

Abstract
Metastasis is the cause of 90% of mortality in cancer patients. For metastatic colorectal cancer (mCRC), the standard of care drug therapies only palliate the symptoms but are ineffective, evidenced by a low survival rate of ~11%. T-cell factor (TCF) transcription is a major driving force in CRC and we have characterized it to be a master regulator of epithelial-mesenchymal transition (EMT). EMT transforms relatively benign epithelial tumor cells into quasi-mesenchymal or mesenchymal cells that possess cancer stem cell properties, promoting multidrug resistance and metastasis. We have identified topoisomerase IIα (TOP2A) as a DNA-binding factor required for TCF-transcription. Herein, we describe the design, synthesis, biological evaluation, and in vitro and in vivo pharmacokinetic analysis of TOP2A ATP-competitive inhibitors that prevent TCF-transcription and modulate or reverse EMT in mCRC. Unlike TOP2A poisons, ATP-competitive inhibitors do not damage DNA, potentially limiting adverse effects. This work demonstrates a new therapeutic strategy targeting TOP2A for the treatment of mCRC and potentially other types of cancer.
Graphical Abstract
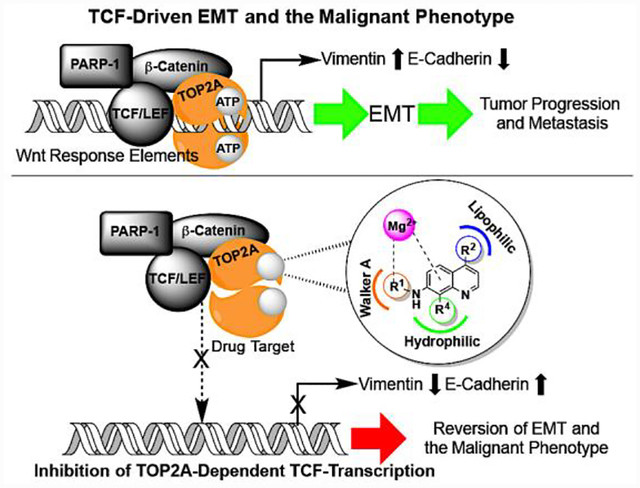
Introduction
Colorectal cancer (CRC) is the third most common cancer among men and second among women, and is the second leading cause of cancer deaths in the United States.1 The majority of CRC initiates through mutations in the Wnt signaling pathway, leading to constitutive activation of T-Cell Factor/Lymphoid Enhancer Factor transcription, denoted as TCF-transcription or TCF-complex.2, 3 Aberrant TCF-transcription is a driving force promoting metastatic CRC (mCRC) by stimulating epithelial-mesenchymal transition (EMT), a mechanism that increases invasive potential, multi-drug resistance (MDR), and cancer stem cell (CSC) stemness in many solid tumors.4–7 Drug development targeting the Wnt/TCF pathway has been hindered by significant challenges in identifying effective drug targets. Some of the approaches include targeting CK1 and CDK8 kinases, the acetyltransferase porcupine, and tankyrases (TNKS).8 A major strategy has been to target protein-protein interactions between β-catenin and other TCF-complex proteins.8, 9 Directly targeting TCF-transcription has proved to be a successful and clinically relevant approach, as exemplified by the investigational new drug, PRI-724, which inhibits protein interactions between β-catenin and CBP (CREB Binding Protein).10 Hence, directly targeting TCF-transcription may be the most effective strategy for the treatment of late stage and mCRC.
We have identified a new therapeutic strategy to directly inhibit aberrant TCF-transcription, by characterizing topoisomerase IIα (TOP2A) as a DNA binding factor required for TCF-transcription.7 TOP2A forms protein-protein interactions with TCF4 and β-catenin, and protein-DNA interactions with Wnt response elements (WRE) and with promoter sites of genes directly regulated by TCF-transcription. TOP2A is an important drug target for the treatment of many types of cancer, especially those that overexpress or amplify the enzyme.7 TOP2A is overexpressed in CRC, particularly mCRC or recurring CRC.11, 12 We have validated TOP2A as a druggable target preventing TCF-transcription by using short hairpin RNA knockdown and small molecule N-terminal ATP-competitive inhibitors7, notably, the marine alkaloid neoamphimedine (neo).13–15 Neo inhibits TOP2A and the TCF-complex from binding to WRE/promoter sites.7 This in turn prevents TCF-transcription, leading to the reversion of EMT and its associated malignant properties. Unlike neo, conventional TOP2A inhibitors that bind to the C-terminal domain such as etoposide and merbarone do not inhibit TOP2A mediated TCF-transcription. We hypothesize that this lack of inhibition is primarily due to alterations in TOP2A C-terminal drug binding sites that arise from TOP2A participation in the TCF-complex7, but also potentially from other TOP2A poison associated MDR mechanisms.16 Furthermore, clinically used TOP2A poisons cause DNA lesions and significant adverse effects.17, 18 Thus, TOP2A poisoning is not a suitable strategy for inhibiting TOP2A-dependent TCF-transcription.
We hypothesize that TOP2A is recruited by the TCF-complex to help dynamically regulate EMT genes and other metastatic genes (e.g. c-Myc) promoting mCRC. TOP2A-DNA interactions essential to this process are facilitated by ATP binding and hydrolysis.19 Therefore, we propose that N-terminal ATP sites are conserved during TOP2A mediated TCF-transcription, providing an Achilles’ heel to MDR and optimal sites for drug design. Although neo displays promising antitumor activity in several models of cancer7, 14, 15, it also displays a suboptimal in vivo pharmacokinetics (PK) profile.7 Inspired by neo’s activity, we describe the design, synthesis, and biological evaluation of novel TOP2A ATP-competitive inhibitors with improved drug-like physicochemical and in vivo PK properties that are effective at preventing TOP2A mediated TCF-transcription and reverse EMT.
Results and Discussion.
Structure Based Drug Design (SBDD).
Previously, we have developed an in silico molecular model using neo and the crystal structure of the N-terminal ATPase domain of TOP2A (PDB:1ZXM, 1.87Å).15, 20, 21 Molecular docking studies using our model show that neo preferentially binds to the TOP2A N-terminal ATP-binding sites compared to other sites in the TOP2A structure (SI Figure 1). Neo is anchored in the ATP-binding site through a network of hydrogen bonds (H-bonds) with Ser148 and Asn150, and through a charge transfer π-cation interaction within the active site magnesium ion (Figure 1a).15 Neo’s binding interactions mimic those of the non-hydrolysable form of ATP, 5’-adenylyl-imidodiphosphate (AMPPNP), including hydrophobic interactions with adenine and H-bonds between the adenine ring and Asn120, Asn91, and between the ribose ring and Ser148 and Asn150.15 Based on our docking studies, we concluded that the flat pyrido acridine ring system of neo is dispensable and that neo’s interactions with Ser148, Asn150, and magnesium are key to its biological activity.15 Therefore, we have characterized the pharmacophore of neo as the substituted quinoline shown as red bonds in the 2D structure of Figure 1a. In addition, we postulated that the flat pyrido acridine ring may be the primary cause of neo’s unfavorable in vivo disposition by hindering diffusion through lipophilic barriers. Using computer aided drug design (CADD) through molecular docking studies with the pharmacophore of neo (Figure 1a inset red bonds) and the N-terminal ATPase domain of TOP2A (PDB:1ZXM, 1.87Å), we set out to design novel TOP2A ATP-competitive inhibitors. Our design goals were to increase interactions between the hydrophobic region (blue), hydrophilic region (green), magnesium ion (pink), and the Walker A motif (orange)22, 23, while maintaining the key H-bonding observed with neo (Figure 1a). In addition, neo is insoluble in water and metabolically unstable7 so our goal was also to improve water solubility and metabolic stability while maintaining biological activity and improve other drug-like properties.
Figure 1. Structure based drug design inspired by neo.
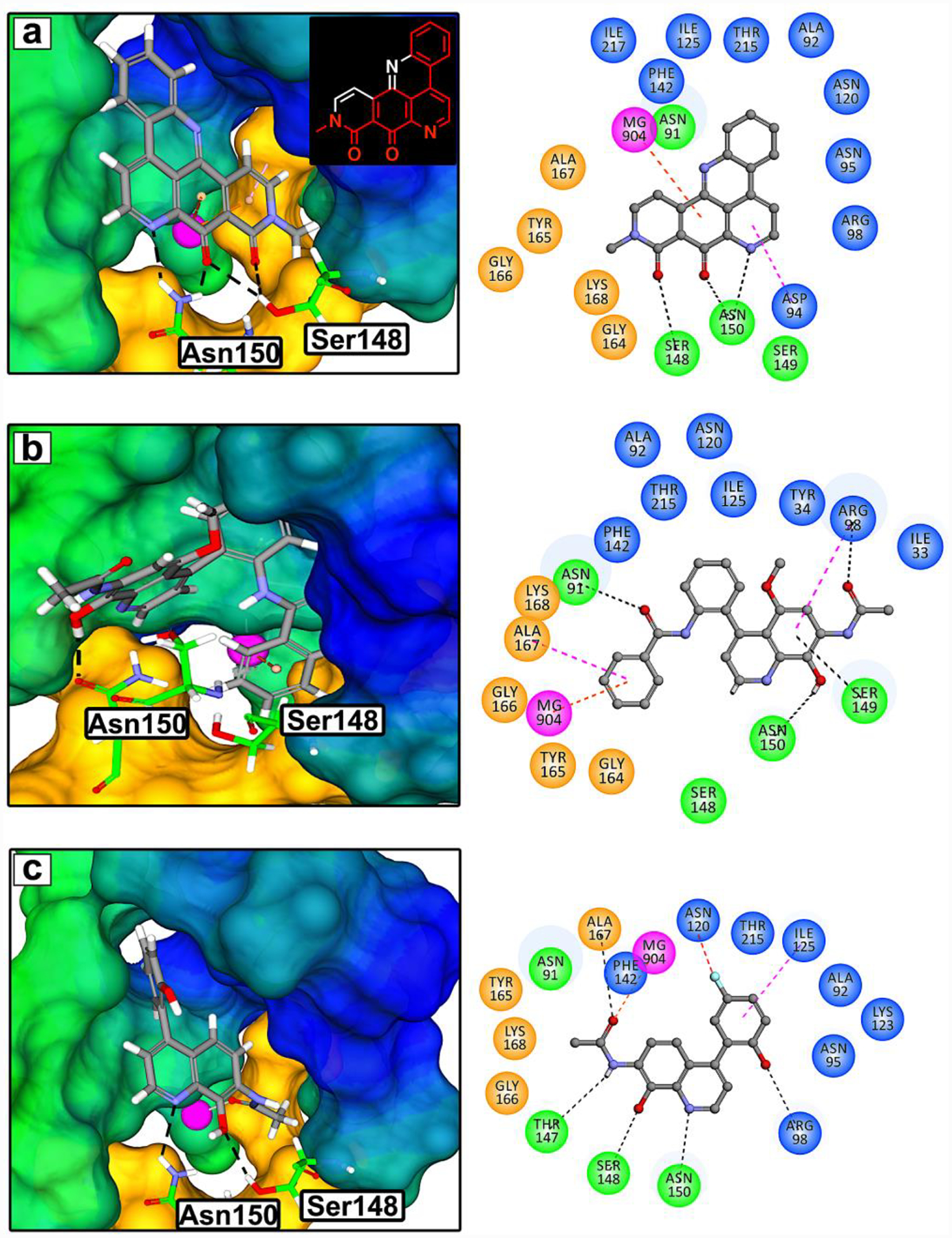
(a) 3D rendering of the molecular model of neo docked into the ATP-binding site of the N-terminal domain of TOP2A (PDB:1ZXM, 1.87Å). The colored regions represent hydrophobic (blue), hydrophilic (green), Walker A motif (orange) residues, and Mg2+ (pink sphere). Based on this model, the pharmacophore of neo was deduced to be a substituted quinoline shown as red bonds in the chemical structure. Novel TOP2A ATP-competitive inhibitor prototypes 3 (b) and 7 (c) were designed using CADD and the pharmacophore of neo. TOP2A ATP-binding site interactions are shown with a 2D rendering as dashed lines: H-bonds (black), Mg2+ π-cation or chelation (orange), π-sigma (pink), π-loan pair (blue), and halogen hydrogen bond (red).
Deconstructing neo’s pyrido acridine ring using retro synthetic analysis provided known intermediate 124, which we used in conjunction with CADD to design and prepare 2 and 3 (Scheme 1a). CADD revealed that the 8-methoxy group of 2 lost key H-bonding with Ser148 and Asn150, which appears to be due to steric effects. However, the 8-hydroxy functionality of compound 3 anchors in the ATP site similar to neo and maintains H-bonding interactions with Asn150 (Figure 1b). In addition, 3 forms a π-lone pair25 interaction with Ser149 and the amide group in the 7-position forms favorable chelation interactions with the active site Mg2+. CADD also revealed that the 4-phenyl ring system of 3 could be functionalized to increase van der Waals and other interactions in the ATP-binding site. Furthermore, the 5-methoxy group of 3 appears to be dispensable with no observed interactions in the ATP-binding site. As a result, compound 3 was further optimized to generate 4-aryl analogs exemplified by compound 7 that is prepared from intermediate 4 (SI Scheme 1) in three steps (Scheme 1b). Compound 7 also anchors in the ATP-binding site similar to neo, maintaining H-bonding with Ser148 and Asn150, but also forms more interactions compared to 3 and neo (Figure 1c). We have designed compound 7 to contain a fluorine group and adding fluorine is known to improve physicochemical properties but can also increase binding affinity through direct protein interactions or indirectly by influencing desolvation and increased hydrophobic interactions.26, 27 Our CADD studies demonstrate that the fluorine group of 7 prefers to orientate in the hydrophobic pocket of TOP2A and all the top ranked docking poses of compound 7 form a fluoro-H-bond with Asn120.
Scheme 1.

Syntheses of quinolines 3 and 7 prototype TOP2A ATP-competitive inhibitors.
The synthesis and biological evaluation of prototypes 3 and 7.
The synthesis of 3 starts with catalytic reduction of the nitro group of 1, followed by acylation using benzoyl chloride to provide 2 in good yield (Scheme 1a). Of note, while reductions with both cyclohexene/Pd on carbon or iron work, we found iron to provide higher yield with no side products. Selective demethylation of 2 using lithium iodide and 2,6-lutidine gave 3 in 90% yield.28 Presumably, this selectivity is facilitated through the coordination of Li with the quinoline nitrogen of 2 and with 2–6-lutidine. However, demethylation using BBr3 is ideal when selectivity is not needed. Compound 7 was synthesized from 4 (SI Scheme 1) by the iron reduction of the nitro group,29 followed by acetylation using acetic acid and acetic anhydride, providing 5 (Scheme 1b). Next, we employed Suzuki cross coupling using phenyl boronic acids in the presence of 1,2-bis(diphenylphosphino)ethane (DPPE), cesium carbonate, palladium(II)acetate and intermediate 5 to obtain coupled products 6 in 95% yield.21, 30 The final product was obtained by demethylation of the methoxy groups using boron tribromide (BBr3), which afforded 7 in 90% yield.
To validate our drug design, we conducted biological studies with 3 and 7 using TOP2A recombinant enzyme inhibition studies and cell-based models, including 3D tumor organoid models7, 31–33 to measure TOP2A-dependent TCF-transcription (TOPflash assay) and the reversion of EMT using biomarker To validate our drug design, we conducted biological studies with 3 and 7 using TOP2A recombinant enzyme inhibition studies and cell-based models, including 3D tumor organoid models7, 31–33 to measure TOP2A-dependent TCF-transcription (TOPflash assay) and the reversion of EMT using biomarker reporters of E-cadherin promoter mCherry red fluorescent protein (EcadPro-RFP) and Vimentin promoter green fluorescent protein (VimPro-GFP). Like neo, and consistent with our drug design, both 3 and 7 maintain a competitive mode of TOP2A inhibition, measured by Michaelis-Menten kinetics of ATP hydrolysis using our established malachite green assay (Figure 2a).15 For enzyme reactions treated with neo, 3 and 7, the Vmax does not change but the KM significantly changes, indicating a competitive mode of inhibition. Despite being TOP2A inhibitors, neo and 7 do not damage DNA compared to the TOP2A poison etoposide, which generates DNA double stranded breaks (DSB) (Figure 2b). We demonstrate this by using three different TOP2A ATP-competitive inhibitors, neo, compound 7, and benzoamino purine (BAP-1, developed by Novartis34, 35), by measuring DSB through γ-H2AX staining. γ-H2AX is a clinical pharmacodynamics (PD) biomarker used to assess DNA damaging chemotherapy in patients.36, 37 Bates et al. proposed that the energy of ATP hydrolysis by TOP2A is used to strengthen both TOP2A homodimer interactions and TOP2A–DNA interactions by inducing favorable binding conformations, which increases binding affinity for DNA.19 This theory agrees with our findings and hypothesis.
Figure 2.
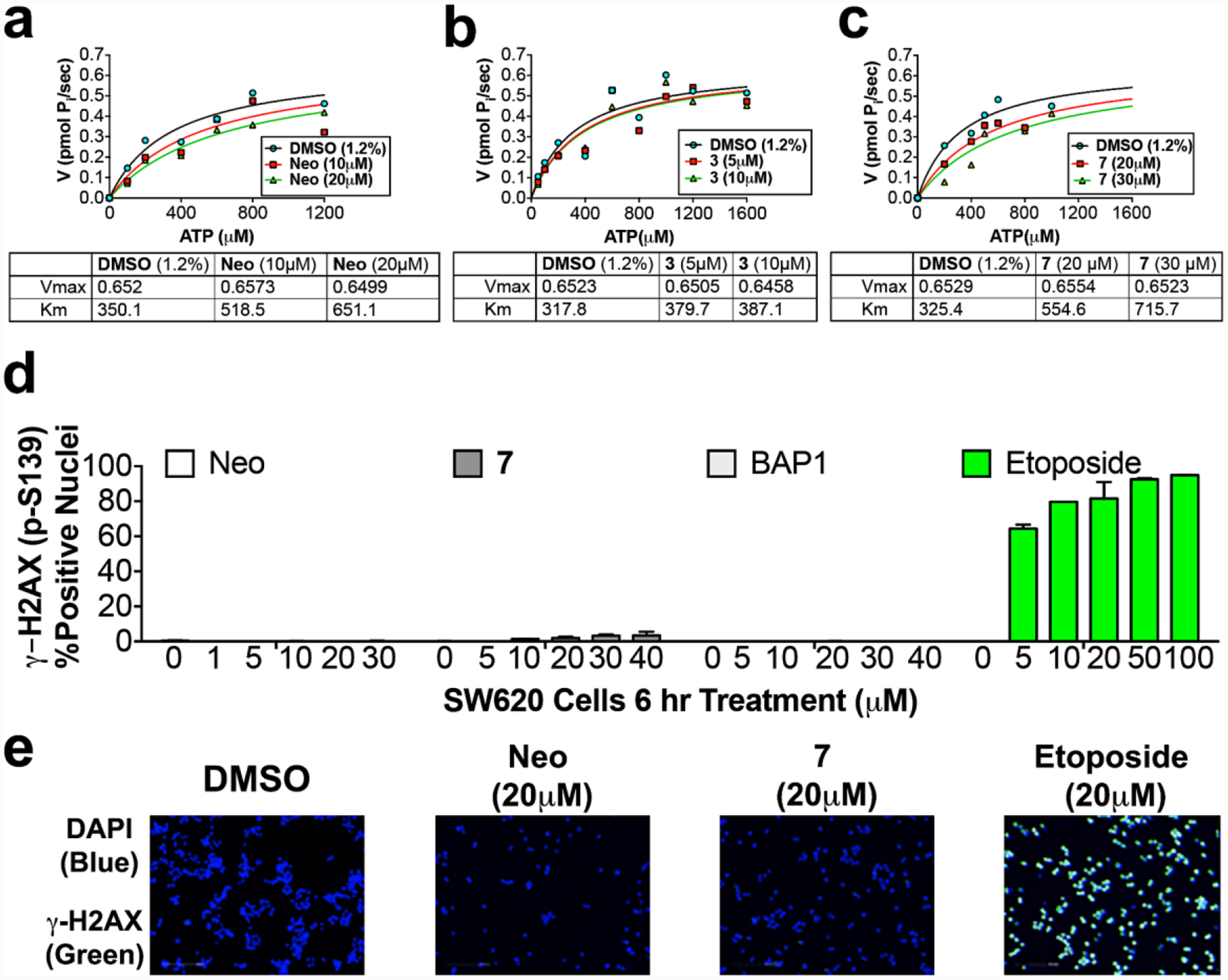
TOP2A enzyme Michaelis-Menten inhibition studies measuring ATP hydrolysis with (a) neo, (b) 3, and (c) 7. Like neo, the quinoline based pharmacophores of 3 and 7 display an ATP-competitive mode of inhibition against TOP2A, evidenced by shifts in KM with no changes in Vmax. (d) TOP2A ATP-competitive inhibitors Neo, 7, and BAP1 do not cause DNA DSBs, over a concentration range from 0–40 μM, measured by H2AX S139 phosphorylation immunofluorescence, while the TOP2A poison, etoposide, caused significant DNA damage over a concentration range from 5 to 100 μM. (e) Representative fluorescence images showing that neo and 7 do not cause induction of γ-HA2X, while etoposide caused significant induction of γ-HA2X.
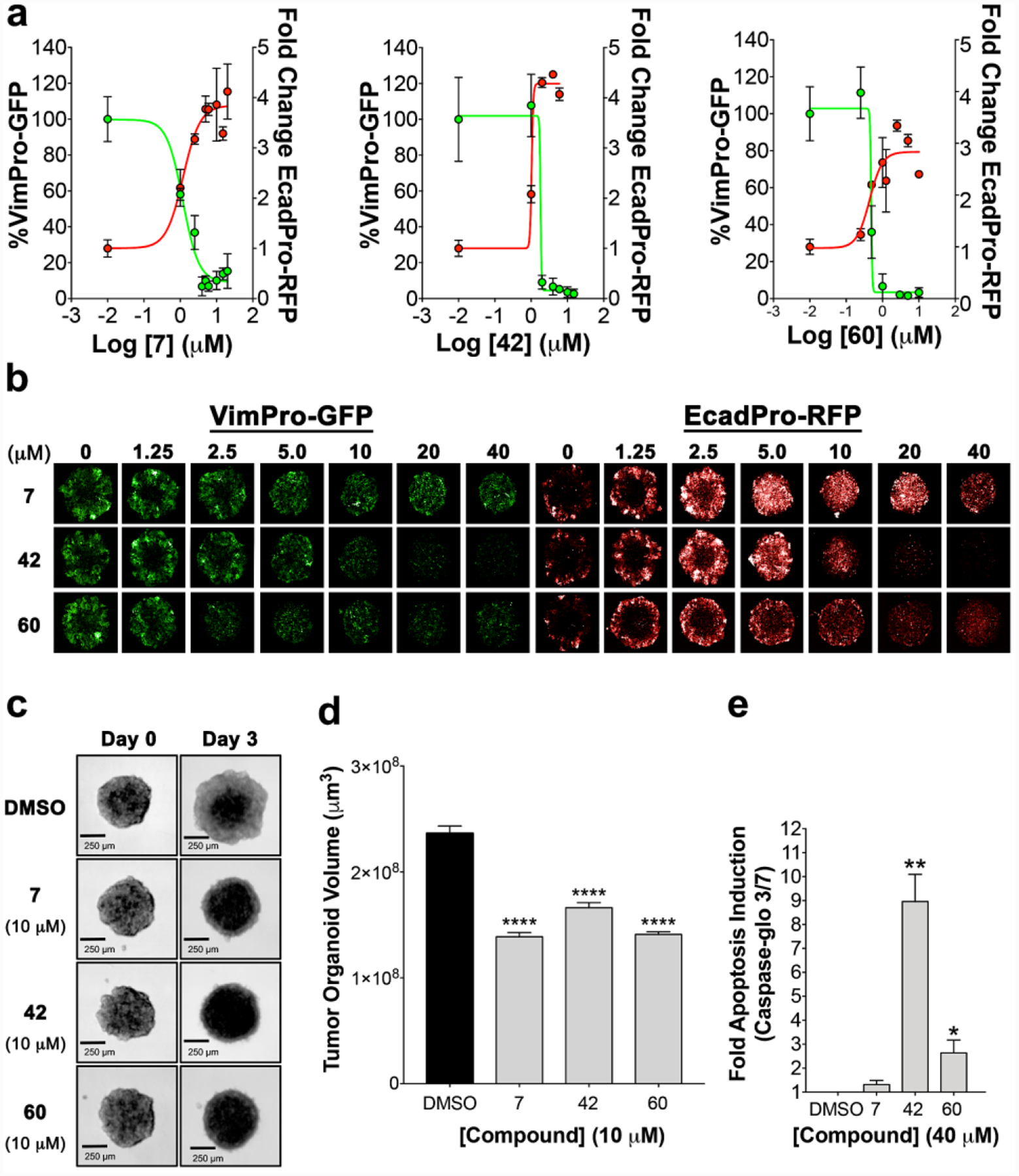
After characterizing 3 and 7 as TOP2A ATP-competitive inhibitors, we next tested their ability to inhibit TOP2A-dependent TCF-transcription using the well characterized TOPflash (TOP) luminescent reporter38, which we previously used to generate a stably transduced SW620-TOP reporter cell line.7 These cells were used to generate single 3D tumor organoids (700 μm in diameter) uniformly arrayed in 96-well plates. Both 3 and 7 significantly inhibit TOP-activity (Figure 3a and b), which correlates with downregulation of vimentin and upregulation of E-cadherin measured by VimPro-GFP and EcadPro-RFP biomarker reporter activity in SW620 tumor organoids (Figure 3c and d). Like neo, the mode of action of 3 and 7 also results in significant antitumor activity (Figure 3e–h). Pretreating SW620 cells with 3 or 7 for 24 h, followed by washing and plating viable cells for the clonogenic assay, is sufficient to inhibit colony formation over seven days compared to the DMSO control. In other words, the activity of 3 and 7 is long lasting, suppressing CSC stemness.39, 40 EMT is a major driving force promoting tumor cell invasion and metastasis. To assess if the reversion of EMT by 3 or 7 also inhibits the invasive potential we treated SW620 tumor organoids as indicated over 72 h. Tumor organoids were dissociated and viable cells were washed and plated into the top chamber of 96-well invasion plates coated with Matrigel™ and incubated for an additional 72 h without adding more compound. The results demonstrate that both 3 and 7 inhibit the invasive potential of SW620 tumor organoid cells. Taken together, the biological activity of 3 and 7 is consistent with the activity of neo, inhibiting TOP2A-dependent TCF-transcription that in turn reverses EMT while inhibiting the associated tumorigenic properties of mCRC.
Figure 3. Antitumor activity of prototypes 3 and 7.
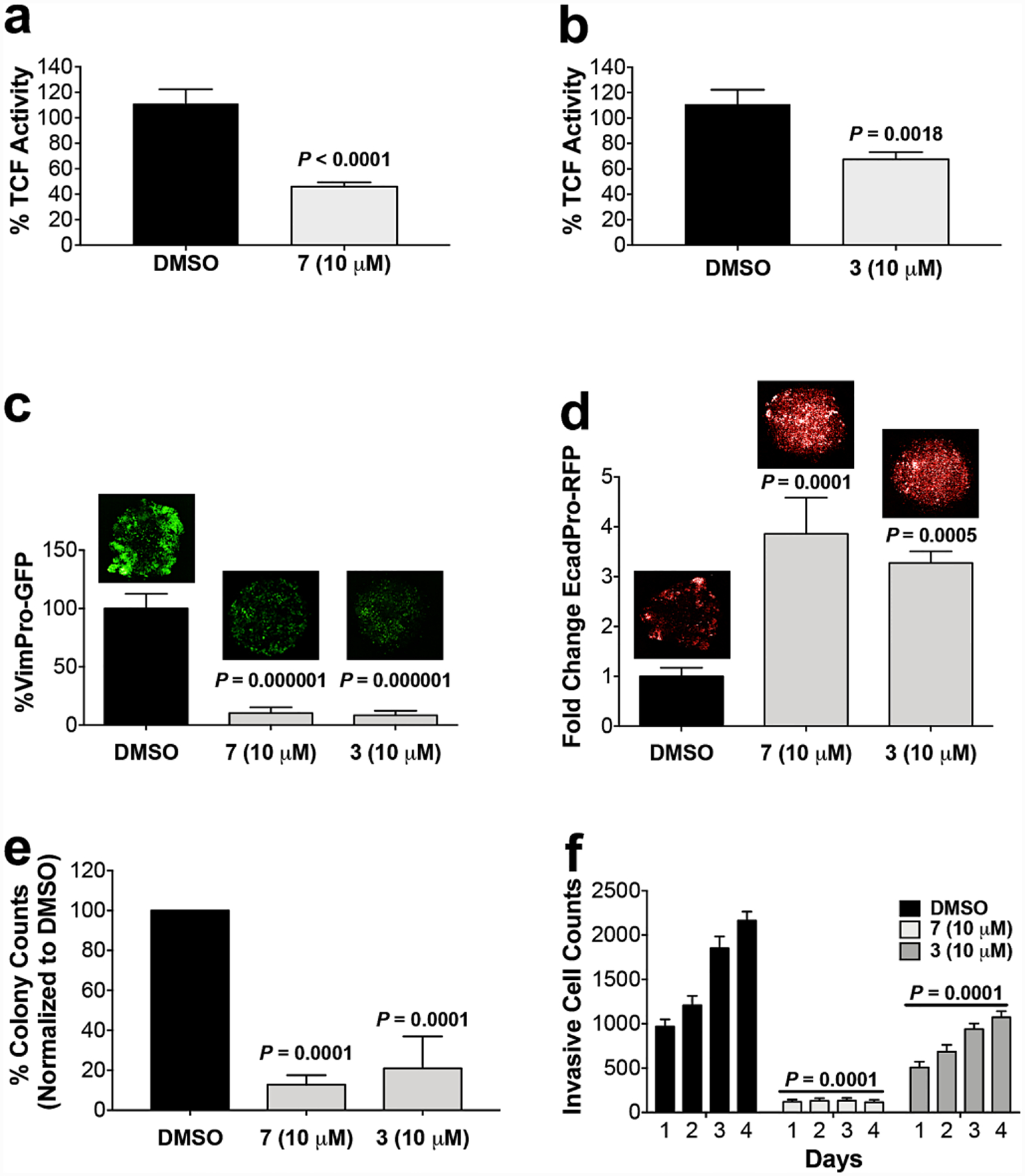
Compounds 7 (a) and 3 (b) demonstrate the ability to significantly inhibit TOP2A-dependent TCF-transcription using SW620 tumor organoids transduced with the TOPflash luminescent reporter. Compounds 7 (c) and 3 (d) also demonstrated the ability to reverse EMT using SW620 tumor organoids transduced with EcadPro-RFP or VimPro-GFP EMT biomarker reporters over a 72 h treatment time course as indicated. Reversion of EMT is characterized by downregulation of vimentin and upregulation of E-cadherin promoter activity. Representative tumor organoid images are shown above each bar graph. (e) Compounds 7 and 3 also significantly inhibit colony formation over seven days, which is a measure of the loss of cancer stem cell stemness. (f) SW620 tumor organoids were treated with 7 and 3 for 72 h, tumor organoids were dissociated, and viable cell invasive potential was measured for additional 72h incubation. Compounds 3 and 7 both significantly inhibit the invasive potential of SW620 cells. Significance was determined using the Student’s t-test analysis.
Further drug design and synthesis of prototype 7 analogs.
Given the promising biological activity of 7, we set out to conduct further drug design optimization using our CADD approach with the goal to improve the physicochemical properties (e.g. water solubility) while improving or maintaining potency against TOP2A and efficacy in cell-based tumor models. These CADD studies led to the design and synthesis of novel intermediates and final analogs (Scheme 2 and 3). CADD indicated that introducing a pivalamide or morpholine propionamide functionality into R1 increased favorable hydrophobic and hydrophilic properties, respectively.
Scheme 2.
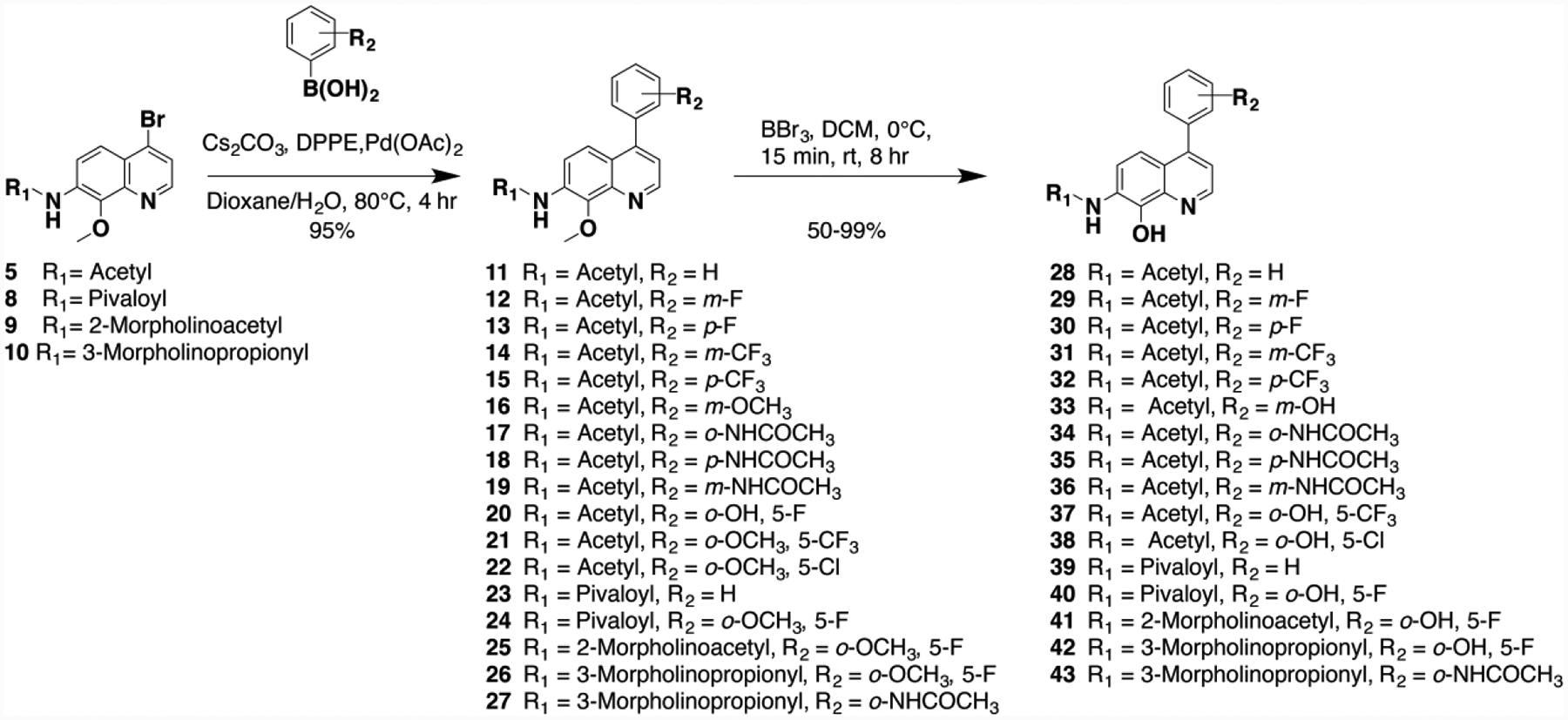
Syntheses of prototype 7 designed analogs.
Scheme 3.
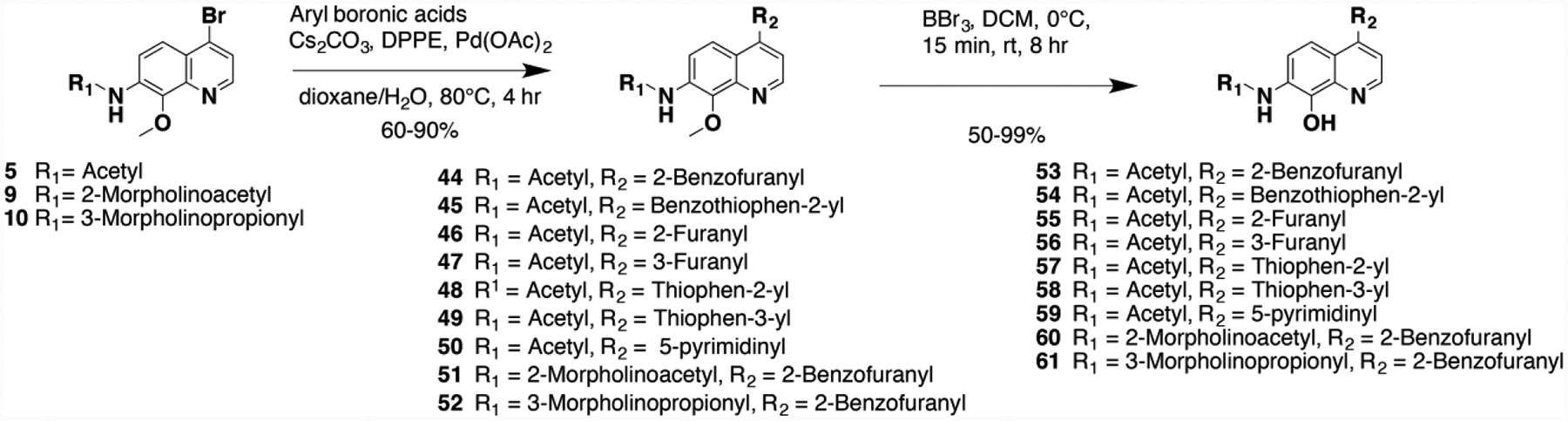
Syntheses of prototype 7 analogs with position 4 aromatic ring variations.
In addition, these functional groups improved interactions with the Walker A motif in the TOP2A ATP-binding pocket. In particular, the morpholine oxygen forms a chelation interaction with the active site magnesium, with a bond length of 2.2Å. Thus, we utilized brominated quinoline 4 to generate intermediates 5 (Scheme 1), 8, 9, and 10. Reduction of 4 followed by acylation with pivaloyl chloride gave intermediate 8, while peptide coupling using morpholine acetic acid or morpholine propionic acid in the presence of propyl phosphonic anhydride (T3P)21 provided 9 and 10, respectively.
Next, to enhance interactions in the lipophilic pocket of the ATP-binding site, we explored a facile Suzuki cross coupling methodology, using phenyl boronic acids with various substituent groups as directed by our CADD, in the presence of 1,2-bis(diphenylphosphino)ethane (DPPE), cesium carbonate, palladium(II)acetate and key intermediate 5, 8–10 to obtain coupled products 11–27.21, 30, 41The final products were obtained by demethylation of the methoxy functionality of the coupled products using boron tribromide (BBr3), which afforded the syntheses of 28–43 in excellent yields (Scheme 2).21, 30, 41 Similarly, we utilized Suzuki coupling with 5, 9 and 10 to introduce a diversity of aromatic ring systems to generate 44–52. As in scheme 2, demethylation using BBr3 afforded compounds 53–61 as the final products (Scheme 3).
TOP2A Inhibition studies and observed structure activity relationships (SAR).
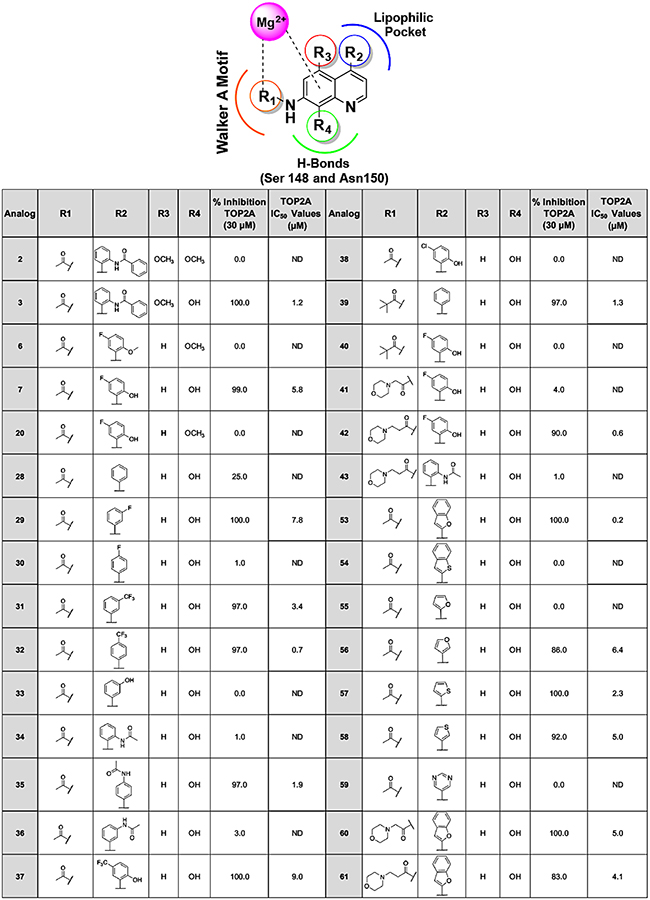
Catenation/decatenation or linking/unlinking of DNA is one of the main TOP2A functions essential for regulating DNA topology, and this is ATP dependent.42, 43 The TOP2A DNA decatenation assay is robust and we have previously demonstrated that neo significantly inhibits TOP2A mediated DNA decatenation.15 As a starting point, we screened our analogs against TOP2A-dependent decatenation using 30 μM fixed concentrations and the DNA from reactions were resolved on 0.8% agarose gels containing ethidium bromide. Our designed analogs displayed potent inhibition of TOP2A decatenation, ranging from 80–100% inhibition (Table 1). Subsequently, we conducted dose response studies using select analogs that displayed ≥ 80% inhibition of TOP2A decatenation to determine the inhibition concentration 50% (IC50) values (Table 1). These studies demonstrate inhibition of TOP2A ranging from nanomolar to low micromolar activity. Notably, compound 53 and 42 proved to be the most potent TOP2A inhibitors with IC50 values of 0.24 μM and 0.6 μM, respectively, compared to prototype 7 (IC50 = 5.81 μM) (Figure 4). Moreover, these studies provided interesting SAR, which shed light on the potential lead drug target binding interactions and affinity, biological activity, and future drug design optimization with this unique class of TOP2A inhibitors.
Table 1.
TOP2A decatenation inhibition studies with prototype 7 analogs. Analogs that did not inhibit TOP2A at 30 μM were not used to determine dose responses indicated as not determined (ND).
|
Figure 4. Dose response studies measuring inhibition of TOP2A decatenation.
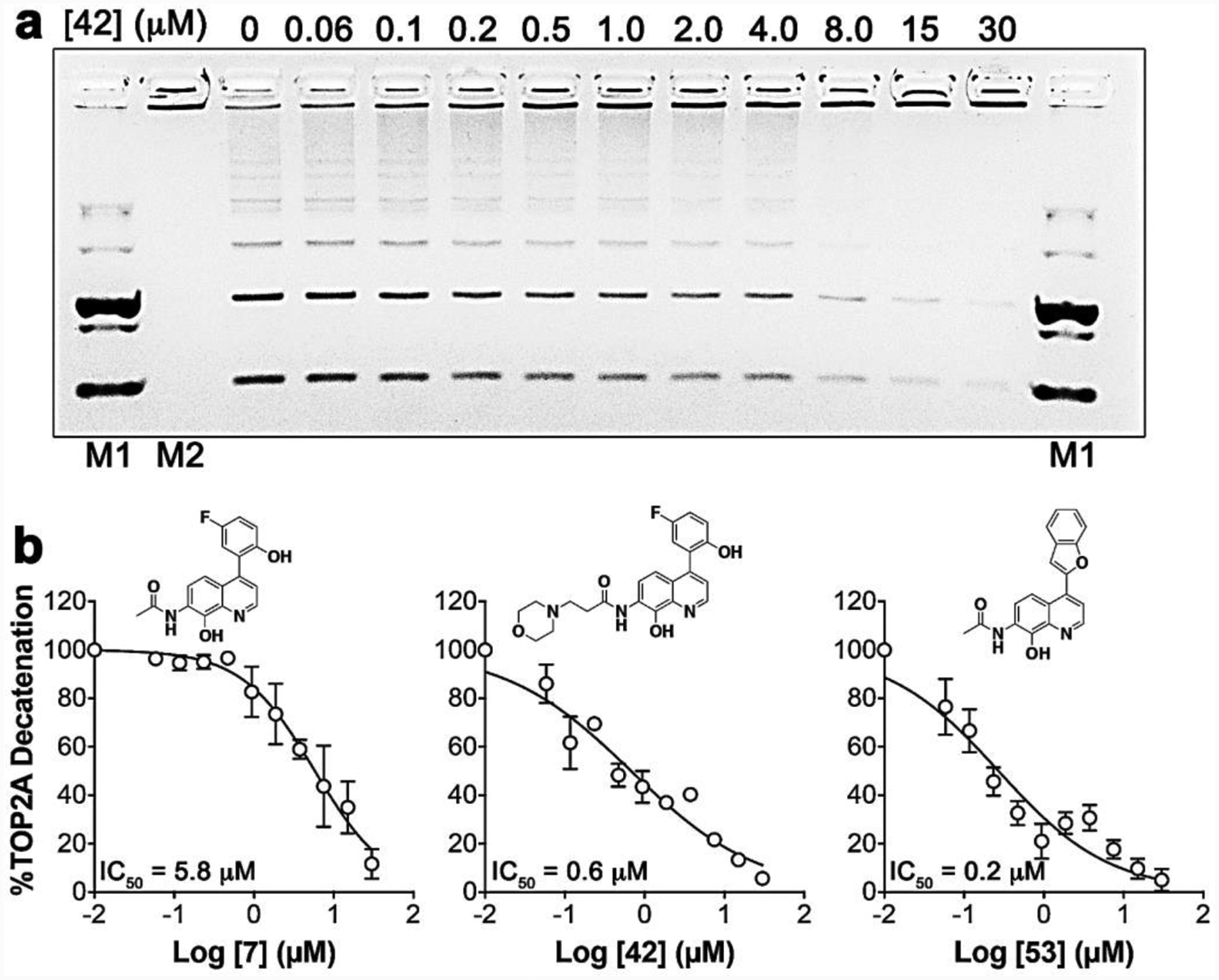
(a) Prototype 7 analog dose response curves were generated using DNA decatenation gels as demonstrated with compound 42. Control lanes with DNA markers, include: decatenated DNA (M1) and Catenated DNA (M2). Dose response studies were conducted using 2-fold dilutions as indicated. (b) Representative dose response curves with analogs 7, 42 and 53.
Compound 7, which contains a 5-fluoro-2-hydroxy phenyl ring showed complete inhibition of TOP2A at 30 μM with an IC50 of 5.8 μM. As we have described above, fluorine can improve binding in various ways both directly and indirectly.26, 27 However, during CADD our docking studies demonstrate favorable halogen H-bonding with compound 7 between fluorine and Asn120 amino proton (Figure 5a). This interaction was observed in all the top ranked docking poses. Similarly, we observed these fluorine H-bonding interactions with analog 29, which has a meta fluoro-functionality (Figure 5b). When the fluorine is moved to the para position of the ring, as in analog 30, the activity is lost and may be attributed, in part, due to the loss of fluorine bonding interactions. Interestingly, having trifluoromethyl groups in the meta (compounds 31 and 37) or para (compound 32) positions resulted in excellent inhibitory activity, and we observed both halogen bond donor and acceptor interactions (Figure 5b). Taken together, the fluorine interactions we observe may be important for TOP2A inhibition. The chloro-analog 38 did not inhibit TOP2A and the loss of activity is consistent with our molecular docking studies that indicated significant loss of favorable bonding interactions. We also observed that having an acetamide in the ortho and meta position of the 4-phenyl ring, as with analogs 34 and 36, caused significant loss of activity. However, moving the acetamide to the para position in 35 restored inhibitory activity against TOP2A, which we attribute to a conventional H-bond resulting from the para position.
Figure 5. Structure activity relationship summary.
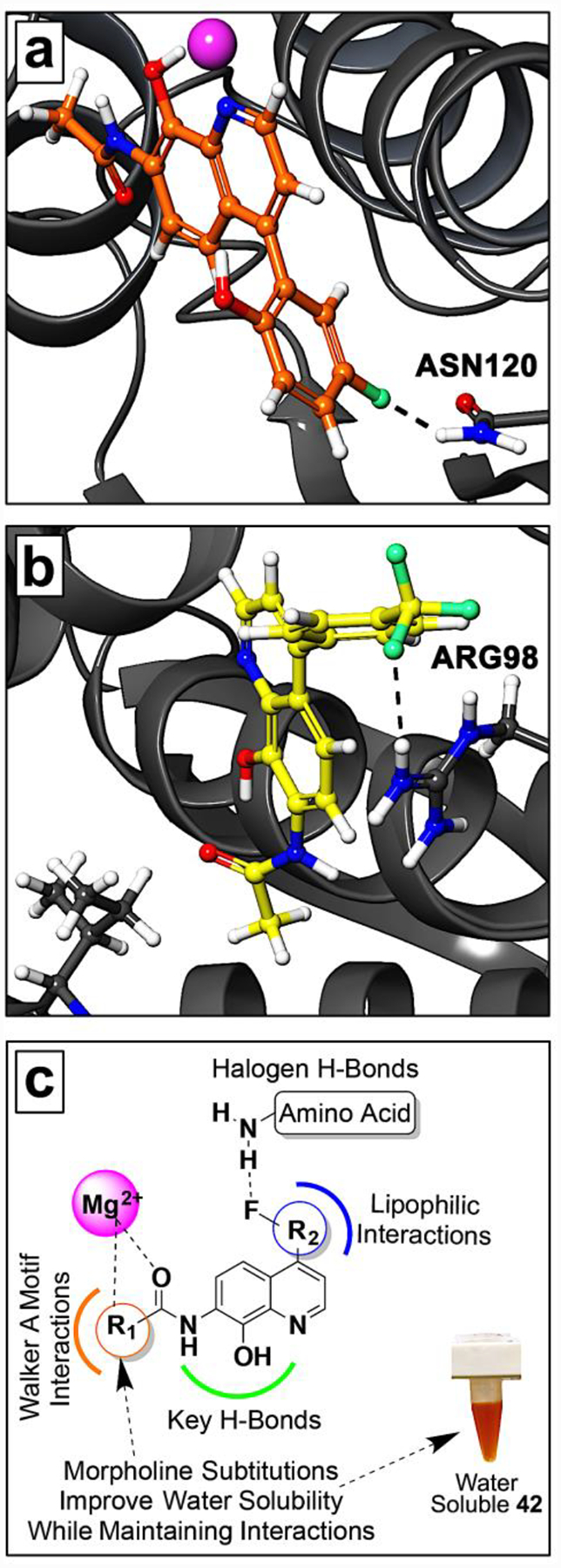
(a) Compound 7 docked into the TOP2A ATP binding site highlighting fluorine H-bond interaction (black dashed line). (b) Similarly, the trifluoromethyl group of 32 forms fluorine H-bonds with Arg98 (black dashed lines). (c) The SAR summary of TOP2A inhibitors, demonstrating water solubility with compound 42.
Substituting the 4-phenyl ring with various aromatic ring systems provided further SAR. Compound 53, containing a benzofuran ring, displayed complete inhibitory activity against TOP2A. Interestingly, inhibition was lost in 54 (benzothiophene ring) due to the minor substitution of oxygen with sulfur. In addition, analogs containing the smaller 5-membered furan rings, 55 and 56, displayed differential inhibitory activity due to the position of the oxygen. When the oxygen is in the 2nd position, as in 55, significant loss of activity was observed compared to when the oxygen is in the 3rd position as with analog 56. Unlike the benzothiophene 54, the 3-thienyl ring of 58 improves activity compared to the 3-furanyl analog 56 (Table 1). Taken together, the stereo- electronic effects of the aromatic heteroatoms appear to play an important role in TOP2A inhibition.
Finally, we explored the SAR resulting from R1 substitutions (Table 1). Molecular modeling shows that the morpholino functionality of 42 allows for favorable binding with TOP2A through H-bonds with Thr215 and the Walker A motif portion of the binding pocket, and through interactions with Mg2+. In addition, morpholine groups promote water solubility while maintaining favorable calculated LogP (e.g 42 CLogP = 2.57). Similarly, 3 and 39 display reasonably good IC50 values (~1 μM) due to apparent increased lipophilic interactions with the TOP2A binding pocket. Compounds 60 and 61, morpholino derivatives of 53, displayed reasonable IC50s of 4 and 5 μM, respectively. These analogs maintain relatively good potency against TOP2A but are more water soluble compared to 53, which is an important drug physicochemical property to consider. The SAR resulting from our TOP2A inhibitor design and TOP2A enzymes studies is summarized in Figure 5d.
Biological evaluation of prototype 7 analogs.
Similar to compound 7, we expected analogs to significantly downregulate TCF-transcription. Thus, we tested the most potent TOP2A inhibitor using SW620-TOP tumor organoids treated for 72 h at various doses to determine dose responses (Table 2). We observed a similar trend in IC50 values compared to TOP2A enzyme studies, further supporting our proposed mechanism of action as inhibition of TOP2A-dependent TCF-transcription. Analogs were also assessed for their ability to modulate or reverse EMT using VimPro-GFP and EcadPro-RFP SW620 reporter cells cultured as tumor organoids, determining EC50 dose responses over 72 h (Table 2). As observed with compound 7, analogs that inhibited TCF transcription also displayed excellent activity at downregulating vimentin (mesenchymal biomarker) and upregulating E-cadherin (epithelial biomarker) promoter activity, indicating the reversion of EMT. Notably, compared to prototype 7, analogs 42 and 60 displayed the most potent effects on TCF-transcription and reversing EMT (Figure 6a and b). At higher treatment doses all three analogs appeared to cause tumor organoid cell death, evidenced by loss of EcadPro-RFP fluorescence (Figure 6b).
Table 2.
The biological activity of TOP2A inhibitors using SW620 tumor organoid models of TCF-transcription, 3D cell viability, and EMT reporters of vimentin and E-cadherin.
| Analog | TCF Inhibition IC50 (μM) | Cytotoxicity (CellTox Green) EC50 (μM) | Vimentin downregulation EC50 (μM) | E-cadherin upregulation EC50 (μM) |
|---|---|---|---|---|
| 3 | 2.4 | >30 | 4 | 9 |
| 7 | 2.4 | >30 | 1.2 | 1.6 |
| 35 | 2.0 | >30 | 9.5 | 5.5 |
| 39 | 4.7 | >30 | 4.7 | 3.1 |
| 42 | 0.7 | 16 | 1.9 | 1.2 |
| 53 | 1.0 | > 30 | 3.5 | 5.4 |
| 56 | 1.7 | >30 | 1.7 | 3.4 |
| 57 | 5.0 | >30 | 1.2 | 1.1 |
| 60 | 1.1 | 22 | 1.0 | 3.3 |
| 61 | 2.5 | 17.4 | 2.4 | 3.0 |
Figure 6.
(a) dose responses for the reversion of EMT biomarker reporter activity in SW620 tumor organoids. (b) Representative dose response images for both vimentin and E-cadherin reporter activity. At higher doses we observed potential early stages of cell death indicated by loss of RFP reporter activity. (c) representative brightfield images indicating TOP2A inhibitors prevent SW620 tumor organoid growth compared to DMSO (scale bars = 250 μm). (d) SW620 tumor organoid volumes were measured at 72 h quantifying compound effects on tumor growth. (e) At the highest doses, all compounds display an induction of apoptosis based on cleaved caspase 3 and 7 activity with 42 and 60 at ~9-fold and ~3-, respectively.
To further assess analog 7, 42, and 60 effects on tumor cell growth and cell death, we first measured the growth in volume (μm3) of SW620 tumor organoids over 72 h. All three analogs significantly inhibited tumor organoid growth compared to DMSO (Figure 6c and d). To shed light on a possible mechanism of cell death we measured caspase 3 and 7 cleavage activation, which are prominent markers of apoptosis (Figure 6e).44 At higher doses all three analogs induce apoptosis compared to DMSO but 42 and 60 displayed significant induction of apoptosis compared to 7. Finally, we conducted SW620 tumor organoid cytotoxicity experiments with analogs in table 2, using the CellTox Green assay, which measures changes in cell membrane integrity associated with the end stages of cell death (Table 2). In general, most analogs tested displayed EC50 values in the CellTox Green assay >30 μM. Taken together, analog effects on growth and tumor cell death appear to be the consequence of inhibiting TOP2A, TCF-transcription, and the reversion of EMT. Furthermore, these results are consistent with our previous report of neo’s effects on TCF-transcription and EMT.7
Clonogenic and invasion assays.
Common features of EMT positive cell phenotypes are increased invasive potential and CSC stemness, tumor cell capabilities promoting metastatic dissemination and drug resistance.5, 45 SW620 tumor organoids were pretreated with analogs at 10 μM over 72 h, followed by harvesting viable cells from 3D culture and plating these cells into invasion or clonogenic colony formation assays. We observed significant potent inhibition of invasive potential and CSC stemness by all analogs tested (Figure 7). Interestingly, we observed a trend that analogs with more potent EC50 for reversion of EMT displayed better activity against colony formation of SW620 cells (Figure 7a). Likewise, the analogs tested also significantly inhibited the invasive potential of viable cells harvested from SW620 tumor organoids (Figure 7b).
Figure 7. TOP2A inhibitor effects on malignant properties of SW620 tumor organoids.
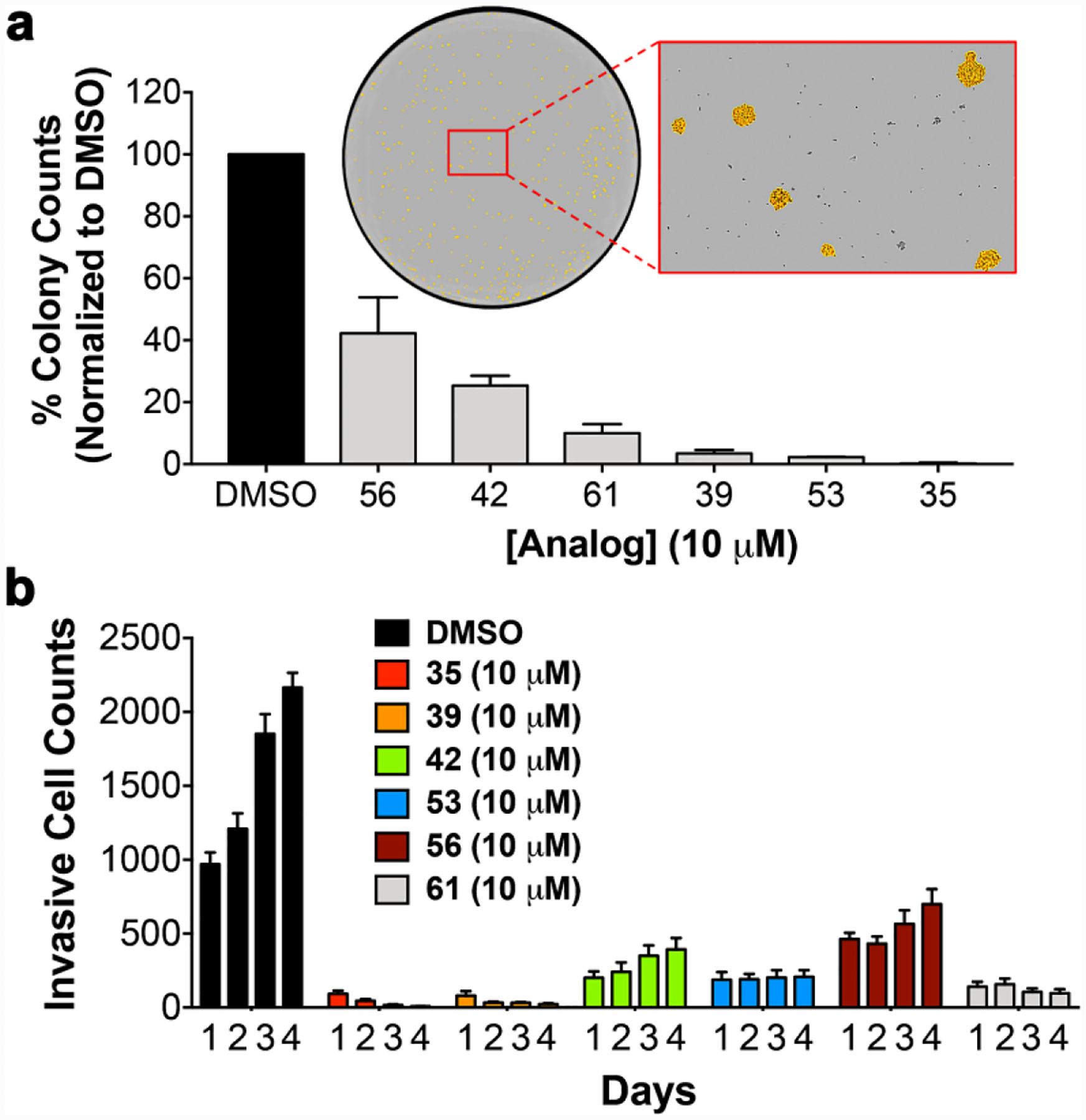
(a) The clonogenic assay results with TOP2A inhibitors demonstrate significant inhibition of CSC colony formation. The inset is a representative image of a DMSO control well of a 6-well plate. The expansion view shows detail of colonies formed, which are identified using area (μm2) min: 1×104 and max: 4×105, and masked in orange color for contrast. (b) The invasion assay results with TOP2A inhibitors demonstrate significant loss of invasive potential over days 1 through 4. The results in panel a and b were determined to be statistically significant using one-way ANOVA analysis, where P ≤ 0.0008 for each analog tested.
Pharmacokinetic analysis and structure property relationships (SPR).
The microsomal stability of prototypes 3 and 7 and analogs 28, 31, 35, 42, 53, 58, 60, and 61 were determined (Table 3 and SI Table 3). These analogs were allowed to react with mouse microsomes and the fraction of remaining parent drug over time was determined. Analog 42 was the least stable as measured by the last time point, but prototypes 3 and 7 and compound 58 were all metabolized faster within the first 30 minutes. The remaining compounds were all appreciably stable in comparison. Analysis of control inactive microsome reactions suggests that all compounds are stable at 37 °C for one hour (SI Table 3). Importantly, loss of parent compound is attributed only to oxidative Phase I metabolism. Formation of Phase II metabolites were not determined.
Table 3.
PK and SPR analysis of prototype 7 and select analogs.
| Pharmacokinetic Parameters | In Vitro | In Vivo | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound Number | Structure | CLogP* | Solubility In PBS (mg/mL) | Microsomal Half Life (min) | CMax (ng/ml) | tmax | Vz (L/kg) | AUC0-t (ng/ml × hr) | CL (L/hr/kg) |
t1/2λ (hr) |
| 7 |  |
2.67 | 0.45 | 6.72 | 18,986 | 15 min | 10.9 | 12,488 | 3.87 | 1.96 |
| 42 | 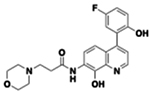 |
2.57 | 1.52 | 8.53 | 10,887 | 15 min | 34 | 13,032 | 3.73 | 6.31 |
| 53 | 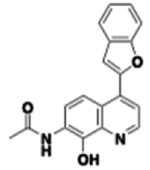 |
3.08 | 0.028 | >60 | 707 | 15 min | 108.9 | 2841 | 16.99 | 4.44 |
| 61 | 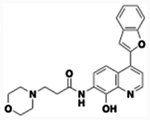 |
2.96 | 1.16 | 14 | 6,830 | 15 min | 32 | 12,871 | 3.71 | 5.97 |
| 60 | 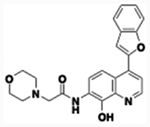 |
2.71 | 0.045 | 30.18 | 1,982 | 15 min | 69.3 | 5,890 | 8.22 | 5.84 |
The consensus LogP (CLogP) values were obtained using the SwissADME website and tools.47
The microsomal stability demonstrates that several of these analogs have high relative stability that could be exploited pharmacologically. The time-course stability of analogs 31, 35, 53, 60, and 61 suggest they are not readily transformed by Phase I metabolic enzymes, particularly when compared to analog 42 and prototypes 3 and 7, and their metabolism can be described by first-order kinetics. Analogs 28 and 58 both diverge from first-order kinetics and the curves suggest biphasic kinetics. When comparing these molecules only to each other, we anticipate all but prototype 3 and 7, and analog 42 to be classified as low liver extraction drugs in vivo. Furthermore, when comparing their structure property relationship (SPR), we observed a trend where the more electron deficient the aromatic ring system at the 4-position of the quinoline the more stable the compounds are in microsomes. For example, 7 and 42 contain a phenolic group in the phenyl ring at the 4-position of the quinoline. The phenol adds potential metabolic liability by adding electron density to the phenyl ring, which can lead to hydroxylation by cytochrome P450s.46 In addition, because there is a Fluorine group para to the phenol (aka ipso-position) this may lead to hydroquinone formation also mediated by cytochrome P450.46 Conversely, analog 35 does not have a phenol but contains an electron withdrawing acetamido group, and it is stable in microsome preparations (SI Table 3). Likewise, analogs 53, containing a benzofuran ring at the 4-position of the quinoline, is metabolically stable (Table 3). Interestingly, when comparing analogs containing morpholine (i.e. 42, 60, and 61) compared to their parent analog (7 and 53) the metabolic instability appears to be associated with the carbon side chain. The propanamide side chain of 61 has a metabolic half-life of 15 min while the ethanamide chain of 60 has a metabolic half-life of 30 min. However, both 60 and 61 still display relatively good metabolic stability. In addition, the morpholine functional group adds valuable aqueous solubility for 42, 60, and 61 compared to the parent analogs 7 and 53. For example, compound 42 is 3.4 times more soluble in phosphate buffered solution (PBS) compared to prototype 7 and (Table 3 and SI Figure S2). However, both 7 and 42 have very similar CLogP values of 2.67 and 2.57, respectively. Hence, the morpholine adds significant water solubility while maintaining lipophilicity, which is a desired drug-like feature. Finally, the phenol at the 8-position of the quinoline ring, which is common to all analogs, appears to not be a metabolic liability during phase 1 metabolism. However, it is currently unknown how stable the 8-hydroxy quinoline moiety will be against Phase II metabolic reactions. The potential for glucuronidation and sulfation of this moiety is an avenue for further pre-clinical research.
Next, using CD-1 mice, we assessed the key PK parameters: maximum plasma drug concentration (CMax), apparent volume of distribution during terminal phase (VZ), area under the plasma concentration-time curve (AUC0-t), apparent total body clearance from plasma (CL), and elimination half-life (t1/2λ) (Table 3). The results showed that compound exposure as measured by Cmax and AUCo-t ranged from 707 to 18,986 ng/ml and 2,841 to 13,031 ng/ml × hr, respectively. This broad range in exposure was not observed in the t1/2 that only ranged from 2 to 6.3 hours. Taken together, the data suggest that the primary factor driving the differences in exposure are based on the distribution of the compounds to tissues which is reflected in the Vz observed values ranging from 10.9 to 108.9 L/kg. In addition, when considering aqueous solubility, analogs 42, 60, and 61 appear to have potential for in vivo pharmacology and efficacy.
Conclusion
The standard of care treatments for unresectable mCRC have limited to no efficacy and only extend life by 20-to-30 months or palliate symptoms, including chemotherapy (e.g. 5FU, oxaliplatin, irinotecan) and biologics (e.g. Avastin).48 Therefore, the conventional approach of inhibiting tumor growth may not be the most effective way to treat mCRC or other late stage metastatic cancers. Herein, we describe a novel therapeutic strategy focused more on targeting the malignant properties of CRC through inhibiting TCF-driven EMT, a process that promotes tumor cell heterogeneity, MDR, and metastasis. Our structure-based drug design has produced novel potent ATP-competitive inhibitors of TOP2A. However, unlike conventional TOP2A poisons (e.g. Etoposide), these inhibitors do not damage DNA and are not potent cytotoxic agents. Instead, they effectively inhibit TOP2A dependent TCF-transcription, leading to the reversion of EMT and associated malignant properties. Future studies will focus on in vivo models to assess pharmacokinetics and anticancer efficacy. EMT targeting agents have the potential to intervene as therapies for multiple types of cancer at various clinical stages, including: (1) inhibiting the less malignant epithelial tumor cell from becoming mesenchymal, (2) reversing the mesenchymal tumor cell to the more benign epithelial state, and (3) sensitizing the mesenchymal cell to conventional therapies.49 Therefore, we anticipate that this novel class of EMT targeting compounds may be an effective combination treatment by sensitizing both primary tumors and metastatic lesions to clinically relevant therapies, and potentially inhibit tumor cell metastasis.
Experimental Section
General Experimental Methods.
All commercial chemicals were used as supplied unless otherwise stated. All solvents used were dried and distilled using standard procedures. Thin layer chromatography (TLC) was performed using Aluminum backed plates coated with 60Å Silica gel F254 (Sorbent Technologies, Norcross, GA, USA). Plates were visualized using a UV lamp (λmax = 254 nm). Column chromatography was carried out using 230–400 mesh 60Å silica gel. NMR spectra were recorded on a Bruker Avance III 400 (1H 400 MHz, 13C 100 MHz) or a Varian 500 MHz spectrometer (500 MHz proton, 125.7 MHz carbon). All chemical shifts are recorded in parts per million (ppm), referenced to residual solvent frequencies (1H NMR: Me4Si = 0, CDCl3 = 7.26, D2O = 4.79, CD3OD = 4.87 or 3.31, DMSO-d6 = 2.50, Acetone-d6 = 2.05 and 13C NMR: CDCl3 = 77.16; CD3OD = 49.0, DMSO-d6 = 39.5, Acetone-d6 = 29.9 Coupling constants (J) values are expressed in hertz (Hz). The following splitting abbreviations were used: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets. Melting points (m.p.) were determined using a Stuart melting point apparatus (SMP20). Infrared (IR) spectra were recorded on a Bruker ALPHA platinum ATR (oils and solids were examined neat). Compounds purity (≥ 95%) was measured using a Shimadzu prominence HPLC equipped with a photodiode array detector (PDA) and LunaR Omega Polar C18 column (5 μm, 100 Å, 250 mm × 4.6 mm). Using a flow rate of 1.0 mL/min, compounds were eluted with a gradient of water/methanol or acetonitrile (ACN), and 0.1%TFA in water/0.1TFA in ACN over 0 to 45 min.
General Mass Spectrometry Methods.
HRMS were recorded using Q Exactive mass spectrometer (ThermoFisher, San Jose, CA) operated independently in positive or negative ion mode, scanning in full MS mode (2 μscans) from 150 to 1500 m/z at 140,000 resolution, with 4 kV spray voltage, 45 sheath gas, 15 auxiliary gas. Acquired data were then converted from raw to mzXML file format using Mass Matrix (Cleveland, OH). Microsomal stability and in vivo PK analysis were evaluated by LC-MS/MS and positive ion electrospray ionization were obtained with an Applied Biosystems SCIEX 3200 QTRAP triple quadrupole mass spectrometer (SCIEX LLC, Framingham, MA) with a Turbo Ion Spray source interfaced to an Agilent 1200 Series Binary Pump SL HPLC system (Agilent Technologies, Santa Clara, CA) and a CTC Analytics HTC PAL System autosampler (CTC Analytics, Lake Elmo, MN). Samples were chromatographed with a Waters SunFire C18 5 μm, 4.6×50 mm column (Waters Corp., Milford, MA). Ultrapure water was solvent A, and acetonitrile acidified with 0.1% formic acid was solvent B. Chromatographic separation was achieved in 6 min at a flow rate of 1 mL/min using the following gradient: min 0.0, 10% B; min 1.0, 10% B; minute 2.0, 98% B; minute 4.0, 98% B; minute 5.0, 10% B; and minute 6.0, 10% B. Analytes were quantified via multiple reaction monitoring of the transitions for each analog plus internal standard.
TOP2A ATPase Assay.
The mode of inhibition of TOP2A analogs were tested with a DNA relaxation assay and the inorganic phosphate (pi) generated was measured with malachite green phosphate reagent (Abcam, MA) in polystyrene 384-well assay plates (Thermo Fisher Scientific, MA). ATP-dependent DNA relaxation assays in 15 μL contained 2U hTopoIIα (Topogen, CO), 85 μg/mL (10 nM) supercoiled plasmid (a gift from AstraZeneca), and various concentrations of ATP (between 0 and 1800 μM) and analog 3 and 7 (between 0 and 20 μM) in 50 mM Tris–HCl (pH 7.5), 150 mM KCl, 5% (v/v) glycerol, 10 mM MgCl2, 1mM dithiothreitol, 0.002% (w/v) Brij-35, and 200 nM bovine serum albumin (BSA). A duplicate plate was prepared in the same way except for the omission of the enzyme to account for the background pi. Reactions were conducted at 37 °C for 1 h. An addition of 70 μL of ddH2O and 15 μL of phosphate reagent were added to each well and the plates were incubated in dark at room temp for 30 min before OD600 was read with a BioTek plate reader. The inorganic phosphate standard curve was generated based on the manufacturer’s recommendation and used to determine the concentration of Pi produced in each well as a result of TOP2A-dependent DNA relaxation. The Michaelis-Menten curves were generated using GraphPad Prism 7.0 (GraphPad, CA).
TOP2A decatenation assay.
Human TOP2A, kDNA, and assay buffers were obtained from TopoGEN (Buena Vista, CO). The synthesized compounds were screened for their TOP2A inhibitory activity by the intensity of DNA decatenation bands on a DNA gel when compared to the vehicle. Compounds were initially incubated with TOP2A (3U/reaction) for 20 min at a 30 μM and 1.2% DMSO concentration in a 37°C incubator without CO2. The decatenation reaction was initiated immediately after by adding 2 mM ATP and 35 ng/reaction of kDNA, then incubated for 1 h in a 37 °C water bath. The samples were loaded into a 0.8% agarose gel in 1X Tris -Borate-EDTA (TBE) at a 0.2 μg/mL ethidium bromide concentration and allowed to run in an electrophoretic chamber for 1 h at 100V. Compounds with the highest inhibitory activity were selected for 2-fold serial dilution dose-response assays (58.6 nM-30 μM). The dose-response assays followed the same protocol with the exception of 4U/reaction of TOP2A.
γ-H2AX DNA damage assay.
Colon cancer cell lines SW620 were plated as monolayers in black clear bottom 96 well plates at 20,000 cells/well in 100 μL of phenol red free medium. After cells attached, various concentrations of neo, 7 and etoposide were added for 6 or 24 h. Cells were fixed with 4% paraformaldehyde at room temp for 20 min, washed 3 times with 400 μL/well of PBS, permeabilized and blocked with 100 μL of 3% BSA and 0.1% IGEPAL in PBS for 20 min. Cells were then incubated in γ-H2AX primary antibody (Cell Signaling, MA, 1:400 dilution) at 4 °C overnight, washed 4 times with PBS, and incubated with FITC conjugated anti-rabbit secondary antibody (Cell Signaling, MA, 1:200 dilution) at room temp for 1 h, and washed 4 times with PBS. Cells were stained with 10 μM of Hoechst 33342 at room temp for 10 min, washed 4 times with PBS before imaged with a 20x objective using PerkinElmer Operetta imager.
3D cell culture of tumor organoids.
Cell lines were cultured as tumor organoid using phenol red free RPMI-1640 containing 5% FBS. Tumor organoid were generated by seeding 5,000 cells/well (or as indicated) into un-coated 96-well U-bottom Ultra Low Attachment Microplates (PerkinElmer, MA) followed by centrifugation for 15 min at 1000 rpm to promote cells aggregation. A final concentration of 2% Matrigel was then added and tumor organoid were formed for 72 h under incubation (5% CO2, 37 °C, humidity) before treatment, and maintained under standard cell culture conditions during treatment time courses.
3D Tumor organoid TOPflash reporter assay.
Stable engineered cells containing pCDH-TOPflash-luc-EF1-puro7 were used to generate tumor organoids arrayed in 96-well plates and treated for 72 h with TOP2A inhibitors. One-Glo™ luciferase reagent and CellTiter-Glo™ 3D cell viability reagent (Promega, WI) were used to determine the TCF reporter activity in viable cells. Briefly, reagents were added to the tumor organoids at 1:1 ratio (v:v) and the plates were placed on an orbital shaker (300 rpm) for 10 min at room temperature before the luminescence was read with Envision plate reader (PerkinElmer, MA). TCF reporter activity was normalized to organoid viability and inhibitors treated conditions were normalized to DMSO treated controls.
pCDH-EcadherinPromoter-mCherry-EF1-Puro (EcadPro-RFP) plasmid cloning.
The CMV promoter in pCDH-CMV-MCS-EF1-puro plasmid was digested out and replaced with E-cadherin promoter using ClaI and XbaI restriction enzymes. The mCherry cassette was then inserted using EcoRI and NotI downstream of Ecadherin promoter. We thank Dr. Jennifer Richer, University of Colorado AMC SOM, for providing us with the Ecadherin promoter luc-zeo plasmid.
VimPro-GFP and EcadPro-RFP reporter assays.
Stable VimPro-GFP7 or EcadPro-RFP SW620 reporter cells were generated using pCDH-VimPro-GFP-EF1-puro virus or pCDH-EcadPro-mCherry-EF1-puro virus as previously reported.7 Transduced cells were selected with puromycin at 2 μg/mL. The stable fluorescent labeled reporter cells were then used to generate tumor organoids as described. Tumor organoids were then treated with TOP2A inhibitors at 10 μM for an additional 72 h. Following treatment, tumor organoids were stained with 16 μM of Hoechst 33342 for 1 h, which functions as a nuclear stain for imaging segmentation. Images were taken with a 5x air objective. Z stacks were set at 26.5 μm apart for a total of 15 layers. Imaging analysis was performed using an Opera Phenix high content screening (HCS) system and Harmony high-content imaging and analysis software interface (PerkinElmer, MA). Nuclei were identified with Hoechst 33342 stain within each layer, and cells were found with either GFP or mCherry channel. The intensities of each fluorescence channel within the cells were calculated and thresholds were set based on the background intensities. Percentages of GFP or mCherry positive cells were calculated and normalized to the DMSO treated group.
Tumor organoid cytotoxicity.
SW620 tumor organoids were grown as previously outlined. CellTox™-Green-Express Cytotoxicity Assay (Promega, WI) solution was prepared per manufacturer’s protocol. Briefly, organoids were treated for 72 h with CellTox™ Green Express (0.5X) reagent and various doses of TOP2A inhibitors over a range of 0-to-40 μM. Organoids were then imaged using the Opera Phenix HCS system (PerkinElmer, MA). CellTox™ Green Express was excited at 488 nm and emission was detected at 500–550 nm. Mean intensity of the whole well was utilized for calculating cytotoxicity with Lysis Buffer (Promega, WI, 0.025X) as the 0% viability control and 0.4% DMSO as the 100% viability control. Intensity values were normalized to these controls using GraphPad Prism 7.0 (GraphPad, CA).
Tumor organoid CSC stemness assay (3D clonogenic assay).
SW620 cells were grown in monolayer in RPMI-1640 media supplemented with 5% FBS, then seeded at 20,000 cells/well in an ultra-low attachment 96-well plate (PerkinElmer, MA) supplemented with Matrigel (Corning, NY). The SW620 organoids were cultured for 72 h, then treated with 10 μM of analogs in 0.1% DMSO for an additional 72 h. Afterwards, the organoids were collected and dissociated using 0.25% Trypsin-EDTA for 5–10 min, then neutralized in FBS containing media, centrifuged at 500 g and resuspended in media. Suspended cells were then counted, and only live cells were plated at 1,000 cells/well in a 6-well plate in 2 mL of growth medium. After 7 days of culture, a whole well image was obtained using the Incucyte S3 (Sartorius, Germany). Colonies were counted using the Incucyte S3 2018A software with the following parameters: “Segmentation Adjustment 0”; “Hole Fill (μm2) = 0”; “Area (μm2) min: 1×104 max: 4×105”; No filter restrictions for “Eccentricity”.
Tumor organoid cell invasive potential assay.
SW620 cells were transduced with NucLight®-Red virus (Essen BioSciences, MI) and selected with Zeocin (200 μg/mL).7 Transduced cells were cultured in RPMI-1640 media supplemented with 5% FBS and allowed to grow in a 10 cm2 dish until ~80% confluent. Cells were then used to generate tumor organoids as described. Tumor organoids were treated with 10 μM of TOP2A inhibitors at 0.1% DMSO and incubated for an additional 72 h. Following treatment incubation, 48 organoids per condition were collected into a 1.5 mL tube and centrifuged at 500 g, washed with ice cold PBS, and dissociated with 500 μL of Cultrex® organoid harvesting solution (Trevigen, MD) for 30 min on ice with moderate mixing. Single cells were collected by centrifugation at 500 g, the supernatant was discarded, cells were washed with ice cold PBS, re-centrifuged and resuspended in RPMI-1640 media supplemented with 0.5% FBS. Live cells were counted (5,000 cells/well) and then suspended in a 1:1 ratio of Matrigel™ (Corning, NY) and RPMI-1640 media (supplemented with 0.5% FBS and 1% penicillin/streptomycin). The 50% Matrigel™ cell suspensions were then plated in the upper chamber of an Incucyte® ClearView 96-well cell migration plate (Essen BioSciences, MI) and centrifuged at 50 g for 3 min at 4 °C. The Incucyte® ClearView 96-well cell migration plate was prepared as per manufacturer’s protocol. Briefly, the plate was maintained in a CoolBox™ 96F system with a frozen gel pack for uniform cooling and the membrane was hydrated in ice cold PBS for 20 min prior to the addition of cells. All cell handling was performed on ice or at 4 °C to avoid polymerization of Matrigel. Cell seeded invasion plates were placed into a pre-warmed CoolSink 96F plate from Essen BioSciences and were incubated for 1 h at 37 °C under standard tissue culture conditions. Finally, 200 μL of RPMI-1640 media supplemented with 20% FBS and 1% penicillin/streptomycin was added to the bottom chamber, and 40 μL of media (0.5% FBS) was added to the top chamber to generate the chemoattractant gradient. Cell invasion was monitored using the Incucyte® ZOOM imaging system (Essen BioSciences, MI) for bottom fluorescence of invading cells per manufacturer’s recommendations with six replicates per condition.
Tumor organoid caspase 3/7 activity.
SW620 cells were seeded at 2,000 cells/well in a round bottom ultra-low attachment 384-well black plate (Corning, NY) in RPMI-1640 media (5% FBS) and supplemented with 2% Matrigel (Corning, NY). The SW620 organoids were cultured for 72 h, then treated with various doses of analogs for an additional 72 h. Caspase 3/7 activity was measured using the Caspase-Glo® 3/7 assay kit (Promega, WI) per manufacturer’s recommendations. Briefly, Caspase-Glo® 3/7 reagent was added at a 1:1 ratio (v:v) and the plates were placed on an orbital shaker (300 rpm) for 30 min before luminescence was read with the Envision plate reader (PerkinElmer, MA). Staurosporine (VWR International, PA) was used as an apoptosis positive control and 0.4% DMSO was used as a negative control to statistically validate this assay using Z’ factor analysis of 0.788 at 20 μM.50
Microsome stability studies.
The microsomal stability TOP2A inhibitors were tested using female CD-1 mouse microsomes (M1500) purchased from Sekisui XenoTech LLC (Kansas City, KS). Reactions were prepared with microsomes (0.5 mg/mL), 1 μg/mL substrate, and 1.5 mM NADPH in 0.1 M phosphate buffer (44mM KH2PO4, 56 mM K2HPO4, pH 7.4 and adjusted with NaOH). A reaction master mixture was prepared without compound and pre-incubated at 37 °C for five min, and then was initiated by the addition of compound and incubated at 37 °C for the duration of the reaction. At time points throughout a range of 0 to 60 min, 100 μL of sample was removed from the master mix and stopped with an equal volume of acetonitrile and vortexed briefly. Microsome control samples were also prepared to test the stability of each analog in the reaction conditions. Samples were centrifuged at 20,000g for 10 min and the supernatant was transferred to an autosampler vial for LCMS analysis.
The following mass transitions (m/z, amu) were monitored for each analog: 3 (428.2 – 386.3, 428.2 – 410.3, 428.2 – 306.2); 7 (313.2 – 295.3, 313.2 – 270.6); 28 (279.1 – 237.3, 279.1 – 261.1); 31 (347 – 305.3, 347 – 329.3); 35 (336.2 – 294.6, 336.2–318.5); 42 (412.3 – 325.2); 53 (319.5 – 301.4, 319.5 – 277.3); 58 (285.2 – 242.9, 285.2 – 267); 60 (404.3 – 352.4). A unique LC-MS/MS method was generated for each analog. Multiple peaks for each analog were incorporated into the final method, as opposed to a single transition, as the sum of the peaks greatly improved sensitivity and detection. The chromatographic peaks associated with each analog were integrated and the concentrations of the samples were calculated using Analyst® (AB SCIEX LLC, Framingham, MA) software. Internal standards were not used in any of the MS methods.
In vivo PK studies.
The following animal studies were approved by the Institutional Animal Care and Use Committee at Colorado State University. Female CD-1 mice, purchased from Charles River, were used for all studies. The PK studies were designed to cover a range of 0.25–24 h with three mice per time point for a total of 21 mice per compound. Each mouse was dosed with a single IP injection of drug at 50 mg/kg prepared in 100% DMSO. Whole blood was harvested at specific time points and was transferred to tubes containing sodium heparin. Blood was spun at 1800xg for 10 min at 4 °C and the separated plasma was transferred to a new tube and frozen at −80 °C prior to LC-MS/MS analysis. In preparation for LC-MS/MS analysis, samples were thawed on ice. Plasma was mixed with acidified ACN at a 1:1 ratio and vortexed for 5 min at room temperatures. For compounds 7, 42, and 53 samples were mixed with ACN acidified with 0.1% formic acid, and for compounds 60 and 61 samples were mixed with ACN acidified with 0.1% trifluoroacetic acid. Samples were then centrifuged at 20,000xg for 10 min and the supernatant was transferred to an autosampler vial for analysis.
Compound aqueous solubility in PBS.
Aqueous solubility was tested using the methodology published by the National Institutes of Health (NIH), National Center for Advancing Translational Sciences (NCATS) in collaboration with Eli Lilly and Company.51 One mg of each compound was dissolved in 200 μL of 1-propanol and allowed to reach thermodynamic equilibrium by incubating for 18 hours at 25 °C. After incubation, undissolved compound and saturated solution were separated by centrifugation at 5,000g for 45 min at room temperature. Compound UV absorption of the fully saturated solution was read along with the background (1-propanol only), dissolved and undissolved compounds were weighed after concentrating and drying on a speedvac equipped with a high vacuum pump. Using the same method as with 1-propanol solutions, 1 mg of each compound was dissolved in 200 μL of 10% DMSO in phosphate buffered solution (PBS, pH = 7.4). The solution was allowed to reach thermodynamic equilibrium and separated by centrifugation. The PBS UV absorption spectra were compared to fully saturated solution in 1-propanol and the solubility of each compound in 10% DMSO in PBS (pH7.4) was determined using linear regression analysis. The solubility in PBS for each compound was conducted in duplicate experiments.
Structure based drug design.
CADD were performed using the Discovery Studio (Dassault Systemés Biovia). The crystal structure of TopoIIα (PDB:1ZXM, 1.87Å) was obtained from the protein data bank and prepared using the CHARM force field in the Discovery Studio. Water molecules were removed, and the residues were corrected for physiological pH. Binding sites were identified as stated in the discovery studio protocol and were defined as whole residues within a 10 Å. Neo and other analogs were prepared using discovery studio and preferred to not generate isomers of the analogs prepared. The receptor and the analogs were minimized based on Discovery Studio algorithm using a root-mean-square gradient tolerance of 3. The CDOCKER and Libdock protocol were used for the docking studies of neo and other analogs in the ATP binding site. The CDOKER approach was set to generate 10 poses of each analog, while the Libdock was set to generate 100 poses of each analog. Top ranked poses were analyzed for binding interactions.
Statistical analysis.
Biological data were subjected to one-way ANOVA or using the Student’s t-test analysis with Prism v7.0 (GraphPad Software Inc, La Jolla, CA, USA). All experiments were conducted using three replicates and experiments were also repeated on separate days two or three times unless otherwise described.
Chemical syntheses.
General Method for Suzuki Coupling.
To 1 molar equivalent of intermediates 5, 8-10, were added 2 molar equivalents of boronic acids, 3 molar equivalents of K2CO3 or Cs2CO3, 0.1 molar equivalent of Pd (II) acetate, 0.1 molar equivalent of DPPE (ligand) and dioxane/H2O (10:1). The mixture was heated at 80–85 °C under N2 for 1 – 5 h. The reaction was monitored by TLC. The reaction was cooled to room temperature, filtered over a pad of celite and washed with ethyl acetate. The filtrate was concentrated in vacuo and the crude product purified by gravity drip chromatography on silica gel, using a gradient of hexanes (0–30%) in ethyl acetate to afford pure product from 60–90% yield.
7-Acetamido-4-(2-nitrophenyl)-5,8-dimethoxyquinoline (1).
To 6.5 g (12.6 mmol) of 7-Acetamido-5,8-dimethoxy-4-(2-nitrophenyl)-2-(trifluoromethane sulfonoxy) quinoline, 11.1 mL (80 mmol) of triethylamine, 600 mg (2.7 mmol) of palladium (II) acetate and 3.0 g (5.4 mmol) of 1,1-bis(diphosphino)ferrocene dissolved in 130 mL of dry dimethylformamide (DMF) at 0 °C was added formic acid (99%) in a dropwise manner, under nitrogen over 10 min. The reaction mixture was slowly warmed to 60 °C, over 2 h and the reaction mixture was poured into 1.5 L of water and made basic with saturated aqueous NaHCO3. The product was extracted with 3 × (400 mL) with dichloromethane. The organic layers were combined, washed with 200 mL of saturated aqueous sodium chloride (NaCl), dried with sodium sulfate (Na2SO4), and concentrated in vacuo to give a crude product that was purified by silica chromatography eluting with 1:1 dichloromethane/ethyl acetate to afford 1 as a yellow crystal, which was further recrystallized in ethyl acetate to obtain 3.47g (75% yield). TLC (ethyl acetate) Rf = 0.20. 1H NMR (400 MHz, CDCl3) δ 8.90 (d, J = 4.4 Hz, 1H), 8.17 (d, J = 9.2 Hz, 1H), 8.13 (s, 1H), 8.09 (s, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.55 (t, J = 8.6 Hz, 1H), 7.29 (d, J = 8.8, 1H), 7.06 (d, J = 4.4 Hz, 1H), 3.14 (s, 3H), 3.44 (s, 3H), 2.28 (s, 3H).
7-Acetamido-4-(2-benzamidophenyl)-5,8-dimethoxyquinoline (2).
120 mg (0.32 mmol) of 1 was dissolved in 13.2 mL of ethanol, to which were added 260 mg of palladium on carbon, and 6.6 mL of cyclohexene. The mixture was refluxed for 2 h to generate the amine product, which was filtered through a pad of celite. The yellow solution was concentrated in vacuo and without purification was dissolved in 3 mL of pyridine under nitrogen, to this solution was added 46 μL (1.2 equivalent) of benzoyl chloride. The reaction was stirred at room temperature for 8 h. The orange solution was concentrated in vacuo to give a crude product which was purified by column chromatography using ethyl acetate/dichloromethane (1:1) as the eluent and silica gel as the adsorbent material to give a tan color solid 126.5 mg (88% yield); TLC (ethyl acetate / dichloromethane (1:1)), Rf = 0.41; Melting Point 204 – 206 °C; 1H NMR (400 MHz, CDCl3) δ 8.93 (d, J = 4.0 Hz, 1H), 8.34 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 4.4 Hz, 2H), 7.45 (t, J = 7.6 1H), 7.39 (s, 1H), 7.36 (t, J = 4.2 Hz, 1H), 7.13 (s, 2H), 7.21 – 7.17 (m, 7 H), 4.14 (s, 3H), 3.52 (s, 3H), 2.29 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 169.2, 165.4, 152.6, 150.5, 144.1, 143.3, 136.6, 135.1, 135.0, 134.0, 133.6, 132.4, 131.9, 129.0, 128.7, 128.5, 126.9, 124.3, 122.9, 121.4, 116.3, 100.8, 62.6, 56.4, 25.6; IR (neat) νmax 3406.5, 3301.8, 2971.2, 1652.0, 1520.7, 1498.5, 1446.3, 1385.1, 1257.4; ESI-HRMS [M + H]+ calculated for C26H24N3O4, 442.1761, found 442.1754.
7-Acetamido-4-(2-benzamidophenyl)-8-hydroxyquinoline (3).
To 160 mg (0.36 mmol) of 2 were added 4 mL of 2,6-lutidine under N2, and lithium iodide (67.8 mg, 0.50 mmol, 1.4 equivalents), The mixture was refluxed for 4 h, and quenched by the addition of ice-cold water. The pH of the solution was adjusted with HCl to pH = 7. The product was extracted with chloroform (50 mL × 3) and dried over sodium sulfate. The product was concentrated and dried in vacuo to give a crude product, which was recrystallized using ethanol to give 92.3 mg, the filtrate was recover and purified by flash chromatography using 10% methanol in ethyl acetate and silica gel to give a bright yellow solid 47.7 mg, with the total yield of 140 mg (90% yield) of the product. TLC (ethyl acetate /acetone (1:1)), Rf = 0.34; Melting Point 218–219 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 9.43(broad s, 1H), 9.33 (s, 1H), 8.79 (d, J = 4.4 Hz, 1H), 7.63 (t, J = 5.8Hz, 2H), 7.45 −7.38 (m, 2H), 7.303 −7.26 (m, 6H), 7.21 (d, J = 4.4Hz, 1H), 3.35 (s, 3H), 2.14 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 165.3, 148.7, 148.3, 144.1, 138.3, 135.1 134.7, 133.7, 133.5, 131.7, 128.8, 128.6, 126.7, 124.3, 123.3, 123.1, 121.6, 115.3, 102.4, 56.5, 25.1; IR (neat) νmax 3305.8, 2948.6, 2832.3, 2361.8, 2325.2, 1670.6, 1622.7, 1582.6, 1525.8, 1448.6; ESI-HRMS [M + Na]+ calculated for C25H21N3NaO4, 450.1424, found 450.1420.
4-Bromo-8-methoxy-7-nitroquinoline (4).
To 164 mg (0.74 mmol) of 8-methoxy-7-nitroquinolin-4(1H)-one (S6) in 2 mL of dimethylformamide was added 78 μL of phosphorus tribromide drop wise at 0 °C under N2. The reaction solution was warmed to room temperature and stirred at that temperature for 1 h. The reaction was quenched by the addition of ice-cold water and the pH of the solution was adjusted by the addition of saturated NaHCO3 to pH = 8. The product was extracted with ethyl acetate 3 × (20 mL). The organic extracts were combined and dried over sodium sulfate. The solvent was concentrated in vacuo to give a crude product, which was chromatographed on silica gel, using ethyl acetate as the eluent to afford a light yellow solid 188 mg (89% yield). TLC (ethyl acetate) Rf = 0.61; Melting Point 142–144 °C; 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 4.0 Hz, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.93 (d, J = 9.2 Hz, 1H), 7.86 (d, J = 4.0 Hz, 1H) 4.35 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 150.5, 150.3, 150.2, 144.0, 142.9, 134.3, 130.9, 127.3, 127.3, 122.3, 122.1, 64.4; IR (neat) νmax 3080.9, 3024.7, 2956.7, 2922.6, 2852.0, 1603.4, 1571.8, 1523.9; ESI-HRMS [M + Na]+ calculated for C10H7N2O3Na 306.9517, found 306.9519.
7-Acetamido-4-bromo-8-methoxyquinoline (5).
To 178 mg (0.63 mmol) of 4-bromo-8-methoxy-7-nitroquinoline were added 210 mg (6 equivalents) of iron powder, 2 mL (water/ethanol (2:8), 67 mg (2 equivalents) of ammonium chloride. The mixture was refluxed for 1.5 h and allowed to cool to room temperature. The mixture was filtered over a pad of celite and washed with ethyl acetate. The filtrate was diluted with water, and the pH was adjusted with NaHCO3 to pH = 8. The product was extracted with ethyl acetate, dried over sodium sulfate and concentrated in vacuo to generate the amine intermediate. Without purification, the amine was dissolved in 2 mL acetic acid and 2 mL acetic anhydride under drying tube for 8 h. The solvent was concentrated in vacuo to give a crude product, which was purified by flash chromatography using silica gel as the solid phase, 35% ethyl acetate in hexane to afford 168 mg (91% yield). TLC (ethyl acetate) Rf = 0.21; Melting Point 132–133 °C; 1H NMR (500 MHz, CDCl3) δ 8.80 (d, J = 9.0 Hz, 1H), 8.64 (d, J = 4.5 Hz, 1H), 8.14 (broad s, 1H), 7.95 (d, J = 9.5 Hz, 1H), 7.63 (d, J = 4.5, 1H), 4.21 (s, 3H), 2.32 (s, 3H), 13C NMR (125.7 MHz, CDCl3) δ 171.3, 151.8, 145.2, 136.9, 134.9, 127.9, 126.6, 125.2, 123.8, 105.0, 65.1, 27.7; IR (neat) νmax 3217.9, 3074.8, 2973.2, 2931.9, 2896.1, 1650.1, 1606.0, 1573.2; ESI-HRMS [M + Na]+ calculated for C12H11BrN2NaO2 318.9876, found 318.9886.
7-Acetamido-4-(5-fluorophenyl-2-methoxy)-8-methoxyquinoline (6).
The compound was prepared from 65 mg (0.22 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 74.7 mg (0.44 mmol) of 5-fluoro-2-methoxyphenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 50.6 mg (68% yield). TLC (ethyl acetate) Rf = 0.21; Melting Point 180–182 °C; 1H NMR (500 MHz, CDCl3) δ 8.91 (d, J = 4.0 Hz, 1H), 8.56 (d, J = 9.5 Hz, 1H), 8.14 (s, 1H), 7.32 (d, J = 9.5 Hz, 1H), 7.21 (d, J = 4.5 Hz, 1H), 7.13 (td, J = 3.5, 9.0 Hz, 1H), 6.98–6.95 (m, 2H), 4.21 (s, 3H), 3.66 (s, 3H), 2.27 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 168.7, 157.8, 155.9, 153.0, 149.6, 144.9, 142.5, 142.2, 131.2, 128.0 (d, 3JC-F = 7.8 Hz), 124.8, 122.0 (d, 3JC-F = 7.7 Hz), 121.0 (d, 1JC-F = 242.3 Hz), 118.0 (d, 2JC-F = 23.6 Hz), 116.0 (d, 2JC-F = 23.3 Hz), 112.2, 62.4, 56.2, 25.41; IR (neat) νmax 3361.4, 3069.8, 3051.0, 3021.8, 2963.2, 2926.62849.4, 1685.7, 1611.8, 1586.4, 1501.8; ESI-HRMS [M + Na]+ calculated for C19H17FN2O3Na 363.1115, found 363.1105.
7-Acetamido-4-(5-fluoro-2-hydroxyphenyl)-8-hydroxyquinoline (7).
The compound was prepared from 33.0 mg (0.10 mmol) of 7-acetamido-4-(5-fluoro-2-methoxyphenyl)-8-methoxyqunoline and 1.9 mL of 1M BBr3 in DCM using the general method of demethylation to afford the product, 26 mg (86% yield). TLC (ethyl acetate) Rf = 0.21; Melting Point 215–217 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.58 (broad s, 1H), 9.56 (broad s, 1H), 8.85 (d, J = 4.4 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.36 (d, J = 4.4 Hz, 1H), 7.17 (dt, J = 3.2, 8.8 Hz, 1H), 7.07 (dd, J = 3.2, 8.8 Hz, 1H), 7.01 – 6.98 (m, 2H), 2.26 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 168.5, 155.1 (d, 1JC-F =234.5 Hz), 151.1, 147.4, 144.4, 143.8, 138.9, 125.6 (d, 3JC-F = 7.54 Hz), 123.9,123.1, 123.0, 121.4, 116.8, (d, 2JC-F = 24.5 Hz) 116.7(d, 1JC-F = 8.54 Hz), 115.9 (d, 2JC-F = 22.6 Hz), 114.0, 113.9, 23.7; IR (neat) νmax 3294.3, 3082.1, 2709.8, 2359.4, 1610.5, 1505.4, 1432.8, 1306.5; ESI-HRMS [M + Na]+ calculated for C17H13FN2NaO3 335.0802, found 335.0797.
4-Bromo-8-methoxy-7-pivalamidoquinoline (8).
To 160 mg (0.57 mmol) of 4-bromo-8-methoxy-7-nitroquinoline were added 220.6 mg (7 equivalents) of iron powder, 60.3 mg (2 equivalents) of ammonium chloride, 8.3 mL water/ethanol (2:8). The mixture was refluxed for 2 h and allowed to cool to room temperature. The mixture was filtered over a pad of celite and washed with ethylacetate. The filtrate was diluted with water, and the pH was adjusted with NaHCO3 to pH = 8. The product was extracted with ethyl acetate, dried over sodium sulfate and concentrated in vacuo to generate the aniline intermediate (141.2 mg). Without purification, to the aniline were added 6.81 mg (0.1 equivalent, 0.05 mmol) of dimethyl amino pyridine (DMAP), 155.7 μL (2 equivalents, 1.11 mmol) of triethylamine, 82 μL (0.67 mmol,1.2 equivalents) of pivaloyl chloride, 5 mL of dry dichloromethane at 0 °C and stirred for 40 min. The resulting dark orange solution became yellow after 5 min of stirring and was allowed to warm to room temperature. The reaction was stirred at room temperature for 18 h under nitrogen, quenched by the addition of water, and the organic layer was extracted using dichloromethane 3 × (30 mL). The organic layers were combined and dried over sodium sulfate, concentrated in vacuo to give crude product, which was chromatographed using silica gel and 15% ethyl acetate in dichloromethane to give 189 mg (98% yield) as a syrupy product. TLC (20% ethyl acetate in dichloromethane) Rf = 0.21; 1H NMR (400 MHz, CDCl3) δ 8.80 (d, J = 9.2 Hz, 1H), 8.75 (d, J = 4.8 Hz, 1H), 8.48 (s, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.41 (d, J = 4.8 Hz, 1H), 4.19 (s, 3H), 1.39 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 177.1, 149.4, 142.6, 142.5, 134.4, 132.7, 125.3, 124.0, 122.8, 121.3, 62.3, 40.3, 27.7; IR (neat) νmax 3426.6, 2961.7, 2870.2, 1683.9, 1608.9, 1497.5, 1454.0, 1407.7; ESI-HRMS [M + H]+ calculated for C15H17BrN2O2 336.0473, found 337.0546.
4-Bromo-8-methoxy-7-(2-(morpholin-4-yl)acetamido)quinoline (9).
To 250 mg (0.883 mmol) of 4-bromo-8-methoxy-7-nitroquinoline were added 346 mg (7 equivalents) of iron powder, 12.5 mL of water:ethanol (2:8) solution, 94.3 mg (2 equivalents) of ammonium chloride. The mixture was refluxed for 2 h and allowed to cool to room temperature. The mixture was filtered over a pad of celite and washed with methanol. The filtrate was diluted with water, and the pH was adjusted with NaHCO3 to pH = 8. The product was extracted with ethyl acetate, dried over sodium sulfate and concentrated in vacuo to generate the aniline intermediate. Without purification, the aniline was dissolved in 5 mL of dry dichloromethane, to which were added 286.7 mg (2 equivalents) of morpholine acetic acid, 344 μL (2.5 equivalents, 249.7 mg) of triethylamine, and 1.047 mL (628.6 mg, 2.0 equivalents) of T3P. The resulting solution was stirred for 24 h under nitrogen. The reaction was quenched by the addition of water and the pH was adjusted with saturated NaHCO3 until pH = 8. The product was extracted using of dichloromethane 3 × (50 mL) and dried over sodium sulfate and concentrated in vacuo to generate the crude product. The compound was purified by flash chromatography using silica gel and 50% hexane in acetone to afford 282 mg (75% yield) of 9 that has the consistency of syrup. TLC (50% hexane in acetone) Rf = 0.21; 1H NMR (400 MHz, CDCl3) δ 10.18 (s, 1H), 8.82 (d, J = 9.2 Hz, 1H), 8.62 (d, J = 4.4 Hz, 1H), 7.93 (d, J = 9.2Hz, 1H), 7.61 (d, J = 4.4 Hz, 1H), 4.20 (s, 3H), 3.83 (t, J = 4.4 Hz, 4H), 3.23 (s, 2H), 2.68 (t, J = 4.2 Hz, 4H); 13C NMR (125 MHz, CDCl3) δ 168.6, 149.4, 142.9, 142.5, 134.4, 131.9, 125.5, 124.1, 122.7, 122.8, 120.9, 67.4, 62.7, 62.5, 53.9; IR (neat) νmax 3303.5, 2924.3, 2854.14, 1696.01, 1606.4, 1567.7, 1495.1, 1449.3, 1409.2; ESI-HRMS [M + H]+ calculated for C16H18BrN3O3 379.0532, found 380.0609.
4-Bromo-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline (10).
To 400 mg (1.41 mmol) of 4-bromo-8-methoxy-7-nitroquinoline were added 551.7 mg (7 equivalents) of iron powder, 20.5 mL water/ethanol (2:8), 150.9 mg (2 equivalents) ammonium chloride. The mixture was refluxed for 2 h and allowed to cool to room temperature. The mixture was filtered over a pad of celite and washed with methanol. The filtrate was diluted with water, and the pH was adjusted with NaHCO3 to pH = 8. The product was extracted with ethyl acetate, dried over sodium sulfate and concentrated in vacuo to generate the amine intermediate. Without purification, the amine was dissolved in 5 mL of dry dichloromethane, to which were added 553 mg (2 molar equivalents) of morpholine propionic acid hydrochloride 490.6 μL triethylamine (2.5 equivalents, 357.3 mg), 1.1 mL (1.08g, 1.2 equivalents) of T3P. The solution was stirred for 24 h under N2. The reaction was quenched by the addition of water and basified with saturated NaHCO3 until pH = 8. The product was isolated using of dichloromethane 3 × (50 mL) and dried over sodium sulfate. The solvents were removed by concentrating the product in vacuo to generate the crude product, which was purified by flash chromatography using silica gel, and 50% hexane in acetone to afford 496 mg (89% yield) of syrupy product. TLC (50% hexane in acetone) Rf = 0.21; 1H NMR (500 MHz, CDCl3) δ 10.96 (s, 1H), 8.79 (d, J = 11.5 Hz, 1H), 8.63 (d, J = 5.5 Hz, 1H), 7.92 (d, J = 12.0 Hz, 1H), 7.60 (d, J = 5.5 Hz, 1H), 4.12 (s, 3H), 3.87 (s, 4H), 2.77 (t, J = 6.8 Hz, 2H), 2.65 (t, J = 7.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 171.37, 149.3, 143.0, 143.0, 134.3, 132.9, 125.5, 122.7, 122.4, 66.7, 62.3, 54.5, 53.4, 33.2; IR (neat) νmax 3508.6, 3313.3, 2966.0, 2932.4, 2854.0, 2820.0, 1676.8, 1608.5, 1517.8, 1494.5; ESI-HRMS [M + H]+ calculated for C17H21BrN3O3 394.0761, found 394.0759.
7-Acetamido-4-(phenyl)-8-methoxyquinoline (11).
The compound was prepared from 40 mg, (0.14 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 34.1 mg (0.28 mmol) of phenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 30.0 mg (74% yield). TLC (ethyl acetate) Rf = 0.22; Melting Point 175–177 °C; 1H NMR (500 MHz, CDCl3) δ 8.9 (broad s, 1H), 8.59 (d, J = 9.0 Hz, 1H), 8.13 (broad s, 1H) 7.67 (d, J = 9.0 Hz, 1H), 7.60 (d, J = 9.2 Hz, 1H), 7.51–7.47 (m, 5H), 7.26 (d, J = 4.5 Hz, 1H), 4.23 (s, 3H), 2.29 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 169.0, 149.9, 149.1, 142.8, 131.6, 129.9, 124.8, 122.2, 120.8, 120.5, 62.7, 25.4; IR (neat) νmax 3378.5, 2924.0, 2851.9, 1696.0, 1610.9, 1499.3, 1453.1; ESI-HRMS [M + Na]+ calculated for C18H16N2O2Na 315.1104, found 315.0993.
7-Acetamido-4-(3-fluorophenyl)-8-methoxyquinoline (12).
The compound was prepared from 257.6 mg (0.872 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 183.2 mg (1.31 mmol) of 3-fluorophenyl boronic acid using the general coupling procedure to give the title compound as an off white solid (256.4 mg, 95% yield). TLC (ethyl acetate) Rf = 0.22; Melting Point 129–131 °C; 1H NMR (500 MHz, CDCl3) δ 8.83 (d, J = 4.0 Hz, 1H), 8.54 (d, J = 9.5 Hz, 1H), 8.27 (broad s, 1H), 7.54 (d, J = 9.5 Hz, 1H), 7.38 (q, J = 7.5 Hz, 1H), 7.16 – 7.07 (m, 4H), 4.14(s, 3H) 2.23 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 168.8, 162.5 (d, 1JC-F = 247.3), 149.3, 147.1, 142.6, 142.4, 139.8, 131.3, 130.0, 125.1, 123.9, 121.2, 120.5, 120.1, 116.3 (d, 2JC-F = 22.0), 115 (d, 2JC-F = 20.7), 62.1, 24.6; IR (neat) νmax 3279.1, 2920.7, 2854.1, 1685.5, 1607.9, 1576.1, 1498.2, 144.1, 1408.5, 1282.1, 1229.9; ESI-HRMS [M + H]+ calculated for C18H16FN2O2 311.1190, found 311.1195.
7-Acetamido-4-(4-fluorophenyl)-8-methoxyquinoline (13).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 47.4 mg (0.33 mmol) of 4-fluorophenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 45.5 mg (88% yield). TLC (50% hexanes in acetone) Rf = 0.32; Melting Point 159–161 °C; 1H NMR (500 MHz, CDCl3) δ 8.91 (d, J = 4.0 Hz, 1H), 8.61 (d, J = 9.0 Hz, 1H), 8.13 (broad s, 1H), 7.62 (d, J = 9.5 Hz, 1H), 7.46 (dd, J = 5.5, 8.5 Hz, 2H), 7.26 – 7.20 (m, 3H), 4.23 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 163.1 (d, 1JC-F = 246.9 Hz), 149.6, 147.8, 142.6, 134.0, 131.4 (d, 3JC-F = 7.8 Hz), 131.3 (d, 3JC-F = 8.2 Hz), 131.3 (d, 2JC-F = 16.0 Hz), 124.5, 121.6, 120.4, 115.8 (d, 2JC-F = 21.5 Hz), 62.4, 25.2; IR (neat) νmax 3356.2,, 2990.6, 2943.2, 2919.1, 2849.6, 1689.2, 1613.8, 1506.8, 1494.4, 1453.3, 1415.7, 1383.4, 1291.1; ESI-HRMS [M + H]+ calculated for C18H16FN2O2 311.1190, found 311.1195.
7-Acetamido-4-((3-trifluoromethyl)phenyl)-8-methoxyquinoline (14).
The compound was prepared from 63 mg (0.21 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 77.2 mg (0.41 mmol) of 3-trifluoromethylphenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 76 mg (99% yield). TLC (ethyl acetate) Rf = 0.15; Melting Point 130–131 °C; 1H NMR (500 MHz, CDCl3) δ 8.95 (d, J = 4.0 Hz, 1H), 8.64 (d, J = 9.0 Hz, 1H), 8.15 (broad s, 1H), 7.76 (d, J = 10.0 Hz, 2H), 7.66 (q, J = 7.6 Hz, 2H), 7.55 (d, J = 9.0 Hz, 1H), 7.27 (d, J = 4.5 Hz, 1H), 4.24 (s, 3H), 230 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 168.8, 148.4 (d, 1JC-F = 303.7 Hz), 142.6 (d, 4JC-F = 18.1 Hz), 138.8, 132.9, 131.6, 131.3 (d, 3JC-F = 32.6 Hz), 129.2, 126.3, 125.5, 125.1, 122.9, 121.0 (d, 2JC-F = 70.7 Hz), 120.6 (d, 1JC-F = 31.4 Hz), 62.5, 25.2; IR (neat) νmax 3328.3, 2981.1, 2939.3,1668.1, 1616.3, 1509.6, 1442.9; ESI-HRMS [M + H]+ calculated for C19H16F3N2O2 361.1158, found 361.1154.
7-Acetamido-4-((4-trifluoromethyl)phenyl)-8-methoxyquinoline (15).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 64.3 mg (0.34 mmol) of 4-trifluoromethylphenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 58 mg (95% yield). TLC (50% hexanes in acetone) Rf = 0.20; Melting Point 159–160 °C 1H NMR (500 MHz, CDCl3) δ 8.95 (d, J = 4.5 Hz, 1H), δ 8.63 (d, J = 9.5 Hz, 1H), 8.16 (broad s, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.56 (d, J = 9.0 Hz, 1H), 7.26 (d, J = 4.5 Hz, 1H), 4.24 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 148.4 (d, 1JC-F = 229.4 Hz), 142.6 (d, 4JC-F = 16.7 Hz), 142.1 (d, 2JC-F = 90.5 Hz), 130.8 (d, 3JC-F = 32.5 Hz), 129.9, 125.7 (q, 5JC-F = 3.7 Hz), 125.5, 124.0, 122.8, 121.3, 120.5 (d, 3JC-F = 35.0 Hz), 62.5, 25.1; IR (neat) νmax 3597.9, 3418.8, 3291.1, 2946.0,1701.5, 1686.0, 1612.3, 1497.6,1456.7,1386.1; ESI-HRMS [M + H]+ calculated for C19H16F3N2O2 361.1158, found 361.1160.
7-Acetamido-4-(3-methoxyphenyl)-8-methoxyquinoline (16).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 51.5 mg (0.34 mmol) of 3-methoxyphenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 59.4 mg (99% yield). TLC (ethyl acetate), Rf = 0.26; Melting Point 185–187 °C; 1H NMR (500 MHz, CDCl3) δ 8.91 (d, J = 4.0 Hz, 1H), 8.60 (d, J = 9.5 Hz, 1H), 8.15 (broad s, 1H), 7.70 (d, J = 9.5 Hz, 1H), 7.43 (t, J = 7.7 Hz, 1H), 7.27 (s, 1H), 7.26 – 7.01 (m, 3H), 4.23 (s, 3H), 3.86 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 159.7, 149.6, 148.7, 142.6, 142.5, 139.3, 131.3, 129.7, 124.5, 122.0, 120.3, 120.2, 115.2, 114.2, 62.4, 55.5, 25.2; IR (neat) νmax 3322.6, 2920.2, 2849.2, 2836.7, 1687.3, 1612.2, 1579.1, 1500.1; ESI-HRMS [M + H]+ calculated for C19H19N2O3 323.1390, found 323.1393.
7-Acetamido-4-(2-acetamidophenyl)-8-methoxyquinoline (17).
To 40 mg (0.14 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline in toluene/ethanol were added 45.24 mg (0.27 mmol) of 2-nitrophenyl boronic acid, 135.5 μL (2.0 M) potassium carbonate and 30.23 mg of tetrakis(triphenylphosphine) palladium (0) ligand. The mixture was heated at 85°C for 16 h. The reaction was allowed to cool to room temperature. The product was filtered over a pad of celite to recover the product. The celite washed with ethylacetate, and the filtrate was concentrated and chromatographed on a silica gel using ethyl acetate to afford 7-acetamido-4-(2-nitrophenyl)-8-methoxyquinoline 56.9 mg. To (40.0 mg (0.12 mmol) of this product dissolved in 4.0 mL of ethanol, were added 94.0 mg of 10% palladium on carbon and 2.5 mL of cyclohexene, and the mixture was refluxed for 2.5 h to generate the amine. The reaction was filtered hot through a pad of celite, concentrated and dried in vacuo to give a red residue. Without purification, the red residue obtained was dissolved in 3 mL of acetic acid and 0.2 mL of acetic anhydride. The resulting orange solution was stirred at room temperature under drying tube for 8 h. The solvents were removed in vacuo to give a crude product, which was purified by flash column chromatography using silica gel and 50% ethyl acetate in acetone as the eluent to give an off white solid 27.7 mg (67% yield). TLC (50% ethyl acetate in acetone) Rf = 0.2; Melting Point 232–234 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.66 (s, 1H), δ 9.08 (s, 1H), 8.91 (d, J = 4.5 Hz, 1H), 8.17 (d, J = 11.5 Hz, 1H), 7.66 (d, J = 10.0 Hz, 1H), 7.49 (t, J = 9.3 Hz, 1H), 7.35– 7.29 (m, 3H), 7.17 (d, J = 11.5 Hz, 1H), 4.09 (s, 3H), 2.17 (s, 3H), 1.67 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.1, 1.68.5, 149.2, 145.2, 142.5, 135.7, 131.5, 131.0, 130.7, 128.8, 126.3, 125.2, 124.2, 122.0, 121.3, 120.4, 61.7, 24.0, 23.0; IR (neat) νmax 3249.8, 3218.0, 2925.5, 2360.6, 232978, 1671.5, 1612.2, 1523.4, 1503.6; ESI-HRMS [M + Na]+ calculated for C20H19N3O3Na 372.1319, found 372.1337.
7-Acetamido-4-(4-acetamidophenyl)-8-methoxyquinoline (18)
The compound was prepared from 100 mg (0.34 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 121.3 mg (0.68 mmol) of 3-acetamidophenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 78.5 mg (66% yield). TLC (ethyl acetate) Rf = 0.22; Melting Point 186–188 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), δ 9.66 (s, 1H), 8.87 (d, J = 4.5 Hz, 1H), 8.27 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 9.0 Hz, 1H), 7.47 (d, J = 6.5 Hz, 2H), 7.34 (d, J = 4.5 Hz, 1H), 4.07 (s, 3H), 2.18 (s, 3H), 2.09 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 169.2, 168.6, 149.3, 147.3, 144.3, 142.7, 139.7, 131.8, 131.1, 129.9, 123.8, 122.2, 120.3, 119.0, 61.7, 23.9; IR (neat) νmax 3387.2, 3303.9, 3233.0, 3187.8, 3103.6, 3041.4, 1665.4, 1603.1, 1499.4; ESI-HRMS [M + Na]+ calculated for C20H19N3O3Na 372.1319, found 372.1323.
7-Acetamido-4-(3-acetamidophenyl)-8-methoxyquinoline (19).
The compound was prepared from 100 mg (0.34 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 121.3 mg (0.68 mmol) of 3-acetamidophenyl boronic acid using the general coupling procedure to give the title compound as an off-white solid 74.3 mg (63% yield). TLC (ethyl acetate) Rf = 0.22; Melting Point 228–230 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.15 (s, 1H), δ 9.66 (s, 1H), 8.88 (d, J = 4.0 Hz, 1H), 8.27 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 9.5 Hz, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.34 (d, J = 4.0 Hz, 1H), 4.07 (s, 3H), 2.18 (s, 3H), 2.09 (s, 3H), 13C NMR (125 MHz, DMSO-d6) δ 169.3, 168.7, 149.4, 147.5, 142.7, 139.5, 137.8, 131.2, 129.2, 123.9, 122.5, 120.2, 119.8, 119.0, 61.8 24.0; IR (neat) νmax 3302.9, 3228.4, 2928.3, 2851.4, 1693.4, 1673.1, 1585.2, 1504.7; ESI-HRMS [M + Na]+ calculated for C20H19N3O3Na 372.1319, found 372.1320.
7-Acetamido-4-(5-fluoro-2-hydroxyphenyl)-8-methoxyquinoline (20).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 52.8 mg (0.34 mmol) of 5-fluoro-2-hydroxyphenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 38 mg (69% yield). TLC (ethyl acetate), Rf = 0.20; Melting Point 150–152 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.65 (broad s, 1H), 9.59 (s, 1H), 8.89 (d, J = 4.5 Hz, 1H), 8.20 (d, J = 9.0 Hz, 1H), 7.33 (d, J = 4.0 Hz, 1H), 7.28 (d, J = 9.0 Hz, 1H), 7.17 (td, J = 3.5, 8.5 Hz, 1H), 7.00 (dd, J = 5.0, 9.0 Hz, 1H), 7.17 (dd, J = 3.0, 9.0 Hz, 1H), 4.09 (s, 3H), 2.18 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 169.1, 155.2 (d, 1JC-F = 235.1Hz), 150.9, 149.2, 144.5, 144.2, 130.9, 125.4, 125.3, 124.3, 122.1, 121.2, 120.8, 116.9 (d, 1JC-F = 23.3Hz), 116.7 (d, 3JC-F = 8.0 Hz), 116.1(d, 2JC-F = 22.5Hz), 79.1, 61.7, 23.9; IR (neat) νmax 3052.8, 2935.7, 2328.8, 1666.9, 1613.5, 1501.4; ESI-HRMS [M + H]+ calculated for C18H16FN2O3 327.1139, found 327.1143.
7-Acetamido-4-(2-methoxy-5-trifluoromethylphenyl)-8-methoxyquinoline (21).
The compound was prepared from 100 mg (0.34 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 149 mg (0.68 mmol) of 2-methoxy-5-trifluoromethylphenyl boronic acid using the general coupling procedure to give the product as an off white solid 130 mg (98% yield). TLC (ethyl acetate) Rf = 0.21; Melting Point 102–104 °C; 1H NMR (400 MHz, CDCl3) δ 8.95 (d, J = 4.4 Hz, 1H), 8.59 (d, J = 9.5 Hz, 1H), 8.16 (broad s, 1H), 7.74 (dd, J = 2.0, 8.8 Hz, 1H), 7.52 (d, J = 2.0 Hz, 1H), 7.28 – 7.24 (m, 2H), 7.12 (d, J = 8.8 Hz, 1H), 4.24 (s, 3H), 3.77 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 159.3, 149.6, 144.6, 142.5, 142.2, 128.3 (q, 4JC-F = 3.5 Hz), 127.9 (q, 2JC-F = 71.4 Hz), 127.6 (q, 4JC-F = 3.7 Hz), 127.3, 125.6, 124.8, 124.3 (q, 1JC-F = 263.3 Hz), 123.5 (q, 3JC-F = 32.8 Hz), 123.2 (q, 3JC-F = 32.8 Hz), 122.9, 122.7, 121.8, 121.2, 120.2, 111.1, 62.5, 25; IR (neat) νmax 3315.2, 2931.7, 2848.8, 1669.8, 1613.5, 1503.1, 1454.4; ESI-HRMS [M + H]+ calculated for C20H18FN3O2 391.1264, found 391.1270.
7-Acetamido-4-(5-chloro-2-methoxyphenyl)-8-methoxyquinoline (22).
The compound was prepared from 100 mg (0.34 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 126.3 mg, (0.68 mmol) of 5-chloro-2-methoxyphenyl boronic acid using the general coupling procedure to give the title compound as an off white solid 120 mg (99% yield). TLC (30% hexane in ethyl acetate) Rf = 0.21; Melting Point 198–200 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.67 (broad s, 1H), 8.90 (d, J = 4.4 Hz, 1H), 8.22 (d, J = 9.2 Hz, 1H), 7.74 (dd, J = 2.4, 8.8 Hz, 1H), 7.35 – 7.33 (m, 2H), 7.25 (d, J = 8.8 Hz, 1H), 7.17 (d, J = 9.2 Hz, 1H), 4.10 (s, 3H), 3.68 (s, 3H), 2.18(s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.1, 155.3, 149.3, 144.2, 143.8, 142.1, 131.1, 130.0, 129.7, 127.9, 124.3, 122.4, 121.1, 120.4, 113.4, 61.7, 55.9, 23.9; IR (neat) νmax 3364.0, 2921.3, 2850.6, 168.5, 1608.5, 1494.1, 1451.1, 1411.2, 1373.0; ESI-HRMS [M + H]+ calculated for C19H17ClN2O3 356.0928, found 357.1005.
8-Methoxy-4-(phenyl)-7-pivalamidoquinoline (23).
The compound was prepared from 70 mg (0.20 mmol) of 4-bromo-8-methoxy-7-pivalamidoquinoline, and 50.6 mg (0.41 mmol) of phenyl boronic acid using the general coupling procedure to give the pure title compound as an off white solid 52 mg (75%). TLC (30% hexane in ethyl acetate) Rf = 0.41; Melting Point 139–140 °C; 1H NMR (500 MHz, CDCl3) δ 8.92 (broad s, 1H), 8.61 (d, J = 9.5 Hz, 1H), 8.48 (s, 1H), 7.67 (d, J = 9.5 Hz, 1H), 7.53 – 747 (m, 5H), 7.26 (s, 1H), 4.20 (s, 3H), 1.39 (s, 9H); 13C NMR (125.7 MHz, CDCl3) δ 177.1, 149.7, 148.9, 142.9, 142.5, 138.0, 131.7, 129.6, 128.7, 128.6, 122.0, 120.2, 62.1, 40.3, 27.7; IR (neat) νmax 3429.1, 3030.7, 2943.4, 2862.0, 1685.3, 1611.3, 1499.7, 1449.4; ESI-HRMS [M + H]+ calculated for C21H23N2O2 335.1754, found 335.1759.
4-(5-fluoro-2-methoxyphenyl)-8-methoxy-7-pivalamidoquinoline (24).
The compound was prepared from 80 mg (0.23 mmol) of 4-bromo-8-methoxy-7-pivalamidoquinoline and 78 mg (0.46 mmol) of 5-fluoro-2-methoxyphenyl boronic acid using the general coupling procedure to give the pure title compound as an off white solid 57 mg (63% yield). TLC (30% hexanes in ethyl acetate), Rf = 0.36; Melting Point 149–151 °C; 1H NMR (500 MHz, CDCl3) δ 8.92 (d, J = 4.5 Hz, 1H), 8.59 (d, J = 9.5 Hz, 1H) 8.45 (broad s, 1H), 7.33 (d, J = 9.5 Hz, 1H), 7.22 (d, J = 4.0 Hz, 1H), 7.14 (td, J = 3.0, 8.5 Hz, 1H), 6.99 – 6.96 (m, 2H), 4.20 (s, 3H), 3.66 (s, 3H), 1.38 (s, 9H); 13C NMR (125.7 MHz, CDCl3) δ 177.1, 156.9 (d, 1JC-F = 240.0 Hz), 153.0, 149.6, 144.9, 142.7, 142.1, 131.6, 128.0 (d, 3JC-F = 7.5 Hz), 124.7, 124.2, 122.1, 121.0, 118.0 (d, 2JC-F = 23.8 Hz), 117.0 (d, 1JC-F = 243.8 Hz), 116.1 (d, 2JC-F = 22.3 Hz), 112.3 (d, 3JC-F = 8.1), 62.2, 56.2, 27.7; IR (neat) νmax 3426.2, 2956.6, 2924.5, 2856.9, 1674.8, 1612.1, 1497.5, 1455.3; ESI-HRMS [M + H]+ calculated for C22H24FN2O3 383.1765, found 383.1769.
4-(5-fluoro-2-methoxyphenyl)-8-methoxy-7-(2-(morpholin-4-yl)acetamido)quinoline (25).
The compound was prepared from 70 mg (0.18 mmol) of 4-bromo-8-methoxy-7-(2-(morpholin-4-yl)acetamido)quinoline and 62.5 mg (0.37 mmol) of 5-fluoro-2-methoxyphenylboronic acid using the general coupling procedure to give the crude product which was purified using 20% dichloromethane in ethyl acetate to give the pure title compound as an off white solid 71.4 mg (91% yield). TLC (20% dichloromethane in ethyl acetate) Rf = 0.24; Melting Point 175–177 °C; 1H NMR (400 MHz, CDCl3) δ 10.15 (broad s, 1H), 8.92 (d, J = 4.4 Hz, 1H), 8.62 (d, J = 9.2 Hz, 1H), 7.34 (d, J = 9.2 Hz, 1H), 7.22 (d, J = 4.4 Hz, 1H), 7.13 (dt, J = 3.2 Hz, 8.6 Hz, 1H), 6.99 – 6.95 (m, 2H), 4.24 (s, 3H), 3.85 (broad t, J = 4.4 Hz, 4H), 3.67 (s, 3H), 3.22 (s, 3H), 2.69 (broad t, J = 4.4 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ 168.5, 156.9 (d, 1JC-F = 232.4 Hz), 153.0, 149.7, 144.9, 142.8, 142.4, 130.9, 128.0 (d, 3JC-F = 7.5 Hz), 124.9, 122.2, 121.1, 119.7, 118.0 (d, 2JC-F = 23.6 Hz), 116.0 (d, 2JC-F = 22.5 Hz), 112.3 (d, 3JC-F = 8.2 Hz), 67.4, 62.8, 62.3, 56.2, 53.9; IR (neat) νmax 3302.4, 2953.7, 2926.2, 2862.2, 2816.6, 1685.4, 1613.3, 1500.4, 1454.7, 1414.9; ESI-HRMS [M + H]+ calculated for C23H24FN3O4 425.1751, found 426.1832.
4-(5-fluoro-2-methoxyphenyl)-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline (26).
The analog was prepared from 58 mg (0.15 mmol) of 4-bromo-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline and 50 mg (0.30 mmol) of 5-fluoro-2-methoxyphenylboronic acid using the general coupling procedure to give the crude product which was purified using 40% hexanes in acetone, to give the pure title compound as an off white solid 60.2 mg (93% yield). TLC (40% hexane in acetone) Rf = 0.17; Melting Point 166–168 °C; 1H NMR (400 MHz, CDCl3) δ 10.90 (broad s, 1H), 8.92 (d, J = 3.2 Hz, 1H), 8.57 (d, J = 9.2 Hz, 1H), 7.32 (d, J = 9.2 Hz, 1H), 7.22 (d, J = 2.8 Hz, 1H), 7.13 (t, J = 8.4 Hz, 1H), 6.98 (d, J = 6.0 Hz, 1H), 4.16 (s, 3H), 3.88 (s, 4H), 3.66 (s, 3H), 2.77 (t, J = 5.2Hz, 2H), 2.64 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 171.2, 156.8 (d, 1JC-F = 238.5 Hz),, 153.0, 149.6, 144.8, 143.3, 142.5, 131.8, 128.1 (d, 3JC-F = 7.6 Hz), 124.8, 121.9, 121.1, 121.0, 118.0 (d, 2JC-F = 23.6 Hz), 116.0 (d, 2JC-F = 22.5 Hz), 112.2 (d, 3JC-F = 8.2 Hz), 66.7, 62.1, 56.2, 54.6, 53.3, 33.2; IR (neat) νmax 3042.0, 2921.5, 2850.5, 2815.2, 1678.0, 1610.0, 1582.8, 1500.3; ESI-HRMS [M + H]+ calculated for C24H27FN3O4 440.1980 found 440.1985.
4-(2-Acetamidophenyl)-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline (27).
The analog was prepared from 102.6 mg (0.26 mmol) of 4-bromo-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline and 86.9 mg (0.52 mmol) of 2-nitrophenylboronic acid using the general coupling procedure to give 8-methoxy-7-(3-(morpholin-4-yl)propanamido)-4-(2-nitrophenyl) quinoline as a yellow solid 104.6 mg (92 %) TLC (40% hexanes in acetone) Rf = 0.19; Melting Point 178–180 °C; NMR (400 MHz, CDCl3) δ 10.99 (broad s, 1H),(8.77 (d, J = 4.4 Hz, 1H), 8.67 (d, J = 9.6 Hz, 1H), 8.36 – 8.34 (m, 2H), 7.82 (d, J = 7.6 Hz, 1H), 7.71 (t, J = 8.2 Hz, 1H), 7.50 (d, J = 9.2 Hz, 1H), 7.28 (d, J = 4.4 Hz, 1H), 4.184 (s, 3H), 3.89 (t, J = 4.6, 4H), 2.79 – 2.77 (m, 2H), 2.67 – 2.64 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 171.2, 149.4, 148.8, 145.0, 143.6, 142.4, 133.2 132.9, 132.3, 129.8, 124.8, 124.2, 121.9, 120.5, 119.4, 66.72, 62.2, 54.5, 53.3, 33.1; IR (neat) νmax 2923, 2852.5, 2817.7, 2362.3, 2334.5, 1677.9, 1609.9, 1578.3, 1504.9, 1454.1; ESI-HRMS [M + H]+ calculated for C23H25N4O5 437.1819, found 437.1825. To 38.4 mg (0.09 mmol) of this product dissolved in 3.52 mL of ethanol, were added 69 mg of 10% palladium on carbon and 1.76 mL of cyclohexene and refluxed for 2.5 h to generate the amine. The solution was filtered through a pad of celite to recover the product. The filtrate was concentrated and dried in vacuo to give a yellow residue. Without purification, the yellow residue obtained was dissolved in 1.5 mL of acetic acid and 1.5 mL of acetic anhydride. The resulting orange solution was stirred at room temperature under drying tube for 8 h. The solvents were removed in vacuo to give a crude product. The crude product was purified by flash column chromatography using silica gel and 10 % chloroform in acetone as the eluent to give an off white solid 20 mg (20% yield) TLC (10% methanol in acetone) Rf = 0.32; 1H NMR (400 MHz, CDCl3) δ 10.9 (broad s, 1H), 8.89 (d, J = 3.6 Hz, 1H), 8.60 (d, J = 9.2 Hz, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.32 – 725 (m, 4H), 7.02 (s, 1H), 4.15 (s, 3H), 3.86 (s, 4H), 2.78 (d, J = 5.2Hz, 2H) 1.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.3, 168.5, 149.7, 144.9, 143.3, 142.7, 135.3, 132.5, 130.4, 129.7, 128.2, 124.7, 124.5, 122.9, 122.0, 121.4, 121.2, 66.7, 62.1, 54.6, 53.4, 33.1, 29.8; IR (neat) νmax 2920.8, 2852.5, 2358.6 1678.6, 1611.7, 1505.8, 1452.5, 1413.4; ESI-HRMS [M + H]+ calculated for C25H29N4O4 449.2183, found 449.2188.
General method for demethylation.
To a solution of 8-methoxyquinoline derivative in dry DCM (0.08 M) on ice, was added 1 M BBr3 in DCM (6–10 equivalents) and the reaction was stirred at 0 °C for 15 min. The orange solution was allowed to warm to room temperature and stirred for 8 h. The reaction was quenched by dropwise addition of ice-cold water and the neutralized with saturated NaHCO3 solution. The solution was extracted with chloroform 3 × (20 mL) and concentrated in vacuo. The crude product was purified by gravity drip reverse phase C18 chromatography by first using a step gradient of methanol (0 to 70%) in H2O. The fractions were pooled and concentrated to afford pure product.
7-Acetamido-4-(phenyl)-8-hydroxyquinoline (28).
The compound was prepared from 250 mg (0.85 mmol) of 7-acetamido-4-(phenyl)-8-methoxyquinoline and 8.56 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 237 mg (99% yield).TLC (ethyl acetate) Rf = 0.32; Melting Point 177–179 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.58 (broad s, 1H), 8.87 (d, J = 4.0 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.95 – 7.53 (m, 5H), 7.40 (d, J = 4.0 Hz, 1H), 7.26 (d, J = 9.0 Hz, 1H), 2.15 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 168.7, 152.9, 147.9, 147.5, 142.9, 138.8, 137.5, 129.3, 128.7, 128.5, 123.5, 123.2, 120.7, 114.2, 23.6; IR (neat) νmax 3427.9, 3278.6, 1702.9, 1624.8, 1503.0, 1453.9, 1403.1, 1296.1.
7-Acetamido-4-(3-fluorophenyl)-8-hydroxyquinoline (29).
The compound was prepared from 244.4 mg (0.79 mmol) of 7-acetamido-4-(3-fluorophenyl)-8-methoxyquinoline and 7.9 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 230 mg (98% yield). TLC (ethyl acetate) Rf = 0.25; Melting Point 151–153 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.21 (broad s, 1H), 9.59 (broad s, 1H), 8.88 (d, J = 3.1 Hz, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.61 (q, J = 7.5 Hz, H), 7.44 – 7.36 (m, 4H) 7.24 (d, J = 9.0 Hz, 1H), 2.15 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 168.8, 162.1 (d, 1JC-F = 244.6 Hz), 147.9, 146.0, 142.8, 139.8 (d, 3JC-F = 7.5 Hz), 138.8, 130.7 (d, 2JC-F = 8.4 Hz), 125.6, 123.3, 122.9, 120.8, 116.2 (d, 2JC-F = 22.1 Hz), 115.4 (d, 2JC-F = 20.8 Hz), 114.0, 23.6; IR (neat) νmax, 3253.7, 3026.6, 1657.2, 1537.4, 1506.5; ESI-HRMS [M + H]+ calculated for C17H14FN2O2 297.1034, found 297.1043.
7-Acetamido-4-(4-fluorophenyl)-8-hydroxyquinoline (30).
The compound was prepared from 29.0 mg (0.09 mmol) of 7-acetamido-4-(4-fluorophenyl)-8-methoxyquinoline and 0.93 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 27.0 mg (98 % yield). TLC (ethyl acetate) Rf = 0.25; Melting Point 161–163 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.59 (broad s, 1H), 8.87 (d, J = 4.0 Hz, 1H), 8.10 (d, J = 9.0 Hz, 1H), 7.59 (dd, J = 5.5, 8.0 Hz, 2H), 7.42 – 7.38 (m, 3H) 7.23 (d, J = 9.0 Hz, 1H), 2.15 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 168.9, 162.3 (d, 1JC-F = 245.7 Hz), 148.0, 146.4, 142.9, 138.9, 133.8, 131.5 (d, 1JC-F = 8.17 Hz), 123.7, 123.3, 120.9, 115.7 (d, 1JC-F = 21.7 Hz), 114.2, 23.7; IR (neat) νmax, 3315.4, 3071.9, 2918.1, 2851.2, 1683.1, 1658,5, 1622.9, 1536.8, 1461.8; ESI-HRMS [M + H]+ calculated for C17H14FN2O2 297.1034, found 297.1040.
7-Acetamido-4-((3-trifluoromethyl)phenyl)-8-hydroxyquinoline (31).
The compound was prepared from 50.0 mg (0.14 mmol) of 7-acetamido-4-(3-trifluoromethylphenyl)-8-methoxyquinoline and 1.4 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 40 mg (83% yield). TLC (ethyl acetate) Rf = 0.22; Melting Point 133–135°C; 1H NMR (400 MHz, DMSO-d6) δ 9.77 (broad s, 1H), 8.94 (d, J = 4.4 Hz, 1H), 8.09 (d, J = 9.2 Hz, 1H), 7.94 – 7.08 (m, 4H), 7.78 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 4.0 Hz, 1H), 7.22 (d, J = 9.2 Hz, 1H), 2.17 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.2, 157.8, 147.4, 147.0, 142.2, 138.2, 137.7, 137.5, 133.5, 132.9, 129.9, 129.7, 129.4, 125.7, 125.5, 125.4, 124.6, 124.2, 124.1, 123.3, 122.8, 121.2, 114.3, 23.6; IR (neat) νmax 2921.0, 2851.0, 1659.3, 1626.4, 1531.6, 1508.2, 1405.1; ESI-HRMS [M + H] + calculated for C18H14F3N2O2 347.1002 found 347.1010.
7-Acetamido-4-((4-trifluoromethyl)phenyl)-8-hydroxyquinoline (32).
The compound was prepared from 26.0 mg (0.07 mmol) of 7-acetamido-4-(4-trifluoromethylphenyl)-8-methoxyquinoline and 0.7 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 23.0 mg (92% yield). TLC (ethyl acetate) Rf = 0.26; Melting Point 178–180 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.68 (broad s, 1H), 8.93 (d, J = 4.0 Hz, 1H), 8.09 (d, J = 9.0 Hz, 1H), 7.94 (d, J = 8.0 Hz, 2H), 7.78 (d, J = 7.5 Hz, 2H), 7.49 (d, J = 4.0 Hz, 1H) 7.22 (d, J = 9.5 Hz, 1H), 2.16 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.5, 147.0, 141.4 (d, 2JC-F = 69.1 Hz), 130.3, 130.2 (d, 4JC-F = 9.2 Hz), 129.9, 129.5 (d, 4JC-F = 10.1 Hz), 129.3 (d, 3JC-F = 36.9 Hz), 125.6 (q, 5JC-F = 3.6 Hz), 124.7, 124.1 (d, 1JC-F = 270 Hz), 124.6, 124.5, 124.4, 124.9, 124.2, 123.3, 122.8, 121.0, 114.8, 23.5; IR (neat) νmax 3743.3, 3405.2, 2918.7, 2366.4, 2320.9, 1694.9, 1620.8, 1519.5, 1499.5, 1411.8; ESI-HRMS [M + H]+ calculated for C18H14F3N2O2 347.1002 found, 347.1005.
7-Acetamido-4-(3-hydroxyphenyl)-8-hydroxyquinoline (33).
The compound was prepared from 29.0 mg (0.09 mmol) of 7-acetamido-4-(3-methoxyphenyl)-8-methoxyquinoline and 1.8 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a syrupy product, 21.3 mg (80% yield). TLC (ethyl acetate) Rf = 0.13; 1H NMR (500 MHz, DMSO-d6) δ 9.57 (broad s, 1H), 8.30 (d, J = 4.5 Hz, 1H), 8.08 (d, J = 9.0 Hz, 1H), 7.35 (m, 2H), 7.26 (d, J = 9.0 Hz, 1H), 7.35 (m, 3H), 2.14 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.7, 157.6, 147.8, 147.6, 143.3, 139.0, 138.8, 129.8, 123.3, 123.2, 120.4, 119.9, 116.1, 115.5, 113.9, 79.1, 23.7; IR (neat) νmax 3202.2, 2919.1, 2850.2, 1659.0, 1625.7, 1582.1, 1534.0, 1505.81457.91404.0; ESI-HRMS [M + H]+ calculated for C17H15FN2O3 295.1077, found 295.1082.
7-Acetamido-4-(2-acetamidophenyl)-8-hydroxyquinoline (34).
The compound was prepared from 21.5 mg (0.06 mmol) of 7-acetamido-4-(2-acetamidophenyl)-8-methoxyquinoline and 0.6 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 19.2 mg (93% yield). TLC (50% acetone in ethyl acetate) Rf = 0.24; Melting Point 214–216 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.55 (broad s, 1H), 9.03 (broad s, 1H), 8.87 (d, J = 4.0 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 7.5 Hz, 1H), 7.49 (t, J = 6.5 Hz, 1 H), 7.35 – 7.32 (m, 3H), 6.89 (d, J = 9.0 Hz, 1H), 2.14 (s, 3H), 1.67 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 168.7, 168.4, 147.8, 145.0, 142.7, 138.8, 135.7, 131.5, 130.6, 128.7, 126.1, 1250.1, 123.7, 123.1, 122.9, 121.5, 114.4, 23.6, 22.9; IR (neat) νmax 32228.3, 2925.6, 2853.5, 2359.3, 2330.6, 1658.8, 15006.3, 1442.4, 1366.1, 1294.9, 1025.8; ESI-HRMS [M + Na]+ calculated for C19H17N3NaO3 358.1162 found, 358.1156.
7-Acetamido-4-(4-acetamidophenyl)-8-hydroxyquinoline (35).
The compound was prepared from 26.4 mg (0.07 mmol) of 7-acetamido-4-(4-acetamidophenyl)-8-methoxyquinoline and 0.76 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 15.0 mg (60% yield). TLC (50% acetone in ethyl acetate); Rf = 0.22; 1H NMR (500 MHz, DMSO-d6) δ 10.1 (broad s, 1H), 9.56 (broad s, 1H), 8.83 (d, J = 4.5 Hz, 1H), 8.09 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 8.5 Hz, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 4.5 Hz, 1H), 7.29 (d, J = 9.0 Hz, 1H), 2.14 (s, 3H), 2.10 (s, 3H); 13C NMR (125.7 MHz, DMSO-d6) δ 168.7, 168.5, 147.9, 147.1, 142.8, 139.7, 138.9, 131.9, 129.8, 123.3, 123.2, 123.1, 120.6, 118.9, 114.2, 24.1, 23.6; IR (neat) νmax 3429.8, 3308.0, 2924.6, 1629.5, 1596.3, 1520.1, 1456.8; ESI-HRMS [M + H]+ calculated for C19H18N3O3 336.1343, found 336.1347.
7-Acetamido-4-(3-acetamidophenyl)-8-hydroxyquinoline (36).
The compound was prepared from 20.0 mg (0.05 mmol) of 7-acetamido-4-(3-acetamidophenyl)-8-methoxyquinoline 0.5 mL of 1.0 M BBr3 in DCM to afford a solid product 10 mg (52% yield). TLC (20% methanol in ethyl acetate) Rf = 0.21; Melting Point 156–158 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.32 (broad s, 1H), 9.62 (broad s, 1H), 8.07 (d, J = 9.2 Hz, 1H), 7.83 (s, 1H), 7.74 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 7.4 Hz, 1H), 7.37 (d, J = 3.6 Hz, 1H), 7.25 (d, J = 9.2 Hz 1H), 7.19 (d, J = 7.2 Hz, 1H), 2.15 (s, 3H), 2.07 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.7, 168.6, 147.9, 147.4, 142.3, 139.6, 139.1, 137.9, 134.1, 129.1, 123.8, 123.4, 123.3, 120.5, 119.7, 113.8, 24.0, 23.6; IR (neat) νmax 3233.4, 3057.9, 2920.7, 2850.9, 1665.0, 1543.6, 1428.4; ESI-HRMS [M + Na]+ calculated for C19H17N3NaO3 358.1162, found 358.1149.
7-Acetamido-4-(2-hydroxy-5-trifluoromethylphenyl)-8-hydroxyquinoline (37).
The compound was prepared from 51 mg (0.13 mmol) of 7-acetamido-4-(2-methoxy-5-trifluoromethyl phenyl)-8-methoxyquinoline and 3.0 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a syrupy product, 20 mg (42% yield). TLC (ethyl acetate) Rf = 0.19; 1H NMR (400 MHz, DMSO-d6) δ 10.59 (broad s, 1H), 9.67 (broad s, 1H), 8.89 (d, J = 4.4 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.71 (dd, J = 2.4, 8.8 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8.8 Hz, 1H), 2.08 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.3, 158.0, 147.1, 127.8 (d, 4JC-F = 3.8 Hz), 127.6 (d, 3JC-F = 37.5 Hz), 127.4 (d, 4JC-F = 3.1 Hz), 124.5 (d, 1JC-F = 269.4Hz), 124.3, (d, 2JC-F = 78.1 Hz), 123.3 (d, 1JC-F = 286.6 Hz), 119.7 (d, 3JC-F = 32 Hz), 116.4, 115.3, 23.5; IR (neat) νmax. 3049.0, 2928.9, 2250.0, 2123.6, 1673.3, 1618.8, 1509.7, 1458.3, 1409.1; ESI-HRMS [M + H]+ calculated for C18H14F3N2O3 363.0951, found 363.0946.
7-Acetamido-4-(5-chloro-2-hydroxyphenyl)-8-hydroxyquinoline (38).
The compound was prepared from 53 mg (0.15 mmol) of 7-acetamido-4-(5-chloro-2-methoxyphenyl)-8-methoxyquinoline and 3.4 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 43.6 mg (89% yield). TLC (ethyl acetate) Rf = 0.11; Melting Point 234–236 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.90 (broad s, 1H), 9.56 (broad s, 1H), 8.86 (d, J = 4.4 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.39 – 7.36 (m, 2H), 7.24 (d, J = 2.8 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H) 6.97 (d, J = 9.2 Hz, 1H), 2.14 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.7, 153.6, 147.8, 144.1, 142.8, 138.6, 130.0, 129.4, 126.3, 123.8, 123.4, 123.0, 122.5, 121.6, 117.4, 114.9, 23.6; IR (neat) νmax 3343.6, 3123.0, 22920.3, 2851.4, 1675.5, 1625.7, 1500.7 1468.2, 1410.3; ESI-HRMS [M + H]+ calculated for C17H14ClN2O3 329.0687, found 329.0687.
8-Hydroxy-4-(phenyl)-7-pivalamidoquinoline (39).
The compound was prepared from 25.0 mg (0.07 mmol) of 4-(phenyl)-7-pivalamido-8-methoxyquinoline and 0.75 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 22.9 mg (96% yield). TLC (ethyl acetate) Rf = 0.52; Melting Point 134–136 °C; 1H NMR (500 MHz, CD3OD) δ 8.80 (broad s, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.53 – 7.51 (m, 5H), 7.35 (d, J = 9.0 Hz, 2H), 2.26 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 179.9, 150.5, 129.0, 144.4, 139.1, 130.5, 129.7, 125.7, 124.2, 122.2, 116.5, 40.4, 27.9; IR (neat) νmax 3428.8, 3053.2, 2956.6, 2919.6, 2866.1, 1673.0, 1625.2, 1502.8, 1446.5, 1402.2; ESI-HRMS [M + H]+ calculated for C20H21N2O2 321.1598, found 321.1602.
4-(5-fluoro-2-hydroxyphenyl)-8-hydroxy-7-pivalamido-quinoline (40).
The compound was prepared from 47.0 mg (0.12 mmol) of 4-(5-fluoro-2-hydroxyphenyl)-7-pivalamido-8-methoxyquinoline and 2.40 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 43.4 mg (99% yield). TLC (ethyl acetate) Rf = 0.52; Melting Point 161–163°C; 1H NMR (500 MHz, CD3OD) δ 8.82 (broad s, 1H), 7.91 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 9.2 Hz, 1H), 7.36 (s, 1H), 7.14 (d, J = 7.0 Hz, 1H), 6.97 (s, 2H), 1.37 (s, 9H); 13C NMR (125.7 MHz, CD3OD) δ 180.0, 157.5 (d, 1JC-F = 237.4 Hz), 152.0, 149.2, 146.6, 140.4, 127.1, 126.3, 123.9 (d, 2JC-F = 24.6 Hz), 123.1, 118.2, 117.9, 117.1 (d, 2JC-F = 22.3 Hz), 40.7, 27.9; IR (neat) νmax 3393.1, 3196.5, 2957.3, 2919.6, 2854.6, 1642.3, 1579.2, 1452.9; ESI-HRMS [M + H]+ calculated for C20H20N2O3 355.1452, found 355.1456.
4-(5-fluoro-2-hydroxyphenyl)-8-hydroxy-7-(2-(morpholin-4-yl)acetamido)quinoline (41).
The compound was prepared from 33.5 mg (0.08 mmol) of compound 4-(5-fluoro-2-methoxyphenyl)-8-methoxy-7-(2-(morpholin-4-yl)acetamido)quinoline and 1.9 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 14 mg (45% yield). TLC (50% hexane in acetone) Rf = 0.21; Melting Point 235–237 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.98 (broad s, 1H), 9.57 (broad s, 1H), 9.02 (d, J = 4.4 Hz, 1H), 7.91 (d, J = 9.2 Hz, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 4.4 Hz, 1H), 7.45 (broad s, 1H), 7.33 (broad s, 1H), 7.23 – 7.10 (m, 3H), 3.70 (s, 4H), 3.38 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 155.2 (d, 1JC-F = 233.5 Hz), 151.1, 150.5, 145.6 (d, 2JC-F = 26.6 Hz), 140.3, 135.4, 128.8, 125.2 (d, 3JC-F = 7.7 Hz), 124.9, 123.4, 122.4, 119.0, 117.0 (d, 3JC-F = 5.9 Hz), 116.8 (d, 3JC-F = 9.4 Hz), 116.3 (d, 2JC-F = 22.2 Hz), 99.5, 65.3, 52.4, 51.6, 28.9; IR (neat) νmax 3367.1, 3241.6, 3079.7, 2936.5, 2848.3, 1635.3, 1559.0, 1484.7, 1452.3, 1401.3; ESI-HRMS [M + H]+ calculated for C21H21FN3O4 398.1511, found 398.1523.
4-(5-fluoro-2-hydroxyphenyl)-8-hydroxy-7-(3-(morpholin-4-yl)propanamido)quinoline (42).
The compound was prepared from 400 mg (0.91 mmol) of 4-(5-fluoro-2-methoxyphenyl)-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline and 18.2 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford tan solid 314 mg (82% yield). TLC (10% methanol in ethyl acetate) Rf = 0.18; Melting Point 247–248 °C; 1H NMR (500 MHz, CD3OD) δ 10.57 (broad s, 1H), 9.57 (broad s, 1H), 8.85 (d, J = 4.0 Hz, 1H), 8.22 (d, J = 9.0 Hz, 1H), 7.34 (d, J = 4.5 Hz, 1H), 7.17 (dt, J = 3.0, 7.5 Hz, 1H), 7.06 (dd, J = 3.0, 9.0 Hz, 1H), 7.02 – 6.99 (m, 2H), 3.69 (t, J = 3.75 Hz, 4H), 2.66–2.58 (m, 4H), 2.50 (s, 6H); 13C NMR (100 MHz, CD3OD) δ 168.1, 155.2 (d, 1JC-F = 233 Hz), 150.9, 147.8, 144.5, 143.1, 138.4, 125.3 (d, 3JC-F = 7.8 Hz), 124.1, 123.2, 122.4, 121.8, 116.9 (d, 2JC-F = 32.5 Hz), 116.8, 116.1 (d, 2JC-F = 22.6 Hz), 115.0, 63.3, 52.2, 51.3, 29.9; IR (neat) νmax 3329.5, 3156.0, 2678.0, 2558.9, 2451.1, 2359.7, 1682.4, 1628.5, 1509.2, 1466.4, 1414.5; ESI-HRMS [M + H]+ calculated for C20H17N2O3 333.1234 found 333.1235; ESI-HRMS [M + H]+ calculated for C22H23FN3O4 412.1667 found 412.1676.
4-(2-Acetamidophenyl)-8-hydroxy-7-(3-(morpholin-4-yl)propanamido)quinoline (43).
The compound was prepared from 21.8 mg (0.05 mmol) of 4-(2-acetamido)-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline and 0.5 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 17.1 mg (81% yield). TLC (10% acetone in ethyl acetate) Rf = 0.15; Melting Point 260–262 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.60 (broad s, 1H), 9.03 (broad s, 1H), 8.87 (d, J = 4.0 Hz, 1H), 8.26 (d, J = 9.0 Hz, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 7.35 – 7.30 (m, 3H), 6.89 (d, J = 9.5 Hz, 1H), 4.10 (s, 4H), 2.64 (d, J = 5.0 Hz, 2H), 2.58 (d, (d, J = 5.0 Hz, 2H), 2.50 (s, 6H), 1.66 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.6, 168.4, 144.7, 141.3, 135.7, 130.5, 128.5, 126.0, 125.0, 124.2, 123.9, 120.8, 120.7, 66.0, 54.2, 52.9, 48.6, 33.4, 23.0; IR (neat) νmax, 3295.6, 2948.0, 2923.7, 2855.4, 2821.6, 1629.1, 1581.6, 1513.0, 1445.7; ESI-HRMS [M + H]+ calculated for C20H17N2O3 333.1234, found 333.1235.
7-Acetamido-4-(2-benzofuranyl)-8-methoxyquinoline (44).
The compound was prepared from 50 mg (0.19 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 61.5 mg (0.38 mmol) of 2-benzofuranylboronic acid using the general coupling procedure to give the title compound as an off white solid 56.2 mg (85% yield). TLC (30% hexanes in ethyl acetate), Rf = 0.25; Melting Point 139–141 °C; 1H NMR (500 MHz, CDCl3) δ 8.96 (d, J = 6.0 Hz, 1H), 8.76 (d, J = 9.0 Hz, 1H), 8.31 (d, J = 10 Hz, 1H), 8.16 (broad s, 1H), 7.72 – 7.69 (m, 2H), 7.62 (d, J = 8.5 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.33 – 7.30 (m, 2H), 4.23 (s, 3H), 2.32 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 155.3, 152.6, 149.6, 143.0, 142.7, 136.1, 131.6, 128.6, 125.7, 123.5, 122.5, 121.7, 121.4, 120.9, 118.8, 111.7, 108.9, 62.5, 25.2; IR (neat) νmax 3403.2, 3296.6, 2925.5, 2852.5, 1673.7, 1610.2, 1498.0, 1451.8; ESI-HRMS [M + H]+ calculated for C20H18N2O3 333.1234, found 333.1235.
7-Acetamido-4-(benzo[b]thiophen-2-yl)-8-methoxyquinoline (45).
The compound was prepared from 150 mg (0.51 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 181 mg (1.01 mmol) of benzo[b]thiophen-2-yl boronic acid using the general coupling procedure to give the title compound as an off white solid 166.1 mg (94% yield). TLC (30% hexanes in ethyl acetate), Rf = 0.10; Melting Point 201–203 °C; 1H NMR (400 MHz, CDCl3) δ 8.93 (d, J = 4.4 Hz, 1H), 8.69 (d, J = 9.2 Hz, 1H), 8.16 (broad s, 1H), 8.10 (d, J = 9.2 Hz, 1H) 7.92 −7.87 (m, 2 H), 7.57 (s, 1H), 7.46 – 7.40 (m, 3H), 4.23 (s, 3H), 2.32 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 169.2, 149.3, 144.2, 142.7, 139.9, 139.6, 131.7, 126.0, 124.4, 123.1, 122.8, 122.4, 121.0, 120.0,, 108.9, 61.8, 24.0; IR (neat) νmax 3369.7, 2920.6, 2851.4, 2851.4, 1698.3, 1610.9, 1491.7, 1434.4; ESI-HRMS [M + H]+ calculated for C20H18N2O2S 349.1005, found 349.101.
7-Acetamido-4-(2-furanyl)-8-methoxyquinoline (46).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 40.3 mg (0.34 mmol) of 2-furanylboronic acid using the general coupling procedure to give the title compound as an off white solid 47.2 mg (99% yield). TLC (ethyl acetate), Rf = 0.28; Melting Point 111–113 °C 1H NMR (500 MHz, CDCl3) δ 8.90 (d, J = 4.5 Hz, 1H), 8.71 (d, J = 9.0 Hz, 1H), 8.25 (d, J = 9.5 Hz, 1H), 8.14 (broad s, 1H) 7.69 (s, 1H), 7.56 (d, J = 4.5 Hz, 1H), 6.98 (d, J = 3.0 Hz, 1H), 6.64 (t, J = 2.0 Hz, 1H), 4.19 (s, 3H), 2.31 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 171.4, 153.5, 152.1, 146.9, 146.7, 145.4, 138.8, 134.1, 124.6, 124.1, 123.1, 120.2, 120.1, 115.2, 114.7, 65.0, 27.7; IR (neat) νmax 3331.2, 3217.7, 3074.4, 2923.2, 2852.8, 2361.4, 1694.2, 1650.8, 1607.6, 1492.1; ESI-HRMS [M + H]+ calculated for C16H15N2O3 283.1077, found 283.1034.
7-Acetamido-4-(3-furanyl)-8-methoxyquinoline (47).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 40.3 mg (0.34 mmol) of 3-furanylboronic acid using the general coupling procedure to give the title compound as an off white solid (52.9. mg, quantitative yield). TLC 50% hexane in acetone), Rf = 0.21; Melting Point 130–132 °C 1H NMR (400 MHz, CDCl3) δ 8.85 (d, J = 4.4 Hz, 1H), 8.64 (d, J = 9.2 Hz, 1H), 8.15 (broad s, 1H), 7.89 (d, J = 9.2 Hz, 1H), 7.74 (s, 1H), 7.59(t, J = 1.6 Hz, 1H), 7.27 (d, J = 4.4 Hz, 1H), 6.71 (d, J = 1.2 Hz, 1H), 4.19 (s, 3H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.8, 149.6, 143.7, 142.7, 142.5, 141.4, 139.8, 131.4, 124.3, 122.8, 121.3, 120.4, 119.9, 116.6, 62.4, 25.1; IR (neat) νmax 3276.2, 3125.6, 2926.7, 2855.6, 1754.7, 1654.3, 1609.6, 1500.0, 1454.7; ESI-HRMS [M + H]+ calculated for C16H15N2O3 283.1077, found 283.1083.
7-Acetamido-4-(thiophen-2-yl)-8-methoxyquinoline (48).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 43.4 mg (0.34 mmol) of 2-thiopheneboronic acid using the general coupling procedure to give the title compound as an off-white solid 59.0 mg (quantitative yield). TLC (50% hexane in acetone Rf = 0.32; Melting Point 128–130 °C; 1H NMR (400 MHz, CDCl3) δ 8.85 (d, J = 4.4 Hz, 1H), 8.64 (d, J = 8.8 Hz, 1H), 8.19 (broad s, 1H), 8.01 (d, J = 9.6 Hz, 1H), 8.30 (d, J = 9.2 Hz, 1H), 7.48 (d, J = 4.8 Hz, 1H), 7.34 (d, J = 4.0 Hz, 1H), 7.18 (t, J = 4.4 Hz, 1H), 4.18 (s, 3H), 2.28 (s, 3H); IR (neat) νmax 3339.2, 3060.0, 2920.6, 2850.5, 1692.5, 1668.9, 1613.8, 1579.6, 1500.7, 1455.9; ESI-HRMS [M + H]+ calculated for C16H15N2O2S 299.0849, found 299.0855.
7-Acetamido-4-(thiophen-3-yl)-8-methoxyquinoline (49).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 43.4 mg (0.34 mmol) of 3-thiopheneboronic acid using the general coupling procedure to give the title compound as an off white solid 48.8 mg (97% yield). TLC (ethyl acetate); Melting Point 130–132 °C; 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 4.4 Hz, 1H), 8.62 (d, J = 9.2 Hz, 1H) 8.13 (broad s, 1H), 7.83 (d, J = 9.2 Hz, 1H), 7.50 – 7.49 (m, 2H), 7.32 – 7.30 (m, 2H), 4.21 (s, 3H), 2.3 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.5, 149.4, 143.25, 142.4, 138.2, 131.1, 128.6, 126.1, 124.9, 124.3,121.4, 119.9, 62.1, 24.9; IR (neat) νmax 3349.6, 3310.6, 3098.7, 3068.6, 2991.3, 2967.0, 2935.9, 2850.8, 1692.7, 1667.5, 1612.8 1514.0, 1501.4; ESI-HRMS [M + H]+ calculated for C16H15N2O2S 299.0849, found 299.0855.
7-Acetamido-4-(5-pyrimidinyl)-8-methoxyquinoline (50).
The compound was prepared from 50 mg (0.17 mmol) of 7-acetamido-4-bromo-8-methoxyquinoline and 42 mg (0.34 mmol) of pyrimidine-5-boronic acid using the general coupling procedure to give the title compound as an off white solid (72.3 mg, 81% yield). TLC (ethyl acetate), Rf = 0.11; Melting Point 202–204 °C; 1H NMR (400 MHz, CDCl3) δ 9.37 (s, 1H), 8.99 (d, J = 4.4 Hz, 1H), 8.91 (s, 2H), 8.17 (s, 1H), 7.51 (d, J = 9.2 Hz, 1H), 7.28 (d, J = 4.0 Hz, 1H), 4.26 (s, 3H), 2.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.9, 158.8, 156.9, 154.9, 142.8, 142.5, 141.3, 132.0, 123.8, 121.3, 1207, 120.3, 62.6, 25.2; IR (neat) νmax 3328.4, 3054.5, 2923.5, 2851.8, 1691.7, 1611.2, 1497.5, 1436.8; ESI-HRMS [M + H]+ calculated for C16H15N4O2 295.1190, found 295.1195.
4-(2-benzofuranyl)-8-methoxy-7-(2-(morpholin-4-yl)acetamido)quinoline (51).
The compound was prepared from 100 mg (0.26 mmol) of 4-bromo-8-methoxy-7-(3-(morpholin-4-yl)acetamido)quinoline and 85 mg (0.53 mmol) of 2-benzofuranyl boronic acid using the general coupling procedure to give the title compound as an off white solid 109 mg (99% yield). TLC (Ethyl acetate) Rf = 0.15; Melting Point 154–156 °C; 1H NMR (400 MHz, CDCl3) δ 10.21 (broad s, 1H), 8.97 (d, J = 4.4 Hz, 1H), 8.81 (d, J = 9.2 Hz, 1H), 8.34 (d, J = 9.6 Hz, 1H), 7.73–7.69 (m, 2H), 7.62 (d, J = 1.4, 7.2 Hz, 1H), 7.40 (dt, J = 4.4 Hz, 1H), 7.34 – 7.30 (m, 2H), 4.25 (s, 3H), 3.86 (broad t, J = 4.6 Hz, 4H), 3.25 (s, 2H), 2.71 (broad t, J = 4.4 Hz, 4H); 13C NMR (100 MHz, CDCl3) δ 168.7, 153.4, 152.7, 149.7, 143.2, 143.0, 136.2, 131.2, 128.6, 125.8, 123.6, 122.6, 121.8, 121.6, 120.5, 118.8, 111.7, 108.9, 67.4, 62.8, 62.4, 54.0; IR (neat) νmax 3305.0, 2961.4, 2924.7, 2860.1, 2809.3, 1691.6, 1610.05, 1507.9,1448.61415.8; ESI-HRMS [M + H]+ calculated for C24H24N3O4 418.1761, found 418.1763.
4-(2-benzofuranyl)-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline (52).
The compound was prepared from 80.3 mg (0.20 mmol) of 4-bromo-8-methoxy-7-(3-(morpholin-4-yl)propanamido)quinoline and 66.0 mg (0.4 mmol) of 2-benzofuranyl boronic acid using the general coupling procedure to give the pure title compound as an off white solid 84 mg (96% yield). TLC (ethyl acetate) Rf = 0.17; Melting Point 105–107 °C; 1H NMR (400 MHz, CDCl3) δ 10.97 (broad s, 1H), 8.97 (d, J = 4.4 Hz, 1H), 8.78 (d, J = 9.6 Hz, 1H), 8.32 (d, J = 9.6 Hz, 1H), 7.72 (d, J = 4.4 Hz, 1H), 7.70 (d, J = 7.2 Hz, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.40 (dt, J = 1.4, 8.0 Hz, 1H), 7.33 – 7.30 (m, 2H), 4.17 (s, 3H), 3.91 (broad t, J = 4.4 Hz, 4H), 2.80 (broad t, J = 5.6 Hz, 4H), 2.69 – 2.63 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 171.4, 155.4, 152.7, 149.6, 143.6, 143.3, 136.1, 132.2, 128.6, 125.8, 123.6, 122.6, 122.0, 121.8, 121.4, 118.8, 111.7, 108.9, 66.8, 62.2, 54.6, 53.4, 33.2; IR (neat) νmax 2922.7, 2852.6, 2816.3, 1676.4, 1608.9, 1501.0, 1451.9; ESI-HRMS [M + H]+ calculated for C25H26N3O4 432.1918, found 432.1929.
7-Acetamido-4-(2-benzofuranyl)-8-hydroxyquinoline (53).
The compound was prepared from 58.2 mg (0.18 mmol) of 7-acetamido-4-(2-benzofuranyl)-8-methoxyquionline and 1.8 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a yellow solid, 40 mg (72% yield). TLC (ethyl acetate) Rf = 0.23; Melting Point 222–224 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.69 (broad s, 1H), 8.94 (d, J = 4.5 Hz, 1H), 8.26 (d, J = 9.5 Hz, 1H), 8.05 (d, J = 9.5 Hz, 1H), 7.94 (d, J = 4.5 Hz, 1H), 7.81 – 7.76 (m, 3H), 7.46 (t, J = 8.5 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 2.18 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.5, 154.5, 152.4, 147.1, 140.0, 134.3, 128.3, 125.7, 124.1, 123.5, 121.8, 121.2, 118.7, 111.4, 108.8, 23.8; IR (neat) νmax 3306.1, 1656.4, 1633.4, 1582.5, 1540.5, 1511.5, 1465.4, 1406.9; ESI-HRMS [M + H]+ calculated for C19H15N2O3 319.1077, found 319.1080.
7-Acetamido-4-(benzo[b]thiophen-2-yl)-8-hydroxyquinoline (54).
The compound was prepared from 33.0 mg (0.10 mmol) of 7-acetamido-4-(benzo[b]thiophen-2-yl)-8-methoxyquinoline and 1.0 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 31.0 mg (98% yield) of the product. TLC (ethyl acetate) Rf = 0.25; Melting Point 170–172 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.63 (broad s, 1H), 8.90 (d, J = 4.4 Hz, 1H), 8.21 (d, J = 9.2 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.89(s, 1H), 7.76 (d, J = 9.2 Hz, 1H), 7.63 (d, J = 4.0 Hz, 1H) 7.50 – 7.45 (m, 2H), 2.16 (s, 3H) 13C NMR (100 MHz, DMSO-d6) δ 168.8, 147.9, 142.8, 139.8, 139.7, 139.0, 138.2, 125.9, 125.3, 124.9, 124.4, 124.0, 123.7, 122.5, 122.4, 121.3, 114.0, 23.7; IR (neat) νmax 3319.3, 3053.1, 2919.82851.8, 1681.8, 1658.6, 1623.2, 1529.9, 1500.1, 1460.5; ESI-HRMS [M + H]+ calculated for C19H15N2O2S 335.0849, found 335.0849.
7-Acetamido-4-(2-furanyl)-8-hydroxyquinoline (55).
The compound was prepared from 32 mg (0.10 mmol) of 7-acetamido-4-(2-furanyl)-8-methoxyquinoline and 1.0 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 30.3 mg (99% yield). TLC (ethyl acetate) Rf = 0.15; Melting Point 185–187 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.13 (broad s, 1H), 9.58 (broad s, 1H), 8.85 (d, J = 4.8 Hz, 1H), 8.21 (d, J = 9.2 Hz, 1H), 8.03 (s, 1H), 7.94 (d, J = 9.2 Hz, 1H), 7.73 (d, J = 3.2 Hz, 1H), 6.79 (d, J = 1.2 Hz, 1H), 2.16 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.8, 150.2, 147.9, 145.0, 144.7, 139.3, 134.6, 123.8, 120.4, 117.5, 114.2, 113.0, 112.5, 23.7; IR (neat) νmax 3291.2, 2918.9, 2850.6, 1651.8, 1511.5, 1466.1, 1402.5; ESI-HRMS [M + H]+ calculated for C15H13N2O3 269.0921, found 269.0925.
7-Acetamido-4-(3-furanyl))-8-hydroxyquinoline (56).
The compound was prepared from 38.8 mg (0.14 mmol) of 7-acetamido-4-(3-furanyl)-8-methoxyquinoline and 1.4 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 22.0 mg (60% yield). TLC (ethyl acetate) Rf = 0.22; Melting Point 148–150 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.57 (broad s, 1H), 8.82 (d, J = 4.4 Hz, 1H), 8.26 (s, 1H), 8.16 (d, J = 9.2 Hz, 1H), 7.92 (s, 1H), 7.60 (d, J = 9.2 Hz, 1H), 7.49 (d, J = 4.4 Hz, 1H) 6.99 (d, J = 1.2 Hz, 1H), 2.15 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.8, 148.4, 147.9, 144.2, 142.2, 138.9, 138.4, 123.4, 123.3, 122.8, 120.0, 111.4, 23.7; IR (neat) νmax 3285.5, 2921.3, 2851.9, 1652.0, 1629.0, 1508.81463.4, 1404.9; ESI-HRMS [M + H]+ calculated for C15H13N2O3 269.0921, found 269.0927.
7-Acetamido-4-(thiophen-2-yl)-8-hydroxyquinoline (57).
The compound was prepared from 32 mg (0.10 mmol) of 7-Acetamido-4-(thiophen-2-yl)-8-methoxyquinoline and 1.0 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 29 mg (95% yield). TLC (10% methanol in ethyl acetate) Rf = 0.21; Melting Point 118–120 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.60 (broad s, 1H), 8.84 (d, J = 4.4 Hz, 1H), 8.19 (d, J = 9.2 Hz, 1H), 7.85 (dd, J = 1.2, 5.2 Hz, 1H), 7.70 (d, J = 9.2 Hz, 1H), 7.57 (dd, J = 1.2, 3.6 Hz, 1H), 7.53 (d, J = 4.4 Hz, 1H), 7.32 – 7.302 (m, 1H), 2.16 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.9, 147.8, 142.8, 139.7, 139.1, 138.1, 129.1, 128.5, 128.3, 123.5, 122.5, 120.7, 113.9, 23.7; IR (neat) νmax 3228.2, 2918.9, 2850, 1666.6, 1622.4.2, 1525.0, 1498.0, 1457.3, 1400.3; ESI-HRMS [M + H]+ calculated for C15H13N2O2S 285.0692, found 285.0686.
7-Acetamido-4-(thiophen-3-yl)-8-hydroxyquinoline (58).
The compound was prepared from 25.0 mg (0.08 mmol) of 7-acetamido-4-(thiophen-3-yl)-8-methoxyquinoline and 0.8 mL of 1.0 M BBr3 in DCM to afford a solid product, 23.0 mg (96% yield. TLC (ethyl acetate) Rf = 0.25; Melting Point 152–153 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.58 (broad s, 1H), 8.84 (d, J = 4.4 Hz, 1H), 8.13 (d, J = 9.2 Hz, 1H), 7.91 (d, J = 1.6 Hz, 1H), 7.80 – 7.78 (m, 1H), 7.49 – 7.44 (m, 3H), 2.15 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.8, 147.9, 142.7, 142.2, 138.9, 137.8, 128.8, 127.1, 126.0, 123.5, 123.2, 123.1, 120.5, 114.2, 23.7; IR (neat) νmax 3297.4, 3108.5, 2958.2, 2920.1, 1665.6, 1622.6, 1525.7, 1497.9, 1460.7, 1411.8; ESI-HRMS [M + H]+ calculated for C15H13N2O2S 285.0692, found 285.0697.
7-Acetamido-4-(5-pyrimidinyl)-8-hydroxyquinoline (59).
The compound was prepared from 26.2 mg (0.09 mmol) of 7-acetamido-4-(5-pyrimidinyl)-8-hydroxyquionline and 0.90 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 23.0 mg (92% yield). TLC (ethyl acetate) Rf = 0.13; Melting Point 211–213 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.62 (broad s, 1H), 9.36 (s, 1H), 9.04 (s, 2H), 8.94 (d, J = 4.4 Hz, 1H), 8.16 (d, J = 9.2 Hz, 1H), 7.56 (d, J = 4.0 Hz, 1H), 7.21 (d, J = 9.2 Hz, 1H), 2.15 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.8, 158.2, 156.7, 155.1, 147.9, 142.8, 140.6, 138.7, 131.4, 124.1, 123.8, 122.9, 121.4, 113.6, 23.7; IR (neat) νmax 3518.2, 3197.1, 3048.3, 1695.6, 1624.6, 1507.8, 1459.8, 1425.2; ESI-HRMS [M + H]+ calculated for C15H13N4O2 281.1033, found 281.1036.
4-(2-benzofuranyl)-8-hydroxy-7-(2-(morpholin-4-yl)acetamido)quinoline (60).
The compound was prepared from 50 mg (0.12 mmol) of analog 4-(2-benzofuranyl)-8-methoxy-7-(3-(morpholin-4-yl)acetamido)quinoline and 1.2 mL of 1.0 M BBr3 DCM using the general method for demethylation to afford a solid product, 36.3 mg (75% yield). TLC (50% hexane in acetone) Rf = 0.21; Melting Point 240–242 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.90 (broad s, 1H), 9.03 (s, 1H), 8.31– 8.26 (m, 2H), 813 (d, J = 7.2 Hz, 1H), 8.00 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.40 (t, J = 6.8 Hz, 1H), 4.38 (s, 2H), 3.95 – 3.91 (m, 4H), 3.56 – 3.50 (m, 2H), 3.37(s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 163.4, 154.9, 128.0, 126.7, 125.0, 123.9, 122.3, 115.5, 111.7, 63.0, 56.9, 51.9, 48.5; IR (neat) νmax 3384.8, 3337.9, 3214.8, 2978.5, 2921.8, 2853.8, 1704.9, 1628.3, 1591.8, 1547.1, 1520.5, 1453.4, 1418.2; ESI-HRMS [M + H]+ calculated for C23H24N3O4 404.1605, found 404.1613.
4-(2-benzofuranyl)-8-hydroxy-7-(3-(morpholin-4-yl)propanamido)quinoline (61).
The compound was prepared from 33.4 mg (0.08 mmol) of analog 4-(2-benzofuranyl)-8-methoxy-7-(3-(morpholin-1-yl)propanamido)quinoline and 0.8 mL of 1.0 M BBr3 in DCM using the general method for demethylation to afford a solid product, 30 mg (93% yield). TLC (30% hexane in acetone) Rf = 0.22; Melting Point 222–224 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.73 (broad s, 1H)), 8.94 (d, J = 4.4 Hz, 1H), 8.56 (d, J = 9.2 Hz, 1H), 8.05 (d, J = 9.2 Hz, 1H), 7.89 (d, J = 4.4 Hz, 1H), 7.82 – 7.76 (m, 4H), 7.45 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 3.72 (s, 4H), 3.32 (s, 4H), 2.68 – 2.62 (m, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.6, 152.2, 148.0, 139.0, 134.4, 128.2, 125.9, 123.6, 122.6, 118.7, 114.4, 111.5, 109.1, 65.9, 57.5, 53.9, 52.8; IR (neat) νmax 3296.1, 2921.3, 2852.1, 2816.6, 2768.2, 1653.2, 1582.8, 1542.8, 1510.5, 1462.4, 1403.3; ESI-HRMS [M + H]+ calculated for C24H24N3O4 418.1761 found 418.1770.
Supplementary Material
Acknowledgements
This research was supported in part by the Department of Defense Peer Reviewed Cancer Research Program (W81XWH-13-1-0344 and W81XWH-18-1-0142) to D.V.L. The Colorado Cancer Translational Research Accelerator awarded (D.V.L PI and D.L.G co-PI). S.R. was supported by the Cancer Foundation of Luxembourg. We thank the high-throughput screening and chemical biology core facility at the University of Colorado AMC Skaggs School of Pharmacy and Pharmaceutical Sciences. We also thank the University of Colorado Cancer Center Shared Resource Laboratories, particularly the Pharmacology core facility for microsomal and PK studies and the MS and NMR core facilities for structural characterization. The University of Colorado Cancer Center is a National Cancer Institute designated cancer center supported by grant number P30CA046934.
Abbreviations Used:
- RFP
mCherry red fluorescent protein
- GFP
Enhanced green fluorescent protein
- mCRC
Colorectal cancer (CRC) or metastatic CRC
- TOP2A
Topoisomerase IIα
- TCF
T-cell factor
- MDR
Multidrug resistance
- DSBs
Double stranded DNA breaks
- CSC
Cancer stem cell
- WRE
Wnt response elements
- CADD
Computer aided drug design
Footnotes
Supporting Information: The supporting information contains SI data figures and tables; the synthesis and methods to prepare key starting materials and intermediates; compound structural characterization data for all novel compounds: NMR, HRMS, and IR spectra; HPLC purity traces for all final compounds demonstrating ≥95% purity. Molecular Formula Strings are available as a separate file.
PDB ID Codes: 1ZXM is N-terminal ATPase domain of TOP2A20
References
- (1).Howlader N; Noone A; Krapcho M; Garshell J; Miller D; Altekruse S; Kosary C; Yu M; Ruhl J; Tatalovich Z; Mariotto A; Lewis D; Chen H; Feuer E; Cronin K. e. SEER Cancer Statistics Review, 1975–2013, National Cancer Institute; Bethesda, MD, http://seer.cancer.gov/csr/1975_2013/, based on November 2015 SEER data submission, posted to the SEER web site, April 2016. ((accessed Nov 7, 2004)). [Google Scholar]
- (2).Clevers H Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [DOI] [PubMed] [Google Scholar]
- (3).Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sánchez-Tilló E; de Barrios O; Siles L; Cuatrecasas M; Castells A; Postigo A β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 19204–19209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chaffer CL; San Juan BP; Lim E; Weinberg RA EMT, cell plasticity and metastasis. Cancer Metast. Rev 2016. [DOI] [PubMed] [Google Scholar]
- (6).Cao H; Xu E; Liu H; Wan L; Lai M Epithelial-mesenchymal transition in colorectal cancer metastasis: A system review. Pathol. Res. Pract 2015, 211, 557–569. [DOI] [PubMed] [Google Scholar]
- (7).Zhou Q; Abraham AD; Li L; Babalmorad A; Bagby S; Arcaroli JJ; Hansen RJ; Valeriote FA; Gustafson DL; Schaack J; Messersmith WA; LaBarbera DV Topoisomerase IIα mediates TCF-dependent epithelial-mesenchymal transition in colon cancer. Oncogene 2016, 35, 4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Polakis P Drugging Wnt signalling in cancer. EMBO J. 2012, 31, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lee H-J; Zhang XX; Zheng JJ Inhibiting the Wnt signaling pathway with small molecules In Targeting the Wnt Pathway in Cancer, Goss KH; Kahn M, Eds. Springer New York: New York NY, 2011; pp 183–209. [Google Scholar]
- (10).Katoh M Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol 2017, 51, 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tsavaris N; Lazaris A; Kosmas C; Gouveris P; Kavantzas N; Kopterides P; Papathomas T; Agrogiannis G; Zorzos H; Kyriakou V; Patsouris E Topoisomerase I and IIalpha protein expression in primary colorectal cancer and recurrences following 5-fluorouracil-based adjuvant chemotherapy. Cancer Chemoth. Pharmacol 2009, 64, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gouveris P; Skopelitis E; Tsavaris N Topoisomerase I and II expression in recurrent colorectal cancer cells: A dubious matter In DNA Replication-current advances, Seligmann H, Ed. InTech: 2011; pp 639–668. [Google Scholar]
- (13).Guzman FSD; Carte B; Troupe N; Faulkner DJ; Harper MK; Concepcion GP; Mangalindan GC; Matsumoto SS; Barrows LR; Ireland CM Neoamphimideine: A new pyridoacridine topoismerase II inhibitor which catenates DNA. J. Org. Chem 1999, 64, 1400–1402. [Google Scholar]
- (14).Marshall KM; Matumoto SS; Holden JA; Concepcion GP; Tasdemir D; Ireland CM; Barrows LR The anti-neoplastic and novel topoisomerase II-mediated cytotoxicity of neoamphimedine, a marine pyridoacridine. Biochem. Pharmacol 2003, 66, 447–458. [DOI] [PubMed] [Google Scholar]
- (15).Ponder J; Yoo BH; Abraham AD; Li Q; Ashley AK; Amerin CL; Zhou Q; Reid BG; Reigan P; Hromas R; Nickoloff JA; LaBarbera DV Neoamphimedine circumvents metnase-enhanced DNA topoisomerase IIalpha activity through ATP-competitive inhibition. Mar. Drugs 2011, 9, 2397–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Beck WT; Morgan SE; Mo YY; Bhat UG Tumor cell resistance to DNA topoisomerase II inhibitors: new developments. Drug Resist Updat 1999, 2, 382–389. [DOI] [PubMed] [Google Scholar]
- (17).Felix CA Leukemias related to treatment with DNA topoisomerase II inhibitors. Med. Pediatr. Oncol 2001, 36, 525–535. [DOI] [PubMed] [Google Scholar]
- (18).Baldwin EL; Osheroff N Etoposide, topoisomerase II and cancer. Anti-Cancer Agent. ME 2005, 5, 363–372. [DOI] [PubMed] [Google Scholar]
- (19).Bates AD; Berger JM; Maxwell A The ancestral role of ATP hydrolysis in type II topoisomerases: prevention of DNA double-strand breaks. Nucleic Acids Res. 2011, 39, 6327–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wei H; Ruthenburg AJ; Bechis SK; Verdine GL Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J. Biol. Chem 2005, 280, 37041–37047. [DOI] [PubMed] [Google Scholar]
- (21).Li L; Abraham AD; Zhou Q; Ali H; O’Brien JV; Hamill BD; Arcaroli JJ; Messersmith WA; LaBarbera DV An improved high yield total synthesis and cytotoxicity study of the marine alkaloid neoamphimedine: an ATP-competitive inhibitor of topoisomerase IIalpha and potent anticancer agent. Mar. Drugs 2014, 12, 4833–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chene P ATPases as drug targets: learning from their structure. Nat. Rev. Drug. Discov 2002, 1, 665–673. [DOI] [PubMed] [Google Scholar]
- (23).Walker JE; Saraste M; Runswick MJ; Gay NJ Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).LaBarbera DV; Bugni TS; Ireland CM The total synthesis of neoamphimedine. J. Org. Chem 2007, 72, 8501–8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Egli M; Sarkhel S Lone pair-aromatic interactions: to stabilize or not to stabilize. Acc. Chem. Res 2007, 40, 197–205. [DOI] [PubMed] [Google Scholar]
- (26).Shah P; Westwell AD The role of fluorine in medicinal chemistry. J. Enzyme. Inhib. Med. Chem 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- (27).Bissantz C; Kuhn B; Stahl M A medicinal chemist’s guide to molecular interactions. J. Med. Chem 2010, 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Echavarren AM; Stille JK Total synthesis of amphimedine. J. Am. Chem. Soc 1988, 110, 4051–4053. [Google Scholar]
- (29).Tietze M; Iglesias A; Merisor E; Conrad J; Klaiber I; Beifuss U Efficient methods for the synthesis of 2-hydroxyphenazine based on the Pd-catalyzed N-arylation of aryl bromides. Org. Lett 2005, 7, 1549–1552. [DOI] [PubMed] [Google Scholar]
- (30).Nakahara S; Kubo A Total synthesis of styelsamine C, and formal synthesis of norsegoline. Heterocycles 2005, 65, 1925–1929. [Google Scholar]
- (31).Reid BG; Jerjian T; Patel P; Zhou Q; Yoo BH; Kabos P; Sartorius CA; Labarbera DV Live multicellular tumor spheroid models for high-content imaging and screening in cancer drug discovery. Curr. Chem. Genomics. Transl. Med 2014, 8, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li Q; Chen C; Kapadia A; Zhou Q; Harper MK; Schaack J; LaBarbera DV 3D Models of epithelial-mesenchymal transition in breast cancer metastasis: high-throughput screening assay development, validation, and pilot screen. J Biomol Screen 2011, 16, 141–154. [DOI] [PubMed] [Google Scholar]
- (33).Li L; LaBarbera DV 2.16 – 3D High-Content Screening of Organoids for Drug Discovery A2 - Chackalamannil, Samuel In Comprehensive Medicinal Chemistry III, Rotella D; Ward SE, Eds. Elsevier: Oxford, 2017; pp 388–415. [Google Scholar]
- (34).Chène P; Rudloff J; Schoepfer J; Furet P; Meier P; Qian Z; Schlaeppi J-M; Schmitz R; Radimerski T Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem. Biol 2009, 9, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Furet P; Schoepfer J; Radimerski T; Chene P Discovery of a new class of catalytic topoisomerase II inhibitors targeting the ATP-binding site by structure based design. Part I. Bioorg. Med. Chem. Lett 2009, 19, 4014–4017. [DOI] [PubMed] [Google Scholar]
- (36).Wang LH; Pfister TD; Parchment RE; Kummar S; Rubinstein L; Evrard YA; Gutierrez ME; Murgo AJ; Tomaszewski JE; Doroshow JH; Kinders RJ Monitoring drug-induced gammaH2AX as a pharmacodynamic biomarker in individual circulating tumor cells. Clin. Cancer. Res 2010, 16, 1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ivashkevich A; Redon CE; Nakamura AJ; Martin RF; Martin OA Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Korinek V; Barker N; Morin PJ; van Wichen D; de Weger R; Kinzler KW; Vogelstein B; Clevers H Constitutive transcriptional activation by a β-catenin-TCF complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [DOI] [PubMed] [Google Scholar]
- (39).Franken NA; Rodermond HM; Stap J; Haveman J; van Bree C Clonogenic assay of cells in vitro. Nat. Protoc 2006, 1, 2315–2319. [DOI] [PubMed] [Google Scholar]
- (40).Beck B; Blanpain C Unravelling cancer stem cell potential. Nat. Rev. Cancer 2013, 13, 727–738. [DOI] [PubMed] [Google Scholar]
- (41).Nakahara S; Mukai Y; Kubo A Synthesis of neoamphimedine. Heterocycles 2012, 85, 993–940. [Google Scholar]
- (42).Champoux JJ DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- (43).Nitiss JL Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Li K; Wu D; Chen X; Zhang T; Zhang L; Yi Y; Miao Z; Jin N; Bi X; Wang H; Xu J; Wang D Current and emerging biomarkers of cell death in human disease. Biomed. Res. Int 2014, 2014, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McDonald PC; Dedhar S The role of epithelial– mesenchymal transition in cancer metastasis In Principles of stem cell biology and cancer future applications and therapeutics, 1st ed.; Regad T; Sayers TJ; Rees RC, Eds. John Wiley & Sons, Ltd: West Sussex, UK, 2015; pp 101–121. [Google Scholar]
- (46).Ohe T; Mashino T; Hirobe M Substituent elimination from p-substituted phenols by cytochrome P450 ipso-substitution by the oxygen atom of the active species. Drug Metab. Dispos 1997, 25, 116–22. [PubMed] [Google Scholar]
- (47).Daina A; Michielin O; Zoete V SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 2017, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fakih MG Metastatic colorectal cancer: current state and future directions. J. Clin. Oncol 2015, 33, 1809–1824. [DOI] [PubMed] [Google Scholar]
- (49).Marcucci F; Stassi G; De Maria R Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat. Rev. Drug. Discov 2016, 15, 311–325. [DOI] [PubMed] [Google Scholar]
- (50).Zhang JH; Chung TDFAUO; Oldenburg KR A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 1999, 67–73. [DOI] [PubMed] [Google Scholar]
- (51).Chung TDY; Terry DB; Smith LH In Vitro and In Vivo Assessment of ADME and PK Properties During Lead Selection and Lead Optimization - Guidelines, Benchmarks and Rules of Thumb In Assay Guidance Manual, Sittampalam GS; Grossman A; Brimacombe K; Arkin M; Auld D; Austin C; Baell J; Bejcek B; Caaveiro JMM; Chung TDY; Coussens NP; Dahlin JL; Devanaryan V; Foley TL; Glicksman M; Hall MD; Haas JV; Hoare SRJ; Inglese J; Iversen PW; Kahl SD; Kales SC; Kirshner S; Lal-Nag M; Li Z; McGee J; McManus O; Riss T; Trask OJ Jr.; Weidner JR; Wildey MJ; Xia M; Xu X, Eds. Bethesda (MD), 2004. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.