. (B) Quantitative analysis of apoptotic cells. Data were analyzed with a student's t-test for two group comparison. Data are presented as the mean ± SD. n=3. *P<0.05 and **P<0.01. IMD, intermedin; PI, propidium iodide; TG, thapsigargin.
Discussion
The results of the present study demonstrated that IMD decreased the thapsigargin-induced upregulation of GRP78, CHOP and caspase-12, the specific markers of ER stress. IMD alleviated thapsigargin-induced ER stress by restoring SERCA activity and Ca2+ overload in cultured cardiomyocytes. PLB phosphorylation may also contribute to IMD-enhanced SERCA activity. Co-treatment with the PAK inhibitor H89 appeared to counteract the protective effects of IMD on PLB phosphorylation and restoration of SERCA activity, suggesting the involvement of the PKA signaling pathway in the effects of IMD.
IMD belongs to a multifunctional calcitonin/calcitonin gene-related peptide superfamily and shares common receptors with calcitonin gene-related peptide, adrenomedullin and amylin, with unique and important cardioprotective functions, including improving cardiac function, pro-angiogenesis, anti-oxidation and anti-ER stress (5). ER stress is increasingly recognized as an important contributor to myocardial injury (27). ER, as the primary site of proper folding and sorting of proteins, is vulnerable to ischemia, hypoxia and oxidative stress. Various pathophysiological conditions disturb the ER function and initiate the unfolded protein response to promote cell survival (24). However, persistent and excessive ER stress triggers apoptosis and aggravates cardiovascular diseases, including heart failure, cardiac hypertrophy and ischemic heart disease. ER stress and its induced apoptosis have been highlighted as important mechanisms underlying myocardial injury (28,29). Alleviating ER stress is accepted as a promising therapeutic approach for the treatment of cardiovascular diseases (30).
In previous studies, IMD has been demonstrated to exert a cardioprotective effect against ER stress induced by tunicamycin and dithiothreitol (6,7). The pathways by which IMD inhibits ER stress and protects myocardial injury are not well understood. Thapsigargin is also a classic ER stress inducer. Tunicamycin and dithiothreitol induce ER stress via inhibition of protein glycosylation or disulfide bond formation, respectively (31), whereas thapsigargin possesses a unique mechanism to induce ER stress. To date, there have been no published reports on the effects of IMD on thapsigargin-induced cardiomyocyte injury. The present study assessed the protein expression levels of ER stress-related markers GRP78, CHOP and caspase-12 in cardiomyocytes by western blotting; thapsigargin treatment significantly upregulated the expression levels of GRP78, CHOP and caspase-12, whereas IMD pretreatment significantly ameliorated these changes in a dose-dependent manner, suggesting that IMD inhibited thapsigargin-induced ER stress. To the best of our knowledge, the present study demonstrated for the first time that IMD may rescue cardiomyocytes from thapsigargin-triggered ER stress.
Experimental and clinical studies have indicated that IMD suppresses cardiac hypertrophy and heart failure, a major health issue that is a leading cause of death worldwide (32–34). During this process, cytosolic Ca2+ overload serves a crucial role in the development of pathological cardiac hypertrophy and heart failure. Cytosolic Ca2+ homeostasis is tightly controlled by Ca2+-handling enzymes, proteins, channels and transporters in the plasma membrane and Ca2+ storage organelles (35). The ER is the major Ca2+ storage organelle that releases Ca2+ predominately via the Inositol 1,4,5-thriphosphate and the ryanodine receptor, and uptakes Ca2+ via SERCA, which is the only active Ca2+ transporter from the cytosol to the ER in the heart (36). Suppression of SERCA activity and subsequent alteration of cytosolic Ca2+ signaling can severely impair the systolic and diastolic function of the heart, which are major etiologies of cardiac hypertrophy and heart failure (37–39). As the primary function of SERCA is to replenish the sarcoplasmic reticulum Ca2+ load during the contraction-relaxation cycle of the heart, resulting in cytosolic Ca2+ overload, which is associated with heart failure and cardiac hypertrophy, it was hypothesized in the present study that restoration of SERCA activity may mediate the cardioprotective effects of IMD on ER stress and ER stress-related apoptosis. Thapsigargin is a highly selective inhibitor of SERCAs (40). By inhibiting SERCAs, thapsigargin disrupts Ca2+ transport into the ER lumen, leading to an increase in cytoplasmic Ca2+ concentration, and subsequently activates ER stress (41). To further investigate this signaling pathway, thapsigargin was used in the present study, as it is a well-established model to study ER stress and SERCA activity (42). The present results demonstrated that thapsigargin attenuated the protein expression and activity of SERCA2a in cardiomyocytes, and IMD pretreatment reversed this change. Furthermore, [Ca2+]i in thapsigargin-treated cardiomyocytes was significantly increased, whereas IMD pretreatment inhibited this increase, suggesting that modulating SERCA function and Ca2+ homeostasis may contribute to IMD-mediated cardioprotection. Thus, the results of the present study indicated that the regulation of SERCA activity may serve an important role in the IMD-mediated protection of cardiomyocytes against ER stress, which in part explains the underlying mechanism of IMD improving cardiac hypertrophy and heart failure.
PLB inhibits SERCA activity by reducing its affinity for Ca2+ (42). Unphosphorylated PLB binds SERCA to inhibit SERCA activity; this inhibition is abolished upon PLB phosphorylation (25). PLB is a key regulator of SERCA (42). Considering the relationship between PLB and SERCA, the present study assessed the protein levels of p-PLB and t-PLB in cardiomyocytes treated with thapsigargin in the presence or absence of IMD to further investigate the underlying mechanisms of IMD. The results demonstrated that the expression of p-PLB was decreased in the thapsigargin group compared with those in the vehicle group, whereas IMD reversed this change, suggesting that PLB phosphorylation may be associated with the protective effects of IMD on SERCA activity and cytosolic Ca2+ influx.
IMD stimulates cardiomyocyte PKA activity (43), and SERCA activity can be increased by PKA (20), which provides a potential mechanism by which IMD stimulates SERCA function and subsequently attenuates ER stress-related apoptosis in thapsigargin-treated cardiomyocytes. To investigate this in the present study, cardiomyocytes were co-pretreated with the PKA inhibitor H-89 and/or IMD, and incubated with thapsigargin. The results demonstrated that the PKA inhibitor H-89 inhibited the protective effect of IMD on ER stress and SERCA function, as indicated by the changes in the expression levels of GRP78, CHOP and caspase-12, and SERCA activity. H-89 pretreatment also abrogated the antiapoptotic effects of IMD. These results suggested that IMD may exert antiapoptotic effects at least partly via the regulation of the PKA signaling pathway in cardiomyocytes treated with thapsigargin.
H-89 is a highly selective, but not exclusive inhibitor of PKA, and reportedly also binds to other protein kinases, including ribosomal protein S6 kinase β1, ribosomal protein S6 kinase α5, rho-associated protein kinase 2, RAC-α serine/threonine-protein kinase and ribosomal protein S6 kinase alpha-1, with a low binding affinity (44,45). Thus, the possibility that these protein kinases may in part contribute to the thapsigargin-induced cardiomyocyte apoptosis cannot be eliminated. In addition, the present study did not investigate whether other Ca2+-handling enzymes, proteins, channels and transporters, such as inositol 1,4,5-trisphosphate receptor type, ryanodine receptor 1 and stromal interaction molecule 1, are involved in the protection of IMD on thapsigargin-induced cardiomyocyte apoptosis. Accumulating evidence has suggested that thapsigargin-induced apoptosis is mediated by autophagy in cardiomyocytes (46,47). The present results indicated that IMD attenuated thapsigargin-induced cardiomyocyte apoptosis by inhibiting ER stress with the possible involvement of the PKA/SERCA signaling pathway. The present study focused on the mechanisms upstream of ER stress, but did not explore the involvement of autophagy, which is downstream of ER stress. This is a limitation of this study, and future studies should evaluate whether autophagy contributes to the effects of IMD on ER stress-related apoptosis. In addition, PLB has been demonstrated to be a major regulator of SERCA activity. It is the only SERCA-associated protein directly involved in the development of cardiac disease, including heart failure (18). Thus, the present study focused on the PLB/SERCA signaling pathway. Accumulating evidence has indicated that sarcolipin also inhibits the affinity of SERCA for Ca2+ (48,49). In future studies, we will explore whether the effect of IMD on cardiomyocyte apoptosis is associated with sarcolipin and other SERCA regulators.
In conclusion, the results of the present study identified an additional signaling pathway through which IMD responds to ER stress. The protective role of IMD in attenuating thapsigargin-induced cardiomyocyte apoptosis may be mediated by inhibiting ER stress with the possible involvement of the PKA/SERCA signaling pathway. These findings provided a novel insight into the mechanisms underlying the cardioprotective effects of IMD.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- IMD
intermedin
- ER
endoplasmic reticulum
- GRP78
glucose-regulated protein 78
- CHOP
C/EBP-homologous protein
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- SERCA
sarco/endoplasmic reticulum calcium ATPase
- PKA
cAMP-dependent protein kinase A
- PLB
phospholamban
Funding
The present work was supported by the National Natural Science Foundation of China (grant no. 81600256), the Natural Science Foundation of Shanxi Province (grant no. 2014021038-2) and the Postdoctoral Research Startup Fund of the First Hospital of Shanxi Medical University.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
MZ and ZL designed the experiments. ZL and JG performed the experiments. YB and MZ analyzed the data. ZL and JG wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The experimental protocols were approved by the Ethical Committee of Shanxi Medical University (approval no. SYXk:2015-0507) and complied with the internationally accepted principles of laboratory animal care and use.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Takei Y, Inoue K, Ogoshi M, Kawahara T, Bannai H, Miyano S. Identification of novel adrenomedullin in mammals: A potent cardiovascular and renal regulator. FEBS Lett. 2004;556:53–58. doi: 10.1016/S0014-5793(03)01368-1. [DOI] [PubMed] [Google Scholar]
- 2.Taylor MM, Samson WK. Stress hormone secretion is altered by central administration of intermedin/adrenomedullin-2. Brain Res. 2005;1045:199–205. doi: 10.1016/j.brainres.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 3.Teng X, Song J, Zhang G, Cai Y, Yuan F, Du J, Tang C, Qi YF. Inhibition of endoplasmic reticulum stress by intermedin(1–53) protects against myocardial injury through a PI3 kinase-Akt signaling pathway. J Mol Med (Berl) 2011;89:1195–1205. doi: 10.1007/s00109-011-0808-5. [DOI] [PubMed] [Google Scholar]
- 4.Tang B, Zhong Z, Shen HW, Wu HP, Xiang P, Hu B. Intermedin as a prognostic factor for major adverse cardiovascular events in patients with ST-segment elevation acute myocardial infarction. Peptides. 2014;58:98–102. doi: 10.1016/j.peptides.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 5.Yang SM, Liu J, Li CX. Intermedin protects against myocardial ischemia-reperfusion injury in hyperlipidemia rats. Genet Mol Res. 2014;13:8309–8319. doi: 10.4238/2014.October.20.7. [DOI] [PubMed] [Google Scholar]
- 6.Ni X, Zhang J, Tang CX, Qi YF. Intermedin/adrenomedullin2: An autocrine/paracrine factor in vascular homeostasis and disease. Sci China Life Sci. 2014;57:781–789. doi: 10.1007/s11427-014-4701-7. [DOI] [PubMed] [Google Scholar]
- 7.Bell D, Gordon BJ, Lavery A, Megaw K, Kinney MO, Harbinson MT. Plasma levels of intermedin (adrenomedullin-2) in healthy human volunteers and patients with heart failure. Peptides. 2016;76:19–29. doi: 10.1016/j.peptides.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Lv Z, Wu K, Chen X, Zhang X, Hong B. Plasma intermedin levels in patients with acute myocardial infarction. Peptides. 2013;43:121–125. doi: 10.1016/j.peptides.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Zhang JS, Hou YL, Lu WW, Ni XQ, Lin F, Yu YR, Tang CS, Qi YF. Intermedin1-53 protects against myocardial fibrosis by inhibiting endoplasmic reticulum stress and inflammation induced by homocysteine in apolipoprotein E-deficient mice. J Atheroscler Thromb. 2016;23:1294–1306. doi: 10.5551/jat.34082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canova NK, Kmonickova E, Martinek J, Zidek Z, Farghali H. Thapsigargin, a selective inhibitor of sarco-endoplasmic reticulum Ca2+ -ATPases, modulates nitric oxide production and cell death of primary rat hepatocytes in culture. Cell Biol Toxicol. 2007;23:337–354. doi: 10.1007/s10565-007-0185-6. [DOI] [PubMed] [Google Scholar]
- 11.Chen G, Shen Y, Li X, Jiang Q, Cheng S, Gu Y, Liu L, Cao Y. The endoplasmic reticulum stress inducer thapsigargin enhances the toxicity of ZnO nanoparticles to macrophages and macrophage-endothelial co-culture. Environ Toxicol Pharmacol. 2017;50:103–110. doi: 10.1016/j.etap.2017.01.020. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Zhang X, Gross S, Houser SR, Soboloff J. Acetylation of SERCA2a, another target for heart failure treatment? Circ Res. 2019;124:1285–1287. doi: 10.1161/CIRCRESAHA.119.315017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad AM, Ma H, Sumbilla C, Lee DI, Klein MG, Inesi G. Phenylephrine hypertrophy, Ca2+-ATPase (SERCA2), and Ca2+ signaling in neonatal rat cardiac myocytes. Am J Physiol Cell Physiol. 2007;292:C2269–C2275. doi: 10.1152/ajpcell.00441.2006. [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Xue M, Wang XR, Tao TQ, Xu FF, Liu XH, Shi DZ. Panax quinquefolium saponin attenuates cardiomyocyte apoptosis induced by thapsigargin through inhibition of endoplasmic reticulum stress. J Geriatr Cardiol. 2015;12:540–546. doi: 10.11909/j.issn.1671-5411.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adachi T. Modulation of vascular sarco/endoplasmic reticulum calcium ATPase in cardiovascular pathophysiology. Adv Pharmacol. 2010;59:165–195. doi: 10.1016/S1054-3589(10)59006-9. [DOI] [PubMed] [Google Scholar]
- 16.Cook NL, Viola HM, Sharov VS, Hool LC, Schoneich C, Davies MJ. Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity and perturb Ca2+ homeostasis in human coronary artery endothelial cells. Free Radic Biol Med. 2012;52:951–961. doi: 10.1016/j.freeradbiomed.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, Bose DD, Thomas DW. Paradoxical effects of sarco/endoplasmic reticulum Ca(2+)-ATPase (SERCA) activator gingerol on NG115-401L neuronal cells: Failure to augment ER Ca(2+) uptake and protect against ER stress-induced cell death. Eur J Pharmacol. 2015;762:165–173. doi: 10.1016/j.ejphar.2015.05.055. [DOI] [PubMed] [Google Scholar]
- 18.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorski PA, Ceholski DK, Young HS. Structure-function relationship of the SERCA pump and its regulation by phospholamban and sarcolipin. Adv Exp Med Biol. 2017;981:77–119. doi: 10.1007/978-3-319-55858-5_5. [DOI] [PubMed] [Google Scholar]
- 20.Cerra MC, Imbrogno S. Phospholamban and cardiac function: A comparative perspective in vertebrates. Acta Physiol (Oxf) 2012;205:9–25. doi: 10.1111/j.1748-1716.2011.02389.x. [DOI] [PubMed] [Google Scholar]
- 21.Xu J, Han Q, Shi H, Liu W, Chu T, Li H. Role of PKA in the process of neonatal cardiomyocyte hypertrophy induced by urotensin II. Int J Mol Med. 2017;40:499–504. doi: 10.3892/ijmm.2017.3038. [DOI] [PubMed] [Google Scholar]
- 22.Ogden BE, Pang William W, Agui T, Lee BH. Laboratory Animal Laws, Regulations, Guidelines and Standards in China Mainland, Japan, and Korea. ILAR J. 2016;57:301–311. doi: 10.1093/ilar/ilw018. [DOI] [PubMed] [Google Scholar]
- 23.Bian YF, Hao XY, Gao F, Yang HY, Zhang N, Xiao CS. Adiponectin attenuates hypoxia/reoxygenation-induced cardiomyocyte injury through inhibition of endoplasmic reticulum stress. J Investiq Med. 2011;59:921–925. doi: 10.2310/JIM.0b013e318216ad04. [DOI] [PubMed] [Google Scholar]
- 24.Sozen E, Karademir B, Ozer NK. Basic mechanisms in endoplasmic reticulum stress andrelation to cardiovascular diseases. Free Radic Biol Med. 2015;78:30–41. doi: 10.1016/j.freeradbiomed.2014.09.031. [DOI] [PubMed] [Google Scholar]
- 25.Gustavsson M, Traaseth NJ, Karim CB, Lockamy EL, Thomas DD, Veglia G. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum Ca2+-ATPase. J Mol Biol. 2011;408:755–765. doi: 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Periasamy M, Bhupathy P, Babu GJ. Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res. 2008;77:265–273. doi: 10.1093/cvr/cvm056. [DOI] [PubMed] [Google Scholar]
- 27.Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: Endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Hu X, Jiang H. ER stress-induced apoptosis: A novel therapeutic target in myocardial ischemia and reperfusion injury. Int J Cardiol. 2016;214:233–234. doi: 10.1016/j.ijcard.2016.03.176. [DOI] [PubMed] [Google Scholar]
- 29.Wang M, Meng XB, Yu YL, Sun GB, Xu XD, Zhang XP, Dong X, Ye JX, Xu HB, Sun YF, Sun XB. Elatoside C protects against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes through the reduction of endoplasmic reticulum stress partially depending on STAT3 activation. Apoptosis. 2014;19:1727–1735. doi: 10.1007/s10495-014-1039-3. [DOI] [PubMed] [Google Scholar]
- 30.Hong J, Kim K, Kim JH, Park Y. The role of endoplasmic reticulum stress in cardiovascular disease and exercise. Int J Vasc Med. 2017;2017:2049217. doi: 10.1155/2017/2049217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Yi P, Zhang B, Xu C, Liu Q, Pi Z, Xu X, Chevet E, Liu J. Differences in endoplasmic reticulum stress signalling kinetics determine cell survival outcome through activation of MKP-1. Cell Signal. 2011;23:35–45. doi: 10.1016/j.cellsig.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 32.Chen H, Wang X, Tong M, Wu D, Wu S, Chen J, Wang X, Wang X, Kang Y, Tang H, Tang C, Jiang W. Intermedin suppresses pressure overload cardiac hypertrophy through activation of autophagy. PLoS One. 2013;8:e64757. doi: 10.1371/journal.pone.0064757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu K, Deng X, Gong L, Chen X, Wang S, Chen H, Chen X, Amrit B, He S. The effect of intermedin on angiotensin II and endothelin-1 induced ventricular myocyte hypertrophy in neonatal rat. Clin Lab. 2013;59:589–596. doi: 10.7754/Clin.Lab.2012.120708. [DOI] [PubMed] [Google Scholar]
- 34.Hirose T, Totsune K, Mori N, Morimoto R, Hashimoto M, Nakashige Y, Metoki H, Asayama K, Kikuya M, Ohkubo T, et al. Increased expression of adrenomedullin 2/intermedin in rat hearts with congestive heart failure. Eur J Heart Fail. 2008;10:840–849. doi: 10.1016/j.ejheart.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 35.Reddish FN, Miller CL, Gorkhali R, Yang JJ. Calcium dynamics mediated by the endoplasmic/sarcoplasmic reticulum and related diseases. Int J Mol Sci. 2017;18:1024. doi: 10.3390/ijms18051024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chemaly ER, Troncone L, Lebeche D. SERCA control of cell death and survival. Cell Calcium. 2018;69:46–61. doi: 10.1016/j.ceca.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Louch WE, Niederer SA, Aronsen JM, Christensen G, Sejersted OM, Smith NP. Sodium accumulation in SERCA knockout-induced heart failure. Biophys J. 2012;102:2039–2048. doi: 10.1016/j.bpj.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roe AT, Ruud M, Espe EK, Manfra O, Longobardi S, Aronsen JM, Norden ES, Husebye T, Kolstad TRS, Cataliotti A, et al. Regional diastolic dysfunction in post-infarction heart failure: Role of local mechanical load and SERCA expression. Cardiovasc Res. 2019;115:752–764. doi: 10.1093/cvr/cvy257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi H, Han Q, Xu J, Liu W, Chu T, Zhao L. Urotensin II induction of neonatal cardiomyocyte hypertrophy involves the CaMKII/PLN/SERCA 2a signaling pathway. Gene. 2016;583:8–14. doi: 10.1016/j.gene.2016.02.039. [DOI] [PubMed] [Google Scholar]
- 40.Kamiya T, Hara H, Adachi T. Effect of endoplasmic reticulum (ER) stress inducer thapsigargin on the expression of extracellular-superoxide dismutase in mouse 3T3-L1 adipocytes. J Clin Biochem Nutr. 2013;52:101–105. doi: 10.3164/jcbn.12-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- 42.Schmitt JP, Ahmad F, Lorenz K, Hein L, Schulz S, Asahi M, Maclennan DH, Seidman CE, Seidman JG, Lohse MJ. Alterations of phospholamban function can exhibit cardiotoxic effects independent of excessive sarcoplasmic reticulum Ca2+-ATPase inhibition. Circulation. 2009;119:436–444. doi: 10.1161/CIRCULATIONAHA.108.783506. [DOI] [PubMed] [Google Scholar]
- 43.Bell D, McDermott BJ. Intermedin (adrenomedullin-2): A novel counter-regulatory peptide in the cardiovascular and renal systems. Br J Pharmacol. 2008;153(Suppl 1):S247–S262. doi: 10.1038/sj.bjp.0707494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lochner A, Moolman JA. The many faces of H89: A review. Cardiovasc Drug Rev. 2006;24:261–274. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 45.Saad NS, Elnakish MT, Ahmed AAE, Janssen PML. Protein kinase a as a promising target for heart failure drug development. Arch Med Res. 2018;49:530–537. doi: 10.1016/j.arcmed.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Yuan Y, Jiang L, Zhang J, Gao J, Shen Z, Zheng Y, Deng T, Yan H, Li W, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy. 2014;10:1801–1013. doi: 10.4161/auto.32136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindner P, Christensen B, Nissen P, Møller JV, Engedal N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun Signal. 2020;18:12. doi: 10.1186/s12964-019-0499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhupathy P, Babu GJ, Periasamy M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol. 2007;42:903–911. doi: 10.1016/j.yjmcc.2007.03.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asahi M, Nakayama H, Tada M, Otsu K. Regulation of sarco(endo)plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: Implication for cardiac hypertrophy and failure. Trends Cardiovasc Med. 2003;13:152–157. doi: 10.1016/S1050-1738(03)00037-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.